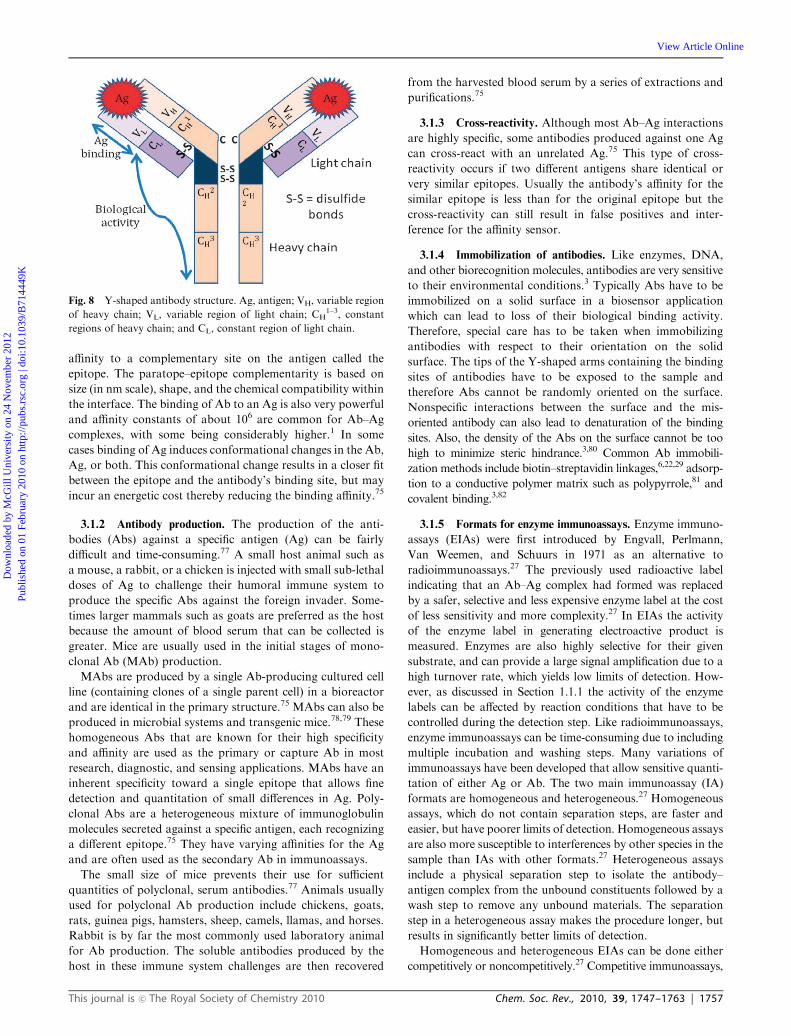
Electrochemical biosensors Niina J. Ronkainen,* a H. Brian Halsall b and William R. Heineman b Received 3rd November 2008 First published as an Advance Article on the web 1st February 2010 DOI: 10.1039/b714449k Electrochemical biosensors combine the sensitivity of electroanalytical methods with the inherent bioselectivity of the biological component. The biological component in the sensor recognizes its analyte resulting in a catalytic or binding event that ultimately produces an electrical signal monitored by a transducer that is proportional to analyte concentration. Some of these sensor devices have reached the commercial stage and are routinely used in clinical, environmental, industrial, and agricultural applications. The two classes of electrochemical biosensors, biocatalytic devices and affinity sensors, will be discussed in this critical review to provide an accessible introduction to electrochemical biosensors for any scientist (110 references). 1. Introduction 1.1 Background Sensors are devices that register a physical, chemical, or bio- logical change and convert that into a measurable signal. 1 The sensor contains a recognition element that enables the selective response to a particular analyte or a group of analytes, thus minimizing interferences from other sample components (Fig. 1). Another main component of a sensor is the transducer or the detector device that produces a signal. A signal processor collects, amplifies, and displays the signal. Electrochemical biosensors, a subclass of chemical sensors, combine the sensitivity, as indicated by low detection limits, of electrochemical transducers with the high specificity of bio- logical recognition processes. These devices contain a biological recognition element (enzymes, proteins, antibodies, nucleic acids, cells, tissues or receptors) that selectively reacts with the target analyte and produces an electrical signal that is related to the concentration of the analyte being studied. Electrochemical biosensors can be divided into two main categories based on the nature of the biological recognition process i.e. biocatalytic devices and affinity sensors. 2 Bio- catalytic devices incorporate enzymes, whole cells or tissue slices that recognize the target analyte and produce electro- active species. Special emphasis will be placed on enzyme electrodes for the detection of glucose, lactose, and xanthine. Affinity sensors rely on a selective binding interaction between the analyte and a biological component such as an antibody, Fig. 1 A schematic of a biosensor with electrochemical transducer. a Department of Chemistry, Benedictine University, 5700 College Road, Lisle, IL 60532-0900, USA. E-mail: [email protected]; Fax: +1 630 829 6547; Tel: +1 630 829 6549 b Department of Chemistry, University of Cincinnati, P.O. Box 210172, Cincinnati, OH 45221-0172, USA. E-mail: [email protected], [email protected]; Fax: +1 513 556 9239; Tel: +1 513 556 9274, +1 513 556 9210 Niina J. Ronkainen Niina J. Ronkainen received her BS in chemistry and biology at Butler University (Indianapolis, USA) in 1997 and her PhD at the University of Cincinnati (USA) in 2003 where she specialized in bio- analytical chemistry. From 2003–2004 she taught chemis- try as a visiting assistant professor at Tulane University (New Orleans, USA). In 2004 she joined Benedictine Univer- sity as an assistant professor of chemistry. She currently does basic research in bio- sensors and electrochemistry. She is an active member of the Chemical Education division of the American Chemical Society. H. Brian Halsall H. Brian Halsall is a professor of chemistry, and a member of the Sensors & Biosensors Group in the Department of Chemistry at the University of Cincinnati. He received a BSc (Hons) and PhD in chemistry at the Uni- versity of Birmingham, UK. This was followed by post- doctoral work at UCLA, after which he joined the staff of the MAN Program at Oak Ridge National Laboratory before settling in Cincinnati. His principal research interests include biosensors, electro- chemical immunoassay, and glycoprotein biochemistry. This journal is c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 1747–1763 | 1747 CRITICAL REVIEW www.rsc.org/csr | Chemical Society Reviews Downloaded by McGill University on 24 November 2012 Published on 01 February 2010 on http://pubs.rsc.org | doi:10.1039/B714449K View Article Online / Journal Homepage / Table of Contents for this issue
Transcript
Electrochemical biosensors
Niina J. Ronkainen,*aH. Brian Halsall
band William R. Heineman
b
Received 3rd November 2008
First published as an Advance Article on the web 1st February 2010
DOI: 10.1039/b714449k
Electrochemical biosensors combine the sensitivity of electroanalytical methods with the inherent
bioselectivity of the biological component. The biological component in the sensor recognizes its
analyte resulting in a catalytic or binding event that ultimately produces an electrical signal
monitored by a transducer that is proportional to analyte concentration. Some of these sensor
devices have reached the commercial stage and are routinely used in clinical, environmental,
industrial, and agricultural applications. The two classes of electrochemical biosensors,
biocatalytic devices and affinity sensors, will be discussed in this critical review to provide an
accessible introduction to electrochemical biosensors for any scientist (110 references).
1. Introduction
1.1 Background
Sensors are devices that register a physical, chemical, or bio-
logical change and convert that into a measurable signal.1 The
sensor contains a recognition element that enables the selective
response to a particular analyte or a group of analytes, thus
minimizing interferences from other sample components
(Fig. 1). Another main component of a sensor is the transducer
or the detector device that produces a signal. A signal processor
collects, amplifies, and displays the signal.
Electrochemical biosensors, a subclass of chemical sensors,
combine the sensitivity, as indicated by low detection limits, of
electrochemical transducers with the high specificity of bio-
logical recognition processes. These devices contain a biological
recognition element (enzymes, proteins, antibodies, nucleic
acids, cells, tissues or receptors) that selectively reacts with
the target analyte and produces an electrical signal that is
related to the concentration of the analyte being studied.
Electrochemical biosensors can be divided into two main
categories based on the nature of the biological recognition
process i.e. biocatalytic devices and affinity sensors.2 Bio-
catalytic devices incorporate enzymes, whole cells or tissue
slices that recognize the target analyte and produce electro-
active species. Special emphasis will be placed on enzyme
electrodes for the detection of glucose, lactose, and xanthine.
Affinity sensors rely on a selective binding interaction between
the analyte and a biological component such as an antibody,
Fig. 1 A schematic of a biosensor with electrochemical transducer.
aDepartment of Chemistry, Benedictine University, 5700 College Road,Lisle, IL 60532-0900, USA. E-mail: [email protected];Fax: +1 630 829 6547; Tel: +1 630 829 6549
bDepartment of Chemistry, University of Cincinnati,P.O. Box 210172, Cincinnati, OH 45221-0172, USA.E-mail: [email protected], [email protected];Fax: +1 513 556 9239; Tel: +1 513 556 9274, +1 513 556 9210
Niina J. Ronkainen
Niina J. Ronkainen receivedher BS in chemistry andbiology at Butler University(Indianapolis, USA) in 1997and her PhD at the Universityof Cincinnati (USA) in 2003where she specialized in bio-analytical chemistry. From2003–2004 she taught chemis-try as a visiting assistantprofessor at Tulane University(New Orleans, USA). In 2004she joined Benedictine Univer-sity as an assistant professorof chemistry. She currentlydoes basic research in bio-
sensors and electrochemistry. She is an active member of theChemical Education division of the American Chemical Society.
H. Brian Halsall
H. Brian Halsall is a professorof chemistry, and a member ofthe Sensors & Biosensors Groupin the Department of Chemistryat the University of Cincinnati.He received a BSc (Hons) andPhD in chemistry at the Uni-versity of Birmingham, UK.This was followed by post-doctoral work at UCLA, afterwhich he joined the staff of theMAN Program at Oak RidgeNational Laboratory beforesettling in Cincinnati. Hisprincipal research interestsinclude biosensors, electro-chemical immunoassay, andglycoprotein biochemistry.
This journal is �c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 1747–1763 | 1747
CRITICAL REVIEW www.rsc.org/csr | Chemical Society Reviews
Dow
nloa
ded
by M
cGill
Uni
vers
ity o
n 24
Nov
embe
r 20
12Pu
blis
hed
on 0
1 Fe
brua
ry 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B71
4449
KView Article Online / Journal Homepage / Table of Contents for this issue
such as enzymes, whole cells or tissue slices that recognize
the target analyte and produce electroactive species or some
other detectable outcome.2 Enzymes, globular proteins com-
posed mainly of the 20 naturally occurring amino acids that
catalyze biochemical reactions, are the oldest and still most
commonly used biorecognition element in biosensors.2,4
Enzymes can increase the rate of a reaction significantly
relative to an uncatalyzed reaction. The enzyme–substrate
interactions can be characterized by kinetic studies. Para-
meters such as origin and availability of the biological com-
ponent, its operational and storage stability as well as
immobilization procedure should be considered when preparing
a biocatalytic sensor.5 Also, sensitivity of the biorecognition
element to experimental conditions such as pH, temperature,
and stirring should be minimal and variation between measure-
ments should be as low as possible.3 Because of their complex
molecular structures, enzymes often have exquisite specificity
for their substrate molecule and can detect individual sub-
stances in a complex mixture, such as urine or blood, very
selectively. This removes the need for time-consuming, labor-
intensive, and interference-prone sample pretreatment and
separation steps used in composite methods. The arrangement
of amino acids at the active site of the enzyme, often found at
the centroid of the protein, bind with the specific sub-
strate making the enzyme selective for one type of substrate
molecule.4 Many enzymes also incorporate small nonprotein
chemical groups, such as cofactors or prosthetic groups, into
the structures of their active site that help determine substrate
specificity.4 The inherent selectivity of enzymes often circumvents
the signals produced by interfering species that are sometimes
found in complex samples. However, enzyme activity is often
further modulated by other components such as activators and
inhibitors.4 Researchers also had to find ways to manage the
enzyme adsorption that could lead to electrode fouling as well
as denaturation and loss of enzyme’s catalytic activity on the
electrode surface.5 Biocatalytic biosensors will be described in
more detail in Section 2.
Many biochemical analytes of interest are not amenable to
detection by enzyme electrodes due to the lack of sufficiently
selective enzymes being available for the analyte or the analyte
not being commonly found in living systems.1,5 That is when
affinity biosensors are considered as an alternative method.
1.1.2 Affinity biosensors. Affinity sensors use the selective
and strong binding of biomolecules such as antibodies (Ab),
membrane receptors, or oligonucleotides, with a target analyte
to produce a measurable electrical signal.2 The molecular
recognition in affinity biosensors is mainly determined by
the complementary size and shape of the binding site to the
analyte of interest.2 The high affinity and specificity of the
biomolecule for its ligand make these sensors very sensitive
and selective.1 The binding process such as DNA hybridization
or antibody–antigen (Ab–Ag) complexation is governed by
thermodynamic considerations.2
Immunosensors are Ab-based affinity biosensors where the
detection of an analyte, an antigen or hapten, is brought about
by its binding to a region of an Ab.6 The electrochemical
transducer responds to the binding event and converts the
electrical response to an output that can be amplified, stored,
and displayed. Complementary regions of the Ab bind to an
Ag that was used to produce the antibodies in a host organism
William R. Heineman
William R. Heineman is a Dis-tinguished Research Professorin the Department of Chemis-try at the University ofCincinnati. He received a BSin chemistry at Texas TechUniversity and a PhD at theUniversity of North Carolinain Chapel Hill and was a post-doctoral associate at CaseWestern Reserve Universityand The Ohio State Univer-sity. His research interestsinclude spectroelectrochemistry,electrochemical immunoassay,sensors, and bioanalytical
chemistry. He is a recipient of the Charles N. Reilley Awardin Electroanalytical Chemistry and the Torbern Bergman Medalfrom the Analytical Section of the Swedish Chemical Society.
1748 | Chem. Soc. Rev., 2010, 39, 1747–1763 This journal is �c The Royal Society of Chemistry 2010
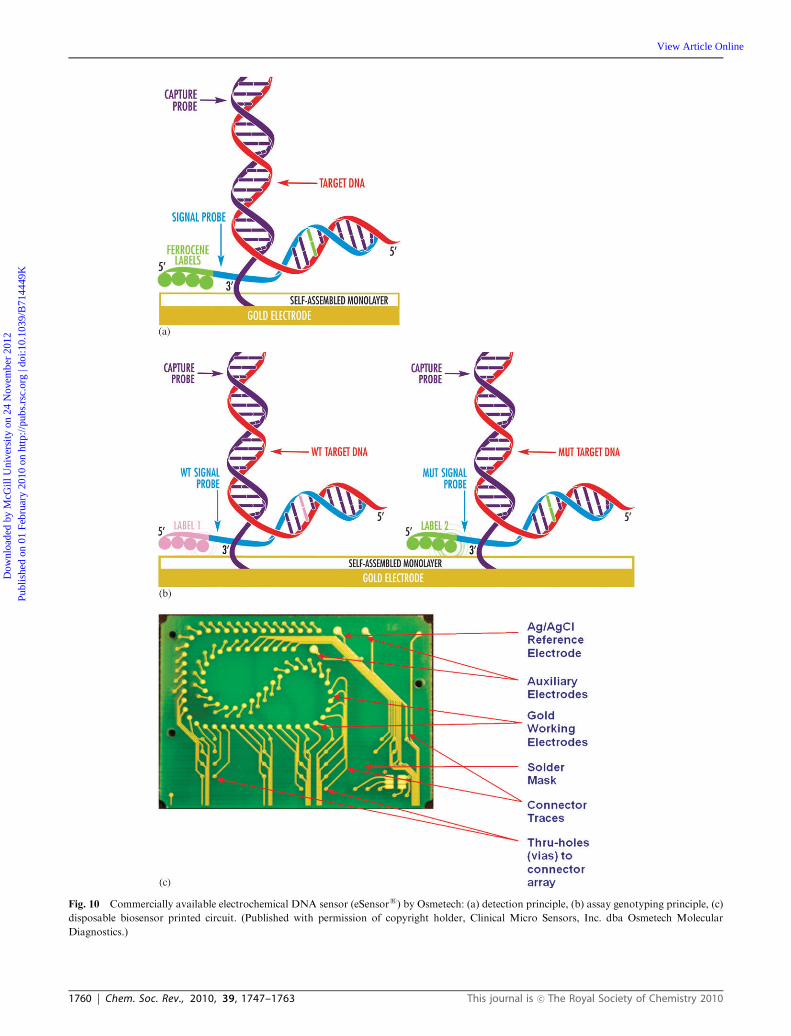
electrode to the sample results in hybridization between the
capture probe and the complementary strand of the target
DNA. The capture probe is designed to be shorter than the
complementary target strand, leaving a segment on the target
DNA where a signal probe containing an electroactive label
can bind. The label, ferrocene, is detected by measuring the
peak current for its oxidation by a positive potential scan in ac
voltammetry. The layer of alkane thiols is sufficiently thin as to
not interfere with the electrochemistry. The current is propor-
tional to the target DNA concentration in the sample. As
shown in Fig. 10B, genotyping can be done using different
ferrocene labels with distinguishable electrochemical poten-
tials for each label. The biochip consists of a microarray of
72 working electrodes, a Ag/AgCl reference electrode and two
auxiliary electrodes (Fig. 10C). Each working electrode of the
array can be interrogated independently which allows multiple
measurements to be made on the same chip. The chip is used
with a cartridge that features an auto-fill sample chamber,
microfluidic circulation to accelerate hybridization, and con-
tact with a resistive heating element. Osmetech has received
FDA clearance for eSensors
assays for detecting cystic fibrosis
carriers, and for identifying single-nucleotide polymorphisms
(SNPs) which result in increased sensitivity to warfarin, a
commonly prescribed blood anticoagulant.
3.3 Biosensors based on receptors
Receptors are proteins embedded in the cellular membrane
that specifically bind to their target analytes resulting in
physiological changes. The physiological response can be
opening ion-channels, producing second messenger systems,
or activating enzymes.1 A binding event at the receptor usually
causes the conformation of the receptor to change, which is
translated into an amplified electrochemical potential change.5
Unlike Abs that bind tightly with their complementary Ag,
receptors are like messengers that transmit signals upon ligand
binding between different parts of a biological system.1 Most
receptors are difficult to isolate and tend to bind to classes of
compounds having common chemical properties rather than
being highly specific for a given analyte like Abs.1,2 Therefore,
receptor-based biosensors are usually class-specific affinity
sensors that may not be a good feature for some biosensor
applications. Examples of receptor-based sensors include ion-
channel sensors where receptors in a lipid bilayer open or close
in response to a binding event with a ligand resulting in a rapid
ion flux through the membrane protein that causes a change in
the transmembrane conduction.109 The ion-channel membrane
proteins contain pores that allow ions such as Na+, K+ or
Ca2+ to flow through the channel until the potential difference
reaches equilibrium or the channel closes in response to a
stimulus. Also, nerve fibers from crayfish have been used to
monitor for local anesthetics and other drugs at low levels
(down to 10�15 M) with fast response times.110 Unfortunately,
these systems relying on axons from crayfish have a lifetime of
only 4 to 8 hours.
4. Conclusions
Catalytic and affinity biosensors with electrochemical detec-
tion continue to play an important role in many clinical,
environmental, industrial, pharmaceutical, defense, and
security applications due to their superior sensitivity and
selectivity. Although many electrochemical sensors are still
in the development and testing phases, some have reached
the consumer market as handheld devices, portable units used
for field measurements or are routinely used in a laboratory
setting. Recent developments in nanotechnology and mate-
rial science as well as being able to custom engineer the
biorecognition component will further push the develop-
ment of useful and reliable biosensor devices. The sometimes
limited shelf life and stability of the biorecognition component
as well as nonspecific binding continue to be the biggest
limitations of biosensors. However, many strategies have
helped with overcoming or minimizing these problems.
Notes and references
1 B. R. Eggins, Chemical Sensors and Biosensors, John Wiley &Sons, West Sussex, England, 2002.
2 J. Wang, Analytical Electrochemistry, John Wiley & Sons VCH,Hoboken, New Jersey, USA, 2006.
3 D. Grieshaber, R. MacKenzie, J. Voros and E. Reimhult,Sensors, 2008, 8, 1400.
4 R. A. Copeland, Enzymes, John Wiley & Sons VCH, New York,NY, USA, 2000.
5 Bioelectrochemistry Fundamentals, Experimental Techniques andApplications, ed. P. N. Bartlett, John Wiley & Sons, West Sussex,England, 2008.
6 N. J. Ronkainen-Matsuno, J. H. Thomas, H. B. Halsall andW. R. Heineman, TrAC, Trends Anal. Chem., 2002, 21(4),213.
7 C. G. Bauer, A. V. Eremenko, E. Ehrentreich-Forster, F. F. Bier,A. Makower, H. B. Halsall, W. R. Heineman and F. W. Scheller,Anal. Chem., 1996, 68(15), 2453.
8 S. H. Jenkins, W. R. Heineman and H. B. Halsall,Anal. Biochem.,1988, 168(2), 292.
9 C. A. Wijayawardhana, H. B. Halsall and W. R. Heineman,Electroanalytical Methods of Biological Materials, ed. A. Brajter-Toth, J. Q. Chambers, Marcel Dekker, New York, 2002,pp. 329–365.
10 H. Yao, S. H. Jenkins, A. J. Pesce, H. B. Halsall andW. R. Heineman, Clin. Chem. (Washington, D. C.), 1993, 39,1432.
11 Laboratory Techniques in Electroanalytical Chemistry, ed.P. T. Kissinger and W. R. Heineman, Marcel Dekker Inc, NewYork, NY, USA, 1996.
12 H. B. Halsall and W. R. Heineman, J. Int. Fed. Clin. Chem., 1990,2, 179–187.
13 M. Trojanowicz, M. Szewczynska and M. Wcislo, Electroanalysis,2003, 15(5–6), 347.
14 C. A. Wijayawardhana, S. Purushothama, M. A. Cousino,H. B. Halsall and W. R. Heineman, J. Electroanal. Chem.,1999, 468, 2.
15 C. A. Wijayawardhana, H. B. Halsall and W. R. Heineman, Anal.Chim. Acta, 1999, 399, 3.
16 R. Puchades and A. Maquieira, Crit. Rev. Anal. Chem., 1996,26(4), 195.
17 T. Jiang, H. B. Halsall and W. R. Heineman, J. Agric. FoodChem., 1995, 43, 1098.
18 R. M. Wightman and D. O. Wipf, Electroanalytical Chemistry,ed. A. J. Bard, Marcel Dekker, New York, USA, 1989,p. 267.
19 R. M. Wightman, Anal. Chem., 1981, 53(9), 1125A.20 J. Heinze, Angew. Chem., Int. Ed. Engl., 1993, 32, 1268.21 J. T. Thomas, N. J. Ronkainen-Matsuno, S. Farrell, H. B. Halsall
and W. R. Heineman, Microchem. J., 2003, 74, 267.22 S. Farrell, N. J. Ronkainen-Matsuno, H. B. Halsall and
W. R. Heineman, Anal. Bioanal. Chem., 2004, 379, 358.23 P. T. Kissinger and W. R. Heineman, J. Chem. Educ., 1983, 60(9),
702.
This journal is �c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 1747–1763 | 1761
24 J. P. Hart, A. Crew, E. Crouch, K. C. Honeychurch andR. M. Pemberton, Anal. Lett., 2005, 37, 789.
25 S. Laschi, I. Palchetti and M. Mascini, Sens. Actuators, B, 2006,114, 460.
26 E. V. Gogol, G. A. Evtugyn, J. L. Marty, H. C. Budnikov andV. G. Winter, Talanta, 2000, 53, 379.
27 Immunoassay and Other Bioanalytical Methods, ed. J. M. vanEmon, CRC Press, Boca Raton, FL, USA, 2007.
28 O. Niwa, M. Morita and H. Tabei, Anal. Chem., 1990, 62,447.
29 J. H. Thomas, S. K. Kim, P. J. Hesketh, H. B. Halsall andW. R. Heineman, Anal. Chem., 2004, 76, 2700.
30 A. Bange, J. Tu, X. Zhu, C. Ahn, H. B. Halsall andW. R. Heineman, Electroanalysis, 2007, 19(21), 2202.
31 W. Lorenz and K. D. Schulze, J. Electroanal. Chem., 1975, 65(1),141.
32 I. I. Suni, TrAC, Trends Anal. Chem., 2008, 27(7), 604.33 T. M. Anh, S. V. Dzyadevych, M. C. Van, N. J. Renault,
C. N. Duc and J.-M. Chovelon, Talanta, 2004, 63, 365.34 Z. Muhammad-Tahir and E. C. Alocilja, Biosens. Bioelectron.,
2003, 18, 813.35 K. Yagiuda, A. Hemmi, S. Ito, Y. Asano, Y. Fushinuki,
C. Y. Chen and I. Karube, Biosens. Bioelectron., 1996, 11(8),703.
36 G. J. Papariello, A. K. Mukherji and C. M. Shearer, Anal. Chem.,1973, 45, 790.
37 C. Colapicchioni, A. Barbaro, F. Porcelli and I. Giannini, Sens.Actuators, B, 1991, 4, 245.
38 A. Barbaro, C. Colapicchioni, E. Davini, G. Mazzamurro,A. Piotto and F. Porcelli, Adv. Mater., 1992, 4(6), 402.
39 S. Caras and J. Janata, Anal. Chem., 1980, 52, 1935.40 D. G. Hafeman, J. W. Parce and H. M. McConnell, Science,
1988, 240, 1182.41 T. V. Shishkanova, R. Volf, M. Krondak and V. Kral, Biosens.
Bioelectron., 2007, 22, 2712.42 P. W. Davies and F. Brink, Rev. Sci. Instrum., 1942, 13,
524.43 J. B. Chien, R. A. Wallingford and A. G. Ewing, J. Neurochem.,
1990, 54, 633.44 T. K. Chen, G. Luo and A. G. Ewing, Anal. Chem., 1994, 66,
3031.45 J. A. Jankowski, T. J. Schroeder, R. W. Holz and
R. M. Wightman, J. Biol. Chem., 1992, 267, 18329.46 D. Bruns and R. Jahn, Nature, 1995, 377, 62.47 L. X. Tang and P. Vadgama, Med. Biol. Eng. Comput., 1990, 28,
B18.48 J. Wang, Anal. Chem., 1995, 67, 487.49 J. Wang, Electroanalysis, 2001, 13, 983.50 L. C. Clark and C. Lyons, Ann. N. Y. Acad. Sci., 1962, 102,
29.51 S. J. Updike and G. P. Hicks, Nature, 1967, 214, 986.52 S. R. Mikkelsen and E. Corton, Bioanalytical Chemistry, John
Wiley & Sons, Hoboken, New Jersey, USA, 2004.53 L. Gorton, A. Lindgren, T. Larsson, F. Muteanu, T. Ruzgas and
I. Gazaryan, Anal. Chim. Acta, 1999, 400, 91.54 A. J. Bard and L. R. Faulkner, Electrochemical Methods Funda-
mentals and Applications, John Wiley & Sons, Hoboken, NewJersey, USA, 2001.
55 Electron Transfer from Isolated Molecules to Biomolecules,Part Two, Advances in Chemical Physics, ed. J. Jortner andM. Bixon, John Wiley & Sons, New York, NY, USA, 1999,vol. 107.
56 B. A. Gregg and A. Heller, Anal. Chem., 1990, 62, 258.57 T. J. Ohara, R. Rajagopalan and A. Heller, Anal. Chem., 1993,
65, 3512.58 E. Lojou and P. Bianco, J. Electroceram., 2006, 16, 79.59 S. Cosnier, Electroanalysis, 2005, 19, 1715.60 L. X. Tang and P. Vadgama, Med. Biol. Eng. Comput., 1990, 28,
B18.61 D. C. Klonoff, Diabetes Care, 2005, 28, 1231.62 D. C. Klonoff, Diabetes Care, 2004, 27, 834.63 F. Arslan, A. Yaar and E. Kilic, Artif. Cells, Blood Substitutes,
Biotechnol., 2006, 34, 113.64 L. Agui, J. Manso, P. Yanez-Sedeno and J. M. Pingarron, Sens.
Actuators, B, 2006, 113, 272.
65 H. Shizuko Nakatani, L. Vieira dos Santos, C. Peralta Pelegrine,S. Terezinha Marques Gomes, M. Matsushita, N. Evelazio deSouza and J. Vergilio Visentainer, Am. J. Biochem. Biotechnol.,2005, 1, 85.
66 S. H. Hu and C. C. Liu, Electroanalysis, 1997, 9, 1229.67 J. Niu and J. Y. Lee, Sens. Actuators, B, 2000, 62, 190.68 H. Okuma and E. Watanabe, Biosens. Bioelectron., 2002, 17,
367.69 G. Palleschi, M. Mascini, L. Bernardi and P. Zeppilli, Med. Biol.
Eng. Comput., 1990, 28, B25.70 J.-W. Mo and W. Smart, Front. Biosci., 2004, 9, 3384.71 J. Boldt, Crit. Care, 2002, 6(1), 52.72 P. W. Alexander and G. A. Rechnitz, Electroanalysis, 2000, 12(5),
343.73 J. S. Sidwell and G. A. Rechnitz, Biotechnol. Lett., 1985, 7(6),
419.74 I. Karube and M. Suzuki, Microbial Biosensors in Biosensors: A
Practical Approach A, ed. E. G. Cass, The Practical ApproachSeries 7, Oxford University Press, Oxford, England, 1990.
75 R. A. Goldsby, T. J. Kindtm, B. A. Osborne and J. Kuby,Immunology, W.H. Freeman and Company, New York, NY,USA, 2003.
76 K. A. Fahnrich, M. Pravda and G. G. Guilbault, Anal. Lett.,2002, 35(8), 1269.
77 Antibodies: Volume 1: Production and Purification, ed.G. Subramanian, Springer, New York, NY, USA, 2004.
78 K. Graumann and A. Premstaller, Biotechnol. J., 2006, 1(2), 111.79 S. A. Kellermann and L. L. Green, Curr. Opin. Biotechnol., 2002,
13(6), 593.80 B. Lu, M. R. Smyth and R. Kennedy, Analyst, 1996, 121(3),
29R.81 W. J. Sung and Y. H. Bae, Sens. Actuators, B, 2006, 114(1),
164.82 R. Danczyk, B. Krieder, A. North, T. Webster, H. HogenEsch
and A. Rundell, Biotechnol. Bioeng., 2003, 84(2), 215.83 A. Kumari, Use of (poly)ethylene glycol modified solid supports in
electrochemical immunoassay, MS thesis, University of Cincinna-ti, Cincinnati, 1991.
84 K. Kato, Y. Umedo, F. Suzuki and A. Kosaka, Clin. Chim. Acta,1980, 102(2–3), 261.
85 R. F. Vogt Jr, D. L. Phillips, L. O. Henderson, W. Whitfield andF. W. Spierto, J. Immunol. Methods, 1987, 101(1), 43.
86 C. D. Hodneland, Y. S. Lee, D. H. Min and M. Mrksich, Proc.Natl. Acad. Sci. U. S. A., 2002, 99, 5048.
87 C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides andD. E. Ingber, Biotechnol. Prog., 1998, 14, 356.
88 M. Mrksich and G. M. Whitesides, Annu. Rev. Biophys. Biomol.Struct., 1996, 25, 55.
89 B. Johnsson, S. Lofas and G. Lindquist, Anal. Biochem., 1991,198, 268.
90 N. Kaneki, Y. Xu, A. Kumari, H. B. Halsall, W. R. Heinemanand P. T. Kissinger, Anal. Chim. Acta, 1994, 287, 253.
91 D. Cunningham, Anal. Chim. Acta, 2001, 429, 1.92 D. D. Cunningham and J. Russell, Abstract 1650–5, Pittcon,
Orlando, Florida, 2006.93 H. Huang, Z. Liu and X. Yang, Anal. Biochem., 2006, 356,
208.94 H. Huang, P. Ran and Z. Liu, Bioelectrochemistry, 2007, 70,
257.95 M. Wang, L. Wang, H. Yuan, X. Ji, C. Sun, L. Ma, Y. Bai, T. Li
and J. Li, Electroanalysis, 2004, 16, 757.96 H. Tang, J. Chen, L. Nie, Y. Kuang and S. Yao, Biosens.
Bioelectron., 2007, 22, 1061.97 Y. H. Yun, V. N. Shanov, M. J. Shulz, Z. Dong, A. Jazieh,
W. R. Heineman, H. B. Halsall, D. K. Y. Wong, A. Bange, Y. Tuand S. Subramanian, Sens. Actuators, B, 2006, 120, 298.
98 Y. H. Yun, A. Bange, W. R. Heineman, H. B. Halsall,V. N. Shanov, Z. Dong, S. Pixley, M. Behbehani, A. Jazieh, D.K. Y. Wong, A. Bhattacharya and M. J. Shulz, Sens. Actuators,B, 2007, 123, 177.
99 Y. H. Yun, Z. Dong, V. N. Shanov andM. J. Shulz,Nanotechnology,2007, 18, 465.
100 S. Takenaka, K. Yamashita, M. Takagi, Y. Uto and H. Kondo,Anal. Chem., 2000, 72, 1334.
101 Y. Zhang, H. Kim and A. Heller, Anal. Chem., 2003, 75, 3267.
1762 | Chem. Soc. Rev., 2010, 39, 1747–1763 This journal is �c The Royal Society of Chemistry 2010
102 J. Wang, D. Xu, R. Polsky and E. Arzum, Talanta, 2002, 56,931.
103 Y. Zhang, H. Kim and A. Heller, Anal. Chem., 2003, 75,3267.
104 A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818.105 C. Tuerk and L. Gold, Science, 1990, 249, 505.106 I. Willner and M. Zayats, Angew. Chem., Int. Ed., 2007, 46,
6408–6418.
107 R. M. Umek, S. W. Lin, J. Vielmetter, R. H. Terbrueggen,B. Irvine, C. J. Yu, J. F. Kayyem, H. Yowanto, G. F. Blackburn,D. H. Farkas and Y.-P. Chen, J. Mol. Diagn., 2001, 3(2), 74.
108 R. H. Liu, W. A. Coty, M. Reed and G. Gust, IVD Technol.,2008, 14(4), 31.
109 N. Matsuno, M. Murawsky, J. Ridgeway and J. Cuppoletti,Biochim. Biophys. Acta, Biomembr., 2004, 1665, 184.
110 R. M. Buch and G. A. Rechnitz, Anal. Chem., 1989, 61, 533A.
This journal is �c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 1747–1763 | 1763