1 2. Biosynthesis of Natural Products - Terpene Biosynthesis 2.1 Introduction Terpenes are a large and varied class of natural products, produced primarily by a wide variety of plants, insects, microoroganisms and animals. They are the major components of resin, and of turpentine produced from resin. The name "terpene" is derived from the word "turpentine". Terpenes are major biosynthetic building blocks within nearly every living creature. Steroids, for example, are derivatives of the triterpene squalene. When terpenes are modified, such as by oxidation or rearrangement of the carbon skeleton, the resulting compounds are generally referred to as terpenoids. Some authors will use the term terpene to include all terpenoids. Terpenoids are also known as Isoprenoids. Terpenes and terpenoids are the primary constituents of the essential oils of many types of plants and flowers. Essential oils are used widely as natural flavor additives for food, as fragrances in perfumery, and in traditional and alternative medicines such as aromatherapy. Synthetic variations and derivatives of natural terpenes and terpenoids also greatly expand the variety of aromas used in perfumery and flavors used in food additives. Recent estimates suggest that over 30'000 different terpenes have been characterized from natural sources. Early on it was recognized that the majority of terpenoid natural products contain a multiple of 5C-atoms. Hemiterpenes consist of a single isoprene unit, whereas the monoterpenes include e.g.: Terpenes with 15 C-atoms are known as sesquiterpenes : The terpenes containing, or originating from precursors, containing 20 C-atoms are known as diterpenes, those with 30 C-atoms as triterpenes and those with 40 C-atoms as tetraterpenes : CH 2 OH CH 2 OH OH CHO CHO O O Camphor α-Pinene Citronellal Menthol Citral Geraniol Nerol Limonens Myrcens Monoterpenes CH 2 OH O Farnesol Bisabolene Cadinene Selinene Vetivone HO Patchoulol (Perfume) O COOH OH Abscisic acid (Phytohormone) O O O COOMe OH Pentalenolactone (Antibiotic) Sesquiterpenes
Transcript
1 2. Biosynthesis of Natural Products - Terpene Biosynthesis 2.1 Introduction Terpenes are a large and varied class of natural products, produced primarily by a wide variety of plants, insects, microoroganisms and animals. They are the major components of resin, and of turpentine produced from resin. The name "terpene" is derived from the word "turpentine". Terpenes are major biosynthetic building blocks within nearly every living creature. Steroids, for example, are derivatives of the triterpene squalene. When terpenes are modified, such as by oxidation or rearrangement of the carbon skeleton, the resulting compounds are generally referred to as terpenoids. Some authors will use the term terpene to include all terpenoids. Terpenoids are also known as Isoprenoids. Terpenes and terpenoids are the primary constituents of the essential oils of many types of plants and flowers. Essential oils are used widely as natural flavor additives for food, as fragrances in perfumery, and in traditional and alternative medicines such as aromatherapy. Synthetic variations and derivatives of natural terpenes and terpenoids also greatly expand the variety of aromas used in perfumery and flavors used in food additives. Recent estimates suggest that over 30'000 different terpenes have been characterized from natural sources. Early on it was recognized that the majority of terpenoid natural products contain a multiple of 5C-atoms. Hemiterpenes consist of a single isoprene unit, whereas the monoterpenes include e.g.:
Terpenes with 15 C-atoms are known as sesquiterpenes :
The terpenes containing, or originating from precursors, containing 20 C-atoms are known as diterpenes, those with 30 C-atoms as triterpenes and those with 40 C-atoms as tetraterpenes :
CH2OH
CH2OH
OH
CHO
CHO OO
Camphorα-PineneCitronellal
MentholCitralGeraniolNerolLimonensMyrcens
Monoterpenes
CH2OH
O
Farnesol Bisabolene Cadinene Selinene Vetivone
HO
Patchoulol(Perfume)
O
COOH
OH Abscisic acid(Phytohormone)
OO
O
COOMeOH
Pentalenolactone(Antibiotic)
Sesquiterpenes
2
In contrast to other classes of terpenes that vary greatly in structure and molecular size, the steroids constitute a family of terpenes with a common structural feature, namely, the steroid ring system:
Ruzicka (ETH-ZH) recognized already in the 1920's that most terpenes appear to be constructed from a multiple of linked isoprene units. This is called the isoprene rule. The isoprene rule (cf. Birch, Polyketide Hypothesis) was of great value also in the structure determination of new terpenoids isolated from Nature. However, isoprene itself is not the building block used by Nature to construct terpenes.
2.2 The Mevalonate Pathway It was only much later (ca. 1955) shown that the biosynthesis of terpenes does indeed occur starting from isoprene-like C5 building blocks. Labelling experiments, using 14C-labelled acetic acid, showed early on that the steroid skeleton is constructed from this building block, but not simply through regular head-to-tail coupling reactions:
Mixed origin
N N
NN
Me
Me
MeMe
Mg
OCOOMeO
O
Chlorophyll-a(Photosynthesis)
O
O 18
Plastoquinone(Electron transport)
O
OH
C5H11
Tetrahydrocannabinol(Cannabis sativa)
PolymerOH
Rubber(Heva brasilensis)
500-5000
CH2OH
OH
O OH
Vitamin ACadinene
Grandisol
Camphor
Menthol
Me COOH
HO
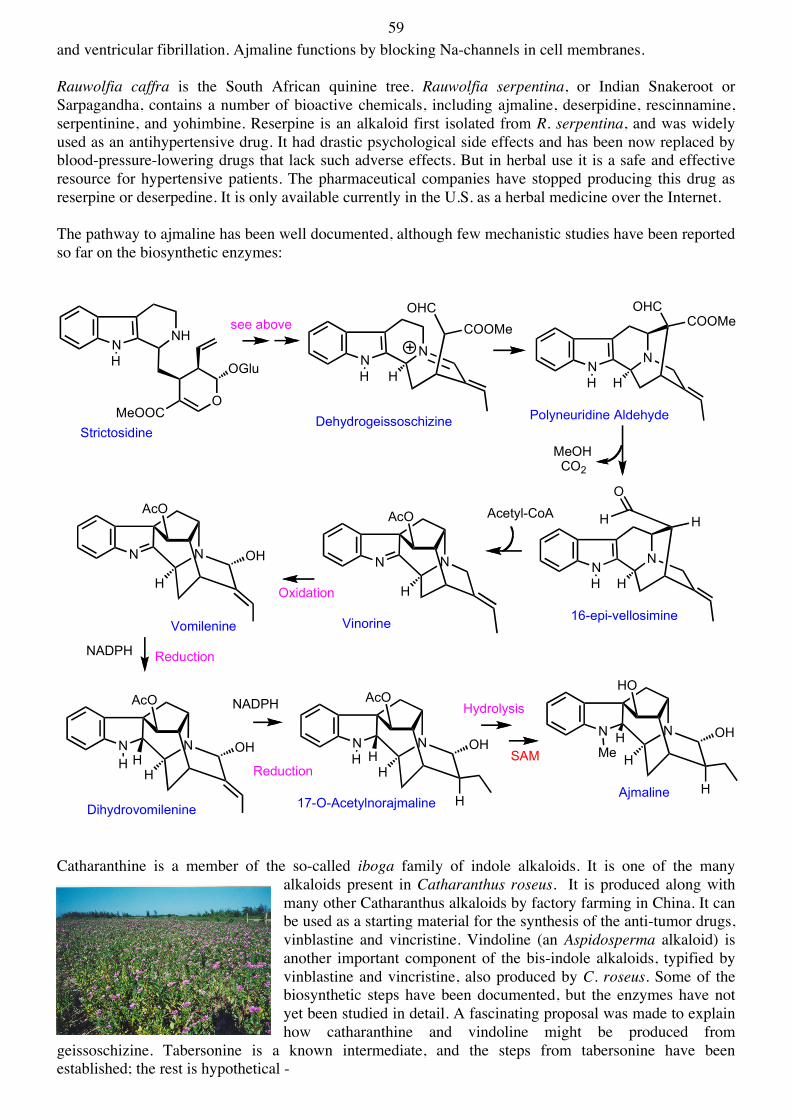
MeH
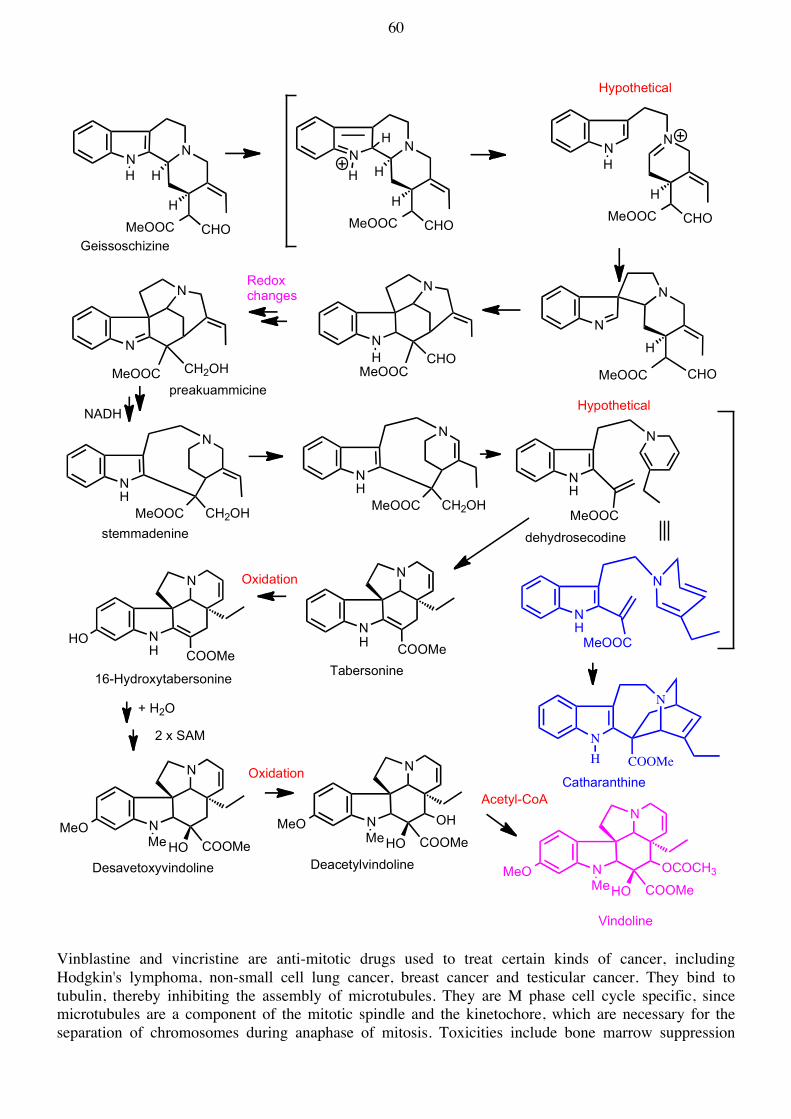
Me
Me Me
Me
HH
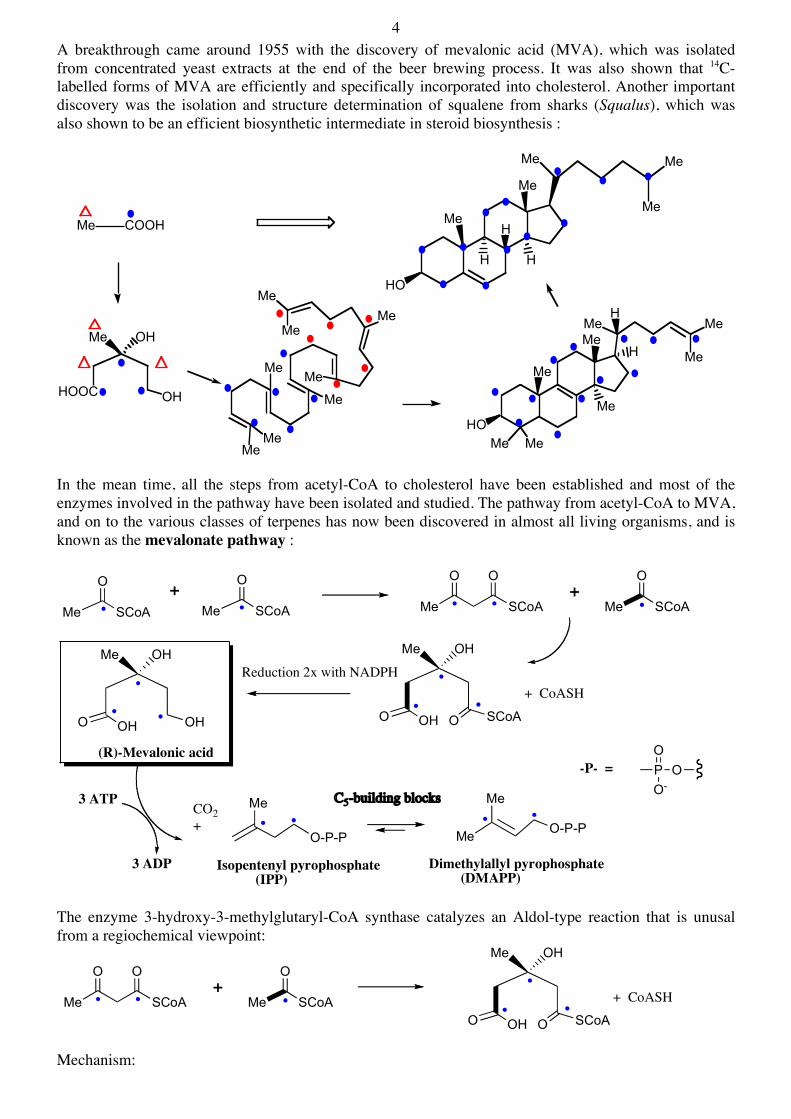
4 A breakthrough came around 1955 with the discovery of mevalonic acid (MVA), which was isolated from concentrated yeast extracts at the end of the beer brewing process. It was also shown that 14C-labelled forms of MVA are efficiently and specifically incorporated into cholesterol. Another important discovery was the isolation and structure determination of squalene from sharks (Squalus), which was also shown to be an efficient biosynthetic intermediate in steroid biosynthesis :
In the mean time, all the steps from acetyl-CoA to cholesterol have been established and most of the enzymes involved in the pathway have been isolated and studied. The pathway from acetyl-CoA to MVA, and on to the various classes of terpenes has now been discovered in almost all living organisms, and is known as the mevalonate pathway :
The enzyme 3-hydroxy-3-methylglutaryl-CoA synthase catalyzes an Aldol-type reaction that is unusal from a regiochemical viewpoint:
Mechanism:
HO
MeH
Me
Me Me
Me
HH
Me COOH
Me OH
HOOC OH
MeMeMe
Me
Me
Me
Me
Me Me
Me
Me
MeHO
Me
MeH
HMe
Me
Me
O
SCoA Me
O
SCoA Me SCoA
O O
Me
O
SCoA
Me OH
O OH O SCoA
Me OH
O OH
Me
OH
Me O-P-P
Me
O-P-P
PO
O-O
CO2+
+ CoASH
3 ADP
3 ATP C5-building blocks
Isopentenyl pyrophosphate (IPP)
Dimethylallyl pyrophosphate (DMAPP)
(R)-Mevalonic acid
Reduction 2x with NADPH
++
-P- =
Me OH
O OH O SCoA
+ CoASHMe SCoA
O O
Me
O
SCoA+
5
Through crystallographic studies, also with substrates bound at the active site, a good model for the reaction mechanism has been established. The structures have also shown which residues at the active site are most likely involved in catalysis (Vgl PNAS 2004, 101, 16442):
A. Acetoacetyl-CoA and Acetyl-Cys, and B. HMG-CoA in the active site
In the next step of the mevalonate pathway, the CoAS-thioester group is reduced in a reaction requiring two equivalents of NADPH. The reaction proceeds in two steps (thioester aldehyde alcohol). Many inhibitors of this enzymic reaction have been discovered, and several of these (called statins) are now important pharmaceutical products. The statins (or HMG-CoA reductase inhibitors) form a class of hypolipidemic drugs used to lower cholesterol levels in people with, or at risk of, cardiovascular disease. They lower cholesterol by inhibiting the enzyme HMG-CoA reductase (HMGR), which is the rate-limiting enzyme of the mevalonate pathway of cholesterol synthesis.
SH SCoA
O
S
O
CoASH
S
O
H
B
S
OSCoA
O OA H
S SCoA
O OHO MeH2O
HO SCoA
O OHO Me+ HMGS
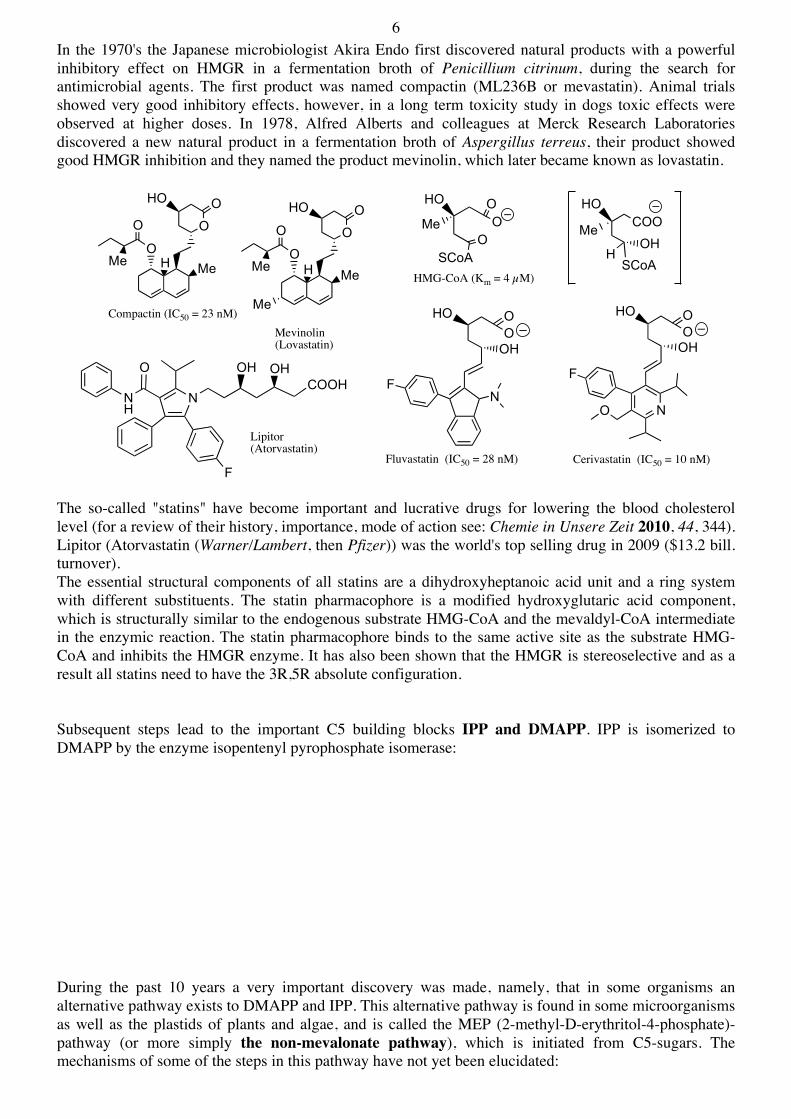
6 In the 1970's the Japanese microbiologist Akira Endo first discovered natural products with a powerful inhibitory effect on HMGR in a fermentation broth of Penicillium citrinum, during the search for antimicrobial agents. The first product was named compactin (ML236B or mevastatin). Animal trials showed very good inhibitory effects, however, in a long term toxicity study in dogs toxic effects were observed at higher doses. In 1978, Alfred Alberts and colleagues at Merck Research Laboratories discovered a new natural product in a fermentation broth of Aspergillus terreus, their product showed good HMGR inhibition and they named the product mevinolin, which later became known as lovastatin.
The so-called "statins" have become important and lucrative drugs for lowering the blood cholesterol level (for a review of their history, importance, mode of action see: Chemie in Unsere Zeit 2010, 44, 344). Lipitor (Atorvastatin (Warner/Lambert, then Pfizer)) was the world's top selling drug in 2009 ($13.2 bill. turnover). The essential structural components of all statins are a dihydroxyheptanoic acid unit and a ring system with different substituents. The statin pharmacophore is a modified hydroxyglutaric acid component, which is structurally similar to the endogenous substrate HMG-CoA and the mevaldyl-CoA intermediate in the enzymic reaction. The statin pharmacophore binds to the same active site as the substrate HMG-CoA and inhibits the HMGR enzyme. It has also been shown that the HMGR is stereoselective and as a result all statins need to have the 3R,5R absolute configuration. Subsequent steps lead to the important C5 building blocks IPP and DMAPP. IPP is isomerized to DMAPP by the enzyme isopentenyl pyrophosphate isomerase: During the past 10 years a very important discovery was made, namely, that in some organisms an alternative pathway exists to DMAPP and IPP. This alternative pathway is found in some microorganisms as well as the plastids of plants and algae, and is called the MEP (2-methyl-D-erythritol-4-phosphate)-pathway (or more simply the non-mevalonate pathway), which is initiated from C5-sugars. The mechanisms of some of the steps in this pathway have not yet been elucidated:
O
O
Me H
O
HO O
Me
Compactin (IC50 = 23 nM)
HO
MeO
SCoA
OO
HMG-CoA (Km = 4 µM)
HO OOOH
NF
Fluvastatin (IC50 = 28 nM)
NO
HO OOOH
F
Cerivastatin (IC50 = 10 nM)
O
O
Me H
O
HO O
Me
Me
Mevinolin(Lovastatin)
SCoA
COOHO
OHH
Me
NH
N
O
F
OHCOOH
OH
Lipitor(Atorvastatin)
7
After the formation of IPP and DMAPP, there exists in all organisms a central route to the universal building blocks needed for mono-, sesqui-, di-, tri and tetra-terpene biosynthesis:
The mechanism and stereochemical course of all these steps was investigated by J. W. Cornforth, who received the Nobel Prize in Chemistry for his work (1975, mit V. Prelog, ETH-ZH). In recent years, direct access to the biosynthetic genes for many of the enzymes in terpene biosynthesis has provided an enormous impulse for structural and mechanistic studies. There is also great interest in the design and development of specific inhibitors, as potential drugs against bacterial and parasitic infections, and in the agrochemical area.
Me
O
COOH
CHO
OH
CH2O-PO3
Me
O
OH
CH2O-PO32-
HO O PO32-
HO
MeHO
OH
IPP
Deoxyxylulose-5-Phosphate
CO2
TPPNADPH
O P2O63-
HO
Me
DMAPP
CTP PPiO P
HO
MeHO
OH
O
O-O CMP
ATP
ADP
O P
HO
Me2-O3PO
OH
O
O-O CMP
CMP
OPO2
HO
Me O
OH
PO2 O
H+ 2e-H2O H+
2e-
H2O Me O P2O63-
Me
O P2O63-
Me
Me
R O-P-P
Me
O-P-P Me
Me Me
O-P-P
Me
Me
R O-P-P Me
RP-P-OMe
Me Me MeMe
MeMeMe
Me
Me O-P-P
Me
O-P-P Me
Me
R
O-P-P
Me Me
O-P-P
Me
O-P-P Me
Me Me Me
O-P-P
Me
+
C20-Building block
Diterpenes
Geranylgeranyl pyrophosphate (GGPP)
+
Steroids
DMAPP
C30-Building block
C15-Building block
C10-Building block
TriterpenesSqualene
Sesquiterpenes
Monoterpenes
Farnesyl pyrophosphate (FPP)
+
GPP
Geranyl pyrophosphate (GPP)
+
IPPFPP
IPP
IPP
FPPFPP
TetraterpenesC40-Building block
Me
R O-P-P Me
RP-P-O+
GGPPGGPP
8 2.3 The Formation of GPP, FPP und GGPP The steps from DMAPP and IPP to GPP, FPP and GGPP are catalyzed by so-called prenyl transferases. These enzymes (35 - 80 kDa) require Metal2+-ions for activity. The Km values are typically <10µM und kcat values are in the range 0.03-0.3 s-1. In the active site of each enzyme, the pyrophosphate group is activated and acts as a leaving group to generate an allylic-tertiary carbocation, stabilized in an ion pair with the pyrophosphate group. This electrophile is then attacked by the double bond in the neighboring substrate
The reaction proceeds stereospecifically - only the pro-R H-atom is lost, and the new C=C double bond has the E configuration. A trifluoromethyl analogue of the substrate GPP reacts 106-times slower, which supports the mechanism involving formation of a carbocation, since formation of this would now be destabilized by a strong inductive effect. 2.4. The Formation of Squalene Here two FPP molecules are combined in a head-to-head coupling, which requires NADPH :
During assays in vitro, if NADPH is not present an intermediate can be detected, which normally does not accumulate:
Me
Me O-P-P
Me
O-P-PHSHR
Me
CF3O-P-P
HH
H H Me
RP-P-O
RR
Me
Me
Me
R O-P-P
H H
H H
NADPH NADP+
R Me
OPPHS
HR
OPP
Me
RMeR
H
OPP
Me
RMeR
H
R Me
HH
RMe
OPP
R Me Me R
Presqualen-pyrophosphat
Me
RMeR
H
FPP
9 2.5. The Formation of Mono-, Sesqui- und Di-Terpenes (see also: Topics in Current Chemistry, Vol 209 Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids. Springer Verlag, 2000) The terpene cyclases form a large family of enzymes that use GPP, FPP or GGPP as substrate and catalyze the formation of a mono-, sesqui- or di-terpenoid products. The cyclases are similar in mode of action to the prenyl transferases, except now mostly intramolecular cyclization reactions are catalyzed. This family of enzymes catalyze a huge variety of different transformations, which may include steps where H-atom migrations, Wagner-Meerwein rearrangements, and related reactions occur. The chemistry is dominated by that of the carbocation intermediates. Some terpene cyclases have been intensively investigated over the past 10 years, and a good understanding of their mechanisms of action is starting to emerge. In some cases, crystal structures of the enzymes are available, also with substrates/products or inhibitors bound at the active site. Before the emergence of gene cloning technologies, the study of terpene cyclases was very difficult, because only very small amounts of enzyme could be isolated from natural sources. Far easier were labelling experiments, in which 14C-labelled precursors were fed to intact plants. Several days to weeks later, the natural products were isolated and examined to determine how the radioactive label had been incorporated (if at all):
The labelling experiments, if designed well, would provide insights into how the precursor GPP must fold and cyclize to produce the natural product. The occurrence of intermediates in the pathway could be sought in extracts of the plant. Over the past 10 years or so, with advances in molecular genetics and genomics, direct access to the genes for the biosynthetic enzymes has been obtained. With recombinant DNA methods the enzymes can be produced in large amounts and their mechanisms can be studied directly in vitro. The monoterpene cyclases (see Chem. Rev. 1987, 87, 929) from plants have been well studied. These enzymes use GPP as substrate, and catalyze typically a unique cyclization reaction. However, sometimes more than one product is observed! The enzymes usually required Mg2+ or Mn2+ as cofactor. One important question is: how can GPP (with an E-double bond) be cyclized to produce a 6-membered ring? In a first step, the GPP is converted into an enzyme bound (3R)- oder (3S)-linalyl pyrophosphate, which, after a change in conformation, then reacts further to cyclic products, e.g.
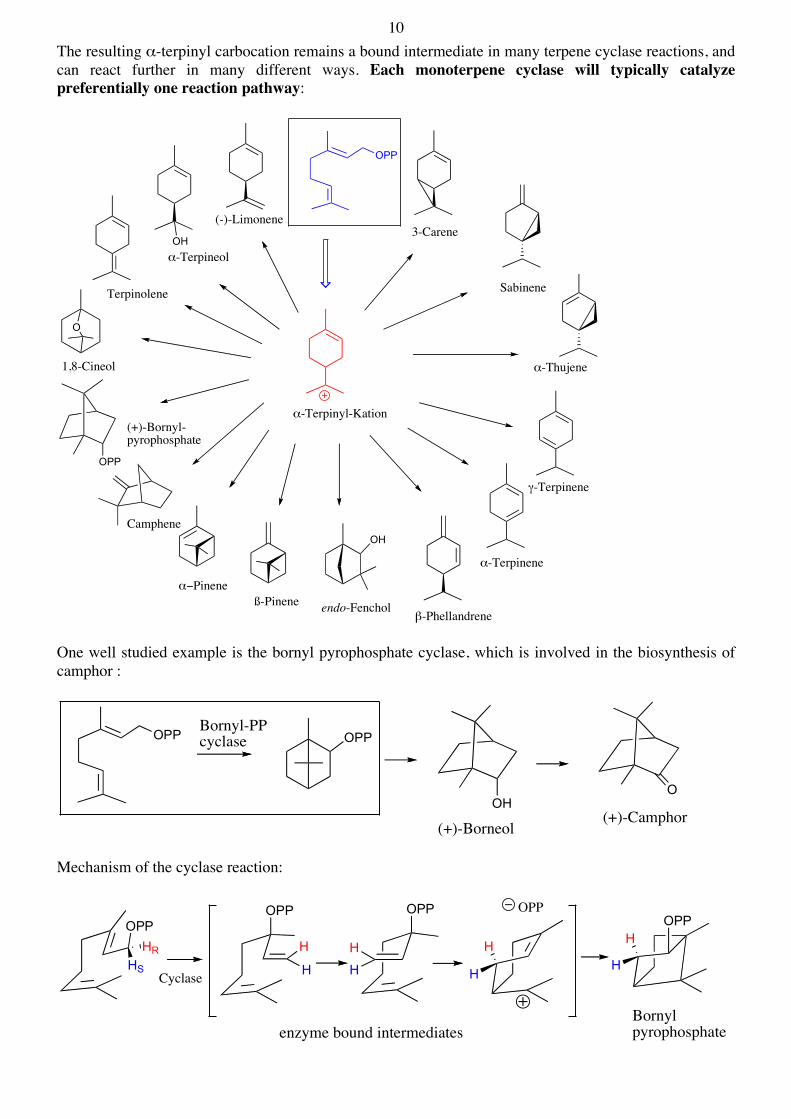
10 The resulting α-terpinyl carbocation remains a bound intermediate in many terpene cyclase reactions, and can react further in many different ways. Each monoterpene cyclase will typically catalyze preferentially one reaction pathway:
One well studied example is the bornyl pyrophosphate cyclase, which is involved in the biosynthesis of camphor :
Mechanism of the cyclase reaction:
OPP
3-Carene
Sabinene
α-Thujene
γ-Terpinene
α-Terpinene
β-Phellandrene
OH
endo-Fencholß-Pineneα−Pinene
Camphene
OPP
(+)-Bornyl-pyrophosphate
O
1,8-Cineol
OHα-Terpineol
(-)-Limonene
Terpinolene
α-Terpinyl-Kation
OPP OPP
OOH
(+)-Camphor(+)-Borneol
Bornyl-PP cyclase
OPPOPP OPP
OPP
Cyclase
⊕
enzyme bound intermediates
HSHR H
H
H
H
H
H
Bornylpyrophosphate
H
H
OPP
11 Limonene synthase is another well-studied enzyme. (-)-Limonene is the precursor of menthol and carvone, which can be isolated from extracts of peppermint, carraway (Carum carvi) and dill.
The main product of the limonene synthase reaction is limonene, but small amounts of myrcene (2%), α-pinene und ß-pinene (4%) can also be detected:
Sesquiterpene Synthases (Curr. Opin. Struct. Biol. 1998, 8, 695; Chem. Rev. 1990, 90, 1089) All sesquiterpenes are formed from FPP. A large variety of different cyclic sesquiterpenes have been discovered in Nature.
12 The sesquiterpene cyclases require Mg2+ as cofactor and use FPP as substrate. The metal is coordinated both to the protein and to the pyrophosphate group of the substrate. An important point is the stereochemical course of the cyclization at C1, with some reactions proceeding with retention and others with inversion of configuration. Mechanism in overview:
A well studied example is the enzyme trichodiene synthase from the fungus Fusarium sporotrichioides, which converts FPP into trichodiene:
The aristolochene synthase isolated from tobacco plants and the vetispiradiene synthase from Hyoscyamus muticus are two phylogenetically closely related enzymes that catalyze also very closely related reactions. In both cases, E,E-germacrene-A is formed as a short-lived enzyme-bound intermediate. Studies reported so far suggest the following mechanism of action:
OPP OPP
OPP
OPP OPP
FPHPH2 E-ß-Farnesene
1,10 cyclization
Aristolochene- H+
γ-Humulene Longifolene
α-Longipinene
α-Ylangene
1,11 cyclization
1,10 cyclizationß-Bisabolene
1,6- 1,11-
OPP
OPP OPP
H⊕
⊕
Trichodiene
⊕H
⊕
H⊕
13
Two other very interesting sesquiterpene cyclases are γ-humulene synthase und δ-selinene synthase, both isolated from fir trees, which can catalyze the formation of a surprisingly large variety of cyclic sesquiterpenes starting from FPP. In in vitro assays monitored by GC, the formation of 34 different products catalyzed by δ-selinene synthase can be observed starting from FPP, whereas γ-humulene synthase catalyzes the formation of 52 different products, although about half so far have unknown structures. Interestingly, the same mixture of terpenoid products can also be found in fir trees (J. Biol. Chem. 1998, 273, 2078). See below. Diterpene Synthases. The diterpene synthases catalyze similar reactions, but use now GGPP as substrate. The same principles of reactivity apply. Thus the allylic pyrophosphate acts as a leaving group (with assistance by Mg2+), and the resulting carbocation can initiate a variety of different reaction paths (carbocation-addition to double bonds, rearrangements (Wagner-Meerwein), hydride shifts, as well as deprotonations) depending upon the bound conformation at the active site of each enzyme. Examples are also seen where the protonation of a double bond is used to initiate a cyclization reaction. A great structural diversity is seen amongst diterpene natural products :
Taxol is an important natural product because of its anti-cancer activity. It was discovered in a National Cancer Institute program at the Research Triangle Institute in 1967 when it was isolated from the bark of
OPP E,E-Germacradienyl cationOPHPH2
+ H
AristolocheneVetispiradiene
Germacrene-A
VetispiradieneSynthase
5-epi-AristolocheneSynthase
FPP
OPP
GGPP
OPP OPP
(-)-Abietadiene
(+)-Copalyl Diphosphate (-)-Copalyl Diphosphate
(-)-Kaurene
TaxadieneCasbene
14 the Pacific yew tree, Taxus brevifolia and named 'taxol'. When developed commercially by Bristol-Myers Squibb (BMS) the generic name was changed to 'paclitaxel'. The BMS compound is sold under the trademark 'Taxol'. Paclitaxel is now used to treat patients with lung, ovarian, breast cancer, head and neck cancer, and advanced forms of Kaposi's sarcoma. Paclitaxel is also used for the prevention of restenosis. Paclitaxel works by interfering with normal microtubule growth during cell division. From 1967 to 1993, almost all the paclitaxel produced was derived from the bark of the Pacific yew, the harvesting of which kills the tree in the process. In 1992 BMS started to manufacture paclitaxel from 10-deacetylbaccatin isolated from the needles of the European yew. By the end of 1995, BMS stopped production from the bark of the Pacific yew, effectively terminating the ecological controversy over its use. Currently, all paclitaxel production for BMS uses plant cell fermentation technology. This starts from a specific taxus cell line propagated in aqueous medium in large fermentation tanks. Paclitaxel is then extracted directly, purified by chromatography and isolated by crystallization. There is now great interest in trying to reconstitute the entire biosynthetic pathway in vitro. Several of the enzymes on the pathway have already been cloned and produced by recombinant DNA techniques. A key step is catalyzed by the taxadiene synthase:
The mechanism of the cyclization has been intensively studied:
2.6. The formation of triterpenes from squalene (Angew. Chem. 2000, 112, 2930; Chem. Revs. 2011, 111, 6423) Squalene is the universal precursor of all triterpenes, including all steroids. In animals, squalene is converted in only two steps into a steroid called lanosterol. The first step is catalyzed by a monooxygenase, which is a flavo-enzyme not a hemoprotein, but uses molecular oxygen and NADPH to epoxidize squalene:
O
NH OH
O
O
Me
MeMe
O OMe
OH
OO
H
HO O
OMe
OMe
OPhTaxol
GGPP
OPP H
D
DD
H
D
D
HH
15
Perhaps of most interest here is how the cyclase enzyme can take squalene epoxide as substrate and release lanosterol as product. What chemical steps take place at the active site of the enzyme and how is the reaction catalyzed? Oxidosqualene-Lanosterol-Cyclase In higher organisms the steroid skeleton is produced through the action of a membrane-bound enzyme. In the course of the transformation, a series of ring-forming steps and rearrangements take place:
O MeMe
Me
Me
Me
MeMe
Me
Me Me
Me
Me
Me
Me
HO
Me
MeH
H
Squalene Epoxide Lanosterol
Steroide
Me
Me Me MeMe
MeMeMe
Squalene
NADPH, O2
NADP+, H2O squalene epoxidase
squalene epoxidecyclase
Me
Me
OMe
Me
Me H
Me
X
Me
Me
MeMe
MeHO
Me
MeH
MeH
MeMe
H
Me
H
MeOMe
MeHO
Me H
MeH H
Lanosterol
X = OEnzym
Me
MeHO
Me H
MeH
Me Me
Me
HH
Me
AH
Me
MeHO
Me H
MeH
Me Me
Me
H
Me
H
MeMe
MeHO
Me
MeH
MeH
MeMe
H
H
B
O
Br
F
ON
Ro 48-8071cyclase inhibitor
16 The cyclase must bind the substrate in the correct folded conformation to allow a stereoelectronically assisted series of rapid ring closure steps, and form the product with the correct relative and absolute configuration. All the intermediate carbocation intermediates must be shielded from reaction either with water or with the protein. Finally, the correct proton must be removed to terminate the reaction. The H-atom shifts and Wagner-Meerwein rearrangements occur along a kinetically and thermodynamically preferred pathway, until the end product is reached. In 2004 a group at Hoffmann-La Roche in Basel succeeded for the first time in crystallizing the enzyme (Nature, 2004, 432, 118).
Left: Ribbon diagram of human OSC. a, The C and N termini and several sequence positions are labelled. The inner barrel helices are coloured yellow. The bound inhibitor (black) indicates the location of the active site. b, The orientation of OSC relative to one leaflet of the membrane, whose polar and nonpolar parts are depicted in light blue and light yellow respectively. Internal surfaces and channels of OSC are shown with the following colour code: blue, positive; red, negative; cyan, hydrogen-bond donor; magenta, other polar. Ro 48-8071 binds in the central active-site cavity. Two channels lead to the enzyme surface: one is hydrophobic to the membrane insertion site and one is polar. The fragment of lipid (blue) binds to the hydrophobic substrate entrance channel. A ß-OG molecule belonging to a crystal neighbour (black) interacts with the membrane-inserting hydrophobic surface. Right: Stereoview of the electron density representing the bound substrate. Residues in the enzyme within 5Å are shown. A/B-Rings: The cationic intermediates may be stabilized by cation-π interactions with the aromatic rings of Trp387, Phe444 and Trp581. The catalytic Asp455 is activated by Cys 456 and Cys 533. The Tyr 98 side chain sterically hinders the B-ring from assuming the favourable chair conformation. C/D-Rings: Phe 696 and His232 can stabilize the positive charge at the C20 cation by cation-π interactions. His 232 is the nearest basic residue that could deprotonate the C8/9 lanosterol cation.
17 From lanosterol, the pathway for steroid biosynthesis continues on to cholesterol (see Chem. Revs. 2011, 111, 6423). Three methyl groups must be removed, one double bond is reduced and another is shifted. Cholesterol then becomes a branch point in steroid biosynthesis, serving as a precursor from which other steroids are produced
In plants the oxidosqualene cyclase does not form lanosterol, but rather cycloartenol, which is then the precursor for the formation of other plant steroids:
Bacterial squalene cyclase catalyzes a different cyclization cascade, which is mechanistically related, but not so complicated. Now squalene (not the epoxide) is bound in a specific conformation, which allows a rapid series of cyclization steps to occur. The process is now started by protonation of the terminal double bond (Chem.Biol. 2000, 7, 643):
This cyclase is a homodimeric, soluble enzyme. The active site is a buried cavity, which binds squalene in the preferred conformation. Most probably the side chain of Asp376 acts as a general acid catalyst to start the cyclization cascade.
Me
Me Me
Me
Me
Me
HO
Me
MeH
H
Lanosterol
Me Me
Me
HO
Me
MeH
H
Cholesterol
HH
H
Me
Me
OMe
Me
MeBH Me Me
Me
MeMe
Me
MeMe
MeHO
Me H
MeH H
H
MeMe
MeHO
Me
MeH
MeH
MeMe
H
H
Me
MeHO
Me
MeH
MeH
Me
Me
H
Cycloartenol
H
H
Squalene-HopeneCyclase
18
19
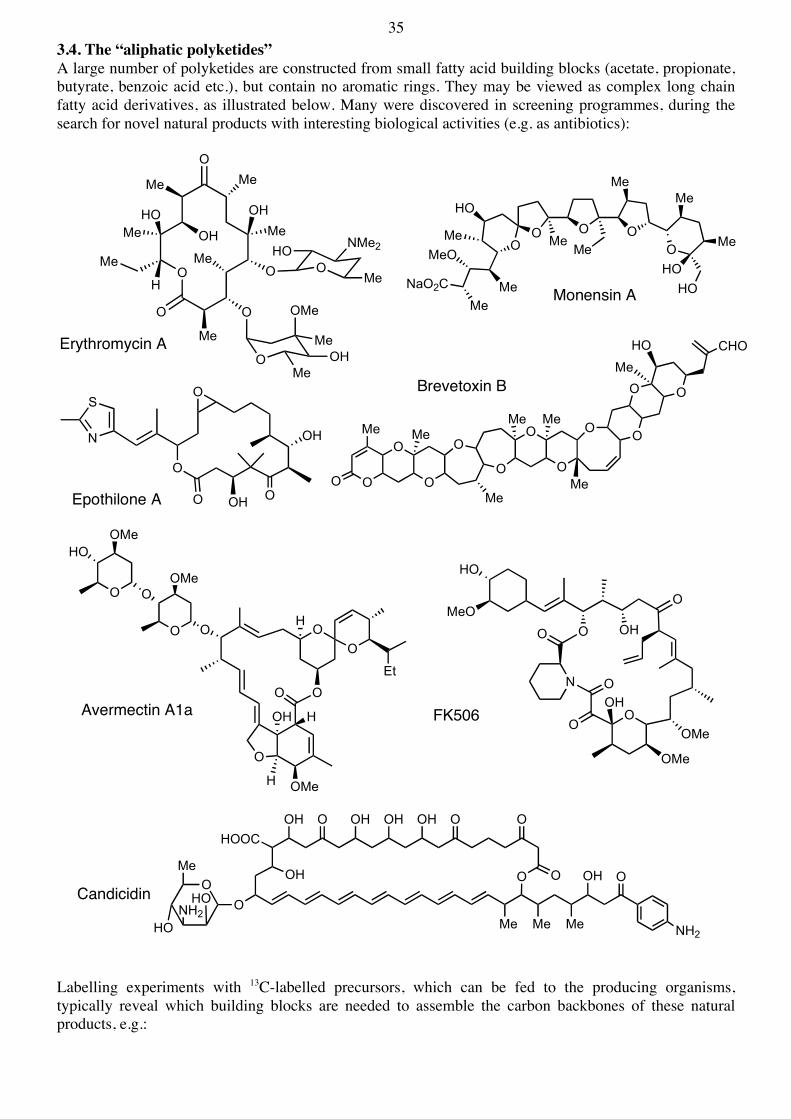
20 3. Biosynthesis of Polyketide Natural Products 3.1 Polyketide Biosynthesis The polyketides constitute a large class of natural products, with widely varying structures. Some are aromatic, others aliphatic, some are cyclic others are acyclic. Polyketide metabolites are found in essentially all organisms, where they have widely differing biological functions, from simple dyestuffs (e.g. in plants) to antibiotics (in microorganisms). The polyketides form one of the largest classes of natural products. Many have been discovered in screening programs aimed at the isolation and discovery of new biologically active compounds, useful in the pharmaceutical industry. Despite the enormous variety of different structures seen whithin the polyketide family, we can nevertheless classify them in two main groups - the aliphatic (or reduced) and the aromatic polyketides, e.g. Aliphatic (reduced) polyketides:
OO O O
O
NaO2CMe
MeO
Me
HO
Me Me
Me
Me
Me
Me
HOHO
OH
O
Me
O
Me
OH
Me
Me
Me
OMe
OO
Me OHO
NMe2
Me
O
OMe
MeOH
Me
H
HO
O
O
O
O
O
O
O
O O
O O
HO
Me
Me
MeMe
Me
MeMe
O
CHO
Monensin A
Erythromycin A
Brevetoxin B
OO
O
O
OOEt
OH
H OMe
O
OOOMe
OMeHO
H
H
Avermectin A1a
O
O OHO
HO
MeO
N O
OO
OMe
OH
OMeFK506
S
N
O
O OOH
O
OH
Epothilon A
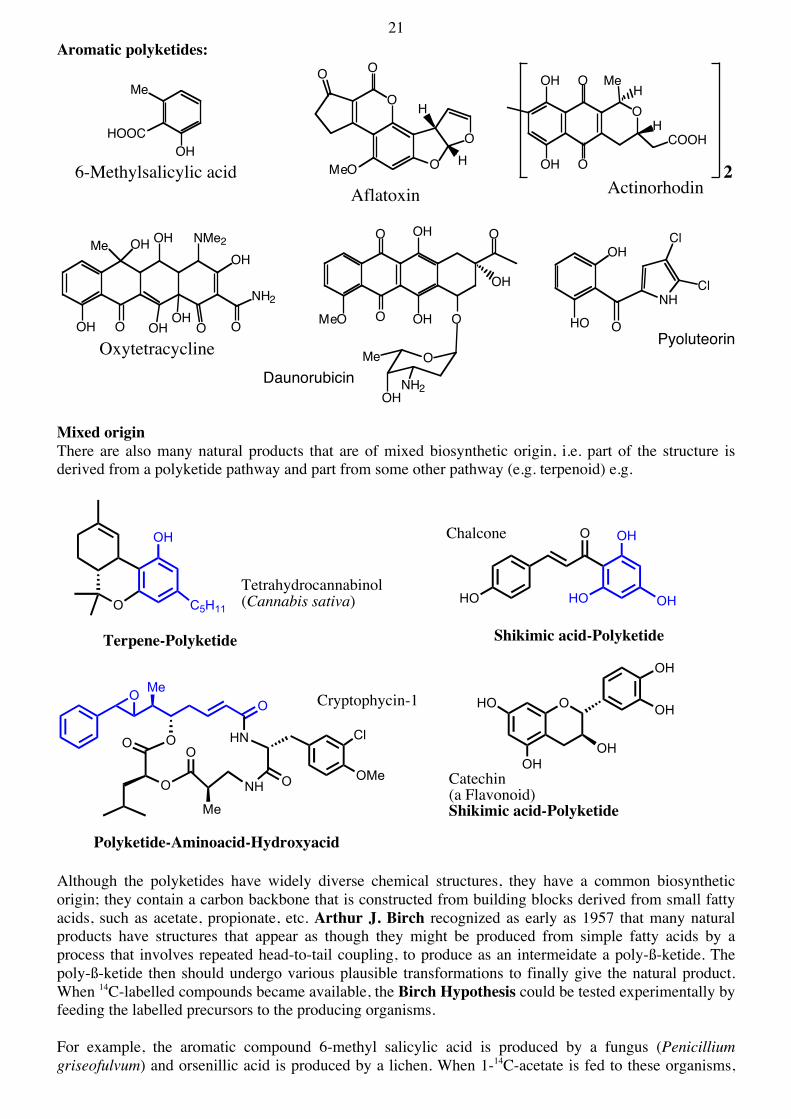
21 Aromatic polyketides:
Mixed origin There are also many natural products that are of mixed biosynthetic origin, i.e. part of the structure is derived from a polyketide pathway and part from some other pathway (e.g. terpenoid) e.g.
Although the polyketides have widely diverse chemical structures, they have a common biosynthetic origin; they contain a carbon backbone that is constructed from building blocks derived from small fatty acids, such as acetate, propionate, etc. Arthur J. Birch recognized as early as 1957 that many natural products have structures that appear as though they might be produced from simple fatty acids by a process that involves repeated head-to-tail coupling, to produce as an intermeidate a poly-ß-ketide. The poly-ß-ketide then should undergo various plausible transformations to finally give the natural product. When 14C-labelled compounds became available, the Birch Hypothesis could be tested experimentally by feeding the labelled precursors to the producing organisms. For example, the aromatic compound 6-methyl salicylic acid is produced by a fungus (Penicillium griseofulvum) and orsenillic acid is produced by a lichen. When 1-14C-acetate is fed to these organisms,
HOOC
Me
OH
O
MeH
H
O
O
OH
OH
COOH
OH
Me
O
O
O
O O
O
H
HMeO
OHOH
O
OHNMe2
O
NH2
OH OH
6-Methylsalicylic acidActinorhodin
2Aflatoxin
Oxytetracycline
MeO
O
O
O
O
OH
OH
OH
OMe
NH2OH
Daunorubicin
O
NH
Cl
Cl
OH
HOPyoluteorin
O
OH
C5H11
Tetrahydrocannabinol(Cannabis sativa)
Terpene-Polyketide
O OH
HO OHHO
Chalcone
Shikimic acid-Polyketide
O
O
MeO
HN
NH O
O
O
O
Me
Cryptophycin-1
Cl
OMe
Polyketide-Aminoacid-Hydroxyacid
O
OH
OH
OHHO
OHCatechin(a Flavonoid)Shikimic acid-Polyketide
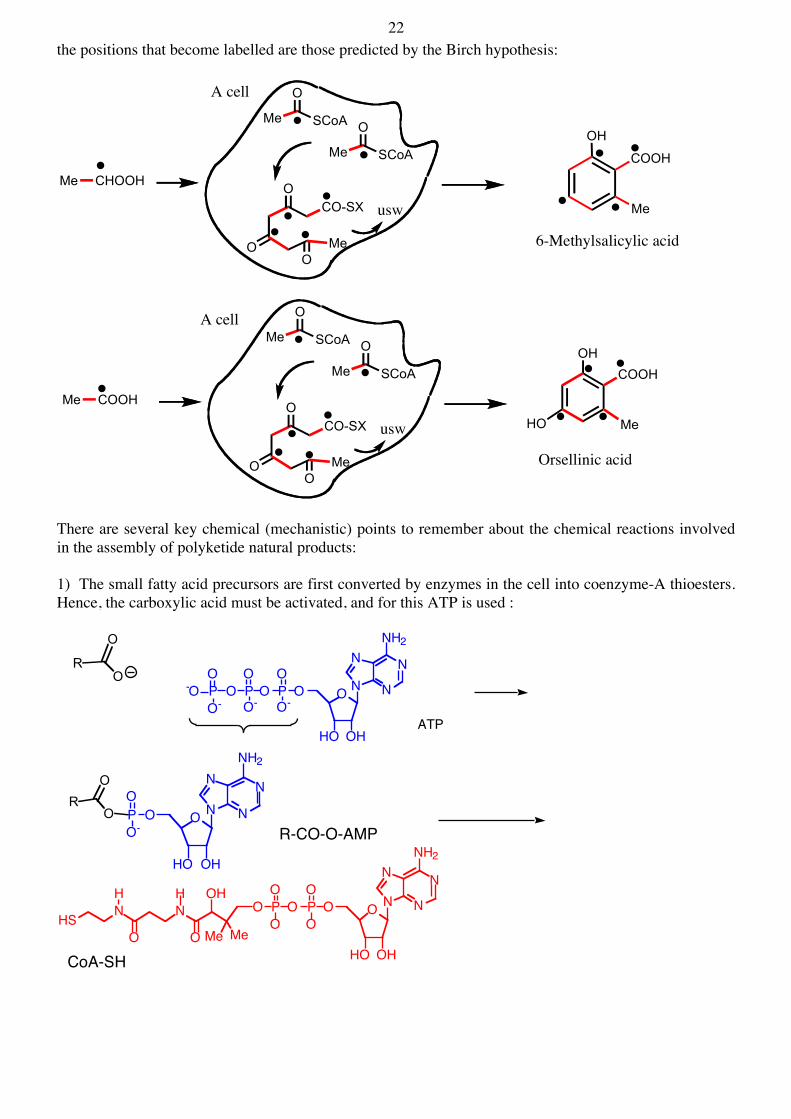
22 the positions that become labelled are those predicted by the Birch hypothesis:
There are several key chemical (mechanistic) points to remember about the chemical reactions involved in the assembly of polyketide natural products: 1) The small fatty acid precursors are first converted by enzymes in the cell into coenzyme-A thioesters. Hence, the carboxylic acid must be activated, and for this ATP is used :
Me COOH
CO-SXO
OO
Me
OHCOOH
MeHO
Me
O
SCoA
Me
O
SCoA
usw
A cell
Orsellinic acid
Me CHOOH
CO-SXO
OO
Me
Me
O
SCoA
Me
O
SCoA
usw
A cell
6-Methylsalicylic acid
COOH
Me
OH
O P O P O O
HO OH
N
N
N
N
NH2
O- O-
O OPO
-OO-
R
O
O
ATP
HS N NH
O
H
O
OH
Me Me
O PO
PO
OO
NH2
N
NN
N
OHHO
OOO
RO
O P O O
HO OH
N
N
N
N
NH2
O-
O
CoA-SH
R-CO-O-AMP
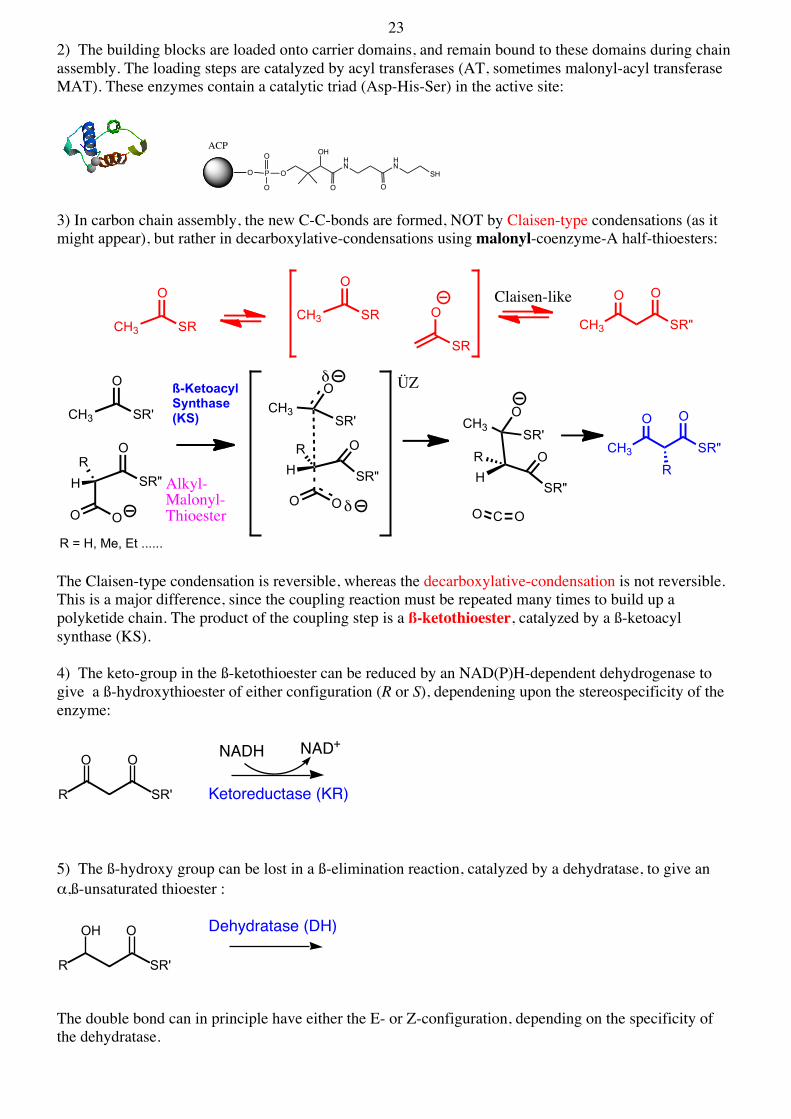
23 2) The building blocks are loaded onto carrier domains, and remain bound to these domains during chain assembly. The loading steps are catalyzed by acyl transferases (AT, sometimes malonyl-acyl transferase MAT). These enzymes contain a catalytic triad (Asp-His-Ser) in the active site:
3) In carbon chain assembly, the new C-C-bonds are formed, NOT by Claisen-type condensations (as it might appear), but rather in decarboxylative-condensations using malonyl-coenzyme-A half-thioesters:
The Claisen-type condensation is reversible, whereas the decarboxylative-condensation is not reversible. This is a major difference, since the coupling reaction must be repeated many times to build up a polyketide chain. The product of the coupling step is a ß-ketothioester, catalyzed by a ß-ketoacyl synthase (KS). 4) The keto-group in the ß-ketothioester can be reduced by an NAD(P)H-dependent dehydrogenase to give a ß-hydroxythioester of either configuration (R or S), dependening upon the stereospecificity of the enzyme:
5) The ß-hydroxy group can be lost in a ß-elimination reaction, catalyzed by a dehydratase, to give an α,ß-unsaturated thioester :
The double bond can in principle have either the E- or Z-configuration, depending on the specificity of the dehydratase.
P
O
O
O O
HN
HN
OH
O OSH
ACP
CH3
O
SRO
SR
CH3
O
SR
CH3
O
SR'
O
SR"
O O
CH3
O
SR'
O
SR"
O O
HR
CH3O
SR'
O
SR"
CO O
HR
HR
R = H, Me, Et ......
ß-KetoacylSynthase (KS)
δ
δ ÜZ
CH3 SR"
O O
R
CH3 SR"
O O
Alkyl-Malonyl-Thioester
Claisen-like
R SR'
O O NADH NAD+
Ketoreductase (KR)
R SR'
OH O Dehydratase (DH)
24 6) The double bond in the α,ß-unsaturated thioester can be reduced to a fully saturated thioester, again catalyzed by a NAD(P)H-dependent enoyl reductase (dehydrogenase):
With this set of reactions it is possible to install a keto-group, an alcohol, a double bond, or a fully saturated unit, in the growing polyketide chain:
7) As a last step in the chain assembly, the thioester is typically hydrolyzed to a free carboxylic acid, or cyclized to a lactone or lactam, catalyzed by a so-called thioesterase. The thioesterases belong mechanistically to the serine-protease class of enzymes, and so have a catalytic triad in the active site (Asp-His-Ser), with a catalytically important Ser acting as nucleophile :
We now have a small library of catalytic activities that can be used for fatty acid and polyketide assembly. These activities can be combined in various ways to generate the large class of polyketide natural products, as shown below. One important example is the multienzyme complex involved in fatty acid biosynthesis. Fatty acid synthases (FASs) function very much like the polyketide synthases (PKSs). 3.2. Fatty acid biosynthesis The biosynthesis of long chain saturated fatty acids (e.g. palmitic and stearic acids) is catalyzed by a large multi-enzyme complex, called the fatty acid synthase (FAS) complex. Typically, the FASs take acetyl-CoA as starter unit, and then extend the chain in a step-wise manner, using malonyl-CoA as extender units. The malonyl-CoA is produced in a biotin-dependent enzymic reaction from acetyl-CoA (see: Biochemistry, 2004, 43, 14035): overall:
R SR'
O NADH NAD+
Enolyreductase (ER)
R
O
SR'
O
SR"
O O
R SR'
O O
R SR'
OH O
R SR'
O
R SR'
OKS KR DH ER
R'"R'" R'" R'" R'"
R'" = H, Me. Et oder ........
R SR'
O Thioesterase (TE)
CH3
O
S-CoA
Acetyl-CoA Carboxylase
ATPHCO3
ADPPi
O
S-CoA
COO
HN NH
S
O
HN
LYSO
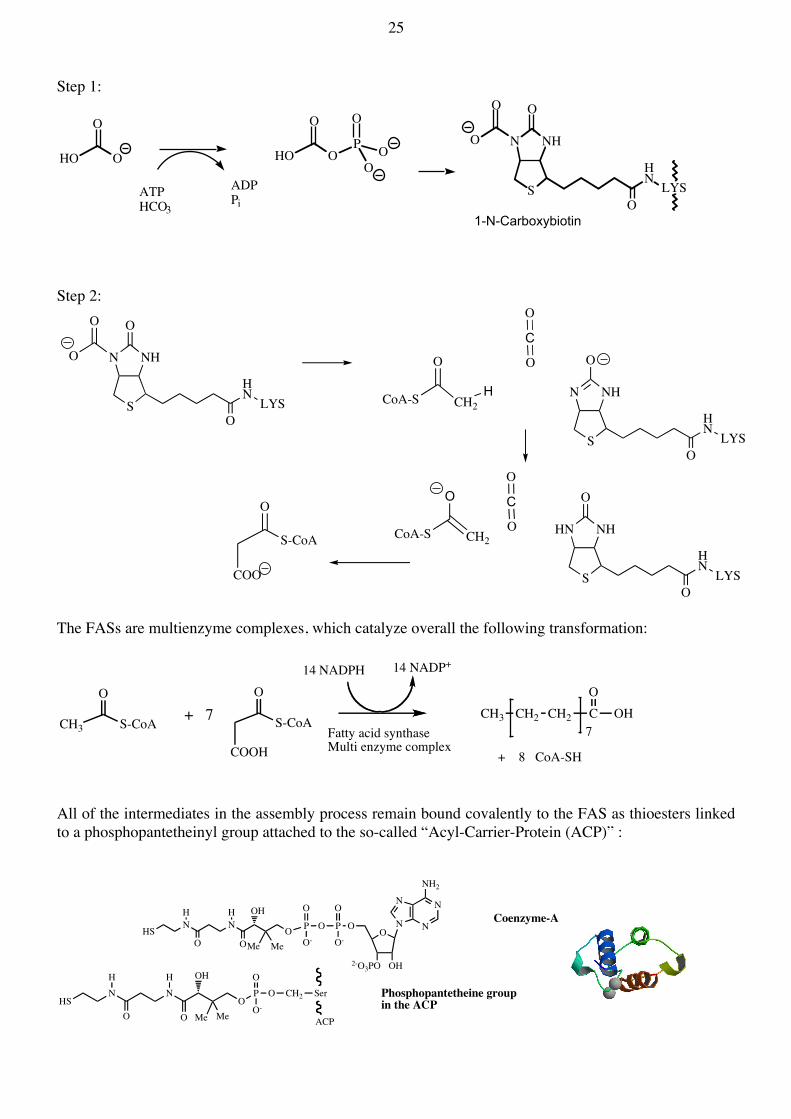
25 Step 1:
Step 2:
The FASs are multienzyme complexes, which catalyze overall the following transformation:
All of the intermediates in the assembly process remain bound covalently to the FAS as thioesters linked to a phosphopantetheinyl group attached to the so-called “Acyl-Carrier-Protein (ACP)” :
HO
O
O HO
O
OP
O
OO
N NH
S
O
HNLYS
O
O
O
ATPHCO3
ADPPi
1-N-Carboxybiotin
N NH
S
O
HNLYS
O
O
O
CH2
O
CoA-S
O
S-CoA
COO
C
O
O
N NH
S
O
HNLYS
O
H
CH2
O
CoA-S
C
O
O HN NH
S
O
HNLYS
O
CH3
O
S-CoA
O
S-CoA
COOH
CH3 CH2 CH2 CO
OH
+ 8 CoA-SH
14 NADP+14 NADPH
+ 77Fatty acid synthase
Multi enzyme complex
HSN N
O PH
O
H
O
OH
Me Me
O
O-O P
O
O-O
ON
2-O3PO OH
N
N
N
NH2
HSN N
OP
H
O
H
O
OH
Me Me
O
O-O CH2
ACP
Ser Phosphopantetheine group in the ACP
Coenzyme-A
26 In plants and most procaryotes 8 distinct proteins are required for fatty acid biosynthesis (the so-called type-II FASs). These 8 proteins work together to assemble fatty acid molecules. The ACP is a small carrier protein containing about 80 amino acids. The active site Ser (modified with the pantetheinyl group) is strictly conserved. Its function is to carry the growing fatty acid chain from one enzyme to the next, in each catalytic cycle. In animals and fungi, Nature has combined these separate proteins into one or two giant proteins, which fold into discrete domains, where each domain then catalyzes one step in the assembly process (the type-I FASs). The complete catalytic cycle is shown below:
In 2006 the first crystal structures of both mammalian (α2) and fungal (e.g. yeast) FASs (α6ß6) were published (Science 2006, 311, 1263). Later a higher resolution structure of the mammalian enzyme was published (see picture below taken from Science 2008, 321, 1315). The 3D organization of the domains differs from their linear arrangement in the sequence:
Ψ-Linker domains showing similarity to methyltransferase (Ψ-ME) and KR (Ψ-KR) domains are also shown. These should have structural but not catalytic functions in the complex.
HOOC(CH2CH2)nCH3
Thioesterase (TE)
Enoyl-Reductase(ER)
NADP+
NADPH
Dehydratase(DH)
NADP+
NADPH
ß-Ketoacylreductase (KR)
ß-Ketoacyl synthase (KS)
Malonyl-CoAAcetyl-CoA
SH
S.COCH3
SH
S.COCH3
S
S
S
OH
S
S
SCO(CH2CH2)nCH3
CH3O
OCOOH
O OMe
Me
O
O
S
Me
O
Me
ACP
ACP
SH
Malonyl-AcetylTransferase(MAT)
ACP
ACP
ACP
ACP
SH
SHACP
ACP
ACP
H2O
CO2
CoASH
CoASH
KS
KS
KS
KS
KS
Malonyl-AcetylTransferase(MAT)
27 3.3. The “aromatic polyketides” (Nat. Prod. Rep. 1999, 16, 425; Accts. Chem. Res. 2009, 42, 631) Only in the past few years have detailed structural and mechanistic studies with PKSs been published. This has become possible through advances in molecular genetics, which provided access to the biosynthetic genes for the individual biosynthetic enzymes. Before this, labelling experiments with intact organisms were possible, which often gave important insights into how a polyketide chain is assembled and cyclized in the producing organisms. The starter unit could be identified in this way, as well as the extender units. Acetyl-CoA is used frequently as starter unit, but in principle any small molecule CoAS-thioester can be used (e.g. benzoic acid-SCoA thioester). As extender units, malonyl-CoA or other malonyl-CoA derivatives may be used. The growing polyketide chains remain bound to the PKS (compare FAS above), and so can never be detected as free intermediates. E.g. three different pathways:
By feeding 13C-labelled precursors to the producing organism, and using 13C NMR spectroscopy to detect sites of enrichment, it was possible to deduce how the polyketide chain is assembled. Of special interest is the use of doubly labelled precursors, like 13C2-acetate (each C-Atom 99% 13C), whose intact incorporation into the end product can be detected through an analysis of 13C-13C couplings e.g.:
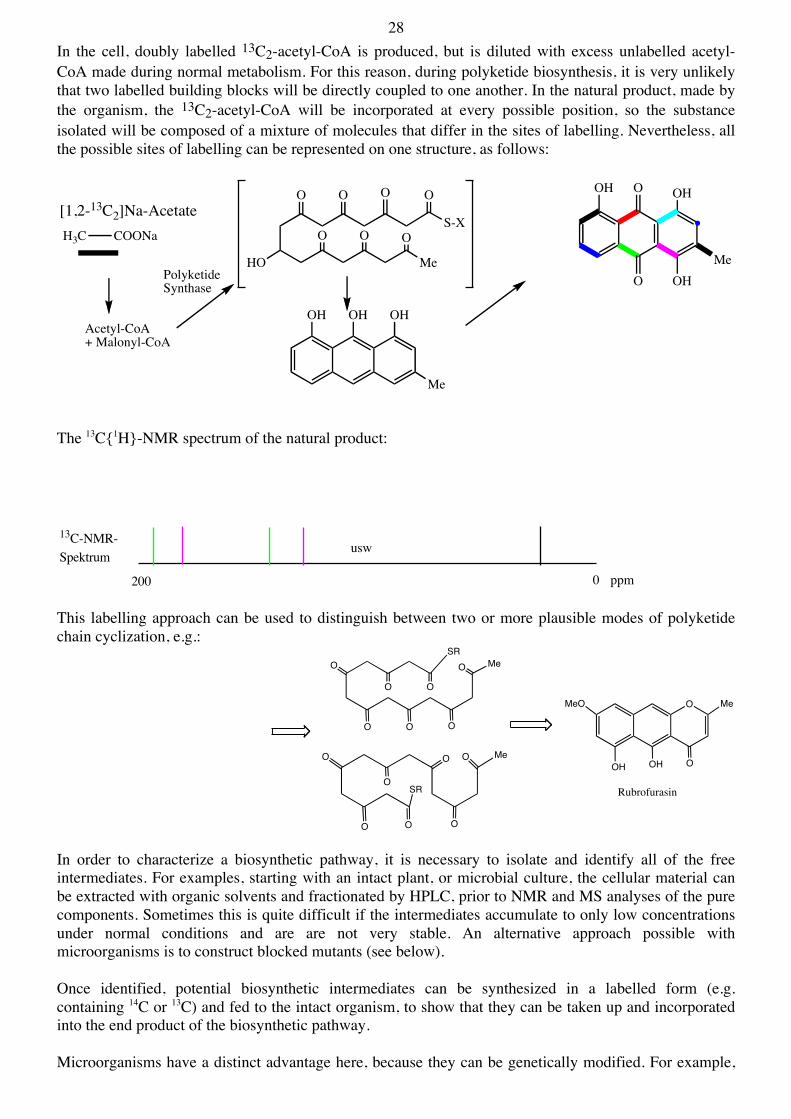
28 In the cell, doubly labelled 13C2-acetyl-CoA is produced, but is diluted with excess unlabelled acetyl-CoA made during normal metabolism. For this reason, during polyketide biosynthesis, it is very unlikely that two labelled building blocks will be directly coupled to one another. In the natural product, made by the organism, the 13C2-acetyl-CoA will be incorporated at every possible position, so the substance isolated will be composed of a mixture of molecules that differ in the sites of labelling. Nevertheless, all the possible sites of labelling can be represented on one structure, as follows:
The 13C{1H}-NMR spectrum of the natural product:
This labelling approach can be used to distinguish between two or more plausible modes of polyketide chain cyclization, e.g.:
In order to characterize a biosynthetic pathway, it is necessary to isolate and identify all of the free intermediates. For examples, starting with an intact plant, or microbial culture, the cellular material can be extracted with organic solvents and fractionated by HPLC, prior to NMR and MS analyses of the pure components. Sometimes this is quite difficult if the intermediates accumulate to only low concentrations under normal conditions and are are not very stable. An alternative approach possible with microorganisms is to construct blocked mutants (see below). Once identified, potential biosynthetic intermediates can be synthesized in a labelled form (e.g. containing 14C or 13C) and fed to the intact organism, to show that they can be taken up and incorporated into the end product of the biosynthetic pathway. Microorganisms have a distinct advantage here, because they can be genetically modified. For example,
H3C COONa
O
O
OH
OHMe
OH
[1,2-13C2]Na-AcetateO O O
HO
O O O
O
S-X
Me
Acetyl-CoA+ Malonyl-CoA
OH OH OH
Me
PolyketideSynthase
13C-NMR-Spektrum
200 0 ppm
usw
SRMe
OO
O
O O O
O
Me
OO
O
O
O
O
O
SR
O Me
OOHOH
MeO
Rubrofurasin
29 mutants can be sought, in which one of the steps in the biosynthetic pathway is inoperative, due to a mutation of one of the biosynthetic enzymes, which renders it non-functional. In such mutants, the accumulation of biosynthetic intermediates should then occur: Through screening of blocked mutants it is often possible to isolate and identify biosynthetic intermediates. In recent years, the molecular genetic approach has become a very powerful tool in the study of biosynthetic pathways, especially in microorganisms that can be genetically manipulated. Molecular genetic approaches can provide access to the genes for the biosynthetic enzymes, which are usually clustered all together in one (relatively) small region of the chromosome. Once one gene has been isolated, the other biosynthetic genes can be found in the flanking DNA, which is easy to isolate and sequence. Once the biosynthetic genes are available, then the biosynthetic enzymes can normally be overproduced by standard recombinant DNA techniques, and this opens the way for detailed structural and mechanistic studies in vitro. How to proceed ? Consider one of the very first examples of the cloning of a biosynthetic gene cluster - that for the polyketide actinorhodin. Actinorhodin Actinorhodin is a polyketide produced by one species of gram-positive soil microrganism called Streptomyces coelicolor. The production of this natural product was simple to detect, because the compound is a bright blue color at slightly basic pH.
O
O OH
Me
O
HO
OH
OMeO
HO CO.SRO O
OOOH
O
OOH
COOH
Me
H
O
OH O
CO.SR
Me
O
Me
CO2H
OOH
O
OH O
OH
Me
OH H
O
Act VI mutantact I (PKS),act III (KR) act Va, Vb
Actinorhodin
act IV
Aloesaponarin II
Act VIIMutant
1 x Acetyl-CoA
7 x Malonyl-CoA2
act VIact VIIAlkohol
30 The cloning of the actinorhodin biosynthetic genes was made possible because:
• the production of the antibiotic is straightforward to detect (without the need for HPLC, NMR, MS etc.).
• all the genes for the pathway are clustered together in the chromosome, making it possible to isolate a single DNA fragment containing all the genes.
• plasmid cloning vectors had been developed, which allowed cloning experiments in these microorganisms.
• many mutants of the actinorhodin-producing organism were available, each blocked at different steps in the pathway. Below are shown different types of act mutants growing on agar plates :
The mutants could be cultivated in pairs on agar plates. The mutants could be classified as early or late mutants depending upon their ability to cosynthesize acinorhodin; thus a late blocked mutant will produce an intermediate that can be taken up by another mutant (an early blocked mutant) and converted to actinorhodin (blue color): The cloning of the entire biosynthetic pathway could then proceed as follows: Step-1. Starting from whole bacterial cells, the chromosomal DNA is isolated and cleaved into small fragments with a restriction enzyme: The small fragments are then cloned into a plasmid cloning vector, which can be stably maintained in this microorganism. Once ligated into the cloning vector, the library of recombinant plasmids+inserts are
31 introduced into one of the late act mutants. One of the cells will obtain a plasmid containing an intact functional copy of the biosynthetic gene that has been inactivated by mutation. This mutant will then be able to make actinorhodin (see left below): Step-2. From this colony or "clone" the plasmid+insert can be isolated. The insert will contain at least one of the late biosynthetic genes. From two such clones, inserts were isolated that contained different but overlapping pieces of chromosomal DNA. From these, a “cut and paste” strategy could be used to reconstruct a new insert containing a larger and hopefully complete copy of the entire biosynthetic gene cluster: This new plasmid+insert was then introduced into a related microorganism (Streptomyces parvulus) that normally does not make actinorhodin. Only now, once the plasmid+insert was introduced into the cell - it could (see right above). The insert in this plasmid thus contains all the information needed to produce all the enzymes needed for actinorhodin biosynthesis (Nature 1984, 309, 462). In the next step, the DNA insert was sequenced, to reveal the locations and nt sequences of all the biosynthetic genes, and hence the primary sequences of all the biosynthetic enzymes. Through sequence comparisons with data-bases of enzymes with known function, the likely functions of the biosynthetic enzymes could be deduced:
Once the genes had been identified in this way, the enzymes could be produced using recombinant DNA methods, and structural and mechanistic studies could begin. Most efforts recently have focused on the PKS. The picture that has emerged is described below: The construction of the polyketide backbone requires a malonyl-CoA:ACP transacylase (MAT, also used in fatty acid biosynthesis), an ACP (actI-ORF3), a ß-ketoacylsynthase (KS) and another protein called the Chain-Length-Factor (CLF), encoded in 3 genes, shown in black above. These together constitute a "minimal PKS", which can assemble a polyketide chain starting from malonyl-CoA.
32 Surprisingly, it was shown that the starter unit is also derived from malonyl-CoA, by decarboxylation, catalysed by the KS protein. The malonyl-CoA units are loaded onto the ACP by the MAT (red below), and then transported to a heterodimer formed by the KS+CLF proteins (green and yellow below). The polyketide chain is then transferred onto a cysteine -SH group in the active site of KS (just like in fatty acid biosynthesis). The ACP (blue) then departs to collect another malonyl unit. Once this is docked again onto the KS-CLF complex, the chain elongation can occur (see below). The cycle can then be repeated until a chain of 16 C-atoms has been constructed:
A crystal structure of the PKS (Nat. Struct. Mol. Biol. 2004, 11, 888) shows that the growing polyketide sits in a long tunnel buried in the protein. The tunnel can only accept a chain of 16 C atoms, not longer. If no other enzymes are present this minimal PKS will slowly release the polyketide chain, which spontaneously cyclizes in solution to produce SEK4 and SEK4b. These are normally not produced during actinorhodin biosynthesis (only in this in vitro assay). The shape of the tunnel seems to force the polyketide chain to bend, which leads to a cyclization at C7 (shown above). In the normal biosynthetic pathway, when all the enzymes are present, this cyclized form is transported on the ACP to the next enzyme, a KR (actIII), which reduces the carbonyl group at C9. The remaining steps have not been elucidated in detail, but the following should occur:
33
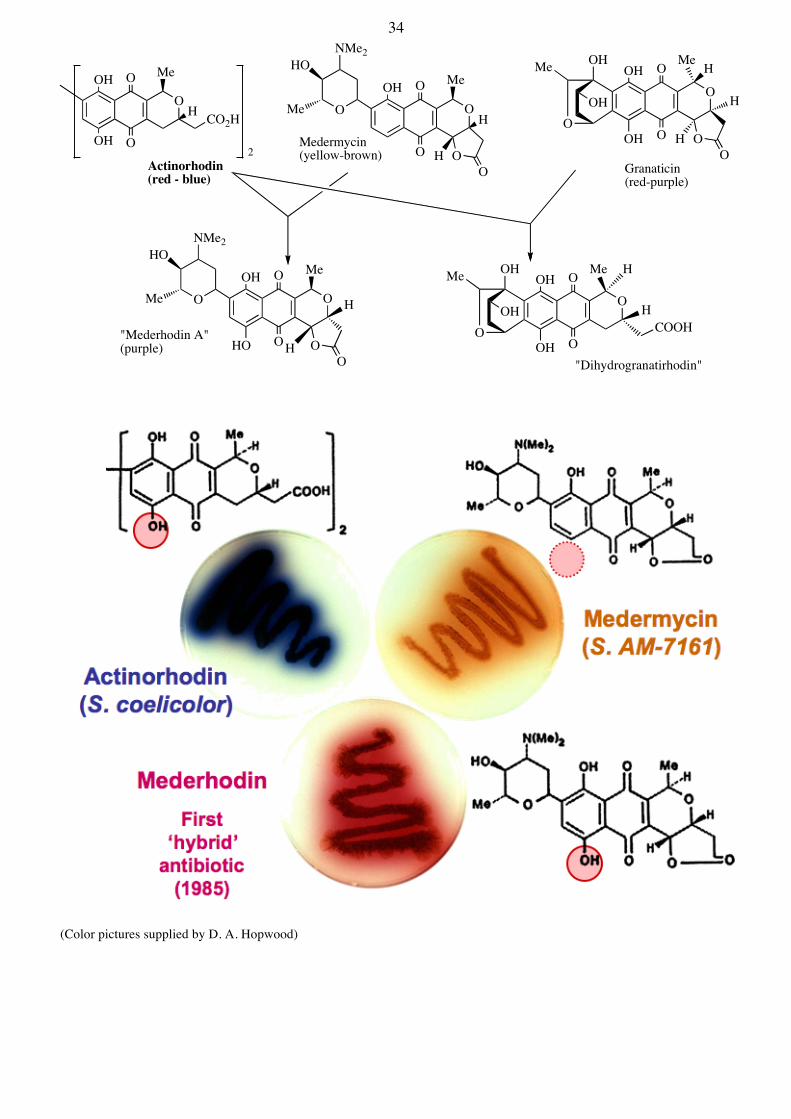
The Biosynthesis of Hybrid-Antibiotics There is now enormous interest in the engineering of novel biosynthetic pathways, by taking genes from different pathways and making new combinations, in an attempt to make novel hybrid natural products. This requires a detailed knowledge of what the individual genes do (i.e. what reactions do the biosynthetic enzymes catalyze?) and the specificity and mechanisms of action of the biosynthetic enzymes. A proof of principle that such experiments are possible came shortly after the cloning of the actinorhodin pathway (Nature, 1985, 314, 642). As shown below, the act genes were introduced into different strains that normally produce granaticin or medermycin. The strains then acquired the ability to generate new natural products:
O
O OH
Me
O
HO
OH
OMeO
OCO.SR
O O
O
OO
O
OOH
COOH
Me
H
O
OH O
CO.SR
Me
O
Me
CO2H
OOH
O
OH O
OH
Me
OH H
O
Actinorhodin
Aloesaponarin II
8 x Malonyl-CoA
2
Mutactin
min. PKS KS, CLF, ACP
OMeO
HO CO.SRO O
O
OH
min. PKS
min. PKS+ KR
OO
OH O
COOH
Me
O
AROmin. PKS+ KR + actVII ARO
CYC2/3
OOH
OH O
COOH
Me
act VI (ORF1)act VI (ORFA)act VI (ORF3)
O
Me
CO2H
OOH
H
act VI (ORF2)act VI (ORF4)
O
Me
CO2H
OOH
H
act VA + other ORFs
O
act VA+VB
O
OHO
OOH
O
O
OHO
O
O+
SEK34 SEK34b
O
OHO
HO
OOH
O O
O
OHO
OH
OH
Me
OSEK4 SEK4b
+
min. PKS+ KR + actVII ARO + ActIV(CYC2/3)
KR
34
(Color pictures supplied by D. A. Hopwood)
O
Me
CO2H
OOH
OOH
H O
MeOOH
O
HO
NMe2HO
Me
OHO
O
MeOOH
O
H
OHO
O
OHMe
OH
H
OH
O
MeOOH
O
HO
NMe2HO
Me
OHO
HO
O
MeOOH
O
H
O
OHMe
OH
H
OHCOOH
Medermycin(yellow-brown)2
Actinorhodin(red - blue)
Granaticin(red-purple)
"Mederhodin A"(purple)
"Dihydrogranatirhodin"
35 3.4. The “aliphatic polyketides” A large number of polyketides are constructed from small fatty acid building blocks (acetate, propionate, butyrate, benzoic acid etc.), but contain no aromatic rings. They may be viewed as complex long chain fatty acid derivatives, as illustrated below. Many were discovered in screening programmes, during the search for novel natural products with interesting biological activities (e.g. as antibiotics):
Labelling experiments with 13C-labelled precursors, which can be fed to the producing organisms, typically reveal which building blocks are needed to assemble the carbon backbones of these natural products, e.g.:
OO O O
O
NaO2C
Me
MeO
Me
HO
Me Me
Me
Me
Me
Me
HOHO
OH
O
Me
O
Me
OH
Me
Me
Me
OMe
OO
Me OHO NMe2
Me
O
OMe
MeOH
Me
H
HO
O
O
O
O
O
O
O
O O
O O
HO
Me
Me
MeMe
Me
MeMe
O
CHO
Monensin A
Erythromycin A
Brevetoxin B
OO
O
O
OOEt
OH
H OMe
O
OOOMe
OMeHO
H
H
Avermectin A1a
O
O OHO
HO
MeO
N O
OO
OMe
OH
OMe
S
N
O
O OOH
O
OH
Epothilone A
FK506
OMe Me Me
OH O
HOOC
OH
OH O OH OH OH O O
O O
NH2
OMe
HONH2HOCandicidin
36
The biosynthesis of polyketides involves, typically, 1) assembly of the carbon backbone by a polyketide synthase (PKS) multienzyme complex; 2) so-called tailoring reactions, which may involve, oxidation, methylation, glycosylation etc. of the carbon backbone. Methylation reactions can occur, and require the use of the coenzyme S-adenosyl methionine (SAM):
As a typical example of an aliphatic, or reduced, polyketide, consider the macrolide antibiotic erythromycin A. The building blocks needed for the assembly of the antibiotic can be identified by labelling experiments. The first free intermediate on the pathway, however, is 6-deoxyerythronolide B. Thereafter, multiple "tailoring reactions" finally lead to the natural product:
An important question is how does the PKS function? Here a poly-ß-ketide is not produced. After most coupling reactions using methylmalonyl-CoA the resulting ß-ketothioester must be reduced (hence "reduced polyketide"). But in some cases an alcohol is left in the polyketide chain, sometimes a fully saturated unit is formed (e.g. at C7), but sometimes the ß-keto group is not reduced (C9). How are these steps controlled, or programmed ? (reviewed in J. Biol. Chem. 2010, 285, 27517). The construction of the backbone is catalyzed by a multienzyme complex. The PKS catalyzes overall, the following process:
OO O O
O
NaOOC Me
*MeO Me
HO
MeMe
Me
Me
Me
Me
HOHO
Acetate
Propionate
Butyrate
*Methionine
O N
N
N
N
NH2
OH OH
SHOOC O
Me
NH2N
N
N
N
NH2
OH OH
SHOOC
NH2
S-Adenosylmethionine
⊕
O
O
O
OH
Me
Me
Me
Me
Me
OH
OH
MeMeOH
O
Me
OMe
OHMe
Me
Me
OMe
OO
MeO
HO NMe2
Me
O
OMe
MeOH
Me
H
HO
Erythromycin A
eryA eryB, C, D, G, HPropionyl-CoA+
6 Methylmalonyl-CoA
6-Deoxyerythronolide B
Propionyl-CoAMethylmalonyl-CoA
Et O
O
Me
OH
Me
O
Me Me
OH
Me
OH
MeEt
O
SCoAMeCOO
O
SCoA
6-Deoxyerythronolide B
NADH NAD+
PKS
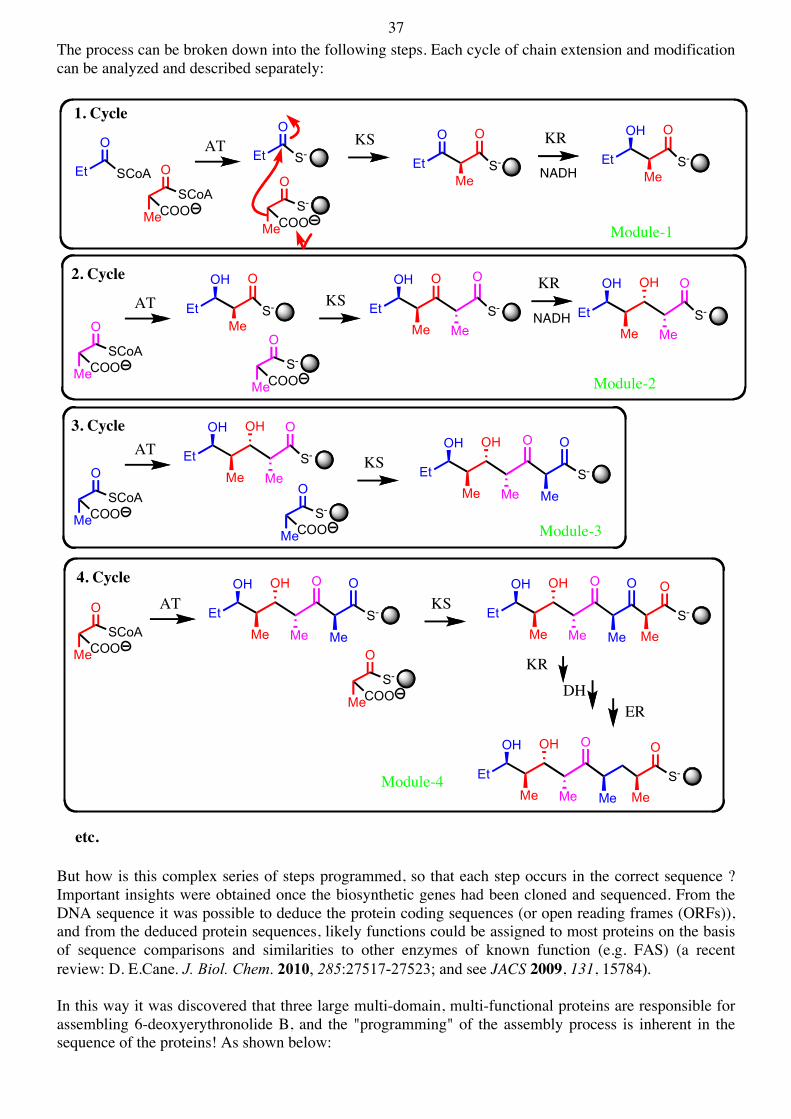
37 The process can be broken down into the following steps. Each cycle of chain extension and modification can be analyzed and described separately:
But how is this complex series of steps programmed, so that each step occurs in the correct sequence ? Important insights were obtained once the biosynthetic genes had been cloned and sequenced. From the DNA sequence it was possible to deduce the protein coding sequences (or open reading frames (ORFs)), and from the deduced protein sequences, likely functions could be assigned to most proteins on the basis of sequence comparisons and similarities to other enzymes of known function (e.g. FAS) (a recent review: D. E.Cane. J. Biol. Chem. 2010, 285:27517-27523; and see JACS 2009, 131, 15784). In this way it was discovered that three large multi-domain, multi-functional proteins are responsible for assembling 6-deoxyerythronolide B, and the "programming" of the assembly process is inherent in the sequence of the proteins! As shown below:
Et
O
SCoA
MeCOO
O
SCoA
1. Cycle
Et
O
S-
MeCOO
O
S-
Et
O
Me
O
S- Et
OH
Me
O
S-
Module-1
2. Cycle
MeCOO
O
SCoA
MeCOO
O
S-
Et
OH
Me
O
S- Et
OH
Me
O O
MeS- Et
OH
Me
OH O
MeS-
3. Cycle
MeCOO
O
SCoA
MeCOO
O
S-
Et
OH
Me
OH O
MeS-
Et
OH
Me
OH O
Me
O
S-
Me
4. Cycle
MeCOO
O
SCoAEt
OH
Me
OH O
Me
O
S-
Me
MeCOO
O
S-
Et
OH
Me
OH O
Me
O
Me
O
S-
Me
ER
Et
OH
Me
OH O
Me Me
O
S-
Me
etc.
Module-2
Module-3
Module-4
AT KS KR
NADH
AT KSKR
NADH
ATKS
AT KS
KR
DH
38
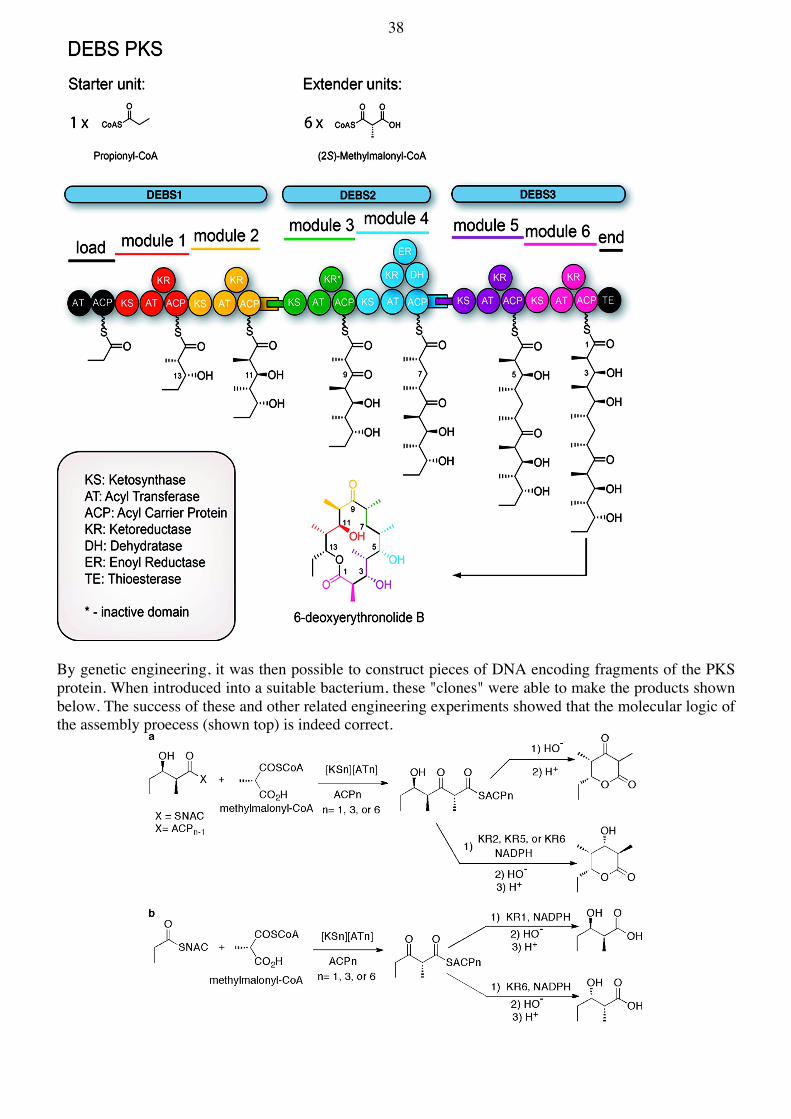
By genetic engineering, it was then possible to construct pieces of DNA encoding fragments of the PKS protein. When introduced into a suitable bacterium, these "clones" were able to make the products shown below. The success of these and other related engineering experiments showed that the molecular logic of the assembly proecess (shown top) is indeed correct.
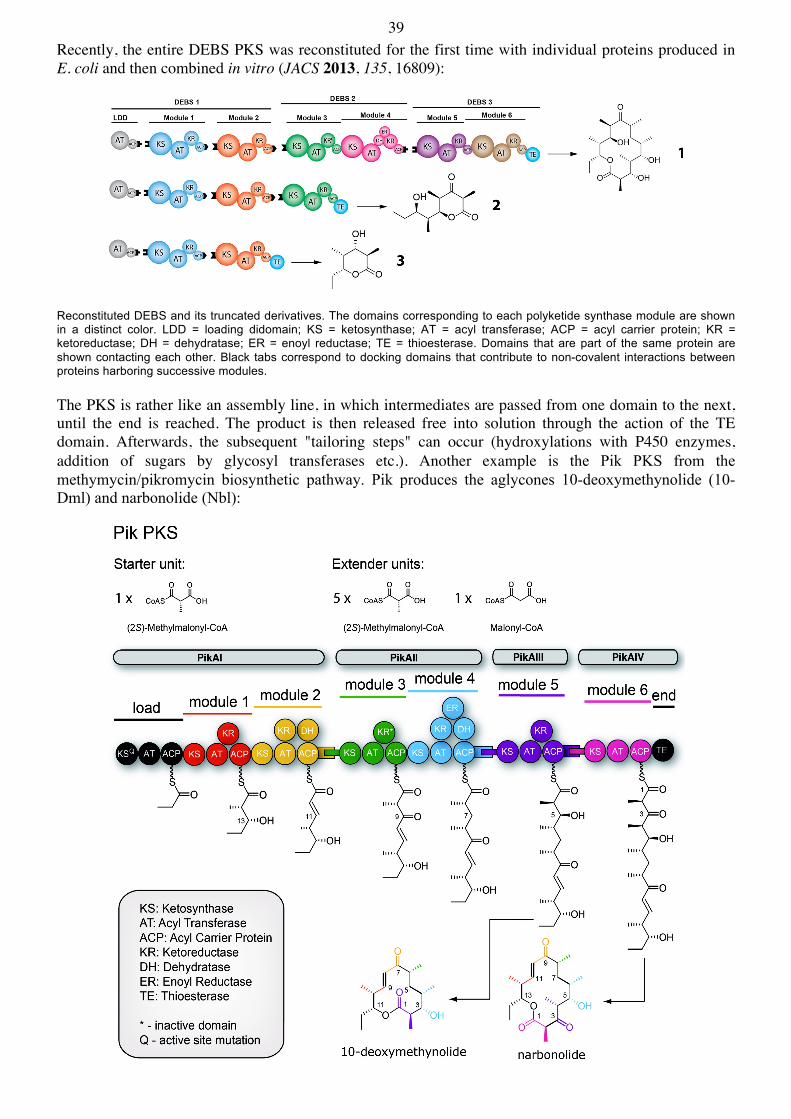
39 Recently, the entire DEBS PKS was reconstituted for the first time with individual proteins produced in E. coli and then combined in vitro (JACS 2013, 135, 16809):
Reconstituted DEBS and its truncated derivatives. The domains corresponding to each polyketide synthase module are shown in a distinct color. LDD = loading didomain; KS = ketosynthase; AT = acyl transferase; ACP = acyl carrier protein; KR = ketoreductase; DH = dehydratase; ER = enoyl reductase; TE = thioesterase. Domains that are part of the same protein are shown contacting each other. Black tabs correspond to docking domains that contribute to non-covalent interactions between proteins harboring successive modules. The PKS is rather like an assembly line, in which intermediates are passed from one domain to the next, until the end is reached. The product is then released free into solution through the action of the TE domain. Afterwards, the subsequent "tailoring steps" can occur (hydroxylations with P450 enzymes, addition of sugars by glycosyl transferases etc.). Another example is the Pik PKS from the methymycin/pikromycin biosynthetic pathway. Pik produces the aglycones 10-deoxymethynolide (10-Dml) and narbonolide (Nbl):
40 Another interesting example is seen in the pathway to the polyether antibiotic monensin A. The monensin PKS assembles the reduced polyketide chain shown below. Note the positions and configurations of the double bonds !
Three monooxygenases then act to produce a triepoxide. It has been shown that four 18O atoms are incorporated into monensin, when the producing bacterium is grown under 18O2. The sites of labelling were determined by 13C NMR spectroscopy. The sites of labelling are consistent with the following cascade cyclization process:
Each cyclization steps occurs stereospecifically with inversion of configuration. The configuration of the triepoxide shown then leads to the correct relative and absolute configuration in monensin A. Similar schemes can be drawn to account for the formation of several of the ether rings (shown in blue) in other polyether antibiotics, such as those shown below, as well as in the complex marine natural product brevetoxin (see page-96):
Me
HO OHMe
Me Me
HOMe
CO2-
CO2-
CO2-
Me
Me
MeMe
Enz-S-OC
OMe
O
SCoA
SCoA
MeOSCoA
O
Et SCoA
O
O
Polyketid-SynthaseMultienzym-Komplex
Me
MeOMeO
O O OO
NaO2C
Me
Me
HO
Me
Me
Me
Me
HOHO
Me
HO OHMe
Me Me
HO
MeMe
Me Me
X.OC
OMe
OO
O
O
Monensin
O2
COOH
O
Me
H HO
MeMe
H
OMe
O OHOHEt
Me
Et
O
Me
OH
Me
Et
Me
OH
Narasin
Me
OH
COOH
Me
OH
Me
O
Et
OO
Me
EtH
Et
Me
H
OH
Lasalocid A
OOOOOOMe
HOOC
Me
Me
MeH
OH
Me
Me H
O Me Me
H H
Me Me
OH OH
O
HMeO
Me
Dianemycin
OO O O
O
H
MeO
MeMe H
Me
H
Me
Me
HO Me
OMe
H H
H
HO
OMe
MeO
OHHOOC Me
OHMe
MeO Me
Septamycin
41 4. Biosynthesis of Natural Products Derived from Shikimic Acid 4.1. Phenyl-Propanoid Natural Products (C6-C3) The biosynthesis of the aromatic amino acids occurs through the shikimic acid pathway, which is found in plants and microorganisms (but not in animals). We (humans) require these amino acids in our diet, since we are unable to produce them. For this reason, molecules that can inhibit enzymes on the shikimate pathway are potentially useful as antibiotics or herbicides, since they should not be toxic for humans.
The aromatic amino acids also serve as starting materials for the biosynthesis of many interesting natural products. Here we will focus on the so-called phenyl-propanoide (C6-C3) natural products, e.g.:
4.2. Shikimic acid biosynthesis The shikimic acid pathway starts in carbohydrate metabolism. Given the great social and industrial significance of this pathway, the enzymes have been intensively investigated. Here we will focus on the mechanisms of action of several key enzymes in the pathway. The following Scheme shows the pathway to shikimic acid:
NH3
COO
R NH
NH3
COO
TryptophanR = H PhenylalanineR = OH Tyrosine
OHO
OH
OH
O a Flavone
ORO
OR
Ar
OH
Anthocyanine
OHO
OH
OH
O a FlavonolOH
O OHO
Umbellierfone a Coumarin)
MeO
HO
OH OH
OOH
OH
O
O
OH
OMeMeO
HO
WoodPolymerization
Cinnamyl alcohol
COOHOH
Cinnamic acid(Zimtsäure)
OHHO
OH
OH
OChalcone
OHO
OH
OH
O a Flavonone
O
O
OHO
O
MeOOMe
OMePodophyllotoxin
COOH
HO
OH
OH
Shikimic acid
42
DAHP-Synthase At first sight this seems to be a straightforward Aldol-like reaction between phosphoenolpyruvate (PEP) und erythrose-4-phosphate. However, for unknown reasons, Nature has made this more complicated than it appears:
Experiments with 18O-labelled PEP have shown that all of the 18O label is lost with phosphate - none is incorportated into the aldol-product. Other labelling experiments with Z-[3-3H]-PEP have shown that the reaction proceeds stereospecifically, even with respect to the new prochiral center in the product. The Si-face of the PEP must add to the Re-face der carbonyl group. A likely mechanism is :
3-Dehydroquinate Synthase This is a very interesting enzymic reaction. At first sight, it is not clear what the reaction mechanism is. The enzyme needs NAD+ as coenzyme, but this is not consumed during the reaction (no net redox change):
It was shown that when DAHP is labelled at C5 or C6 with 2H (deuterium), then a significant kinetic isotope effect on the reaction rate can be observed (i.e. slower with the deuterated substrates). This implies that both the C(6)-H and the C(5)-H bonds are cleaved during the reaction. The following mechanism was suggested:
This mechanism has been suggested, on the basis of studies carried out over many years. At first sight the enzyme appears to catalyze: 1) a redox reaction, 2) an elimination, 3) another redox reaction, 4) an aldol-like reaction. At least the chemical logic of oxidizing the alcohol group then becomes clear. How does one active site achieve all this ??
COO-
O
O
2-O3P-O
HOOH
COO-
O
OH
OH
HO
2-O3P-O
PO-O
-OHA
HB
H
COO-
O
OH
2-O3P-O
HOOH
PO-O
-OHA
HB
H
H
OO
COO-
O
OHOH
HO
2-O3P-O
COO-
O OHOH
HO
DHQ-Synthase
Dehydroquinate(DHQ)3-Deoxy-D-arabinoheptulo-
sonate-7-phosphate (DAHP)
1
2
456 NAD+
O
HO
HOOC
OHO-P
H
O HO
HO
HOOC
OHO
O
H
O
HO
HOOC
OH
O
O
HO
HOOC
O
H
OH
H
-O
HO
HOOC
O
H
OHHO
HOOC
OHO
H
O H
P-OO-
O
DHQ
NAD+ NADH
44 Modifications to the phosphate at C-7 have a dramatic effect on rate, suggesting that it plays an active role in the elimination step. It is known that the labelled substrates 7S- und 7R-[7-3H]-DAHP are converted into labelled products with overall inversion of configuration at C7. So the C-C bond-forming step also proceeds stereospecifically (Proc. Natl. Acad. Sci.USA 1970, 67, 1669). In a model study, however, it was also shown that the the aldol-like reaction can proceed rapidly and also stereospecifically without catalysis by the enzyme (JACS, 1988, 110, 1628):
Apparently, the steps that really need the catalytic action of the enzyme, in order to achieve rapid turnover, are those involving the redox changes (alcohol ketone) with the coenzyme NAD. The catalytic power of the enzyme appears to be focused on making these steps fast, and perhaps is less crucial for providing catalysis for the elimination and aldol-like reactions, which proceed fast anyway if the substrate is bound in an optimal conformation. EPSP-Synthase The sixth step in shikimic acid biosynthesis is the EPSP-synthase reaction. This enzyme has been intensively investigated, not least because it is the target of the commercially important herbicide Glyphosate, which inhibits the enzyme :
Glyphosate is effective in killing a wide variety of plants, including grasses, broadleaf, and woody plants. It has a relatively small effect on some clover species. By volume, it is one of the most widely used herbicides. It is commonly used for agriculture, horticulture, and silviculture, as well as garden maintenance (including home use). Some crops have been genetically engineered to be resistant to glyphosate. Glyphosate was first sold by Monsanto under the tradename "Roundup". Mechanism of the EPSP synthase reaction ? -- the phosphate group is lost from PEP with cleavage of the C-O bond, not the P-O bond. -- If the enzymic reaction is carried out in D2O, then deuterium is incorporated into the product, and is
found equally distributed between the E- and Z-positions in the enolpyruvyl group. -- If [3-2H2]PEP is used as substrate in H2O then 2H is lost in equal amounts from the E- und Z-
positions in the enolpyruvyl group in the product. These observations have led to the proposal of an addition-elimination sequence, as shown below:
In one key experiment, the existence of the tetrahedral intermediate was proven. The enzyme (800µM) +S3P (800µM) + 2-[13C]-PEP (1mM) was mixed for 5s, and then quenched with Et3N. Ion exchange chromatography of the resulting products gave a small amount of the intermediate that could be characterized. Glyphosate is a potent inhibitor of EPSP synthase. The inhibition ist competitive with respect to PEP (Ki = 1µM) but non-competitive with respect to S3P (Eur. J. Biochem. 1984, 143, 351).
Crystallographic studies have revealed how the substrate, intermediate, and glyphosate bind at the active site of the enzyme. A substrate analogue Z-3-fluoro-PEP acts as a pseudosubstrate and forms a relatively stable tetrahedral intermediate that could be crystallized on the enzyme (Mol. Microbiol. 2004, 51, 963).
Chorismate Mutase The chorismate mutase reaction involves formally a Claisen rearrangement. This reaction occurs at a measurable rate in aqueous solution even in the absence of the enzyme (t1/2 in water at 50oC ≈ 90 min), but the reaction is accelarated about ≈106 fold by the enzyme :
COO
2-O3P-O OH
OH
COO
2-O3P-O
COO
2-O3P-O OOH
CH2
COOEPSP-Synthase
OPO32
H
E + S ES E + P
EI E + S ES E + P
EI + S ESI
E + S ES E + P
ESI
46
The enzymic and the spontaneous reactions could proceed through either chair-like or boat-like transition states. The stereochemical consequences, however, are different:
The stereochemical course of both enzymic and spontaneous reactions has been studied, and both have been shown to proceed through chair-like transitions states (JACS, 1984, 106, 2701; JACS 1985, 107, 5306). Other kinetic and spectroscopic studies have shown that the enzymic reaction most likely is a more-or-less concerted pericyclic reaction. The slowest step appears to be release of product (prephenate) from the enzyme (Biochemistry, 1990, 29, 8872). Prephenate dehydrogenase and prephenate dehydratase The conversion of prephenate to p-hydroxyphenylpyruvate is catalyzed by the enzyme prephenate dehydrogenase, which requires NAD+. Kinetic isotope studies have suggested that the reaction proceeds in a concerted manner, as shown below :
Finally a transaminase (PLP-dependent) converts the α-ketoacid into the amino acid tyrosine. For the production of phenylalanine, the enzyme prephenate dehydratase produces first phenylpyruvate, and then again by transamination the amino acid Phe :
HOOCO
COOH
OH
COO-
OOH
COO-
Prephenic acidChorismate
ChorismateMutase
HO
COO-
O
COO-
OCOO-
COO-
OH
O-OOC
COO-
OH
O
COO
COO-
OH
COO-
OH
O
COO-
boat-like TS
chair-like TS
OOC
HO H
O
COOH COOH
OHO
N R
H2NOC
Prephenate dehydrogenase
NADHNAD+
OOC
HO H
O
COO COOH
O
Prephenatedehydratase
COOHTransaminase
NH2
47 Chorismate also plays a key role as precursor to several other very important natural products, including the amino acids tryptophan, p-aminobenzoic acid, as well as p-hydroxybenzoic acid and salicyclic acid. 4.3. Coumarins, Flavonoids und Lignans Phenylalanine and tyrosine also act as precursors to a large variety of C6C3-Phenylpropanoide natural products in plants:
Two interesting coumarin derivatives are dicumarol und warfarin, which can prevent blood clotting and are used clinically to treat thrombosis :
Flavonoids and stilbenes are products from a pathway that uses cinnamoyl-CoA as starter unit, and extends the chain with malonyl-CoA extender units, just like in polyketide biosynthesis. Flavonoids such as Quercitin (in red wine) and catechins (in tea) can act as anti-oxidants. Flavonoids contribute to plant flower colours; yellow from chalcones and flavonols; red, blue and violet from anthocyanidins. Many of these are also found in glycosylated forms in plants. Resveratrol (red wine) has recently been shown to promote longevity in animals:
Cinnamic acid is also used for the biosynthesis of lignin. Apart from cellulose, lignin is the main component of wood. Lignin is a high molecular weight polymeric material, produced by polymerization of coniferyl alcohol.
OH
O
XNH2
COOH
cinnamic acid
HO
MeO OHconiferyl alcohol
HO
OH
p-coumaryl alcohol
O O O
OH HO
O O O
OH OPh
WarfarinDicumarol
O O
coumarin
OHO
OH O
OH
Naringenin(a flavanone)
OHO
OH O
OH
OH
OH
Quercetin(a flavonol)
OHO
OH
OH
OH
OH
Cyanidin(an anthocycanidin)
OH
HO
OH Resveratrol(a stilbene)
OHOMe
OH
O
O
OMeOH
HOOMe
(E)-Coniferyl alcohol
Pinoresinol
OH
OH
Tyrosine
NH2
O
MeO
O
HO
O
MeOO
OH
HO
OMeO
HO
OOMe
OHO
O
MeO
O
MeOO
OH
O
HO
O
OH
O
HO
MeO
OO
OMeMeO
OOH
OMeO
HO
O
HO
OH
O
OMe
MeO
OMeMeO
representative section of a molecule of lignin
48 Plant cell walls are complex structures composed mostly of lignocellulose — the most abundant organic material on Earth — a matrix of cross-linked polysaccharide networks, glycosylated proteins, and lignin. This matrix has three main components: cellulose (38–50%), hemicellulose (17–32%), and lignin (15–30%). Cellulose is a polysaccharide consisting of a linear chain of several hundred to more than 10,000 D-glucose units linked by β-1,4 bonds. This bonding motif differs from the α-1,4 glucose linkage of starch, such as corn starch that comes from corn kernels. This structural difference proves to be quite significant. Cellulose chains are linear and somewhat rigid, but starch takes on a coiled chain structure. That makes the cellulose chains amenable to forming numerous hydrogen bonds, which, unlike starch, leads the cellulose chains to assemble into cablelike bundles of crystalline fibrils that have high tensile strength and are resistant to hydrolysis to glucose. Hemicellulose is also a polysaccharide, but it is typically made up of chains of xylose interspersed with side chains containing arabinose, galactose, mannose, glucose, acetyl, and other sugar groups, depending on plant type. Hemicellulose contains 500 to 3,000 sugar units and includes a small amount of pectin, another polysaccharide, with which it forms a cross-linked network. Lignin is a cross-linked macromolecule composed of three types of substituted phenols (phenylpropanoids). It fills the spaces in the cell wall between cellulose, hemicellulose, and pectin and is covalently linked to hemicellulose. Lignin resembles a kind of phenol-formaldehyde resin that acts like glue to hold the lignocellulose matrix together. Lignin helps provide additional strength to cell walls and resistance to insects and diseases (C & E News, 2008, Dec. 8, p.15).
49 5. Biosynthesis of Alkaloid Natural Products 5.1. Alkaloids are derived from amino acids Nitrogen-containing compounds, with a slightly basic character, have been isolated from many different organisms, mostly plants and microorganisms, and are biosynthesized from amino acids - these are called alkaloids. There are probably over 10'000 known alkaloids, having very diverse structures. They can nevertheless be classified into families, on the basis of structural similarities and the amino acids that are used for their biosynthesis Some alkaloids are also produced using building blocks derived from other secondary metabolic pathways, such as terpenoids, polyketides and peptides. Some of the important classes of alkaloid are shown below:
NMe
O
N ON
N
MeN
O
OO
Ph
OHN
HO
NH
NH2
NH
N
COOMe
NH N
Me
N
MeOOCOH
N
NH
OAcOH
N
OH
HO
HONH2
N
MeO
MeOMeO
MeO
NMe
HO
O
HO
NHAc
OMe
OMeO
MeO
MeO
COOMe
MeOOC
NMe
N
OAcOH
MeOOC
OH
MeO
N-Methylpelletierine
Lycopodine
Scopolamine Retronecine
Dopamine
Papaverine
Colchicine
Catharanthine
Vindoline
Geissoschizine
Vinblastine
H3N
NH3
COO
Lysine
H3NNH3
COOOrnithine
NH3
COO
RPhenylalanineTyrosine
NH
NH3
COO
Tryptophan
N
Me O
MeHygrine
e.g. Pyrrolidine, Pyrrolizidine and Tropane Alkaloids
MeN
OTropinone
Sparteine
e.g. Piperidine, Pyridine und Quinolizidine Alkaloids
N
OH
Lupinine
NH
MeConiine N
NMe
Nicotine
z.B. Isoquinoline Alkaloids
NMe
MeO
HO
OMe
OMe
OH
Autumnaline
Morphine
z.B.Indole Alkaloids
Tryptamine+ Terpene
MeO
50 5.2. Benzylisoquinoline Alkaloids Of special interest within the family of isoquinoline alkaloids are those containing the 1-benzyl(tetrahydro)isoquinoline skeleton, which are found in many different plants. Studies on the biosynthesis of these compounds made progress as soon as radioactively labelled compounds (14C and 3H) became available. Potential precursors could be fed to intact plants, and later the natural prodicts could be isolated from the plants, and then analyzed chemically to detemine whether, and if so, where the radioactive labels had been incorporated. In this way, it was shown that the benzylisoquioline alkaloids are constructed from two molecules of tyrosine:
The formation of norcoclaurine is catalyzed by an enzyme, which in effect catalyzes a Pictet-Spengler-Reaction (see Angew.Chem.Int.Ed 2011,50,8538). The reaction shown actually occurs spontaneously in aqueous solution, but then slowly gives racemic product, whereas the enzymic reaction runs much faster and gives optically pure product:
Next, the norcoclaurine is converted into (S)-reticuline :
Reticuline is used for the biosynthesis of many other benzylisoquinoline alkaloids, amongst others, the so-called aporphine alkaloids, e.g.:
HO NH2
HO
NH
HO
HO
HO
HHO NH2
COOH
Tyrosin
HO
HO NH2
HO
CHOO COOH
Norcoclaurine
Decarboxylase(PLP)
Transaminase(PLP)
Hydroxylase
Decarboxylase(TPP)
HO
HO NH2
HO
CHO
NH
HO
HO
HO
H
Norcoclaurine
NH
HO
HO
HO
NH
O
HO
HO
H
NH
HO
HO
HO
H
Norcoclaurine
HydroxylaseN-Me
MeO
HO
Me-O
HHO
Reticuline
SAM SAM
SAM
NMe
MeO
HO
HO
MeO
H
NMe
MeO
HO
HO
MeO
H
NMe
MeO
MeO
MeO
MeO
H
Glaucine
51 An important step here is the formation of a direct aryl-aryl bond. This occurs in an oxidative phenol coupling reaction. Nature has evolved a series of hemoproteins of the cytochrome P450 family that catalyze specific oxidative phenol coupling reactions (not hydroxylations, compare earlier). Such coupling reactions are well known in synthetic chemistry, where they can be carried out with phenolic compounds, under basic conditions, using K3Fe(CN)6 as oxidizing agent, e.g.:
Such reactions tend to produce mixtures of products, because the free radical intermediates can often couple in more than one way. The enzymes, however, catalyze only one pathway specifically. The mechanisms of the enzymic reactions are not well understood, but require molecular oxygen as well as the hemoprotein (P450). The oxidizing power of compound-I is used to drive the coupling reaction, e.g.:
Oxidative phenol coupling reactions are often found in alkaloid biosynthesis. Perhaps the best-known example occurs during the biosynthesis of morphine. Morphine is a highly-potent opiate analgesic drug and is the principal active agent in opium and the prototypical opioid. It is also a natural endocrine product in humans and other animals. Like other opiates, e.g., diacetylmorphine (heroin), morphine acts directly on the central nervous system (CNS) to relieve pain, and at synapses of the nucleus accumbens in particular. Studies done on the efficacy of various opioids have indicated that, in the management of severe pain, no other narcotic analgesic is more effective or superior to morphine. Morphine is highly addictive when compared to other substances; tolerance, physical and psychological dependences develop very rapidly. The word "morphine" is derived from Morpheus, one of the Greek gods of dreams. The opium poppy is Papaver somniferum.
NMe
MeO
HO
MeO
H
OH
NMe
MeO
O
MeO
H
O
NMe
MeO
O
MeO
H
O
NMe
MeO
O
MeO
H
O
NMe
MeO
HO
MeO
H
OH
2 FeIII
-2H+2 FeII
NMe
MeO
HO
HO
MeO
H+
ortho-para ortho-ortho
K3Fe(CN)6
NMe
MeO
O
MeO
H
O
HH
OH2
S-Cys
FeIII
0
P450 enzyme(resting state)
electrons+ O2 O
FeIV
S-Cys
OH HOOH
FeS-Cys
O HO
FeS-Cys
O O
+ H2O
H2Ocompound-I
52
(R)-Reticuline is an important intermediate in the biosynthesis of morphine, and is produced by racemization of (S)-reticuline in a redox process, as shown below:
Salutaridine is found as a minor alkaloid constituent in the opium poppy:
NMe
MeO
HO
HO
MeO
H
MeO
HO
MeOOH
N-Me
MeO
HO
MeOO
N-Me
(S)-Reticuline(R)-Reticuline
Salutaridine
oxid.Phenol-Coupling
Oxid.
Red.
MeO
O
MeO
N-Me
HO
O
HO
N-Me
MeO
O
HO
N-Me
MeO
HO
MeO
N-Me
MeO
HO
MeO
N-Me
O OH
MeO
O
O
N-Me
Codeine Morphine
Thebaine
Reduction
Salutaridinol
Acetyl-CoA
AcOHCoASH
Neopinone
MeO
O
O
N-Me
OH
MeO
O
O
N-Me
Codeinone
53 The biosynthesis of morphine in the opium poppy was one of the first alkaloid pathways to be elucidated with the aid of 14C-labelled precursors. It was shown that [2-14C]-tyrosine is incorporated into morphine, with the 14C label appearing at the positions indicated above. This was proven, by degrading the 14C-labelled morphine in the following way:
Another interesting benzylisoquinoline alkaloid is colchicine. Colchicine was originally extracted from plants of the genus Colchicum (Autumn crocus, Colchicum autumnale, also known as the "Meadow saffron"). Originally used to treat rheumatic complaints and especially gout, it was also prescribed for its cathartic and emetic effects. Its present medicinal use is mainly in the treatment of gout; it is also being investigated for its potential use as an anti-cancer drug. Colchicine inhibits microtubule polymerization by binding to tubulin, one of the main constituents of microtubules. Tubulin is essential for mitosis, and therefore colchicine effectively functions as a "mitotic poison" or spindle poison. Since one of the defining characteristics of cancer cells is a significantly increased rate of mitosis, this means that cancer cells are significantly more vulnerable to colchicine poisoning than are normal cells. However, the therapeutic value of colchicine against cancer is (as is typical with chemotherapy agents) limited by its toxicity against normal cells. In 2008, the Botanic Gardens Conservation International (representing botanic gardens in 120 countries)
stated that "400 medicinal plants are at risk of extinction, from over-collection and deforestation, threatening the discovery of future cures for disease." These included Yew trees (the bark is used for the cancer drug taxol (paclitaxel)); Hoodia (from Namibia, source of weight loss drugs); half of Magnolias (used as Chinese medicine for 5,000 years to fight cancer, dementia and heart disease); and Autumn crocus (for gout). The group also found that 5 billion people benefit from traditional plant-based medicine for
health care. Early labelling experiments showed that tyrosine and phenylalanine are required for colchicine biosynthesis, and that autumnaline is a key intermediate. However, the Phe provides a C6C3 unit rather than a C6C2 fragment:
HO
O
HO
N-Me
Morphine
1) MeI / K2CO3 / MeOH2) Ag2O, then pyrolysis
MeO
O
HO
NMe2
EtONa / EtOH, Δ
MeO
HO
EtONMe2
+
1) Ac2O2) CrO3
MeO
AcO O
O
1) H2O22) NaOH/H2O
3) H3O
COOH
MeO
O
O
MeO
O
O
H2SO4Δ
NaOH/Me2SO4 MeO
MeO
HOOC
heat/ H+MeO
MeO + CO2
54
The seven membered tropolone ring was shown by labelling experiments to originate by ring expansion of the tyrosine-derived aromatic ring, including the adjacent benzylic carbon atom.
O-Methylandrocymbine has been isolated from Androcymbium melanthioides. The later steps have not been proven, but may involve the following reactions:
NHAc
OMe
OMeO
MeO
MeO
Colchicum
Colchicine
Tyrosine
Phenylalanine
NMe
MeO
HO
MeO
MeOOH
H
(S)-Autumnaline
NH2HO
COOH
COOHH2N
MeON-Me
MeO OH
MeO
HO
NH2
CHO
HO
HO
OH
NH
HO
HO
OH
H
(S)-Autumnaline
Phenylalanine
Tyrosine
Dopamine
cf. above
N
HO
HO
OH
MeON-Me
MeO O
MeO
HO
Isoandrocymbine
NH
OMe
OMeO
MeO
MeO
MeONH-Me
MeO O
MeO
MeO
MeONHMe
MeO O
MeO
MeO
NH-Me
OMe
OMeO
MeO
MeO
H
MeON-Me
MeO O
MeO
MeO
O-Methylandrocymbine
Colchicine
Oxidation
HO
OMe
HCHO
Demethylation
Acetylation
55 Various types of alkaloids are encountered in the daffodil family, called the Amaryllidaceae alkaloids (Amaryllidaceae is the botanical name of a family of flowering plants. The plants are herbaceous perennials that grow from bulbs, often with showy flowers). The Amaryllidaceae family includes
Amarylis, Narcissus and Galanthus, and the alkaloid content of bulbs from most members makes them toxic. However, galanthamine from daffoldils and snowdrops is currently an important drug for the treatment of the symptoms of Alzheimer's disease. The natural sources of galanthamine are certain species of daffodil and because these species are scarce and because the isolation of
galanthamine from daffodil is expensive (a 1996 figure specifies 50,000 US $ per kilogram; the yield from daffodil is 0.1-0.2% dry weight) alternative synthetic sources have been developed. Galanthamine acts as a competitive inhibitor of acetylcholinesterase, and enhances cognitive functions by raising acetylcholine levels in brain areas lacking cholinergic neurons. It does not cure the condition, but merely slows the rate of cognitive decline. Phe and Tyr are again the starting materials used for the biosynthesis of the Amaryllidaceae alkaloids:
Thereafter, three different modes of phenol coupling are seen:
L-Phe
HO
HO CHO
H2N
OH
L-Tyr NH
HO
HO
HO
Norbelladine
SAM
NH
MeO
HO
HO
4'-O-methylnorbelladine
NH
MeO
HO
HO
N
MeO
HO
HO
N
HOOH
O
O
Norpluvine
Lycorine
NH
MeO
HO
OH
para-ortho-coupling
NH
MeO
HO
O
N
MeO
HO
O
N
MeO
HO
OMeOHOxocrine
Haemanthamine
para-para-coupling
OH
NMe
HO
MeO
O
NMeHO
MeO
ortho-para-coupling
NMeO
MeO
OH
Galanthamine
56 5.3. Indole Alkaloids (see Nat. Prod. Rep. 2006, 23, 532) The simplest representative of the indole alkaloids are the natural amines tryptamine und serotonin, which are biosynthesized from the amino acid tryptophan (Trp):
Serotonin is a monoamine neurotransmitter synthesized in serotonergic neurons in the central nervous system (CNS), and enterochromaffin cells in the gastrointestinal tract of animals including humans. Serotonin is also found in many mushrooms and plants, including fruits and vegetables. Serotonin is believed to play an important role as a neurotransmitter, in the modulation of anger, aggression, body temperature, mood, sleep, sexuality, and appetite as well as stimulating vomiting. The vinca alkaloids are a very interesting class of indole alkaloids, and include vinblastine, vincristine, vindesine and vinorelbine. These alkaloids are produced by plants of the genus Catharanthus. Catharanthus (Madagascar Periwinkle) is a genus of eight species of herbaceous perennial plants, seven endemic to the island of Madagascar, the eighth native to the Indian subcontinent in southern Asia. One
species, C. roseus, has been widely cultivated, and after introduction has become an invasive species in some areas. C. roseus has also gained interest from the pharmaceutical industry; the alkaloids vincristine and vinblastine from its sap have been shown to be an effective treatment for leukaemia. Although the sap is poisonous if ingested, some 70 useful alkaloids have been identified from it. In Madagascar, extracts have been used for hundreds of years in herbal medicine for the treatment of diabetes, as hemostatics and tranquilizers, to lower blood pressure, and as disinfectants. The extracts are not without their side effects, however, which include loss of hair.
The structures of these alkaloids reveal that not only Trp is required for the biosynthesis. A C10 fragment is also needed, and is provided from terpene metabolism. Strychnine biosynthesis also incorporates one acetate unit (in red above). The important C10 fragment is produced from geraniol, and is called secologanin:
N
NH3
COO
HN
NH2
H
RR = H TryptamineR = OH Serotonin
NMe
N
Vindoline
H
H
NH
N
MeOOC OHStemmadenine
MeOOCOH
NH
N
COOMe
NH
N
MeOOCOH
NH
N
MeOOC
NMe
N
OAcOH
MeOOC
OH
MeO
Catharanthine
Geissoschizine
Vinblastine
N
O
N
O
H
HH
H
Strychnine H
OAcMeO
57
Secologanin is a glucoside, which can be cleaved by hydrolysis under acidic conditions:
The formation of the indole alkaloids begins with the condensation of tryptamine and secologanin, catalyzed by strictosidine synthase (STR, see below) (compare with Pictet-Spengler reaction):
Strictosidine is then a key intermediate in the formation of over 1000 different indole-terpene alkaloids.
Geraniol
OH O
MeHO
MeOOC
O-Glucose
H
H
Loganin
OMeOOC
CHOO-Glu
Secologanin
OMeOOC
CHOO
OHO
OHOH
OH H3O
OMeOOC
CHOO-Glu
NNH2
HN
NH
H
OMeOOC
O-Glu
NNH
H
OMeOOC
OGlu
Strictosidine
58 For example, the Corynanthe alkaloids:
Yohimbine is the principal alkaloid of the bark of the West-African evergreen Pausinystalia yohimbe Pierre (formerly Corynanthe yohimbe), family Rubiaceae (Madder family). There are 31 other yohimbane alkaloids found in Yohimbe. In Africa, yohimbine has traditionally been used as an aphrodisiac. Yohimbine hydrochloride is a standardized form of yohimbine that is available as a prescription drug in the United States, and has been shown to be effective in the treatment of male impotence. Yohimbine hydrochloride has also been used for the treatment of sexual side effects caused by some antidepressants, female hyposexual disorder, as a blood pressure boosting agent in autonomic failure, xerostomia, and as a probe for noradrenergic activity. Ajmaline was first isolated from the roots of Rauwolfia serpentina, a species of flowering plant in the
family Apocynaceae. It is one of the 50 fundamental herbs used in traditional Chinese medicine, where it has the name shégēn mù (蛇根木) or yìndù shémù (印度蛇木). The extract of the plant has also been used for millenia in India — it was reported that Mahatma Gandhi took it as a tranquilizer during his lifetime. Ajmaline is a class Ia antiarrhythmic agent, a group of pharmaceuticals that are used to suppress fast rhythms of the heart (cardiac arrhythmias), such as atrial fibrillation, atrial flutter, ventricular tachycardia,
NNH
H
OMeOOC
O-Glu
Glucose
NNH
H
OHCHO
MeOOC
NN
H
OHMeOOC
N
MeOOCO
NN
H
MeOOCOH
H
Yohimbine
NN
H
OHMeOOC
NN
H
OHMeOOC
H
H
Geissoschizine
NADPH
NN
H
OMeOOC
NADPH
NN
H
OMeOOC
Me
Ajmalicine
H
H
H
AcetalH
NN
H
MeOOCOH
H
H
2 NADPH
(Imine reduction)
59 and ventricular fibrillation. Ajmaline functions by blocking Na-channels in cell membranes. Rauwolfia caffra is the South African quinine tree. Rauwolfia serpentina, or Indian Snakeroot or Sarpagandha, contains a number of bioactive chemicals, including ajmaline, deserpidine, rescinnamine, serpentinine, and yohimbine. Reserpine is an alkaloid first isolated from R. serpentina, and was widely used as an antihypertensive drug. It had drastic psychological side effects and has been now replaced by blood-pressure-lowering drugs that lack such adverse effects. But in herbal use it is a safe and effective resource for hypertensive patients. The pharmaceutical companies have stopped producing this drug as reserpine or deserpedine. It is only available currently in the U.S. as a herbal medicine over the Internet. The pathway to ajmaline has been well documented, although few mechanistic studies have been reported so far on the biosynthetic enzymes:
Catharanthine is a member of the so-called iboga family of indole alkaloids. It is one of the many
alkaloids present in Catharanthus roseus. It is produced along with many other Catharanthus alkaloids by factory farming in China. It can be used as a starting material for the synthesis of the anti-tumor drugs, vinblastine and vincristine. Vindoline (an Aspidosperma alkaloid) is another important component of the bis-indole alkaloids, typified by vinblastine and vincristine, also produced by C. roseus. Some of the biosynthetic steps have been documented, but the enzymes have not yet been studied in detail. A fascinating proposal was made to explain how catharanthine and vindoline might be produced from
geissoschizine. Tabersonine is a known intermediate, and the steps from tabersonine have been established; the rest is hypothetical -
NNH
H
OMeOOC
OGlu
Strictosidine
NN
H H
Dehydrogeissoschizine
OHCCOOMe
NN
H H
OHCCOOMe
MeOHCO2
NN
H H
H
N N
H
AcO
N N
H
AcO
OH
N NH
H
AcO
OHH
H
NADPH
N NMe H
HO
OHH
H
SAM
Polyneuridine Aldehyde
16-epi-vellosimineVomilenine
DihydrovomilenineAjmaline
N N
H
AcO
OHHH
17-O-Acetylnorajmaline
Vinorine
Acetyl-CoA
NADPH
see above
O
H
Oxidation
Reduction
Reduction
Hydrolysis
60
Vinblastine and vincristine are anti-mitotic drugs used to treat certain kinds of cancer, including Hodgkin's lymphoma, non-small cell lung cancer, breast cancer and testicular cancer. They bind to tubulin, thereby inhibiting the assembly of microtubules. They are M phase cell cycle specific, since microtubules are a component of the mitotic spindle and the kinetochore, which are necessary for the separation of chromosomes during anaphase of mitosis. Toxicities include bone marrow suppression
NN
H
CHOMeOOC
H
H
Geissoschizine
NN
H
CHOMeOOCH
N
N
CHOMeOOC
HN
N
MeOOCCHOH
N
N
MeOOC CH2OH
N
N
MeOOC CH2OHH
N
N
COOMeTabersonine
N
N
COOMeHO
N
N
COOMeMeO
HH
Me HO
16-Hydroxytabersonine
Desavetoxyvindoline
N
N
COOMeMeO
MeHO
OH
Deacetylvindoline N
N
COOMeMeO
MeHO
OCOCH3
Vindoline
+ H2O
2 x SAM
Hypothetical
NN
H
CHOMeOOC
H
H
H
Redoxchanges
NH
N
COOMeCatharanthine
N
N
MeOOC
H
N
N
MeOOCH
Oxidation
Acetyl-CoA
Oxidation
preakuammicine
stemmadenine
N
N
MeOOC CH2OHH
dehydrosecodine
NADH Hypothetical
61 (which is dose-limiting), gastrointestinal toxicity, potent vesicant (blister-forming) activity, and extravasation injury (forms deep ulcers). The coupling of catharanthine and vindoline can be catalyzed by a relatively non-specific peroxidase (a hemoprotein). It is possible that a similar enzyme specifically catalyzes this coupling in C. roseus.
Vinblastine is only present at low levels in C. roseus (0.0002% of dry leaf wt). Over 500 kg of catharanthus is needed to produce 1g of pure vincristine. Much effort has been put into the synthesis of the dimeric alkaloids, starting from the monomers, which can be isolated from the plant in much higher yields. One example is shown below:
NH
N
MeOOC
NMe
N
OAcOH
MeOOC
MeO
Vinblastine (R = Me)Vincristine (R = CHO)
NH
N
COOMeCatharanthine
NH
N
COOMe
PeroxidaseH2O2
HO
NH
N
COOMe
N
N
COOMeMeO
MeHO
OCOCH3
Vindoline
NH
N
MeOOC
NMe
N
OAcOH
MeOOC
MeO NH
N
MeOOC
NR
N
OAcOH
MeOOC
OH
MeO
Reduction
[O]
Reduction
Coupling
62
(see also: JACS, 2008, 130, 420; JACS 2009, 131, 4904). Finally, note that strictosidine is also the precursor to the quinoline alkaloids, including the important anti-malarial drug quinine. But that is another story......