72
11 SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
| Date post: | 30-Jan-2018 |
| Category: |
Documents |
| Upload: | duonghuong |
| View: | 219 times |
| Download: | 1 times |

11
SYNTHESISOF INTERMEDIATES
FOR THE PETROCHEMICALINDUSTRY


11.1.1 Gas phase oxidationprocesses
IntroductionSelective oxidation processes, in particular those that
make use of solid catalysts (heterogeneous oxidationprocesses), play a fundamental role in the petrochemicalindustry. About 50% of the principal chemical productsand over 80% of monomers are synthesized by means ofat least one stage of selective heterogeneous catalyticoxidation. Table 1 contains a list of the main selectiveoxidation processes for hydrocarbons using solidcatalysts, with an indication of the conversion andselectivity values obtained. In many commercial selectiveoxidation processes there is still scope for a significantmargin of improvement in performance. For example, thepotential increase in selectivity in the two main processesof selective oxidation (ethylene to ethylene oxide andpropylene to acrylonitrile) could result in annual savingsin reagent costs of around 800 million of euro.
The action of solid catalysts in oxidation processeshad already been noted by the beginning of theNineteenth century, but it was only towards the middleof the Twentieth century that a systematic study ofselective oxidation processes using solid catalysts andof their industrial applications was begun. The firstprocesses to be developed industrially were: oxidation and ammonia oxidation (oxidation in thepresence of ammonia) of propylene to produceacrolein and acrylonitrile respectively, oxidation ofethylene to ethylene oxide and the oxidation ofaromatics to form anhydrides (maleic and phthalicanhydrides). The development of these processes,which was also driven by the growing demand forthese types of products, led to the development offundamental research, with a synergistic effect on boththe development of new applications and theimprovement of those already on the market.
An example of this is the ammonia oxidation processof propylene using air and ammonia, which quicklyreplaced the previous process based on the reactionbetween acetylene and HCN, both because of the lowerraw material cost and the reduced safety issues. Thismade it possible to produce acrylonitrile with asignificant reduction in costs, which resulted in a rapidexpansion in the market for it between 1960-80. On theother hand, the success of this product stimulated thedevelopment of research into the catalysts being used(mixed Bi and Mo based oxides), resulting in theirgradual improvement. The first generation catalysts,based on supported Bi9PMo12O52, gave a yield of 55%,which increased to 65% with the development of secondgeneration systems containing iron as the redox elementand to about 75% with the development of thirdgeneration multi-component catalysts. The currentfourth generation catalysts, containing up to 25elements, allow yields in excess of 80% to be obtained.The development of new catalysts has brought about acomparable evolution in the type of catalytic reactorsused, initially fixed bed, then ‘bubbling’ fluid bed andfinally ‘braked’ fluid bed.
In the period from 1990-2005 development andinnovation in the sector was instead driven by thegrowing importance attached to environmental andsafety issues. However, in the last decade of that periodthe introduction of new processes was heavily influencedby the reduction of investment in petrochemicalsresulting from the restructuring taking place inbusinesses throughout the sector.
Below is a summary of the principal lines ofdevelopment during that time (Centi and Perathoner,2003b).
Use of new raw materials and alternative oxidizingagents. There has been an increasingly wider use ofalkanes as raw materials, instead of aromatics andalkenes; for example, the synthesis of acrylonitrile from
617VOLUME II / REFINING AND PETROCHEMICALS
11.1
Oxidation processes
propane instead of propylene and the synthesis of maleicanhydride from n-butane instead of benzene, aimed atreducing costs and/or improving the eco-sustainability ofthe process. New processes that use alternative oxidantsare being researched. An example is the direct synthesisof phenol from benzene (instead of the multi-stageprocessing of benzene with cumene as an intermediate),using N2O as the oxidizing agent instead of O2. This is inorder to reduce the complexity and the risks associatedwith the process, to avoid the co-production of acetoneand to make use of a by-product such as N2O (therebyalso reducing its disposal costs).
Development of new classes of catalysts andprocesses. The processes that use solid (heterogeneous)
catalysts are increasingly replacing the homogeneoustype, in order to reduce separation costs and theenvironmental impact and/or to use new raw materials,for example in the direct synthesis of acetic acid fromethane. The processes for oxidative dehydrogenation ofalkanes are increasingly more competitive than those fordehydrogenation of alkenes. New processes are alsobeing investigated which will enable the reduction orelimination of the formation of co-products and/or theformation of toxic or dangerous intermediates. Anexample is the synthesis of methacrylic acid throughdirect isobutane oxidation, as an alternative to thecommercial acetone cyanohydrin process, which usesHCN as a reagent and co-produces ammonium sulphate.
618 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
REAGENT Principal product Types of catalystsConversion* Selectivity*
(%) (%)
Methane/O2/NH3 HCN Lattice of Pt-Rh 100 60-70
CH4 or (CH2)x /O2 Syngas (CO/H2) Supported Rh or Ni �99 90-95
Methanol/air Formaldehyde Ag on a-Al2O3,or Fe-Mo oxides 97-99 91-98
Ethylene/O2/acetic acid Vinyl acetate Pd-Cu-K on a-Al2O3 8-12** 92
Ethylene/O2 Ethylene oxide Ag-K-Cl on a-Al2O3 13-18** 72-76
Ethylene/air or O2/HCl 1,2-dichloroethane Oxychlorides of Cu-Mg(K) on g-Al2O3 �95 93-96
Ethanol/O2 Acetaldehyde Ag, Cu 45-50** 94-96
Propylene/air Acrolein Bi-Mo-Fe-Co-Ksupported oxides 92-97 80-88
Propylene/air/NH3 Acrylonitrile Bi-Mo-Fe-Co-Ksupported oxides 98-100 75-83
Acrolein/air Acrylic acid V-Mo-W oxides �95 90-95
n-butane/air Maleic anhydride V-P oxides 75-80 67-72
n-butane/air Butenes/butadiene Bi-Mo-P oxides 55-65 93-95
tert-butyl alcohol Methacrolein Bi-Mo-Fe-Co-K oxides 99 85-90
Isobutene/air Methacrolein Bi-Mo-Fe-Co-K oxides �97 85-90
Methacrolein/air Methacrylic acid V-Mo-W oxides 97-99 95-98
Benzene/air Maleic anhydride V-Mo oxides 98 75
o-xylene/air Phthalic anhydride Oxides of V-P-Cs-Sb on TiO2 98-100 81-87
Naphthalene/air Phthalic anhydride Oxides of V-K on SiO2 100 84
Table 1. Principal processes of selective oxidation of hydrocarbons using solid catalystsand typical results obtained (Arpentinier et al., 2001; Centi et al., 2002)
* Conversion of the reagents and selectivity of the products compared with the hydrocarbon ** In the processes in which the operation includes recycling of the unconverted reagent, the conversion figure is for a single pass

Conversion of processes based on the use of air intoprocesses based on the feeding of pure oxygen. Theseprocesses enable a reduction in polluting emissions; asexamples there are the synthesis of formaldehyde frommethanol, the epoxidation of ethylene and theoxychlorination of ethylene to 1,2-dichloroethane.
Improvement of the productivity of the processes.This is the result of the development of new generationcatalysts with improved properties and/orimprovements in the engineering of the reactors (forexample, the introduction of a monolithic reactor in thesynthesis of formaldehyde, or of structured-bed reactorsin the synthesis of phthalic anhydride). Moreover,during the period from 2000-05 there was a significantincrease of interest in the development of new reactortechnologies (such as, for example, membranereactors), which made it possible to achieve savings inprocessing even for small-medium scale production(scale-down of the processes; Centi and Perathoner,2003a). The goal was to decentralize production andreduce its environmental impact, in contrast with thetrend typical of the Twentieth century of achievingsavings in processing costs through increases in scaleand high integration in large petrochemical facilities.This came about due to the high environmental impactand the strong public opposition to the latter approach,as well as due to problems linked to a sluggish marketwith large fluctuations in demand.
Selective catalytic oxidation processes can be dividedinto three categories. The first relates to oxidation ofinorganic molecules (for example, oxidation of ammoniato NO and of H2S by sulphur). The second class relatesto synthesis of basic chemical products (for example,ammonia oxidation of methane by HCN or the partialoxidation of methane by syngas; CO/H2 mixtures).Finally, the third category relates to conversion ofhydrocarbons by processing in the liquid phase(principally in the homogeneous phase even if there is agrowing interest in the use of heterogeneous catalysts)and processing in the gas phase, which is the mostcommonly used industrially (see again Table 1). Itshould be pointed out that this last class of processesuses air or O2 as the oxidant (other than the cited processof direct hydroxylation of benzene by phenol with N2O),while in the liquid phase processes, in addition to O2,extensive use is also made of other oxidizing agents suchas alkyl peroxides and H2O2 (Centi and Perathoner,2003b).
The different categories of gas phase selectiveoxidation processes (over solid catalysts) and the relatedprincipal industrial reactions are summarized in Table 2(Arpentinier et al., 2001; Centi et al., 2002). Someimportant classes of reactions, which are not mentionedin the table, since they are not yet used commercially,include: oxidative dehydrogenation of C2-C5 alkanes to
their corresponding olefins; selective oxidation ofalkanes such as synthesis of phthalic anhydride andmaleic anhydride from n-pentane, of acrylic acid frompropane and of methacrolein or methacrylic acid fromisobutane; and ammonia oxidation of propane intoacrylonitrile.
The catalysts used for these reactions can beclassified on the basis of their characteristic reactionmechanisms.
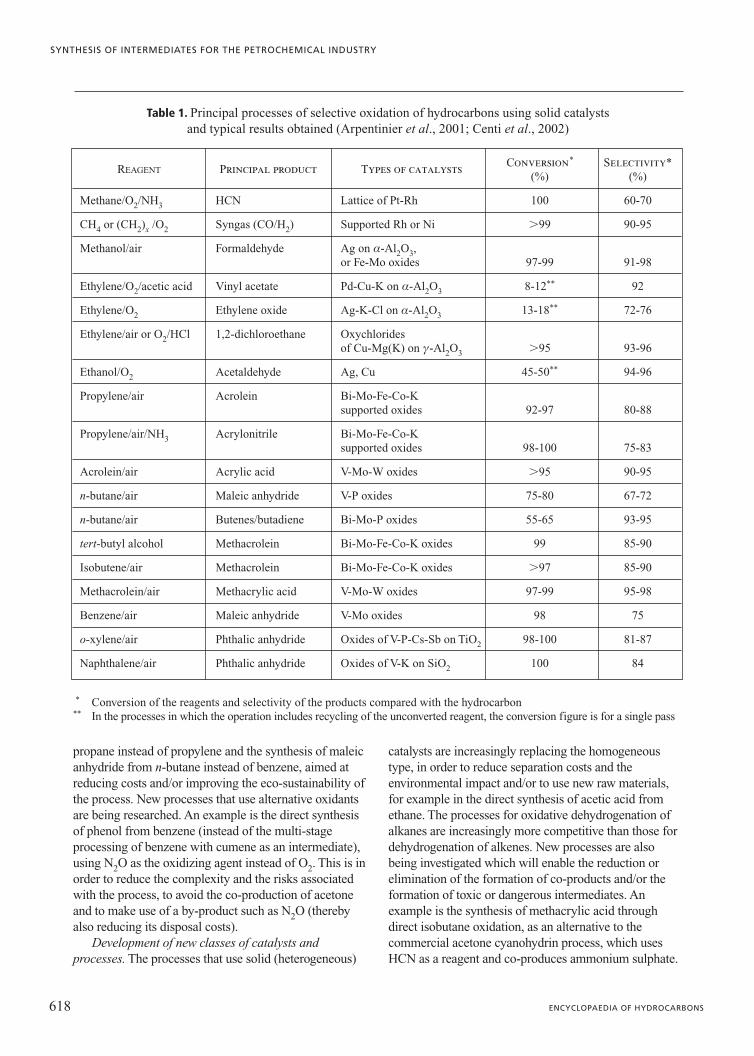
Allylic oxidation. For these reactions catalysts basedon mixed oxides of transition metals are used. Thesecatalysts are capable of selectively extracting a hydrogenatom by breaking a C�H bond in the allyl position andif necessary replacing it with an oxygen atom. Industrialcatalysts are generally multi-component (for example,Bi-Mo oxides, used in the synthesis of acrylonitrile frompropylene, contain various promoters such as Fe, Cu, W,Te, Sb and K), but typically a principal phase can beidentified (Bi-molybdate) which is able to catalysedifferent reactions, such as: the synthesis of acroleinfrom propylene, the ammonia oxidation of propylene toacrylonitrile, the dimerization of propylene tocyclohexene and the oxidative dehydrogenation ofbutenes to butadiene. These reactions are characterizedby a common first stage of allylic oxidation (Fig. 1),where the extraction of a hydrogen atom in the allylposition gives rise to a chemisorbed p-allylic complexon the transition metal. The nature of the subsequentstages determines the type of reaction and product that isobtained. Oxidation and ammonia oxidation of a sidechain of alkyl aromatics (for example, the oxidation oftoluene to benzaldehyde or benzonitrile respectively) inprinciple follow a similar reaction mechanism, but theinteraction of the aromatic ring with the surface isdifferent and therefore different types of catalysts areused, such as vanadium oxides supported on TiO2 orcatalysts based on molybdate of Fe-(V, P, K).
Nucleophilic oxidation to the C�O group (oxidativedehydrogenation of alcohols and oxidation of aldehydesto acids). Although this type of reaction has similaritiesto the mechanism previously described, there are varioustypes of substrates such as alcohols (methanol) oraldehydes (acrolein or methacrolein) which interact toostrongly with the surface of the catalyst when catalystsbelonging to the first category are used. In theconversion of methanol into formaldehyde, the catalystmost often used on an industrial level is iron molybdate(which also contains other components in smallquantities), while multi-component catalysts, based onMo-V oxides or heteropolyacids of P-Mo-V, are used forthe conversion of aldehydes into their correspondingacids.
Electrophilic insertion of an oxygen atom. Thecatalysts for this category of reaction are highly specific.Examples are the systems based on Ag/a-Al2O3 for the
619VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
synthesis of ethylene oxide from ethylene (this catalyst,for example, when applied to the synthesis of propyleneoxide from propylene, is not selective) and Fe/ZSM-5 forthe hydroxylation of phenol with N2O as the oxidant.
Oxidation (or ammonia oxidation) of alkanes. In thiscase the slow stage is the initial selective activation ofthe alkane, for example for the concerted extraction of a
hydrogen atom by a surface Lewis site (a transitionmetal) and of a second hydrogen atom by a base site(oxygen atoms) to give an alkene, which is immediatelyconverted into an oxygenated product through oxidationor allylic ammonia oxidation mechanisms. Catalystswith properties which differ from those of catalystsbelonging to the first reaction category are necessary
620 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
C C
C
HH
HH
H
H
H
H
H
H
C
H
H
H
HC
O
C CC
C
O
O O O
O OBi Mo
OOH
O
O OBi Mo
OOH
O
O
H
OBi Mo
OMe Me O
HH
O
H
OOMe Me Me OMe Me Me
H
p-allylic complex
anionic vacancy
Fig. 1. General outline of the allylic oxidation mechanism and example of the oxidation of propylene on bismuth molybdate to obtain acrolein.
Type of reaction Examples
Allylic oxidation – propylene to acrolein or acrylic acid – isobutene to methacrolein or methacrylic acidSynthesis of the acids can be carried out in a single stage from the alkene,but commercially it is preferred to use two stages for the best possible selectivities
Oxidative dehydrogenation – butenes to butadiene and isopentenes to isoprenes– methanol to formaldehyde – isobutyric acid to methacrylic acid
Electrophilic insertion of an oxygen atom – epoxidation of ethylene to ethylene oxide with O2– direct synthesis of phenol from benzene with N2O
Acetoxylation synthesis of vinyl acetate from ethylene and acetic acid
Oxychlorination synthesis of 1,2-dichloroethane from ethylene and HCl in the presence of O2
Ammonia oxidation – propylene to acrylonitrile– isobutene to methacrylonitrile – a-methylstyrene to atroponitrile
Synthesis of anhydrides – n-butane to maleic anhydride – o-xylene to phthalic anhydride
Table 2. Different classes of gas phase selective oxidation processes (on solid catalysts) and the relativeindustrial reactions (Arpentinier et al., 2001; Centi et al., 2002)

because of the weak interaction of the substrate with thesurface, and the activation mechanism. For example,catalysts based on vanadyl pyrophosphate are used forthe oxidation of n-butane to maleic anhydride, or thosebased on vanadium antimonates are used for theammonia oxidation of propane. In the latter case,antimony oxide is active in the ammonia oxidation ofpropylene, but is not able to activate the propanemolecule; the addition of V gives the system thecapability of oxidizing the alkane.
Wacker-type oxidation mechanism. Vinyl acetate isproduced through acetoxylation of ethylene with aceticacid in the presence of oxygen, with catalysts based onsupported Pd/Au. Pd supported on V2O5/Al2O3 orV2O5/TiO2 is selective in the gas phase synthesis ofacetaldehyde from ethylene or of methylethylketonefrom 1-butene, with a similar reaction mechanism.
Oxychlorination. 1,2-dichloroethane is producedcommercially from ethylene, HC1 and O2 on supportedcopper chloride based catalysts. The mechanism consistsof a direct addition of chlorine atoms by the catalyst ontothe olefin, rather than in oxidation of hydrochloric acidto molecular chlorine, followed by chlorination of thedouble bond.
Addition of oxygen to the aromatic nucleus, with ringopening. The electrophilic attack of oxygen onhydrocarbon substrates typically leads to the formationof carbon oxides, however in the case of the oxidation ofbenzene, selective oxidation to maleic anhydride isobtained. This process, which employs catalysts basedon mixed vanadium and molybdenum oxides, has beenpartially replaced by the synthesis by oxidation ofn-butane. Similar catalysts are used in the selectiveoxidation of polyaromatic compounds.
Non-classic oxidation mechanisms. Ethylbenzenecan be oxidatively dehydrogenated, with high selectivity,to styrene on various catalysts such as oxides andphosphates, but the active phase is constituted by theformation of a thin surface layer of carbon containingthe active sites of the reaction. Recently even some typesof carbon and carbon nanotubes have shown highselectivity in oxidative dehydrogenation of ethylbenzeneto styrene. Another example is the ammoximation ofcyclohexanone (to cyclohexanone oxime) overamorphous silica.
Characteristics of gas phase oxidation processes
General aspects Although the petrochemical industry uses selective
oxidation processes both in the gas phase and in theliquid phase, those in gas phase are more widespread.Typically, oxygen (or air) is used as the oxidizing agent,although the species involved in the selective oxidationreaction are usually made up, not of adsorbed oxygen on
the catalyst, but rather of the structural oxygen of thecatalyst (typically mixed oxides, see again Table 1). TheO2� oxygen ion removes the hydrogen atoms from thehydrocarbon with the subsequent formation of water or,if inserted into the molecular structure of the reagent, itgives rise to the formation of oxygenated compounds(see again Fig. 1). Instead, the gaseous oxygenintervenes in the reoxidation mechanism of the reducedcatalyst, known as the Mars-van Krevelen mechanism(Fig. 2).
The oxidation of the catalyst by O2 comes aboutthrough the formation of intermediate oxygen speciessuch as O2
� and O�, which have electrophiliccharacteristics and tend to make an addition to the
621VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
oxidized catalyst
reduced catalyst
H2O
M2m� M1
n�O2
oxide (catalyst)
oxidizedproduct
hydrocarbon
M(n�1)� O2� O2�O2�
O2� O2�
O2� O22�
O2
O2�O�
O2�
Mn� Mn�
Mn� M(n�1)� O2�O2� O2� Mn�
C
C
H
C
C
products with ruptureof C C bond andformation of COx
electrophilicoxygen species(O2
�, O�, ...)
O2 activation
nucleophilicoxygenspecies(O2�)
products of selectiveoxidation(e.g. aldehydes)
Fig. 3. A, schematic mechanism of the incorporation ofoxygen into oxide based catalysts; B, outline of differenttypes of attack on the hydrocarbon by nucleophilic andelectrophilic species of oxygen(Centi et al., 2002).
A
B
Fig. 2. Mars-van Krevelen mechanism of selective oxidationof hydrocarbons on oxide based catalysts.

unsaturated molecule, breaking the double bond andending with the formation of carbon oxides; in contrast,the structural oxygen of the catalyst (O2�) hasnucleophilic characteristics (Fig. 3).
Hence, in order to be selective, a catalyst must notonly possess activation sites for the hydrocarbon and forselective insertion of the oxygen on the substrate, butmust also be rapidly reoxidizable. This is so as to preventthe non-selective chemisorbed oxygen species fromhaving a lifetime long enough to allow combustionreactions to take place. This mechanism is generallyaccepted for the oxidation of alkenes on mixed oxides,but there are doubts about its validity in the case ofoxidation of other substrates, such as alkanes.
A general characteristic of selective oxidationprocesses of hydrocarbons is the complexity of thereactions involved. For example, the oxidation of n-butane to maleic anhydride is a reaction whichinvolves 14 electrons, the removal of 8 hydrogenatoms and the insertion of 3 oxygen atoms on thesubstrate, with the involvement of another 4 oxygenatoms of the catalyst to form 4 molecules of water.Notwithstanding the complexity of the transformation,the reaction takes place without the formation ofproducts with intermediate levels of oxidation;selectivity of between 70 and 85% is achieved,depending on the reaction conditions. Therefore thecatalyst, made up of a mixed oxide of V and P with thecomposition (VO)2P2O7, possesses characteristics suchas to avoid both the desorption of the reactionintermediates and their non-selective transformationinto carbon oxides.
Finally, another characteristic of selective oxidationcatalysts is their multi-functionality, which is necessaryfor the transformation of the hydrocarbon into the finalproduct; in fact, to bring about the complex mechanismdescribed above, it is necessary that the catalyst becapable of actuating different types of transformationson the substrate (Arpentinier et al., 2001). Moreover, it isnecessary for the different stages involved in thetransformation to have similar rates. Different relativerates could lead to the desorption of intermediateproducts or an increase in the rate of parallel reactions,with a reduction in the selectivity for the desiredproduct. This is well illustrated in the selective oxidationof n-butane (Fig. 4).
Combined design of the catalyst and the reactor Optimizing the yield, productivity and selectivity of
selective oxidation reactions requires not only a detailedknowledge of the nature of the catalyst and themechanism of the interaction of the reagents andproducts with the catalyst itself, but also the optimizationof the reactor used. Recently, new reactor solutions(which enable, for example, the separation of the two
stages of interaction of the catalyst with the hydrocarbonand with oxygen) have led to the development of newclasses of catalysts. The two aspects, involving thedevelopment of the catalyst and the engineering of thereactor, are therefore closely correlated.
Industrial reactors used in the petrochemicalindustry for highly exothermic reactions, such as thosefor selective oxidation, are typically either of themulti-tubular fixed-bed or fluid-bed type.Nevertheless, there is a growing interest in thedevelopment of new reactor solutions, such as forexample, the circulating fluid-bed reactor, recentlyapplied by DuPont to the synthesis of maleicanhydride from n-butane. The ‘uncoupling’ of the tworedox reactions, of oxidation of the hydrocarbon by thecatalyst and the reoxidation of the latter by oxygen(see again Fig. 2), makes it possible to increase theselectivity towards maleic anhydride compared withthe reaction carried out in the simultaneous presenceof both the hydrocarbon and oxygen. Other advantagesof this type of reactor are isothermicity and areduction of the risk of explosion. Nevertheless, alimiting factor is its low productivity; in fact it isnecessary to circulate large quantities of the catalyst(equal to about 1 kg per g of maleic anhydrideproduced) between the two reactor vessels, each ofwhich is adapted to one of the reaction stages.
Another example of a new reactor configuration,adopted for petrochemical processes, is the monolithictype of reactor; these reactors combine the advantages ofthe possibility of autothermic conduction of the reaction
622 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
n-butane
concerted abstractionof 2 H atoms
isomerization
allylic H abstraction
1,4-oxygen insertion
oxygen insertion
allylic dehydrogenationand/or allylic oxygeninsertion
maleicanhydride
COx, H2O
O OO
Fig. 4. Outline of the reaction mechanism in the selectiveoxidation of n-butane to maleic anhydride on catalysts basedon (VO)2P2O7, showing the multifunctional character of thecatalyst.
and a reduction in the loss of pressure. The newgenerations of processes for oxidation of methanol toformaldehyde use a final adiabatic stage (post-reactor),with a catalyst structured in the form of a monolith.
Very interesting results have been obtained usingreactor with extremely short contact times (on theorder of milliseconds, compared with times measuredin seconds in conventional reactor), where the catalystconfiguration is also of a non-conventional type (forexample, in a grid form). Given the high spatial ratesused (that is the high ratio between the input rate ofthe reagents and the quantity of the catalysts) and thetype of mechanism involved, it is possible to avoidsubsequent oxidations and therefore to obtain highselectivity of the intermediate products (for example,in the oxidative dehydrogenation of alkanes intoalkenes).
Finally, it is worth remembering the developments inthe field of catalytic membrane reactors, which allow thecontinuous removal of one of the products or thedifferential addition along the catalytic bed of one of thereagents (for example, oxygen). This makes it possible tomaintain the optimum hydrocarbon/O2 ratio along theentire profile, to limit the formation of hot spots and tocontrol the state of oxidation of the catalyst.Nevertheless, one of the current limiting factors is itslow productivity, apart from the high cost of themembrane itself.
Even conventional fixed-bed reactors can beimproved through greater integration of the design of thecatalyst and that of the reactor. In the process of thesynthesis of phthalic anhydride from o-xylene,specialized catalytic beds are used, that is, containingdifferent layers of catalysts each having a differentcomposition, in order to optimize the axial activity andselectivity profile of the catalyst itself.
A new reactor technique being developed consists ofsystems in which the flow is periodically reversed; thismakes it possible to make the activity and temperatureprofile in the reactor more uniform, even though thereare still some significant problems involving thedifficulty of managing non-stationary operations andtheir potential danger. Also in this case, the design of thecatalyst is different from that for operations in stationaryconditions.
Use of air and pure oxygen as oxidizing agentsCurrently, air is the most widely used reagent in
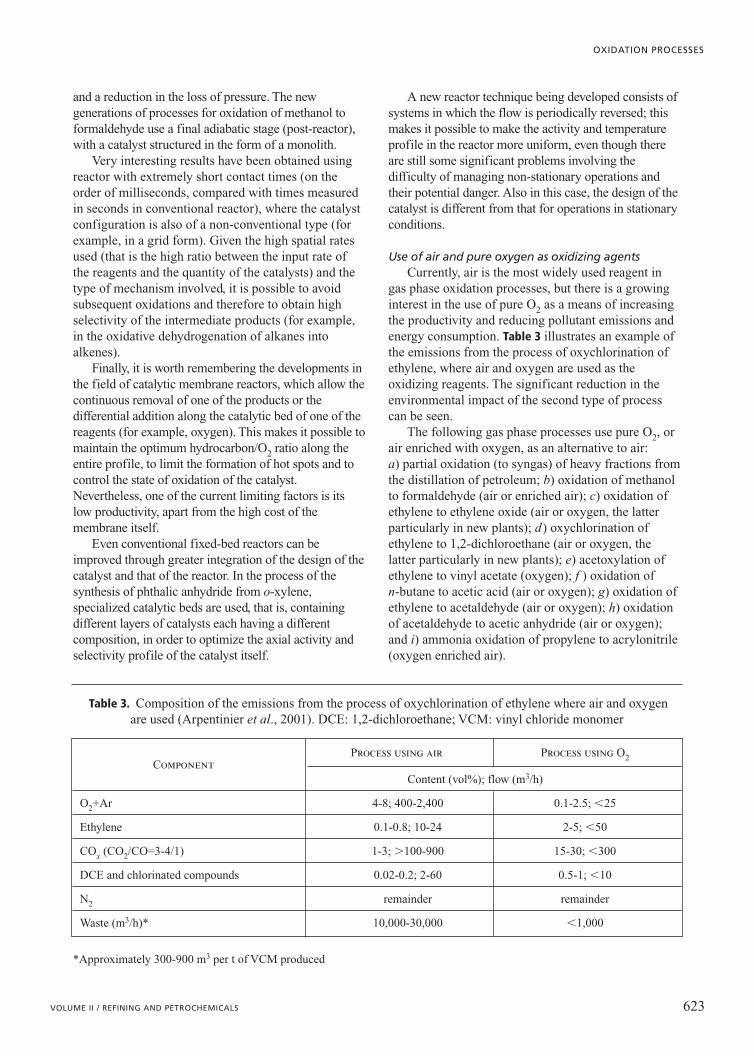
gas phase oxidation processes, but there is a growinginterest in the use of pure O2 as a means of increasingthe productivity and reducing pollutant emissions andenergy consumption. Table 3 illustrates an example ofthe emissions from the process of oxychlorination ofethylene, where air and oxygen are used as theoxidizing reagents. The significant reduction in theenvironmental impact of the second type of processcan be seen.
The following gas phase processes use pure O2, orair enriched with oxygen, as an alternative to air:a) partial oxidation (to syngas) of heavy fractions fromthe distillation of petroleum; b) oxidation of methanolto formaldehyde (air or enriched air); c) oxidation ofethylene to ethylene oxide (air or oxygen, the latterparticularly in new plants); d) oxychlorination ofethylene to 1,2-dichloroethane (air or oxygen, thelatter particularly in new plants); e) acetoxylation ofethylene to vinyl acetate (oxygen); f ) oxidation ofn-butane to acetic acid (air or oxygen); g) oxidation ofethylene to acetaldehyde (air or oxygen); h) oxidationof acetaldehyde to acetic anhydride (air or oxygen);and i) ammonia oxidation of propylene to acrylonitrile(oxygen enriched air).
623VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
ComponentProcess using air Process using O2
Content (vol%); flow (m3/h)
O2+Ar 4-8; 400-2,400 0.1-2.5; �25
Ethylene 0.1-0.8; 10-24 2-5; �50
COx (CO2/CO=3-4/1) 1-3; �100-900 15-30; �300
DCE and chlorinated compounds 0.02-0.2; 2-60 0.5-1; �10
N2 remainder remainder
Waste (m3/h)* 10,000-30,000 �1,000
Table 3. Composition of the emissions from the process of oxychlorination of ethylene where air and oxygenare used (Arpentinier et al., 2001). DCE: 1,2-dichloroethane; VCM: vinyl chloride monomer
*Approximately 300-900 m3 per t of VCM produced

Selective oxidation catalysts for hydrocarbons
Characteristics of oxidation catalystsOxidation catalysts belong to a wider class of
materials having redox or oxidoreductive typecharacteristics; systems which catalyse reactions ofhydrogenation, dehydrogenation, halogenation anddehalogenation also belong to this class. The mostimportant catalysts in the field of petrochemicals for theoxidation of hydrocarbons for processes carried out inthe gas phase are listed in Table 1. In addition to these, itis worth mentioning catalysts used for the oxidation ofinorganic compounds, such as those employed for theoxidation of SO2 to SO3 (based on supported vanadiumoxide), of ammonia to NO (based on Pt/Rh) and ofhydrogen chloride to molecular chlorine (based onsupported copper chloride).
Below is a list of the principal characteristics ofoxidation catalysts for gas phase reactions.
Presence of a transition metal as the principal activecomponent (V, Mo, Cu, Fe, Pd, Pt, Rh, Ag). Often inthese cases, a second element is also present which canbe transition or post transition (for example, P, Sb or Bi),which contributes to establishing the reactivecharacteristics of the catalyst. This effect can beexplained by the formation of a ‘mixed oxide’ (that is, ofa specific compound, such as for example Bi2Mo2O9,possibly only on the surface of another oxide, of a solidsolution or of an oxide doped with the other element),with reactive characteristics different from those of thesingle elements, if present in distinct phases. In somecases the element is initially present in a metallic form,but under reaction conditions it can generate thecorresponding oxide (or chlorides or oxychlorides).
Presence of small quantities of ‘promoter’ (or‘doping’) elements. The purpose of these elements is tooptimize the performance of the principal activeelements. The nature of the promoters can vary and theycan therefore play different roles in the transformation ofthe reagents. The active elements and the promoterelements, constitute the active phase, that is the phasedirectly involved in the transformation of the reagentsinto products.
Presence of a support (usually silica, alumina ortitanium oxide). This support in the catalyst’sformulation can fulfil a variety of tasks. A primary taskis that of dispersing the active elements, conferring alarger surface area to the active phase compared withwhat would have existed in the absence of the support. Itis clear, therefore, that the support must have surfacearea characteristics suitable for the reaction of interest.In selective oxidation, where the selectivity in theformation of the partially oxidized product is heavilydependent on the subsequent reactions to undesiredproducts (for example, carbon oxides, which are
thermodynamically favoured), a support is needed with asurface area which is not too large. In this way the rate ofthe undesired secondary reactions, which are alsodependent on the time needed by the product to diffusefrom the active centre into the gas phase, is limited. Afurther task of the support is that of providing theresistance of the active phase to phenomena which cancause abrasion or disintegration, especially for thoseapplications which involve particular mechanicalstresses on the catalyst (for example, in fluidized-bedreactors), in addition to avoiding powdering during theloading of the catalyst into packed fixed-bed reactors.Finally, in some cases the support serves to alter thecharacteristics of the intrinsic chemical reactivity of theactive phase, through the effects of the interactionbetween the latter and the support itself. This comesabout when the support presents functional groups on itssurface which can lead to the formation of chemicalbonds with the elements of the active phase, or it takesplace as a result of particular crystallographicsimilarities between the surface and the support. Theseinteractive effects can be positive for the reactivity of thecatalyst itself, altering its oxidoreductive characteristicsor reducing its volatility; the undesirable effects of lossby sublimation of components of the active phase arethus reduced.
The most suitable combination of the type andnumber of active phases (including the promoters) andthe type of support is dependent on the characteristics ofthe reaction and the type of reactors used. In particular,below are listed the factors that have the greatestinfluence on the formulation and the morphology of thecatalyst used for oxidation reactions.
Type of chemical transformation involved andmechanism through which it takes place. With increasingcomplexity of the transformation the composition of thecatalyst also becomes the more complex, in terms of thenumber of elements making up the active phase, or ofthe structural complexity (the formation of crystallinephases having multi-functional characteristics). Forexample, catalysts used for oxidation or allylic ammoniaoxidation always contain Mo as the principal element forthe active phase, while catalysts for the synthesis ofanhydrides or of acids almost always contain V.
Optimization of the redox characteristics or of theacidity or basicity properties of the catalyst. Thepromoters (or doping agents) can play a fundamentalrole in the control of these properties. Promoters withbase-type characteristics (alkaline or alkaline earth metaloxides) can reduce the surface acidity of the activephase, with a consequent improvement of selectivitythrough the suppression of the acid-catalysed reactions(cracking, formation of oligomers of unsaturatedcompounds). Promoters with acid-type characteristicscan reduce the interaction between the active phase and
624 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

intermediates of the reaction which have acid-typecharacteristics, thus favouring their desorption into gasphase and limiting the contribution of the subsequentundesired reactions. Other promoters can optimize theoxidoreductive properties of the active phase, by amodification of the overall electronic properties of thesolid.
Reaction scheme. The presence of consecutivereactions (typically, combustion reactions of the desiredproduct, or reactions which lead from the reagent to thedesired product through the formation of intermediateproducts with an increasing state of oxidation) involvesthe use of a catalyst with characteristics such as to limit(or, alternatively, to favour) the contribution of thesereactions. This can be achieved not only by control of theintrinsic activity of the catalyst, but also by amodification of the porosity of the active phase (andtherefore of the support, if present). High surface areaand porosity values entail effective intra-particleresidential times which are much higher that thosecalculable from the feeding capacity of the reactor, andtherefore a significant contribution from the consecutivereactions for a given conversion of the reagent. This canhave a considerable influence on the selectivity of thedesired product.
Reaction heat levels. Highly exothermic reactionsinvolve the need both for fluidized-bed catalytic reactors,which are more efficient in removing heat than multi-tubular reactors, and for catalysts capable of operating inconditions of high mechanical stress. In these casesfluidizable supports are used, which feature particleswith an average diameter of between 50 and 150 mm,resistant to abrasion and with an appropriate density. Formedium-low heat levels, tubular or tube-bundle(multi-tubular) reactors can be used. In these cases thecatalysts have a characteristic morphology for theseapplications and are produced in the form of extrusions(or pellets). When possible, supports with a high heatconduction capacity are used, such as SiC, in order toassist the dissipation of the heat from the reaction.
Spatial rates in the reactor. High spatial rates inpacked catalytic beds can lead to a high loss of pressure,and therefore to the need for heavy compression of theflow upstream of the reactor. It is possible to minimizethe loss of pressure by increasing the vacuum level in thecatalytic bed, through the use of special structures of thecatalyst particles. This can be instrumental inconditioning the performance of the process, as in thecase of oxidative dehydrogenation of methanol toformaldehyde. In this case, the use of cylindrical pelletswith an axial hole enables the loss of pressure to bereduced and therefore, the linear rate in the reactor to beincreased for a given rate of feeding. This involvesshorter contact times, better reaction temperature controland reduced catalyst deactivation effects.
Oxidation catalyst mechanisms in gas phaseThe principal catalytic oxidation mechanisms are
listed in Table 2. The redox type mechanism is the onethat is used in the majority of oxidation reactions. Itworks through a series of successive stages, whichinclude: the adsorption of the reagent (the substrate to beoxidized) on the active centre; the transfer of electronsfrom the reagent to the active centre and thesimultaneous transfer of oxygen ions from this to thereagent (the oxygen is incorporated into the substrate, oralternatively returns in the formation of co-producedwater); and finally, the desorption of the product. Thesame sequence of stages involves the molecule ofoxygen for the catalyst reoxidation stage: co-ordinationat the metallic centre; transfer of electrons (up to 4 foreach oxygen molecule); dissociation of the molecule intotwo atomic species in ionic form; finally incorporationof the oxygen in its ionic form within the active phase.
One or more active centres may be involved for eachreagent molecule, depending on the following factors:a) the overall number of electrons transferred andtherefore of oxygen ions involved in the oxidoreductiveprocess; b) the ionic and electronic conduction capacityof the solid, and therefore of the surface active phaseunder reaction conditions; c) the level of cover of theactive phase by the adsorbed molecules (reagents andproducts); d) the surface mobility of the reactionintermediates; and e) the number of active centres closeto the one in which the activation of the hydrocarbontook place.
On the basis of the redox model, the selectivity ofthe process, that is the relationship between the quantityof the product formed and the total quantity of reagenttransformed, can be traced back to two differentsituations. First and foremost the selectivity depends onthe nature of the oxygen ions present as speciesadsorbed on the active phase and on the interactionbetween them and the reagent or the reactionintermediates. As previously stated, the O2� species,incorporated in the lattice of the oxide, is considered tobe the selective species, while the O2
� and O� specieshave electrophilic characteristics and are considered tobe non-selective species. Since the formation of the firstspecies comes about by the intermediate formation ofthe electrophilic species, it is clear that thetransformation rate of each of them and their reactivitywith the reaction intermediates obtained throughactivation of the substrate determine the selectivity ofthe process (Bielanski and Haber, 1991).
Moreover, the selectivity of the oxidation process istraceable to the concentration of O2� speciesincorporated in the metal oxide lattice, and thereforedirectly to the average state of oxidation of the catalyst(Grasselli, 2002). A strongly oxidized catalyst has a highdensity of active centres capable of receiving electrons
625VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES

from the substrate and of releasing O2� ions, andtherefore it is able to transform that substrate intomolecules with a high state of oxidation (for example,into combustion products). In contrast, a catalyst madeup of a partially reduced oxide has a modest oxidizingcapacity, and therefore is potentially more selective forthe partially oxidized products. According to the redoxmodel, the state of oxidation of a metal oxide in astationary state is dependent on the conditions of thereaction; this implies that the selectivity in its turn isdependent on the operating parameters, such as thecomposition of the feed (that is the relationship betweenthe substrate to be oxidized and the oxidizing agent) orthe reaction temperature.
Both models have been experimentally verified fordifferent oxidation reactions, and still remain valid todayfor the explanation of the selectivity of oxidationprocesses involving reactions with redox type mechanisms.
Principal industrial processes and relevantapplications
Oxidative dehydrogenation of methanol to formaldehyde
Formaldehyde (HCHO) is among the top twentychemical compounds produced on a world scale, and isused in the synthesis of various resins (urea-formaldehyde, phenol-formaldehyde, and polyacetals)which find applications in the construction, automotive,textile and paper sectors. Methanol can be converted intoformaldehyde both by direct oxidative dehydrogenation:
CH3OH�0.5O2�� HCHO�H2O
∆H°=�155 kJ/mol
or by dehydrogenation combined with oxidation of the H2product:
CH3OH�� HCHO�H2 ∆H°= 84 kJ/mol
H2�0.5O2�� H2O ∆H°=�238 kJ/mol
The two processes differ in their operating conditionsand type of catalysts. In the first process lowconcentrations of methanol are used in the feed, in orderto avoid the formation of explosive mixtures and tocontrol the temperature of the reaction. Commercialcatalysts are based on iron molybdate, but also containan excess of molybdenum (Fe2(MoO4)3�MoO3), sincethe presence of molybdenum oxide is a necessarycondition for high selectivity. Typically a ratio of Mo/Fewithin the range of 1.5-3.0 is used; occasionally oxidesof Co and Cr are added as promoters. The excess ofmolybdenum is also necessary because the sublimationof the oxide (particularly at the points of greatestoverheating) cause the progressive depletion of the Moin the catalyst and the condensation of MoO3 in the
coldest parts of the reactor. This induces, not just thedeactivation of the catalyst, but also a progressiveincrease in pressure loss.
Reaction temperatures are typically within the rangeof 310-340°C, with conversions in excess of 98% andselectivity equal to 92-95%. Multi-tubular fixed-bedreactors are generally used. A recent development hasseen the introduction of a final (post-reactor) adiabaticstage.
In dehydrogenation combined with partialcombustion of H2 (the overall process turns out to bepartially exothermic) an understoichiometric oxygencurrent is fed in, to operate in the upper region at thelimit of flammability. Due to the thermodynamic limitsof dehydrogenation, it is necessary to operate at higherreaction temperatures than those for oxidativedehydrogenation. In this process use is made of Agbased catalysts supported on alumina with a low surfacearea, typically in spherical form with a diameter of 1-5 mm. If operating at temperatures in excess of 600°C(particularly 680-720°C), it is possible to obtain analmost total conversion of the methanol, while at lowertemperatures (500-550°C) the conversion is less efficient(65-75%) and it is necessary to recycle the methanolwhich failed to react. Moreover, it is necessary to useshort contact times in order to avoid decomposition ofthe formaldehyde.
Selectivity to formaldehyde of 98-99% is obtained,with the formation of the following by-products:dimethylether ((CH3)2O), whose formation is due to thepresence of acidic sites in the catalyst; methyl formate(HCOOCH3) obtained through disproportionation of theformaldehyde on basic sites; carbon oxides, derivedfrom both parallel and serial reactions. To limit theformation of carbon oxides, rapid cooling of the reactionproducts is necessary when they leave the catalyst bed.High selectivity is achieved through the optimization ofthe acid-base properties of the catalyst, limitation of theoxidation of the formaldehyde to formic acid (a productwhich decomposes easily) and control of the redoxproperties of the catalyst.
Fig. 5 illustrates the reaction mechanism in the caseof direct oxidation of methanol on oxide based catalysts.The methoxy species is the first chemisorbed speciesthat is formed by the contact of methanol with thecatalyst; its subsequent transformation depends both onthe reaction conditions and on the properties of thecatalyst. If the concentration of methanol is high and therate of the subsequent oxidation of the methoxy speciesis low, a condensation reaction takes place that leads todimethylether (typical of the acidic oxides containingnon-reducible cations, such as alumina). Oxidation ofthe methoxy species (extraction of an atom of H andtransfer of an electron) leads to co-ordinatedformaldehyde, which is in equilibrium with the
626 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

dioxymethylene species. The reaction requires anucleophilic attack by the catalyst’s structural oxygen. Ifthe Me�O bond is of a covalent type, the equilibriumbetween the dioxymethylene and the co-ordinatedformaldehyde is shifted towards the latter species, whichin its turn is in equilibrium with the gas phaseformaldehyde. Ionic oxides, instead, favour theformation of the dioxymethylene species. This gives riseto the methoxy and formate species by Cannizzarodisproportionation; the formate can also form throughdirect oxidative dehydrogenation from thedioxymethylene. By reactions with methanol,dioxymethylene forms dimethoxymethane, which candesorb into gas phase. The formate species can also reactwith methanol to give methyl formate. The formation ofthis product requires that the desorption rate of theformaldehyde be low and that theoxidation/disproportionation rate be relatively high. Boththese reaction pathways contribute to the formation ofthe formate species. Their relative rates depend on thedegree of surface coverage of the dioxymethylenespecies and on the reaction conditions. Nevertheless, theformate species can also be converted and therefore theselective formation of methyl formate requires that theconversion rate of the formate species be low and thatthe concentration of methanol be relatively high.
A simplified outline of the commercial processes forconverting methanol to formaldehyde using ironmolybdate and supported Ag as the catalysts is shown inFig. 6.
Oxidative dehydrogenation of methanol on ironmolybdate based catalysts is carried out in a cooledmulti-tubular reactor (see again Fig. 6 A). Due to theprogressive deactivation of the catalyst (the lifespan is
from 1-2 years), it is necessary to gradually increase thetemperature of the reactor in order to keep theproductivity at a constant level. The formaldehyde yieldis about 95-96%. The oldest processes operate with afeed of 6% of methanol in air (a concentration which isless than the lower limit of flammability), but in this caseproductivity is low, the purity of the formaldehyde ispoor because of the formation of formic acid, thecatalyst’s lifespan is limited and it is necessary to operatewith high volumes of inert gas. For this reason, abouthalf of the plants have been converted to a feed withreduced levels of oxygen (�10%), and higherconcentrations of methanol (8.2%). On account of thehigh amount of heat generated, it is necessary to dilutethe catalyst and improve the efficiency of the reactor’scooling system. Some plants also use steam as a diluent.
When the gas leaves the reactor vessel, it undergoesa heat recovery stage, after which it is sent to an absorbercolumn, where water is used as a solvent. Theformaldehyde solution, from the bottom of the absorbercolumn, has a concentration of from 50-60%; it is thensent to an ionic exchange column for removal of theformic acid. On the other hand, the gas from the top ofthe column is recycled; the waste stream keeps thecomposition in the reactor constant.
The process using supported Ag as the catalyst (seeagain Fig. 6 B) has the advantage, in comparison withthe direct oxidation method, of producing a waste streamwhich can be sent directly for incineration, because itcontains H2, methanol and formaldehyde, as well assmall quantities of N2 and CO2. The combustion of thisflow produces the majority of the steam used in theprocess. The outline of the process is similar to thatdiscussed previously, however a purer methanol feed is
627VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
OH
H
�[H] metoxy dioxymethylene
disproportionation
�[H]
CH3OH CH2O
HCOOH H COCH3
�H2O
�H2O
OCH3
OCH3
�CH3OH
CH3OCH3�H2O
O OH
formaldehyde
dimethoxymethaneCH2
OO�
CH2 CH2
O
O
CH2
COx
formate
formic acid
methylformate
oxygen vacancy
O O
CH CH3OH
2CH3OHO OH
Fig. 5. Reaction mechanism of the oxidation of methanol on oxide based catalysts (Centi et al., 2002).

required (there must be no iron carbonyls or sulphurcompounds which would contaminate the catalyst), anda heat exchanger is required to heat the methanol.Furthermore, the reactor must operate with very shortcontact times and have a rapid cooling system (coolingtimes for the discharge flow from the catalyst bed of lessthan 0.02 s), in order to avoid consecutive reactions ofthe formaldehyde, and finally two absorber columns inseries should be used.
Epoxidation of ethylene to ethylene oxide Ethylene oxide is an intermediate product that is
synthesized on a large scale, being used in theproduction of ethylene glycol and polyglycols, ofethanolamine and non-ionic detergents, and of esters ofethylene glycol. There are two technologies: a process
using chlorohydrin, which is no longer used, and directoxidation of ethylene with oxygen over supported Agbased catalysts
CH2�CH2�0.5O2�� H2C�CH2
O∆H°��105 kJ/mol
CsCl and BaCl2 are used as promoters (theconcentration of these doping agents is of 0-10 ppm),since both the alkaline metals and the chloride ionspromote the selectivity.
In addition to CO2, the by-products are acetaldehydeand formaldehyde, but in concentrations no greater than0.1%. Although there are still conflicting opinions on thematter, a significant body of data indicates that the activecatalysing species is made up of Ag�O, withelectrophilic characteristics, while oxygen bridging atomsbetween the silver atoms (AgOAg) have a nucleophiliccharacter and are non-selective, contrary to what happensin the case of selective oxidation on oxides (see againFig. 3 B). In the past it was held that the selective speciesin epoxidation consisted of AgO2, which after theinsertion of oxygen into the ethylene left an adsorbed Oatom and AgO on the surface, which in turn wasresponsible for the non-selective oxidation of ethylene toCO2 and H2O. On the basis of this model, a maximumselectivity of 85.7% was hypothesized, whereas currentcommercial processes operate with greater selectivity (atlow conversion), in the range of 88-94%.
The reaction scheme is of a ‘triangular’ type, withtwo parallel reactions of the transformation of ethyleneinto ethylene oxide and CO2, and a consecutive reactionof ethylene oxide to CO2. The principal factorsinfluencing the reaction kinetics and hence the choice ofthe optimal reaction conditions are summarized here.The ethylene oxide formation rate increases as thepartial pressure of the oxygen increases, while amaximum rate is reached with respect to theconcentration of ethylene, as a result of the competitionbetween the ethylene and the ethylene oxide for the samecatalytic sites. The ethylene oxide formation ratedecreases on increasing the concentration of the chlorideions used as a doping agent (nevertheless, above acertain value traces of chlorinated compounds such asvinyl chloride and dichloroethylene form as by-products)and the concentration of CO2. From a practical point ofview, the concentration of ethylene is determinedessentially by the limits of flammability. The ratio of therate of the two parallel reactions of formation of ethyleneoxide and combustion, that is the initial selectivity, riseswith the concentration of chloride ions, with the contentof alkaline metal promoters and with the partial pressureof the ethylene.
The processes employed for oxidation of ethyleneoperate with air or with pure oxygen. The former are still
628 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
A
B
methanol
methanol
steam
recycle
recycle
catalyst
formaldehydesolution
formaldehydesolution
H2O
H2O
H2O
coolingwater
vent
waste
vent
air
air
A
B
Fig. 6. Simplified outline of the process of the synthesis offormaldehyde from methanol: A, direct oxidativedehydrogenation with catalysts based on iron molybdate; B, a process using catalysts based on supported Ag(Arpentinier et al., 2001).
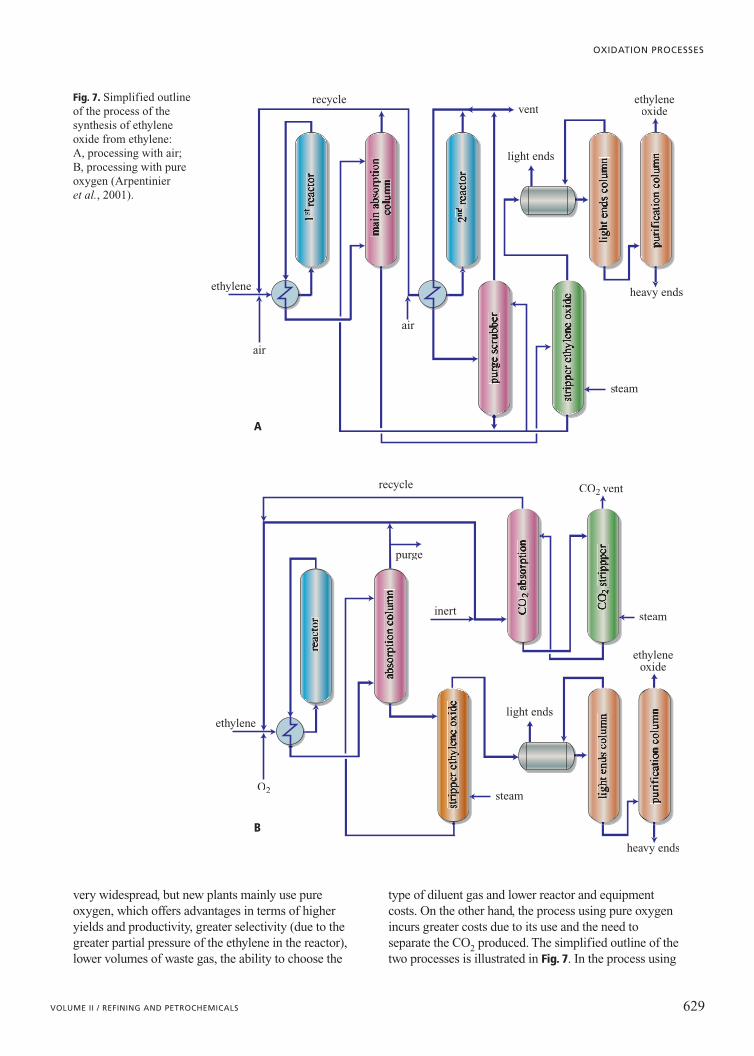
very widespread, but new plants mainly use pureoxygen, which offers advantages in terms of higheryields and productivity, greater selectivity (due to thegreater partial pressure of the ethylene in the reactor),lower volumes of waste gas, the ability to choose the
type of diluent gas and lower reactor and equipmentcosts. On the other hand, the process using pure oxygenincurs greater costs due to its use and the need toseparate the CO2 produced. The simplified outline of thetwo processes is illustrated in Fig. 7. In the process using
629VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
light ends
air
steam
ventrecycle ethylene
oxide
heavy endsethylene
air
light ends
ethyleneoxide
heavy ends
recycle
purge
CO2 vent
ethylene
inert
O2
steam
steam
Fig. 7. Simplified outlineof the process of thesynthesis of ethyleneoxide from ethylene: A, processing with air; B, processing with pureoxygen (Arpentinier et al., 2001).
A
B

air, the ethylene is oxidized in a series of tubular reactors(two, or three in the bigger plants), in order to increasethe conversion and manage the heat from the reactionmore effectively (in the first reactor the conversion isaround 40%). The outflow from the reactor is cooled andthen sent to an absorber column with water, for therecovery of the ethylene oxide. Part of the gasdischarged from the absorber column is recycled, whileanother part is sent to a second reactor where theconversion reaches 80% (95% where there are threereactors in series). Selectivity is about 70% in the firstreactor, but is lower in the subsequent ones.
The process with pure oxygen (purity �97%) issingle stage and uses multi-tubular reactors. Theethylene/O2 ratio is typically 3.0-3.5, while the O2concentration is kept below 9% to avoid the formation ofinflammable mixtures. Conversion through the passageof ethylene is in the range 10-15%, while overall it isgreater than 97%. Selectivity typically is greater than80%. After cooling, the gases leaving the reactor are sentto the ethylene oxide absorber column, while the gasescoming out of the column are compressed and recycled.Part of the gas is sent to a column to eliminate CO2through hot absorption in an aqueous solution ofpotassium carbonate. A small fraction of the gases (lessthan 1%) is discharged, to prevent an accumulation ofinert gases. In some processes a diluent, such as methaneor ethane, is also used. Although CO2 itself can be aneffective diluent, it can contaminate the catalyst andtherefore it is necessary to keep its concentrationlevel low.
Oxychlorination of ethylene The oxychlorination (oxidation in the presence of
HC1) reaction of ethylene to 1,2-dichloroethane(DCE)
CH2�CH2�2HCl�0.5O2�� CH2Cl�CH2Cl
∆H°=�238 kJ/mol
is the basis for the production of the vinyl chloridemonomer (CH2�CHCl), used in the production ofhomo- and copolymers in PVC.
Three different processing options are possible,depending on the technology used and the operatingconditions: the reactors can be based on fixed orfluidized beds; either air or pure O2 can be used; and astoichiometric value or an excess of ethylene may beemployed. Generally, pure oxygen is used whenoperating in a fixed-bed reactor, with a large excess ofethylene (compared with the stoichiometric value) wherethe unconverted ethylene is recycled, or when usingstoichiometric ratios, where high conversion levels ofethylene are achieved, but the gas flow is in any caserecycled so as to reduce the environmental impact of theprocess (see again Table 3).
The use of an excess of ethylene, which has betterthermal conduction properties than nitrogen, offersadvantages such as a more uniform temperature profilein the reactor (which results in better selectivity due toless combustion and reduced formation of secondaryproducts such as trichloroethane), better conversion ofthe HC1 and a longer catalyst life (thanks to a lowersublimation of the active phase and a reduced formationof charcoal on the catalyst’s surface). Since theproductivity of fixed-bed reactor is limited by thecapacity for transferring the heat to the cooling fluid, agas phase with higher conductivity results in an increasein productivity.
The principal problem related to the use of O2 isrepresented by its higher cost compared with air and theneed for more complex systems for operating under safeconditions. Nevertheless, many plants using air have beensatisfactorily modified to operate with oxygen. The mostwidely used reactor is the multi-tubular fixed bed, becauseof the higher productivity attainable; nevertheless, thehigher investment needed (corrosion resistant steel has tobe used) leads to a preference for fluidized-bedtechnology in the construction of new plants.
In Fig. 8 a simplified outline is portrayed of anair-fed fixed-bed process. Three multi-tubular reactorsin series are used; the catalyst is diluted with graphite,in order to reduce the thermal gradients in the reactor;and a fourth reactor is used for direct chlorination ofthe residual ethylene with Cl2. The gases are cooled tocondense the DCE (1,2-dichloroethane), which is thensent to the purification columns: the first removes thewater, the second the light products (ethylene chloride,vinyl chloride, 1,1-dichloroethane anddichloroethylene) and the third the heavy products(trichloroethane, perchloroethane andperchloroethylene).
To optimize the local ethylene/O2 ratio and to obtainbetter control over the temperature in the reactors andhence of the selectivity, ethylene and HC1 are fed intothe first reactor, while the air or oxygen feed isdistributed over the three reactors in series. This alsomakes it possible to operate outside the limits offlammability, to reduce the formation of CO2, to have amore homogeneous temperature profile and finally toincrease the lifespan of the catalyst. The oxygen andethylene are fed at slightly higher levels with respect tothe stoichiometric quantities, in order to achieve aconversion of the HC1 greater than 99.5%. On the otherhand, in processes that operate with oxygen and a largeexcess of ethylene, the unconverted hydrocarbon isseparated and recycled.
The catalysts are based on copper chloride supportedon alumina and the promoters, made up of alkaline oralkaline-earth metals, play a fundamental role. Thereaction temperatures are between 220 and 250°C, with
630 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
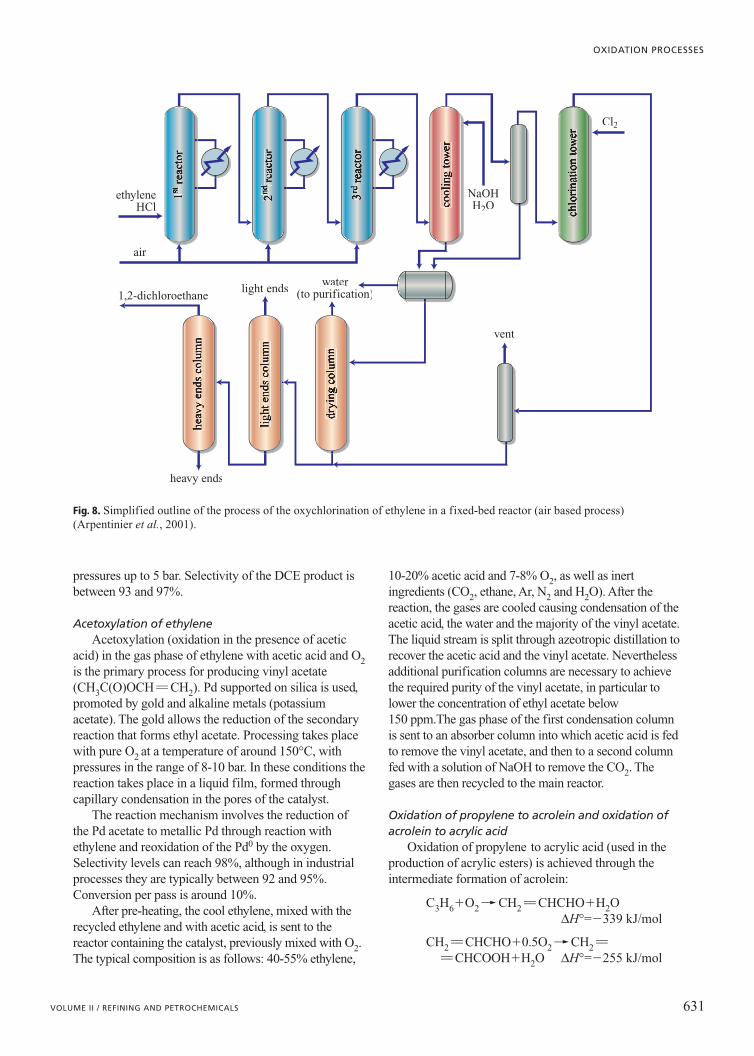
pressures up to 5 bar. Selectivity of the DCE product isbetween 93 and 97%.
Acetoxylation of ethylene Acetoxylation (oxidation in the presence of acetic
acid) in the gas phase of ethylene with acetic acid and O2is the primary process for producing vinyl acetate(CH3C(O)OCH�CH2). Pd supported on silica is used,promoted by gold and alkaline metals (potassiumacetate). The gold allows the reduction of the secondaryreaction that forms ethyl acetate. Processing takes placewith pure O2 at a temperature of around 150°C, withpressures in the range of 8-10 bar. In these conditions thereaction takes place in a liquid film, formed throughcapillary condensation in the pores of the catalyst.
The reaction mechanism involves the reduction ofthe Pd acetate to metallic Pd through reaction withethylene and reoxidation of the Pd0 by the oxygen.Selectivity levels can reach 98%, although in industrialprocesses they are typically between 92 and 95%.Conversion per pass is around 10%.
After pre-heating, the cool ethylene, mixed with therecycled ethylene and with acetic acid, is sent to thereactor containing the catalyst, previously mixed with O2.The typical composition is as follows: 40-55% ethylene,
10-20% acetic acid and 7-8% O2, as well as inertingredients (CO2, ethane, Ar, N2 and H2O). After thereaction, the gases are cooled causing condensation of theacetic acid, the water and the majority of the vinyl acetate.The liquid stream is split through azeotropic distillation torecover the acetic acid and the vinyl acetate. Neverthelessadditional purification columns are necessary to achievethe required purity of the vinyl acetate, in particular tolower the concentration of ethyl acetate below 150 ppm.The gas phase of the first condensation columnis sent to an absorber column into which acetic acid is fedto remove the vinyl acetate, and then to a second columnfed with a solution of NaOH to remove the CO2. Thegases are then recycled to the main reactor.
Oxidation of propylene to acrolein and oxidation ofacrolein to acrylic acid
Oxidation of propylene to acrylic acid (used in theproduction of acrylic esters) is achieved through theintermediate formation of acrolein:
C3H6�O2�� CH2�CHCHO�H2O
∆H°=�339 kJ/mol
CH2�CHCHO�0.5O2�� CH2�
�CHCOOH�H2O ∆H°=�255 kJ/mol
631VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
NaOHH2O
Cl2
vent
light ends
heavy ends
1,2-dichloroethane
ethyleneHCl
air
water(to purification)
Fig. 8. Simplified outline of the process of the oxychlorination of ethylene in a fixed-bed reactor (air based process)(Arpentinier et al., 2001).

By-products of the reaction are carbon oxides, aceticacid, propionic acid, formaldehyde, maleic acid,acetaldehyde and acetone. For the first reactionmulti-component catalysts based on bismuth molybdatesare used (for example, Mo12BiFe3Co4.5Ni2.5Sn0.5K0.1Ox),while for the second reaction the catalysts are based onmolybdenum and vanadium oxides (for example,Mo12V3Cu2.5Fe1.25Mn0.1Mg0.1P0.1Ox). It is possible tocarry out the synthesis in a single stage but, because ofthe strong exothermicity of the reaction the lifespan ofthe catalyst is reduced. Moreover, the overall selectivityis greater in a two stage process, as it is possible tooptimize each independently. In the first stage theselectivity of acrolein is typically greater than 85% andthe conversion of propylene exceeds 90%, while in thesecond stage the selectivity of acrylic acid is greater than95%, with yields of between 90 and 96%.
Multi-tubular fixed-bed reactors are used in series.The first reactor operates with a temperature in the rangeof 330-400°C, and a spatial rate of 1,300-2,600 h�1 (thepressure is 2-2.5 bar), while the second operates at lowertemperatures (250-300°C), higher spatial rates (1,800-3,600 h�1) and lower pressures (due primarily to the lossof pressure in the first reactor).
The concentration of propylene entering the firstreactor is 5-8% in air. Recirculating gas and/or steam areused as diluents, in order to operate outside the limits ofexplosion. The use of steam also allows the reactions inhomogeneous phase to be reduced, the thermal transferto be improved and selectivity to be increased, favouringthe desorption of acrolein and acrylic acid. Nevertheless,excessive concentrations of steam lower theconcentration of the acrylic acid solution.
The gases leaving the reactor, after cooling and heatrecovery, are sent to an absorber column with water. Aninhibitor is added to avoid polymerization of the acrylicacid. The discharge gases are then sent for incinerationand in part are recycled, after the elimination ofcondensable compounds. The solution of acrylic acid issent to the purification section, which consists of a seriesof azeotropic distillation columns (withmethylethylketone as the third component). In the caseof diluted solutions it is possible, as an alternative, tocarry out a separation by extraction, using ethyl acetateor aromatic compounds as solvents. The variousindustrial processes differ in the composition of thecatalyst and in the separation section.
Synthesis of methyl methacrylateMethyl ester of methacrylic acid,
CH2�C(CH3)COOCH3, is used in the production ofvinyl polymers. The acetone cyanohydrin process (whichconsists of a reaction between acetone and HCN,followed by a reaction of acetone cyanohydrin withsulphuric acid and a final hydrolysis of the adduct in the
presence of methanol) is the most widely used for thesynthesis of methyl methacrylate and has manydisadvantages, linked to the toxicity of HCN and theco-formation of high quantities of ammonium sulphate,which is generated in the ratio of 2:1 with methylmethacrylate. The alternative process is direct oxidationof isobutene in the gas phase (with subsequent orintegrated stages of esterification); however, this processgives yields and selectivity which are too low to becompetitive.
Alternative methods of synthesis are: a) directoxidation of isobutane (a process which is still in theresearch phase), which has the advantage of lower rawmaterial costs and lower environmental impact; b)oxidation of isobutyric aldehyde to isobutyric acid,which is then converted into methacrylic acid byoxidative dehydrogenation (Mitsubishi Kasei/Asahimethod); c) oxidation of tert-butyl alcohol tomethacrolein, followed by oxidation to methacrylic acidand esterification; and d) hydroformylation of ethyleneto propionic aldehyde, which is then condensed withformaldehyde to give methacrolein, which finally isoxidized to methacrylic acid and esterified (BASFprocess). Among these alternatives, direct synthesis ofisobutane is the most interesting; nevertheless, theselectivity obtained and the stability of the catalysts arenot sufficient for it to be developed industrially.
The average composition of the catalysts for the lastof the above applications is as follows: (HmY0.2-1.5)(P1-1.2Mo12-nX0.4-1.5Ox), where Y is the ion of an alkalinemetal and X is an element such as V, As and Cu;moreover, various other additives are present. It isnecessary to use high concentrations of isobutane andsteam (up to 65%) to obtain good selectivity andstability of the catalysts.
Synthesis of acrylonitrile by ammonia oxidation ofpropylene
Acrylonitrile is produced on a large scale (over 5million tons per year) by the process of catalysedammonia oxidation (oxidation in the presence ofammonia) of gas phase propylene:
CH2�CH�CH3�NH3�1,5O2��
�� CH2�CH�CN +3H2O ∆H°=�515 kJ/mol
Acrylonitrile finds applications in the synthesis ofvarious homo- and copolymers used as fibres, resins andelastomers; it is also an intermediate in the production ofadiponitrile and acrylamide.
By-products of the reaction are HCN, acetonitrile, N2(from combustion of the ammonia) and carbon oxides.The reaction is strongly exothermic and to control thetemperature most plants use fluid-bed reactors. Thecommercial catalysts are multi-component, based onbismuth molybdate and supported on silica (for example,
632 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

(K, Cs)0.1(Ni, Mg, Mn)7.5(Fe, Cr)2.3Bi0.5Mo12Ox/SiO2). It is not necessary to recycle the propylene, since
conversion rates are as high as 95%, while maintaining ahigh selectivity to acrylonitrile (above 80%). The fluid-bed reactor contains a high quantity of catalyst, up to 70-80 tons, in the form of spherical particles with anaverage diameter of 40-50 mm, with a view to allowingan efficient fluidization. The purity of the feed must bevery high (�90% for the propylene and �99.5% for theNH3). The ratio of the ammonia/propylene feed is equalto 1.05-1.2, and the ratio of O2/propylene is within therange 10-15. The reaction temperature is between 420and 450°C and the pressure between 1.5 and 3 bar.Pressures above 1 bar have a negative effect on theselectivity to acrylonitrile, but are necessary in order toobtain good fluidization and to increase the productivity.
The reagents are fed into the reactor separately, tominimize reactions in the homogeneous phase andprevent the formation of explosive mixtures beforereaching the catalytic bed; the composition of themixture in the reactor is within the limits offlammability, but the presence of the fluid bed inhibitsthe propagation of radical reactions thereby blocking anyflame front. The fluid-bed reactor contains various coilsand systems to minimize the formation of slugs and toreduce the phenomena of retro-mixing of the fluid. Thetop of the reactor has a larger cross section, in order toreduce the velocity of the gas and to diminish theoccurrences of pneumatic transmission and elution ofthe catalyst. Appropriate cyclones allow the recovery ofthe catalyst particles and their reintroduction into thereactor.
The effluents from the reactor are sent to an absorbercolumn with water, while the unconverted ammonia isneutralized with sulphuric acid. The gases leaving thiscolumn, containing N2, carbon oxides and smallquantities of propylene, are sent for incineration. Theacetonitrile/acrylonitrile mixture forms an azeotropewith water that on separation gives rise to an aqueousphase (recycled to the absorber) and an organic phase,rich in acrylonitrile and HCN, that is sent forpurification. The aqueous solution of acetonitrile is
concentrated for azeotropic distillation. The acrylonitrileis purified in two columns in series, to recover thehydrogen cyanide and the impurities (acetone,acetaldehyde, propionaldehyde and acrolein). A finalstage of vacuum distillation is used to obtainacrylonitrile with purity above 99.4%.
Recently a number of studies have been devoted tothe use of propane as an alternative reagent to propylene,by virtue of its lower cost. New classes of catalysts (forexample those developed by Mitsubishi, composed ofquaternary oxides of molybdenum, vanadium, telluriumand niobium, MoV0.3Te0.23Nb0.12Ox) give yields ofacrylonitrile above 50%. This value could be sufficientto justify development of a new process starting withpropane, although a further increase in yield and stabilityof the catalyst would be hoped for.
Ammonia oxidation of alkyl aromaticsNumerous aromatic nitriles such as benzonitrile,
phthalonitrile, isophthalonitrile, terephthalonitrile andnicotinonitrile find applications in the synthesis ofproducts for fine chemicals. For example, nicotinonitrilecan be hydrolysed to the corresponding amine or tonicotinic acid, used for the synthesis of vitamin B.Isophthalonitrile is used in the synthesis of herbicidesand fungicides. Phthalonitrile is an intermediate forpigments based on phthalocyanine. A number ofcatalysts are active in the reaction: vanadium oxidesupported on TiO2 (preferably in the anatase crystallinestructure), doped with Cs, P and W; multi-componentcatalysts based on molybdates; vanadium antimonatesdoped with Bi and Fe; and supported heteropolyacids(PV3Mo12Ox on silica). Apart from maximizing theselectivity to nitrile, it is also important to minimize theoxidizing reaction of the ammonia to N2. Selectivitylevels above 90% are generally possible for conversionswhich are between 50 and 80%, even though the resultsvary considerably according to the type of substrate.
One of the principal problems in the industrialapplication of the process is the need to carry outsuccessive production runs with different types of alkylaromatics, as the market demand for these products is
633VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
CH3
CH3
CH3COx
CN
CN
CN
O2, NH3
V2Ox/TiO2
O2, NH3
V2Ox/TiO2
NH
O
O
tolunitrile
phthalonitrile
phthalimide
Fig. 9. Outline of the synthesis ofphthalimide by catalytic ammoniaoxidation of o-xylene in the gas phase.

insufficient to justify the use of the plant for a singlereaction. The aromatic imides can also be obtained bymeans of catalytic ammonia oxidation (Fig. 9), forexample feeding o-xylene and using a catalyst based onsupported vanadium oxide on titanium oxide.
Maleic anhydride from n-butaneMaleic anhydride is used as an additive in the
synthesis of various polymers, in the synthesis ofchemical products for agriculture and of malic andfumaric acids, as well as being an intermediate for thesynthesis of g-butyrolactone and of tetrahydrofuran.Today, most plants for production of maleic anhydrideuse n-butane as the feed; this process has replaced thatof benzene, by virtue of the smaller number ofby-products, the better atomic efficiency and the lowercosts, and the non-toxicity of the reagent. Theby-products obtained in the oxidation of n-butane arecarbon oxides and acetic acid, whereas numerousby-products are formed by benzene, or when usingbutenes or butadiene as the raw material.
The various commercial processes differ in theirreactor technology (fixed-bed, or fluid or circulating-bedreactor), in the percentage of n-butane in the input (lessthan 2% with fixed-bed reactors, between 2.6 and 5%for processes with fluid-bed reactors, and more than10% in the DuPont process, with circulating-bedreactors, similar to that used in catalytic cracking) and inthe method of recovery and purification of the maleicanhydride (use of organic or aqueous solvents in therecovery and purification method). In all processes thecatalyst consists of pyrophosphate of vanadyl(VO)2P2O7, where necessary promoted with dopingelements, although the methods of preparation,activation and formation and the dimensions of theparticles can vary.
The feed composition is directly related to the choiceof reactor as shown above. An increase in theconcentration of n-butane increases the productivity, butcalls for special precautions for dispersal of the reactionheat and to reduce the risks of explosion. Theunconverted n-butane is not recycled, but is used in theproduction of high temperature steam, since the value ofthis hydrocarbon is close to that of fuels.
The conversion rate of n-butane is between 80 and90% and the yield in maleic anhydride between 55 and65%. The working temperature is 400-450°C. Theprincipal processes are marketed by Denka/ScientificDesign, Amoco, BP-UCB, Lonza/Lummus, MitsubishiKasei, Mitsui Toatsu, Monsanto and DuPont.
Lonza’s ALMA process uses a fluid-bed reactor,while the DuPont process uses a circulating-bed reactor.Fig. 10 shows a simplified outline of the two processes.
In the ALMA process, after the separation of thecatalyst by means of two cyclones in series, the effluents
from the reactor are cooled to 200°C, filtered to removethe finest particles and sent to the maleic anhydriderecovery section. The latter is based on absorption incycloaliphatic solvents. The gases are sent forincineration for the production of high temperaturesteam, while the solution is sent to a stripping unit. Themaleic anhydride is then further purified to remove thelight products (which are sent for incineration), while thesolvent is recycled.
The process gives high productivity, production ofhigh temperature steam, reduced quantity of waste,low formation of fumaric acid and heavy products.The production of steam contributes to the economy ofthe process.
The DuPont process is characterized by the use ofan innovative reactor for the selective oxidation sectorderived from the catalytic cracking, that permitsseparate contact of the hydrocarbon and oxygen withthe catalyst. This brings about a significant increase inthe selectivity, but involves the need for higherrecirculation of the catalyst between the two reactors(the riser reactor for contact with the hydrocarbon andthe fluid-bed reactor for reoxidation of the catalyst).Furthermore it calls for the catalyst to have highmechanical resistance. The DuPont process forsynthesis of maleic anhydride is integrated with thedownstream hydrogenation section, for the productionof tetrahydrofuran. The absorption of the maleicanhydride is, in this case, performed with water.
Phthalic anhydride from o-xylenePhthalic anhydride is used in the preparation of
diesters (plasticizers for PVC), alkylic resins, polyestersand colourings. The original process used naphthalene asthe raw material, but today most plants use o-xylene, byvirtue of the reduced environmental and safetyproblems, as well as the greater purity of the product.
The principal by-products of o-xylene are o-tolualdehyde and phthalide, small quantities of maleicanhydride, benzoic acid, toluic acid, as well as carbonoxides. The formation of phthalide is a critical aspect ofthe process, in that, for applications in the polymersector, the concentration of this compound in phthalicanhydride must be very low. In a multi-tubular reactor,about a third of the catalytic bed enables over 90%conversion of the o-xylene to be obtained, while theremaining two thirds of the bed serves to reduce theconcentration of phthalide.
The catalysts are based on vanadium oxide supportedon TiO2 (in the octahedral crystalline form), with arelatively low surface area (around 10-20 m2/g). K, Cs,Sb, Nb, and P are used as promoters. The latestgeneration of reactors are loaded with two or three layersof catalyst whose compositions differ from one another(above all in terms of type and quantity of promoter
634 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

elements); in this way it is possible to obtain maximumyield and selectivity, by optimizing the activity profileand minimizing ‘hot spots’ along the reactor. Promotersin the gas phase, such as SO2 also have a positive effecton performance, although they are no longer used today.
Various processes exist, among which the principalones are those developed by Wacker, BASF,Lonza-Alusuisse, Atochem and Nippon Shokubai. The processes differ in the type of reactor (fixed or fluidbed, although the former is used in the majority of cases),in the composition of the catalyst and in the
recovery/purification sections. Air is used as an oxidant,although the use of air enriched with oxygen allowshigher productivity. The concentration of o-xylene is notgreater than 1.2% in air, so as to avoid the formation offlammable mixtures and to achieve good control of theheat of the reaction. Recovery of the phthalic anhydride iscarried out by desublimation and then by absorption in oil.
The fixed-bed reactor operates at temperatures ofbetween 350-390°C and with low spatial rates, while theprocess in the fluid-bed reactor operates at highertemperatures (450-550°C) and with lower contact times.The o-xylene and the air, after preheating, are sent to amulti-tubular reactor operating at about 380°C andcooled with molten salts; these salts are cooled externallyin a heat exchanger, producing high temperature steam.
A yield of phthalic anhydride above 80% is obtainedand a conversion of o-xylene greater than 99%. Theeffluents from the reactor are cooled, producing lowpressure vapour, and then sent to an absorber columnwith water. The solution is first sent to a under vacuumsystem to decompose the impurities (polymerisationinhibitors are added) and then to a distillation column(also under vacuum) to separate the maleic anhydrideand the benzoic and toluic acids from the top. Thesolution collected at the bottom of the distillationcolumn is sent to a second column, to separate highpurity phthalic anhydride (�99.5% by weight).
References
Arpentinier P. et al. (2001) The technology of catalyticoxidations, Paris, Technip.
Bielanski A., Haber J. (1991) Oxygen in catalysis, New York,Marcel Dekker.
Centi G., Perathoner S. (2003a) Catalysis and sustainablegreen chemistry, «Catalysis Today», 77, 287-297.
Centi G., Perathoner S. (2003b) Selective oxidation. SectionE (Industrial processes and relevant engineering issues), in:Horvath I.T. (editor in chief) Encyclopedia of catalysis, NewYork, John Wiley, 6v.
Centi G. et al. (2002) Selective oxidation by heterogeneouscatalysis, New York, Kluwer Academic-Plenum.
Grasselli R.K. (2002) Fundamentals principles of selectiveheterogeneous oxidation catalysis, «Topics in Catalysis»,21, 79-88.
Fabrizio CavaniDipartimento di Chimica Industriale e dei Materiali
Università degli Studi di BolognaBologna, Italy
Gabriele CentiDipartimento di Chimica Industriale e
di Ingegneria dei MaterialiUniversità degli Studi di Messina
Messina, Italy
635VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
A
B
maleicanhydride
aqueoussolutionof maleicacid
H2
H2
recy
cle
reac
tor
air
air
ventdry
filter
vent
vent
tetr
ahyd
rofu
ran
fresh solvent
steam
steam
n-butane
n-butanen-butanerecycle
fluid-bedregeneration
reactor
by-p
rodu
cts
Fig. 10. Simplified outline of the process of the synthesisof maleic anhydride from n-butane: A, the ALMA process;B, the DuPont process.
A
B

636 ENCYCLOPAEDIA OF HYDROCARBONS
11.1.2 Oxidation processesin liquid phase with oxygen
Introduction
Liquid phase oxidation is widely applied in thechemical industry for the synthesis of intermediates bothfor the petrochemical industry and for the production ofspecialty chemicals and pharmaceuticals, as well as forwastewater decontamination.
Liquid phase oxidation is chosen rather thanheterogeneous gas phase catalysis in the following cases:when the products are thermally unstable (i.e. in theproduction of hydroperoxides and carboxylic acids, withthe exception of b-unsaturated compounds); when theproducts are so reactive that at high temperature they canbe further oxidized (i.e. epoxides, aldehydes andketones, with the exception of b-unsaturatedcompounds, ethylene oxide and formaldehyde); and, infine chemical production where liquid phase oxidation isespecially suitable due to the thermal instability and/orreactivity of the reagents themselves (i.e. in the oxidationof polyhydroxy alcohols).
In addition to being the largest application ofhomogeneous catalysis, liquid phase oxidation is moreimportant than heterogeneous gas phase oxidationprocesses, on the basis of tonnage and variety ofproducts (Prengle and Barona, 1970a; Lyons, 1980;Sheldon and Kochi, 1981). In homogeneous catalysis,the main technological issues also include selectivity,removal of the heat of reaction and safety considerations.
Liquid phase oxidations can be subdivided into thefollowing five types of catalytic processes, based onthe mechanism involved and the relative catalysts: a)free-radical, chain oxidation (with and withoutcatalyst), with molecular oxygen as the oxidizingagent; b) redox mechanism with Pd or Cu complexes,and molecular oxygen; c) catalytic oxygen transfer,with either alkylhydroperoxide or H2O2 as theoxidizing agent, and with either homogeneous orheterogeneous catalysts; d) oxidative dehydrogenationwith molecular oxygen and with supported, transitionmetal–based catalysts; e) photocatalytic processes.
The first class is the most important from anindustrial point of view and, therefore, the mostimportant technological aspects of this class ofreaction will be examined in detail.
Liquid phase homogeneousmetal–catalysed oxidations by molecular oxygen
The oxidation of organic substrates:substrate + oxygen donor �� oxidized substrate +
reduced donorhas considerable industrial importance and playsfundamental roles in biological processes. Oxotransfer oxygenations of organic substrates (mainlyalkanes and alkenes) are utilized in some processesand are the subject of several investigations. Differentoxygen donors may be used: molecular oxygen,peracids, alkylhydroperoxides, hydrogen peroxide,iodosylarenes, amine N-oxides, hypochlorite andammonium persulfate. Different catalysts areemployed, such as metalloporphyrins,metallophthalocyanines, soluble transition metal saltswith chelating ligands and Schiff base complexes.Polyoxometallates are among the most interestingsystems that have been receiving considerableattention in recent years.
Oxidation of a substrate with molecular oxygenmay involve two electrons (with the involvement ofonly one oxygen atom), or four electrons (with theinvolvement of both oxygen atoms). Coordination withthe metal modifies the features of oxygen (i.e. itsbasicity and radical character), making it moresusceptible to reaction with organic substrates. Asummary of the possible complexes betweenmolecular oxygen and metal ions is presented inTable 1 (Sheldon and Kochi, 1981).
In addition to the superoxo and peroxo species, oxometal species that occur via the transfer of two electronsfrom the metal to the oxygen atom will be addressed.
Hydroperoxo species can also form via hydrolysisof peroxo complexes, or by molecular oxygeninsertion in metal hydrides:
M�O�O�M�H��� M�O�OH��MM�H�O2�
� M�O�OH
The formation of oxo metal species takes place ifthe metal is able to furnish two electrons; such metals
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
M � H� M O OH�
OO

include the redox couples Mn�/M(n�2)�: Co(I)/Co(III),Ir(I)/Ir(III), Pd(0)/Pd(II), Pt(0)/Pt(II). Metals in higheroxidation states (III and IV) can not transfer electronsto molecular oxygen and do not form stable adducts.In this mechanism, the formation of high–valent oxometal species occurs through the O�O bond scissionof an intermediate m-peroxo dinuclear complex, withelectron transfer from the metal and with spin pairingin the molecular oxygen. A more suitablerepresentation would take into account the tworesonance structures:
M(IV)�O2����M(II)�O
The most usual representation of this moiety is M�O.Different classes of metal-catalysed oxidations by
O2 can be distinguished, according to the nature of thecomplex formed with the oxygen and the metaloxidation states formed (i.e. to the role played by themetal atom in the oxygen activation; Sheldon andKochi, 1981; Drago and Beer, 1992). Particularattention will be paid to the oxygenations by oxometals, which are oxidations of relevant importance inthe functionality of organic substrates.
Oxo metal species formed by interaction with peroxides and molecular oxygen
The mechanism for the formation of the M�Ospecies can be divided into two steps: the reaction forthe formation of the peroxide and that of the peroxidewith the metal complex to form the oxo metal species.The metal-catalysed oxidation does not occur as aconsequence of the activation of molecular oxygen bythe metal, since the metal characteristics do not allowthe electron transfer to oxygen. The presence of areductant enables the conversion of molecular oxygeninto H2O2 or into an alkylhydroperoxide (which arestronger oxidizing agents than oxygen) to be carriedout; the latter may react with the metal complex toyield the high-valent oxo metal species. In fact, in
liquid phase homogeneous oxidations in mildconditions, the generation of oxo metal species whenusing molecular oxygen requires the presence of areducing agent, which is necessary to furnish electronsto the system. In addition, the catalytic action of theoxo metal species in the oxygenation of the C�Hbond must always compete with very fast autoxidationprocesses, thus with free radical chain reactions thatare largely unselective and indiscriminate. Therefore,the objective is to develop systems that can operate inthe absence of other reducing agents and thatkinetically compete with radical-initiatedautoxidations.
Examples of reductants that reduce molecularoxygen to H2O2 are anthraquinol derivatives, NADHand BH4
�. The metal-peroxo complexesM(n�2)��O2
2� can react with water or an acid, leadingto the formation of H2O2 or of M�OOH species:
M(n�2)��2e��O2��M(n�2)��O2
2�(peroxo complex)M(n�2)��O2
2��2 H���M(n�2)��H2O2M(n�2)��O2
2��H���M(n�2)��O�OH�
Similarly, metal-O2 superoxo adducts (i.e. whereonly one-electron charge transfer from the metal tomolecular oxygen has occurred) can be reduced tometal peroxo species in the presence of a one–electronreducing agent (sacrificial reductant):
M(n�1)��O�O�� e���M(n�1)��O22�
(peroxo complex)
The second step is the oxidation of metalcomplexes to oxo metal species with H2O2, which canproceed as follows:
M(II)(H2O) �H2O2��M(IV)�O �2H2O
or through ligand substitution and formation of thedinuclear m-peroxo complex, which then decomposesto the oxo species.
The above is the same as the well-known reactionbetween hydrogen peroxide (or alkylhydroperoxides)with metal ions, with oxidation of the metal ion andreduction of H2O2, catalysed by ions such as Fe2� orCu�:
2M2��2H��H2O2��2M3��2H2O
2M3��H2O2��2M2��O2�2H�
In this case, however, molecular oxygen is alsogenerated, since the metal ion with higher oxidationstate formed by the reaction catalyses the oxidation ofH2O2 to O2. This occurs because the metal ion has twostable oxidation states – that is M(II)/M(III) – andhydrogen peroxide dismutation is preferred rather thanthe formation of an even higher oxidation state M(IV).
The formation of the oxo metal speciestheoretically may involve either a homolytic or
637VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
Table 1. Summary of the possible species formed by complexation of oxygen to metal ions
Mn��O2�� M(n�1)��O�
2� (superoxo)
M(n�1)��O�2��Mn��� M(n�1)��(O�O)2��
�M(n�1)�(m-peroxo)
M(n�1)��(O�O)2��M(n�1)��� 2 M(n�2)��O2�(oxo)
M(n�2)��O2��Mn��� M(n�1)��O2��M(n�1)� (m-oxo)
M�O�H2O2�� M(OH)�O�OH (metal peracid)
M(OH)�O�OH�� H2O�M (peroxo)
M(n�1)��O�2��� peroxo

heterolytic rupture of the O�O bond in thehydroperoxide. The heterolytic process involves atwo-electron transfer from the metal (increasing inoxidation state from Mn� a M(n�2)�), while thehomolytic process would evidently imply oxidation toM(n�1)�. In this case, a radical HO� (or RO� in the caseof alkylhydroperoxides) is also generated, whichwould be responsible for undesired side reactions dueto its high intrinsic reactivity, as occurs inmetal-initiated autoxidations.
The formation of highly valent oxo metal species hasbeen proposed to occur in monooxygenase enzymes,among which the cytochrome P-450 is the most widelystudied and characterized. Such enzymes catalysereactions in which one atom of oxygen is incorporatedinto an organic substrate, while the second atom ofoxygen is reduced and forms water (Hayaishi, 1974):
R�H �O2�2e��2H���R�OH �H2O
Fe-porphyrin complexes, analogues of cytochromeP-450 (biomimetic systems), form Fe oxo species viapreliminary reduction of molecular oxygen by meansof a sacrificial co-reductant, such as NaBH4, LiBH4,ascorbic acid, or Pt/H2 (Groves, 1985; Ortiz deMontellano, 1985; Dawson, 1988; Mansuy andBattioni, 1989; Shilov, 1989). The mechanism involvesthe reduction of molecular oxygen and the formationof a Fe(III)-peroxo complex, which decomposes to(P��)Fe(IV)O (P�porphyrin), and which, in turn, actsas the active oxidizing agent. After oxidation, theFe(III) formed is reduced back to Fe(II). The use ofH2O2, instead of O2, renders the sacrificial reductantuseless, since the hydrogen peroxide directly forms theoxo metal species (in enzymes, this is referred to asthe hydrogen peroxide shunt).
Oxo metal centres have been claimed to be theactive oxidizing site in Fe(TFPP)N3, {Fe(TFPP)}2Oand Mn(TFPP)N3 catalysts (TFPP�meso-tetraphenylporphyrinato) for the oxidation ofisobutane to t-butyl alcohol with molecular oxygen(Ellis and Lyons, 1989a e 1989b). It is claimed that thecharacteristics of the complexes used make thepresence of the co-reductant unnecessary.
In the reaction of C�H bond hydroxylation inalkanes or in other substrates containing C�H bonds,different mechanisms are possible (Hill, 1989),depending on the nature of the intermediate species thatis formed. The type of reaction pathway is generally afunction of the electron acceptor properties of the oxometal bond. The following mechanisms are possible:• A mechanism of H� abstraction:
Mn��O2��R�H��[M(n�1)��OH� R�]
with the formation of a radical pair, which thenevolves to:
[M(n�1)��OH� R�]��M(n�1)��ROH
Under mild reaction conditions, this mechanism isgenerally accepted as the operating one in liquidphase alkane hydroxylations by oxo metal species.
• H� abstraction, where an ion pair is formed,[M(n�2)��OH� R�], which successively evolvesto the ROH product and to M(n�2)�.
• Electron transfer with formation of a radical ionpair [M(n�1)�]�� [R�H]��.
• H� abstraction, with the formation of an ion pair[(M�OH)� R�].
• Concerted oxygen atom insertion, involving C�Obond formation, which resembles a SN2 transitionstate, where the M�O bond interactsperpendicular to the C�H bond.The radical pair [LxM
(n�1)��OH R�] (L�ligand)of the first mechanism can evolve differently (Hill,1989). Aside from the possibility of acontemporaneous breaking of the M�O bond and theformation of the C�O bond (with formation of thealcohol product), a possible fate of the radical pair isconversion to an hydroxy-organometallic intermediate:(HO)(Lx)M
n��R (Kochi, 1973), which may evolve tothe ROH and LxM
(n�2)� via reductive elimination.Another possible evolution of the radical pairintermediate is conversion into an ion pair by electrontransfer: [LxM
(n�2)��OH R�]. Finally, as pointed outby Hill (1989), formation of the alcohol by hydrolysisof an intermediate ester may also occur, the ester beingformed by the attack of a freely diffusing radical at thependant oxygen. Diffusion of the radicals out of thepair is possible especially in those cases where theradical is particularly resistant towards oxidation (i.e.in primary radicals). Therefore, the fate of theintermediate radical pair is a function of the nature ofthe complex and the type of the C�H bond, which isto be oxyfunctionalized.
Theoretically, an organometallic intermediatecould be obtained via an electrophilic processinvolving the contemporaneous formation of theC�Mn� bond and the O�H bond by cleavage of theC�H bond in the substrate. However, no evidenceexists for such a mechanism in C�H oxygenation byoxo metal species. Instead, this mechanism has beenclaimed in the case of the oxidation of �CH2�groups to the corresponding ketones catalysed by theso-called Gif(III) system, and the subsequentlydeveloped Gif(IV), GO, GoAgg(I) and GoAgg(III)systems (Barton et al., 1989; Sheu et al., 1990; Bartonand Doller, 1991). The ketone can be produced withvery high selectivity.
These systems are essentially constituted of Fe(II),or Fe(III), pyridine and acetic acid, aside from thesubstrate to be oxidized, while the oxidizing agent may
638 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

be either a peroxide or molecular oxygen (with areducing agent). The highly–valent Fe(V)�O speciesis generated, which inserts into the C�H bond(Barton and Doller, 1991):
Fe(III)�O�Fe(V)�O �RCH2R���
�� Fe(III)�O�Fe(V)(OH)�CRR�H
followed by evolution through several steps to theformation of the ketone and to minor amounts of thesecondary alcohol.
Metal peroxo speciesAs mentioned above, free-radical mechanisms are
very important in the metal-catalysed oxidation oforganic substrates, and often they are kinetically themost important process in competition with oxo metaltransfer reactions. Usually, these processes areindiscriminate and necessarily lead to poorselectivities when the substrate has different sitessusceptible to attack. Even so, in some cases, theseprocesses have industrial applications.
Reactions of peroxides (i.e. hydrogen peroxide,alkyl hydroperoxides) with organic substrates, catalysedby metal ions, can be divided into two classes:homolytic, one-electron processes, which involve theformation of free radical intermediates, and heterolytic,two-electrons processes, where the metal forms metalhydroperoxide or metal alkylperoxide complexes, ableto attack the organic substrate.
Homolytic processesReaction between the metal species and the organic
substrate generates the free radical R�:
Mn�+ R�H��M(n�1)�+ R�H�
R�H��B��R��BH�, B � base
The formation of R� by abstraction of H� by the metal,with the formation of a hydride species, can bedisregarded due to the fact that R–H bonds aregenerally stronger than M�H bonds.
The free radical R� can then evolve either to theformation of a carbocation (electron transfer process):
R��Mn���R��M(n�1)�
or via a ligand transfer mechanism:
R��Mn�X��RX �Mn�
The prevailing mechanism is a function of the natureof the ligand in the metal complex, and bothmechanisms can indeed operate at the same time.
The reactivity of alkylperoxides and hydroperoxideswith metal ions is of the above-mentioned type. Metalions, such as Co(III), decompose ROOH, generatingalkylperoxy radicals, ROO�, which initiate autoxidationreactions of organic substrates. A general scheme for
the metal-catalysed radical chain reaction, including thewell-known Haber-Weiss mechanism of ROOHdecomposition, is the following:
M(II)�ROOH��M(III)OH�RO�(reduction step)M(III)OH �ROOH��M(II) �ROO��H2O
(oxidation step)
and the chain reaction is propagated:
RO��ROOH��ROH �ROO�2ROO���2RO��O2ROO��RH��ROOH �R�
In the free radical autoxidation process, metalcatalysis involves the formation of chain-initiatingradicals via reaction with ROOH; therefore, the metalion is more an initiator than a catalyst.
When the metal compound has two oxidation statesof comparable stability, the reduction and theoxidation reactions occur concurrently. Cobalt, iron,copper and manganese complexes are generallyeffective compounds for the homolysis of peroxidiccompounds. Correspondingly, Mn(II)/Mn(III) andFe(II)/Fe(III) are effective catalysts for thedecomposition of H2O2, where the metal acts as bothreductant and oxidant.
Since alkylperoxy radicals are strong oxidants,they oxidize the reduced form of the metal:
M(II) �ROO���M(III)�O�O�R
Therefore, chain propagation can be inhibited bythe formation of this metal alkylperoxo compound,which corresponds to an induction period that isobserved in metal-catalysed autoxidations, especiallyin media of low polarity. The induction period can beeliminated by the addition of small amounts of analkylhydroperoxide to the reaction medium.
Termination of the chain reaction occurs throughthe following reaction:
2ROO���R�O�O�O�O�R��O2�R�2C�O
and again by the above-mentioned reaction, at highmetal concentration, the prevailing mechanism oftermination is M(II)�ROO���M(III)�O�O�R.
The activating effect of some metal ions on theoxidizing activity of H2O2 is well known, as inFenton’s reagent:
Fe(II) �H2O2��Fe(III)OH �OH�
OH��H2O2��H2O �HO2�
(undesired reaction of unproductive H2O2decomposition, which lowers the yield to oxidizedproducts)
Fe(III)OH �HO2���Fe(II) �O2�H2O
The OH� produced generates R� by reaction with
639VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES

organic substrates, finally converted to oxidizedcompounds:
RH �OH���R��H2OR��Fe3���Fe2��R��� productsR��Fe2���Fe3��R�
R��H���RH
Several examples of oxidation reactions performed byperoxides in the presence of metal ions are describedin the book of Sheldon and Kochi (1981).
Some of the most important industrial liquid phaseoxidation processes proceed via an autoxidationmechanism in the presence of molecular oxygen, suchas: oxidation of cyclohexane to adipic acid, oxidationof p-xylene to terephthalic acid, oxidation of toluene tobenzoic acid, oxidation of cumene to phenol viaintermediate cumylhydroperoxide, and oxidation oflinear aldehydes to linear carboxylic acids.Autoxidation processes are indiscriminate and usuallylow selectivities are achieved. Nevertheless, withmolecules that contain only one reactive C�H bond,it is possible to obtain high selectivity, especially iflow conversions are maintained.
In industrial practice, usually an initiator (In) isadded, necessary to start the chain reaction and toform ROOH:
In2��2In� (by thermal decomposition)
In��RH��InH �R�
In the presence of molecular oxygen, the radicalspecies is transformed to a peroxy radical, which thenevolves propagating the chain (rate-determining stepunder usual reaction conditions, at a partial pressure ofmolecular oxygen higher than 0.13 bar):
R��O2��RO2�
RO2��RH��RO2H �R�
The reaction is terminated by the coupling of theRO2� radical. Therefore, hydroperoxides are theprimary products of the reaction network.
Table 2 reports the oxidizability of various organicsubstrates (Howard, 1972), while Table 3 presentssome C�H bond energies (Kerr, 1966; Benson andShaw, 1970). The oxidizability of organic substrates toundergo autoxidation reactions is expressed in [10](see below) by the ratio between the rate constantrelative to the propagation reaction (kpr in Table 2),and the square root of twice the rate constant relativeto the termination reaction via the mutual coupling oftwo alkylperoxy radicals (kter in Table 2). At highsubstrate conversion, the formation of secondaryproducts (aldehydes and ketones) considerably lowersthe selectivity, and thus the conversion is usuallymaintained lower than 20%. Oxidation is rapid if thebond that will be formed in the rate determining step,
ROO�H (bond strength 90 kcal/mol; Benson, 1965),is stronger than the broken bond R�H.
The overall mechanism in the case of the oxidationof alkylbenzenes under oxygen pressure, in acetic acid,and with Co(III) (CH3COO)3 as the catalyst (and alsotraces of an activator, to reconvert Co(II) to Co(III),avoiding long induction periods) is shown in Table 4.The last reaction is inhibited by the high concentrationof Co2�, which traps the benzylperoxy radical.
Nucleophilic attack on peroxo complexesTransition metals such as vanadium, molybdenum,
tungsten, selenium and titanium in their high oxidationstate promote the heterolysis of the O�O bond inH2O2 and in alkyl hydroperoxides; these metals havethe role of improving the electrophilicity of theperoxide complex. An attack on a coordinated peroxoor alkylperoxo ligand by the substrate occurs, usuallywith high specificity. The most important example isthe industrial production of propylene oxide frompropylene and alkyl hydroperoxide, catalysed in boththe homogeneous and heterogeneous phase.
The above-mentioned metal oxides catalyse theH2O2 reactions through the formation of inorganic
640 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
Table 2. Oxidizability of some organic substances(Howard, 1972)
Substrate 103kpr���2kter
Benzaldehyde 290
2,3-dimethyl-2-butene 3.2
Cyclohexene 2.3
Cumene 1.5
Ethylbenzene 0.2
p-xylene 0.005
Toluene 0.01
Table 3. C�H bond energies
Compound Energy (kcal/mol)
CH3�H 103
C3H7�H(C1) 99
C3H7�H(C2) 94
i-C4H9�H(C3) 90
CH2�CH�H 105
CH2�CH�CH2�H 85
C6H5�H 103
C6H5�CH2�H 85

peracids, which undergo heterolytic cleavage of theO�O bond in the presence of nucleophiles;
more generally:
M�O�ROOH��HO�M�O�OR
In olefin epoxidation:
RCH�CHR�HO�M�O�OR���RCH(O)CHR��HO�M�OR�
HO�MO�R��R�OOH��HO�M�O�OR���R�OH
alkyl hydroperoxides are preferred to H2O2 due to thesuperior activity. The retarding effect of H2O2 has beenattributed to the presence of water. A peculiar featureof this reaction is its stereoselectivity for cis and transepoxides from the corresponding alkenes. Asymmetricepoxidation of prochiral allylic alcohols can be carriedout with optically active diethyltartrate catalysts and t-BuOOH, with more than 90% enantiomeric excess(Finn and Sharpless, 1985). The reason why d0
transition metals are specific for this reaction has beenattributed to the fact that interaction between theoxygen atom and vacant t2g
orbitals of the metal ionprovides a low energy route for the formation of theC�O bond from the peroxometallocycle adduct(Purcell, 1985).
Different mechanisms have been proposed forthe oxygen transfer in the epoxidation of olefins.One mechanism involves nucleophilic attack ofthe double bond on the alkyl hydroperoxide, byone of the different activated complexes, as shownin Fig. 1 (Sheldon, 1973; Chong and Sharpless,1977).
Fig. 2 reports the mechanism proposed by Clericiand Ingallina (1993b) for the epoxidation of alkenesby H2O2 over Ti-silicalite catalysts. A five-memberedring species is the intermediate compound formed byinteraction between the Ti sites in silicalite and H2O2,also including the interaction with the solvent ROH.The epoxidation rate was found to be the highest withmethanol as the solvent, and lowest with the bulky t-butanol, due to the decreased electrophilicity andincreased steric constraints of the cycle (Clerici, 1991;Clerici et al., 1991; Bellussi et al., 1992). Analternative mechanism (Mimoun, 1982) involves theaddition of the alkene double bond to form a
641VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
Table 4. Aerobic oxidation of alkylbenzenes catalyzed by a CoIII complex
ArCH3�Co3���ArCH3���Co2� O�
2�
ArCH3�� ��ArCH�2�H�
ArCH�2�Co3���ArCH2��Co2� (in the absence of O2)
ArCH2��AcOH��ArCH2OAc�H�
ArCH�2�O2��ArCH2O�2 (in the presence of O2)
ArCH2O�2�Co2��� ArCH2O2Co2�
ArCH2O2Co2� �� ArCHO�HOCo3�
HOCo3��HOAc��AcOCo3��H2O
ArCH2O�2�ArCH3��ArCH2OOH�ArCH�2
O O
MO3 � H2O2 HO M O OH
M O
OOH
OR
H
O
O
M OR
OROR
OH
OM
O
MM
O
O
R
� epoxide
� epoxide � ROH
� epoxide
Fig. 1. Proposed mechanisms for olefin epoxidation,involving direct oxygen transfer by the alkyl hydroperoxide, in ROH solvent (adapted from Sheldon and Kochi, 1981).
O
OO
H
R
SiOROH, H2O2
SiOSiO HTi
O
O
H
R
H
Ti
O
OO
H
R
SiO
SiOSiO
HTi
SiO
SiO
SiO
SiOTi
C
C
alkene
� epoxide
Fig. 2. Proposed mechanism for olefin epoxidation withH2O2 catalyzed by Ti-silicalite, in ROH solvent (Clerici and Ingallina, 1993).

peroxometallocycle, thereafter releasing the epoxide(Fig. 3). Another mechanism involves the oxo metalmoiety, which is converted to a metal peroxo (Mimounet al., 1970) complex – the active site.
Metal-centred oxidation of coordinated substratesThis class of reactions involves the oxidation of a
coordinated substrate by a metal ion. Examplesinclude the Pd(II)-catalysed oxidation of olefins(Wacker process), and the synthesis of ketones by theoxidehydrogenation of alcohols. The role of molecularoxygen is to regenerate the high oxidation state of themetal. The substrate is activated towards nucleophilicattack by the metal through the formation of a p-complex with the metal. The overall reaction is anoxidative nucleophilic substitution of hydrogen withtwo-electron reduction of the metal; therefore, theprocess is heterolytic. Group VIII metal salts, such asPt(II), Ir(III), Ru(III) and Rh(III), oxidize olefins inthe same way:
Pd(II)X2�RCH�CH2�H2O��RCOCH3�Pd(0)�2 HX
Nucleophilic attack of water on a Pd(II)-ethylene pcomplex forms ethanol, which remains coordinated bya Pd�C s bond (Fig. 4; Drago and Beer, 1992). Thedissociation of a Cl� ligand from the complex and theelimination of hydride from the ethanol form a vinylalcohol p complex with Pd(II). The formation of thealdehyde is obtained by tautomerization of thedissociated vinyl alcohol from the complex andreduction of Pd(II) to Pd(0). Reoxidation of palladiumis achieved through a Cu(II) co-catalyst, which isreduced to Cu(I), and then finally reoxidized bymolecular oxygen to Cu(II).
Kinetics of liquid phase oxidationof hydrocarbons
Analysis of the kinetics of liquid phase oxidationcan be made according to the nature of the reactionmechanism: reactions that involve the homolyticscission of the C�H bond in the organic substrate,thus involving free radicals (autoxidation), where themetal catalyst acts more like an initiator, and reactionsthat involve the heterolytic scission of the C�H bond,where the substrate coordinates to a metal ion catalyst.
Autoxidation processes are of fundamentalimportance in the chemical industry, and a greatnumber of reactions imply this kind of mechanism, asshown in Table 5 (Sheldon and Kochi, 1976; Carrà andSantacesaria, 1980; Santacesaria and Pimpinelli,1986).
The process is autocatalytic and exhibits aninduction period, an acceleration (controlled by thepropagation and branching reactions) and a
deceleration (controlled by termination reactions). Theinduction period is due to the difficulty in breaking theC�H bond and can be overcome by the addition ofsmall quantities of initiators, which readily decomposein the reaction medium.
In a typical air-sparged system, in the zone near thesparger, the liquid contains higher concentrations ofmolecular oxygen and the latter can rapidly scavengeradicals via the reaction:
R��O2��ROO�
Therefore, the overall rate is controlled by thechemical reaction. Under these conditions, the reactionis zero order with respect to molecular oxygen, and theapparent activation energy is typical of reactions underchemical reaction control. The concentration ofmolecular oxygen in the bubbles progressivelydecreases, going from the bottom to the top of thereactor, and the mass transfer of molecular oxygenfrom the gas to the liquid phase can become ratelimiting, since the reaction between an alkyl radicaland oxygen is extremely fast. Under these conditions,the supply of oxygen can become first order withrespect to O2 partial pressure. In this zone, theapparent activation energy falls to low values, �5 kcal/mol.
The main reactions involved in the process ofhomolytic oxidation are summarized in Table 6. Whenmolecular oxygen is present in relatively high
642 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
MO
O
R
�� MO
O
R
MMOR
O
OR
O
Fig. 3. Mechanism for olefin epoxidation involving aperoxometallocycle intermediate(Mimoun, 1982).
Cl OH2Pd
Cl�H2O
�H�PdCl4
2� � C2H4 � H2O
�Cl�Cl OH2Pd
Cl
CH2CH2OH
OH2Pd
Cl
CH2CH2OH
H
CH3CHO � Pd0 � HCl � H2O
OH2Pd
Cl
OH
Fig. 4. Mechanism of oxidation of ethylene to acetaldehyde(Wacker process).

concentrations (for oxygen pressures usually higherthan 0.13 atm in the gas phase), the rate determiningstep is the propagation reaction, kox�104�105 kpr(Table 6). The RO2� species is thus the predominant oneand mainly contributes to termination reactions. Whenthe contribution of branching reactions is negligible,because ROOH is not decomposed under the reactionconditions (thus its contribution to the generation ofradicals is negligible and it is the main product ofreaction), under these conditions the rate of oxygentransformation may be expressed as the balancebetween oxygen consumption in oxidation and oxygenformation in termination:
d [O2][1] �14421�rox�rter�kox[R�][O2]�kter[RO2�]2
dt
On the other hand, under stationary conditions, therate of R� formation (initiation plus propagation) isequal to the rate of R� disappearance (oxidation),provided that termination reactions involving the R�species can be neglected. Therefore:
[2] kox[R�][O2]� kpr[RO2][RH]�rin
Under stationary conditions, the rate of initiationequals the rate of termination, and therefore:
[3] rin� 2kter[RO2�]2
which gives:
rin[4] [RO2�]��14421�0.5
2kter
By replacing the corresponding terms in theexpression for oxygen disappearance, it is possible toobtain:
d [O2] rin rin[5] �14421�kpr�1242�
0.5
[RH]�1dt 2kter 2
The term relative to the rate of initiation can beneglected, and the final general relationship isobtained for the rate of oxygen (or hydrocarbon)depletion:
d [O2] rin[6] �14421�kpr�1242�0.5
[RH]dt 2kter
This expression is independent of oxygenconcentration, and is thus generally valid underconditions of high oxygen concentration. A moregeneral expression has been proposed (Twigg, 1962;Franz and Sheldon, 1991), which considers all of themore important steps in the reaction scheme (reactions3 and 4 in Table 6 for propagation, reactions 7 and 9for termination, with corresponding kinetic constantskpr3, kpr4, kter7 and kter9):
d [O2][7] �14421�
dt
rin0.5 kpr4kpr3[RH][O2]
�12111111111111124341142{kpr4kter7
0.5 [RH] �kpr4kter90.5 [O2]�kter9
0.5 kter70.5 rin
0.5}
Two limit situations can be considered. The firstregards oxygen concentrations approaching zero.Under these conditions, the rate of reaction isproportional to the oxygen concentration in the liquidphase, since the term at the denominator containing[O2] can be neglected. If, instead, the oxygenconcentration is very high, the rate of reactionbecomes independent of oxygen concentration and therelationship of [6] is valid, where kpr�kpr4 and
643VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
Table 5. Most important industrial processes involving liquid-phase oxidation of hydrocarbons through homolytic mechanisms
Reactant Product Catalyst
n-butane Acetic acid Co acetate/acetic acid
Isobutane t-butylhydroperoxide –
Naphtha Acetic acid Co acetate/acetic acid
Waxes (C18-C30) Fatty acids Mn salts/acetic acid
Cyclohexane Cyclohexanol/cyclohexanone Co naphthenate
Cyclohexanol/o Adipic acid Mn-Cu acetate
Toluene Benzoic acid Mn or Co bromide/acetic
Benzoic acid Phenol Cu-Mg benzoate
p-xylene Terephthalic acid Mn or Co bromide/acetic
Cumene Cumylhydroperoxide –
Acetaldehyde Acetic acid Mn acetate/acetic acid
Acetaldehyde Acetic anhydride Cu–Co acetate/ethyl

2kter�kter9. On the other hand, since the value of kpr4depends to a large extent on the nature of the organicsubstrate, when it is very high (e.g. in the case ofaldehydes), the first term in the denominator of thegeneral equation is dominant and, therefore, theequation becomes:
d [O2][8] �14421�rin0.5kpr3[O2]kter7
�0.5
dt
The rate of the reaction is thus proportional to theoxygen concentration. The value of oxygenconcentration at which the passage from first-orderdependence to zero-order dependence occurs istherefore a function of the nature of the organicsubstrate. For aldehyde oxidation, the reaction dependson oxygen even at relatively high partial pressures,while for cyclohexane oxidation, the reaction dependson oxygen partial pressure up to 0.2-0.3 atm, and forcumene oxidation up to 1.5-1.7 atm (Ladhaboy andSharma, 1969; Manor and Schmitz, 1984; Andrigo etal., 1992).
The ratio [kpr /(2kter)0.5] represents the
susceptibility of the substrate towards oxidation (seeagain Table 2; Sheldon and Kochi, 1976). Thepropagation rate is affected by the C�H bond energy(the homolytic scission of this bond is involved and itis necessary that the ROO�H bond, which is going tobe formed, is at least energetically equivalent to theC�H bond to be broken) and also by the reactivity ofalkylperoxo radicals (Howard, 1973; Sheldon andKochi, 1976; Gates et al., 1979). The latter are morereactive in the presence of an electron-withdrawingsubstituent.
Under conditions at which the hydroperoxide is notstable, decomposition of the latter generates RO� and
OH� radicals, which can lead to very fast reactions:
RO��RH��ROH �R�OH��RH��H2O �R�
The prevailing termination reaction again involvesthe RO2� species, since the RO� and OH� radicals(generated by ROOH decomposition) are readilyconverted. Under these conditions, the rate expressionfor RH transformation becomes:
d [RH] 3k2pr[RH]2
[9] �144231�144114441dt kter
which takes into account that RH is consumed throughreaction with RO2�, RO� and OH�, and that threemolecules of RH are consumed for each molecule ofROOH that has decomposed. This expressionrepresents a limit situation, and hence gives themaximum rate that can be achieved. More generally:
d [RH] n kpr[10] �144231�1 �4411341 [RH]�dt f (2kter)
0.5
where n is the number of radicals produced for eachROOH decomposed and f is the fraction of RHconsumed via attack by RO2�. Once again, the term[kpr /(2kter)
0.5], representing the relative importance ofpropagation and termination reactions, plays animportant role in determining the rate of substratetransformation.
The presence of a metal ion acting as a catalystmay increase the activity of the autoxidation process.The catalytic action is related to the redox property ofthe metal ion, that is, to the redox potential for thecouple Mn�/M(n�1)�. One of the most importanteffects is to favour the decomposition of thehydroperoxides:
ROOH�M(n�1)���RO��OH��Mn�
ROOH�Mn���RO2��H��M(n�1)�
Clearly, only those metals having two oxidationstates of comparable stability (i.e. cobalt andmanganese ions) can perform this catalytic cycle(Sheldon and Kochi, 1976). From the kinetic point ofview, [10] can be used to calculate the maximum rateof reaction, assuming that n�1 and f�2/3. In this case,where the catalyst is only involved in thedecomposition of ROOH, n/f�1.5.
The metal can also interact directly with thesubstrate:
Mn��RH��M(n�1)��H��R�R��O2�
�RO2�M(n�1)��ROOH��Mn��RO��OH�
Also in this case, the kinetic law obeyed is thesame as for the previous case, with n�2 and f�1/3. Inall of these cases, therefore, the metal acts as an
644 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
Table 6. Reaction scheme for autoxidation of organic substrates
1 In2 �� 2 In� (initiation)
2 In��RH�� R��InH (initiation)
3 R��O2�� RO�2 (propagation by oxidation)
4 RO�2 �RH�� ROOH�R� (propagation)
5 2ROOH�� RO��RO�2�H2O (branching by bimolecular decomposition)
6 ROOH�� RO���OH (branching by unimolecolar hydroperoxide decomposition)
7 2R��� non radical products (termination)
8 R��RO�2 �� non radical products (termination)
9 2RO�2 �� O2�non radical products (termination)

initiator, decreasing the induction period andeventually favouring the oxidation process at lowertemperatures.
When the bivalent oxidation state of the catalyst isonly involved in ROOH decomposition and thetrivalent state in the reaction with RH, then n/f�6.Intermediate values of the n/f ratio, between 1.5 and 6,indicate that the catalyst not only takes part in theredox reaction with peroxidic compounds, but alsoreacts with RH through the trivalent oxidation state.
In industrial practice, small amounts of substanceshighly prone to autoxidation are added to the reactionmedium, which will readily give rise to a co-oxidationeffect and provide the hydroperoxides for maintainingthe chain, thus leading to an increase in the reactionrate. In this way, it is possible to avoid undesired co-oxidation on the desired products of partial oxidation(i.e. aldehydes, ketones), which are more easilysubjected to autoxidation than the reactant.
The kinetic expressions described above are oftenin good agreement with the experimental limiting rate.However, this is not the case for the oxidation ofalkylaromatics catalysed by cobalt acetate, where thereaction is first order upon the hydrocarbon and alsodepends on the catalyst concentration (Chester et al.,1977). This is due to the different reaction mechanisminvolved.
Regarding oxidation reactions occurring viaheterolytic dissociations of the C�H bond in thesubstrate, they are catalysed by V, W, Mo and Ti ionsthrough the transfer of two electrons of the substratecoordinated to the metallic complex. In other cases,such as the Wacker process, the action of two redoxcouples – Pd(II)/Pd(0) and Cu(II)/Cu(I) – is necessaryfor the catalytic cycle. In the mechanism, the ratelimiting step is the conversion of the p-complexbetween the olefin and PdCl4
2� into a s complex, theother stages being at equilibrium. The rate expressionfor ethylene disappearance, which better describes theexperimental results, is the following (Moiseev et al.,1974):
d [C2H4] [PdCl42�][C2H4]
[11] �1442231�k 14411144114441dt [Cl�][H3O
�]
Air vs. oxygen in liquid phase oxidationsThe use of molecular oxygen in liquid phase
oxidations may provide a significant improvement inthe reaction kinetics when the overall process iscontrolled by the reaction and the rate depends on theoxygen partial pressure over a wide interval of oxygenconcentration (though not often the case, as describedabove). Indeed, the oxygen concentration in thebubbles (in addition to the continuous gaseous phaseat the top of the reactor) is 100% (neglecting the
vapour pressure of the liquid), against an average 5mol% in the case of air as the oxidizing agent, thusimproving the transfer of molecular oxygen as well asthe rate of reaction. As a consequence, similarproductivities as with the air-based process can beachieved at significantly lower temperatures andpressures. This may lead to a considerableimprovement in selectivity, when the reaction networkconsists of consecutive reactions of oxidativedegradation (usually characterized by higher activationenergy than the selective reaction) to the desiredproduct.
Lower temperatures also entail less solvent loss. Itmay also happen that an increase in the oxygen partialpressure, as a consequence of feeding oxygen insteadof air, leads to a considerable improvement in theoxygen transfer rate, while the rate of reaction is notmuch affected by the variation in oxygenconcentration. Under these conditions, a passage froma mass-transfer limited process (as is usually the casefor the oxidation of liquid hydrocarbons) to akinetically controlled process can occur, and thus thereaction is transferred from the film surrounding thebubble (at the interface between the gas and the liquidphase) to the continuous liquid phase. This may alsochange the temperature distribution from aconsiderable temperature gradient in the film in theformer case (where the heat of reaction develops in thethin film) to a lower, more uniform temperature in thelatter case. The above may considerably improve theselectivity.
The presence of a high oxygen concentration in thereaction zone is also important for favouring theoxidation reactions against the reactions betweenintermediates, leading to undesired high-molecularweight compounds. A high oxygen concentration inthe reaction zone is obtained when the process iskinetically controlled or when, under mass-transferlimiting conditions, the rate of reaction is not verymuch higher than the rate of diffusion (therefore,concentration gradients in the film are not toopronounced). Higher oxygen concentrations may thuslead to a higher oxygen concentration in the reactionzone. Another advantage in the use of oxygen insteadof air is the reduction in total gas throughput, with aconsequent reduction in the energy required for gascompression as well as in the amount of vent gas(which is also much more concentrated in the organiccontaminants, due to the absence of diluent nitrogen)to be treated, with large gas treatment equipment.Therefore, the nitrogen favours stripping of the solventand volatile organic compounds from the solution,compounds which have to be removed to conform withthe regulations for environmental protection.
645VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
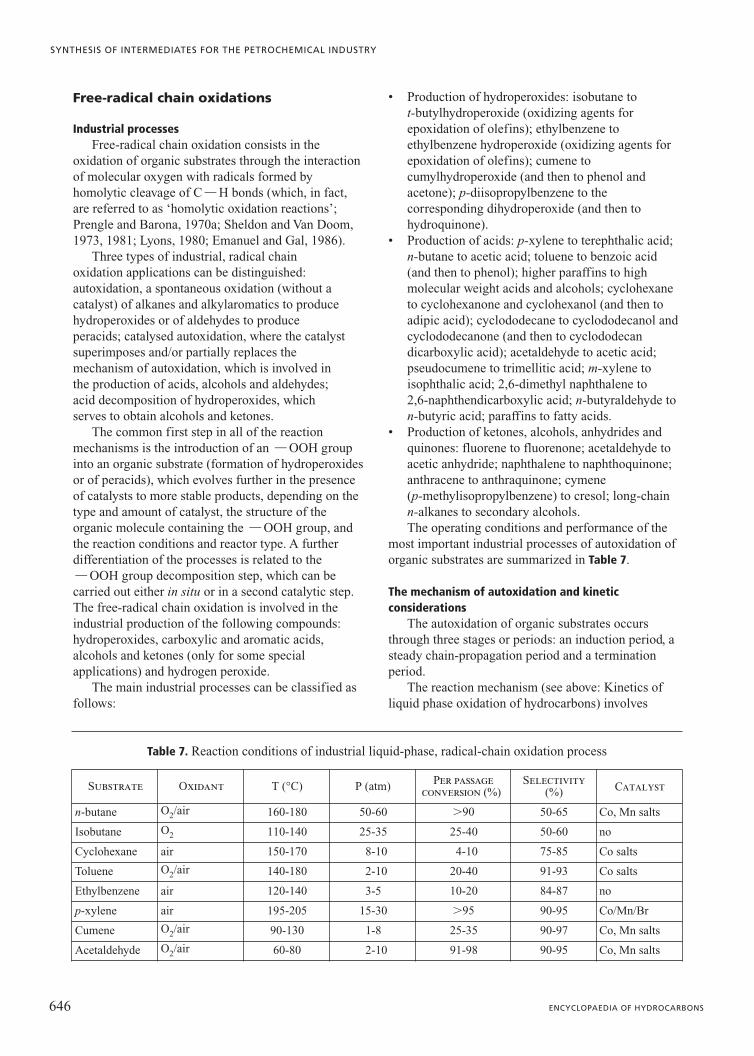
Free-radical chain oxidations
Industrial processesFree-radical chain oxidation consists in the
oxidation of organic substrates through the interactionof molecular oxygen with radicals formed byhomolytic cleavage of C�H bonds (which, in fact,are referred to as ‘homolytic oxidation reactions’;Prengle and Barona, 1970a; Sheldon and Van Doom,1973, 1981; Lyons, 1980; Emanuel and Gal, 1986).
Three types of industrial, radical chainoxidation applications can be distinguished:autoxidation, a spontaneous oxidation (without acatalyst) of alkanes and alkylaromatics to producehydroperoxides or of aldehydes to produceperacids; catalysed autoxidation, where the catalystsuperimposes and/or partially replaces themechanism of autoxidation, which is involved inthe production of acids, alcohols and aldehydes;acid decomposition of hydroperoxides, whichserves to obtain alcohols and ketones.
The common first step in all of the reactionmechanisms is the introduction of an �OOH groupinto an organic substrate (formation of hydroperoxidesor of peracids), which evolves further in the presenceof catalysts to more stable products, depending on thetype and amount of catalyst, the structure of theorganic molecule containing the �OOH group, andthe reaction conditions and reactor type. A furtherdifferentiation of the processes is related to the�OOH group decomposition step, which can becarried out either in situ or in a second catalytic step.The free-radical chain oxidation is involved in theindustrial production of the following compounds:hydroperoxides, carboxylic and aromatic acids,alcohols and ketones (only for some specialapplications) and hydrogen peroxide.
The main industrial processes can be classified asfollows:
• Production of hydroperoxides: isobutane tot-butylhydroperoxide (oxidizing agents forepoxidation of olefins); ethylbenzene toethylbenzene hydroperoxide (oxidizing agents forepoxidation of olefins); cumene tocumylhydroperoxide (and then to phenol andacetone); p-diisopropylbenzene to thecorresponding dihydroperoxide (and then tohydroquinone).
• Production of acids: p-xylene to terephthalic acid;n-butane to acetic acid; toluene to benzoic acid(and then to phenol); higher paraffins to highmolecular weight acids and alcohols; cyclohexaneto cyclohexanone and cyclohexanol (and then toadipic acid); cyclododecane to cyclododecanol andcyclododecanone (and then to cyclododecandicarboxylic acid); acetaldehyde to acetic acid;pseudocumene to trimellitic acid; m-xylene toisophthalic acid; 2,6-dimethyl naphthalene to2,6-naphthendicarboxylic acid; n-butyraldehyde ton-butyric acid; paraffins to fatty acids.
• Production of ketones, alcohols, anhydrides andquinones: fluorene to fluorenone; acetaldehyde toacetic anhydride; naphthalene to naphthoquinone;anthracene to anthraquinone; cymene(p-methylisopropylbenzene) to cresol; long-chainn-alkanes to secondary alcohols.The operating conditions and performance of the
most important industrial processes of autoxidation oforganic substrates are summarized in Table 7.
The mechanism of autoxidation and kineticconsiderations
The autoxidation of organic substrates occursthrough three stages or periods: an induction period, asteady chain-propagation period and a terminationperiod.
The reaction mechanism (see above: Kinetics ofliquid phase oxidation of hydrocarbons) involves
646 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
Table 7. Reaction conditions of industrial liquid-phase, radical-chain oxidation process
Substrate Oxidant T (°C) P (atm) Per passageconversion (%)
Selectivity(%) Catalyst
n-butane O2/air 160-180 50-60 90 50-65 Co, Mn salts
Isobutane O2 110-140 25-35 25-40 50-60 no
Cyclohexane air 150-170 8-10 4-10 75-85 Co salts
Toluene O2/air 140-180 2-10 20-40 91-93 Co salts
Ethylbenzene air 120-140 3-5 10-20 84-87 no
p-xylene air 195-205 15-30 95 90-95 Co/Mn/Br
Cumene O2/air 90-130 1-8 25-35 90-97 Co, Mn salts
Acetaldehyde O2/air 60-80 2-10 91-98 90-95 Co, Mn salts

initiation, propagation, branching and terminationreactions.
Initiation reactions. The initiator reacts with thesubstrate RH, generating radicals R�:
In2��2In�
In��RH��R��InH
or where the organic substrate is itself an initiator:
RH��R��H�RH�O2�
�R��HO2�2RH�O2��
�2R��H2O2
The initiation can be brought about by theintroduction of thermally unstable compounds, such asperoxides or azocompounds, generating radicals ormetal ions, which autoxidize more readily than RH.The initiation step requires a finite time to develop asufficient concentration of radicals to support thepropagation step. Usually, the decomposition of thehydroperoxide formed with the reaction is the sourceof radicals (i.e. branching reactions, see below). In thiscase, the reaction temperature is related to thetemperature of decomposition of the hydroperoxide.For this reason, in industrial processes, usually theproduct is partially recycled to the first reactor in orderto furnish the necessary concentration of initiator.
Propagation reactions. The radicals generatedreact with molecular oxygen forming RO2�, which inturn reacts with RH, generating other R�:
R��O2��RO2�
RO2��RH��R��ROOH
Branching reactions. These reactions are due to thedecomposition of alkylhydroperoxy species togenerate RO�, ROO�, OH� radicals. The nature of theproducts formed in the branching reactions depends onthe type of substrate.
ROOH��RO���OH2ROOH��ROO��RO��H2ORO��RH��R��ROHOH��RH��R��H2OROOH�ROH��2RO��H2O
Termination reactions. The radicals generatenon-radical products:
2R���R2R��RO2��
�ROOR2RO2��
�ROOR�O2RO��R���ROR
The recombination of radicals to terminate thechain reaction occurs more easily with large radicals,whose lifetime is longer than for small radicals. Infact, large radicals are less reactive because theydissipate energy by atomic vibrations. The termination
rate depends on the structure of the radical and also onthe oxygen partial pressure. In all industrial processes,at the end of the reactor, the partial pressure of oxygenis maintained practically nil, mainly for safety reasons.Therefore, radicals accumulate and termination occursby their recombination. The presence of foreignmolecules (impurities) that interact with radicals canalso be the source for termination reactions.
The formulation of kinetic expressions for theautoxidation of organic substrates must not only takeaccount of the reactions occuring in the liquid phase,but also of the transfer of molecular oxygen from thegaseous to the liquid phase (Prengle and Barona,1970b; Hobbs et al., 1972a; Astarita et al., 1983;Doraiswamy and Sharma, 1984). When the entireprocess is either mass-transfer limited or reactionlimited, more simple rate expressions can be used,which are representative of the rate-determining step.This is usually the case for liquid phase autoxidationprocesses, where the diffusion of oxygen is very quickfor oxygen partial pressures above 0.1-1.0 atm (thelimit value depending mostly on reaction conditions),while it becomes rate-controlling when the oxygenpartial pressure is lower than these values (thistypically occurs in continuous stirred reactors; Uri,1961; Bamford and Tipper, 1980).
However, general rate expressions are necessarywhen the rate determining-step changes in the samereactor. For instance, in bubble reactors where theoxygen partial pressure progressively diminishes alongthe reactor, one can move from a reaction-limitedregion (where the oxygen partial pressure is high) to aregion where the process is oxygen-transfer-limited(Hobbs et al., 1972a; Jacobi and Baerns, 1983). Incorrespondence, one may move from a region wherethe apparent activation energy is higher than 20kcal/mol to a region where it is very low, typical ofdiffusional phenomena (less than 5 kcal/mol).
The situation becomes even more complex whentaking into account the very complex reactionmechanism, with the formation of many by-productsand the participation of several intermediate chemicalspecies (i.e. radical compounds). However, it is usuallypossible to adopt simplified kinetic models, where theconcentrations of radical intermediates are not takeninto consideration (i.e. lumped models). This type ofapproach has been utilized for several differentexamples of liquid phase catalytic oxidation reactions(Cavalieri d’Oro et al., 1980; Chen et al., 1985;Krzysztoforski et al., 1986; Morbidelli et al., 1986;Cao et al., 1994).
The direct reaction between RH and O2 to yieldROOH is thermally neutral and is characterized by ahigh activation energy (�35 kcal/mol). Therefore, it isvery slow at temperatures below 150°C. The cleavage
647VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES

of RH by reaction with RO2� is 104 to 106 times slowerthan the reaction between R� and O2 , and thus theformer reaction is usually the rate-determining step.The rate expression then becomes (in a chemically-controlled regime):
d [RH][12] �144231�rpr�kpr[RO�][RH]
dt
Under these conditions, the rate expression doesnot depend on oxygen partial pressure. Since thereaction between the R� species and oxygen is veryquick, often total molecular oxygen consumption isalready achieved in the liquid film.
The reaction between O2 and R� becomesrate-determining only when the partial pressure ofoxygen is very low (below approximately 0.15 atm);the rate of reaction then becomes dependent uponmolecular oxygen and is strongly limited by oxygentransfer into the liquid phase:
d [O2][13] �144231�rox�kox[R�][O2]dt
Therefore, both propagation reactions andoxidation reactions must be taken into account whendeveloping the rate expression. Termination occursmainly by combination of ROO� and R�; thecorresponding rate expression is:
[14] rter�kter[R�][ROO�]
By applying the steady-state approximation toradical compounds ROO� and R�, the followingexpression can be obtained (for low oxygen partialpressure):
d [O2] koxkpr[15] �144231�1313 [O2][RH]dt kter
Under these steady-state conditions, the mass-transfer rate of molecular oxygen (OTR):
[16] OTR �akL([O2]*�[O2])
is equal to the rate of oxygen consumption in theliquid phase, where a is the specific interfacial area, kLis the liquid film mass-transfer coefficient formolecular oxygen and [O2]* is the oxygenconcentration at the gas-liquid inter-phase. A generalrate expression that takes into account both thechemical and diffusional steps is achieved bycombining the two expressions:
koxkpr[17] 144231 [O2][RH]�akL([O2]*�[O2])kter
d [O2] koxkpr[18] �144231�1313 [O2][RH]dt kter
and by assuming a negligible gradient in the gaseous
film, and thus [O2]*= PO2*
/H = PO2/H (where H is the
Henry constant for molecular oxygen and PO2is the
partial pressure of molecular oxygen in the gas phase),the following expression is finally obtained (Jacobiand Baerns, 1983):
d [O2] PO21 kter
[19] �144231�123 �133�11211��1
dt H akL koxkpr[RH]
Higher rates of hydroperoxide formation areobserved from substrates, which present either morelabile C�H bonds or form more stable radicals. Inthe alkane series, the rate is higher for tertiary C�Hbonds, followed by bonds in secondary and primarycarbon atoms. In arylalkanes, the order is thefollowing: tertiary secondary primary benzylic. This order is due more to the instability ofthe radicals formed than to the lability of the C�Hbonds (which is the reason for the lower oxidizabilityof toluene).
The role of catalystTwo types of catalysts are used in the industrial
processes of autoxidation. The first type consists ofcatalysts that superimpose their action on that of theclassical autoxidation by favouring the decompositionof �OOH groups and by addressing thedecomposition profile selectively towards theformation of different, more stable products. Thesecatalysts are either transition element ions or acid/basesystems. The second type includes catalysts that areable not only to decompose the �OOH group, butalso to directly activate the substrate. Therefore, thesecatalysts also participate in the initiation step. They areCo(III) in acetic acid (unique amongst the transitionelements), the MIC (Amoco Mid Century) catalystbased on Co/Mn/Br ions in acetic acid, and Co(III)with a co-oxidant.
Redox catalysts active in the decomposition ofhydroperoxide
Salts of elements of the first row of the transitionseries, which present one-electron redox couples suchas Co(II)/Co(III), Mn(II)/Mn(III), Fe(II)/Fe(III),Cu(I)/Cu(II) (Co salts are the most commonly used),or metal chelates, which are soluble in the reactionmedium, are used to accelerate the rate ofdecomposition of hydroperoxides through the Haber-Weiss mechanism:
M(n�1)��ROOH��RO2��H��Mn�
Mn��ROOH��RO��OH��M(n�1)�
The total reaction is:
2ROOH��ROO��H2O�RO�
In the first reaction, the electron transfer occurs
648 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

between the hydroperoxide and the Co(III) species,through complexation of the hydroperoxide, whichweakens the O�O bond and facilitates thebreakdown of the complex and the formation of theRO2� radical. The second reaction is a reduction byCo(II) with the formation of a RO� radical. The secondstep is very rapid, due to the high oxidation power ofhydroperoxides, while the first step has a very slowrate of reaction since hydroperoxides are weaklyreducing agents. Therefore, optimal catalysts are thosewhich contain ions with high oxidation power in orderto increase the rate of the first step. In fact, Co and Mnions, which are present in the best catalytic systems,have a very high redox potential: 1.82 eV forCo(III)�e���Co(II); 1.51 eV for Mn(III)�e���Mn(II).
Only one-electron redox couples are to be used inorder to avoid side reactions. The transition elementmust be used in an organic solvent; in order to increasethe solubility of the catalyst, carboxylic acids ornaftenic acid must be used as the solvent.
In the oxidation of hydrocarbons, the concentrationof metal ranges from 1 to 500 ppm, while in the caseof aldehyde oxidations, it ranges from 0.001 to 0.1ppm. Higher concentrations of metal ions have anegative effect, since the catalyst decomposes all ofthe hydroperoxide and the rate of oxidation decreasesbecause there are not enough radicals for the initiationstep. Therefore, the role of the transition element is todecrease the activation energy relative to thedecomposition of the hydroperoxide, thus allowing thereaction to occur at lower temperatures and with highenough rates for industrial application. Moreover, byoperating at lower temperatures, the selectivity can bebetter controlled.
The type of products obtained in the catalyticdecomposition of hydroperoxides depends mainly ontheir structure, but also on the type of catalyst usedand the reaction conditions. For example, t-butylhydroperoxide is decomposed at 45°C in thepresence of Co(II) octanoate to yield 88% t-butylalcohol and only 1% acetone (besides molecularoxygen and the dimer of the t-butoxy radical; Hiatt etal., 1968). A similar distribution of products isobtained by decomposition of cumylhydroperoxide,with the formation of the tertiary alcohol (95%) and aproduct of ketonic cleavage (5%). Under the sameconditions, sec-butylhydroperoxide decomposes to61% sec-butylalcohol and 36% methylethylketone,while n-butylhydroperoxide yields 67% n-butanol and32% butyraldehyde. For all hydroperoxides, alcohol isthe main product of decomposition, but other by-products form depending on the nature of thehydroperoxide. The cleavage of a C�H bond in asecondary or primary alkoxy radical to yield thecorresponding carbonyl compound is easier than the
cleavage of a C�CH3 bond in a tertiary radical(Lyons, 1980). The C�C cleavage reaction occurswhen methylenic groups are near the carbon atom, asin 3-methylpentane (Lyons, 1980). An important roleis also played by the transition metal in directing thedecomposition of the hydroperoxide to differentproducts.
The decomposition product of hydroperoxides orperacids can be further transformed to different typesof compounds, depending on the type of transitionmetal used.
Acid and basic catalysts active in the decompositionof the hydroperoxide
Acid can also cause the catalytic decomposition ofthe hydroperoxide. This is the case with phenol andacetone production by acid decomposition ofcumylhydroperoxide. For this reason, an emulsionwith a base is usually used as the reacting medium inthe production of the hydroperoxide, in order to avoidits decomposition due to the occasional presence ofacidity.
Catalysts for the direct activation of the organicsubstrate
Co(III) in acetic acid acts as an initiating speciesreacting with the hydrocarbon and forming, through aone electron exchange, a radical carbocation, whichsuccessively releases a proton forming a radicalspecies. Co(III) is the unique element able to activate ahydrocarbon, especially benzylic hydrogens andC�H bonds in aldehydes. Through this mechanism ispossible to carry out an autoxidation reaction at lowertemperatures, as compared to the classical ones.
A second mechanism of activation by Co(III) isindirect and operates through a co-oxidant, thus in thepresence of Mn and Br ions. In this type of catalyticsystem (MIC catalyst), the role of Co(III) is to oxidizemanganese; the latter oxidizes Br� to Br� and is ableto abstract H from less reactive C�H bonds, such asmethyl groups in polymethylated aromatics. Theadvantages of the MIC catalyst with respect toclassical Co-based catalysts include the rate ofreaction is higher, and therefore it is possible to usesmaller amounts of catalyst or to oxidize groups thatpresent lower reactivity, as well as many aromaticacids, and it precipitates in acetic acid, making theseparation of the products easier. Disadvantagesinclude the decarboxylation of acetic acid at hightemperature and the problems tied to materialcorrosion, which make the use of lined apparatusnecessary. An alternative synthesis consists in the useof an organic co-oxidant, as in the case of n-butaneoxidation to acetic acid, where methylethylketone(obtained as a by-product of the reaction and recycled
649VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES

to the oxidation reactor) is the co-oxidant. The latter isfirst activated by cobalt, and then the radical formedactivates n-butane at low temperatures and with highselectivity.
Co-based catalysts can compete with the MICcatalyst, provided that higher amounts of catalyst and aco-oxidant (either acetaldehyde or methylethylketone)are used.
Reaction conditions and reactor typesAll reactions are carried out in the liquid phase at
temperatures between 75 and 200°C, and at a pressureranging from 3 to 70 atm (to keep oxygen dissolved inthe liquid phase). The pressure must be high enough tomaintain the boiling temperature of the liquid phasehigher than the reaction temperature, since highvapour concentrations in the bubble phase decrease thesolubility of oxygen.
When the substrate is a gas or a low-boilingliquid, high-boiling solvents are used, which in somecases can be the product itself. Acetic acid is oftenused as the solvent because it is not easily oxidizable,and it has both an optimal boiling temperature (116-118°C) and freezing temperature, which makesit easy to handle. It is easily available, low in cost andit dissolves the majority of aromatics at lowtemperature. In some cases, mixtures of two solventsare used. In the oxidation of m-xylene to isophthalicacid, a mixture of acetic acid and dichlorobenzene isthe solvent for the reaction. With this solventmixture, an increase in selectivity is achieved,ranging from 89 to 91%.
The reactors used can be either towers or stirredtanks. The choice essentially depends on the rate-determining step of the reaction. Often in-seriesreactors are set up, in order to limit the heat of reactionevolved or to minimize the overall volume necessaryfor achieving a defined conversion. Sometimes, batchoperation is preferred over continuous operation instirred reactors, since backmixing phenomena canhave negative effects on selectivity, favouringundesired overoxidation reactions. On the other hand,a certain extent of backmixing can be necessary tokeep the correct concentration of radicals in thereaction medium. A compromize can be reached bystirred reactors in series (Prengle and Barona, 1970b).
An advantage arising from the absence ofdiffusional limitations roots in the non-necessity ofstirring inside the reactor in order to improve the gas-liquid interfacial area and hence the mass-transferrate; therefore, less costly reactors sometimes can be used.
Particular attention must be paid in the design of these reactors to achieve efficient gas-liquid contact,realized either through mechanical stirring or by
gas-spraying devices, or through rapid circulation ofthe reactor content by pumping, as well as efficientheat removal for strongly exothermal reactions,realized either via heat exchangers inside the reactoror by the recirculation of the reacting mixture afterexternal refrigeration and the evaporation of thesubstrate, the product or the solvent.
In all of the processes operating in continuousstirred apparatus or in semi-batch reactors, the oxygendiffusion is the rate-limiting step, due to theconcentration of oxygen in the gas phase, which is keptlow for safety reasons (at the expense of selectivity).The rate of oxygen diffusion is a function of its partialpressure in the bubble phase and of the interfacial area(i.e. of bubble size). Operation under conditions ofmass-transfer resistance may be more favourable, sincethe rate of reaction is practically unaffected byvariations in the temperature (the diffusional constantis characterized by a very low activation energy; Hobbset al., 1972a). Instead, under conditions of chemicalcontrol, fluctuations in temperature can lead to theextinction of the reaction. Complete mass-transfercontrol, on the other hand, may cause a lack of oxygenin the liquid phase, favouring bimolecular reactionsbetween radicals and the termination of the chainreaction, thus decreasing the process efficiency. Incontinuous columns, a stable situation consists inhaving a region where the rate is chemically controlledand another where the process is under mass-transfercontrol. This ensures stable operation, being that theoverall system shows very little dependency ontemperature (Hobbs et al., 1972a).
It is necessary to avoid coalescence of the bubbles,which can be induced by high, turbulent flow rates andheavy agitation. The gas must be introduced throughlarge-area devices. The reactor surface-to-volume ratiohas to be minimized in order to reduce reactions ofradical combination at the wall.
Safety considerations include good control of heatremoval to prevent thermal decomposition of thehydroperoxide and operation at low conversion, solimiting the concentration of hydroperoxide and theextent of consecutive reaction of decomposition. Thiscan be realized by using staged reactors, withintermediate feeding of oxygen. In addition, one mustensure that in the gas phase, the composition is alwaysabove the upper flammability limit of the organicvapour by inertizing with nitrogen and with watervapour. Particular care must be taken in the start-up,when no oxygen is consumed along the reactor, andduring the shutdown of the reactor.
The control of selectivityWhen the hydroperoxide is the final product, the
lowering of selectivity is due to its decomposition
650 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

(with formation of alcohols and ketones). Therefore,low conversions and low temperatures allow higherselectivity to be obtained. Usually, when choosingexperimental conditions, a compromise between purityof the hydroperoxide and productivity (conversion)must be reached. An addition of bases can increase theselectivity by decreasing the extent of hydroperoxidedecomposition, which is catalysed by acids present inthe reaction medium.
When the hydroperoxide is not the final productbut only an intermediate, the presence of the catalystdetermines the selectivity. Also in this case, theconversion can be a key factor to control theselectivity for those products which are more easilyoxidizable than the substrate. This is the case with theoxidation of cyclohexane to cyclohexanol andcyclohexanone. Higher conversion and selectivity canbe reached by the addition of boric acid, whichinteracts with cyclohexanol, forming an ester andpreventing its further oxidation. The ester is finallyhydrolysed to recover cyclohexanol.
A change from a chemically limited rate to a mass-transfer limited rate, which may occuroccasionally due to an increase in the reactiontemperature, can lead to significant changes in thedistribution of products. For instance, in the oxidationof methylethylketone to acetic acid, this leads to achange in the average oxidation state of the cobaltcatalyst, which in the absence of dissolved oxygen isreduced, and to an increase in the formation of CO,methanol and formic acid, due to the followingreactions starting from the acetyl radical undermolecular oxygen starvation (Hobbs et al., 1972a):
CH3CO����CH3�CO�CH3��OH��CH3OHCH3OH�1/2O2�
�HCOOH�H2O
Molecular oxygen or air as the oxidizing agentMolecular oxygen, oxygen-enriched air or air are
used as the oxidants. The choice is dictated by bothsafety and economic considerations (Trifirò andCavani, 1994).
Pure molecular oxygen can not be used; a smallfraction of nitrogen must be present, in a concentrationof higher than 3%, in order to keep the gas phase overthe reacting liquid always outside the higher explosionlimit of the organic vapour. The use of molecularoxygen, or oxygen-enriched air, is advantageous whenthe reaction is mass-transfer controlled, and thus therate depends on the partial pressure of oxygen and onefficient mass transfer. Further advantages in the useof oxygen include the non-necessity for separation oflarge amounts of inert (i.e. nitrogen) before recycling,the use of smaller and less expensive apparatus and
lower entrainment of organic vapours in the waste gas.Air is used for its low cost and its safe handling.
Air can be advantageous when the reaction is underchemical control and the partial pressure of oxygenbears no influence on the yields. Further advantagesare related to the stirring effect of nitrogen as well asthe more favoured dissipation of the heat of reaction.When air is used under pressure, energy can berecuperated in a turbine before venting frompressurized nitrogen.
A good example of the importance in choosing anoxidant roots in the processes for the synthesis ofhydroperoxides starting from isobutane and fromethylbenzene. The corresponding hydroperoxides areused as oxidizing agents in propylene epoxidation. Themain difference between the two oxidation processes isthat in the case of isobutene, molecular oxygen ispreferred and the process operates at high pressure,while in the oxidation of ethylbenzene, air is theoxidant of choice and the process operates at lowpressure. In the latter case, the rate is chemicallycontrolled and, therefore, it is not influenced by thepartial pressure of oxygen. In isobutane oxidation,instead, the rate-limiting step is oxygen diffusion.
Main industrial processes of liquid phaseoxidation
In this section, some of the most importantindustrial processes of liquid phase oxidation oforganic substrates are examined, which well representthe chemistry and technology involved in this class ofreactions. Moreover, the industrial technology of thesynthesis of propylene oxide by oxidation is comparedwith the processes of propylene epoxidation understudy or development, in order to exemplify thecurrent trends and future developments in the field ofliquid-phase selective oxidation.
Autoxidation processesAutoxidation processes have a great importance for
the chemical industry. Despite the usual, lowselectivity obtained in radical-like processes, industrialapplications have widely developed for those reactionsinvolving the use of organic substrates that possessonly one site of preferential attack.
The oxidation of cumene to cumylhydroperoxideThe oxidation of cumene to cumylhydroperoxide:
is carried out at 80-120°C, either at 6 atm of pressure(Hercules process; Fleming et al., 1976), in an
651VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
∆H°� �31 kcal/mol
�O2 OOH
∆H°� �31 kcal/mol

homogeneous solution containing a sodium carbonatebuffer (removed by washing with water beforedistillation; Hercules process), or at atmosphericpressure (Allied process; Sifniades et al., 1982),without the addition of a base (pH�3-5). In the lattercase, however, the stream is washed with caustic waterbefore recycling. Per pass cumene conversion is in therange of 25 to 35%.
The reaction mechanism is a typical free-radicalchain, with initiation (usually accelerated bydecomposition of the hydroperoxide itself),propagation and termination steps. Main by-productsinclude acetophenone, dimethylphenylcarbinol(a, a-dimethylbenzyl-alcohol) and a-methylstyrene.The selectivity to cumylhydroperoxide ranges from 90to 96%. The reaction steps responsible for theformation of by-products are presented in Table 8(chain-branching reactions).
In the buffered solution (pH 7-8), the organic acidsformed (i.e. formic acid), which are responsible for thein situ decomposition of the hydroperoxide withformation of phenol (inhibitor of cumene oxidation),are destroyed.
The reaction is carried out in several (typicallyfour) in-series reactors. Air is the preferred oxidizingagent, for both economic and safety considerations.Fresh air is pumped into each reactor. The lower andhigher flammability limits for cumene/air mixtures are0.8 and 8.8%, respectively. The concentration ofmolecular oxygen at the exit of each reactor is keptlower than 4%, in order to avoid the formation offlammable compositions with cumene (Fleming et al.,1976). The vent gas also contains organic compounds,which are condensed by refrigeration and adsorbed onactive charcoal.
The rate equation for cumene oxidation in theconditions which are industrially applied can beexpressed as follows:
11111
d [O2] 2kin[ROOH][20] �144231�kpr[RH]�141111dt kter
where kin, kpr and kter are the rate constants for chaininitiation, propagation and termination, respectively;RH is cumene and ROOH is the hydroperoxide. Therate of reaction is therefore not dependent on oxygenconcentration (Melville and Richards, 1954). However,this expression is only approximate; detailed kineticstudies indeed have demonstrated that the reactiondoes depend on oxygen up to a partial pressure ofapproximately 0.3 atm at 130°C; at 100°C, the ratedoes not depend on oxygen partial pressure for PO2higher than approximately 0.1 atm (Hendry and Amer,1967; Hattori et al., 1970; Andrigo et al., 1992).
Moreover, the rate expression shows that a certainconcentration of the hydroperoxide is necessary to
obtain acceptable rates of cumene oxidation. Thehydroperoxide contributes to initiation, decomposinginto RO� and OH� species. In fact, long inductionperiods are necessary when the reaction is startedwithout the presence of the hydroperoxide. Therefore,the hydroperoxide is in part recycled (together withunconverted cumene) to the first reactor, whichoperates at 8-10 wt% hydroperoxide. The maximumconcentration of hydroperoxide in the last reactor is 30wt%; higher concentrations, in fact, do not lead tofurther increases in the reaction rate, but rather causean increase in the side reactions ofcumylhydroperoxide decomposition.
The oxidation of cyclohexane to cyclohexanol and cyclohexanone
Cyclohexanol and cyclohexanone (the so-called ol� one mixture) are produced by oxidation ofcyclohexane:
and the products are then used for the synthesis ofadipic acid.
The most accepted reaction pattern for thetransformation of cyclohexane to cyclohexanol//cyclohexanone mixtures consists in the oxidation ofthe cycloalkane to cyclohexylhydroperoxide,followed by its decomposition to the alcohol and tothe ketone. Cyclohexanone is also formed by theconsecutive reaction of cyclohexanol oxidation. Theketone is then oxidized to adipic acid and to otherby-products.
The alcohol and the ketone are more reactive thancyclohexane; therefore, they are easily converted toseveral by-products. High selectivity to alcohol �ketone can be reached only at low conversion. Theselectivity is around 90% at very low cyclohexaneconversion (1-2%), while already at 4-5% conversion,the selectivity drops to 77-85%. Therefore, allprocesses operate at low per–pass cyclohexaneconversion, with recycle of unconverted reactant.
A detailed kinetic study has shown that thereaction rate is initially zero order in oxygen partialpressure, and then becomes first order with decreasingoxygen partial pressure in a batch reactor (Suresh etal., 1988).
The main by-product is adipic acid, whoseselectivity reaches high values only at lowtemperatures (T�100°C). The other by-products, suchas glutaric acid or the esters formed by condensationbetween the acids and cyclohexanol are derived fromthe decarboxylation of adipic acid. Although adipicacid is the final desired product, its formation must be
652 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
∆H°� �70 kcal/mol
OH O � H2O2 �3/2O2 �
∆H°� �70 kcal/mol

avoided at this stage, since it is accompanied byseveral other by-products.
Different processes of oxidation of cyclohexanehave been proposed, which yield either a mixture ofcyclohexanone and cycloexanol, or adipic aciddirectly. Only those processes which producecyclohexanol/cyclohexanone have beencommercialized. Common aspects of the variousprocesses include the very low residence times(around 40 min), necessary to limit the consecutivereactions that lead to the formation of by-products, thereaction temperature, which must be high enough(around 150°C) to kinetically favour thedecomposition of the intermediate hydroperoxide andthe pressure, which must be such as to keep thecyclohexane in the liquid phase. A brief description ofthe different processes follows.
Direct synthesis of cyclohexanone andcyclohexanol. This process was developed byStamicarbon. It operates at 155°C. Conversion ofcyclohexane is 4%, with an overall selectivity ol�oneof 77-80%, and a molar ratio ol/one�1. At theseconditions, the consecutive reactions upon the desiredproducts are minimized. The catalyst is cobaltnaphthenate or octeate, both of which are soluble incyclohexane (used as the solvent for the reaction).The concentration of the catalyst is very low, rangingfrom 3 ppm to 300 ppm. The mixture of alcohol andketone is then oxidized with air in the presence of Cuand Mn based catalysts, with a selectivity to adipicacid of 70%.
Process in the presence of boric acid. In order tominimize the formation of by-products, boric acid isadded to the reaction medium. The cyclohexanolformed reacts with boric acid, forming an ester andpreserving it from the consecutive reactions tocyclohexanone and to by-products. Since the waterformed in the reaction of cyclohexanol formation isalso a product in the reversible reaction ofcyclohexanol esterification, stripping of water fromthe reaction medium has been found to positivelyaffect the selectivity to the desired product, being thatit favours the formation of the borate. The addition ofboric acid (H3BO3) allows operation to be carried outat relatively high conversion (12%), with an overallselectivity of 85-87% and a ol/one ratio as high as10/1. The oxidation reaction occurs in the absence of acatalyst. The ester is then hydrolized in a followingstep to alcohol and the boric acid is recycled. Thisprocess has higher investment and operating costs, dueto the recovery and recycle of boric acid.
Process in two steps. In a first step, thehydroperoxide is synthesized in the absence of acatalyst, and in a second step, the hydroperoxide isdecomposed to the alcohol and ketone in the presence
of a Co catalyst. This process operates at 4-5%conversion, with a selectivity in the range of 82 to86% and a ratio ol/one�0.4. In the presence of Cr, theol/one ratio is 0.8.
The direct synthesis of adipic acid. In this process,a large excess of cobalt catalyst is used (also with apromoter) to lower the induction time (Tanaka, 1974).The role of the promoter is to start the oxidation ofCo(II) to Co(III). The solvent is acetic acid, in whichthe ketone is soluble. High concentrations of catalyst(feed ratio cyclohexane/catalyst �100) are necessaryto obtain around 65% selectivity to adipic acid. Lowercatalyst concentrations cause a decrease in selectivity,indicating that the mechanism is a classicalautoxidation in the absence of the catalyst.
The oxidation of n-butane to acetic acidThe synthesis of acetic acid by n-butane or naphtha
liquid phase oxidation, commercialized by Celanese in1962, was the predominant process throughout the1960s.
n-C4H10�5/2O2��2CH3COOH�H2O
DH°��233 kcal/mol
By 1973, approximately 40% of the acetic acidcapacity was based on this process. Currently, only asmall fraction of acetic acid is produced by liquidphase oxidation, due to the superior economics of theMonsanto methanol carbonylation process.
The formation of acetic acid from an alkane isexplained by the fact that acetic acid is the most stableproduct (aside from CO2) and thus tends to accumulatein the reaction medium, which is also the reason why itis used as the solvent for the reaction. The main
653VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
Table 8. Reactions yielding by-products in the oxidation of cumene to cumylhydroperoxide (R�C6H5C(CH3)2�)
RO�2�RO�2��RO��RO��O2
RO���C6H5C(O)�CH3�CH�3 (acetophenone)
CH�3�RH��CH4�R�
CH�3�ROOH��CH4�ROO�
CH�3�O2��CH3OO�
CH3OO��RH��CH3OOH�R�
CH3OO��ROO���CH2O�ROH�O2(ROH�dimethylphenylcarbinol)
CH2O�1�2O2��HCOOH
ROH��C6H5C(CH3)�CH2�H2O (a-methylstyrene)
RO��RH��C6H5C(CH3)�CH2�R��H2O

reaction path for the oxidation of n-butane, accountingfor 70% of converted paraffin, occurs through theabstraction of the methylenic hydrogen and theformation of the sec-butylperoxy radical; theremaining 30% is oxidized with the formation of theprimary peroxy radical. The consumption of these tworadicals is not as fast as the H abstraction from n-butane, and they accumulate in the absence ofcatalyst in the reaction medium.
The peroxy radicals react with the Co(II)-basedcatalyst forming a more reactive secondary or primaryalkoxy radical. The main reaction from the sec-butoxyradical is the b scission, which leads to acetaldehyde(precursor of acetic acid) and the ethyl radical and, to aminor extent, the scission leading to propionic acidand formic acid. Alternatively, sec-BuO� istransformed to methylethylketone. The n-butoxyradical is not subjected to b scission and istransformed to n-butyric acid. The main reactionsoccurring are reported in Table 9 (usual passages areomitted). The ethyl radical is the precursor of ethanol.Once formed, it is quickly oxidized to acetaldehydeand acetic acid (Hobbs et al., 1972b). A second routeto acetic acid is from methylethylketone, throughoxidation at the b position (with respect to the C�Ogroup) and the formation of a ketoperoxyradical thatproduces acetic acid by C�C cleavage.
Two processes have been proposed: the first usesonly cobalt as the catalyst, which operates at 160-220°C and 50-60 atm, and the second, together withcobalt, uses a co-oxidant (methylethylketone) andoperates at milder conditions (120-130°C, 30 atm).Methylethylketone is recycled to the first reactor inorder to introduce a free-radical initiator able toabstract a hydrogen from the sec-CH bond.
Performance varies considerably depending on thereaction conditions and catalyst type – either Mn(III)or Co(III); the reaction may also run in the absence ofcatalyst. Selectivity to acetic acid can be in the rangebetween 50 to 65%; conversion per pass is kept below30%, to avoid over-oxidation. The air is diluted withresidual inert gases up to an oxygen concentration of8-11%, in order to keep its concentration at the exitbelow the explosion limits (Prengle and Barona,1970a).
The oxidation of p-xylene to terephthalic acid or dimethylterephthalate
The oxidation/esterification of p-xylene todimethylterephthalate:
is industrially carried out in several steps (Hercules-
Witten-Dynamit process; Katzschmann, 1966). Thefirst step is the oxidation to p-toluic acid, occurringwith the classic free-radical autoxidation mechanismcatalysed by Co/Mn naphthenates at 150°C and 6 atmpressure, without solvent:
H3C�C6H4�CH3�1.5O2��
��H3C�C6H4�COOH�H2O
The electron-attracting effect of the �COOHgroup prevents the formation of the radical on thesecond methyl group, and thus prevents oxidation tothe diacid. Therefore, the carboxylic group isesterified by reaction with methanol (second step):
H3C�C6H4�COOH�CH3OH��
��H3C�C6H4�C(O)�OCH3�H2O
Then, the second methyl group can be oxidized(third step):
H3C�C6H4�C(O)�OCH3�1.5O2��
��HOOC�C6H4�C(O)�OCH3�H2OThe final step is the esterification:
HOOC�C6H4�C(O)�OCH3�CH3OH��
��H3CO�(O)C�C6H4�C(O)�OCH3
The synthesis of p-toluic acid is accompanied bythe formation of by-products, such as p-toluicalcohol,p-tolualdehyde (these two compounds areintermediates in the pathway leading to p-toluic acid),toluene, terephthalic dialdehyde, 4-carboxybenzaldehyde and polynuclear products (i.e. p-toluylester of p-toluic acid and p-toluic acidanhydride). 4-carboxybenzaldehyde is highlyundesired, since the �CHO group can not undergocondensation with ethylene glycol during PETpolymerization.
The selectivity to by-products (especiallyp-toluylester of p-toluic acid and p-toluic acidanhydride) increases as the partial pressure of oxygen
654 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
Table 9. Reactions occurring in n-butane oxidation
n-C4H10�� sec�BuOO��� sec�BuO�
sec–BuO��� C2H�5�CH3CHO
C2H�5�� C2H5OO��� C2H5O��� C2H5OH
CH3CHO�1�2O2�� CH3COOH
sec–BuO��� CH�3�C2H5CHO
C2H5CHO�1�2O2�� C2H5COOH
sec–BuO��� CH3�C(O)�C2H5
n-C4H10�� n-BuOO��� n-C3H7COOH
∆H°� �340 kcal/mol
� 3O2 � 2CH3OH COOCH3H3COOC
∆H°� �340 kcal/mol

decreases (Jacobi and Baerns, 1983). Therefore, thecomplete depletion of oxygen in the bulk liquid isundesired for selectivity.
The kinetics of liquid-phase oxidation of p-xyleneto p-toluic acid has been studied by several authors(Cavalieri d’Oro et al., 1980; Hronec and Ilavsky,1982; Jacobi and Baerns, 1983; Hronec et al., 1985;Raghavendrachar and Ramachandran, 1992; Cao etal., 1994). When the reaction is carried out in bubblecolumns, regions where the process is controlled bydifferent steps can occur. Typically, at high oxygenpartial pressure, the process is under chemical control(and the rate does not depend on oxygen partialpressure), while at low oxygen partial pressure (for almost total oxygen consumption), the processbecomes mass-transfer controlled. In addition,temperature affects the nature of the rate-determiningstep; at temperatures below 100°C, the reaction isunder chemical control and is zero order when eitherair or oxygen is used. Instead, at 130°C, the process isunder mass-transfer control when air is used (theapparent activation energy falls from 15 to 4kcal/mol), while with pure oxygen, the reaction is stillunder chemical control (Cao et al., 1994; Cincotti etal., 1997).
Under chemical control, the rate equation for theCo-catalysed first step of the process (i.e. oxidation ofp-xylene to p-toluic acid) can be expressed as follows(Jacobi and Baerns, 1983):
d [p-C8H10][21] �144233121�k[ p-C8H10]2
dt
With respect to the hydrocarbon, a second orderreaction arises by combining the rate propagationexpression (Prengle and Barona, 1970b):
d [p-C8H10][22] �144233121�kpr[RO2�][ RH]dt
where R is (CH3)C6H4(CH2) (with the assumption thatthe corresponding step is the rate-limiting reaction),with the termination reaction expression:
d [p-C8H10][23] �144233121�kter[RO2�]dt
(assuming that the termination reaction occurs mainlyby bimolecular reaction between RO2� fragments), andby deriving an expression for [RO2�]. It is also assumedthat the concentration of hydroperoxide is quasi-stationary (Walling, 1969). The kinetic constant k isrelated to kpr and kter through the following expression:
k2pr
[24] k �n1212fkter
where n is the number of radicals produced for eachROOH decomposed, and f is the fraction of RH
consumed via attack by RO2�. An experimental value ofn/f intermediate between the two limit cases (1.5 and6) was found, indicating that the cobalt catalyst notonly was involved in the ROOH decomposition, butalso reacted with p-xylene through the Co(III) species(Jacobi and Baerns, 1983).
Other models have been developed that also takeinto account diffusional constraints and that assume afirst order dependency of rate upon p-xyleneconcentration (Hronec and Ilavsky, 1982; Cao et al.,1994). In all cases, the reaction is independent ofoxygen partial pressure (in conditions of a reaction-controlled process).
An alternative process is one developed by Amoco(and by several other companies), which makes use ofa catalyst (Mid-Century MIC catalyst: Co/Mn/Br ions)in acetic acid as the reaction solvent. Severalvariations of this process exist. The MIC catalyst isable to oxidize the second methyl group of p-toluicacid and, therefore, terephthalic acid is directlyobtained as the final product. The acetic acid keeps theintermediates and by-products in solution, but does notdissolve the products. The temperature is around205°C and the pressure is 12-15 atm. The conversionis almost total. The concentration of oxygen in thewaste gas is around 4%. The advantage of this processlies in the direct availability of terephthalic acid, whichcan be further employed for several differenttransformations. This process is now used to virtuallycover the world production of fibre grade terephthalicacid.
The Teijin process is an improvement of theoriginal Mid-Century process; with respect to thelatter, the conditions are milder (100-130°C, 9 atm); ahigh yield of terephthalic acid (higher than 97%) isachieved by using a cobalt catalyst and a large excessof p-xylene.
Oxidations with oxygen-transfer agentsThe reactions of oxidation via catalysed oxygen
transfer agents can be represented schematically asfollows:
S�ROY�� SO�RY
where S is the substrate and ROY the oxygen transferagent (hydroperoxides, H2O2 and peracetic acid).The hydroperoxides of isobutane, ethylbenzene andcumene are the best oxygen transfer agents forindustrial applications for the following reasons: theyare safer to handle and more stable, soluble in non-polar solvents, not acidic, present high selectivityin the reaction of oxygen transfer to the substrate, andthe by-products of the reaction are easy to separate.
Two main industrial applications can be evidenced:epoxidations of olefins and hydroxylation of aromatic
655VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES

compounds. The former process, which is by far themore important, is industrially carried out in eitherhomogeneous or heterogeneous catalysis. Thehydroxylation of aromatic compounds, on the otherhand, is carried out in heterogeneous catalysis.
The epoxidation of propylene to propylene oxideAll olefins can be epoxidized with a hydroperoxide
and a catalyst (see Section 11.1.3), but the mostimportant process, which has been developed at anindustrial level, is the synthesis of propylene oxide.Two alternative processes have been developed, one inthe homogeneous phase, developed by Halcon andAtlantic Richfield (Oxirane process), with amolybdenum complex as the catalyst (Landau, 1967;Landau et al., 1979), and the other in theheterogeneous phase, with silica-supported titaniumoxide, developed by Shell (Wulff, 1975).
Alkylhydroperoxides used as oxygen-transferagents are either t-butylhydroperoxide or ethylbenzenehydroperoxide, due to the fact that the correspondingcoproduct is valuable and contributes to the economyof the process. t-butylhydroperoxide is converted tot-butylalcohol, and then to isobutene. Ethylbenzenehydroperoxide is decomposed to phenylcarbinol andthe latter is converted to styrene. Cumylhydroperoxide,which could present advantages due its high rate ofepoxidation and higher selectivity, is not used as anoxygen-transfer agent due to the non-practical use ofthe by-product.
All processes, both homogeneous andheterogeneous, operate at temperatures between 90and 120°C and at a pressure of 35 atm, in order tokeep propylene in the liquid phase. The uppertemperature limit is the temperature of hydroperoxidedecomposition and the lower limit is dictated by theactivity of the catalyst. The catalysts for epoxidationreactions are d0 metal complexes: Mo(VI), W(VI),V(V) and Ti(IV) for homogeneous epoxidation(Mimoun, 1980; Jørgensen, 1989), and TiO2 supportedover SiO2 for heterogeneous epoxidation (Sheldon,1980).
The reasons for the superior activity and selectivityof these metal ions can be summarized as follows: theyhave a very low oxidation power and, therefore, do notdecompose the hydroperoxide homolitically; they arein their high oxidation state and have high Lewis-typeacidity, necessary to activate the hydroperoxidetowards nucleophilic attack, through withdrawal ofelectrons from the O�O bond. Indeed, V(V)complexes are worse than those of Mo(VI), becausethey are more active in hydroperoxide decomposition(and thus the selectivity is lower), while Cr(VI)complexes, which have high Lewis acidity, cannot beused due to their high oxidizing power. The mechanism
proposed for homogeneous epoxidation has beendescribed above: Liquidi phase homogeneousmetal-catalysed oxidations by molecular oxygen.
The industrial process consists of three main parts:synthesis of ethylbenzene hydroperoxide; epoxidationof propylene; dehydratation of phenylcarbinol tostyrene.
The synthesis of the hydroperoxide is carried out ina bubble reactor with air as the oxidizing agent. Afterthe removal of volatile compounds, the exit solution issent to a falling film evaporator for concentration. Thehydroperoxide-containing solution is then fed togetherwith fresh propylene to the first reactor of threein-series reactors. The final exit solution, afterseparation of unconverted propylene (recycled to thefirst reactor), is sent to the purification section.
New technologies of epoxidation under study or development
The epoxidation of propylene can also be carriedout catalysed by Ti-silicalite (TS-1), with H2O2 as theoxidizing agent (Neri et al., 1984). The process is notyet commercially applied, though pilot plant operationis operative. The catalyst was developed by Eniresearchers and is made of a zeolite isostructural withsilicalite-1, where titanium substitutes for silicon inthe framework of the zeolite (Taramasso et al., 1983).
The solvent for the reaction is an alcohol(methanol or t-butanol). At 50°C and atmosphericpressure, the yield of epoxide on an H2O2 basis isaround 90%, and the selectivity on a propylene basis is98% (Sheldon, 1973; Chong and Sharpless, 1977).The main by-products observed for the differentolefins are only those derived from the consecutivetransformation of the oxirane ring by reaction withmethanol (yielding glycol ethers), with water (yieldingglycols), or with H2O2 (yielding hydroperoxides).
Several Olin patents claim the synthesis ofpropylene oxide from propylene using molecularoxygen as the oxidizing agent in the presence ofmolten salts (Meyer and Pennington, 1991; Fullingtonand Pennington, 1992). Although it has been claimedthat the salt acts as a catalyst, it is likely that theoxidation is not catalytic and that the salt probablyserves to remove the heat of reaction, allowing bettercontrol of the temperature, and thus allowing the veryexothermal reaction to run more smoothly and moreselectively. Thus the mechanism of reaction is aclassical radical chain reaction, and the molten saltmay possibly serve to facilitate the initiation step.
The feedstock contains propylene at aconcentration of around 50%, with a deficiency ofoxygen of approximately 10%, and the remainderconsists of inert gas (N2 and carbon oxides). Thereaction proceeds at 200-300°C, 5-20 atm pressure and
656 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

at relatively high residence time (higher than 10 s).Gas reactants may be either bubbled through astirred–tank reactor filled with the molten salt, movedcountercurrently in a tower through a downflow ofmolten salt or reinjected into a loop through which thesalt circulates. The molten salt is a mixture of LiNO3,NaNO3 and KNO3. The composition of the mixturedetermines the temperature of the reaction.
Propylene conversion is around 10%, with aselectivity to epoxide in the 55 to 65% range. By-products include carbon oxides (selectivity 22-25%) and acetaldehyde (10-13%); minor amounts offormaldehyde, methanol, acrolein, acetone, allyl alcoholand propylene glycol are also formed. Unreactedpropylene is recycled together with part of the by-product gases. The recycle of acetaldehyde improvesthe yield to propylene oxide, since acetaldehyde isoxidized to peracetic acid, which epoxidizes propylene.Alternatively, acetaldehyde is oxidized to acetic acid ina second reactor in a two-step process.
Another technology under development is the in situ generation of H2O2, which finally acts as theoxygen-transfer agent on propylene (Clerici andIngallina, 1998). This process, proposed byEniTechnologie (Clerici and Ingallina, 1993a),epoxidizes olefins with molecular oxygen in thepresence of Ti-silicalite and a redox couple generatingin situ H2O2 and is discussed in Section 11.1.3.
Redox processes
The oxidation of ethylene to acetaldehydeCurrently, acetaldehyde is mainly obtained through
the process developed by Wacker Chemie andHoechst:
C2H4�1/2O2�� CH3CHO
DH°��58 kcal/mol
The catalyst is made of an aqueous solution ofPdCl2 and CuCl2. Acetaldehyde is formed by reactionbetween ethylene and Pd chloride (i.e. the rate-determining step), which, at the same time, isreduced to metallic Pd. Pd is then reoxidized to PdCl2by CuCl2, with the formation of CuCl, which is finallyreoxidized by molecular oxygen, yielding back CuCl2.(The reaction mechanism was described above: Liquidphase homogeneous metal-catalysed oxidations bymolecular oxygen). By-products of the reaction areacetic acid, oxalic acid, crotonaldehyde, chlorinatedhydrocarbons and chlorinated acetaldehyde.
The process can be carried out either in a singlestep, with pure oxygen as the oxidizing agent, at 130°C and 4 atm, or in a two-step process, wherethe oxidizing agent of choice is air.
In the latter case, the first step consists in the
reaction between catalyst and ethylene (at 110°C and9-10 atm), with the formation of acetaldehyde; thesecond step is the reoxidation of the reduced catalystthrough contact with air.
In the single-step process, ethylene and molecularoxygen are fed into an air-lift tower, where thecatalyst-containing aqueous solution is circulated via a separation vessel. After cooling and washing withwater of the gaseous stream, the liquid fraction,containing water and crude acetaldehyde, is sent to atwo–column separation, while the gaseous stream,containing unreacted ethylene and inerts, is recirculatedto the reactor. The ethylene per pass conversion isbetween 20 and 50%; unconverted ethylene is recycled.
In the two-step process, following the reaction, anacetaldehyde/water mixture is separated from thesolution containing the reduced catalyst in a flashtower. The latter is sent to the reoxidation reactor,where it is put in contact with air. Ethylene andmolecular oxygen almost completely react in the twosteps, respectively. The crude acetaldehyde/watermixture is first concentrated and then sent to a two-stage distillation section. In this processconfiguration, the ethylene conversion is up to 90%,which eliminates the need for recycle of theunconverted olefin. In both process configurations, theyield of acetaldehyde is around 95%.
Bibliography
Bailey C.L., Drago R.S. (1987) Utilization of dioxygen forthe specific oxidation of organic substrates with cobalt (II)catalysts, «Coordination Chemistry Reviews», 79, 321-332.
Balci M. (1981) Bicyclic endoperoxides and syntheticapplications, «Chemical Reviews», 81, 91-108.
Benson S.W., Nangia P.S. (1979) Liquid-phase oxidation ofisobutane. A reanalysis of the data, «International Journalof Chemical Kinetics», 12, 169-181.
Betts A.T., Uri N. (1968) Catalyst-inhibitor conversion inautoxidation reactions, «Advances in Chemistry Series»,76, 160-181.
Burstyn J.N. et al. (1988) Mechanisms of dioxygen activationin metal-containing monooxygenase. Enzimes and modelsystems, in: Martell A.E., Sawyer D.T. (editors) Oxygencomplexes and oxygen activation by transition metals, NewYork, Plenum Press, 175-187.
Choy V.J., O’Cannor C.J. (1972) Chelating dioxygencompounds of the platinum metals, «CoordinationChemistry Reviews», 9, 145-170.
Collman P. et al. (1980) Oxigen binding to heme proteinsand their synthetic analogs, in: Spiro T.G. (editor) Metalions in biology, 2: Metal ion activation of dioxygen, NewYork, John Wiley, 1-72.
Dadyburjor D.B. et al. (1979) Selective oxidation ofhydrocarbons on composite oxides, «Catalysis Reviews.Science and Engineering», 19, 293-350.
657VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES

Fox M.A., Chanon M. (editors) (1988) Photoinducedelectron transfer. Part A: Conceptual basis; Part B:Experimental techniques and medium effects, Amsterdam,Elsevier.
Gubelmann M.H., Williams A.F. (1983) The structures andreactivity of dioxygen complexes of the transition metals,in: Structure and bonding, 55: Transition metal complexes-Structure spectra, Berlin, Springer, 1-65.
Kaufman S., Fisher D.B. (1974) Pterin-requiring aromaticamino acid hydroxilases, in: Hayaishi O. (editor) Molecularmechanisms of oxygen activation, New York, AcademicPress, 285-369.
Meyer T.J. (1988) Metal oxo complexes and oxygenactivation, in: Martell A.E., Sawyer D.T. (edited by)Oxygen complexes and oxygen activation by transitionmetals, New York, Plenum Press, 33-47.
Murray R.W., Wasserman H.E. (edited by) (1979) Organicchemistry, 40: Singlet oxygen, New York, Academic Press.
Patai S. (edited by) (1983) The chemistry of peroxide,Chichester, John Wiley.
Sheldon R.A. (1991) Fine chemicals by catalytic-oxidation,«CHEMTECH», 21, 566-576.
Sheldon R.A. (1991) Heterogeneous catalytic oxidation andfine chemicals, in: Guisnet M. et al. (editors) Studies insurface science and catalysis, 59: Heterogeneous catalysisand fine chemicals II. Proceedings of the 2nd internationalsymposium, Poitiers, 2-5 October, Amsterdam, Elsevier,33-54.
Simandi L.I. (editor) (1991) Dioxygen activation andhomogeneous catalytic oxidation. Proceedings of the 4th
international symposium on dioxygen activation andhomogeneous catalytic oxidation, Amsterdam, Elsevier.
Stevens B. (1973) Kinetics of photoperoxidation in solution,«Accounts of Chemical Research», 6, 90-96.
Tsutsui M., Ugo R. (editors) (1977) Fundamental researchin homogeneous catalysis, New York, Plenum Press.
Vaska L. (1976) Dioxygen-metal complexes. Toward a unifiedview, «Accounts of Chemical Research», 9, 175-183.
References
Andrigo P. et al. (1992) Phenol-acetone process. Cumeneoxidation kinetics and industrial plant simulation,«Chemical Engineering Science», 47, 2511-2516.
Astarita G. et al. (1983) Gas treating with chemical solvents,New York, John Wiley.
Bamford C.H., Tipper C.F.H. (edited by) (1980)Comprehensive chemical kinetics, Amsterdam, Elsevier.
Barton D.H.R., Doller D. (1991) Selective functionalizationof saturated hydrocarbons. Gif and all that, in: SimandiL.I. (edited by) Dioxygen activation and homogeneouscatalytic oxidation. Proceedings of the 4th internationalsymposium on dioxygen activation and homogeneouscatalytic oxidation, Amsterdam, Elsevier, 1-10.
Barton D.H.R. et al. (1989) On the mechanism of the Gif andGif-Orsay systems for the selective substitution of saturatedhydrocarbons, «New Journal of Chemistry», 13, 177-182.
Bellussi G. et al. (1992) Reactions of titanium silicalite with
protic molecules and hydrogen peroxide, «Journal ofCatalysis», 133, 220-230.
Benson S.W. (1965) Effects of resonance and structure on thethermochemistry of organic peroxy radicals and the kineticsof combustion reactions, «Journal of the American ChemicalSociety», 87, 972-979.
Benson S.W., Shaw R. (1970) Thermochemistry of organicperoxides, hydroperoxides, polyoxides, and their radicals,in: Swern D. (edited by) Organic peroxides, New York,John Wiley, 105-139.
Cao G. et al. (1994) Kinetics of p-xylene liquid-phase catalyticoxidation, «American Institute of Chemical EngineersJournal», 40, 1156-1166.
Carrà S., Santacesaria E. (1980) Engineering aspects ofgas-liquid catalytic reactions, «Catalysis Reviews-Scienceand Engineering», 22, 75-140.
Cavalieri d’Oro P. et al. (1980) On the low temperature oxidationof p-xylene, «Oxidation Communications», 1, 153-162.
Chen Z. et al. (1985) Gas-liquid interphase mass transfercoefficients in trickle beds, «Huagong Xuebao», 3, 339-346.
Chester A.W. et al. (1977) Zirconium cocatalysis of the cobalt-catalyzed autoxidation of alkylaromatic hydrocarbons,«Journal of Catalysis», 46, 308-319.
Chong A.O., Sharpless K.B. (1977) Mechanism of themolybdenum and vanadium catalyzed epoxidation of olefinsby alkyl hydroperoxides, «Journal of Organic Chemistry»,42, 1587-1590.
Cincotti A. et al. (1997) Effect of catalyst concentration andsimulation of precipitation processes on liquid-phasecatalytic oxidation of p-xylene to terephthalic acid,«Chemical Engineering Science», 52, 4205-4213.
Clerici M.G. (1991) Oxidation of saturated hydrocarbons withhydrogen peroxide, catalyzed by titanium silicalite, «AppliedCatalysis», 68, 249-261.
Clerici M.G. et al. (1991) Synthesis of propylene oxide frompropylene and hydrogen peroxide catalyzed by titaniumsilicalite, «Journal of Catalysis», 129, 159-167.
Clerici M.G., Ingallina P. (1993a) European Patent 0549013to Eniricerche.
Clerici M.G., Ingallina P. (1993b) Epoxidation of lowerolefins with hydrogen peroxide and titanium silicalite,«Journal of Catalysis», 140, 71-83.
Clerici M.G., Ingallina P. (1998) Oxidation reactions within situ generated oxidants. New concepts in selectiveoxidation over heterogeneous catalysts, «Catalysis Today»,41, 351-364.
Dawson J.H. (1988) Probing structure-function relations inheme-containing oxygenases and peroxidases, «Science»,240, 433-439.
Doraiswamy L.K., Sharma M.M. (1984) Heterogeneousreactions. Analysis, examples and reactor design, NewYork, John Wiley, 2v.
Drago R.S., Beer R.H. (1992) A classification scheme forhomogeneous metal-catalyzed oxidations by molecularoxygen, «Inorganica Chimica Acta», 198, 359-367.
Ellis P.E., Lyons J.E. (1989a) Effect of axial azide on the selective,low temperature metalloporphyrin-catalyzed reactions ofisobutane with molecular oxygen, «Journal of the ChemicalSociety, Chemical Communications», 16, 1187-1189.
658 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

Ellis P.E., Lyons J.E. (1989b) Effect of fluorination of themeso-phenyl groups on selective tetraphenylporphyrinatometal(III)-catalyzed reactions of isobutane with molecularoxygen, «Journal of the Chemical Society, ChemicalCommunications», 16, 1189-1190.
Emanuel N.M., Gal D. (1986) Modelling of oxidationprocesses, Budapest, Akademiai Kiado.
Finn M.G., Sharpless K.B. (1985) On the mechanism ofasymmetric epoxidation with titanium-tartrate catalysts,«Asymmetric Synthesis», 5, 247-308.
Fleming J.B. et al. (1976) Safety in phenol-from-cumeneprocess, «Hydrocarbon Processing», 55, 185-190.
Franz G., Sheldon R.A. (1991) Oxidation, in: Ullmann’sencyclopedia of industrial chemistry, Weinheim, VCH,1985-1996, 37 v.; v. A18, 261-270.
Fullington M.C., Pennington B.T. (1992) WO Patent WO9209588 to Olin Co.
Gates B.C. et al. (1979) Chemistry of catalytic processes, NewYork, McGraw-Hill.
Groves J.T. (1985) Key elements of the chemistry of cytochrome-p-450. The oxygen rebound mechanism,«Journal of Chemical Education», 62, 928-931.
Hattori K. et al. (1970) Kinetics of liquid phase oxidation ofcumene in bubble column, «Journal of ChemicalEngineering of Japan», 3, 72-78.
Hayaishi O. (1974) General properties and biological functionsof oxygenases, in: Hayaishi O. (edited by) Molecularmechanisms of oxygen activation, New York, AcademicPress, 7.
Hendry D.G., Amer J. (1967) Rate constants for oxidation ofcumene, «Journal of the American Chemical Society», 89,5433-5438.
Hiatt R. et al. (1968) Homolytic decompositions ofhydroperoxides. IV: Metal-catalyzed decompositions,«Journal of Organic Chemistry», 33, 1430-1435.
Hill C.L. (editor) (1989) Activation and functionalization ofalkanes, New York, John Wiley, 243.
Hobbs C.C. et al. (1972a) Mass-transfer rate-limitation effects inliquid-phase oxidation, «Industrial & Engineering Chemistry.Product Research and Development», 11, 220-225.
Hobbs C.C. et al. (1972b) Product sequences in liquid-phaseoxidation of paraffins, «Industrial & Engineering Chemistry.Process Design and Development», 11, 59-68.
Howard J.A. (1972) Absolute rate constants for reactions ofoxyl radicals, «Advances in Free-Radical Chemistry», 4,49-173.
Howard J.A. (1973) Homogeneous liquid-phase autoxidations,in: Kochi J.K. (editor) Free radicals, New York, John Wiley,2 v.; v. II, 3-62.
Hronec M., Ilavsky J. (1982) Oxidation of polyalkylaromatichydrocarbons. XII: Technological aspects of p-xyleneoxidation to terephthalic acid in water, «Industrial &Engineering Chemistry. Process Design and Development»,21, 455-460.
Hronec M. et al. (1985) Kinetics and mechanism of cobalt-catalyzed oxidation of p-xylene in the presence of water,«Industrial & Engineering Chemistry. Process Design andDevelopment», 24, 787-794.
Jacobi R., Baerns M. (1983) The effect of oxygen transferlimitation at the gas-liquid interphase. Kinetics and product
distribution of the p-xylene oxidation, «Erdoel Kohle ErdgasPetrochemie», 36, 322-326.
Jørgensen K.A. (1989) Transition-metal-catalyzed epoxidations,«Chemical Reviews», 89, 431-458.
Katzschmann E. (1966) Oxidation of alkyl aromaticcompounds, «Chemie Ingenieur Technik», 38, 1-10.
Kerr J.A. (1966) Bond dissociation energies by kinetic methods,«Chemical Review», 66, 465-500.
Kochi J.K. (1973) Oxidation-reduction reactions of free radicalsand metal complexes, in: Kochi J.K. (editor) Free radicals,New York, John Wiley, 2 v.; v. I.
Krzysztoforski A. et al. (1986) Industrial contribution tothe reaction engineering of cyclohexane oxidation,«Industrial and Engineering Chemistry. Process Designand Development», 25, 894-898.
Ladhaboy M.E., Sharma M.M. (1969) Absorption of oxygenby n-butyraldehyde, «Journal of Applied Chemistry», 19,267-272.
Landau R. et al. (1967) Epoxidation of olefins, in: Proceedingsof the 7th World petroleum congress, Mexico City, April,67-72.
Landau R. et al. (1979) Propylene oxide by the co-productprocesses3, «CHEMTECH», 9, 602-607.
Lyons J.E. (1980) Up petrochemical value by liquid phase catalyticoxidation, «Hydrocarbon Processing», 59, 107-119.
Manor Y., Schmitz R.A. (1984) Gradientless reactor for gas-liquid reactions, «Industrial & Engineering ChemistryFundamentals», 23, 243-252.
Mansuy D., Battioni P. (1989) Catalytically activemetalloporphyrin models for cytochrome P-450, in:Ruckpaul K., Rein H. (edited by) Basis and mechanismsof regulation of cytochrome P–450, London, Taylor &Francis.
Melville H.W., Richards S. (1954) Photochemicalautoxidation of isopropylbenzene, «Journal of the ChemicalSociety», 944-952.
Meyer J.L., Pennington B.T. (1991) US Patent 4992567 toOlin Co.
Mimoun H. (1980) The role of peroxymetalation in selectiveoxidative processes, «Journal of Molecular Catalysis», 1,1-29.
Mimoun H. (1982) Oxygen-transfer from inorganic and organicperoxides to organic substrates. A common mechanism,«Angewandte Chemie-International Edition in English»,21, 734-750.
Mimoun H. et al. (1970) Epoxidation of olefins with covalentperoxomolybdenum(VI) complexes, «Tetrahedron», 26,37-50.
Moiseev I.I. et al. (1974) Kinetics of olefin oxidation bytetrachloropalladate in aqueous solution, «Journal of theAmerican Chemical Society», 96, 1003-1007.
Morbidelli M. et al. (1986) Gas-liquid autoxidation reactors,«Chemical Engineering Science», 41, 2299-2307.
Neri C. et al. (1984) European Patent 0100119 to ANIC.Ortiz de Montellano P.R. (edited by) (1985) Cytochrome
P–450. Structure, mechanism and biochemistry, New York,Plenum Press.
Prengle H.W., Barona N. (1970a) Petrochemicals by liquidphase oxidation, «Hydrocarbon Processing», 49, 106-118.
659VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES

Prengle H.W., Barona N. (1970b) Petrochemicals by liquidphase oxidation. 2: Kinetics, mass transfer, and reactordesign, «Hydrocarbon Processing», 49, 159-175.
Purcell K. (1985) Cycloreversion in metal-assisted olefinoxidation by peroxide. Molybdenum(VI) vs. rhodium(III),«Organometallic», 4, 509-514.
Raghavendrachar P., Ramachandran S. (1992) Liquid-phase catalytic oxidation of p-xylene, «Industrial &Engineering Chemistry Research», 31, 453-462.
Santacesaria E., Pimpinelli N. (1986) Oxidation in the gas-liquid phase in industry. I: Kinetics and mechanical aspects,«La Chimica e L’Industria», 68, 69-79.
Sheldon R.A. (1973) Molybdenum-catalyzed epoxidation ofolefins with alkyl hydroperoxides. I: Kinetic and productstudies, «Recueil des Travaux Chimiques des Pays-Bas»,92, 253-266.
Sheldon R.A. (1980) Synthetic and mechanistic aspects ofmetal-catalyzed epoxidations with hydroperoxides, «Journalof Molecular Catalysis», 7, 107-126.
Sheldon R.A., Doom J.A. van (1973) Metal-catalyzedepoxidation of olefins with organic hydroperoxides. I:Comparison of various metal catalysts, «Journal ofCatalysis», 31, 427.
Sheldon R.A., Kochi J.K. (1976) Metal-catalyzed oxidationsof organic compounds in the liquid phase. A mechanisticapproach, «Advances in Catalysis», 25, 272-413.
Sheldon R.A., Kochi J.K. (1981) Metal-catalyzed oxidationsof organic compounds, New York, Academic Press.
Sheu C. et al. (1990) Activation of dioxygen by bis(2,6-dicarboxylatopyridine)iron(II) for the ketonization ofmethylenic carbons and the dioxygenation of acetylenes,aryl olefins, and catechols. Reaction mimics for dioxygenase
proteins, «Journal of the American Chemical Society»,112, 879-881.
Shilov A.E. (1989) Historical evolution of homogeneousalkane activation systems, in: Hill C.L. (edited by)Activation and functionalization of alkanes, New York,John Wiley.
Sifniades S. et al. (1982) US Patent 4358618 to Allied Corp.
Suresh A.K. et al. (1988) Mass transfer and solubility inautocatalytic oxidation of cyclohexane, «American Instituteof Chemical Engineers Journal», 34, 55-68.
Tanaka K. (1974) Adipic acid in one-step, «CHEMTECH»,4, 555-559.
Taramasso M. et al. (1983) US Patent 4410501 toSnamprogetti.
Trifirò F., Cavani F. (1994) Selective partial oxidation ofhydrocarbons and related oxidations, Catalytica StudiesDivision, 4193 SO.
Twigg G.H. (1962) Liquid-phase oxidation [of organiccompounds] by molecular oxygen, «Chemistry andIndustry», 1, 4-11.
Uri N. (1961) Physico-chemical aspects of autoxidation, in:Lundberg W.O. (editor) Autoxidation and antioxidants, NewYork, John Wiley, 55-106.
Walling C. (1969) Limiting rates of hydrocarbonautoxidations, «Journal of the American Chemical Society»,91, 7590-7594.
Wulff H.P. (1975) US Patent 3923843 to Shell Oil.
Philippe Arpentinier
Air Liquide - Centre de Recherche Claude-DelormeJouy-en-Josas, France
660 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

11.1.3 Oxidation processes with hydrogen peroxide and hydroperoxides
Introduction
Oxidants in industrial chemical processesOxidation reactions are of particular importance in
petrochemicals because, while the traditional rawmaterials are hydrocarbons formed solely by carbonand hydrogen, the majority of the derivatives producedfrom them consist of carbon, hydrogen and oxygen.
From both an economic and environmentalstandpoint, theoretically, the most preferable way byfar to oxidize any substrate should be its reaction withmolecular oxygen. In practice, however, this reactionpresents certain negative aspects. In fact, althoughoxygen is a formidable oxidant from thethermodynamic point of view, it is not very reactive.This scanty reactivity (enabling all living organisms toremain alive for a reasonable time, instead of burningmore or less slowly, forming water and CO2) is due tothe fact that the ground state of the O2 molecule is atriplet state (with two unpaired electrons). On thecontrary, the ground states of the O2 reductionproducts (i.e. water or hydrogen peroxide), as in themajority of the stable organic compounds obtained byoxidation, are all singlet states (with paired electrons).As in a chemical reaction the angular moment must beconserved, the reactions of molecules in a triplet statewith products in a singlet state are spin-prohibited andcan occur only with extremely slow kinetics. Theobstacle can be overcome by supplying energy to thesystem, that is by conducting the reaction at hightemperatures and pressures or by operating inconditions that favour the onset of radical typemechanisms (compatible with the triplet state of themolecular oxygen). Both solutions, however, aregenerally adverse to achieving selective oxidations andhence to a certain part of oxidations of industrialinterest.
As an alternative to molecular oxygen, theoxidation of an organic substrate S can be obtained bycausing it to react with an appropriate oxidant XO,whose basic state is a singlet one:
S �XO → SO �X
The reaction shows, though, that together withthe desired product SO a second product(coproduct) X is also formed, derived from thereduction of the oxidant. At the end of the reaction,the coproduct has to be recovered, recycled orupgraded, or – in the worst case – disposed of.Different oxidants form different coproducts (Table 1), however, in every case, the formation ofa coproduct increases the costs of the process. Inparticular, its mere disposal can have anenvironmental impact, which, in the light of thegrowing sensitivity to problems of pollution, isoften unacceptable. For example, the production ofpropylene oxide by the chlorohydrin process (stillin use) entails that, for every ton of epoxide, about2.1 t of calcium chloride is coproduced and mustbe disposed of; furthermore, it may be polluted bychlorinated organic by-products.
Therefore, from the environmental standpoint, onlya few oxidizing agents (hydrogen peroxide, ozone,nitrous oxide) are acceptable, as they lead to theformation of harmless, easily disposable coproducts(water, oxygen, nitrogen).
Hydrogen peroxideOf the oxidizing agents presented in Table 1, the
most versatile and easiest to use by far is hydrogenperoxide, whose molecule has a non-planar structure,with a C2 symmetry (Fig. 1). It should be stressed thatthe bond between the two oxygen atoms is relativelyweak (51 kcal/mol, to be compared, for example, with89.5 kcal/mol for the two O�H bonds).
661VOLUME II / REFINING AND PETROCHEMICALS
Oxidant Coproduct
H2O2 H2O
N2O N2
O3 O2
Hydroperoxides (ROOH) Alcohols (ROH)
RCO3H RCO2H
NaClO NaCl
Table 1. Different coproducts formed by reductionof selected oxidants

Hydrogen peroxide is commonly produced by theanthraquinone process, originally developed inGermany by IG Farbenindustrie between 1935 and1945, and used on an industrial scale as of the 1950s.This process is based on the capacity of the alkylderivatives of anthrahydroquinone to produce hydrogenperoxide under typical autoxidation conditions:
Other organic compounds also possess thisproperty, such as secondary alcohols, hydroquinonederivatives and azobenzenes, which has been exploitedfor the development of alternative productionprocesses. Mention should be made, in this regard, tothe autoxidation of isopropyl alcohol, the Shellprocess, and of the 1-phenylethanol recently developedby ARCO up to the pilot plant stage. Today the onlyprocess truly applied for the production of largevolumes is the anthraquinone process. Anelectrochemical process also exists, which is used forthe production of small quantities at local scale.
Typically, the anthraquinone process is based onthe cyclic hydrogenation and oxidation of a workingsolution consisting of an alkylanthraquinone andvarious derivatives thereof, dissolved in a solvent mix.The recovery of the hydrogen peroxide includes anextraction with water, a series of purificationtreatments of the aqueous solution, removing theorganic impurities, and the vacuum distillation of theexcess water. The commercial solution thus obtainedgenerally has a concentration of 60-70% in weight.
The process is characterized by various criticalfactors, linked to both the reactivity and the differentsolubilities of the oxidized and reduced forms of thealkylanthraquinone. In fact, during the hydrogenation
and oxidation cycle, this can undergo variousdegradation processes with the formation of variousby-products, bringing about complex regeneration andpurification processes of the working solution (apartfrom the partial loss of a relatively costly reagent). Thesolvent should be able to dissolve both the (apolar)anthraquinone and the (polar) anthrahydroquinone,and, in addition, must guarantee immiscibility withwater so as to permit the extraction and recovery of thehydrogen peroxide.
These problems are tackled, in part, by selectingthe proper alkyl substituent of the anthraquinone(generally ethyl, amyl or tert-butyl) and by using amixture of solvents, chosen from two groupscharacterized by their different polarity. The firstgroup, suitable for dissolving alkylanthraquinone,includes a number of aromatic compounds (toluene,methylnaphthalene). The second group, suitable foralkylanthrahydroquinone, includes the organic estersof phosphoric acid, diisobutylcarbinol andalkylsubstituted ureas. Typically, the composition ofthe working solution varies from one producer toanother; it is optimized to maximize the solubility ofalkylanthraquinone and hence the concentration of thehydrogen peroxide produced, to guarantee high ratesof hydrogenation and oxidation, as well as to minimizethe formation of degradation by-products. The othercharacteristics of the plant are generally the result ofspecific choices and developments.
The many problems to be addressed are reflectedin the process’ complex nature and high investmentcosts. In fact, the anthraquinone process is marketedthroughout the world by a relatively small number ofcompanies. The average capacity of the plants is40,000-50,000 t/y, with numerous examples of outputsof under 20,000 t/y. Plants with a productive capacityof between 70,000 and 110,000 t/y are less common. Itis important to note that for certain applications (seebelow), significantly larger supplies of hydrogenperoxide are required.
For all these reasons, considerable efforts havebeen made in recent years to identify alternativesyntheses to hydrogen peroxide, resulting in two maindirections. The first one is the reaction of carbonmonoxide with oxygen and water to form hydrogenperoxide and carbon dioxide:
CO �O2�H2O��H2O2�CO2
This reaction was reported for the first time in1979 by Yuri Yermakov (of the Catalysis Institute ofNovosibirsk, in Russia). The catalytic system used byYermakov (based on soluble palladium salts andtriphenylphosphine) was rather inefficient, butrecently the substitution of the phosphine withnitrogen ligands (phenanthrolines) has led to
662 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
H
H
O O
96°52'
0.97 Å1.49 Å
93°51'
Fig. 1. The molecule of hydrogen peroxide.
� O2 � H2O2
OH
OH
RO
O
R
� H2
O
O
ROH
OH
R

considerable improvement in the reaction performanceand has enabled it to be conducted continuously(Bianchi et al., 1999).
Of even greater interest is direct synthesis from theelements:
H2�O2��H2O2
This reaction has been known for a long time (thefirst patents date from the 1960s; Hooper, 1967), but ithas not yet been applied industrially due to theproblems linked in particular to safety in managinghydrogen-oxygen mixtures. The two gases are made toreact in a solvent, typically water or methanol(Paparatto et al., 2003a), in the presence of apalladium base catalyst, possibly modified with theaddition of platinum or of other noble metals. At themoment, this direct synthesis seems the mostpromising candidate to replace the traditionalanthraquinone process in the near future.
HydroperoxidesAlkylhydroperoxides, or more briefly
hydroperoxides, formally derive from the substitution ofa hydrogen atom of the hydrogen peroxide moleculewith an alkyl R group, so that their general formula isR�OOH. Hydroperoxides retain the weakness ofhydrogen peroxides oxygen-oxygen bond: in fact, theenergy of this bond is typically 40-44 kcal/mol, evenlower than that of hydrogen peroxide (51 kcal/mol). Theenergy of the O�H bond, instead, is 89-90 kcal/mol,practically coinciding with that of hydrogen peroxide(89.5 kcal/mol). Many hydroperoxides are known –primary, secondary or tertiary – but few have taken onindustrial importance; those produced and used at alarge scale are substantially three: two tertiaryhydroperoxides, i.e. tert-butyl hydroperoxide and cumylhydroperoxide, and a secondary one, i.e. (1-phenyl)ethyl hydroperoxide.
They are prepared by autoxidation of thecorresponding hydrocarbons, a typical radicalreaction, and the hydroperoxide group is formed,mostly, at the expense of the C�H bonds having lessenergy (e.g. tertiary or benzylic ones). These two casesare exploited, respectively, in preparing tert-butylhydroperoxide from isobutane and cumylhydroperoxide from cumene:
and (1-phenyl) ethyl hydroperoxide fromethylbenzene:
For example, the oxidation of isobutane and ofethylbenzene is commonly conducted in the liquidphase at 120-140°C and under moderate pressure(30-40 and about 2 bar, respectively). Analogousconditions (90-130°C and 5-10 bar) are adopted alsoin the oxidation of cumene; however, in this case, thereaction must be conducted in the presence of anemulsified aqueous phase, weakly basic due to theaddition of sodium hydroxide or carbonate. Inevitably,in fact, small quantities of acid by-products areformed, mainly formic acid. These acids favour thedecomposition of cumyl hydroperoxide giving acetoneand phenol; the latter is an excellent inhibitor of manyradical reactions and, in particular, its presence is notcompatible with autoxidation.
In general, the selectivity of all these reactions,between 60 and 95%, is satisfactory and dependsmostly on the conversion of the hydrocarbon, which,however, is rather limited, approximately between 10and 40%.
Some uses of hydroperoxides, exploiting two verydifferent aspects of their reactivity, are describedbelow. On the one hand, their oxidizing properties areused, and in particular their capacity to transfer anoxygen atom to olefins, giving rise to the formation ofan epoxide and an alcohol (which is the product ofhydroperoxide reduction). In this aspect,hydroperoxides (such as hydrogen peroxide) are notvery reactive and are therefore used in the presence ofappropriate catalysts, generally based on transitionmetals (in particular, titanium and molybdenum).
The use of cumyl hydroperoxide in the traditionalcumene process for the production of phenol andacetone is completely different. In this case, thementioned property of cumyl hydroperoxide todecompose in an acid environment, producing the twofinal products, is exploited.
All hydroperoxides, and in particular those oflower molecular weights, tend to give rise todecomposition reactions, which can be of an explosivecharacter.
Oxidation with hydrogen peroxideWithout any catalyst, hydrogen peroxide is not a
particularly reactive oxidizing agent. However, it isemployed in a series of non-catalytic uses, for exampleto bleach paper and wood pulp, in detergency (in thepreparation of perborate and sodium percarbonate) and
663VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
CH
CHCH3H3C
C
OOH
CH3H3C
CH3C OOHCH3
CH3
CH3
� O2
� O2
H3C
H3C
CH2
H3CCH
OOHH3C
� O2

in the environmental field (e.g. for eliminating toxicsubstances in waste water).
A completely different use of hydrogen peroxide ismade in the Ugine Kuhlmann process for theproduction of hydrazine, a process in which H2O2 isused in the presence of methyl ethyl ketone (MEK)and of a nitrile or an amide (Goor, 1992):
With respect to traditional processes (Raschig,Bayer), which are based on the oxidation of ammoniawith chlorine or hypochlorite, the process usinghydrogen peroxide avoids the coproduction of verylarge quantities of sodium chloride.
Regarding the far more numerous catalyzedreactions, the first observation of a catalytic effect inoxidation reactions with peroxides dates from theclassic work by Henry John Horstman Fenton, who, in1876, described the capacity of ferrous salts tocatalyze the oxidation of tartaric acid by hydrogenperoxide (Fenton, 1876, 1894). In its most classicalform, the Fenton reagent consists of a mixture ofhydrogen peroxide and ferrous sulphate, but, moregenerally, the hydrogen peroxide can be activated byany other reducing term of a monoelectronic redoxcouple such as, for example, copper (I). The questionregarding the nature of the active species that comesinto play in oxidations with the Fenton reagent (orwith systems correlated with this) is still beingdebated. The traditional view that it is the hydroxylradical (�OH) has been challenged in recent years and,instead, the intervention of high-valent iron specieswith oxidation state (IV) or (V) has been claimed (atleast for reactions conducted in non-aqueous solvents).In any case, oxidations with the Fenton reagent are notvery selective and, with few exceptions that will bediscussed below, they are of little use in organicsynthesis. They are applied, instead, in theenvironmental field, for example to purify waterspolluted by phenolic compounds.
Long after Fenton’s pioneering studies, in the1930s, Nicholas Athanasius Milas described thecatalytic effect of metals in electronic d0 configurationin oxidation reactions with hydrogen peroxide oforganic and inorganic substrates (Milas and Sussman,
1936; Milas, 1937). The Milas reagents are formed byreaction of metallic oxides with hydrogen peroxide ina tert-butanol solution and were often used foroxidizing olefins to the corresponding vicinal diols, areaction that in most cases leads to the formation of anintermediate epoxide that is successively hydrolyzed todiol by the acid medium. Although Milas usedvanadium(V), chromium(VI) and osmium(VIII), itvery soon became clear that analogous reactions werepossible, often with better results, usingmolybdenum(VI) or tungsten(VI). However, the doubtremains that some of the reactions observed mighthave been caused not so much by the hydrogenperoxide as by the tert-butyl hydroperoxide, which,under the conditions adopted, can be formed byreaction of the tert-butanol with the hydrogen peroxide.
Meanwhile, in the 1960s and 1970s, the reasonswhy it is difficult to use oxygen in selective oxidationsgrew clearer. Thus, many industrial chemists beganassessing with interest the oxidizing properties ofhydrogen peroxide and other peroxides that are partlyreduced forms of molecular oxygen. The alreadymentioned derivatives of tungsten(VI) and, above all,of molybdenum(VI) were obvious candidates for therole of catalyst and, in fact, they formed the basis ofthe first industrial success in this sector: theepoxidation of propylene with hydroperoxides (seebelow).
However, the use of hydrogen peroxide remainedan aim of great interest as it would have made itpossible to decouple the production of propylene oxide(or of other epoxides) from the coproduction oftert-butanol or of styrene. Yet the derivatives ofvanadium(V) or of molybdenum(VI) and tungsten(VI),which catalyze the reactions of hydroperoxidesextremely well, produced very poor results if usedwith hydrogen peroxide. To understand the reason forthis failure, it must be considered that the catalyticallyactive species is formed by reaction of the metalderivative with the oxidizing agent, whether this is ahydroperoxide or hydrogen peroxide. In the presenceof water, however, the latter competes effectively withthe oxidizing agent, giving rise to inactive specieswhose formation eventually inhibits the desiredreaction. However, water is always present inoxidations with hydrogen peroxide, both because thelatter is normally used in the form of an aqueoussolution, and because new water is formed asoxidation takes place and the hydrogen peroxide isconsumed. Therefore, strategies were sought thatwould enable the problem of water to be eliminated or,at least, its consequences to be lessened. Basicallythree approaches were followed: seeking anhydrousreaction conditions, the application of the phase-transfer technique and the development of
664 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
O
CH3C
C N CN
H5C2 CH3
H3C C2H5
C2H5
NH
CH3C C2H5
NH3
�H2O
H2O2, NH3, MEK
�3H2O
H2O2, NH3, MEK
�3H2O
MEK
2H2O
N2H4 + 2MEK
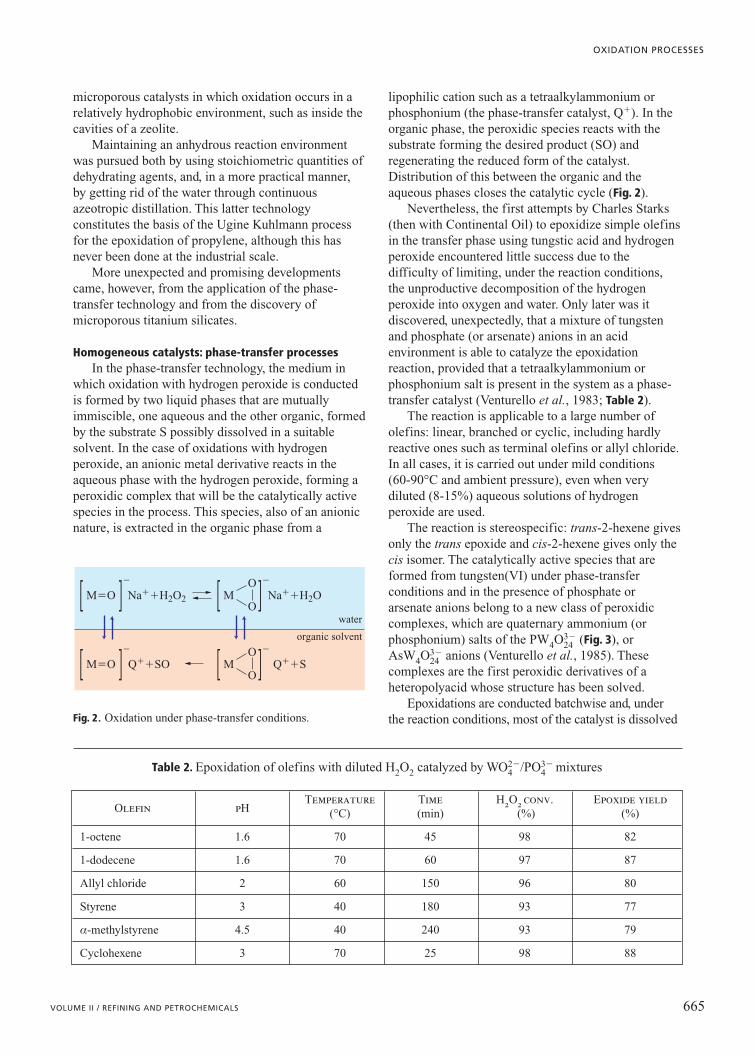
microporous catalysts in which oxidation occurs in arelatively hydrophobic environment, such as inside thecavities of a zeolite.
Maintaining an anhydrous reaction environmentwas pursued both by using stoichiometric quantities ofdehydrating agents, and, in a more practical manner,by getting rid of the water through continuousazeotropic distillation. This latter technologyconstitutes the basis of the Ugine Kuhlmann processfor the epoxidation of propylene, although this hasnever been done at the industrial scale.
More unexpected and promising developmentscame, however, from the application of the phase-transfer technology and from the discovery ofmicroporous titanium silicates.
Homogeneous catalysts: phase-transfer processesIn the phase-transfer technology, the medium in
which oxidation with hydrogen peroxide is conductedis formed by two liquid phases that are mutuallyimmiscible, one aqueous and the other organic, formedby the substrate S possibly dissolved in a suitablesolvent. In the case of oxidations with hydrogenperoxide, an anionic metal derivative reacts in theaqueous phase with the hydrogen peroxide, forming aperoxidic complex that will be the catalytically activespecies in the process. This species, also of an anionicnature, is extracted in the organic phase from a
lipophilic cation such as a tetraalkylammonium orphosphonium (the phase-transfer catalyst, Q�). In theorganic phase, the peroxidic species reacts with thesubstrate forming the desired product (SO) andregenerating the reduced form of the catalyst.Distribution of this between the organic and theaqueous phases closes the catalytic cycle (Fig. 2).
Nevertheless, the first attempts by Charles Starks(then with Continental Oil) to epoxidize simple olefinsin the transfer phase using tungstic acid and hydrogenperoxide encountered little success due to thedifficulty of limiting, under the reaction conditions,the unproductive decomposition of the hydrogenperoxide into oxygen and water. Only later was itdiscovered, unexpectedly, that a mixture of tungstenand phosphate (or arsenate) anions in an acidenvironment is able to catalyze the epoxidationreaction, provided that a tetraalkylammonium orphosphonium salt is present in the system as a phase-transfer catalyst (Venturello et al., 1983; Table 2).
The reaction is applicable to a large number ofolefins: linear, branched or cyclic, including hardlyreactive ones such as terminal olefins or allyl chloride.In all cases, it is carried out under mild conditions (60-90°C and ambient pressure), even when verydiluted (8-15%) aqueous solutions of hydrogenperoxide are used.
The reaction is stereospecific: trans-2-hexene givesonly the trans epoxide and cis-2-hexene gives only thecis isomer. The catalytically active species that areformed from tungsten(VI) under phase-transferconditions and in the presence of phosphate orarsenate anions belong to a new class of peroxidiccomplexes, which are quaternary ammonium (orphosphonium) salts of the PW4O24
3� (Fig. 3), orAsW4O24
3� anions (Venturello et al., 1985). Thesecomplexes are the first peroxidic derivatives of aheteropolyacid whose structure has been solved.
Epoxidations are conducted batchwise and, underthe reaction conditions, most of the catalyst is dissolved
665VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
[ M�O ]�
[ M ]�
Na��H2O2 Na��H2OO
O
[ M�O ]�
[ M ]�
Q��SO Q��SO
O
11.1.3 Clerici fig 02
water
organic solvent
Olefin pHTemperature Time H2O2 conv. Epoxide yield
(°C) (min) (%) (%)
1-octene 1.6 70 45 98 82
1-dodecene 1.6 70 60 97 87
Allyl chloride 2 60 150 96 80
Styrene 3 40 180 93 77
a-methylstyrene 4.5 40 240 93 79
Cyclohexene 3 70 25 98 88
Table 2. Epoxidation of olefins with diluted H2O2 catalyzed by WO42�/PO4
3� mixtures
Fig. 2. Oxidation under phase-transfer conditions.
in the organic phase constituted by the olefin andpossibly by a solvent (generally toluene or a chlorinatedhydrocarbon). At the end of the reaction (indicated bythe more or less complete disappearance of thehydrogen peroxide), the two phases easily separate andthe product is recovered with conventional techniques,generally by distillation. Typical yields, based onhydrogen peroxide, vary between 80 and 95%, and anyexcess olefin is recovered unchanged: generally, in fact,selectivity on the olefin is around 95%. Evensophisticated substrates, of pharmaceutical interest, canbe epoxidized with excellent yields. A block diagram ofthe epoxidation process of a typical olefin is shown inFig. 4.
At the end of the reaction, the catalyst is recovered,with small quantities of by-products, as distillationresidues (or by ultrafiltration of the organic phase) andcan be regenerated and partly recycled.
The epoxidation reaction has been developed on acommercial scale for the production of 1,2-epoxydecane and of isobutyl 3,4-epoxybutyrate.The latter, in particular, has been produced on thescale of 100 t/y by epoxidation of isobutyl vinylacetate
and is a key intermediate in the synthesis of thenootropic drug oxiracetam:
Although epoxidation is the most interesting of thereactions promoted by these new catalysts, they areable to use hydrogen peroxide efficiently for numerousother transformations that are also of significantinterest, including oxidations of primary alcohols andof aldehydes to form carboxylic acids, of secondaryalcohols to give ketones or of sulphides to givesulphoxides (Ricci, 1996). Furthermore, an accuratechoice of reaction conditions makes it possible toobtain, in addition to epoxides, a whole series of otherolefin derivatives, often difficult to prepare by othermethods. Thus it is possible to transform olefins into
666 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
Fig. 3. The PW4O243�
anion.
epoxidation
olefin
H2O2 H2O residue
catalyst epoxide
olefin
phaseseparation distillation
Fig. 4. Epoxidation ofolefins with hydrogenperoxide under phase-transferconditions.
cat.
�H2O� H2O2
O
O OC
O
HO
CH2CONH2
O
O
N
C

vicinal diols or into 1,2-hydroxyketones. Both of theseclasses of compounds can be oxidized, in their turn, togive 1,2-diketones. Lastly, diols or even the originalolefins can be oxidatively cleaved with the formationof carboxylic acids (Fig. 5).
Recently, papers have been published which seemto suggest that, under appropriate conditions (e.g. inthe complete absence of any chlorinated species), theperformances of tungsten and phosphorus basedperoxide catalysts can be equalled by tungstic acidalone or, perhaps, by polymeric species produced bythis in the reaction environment. However, peroxidicphosphotungstates retain the merit of having madepossible the first commercial applications ofoxidations with hydrogen peroxide catalyzed by d0
metals and, more generally, that of having rekindledinterest in their study, then further revitalized by thediscovery of the new rhenium(VII) based catalysts,especially CH3ReO3 (Herrmann et al., 1991).
Heterogeneous catalysts: micro- and mesoporoustitanium-silicates
As already mentioned, another successful approachin the field of oxidation with hydrogen peroxide is that
which followed the discovery of micro- andmesoporous titanium-silicates. In particular, certaintitanium-zeolites are to be included among the mostpromising oxidation catalysts. Their activity andselectivity, often very high, and their intrinsic stabilityagainst oxidative degradation make them idealcatalysts for industrial applications. These titanium-zeolites are part of the more extensive family of mixedoxides containing a transition metal at high dispersion,whose structure can vary from microporous crystalline(redox zeolites) to wholly amorphous, with anintermediate situation represented by amorphous metalsilicates, with an orderly system of pores (MCM-41,MCM-48, HMS).
Among these titanium-silicates, the first, in orderof time and importance, is titanium-silicalite-1 (TS-1),discovered in the late 1970s (Taramasso et al., 1983).This is a microporous crystalline titanium-silicate,with an MFI structure (Baerlocher et al., 2001) whichderives formally from silicalite-1 (S-1), whollysiliceous, by the substitution of part of the silicon withtitanium, up to a maximum of around 2.5%. It isobtained by hydrothermal synthesis, from theprogressive condensation of SiO4 and TiO4 tetrahedraaround the template ion (i-C3H7)4N
� (Fig. 6). Itsporosity is due to a three-dimensional system ofinterconnected channels, having an average diameterof about 0.55 nm. This is an important parameter, as itsets an insuperable limit to the diffusion of reagents inthe active titanium sites and thus to the feasibility ofthe oxidation process.
Materials with an analogous composition, but ofdifferent structure, have been prepared by incorporatingtitanium in the S-2 (titanium-silicalite-2, TS-2), BEA(titanium-b, Ti-b), MOR (titanium-mordenite, Ti-MOR), and MWW (Ti-MCM-22) structures, andothers of less interest for catalysis. Mesoporoustitanium-silicates (Ti-MCM-41, Ti-MCM-48) are alsoknown. Apart from titanium, other metals with redoxcharacteristics have been incorporated into the
667VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
1,2-hydroxyketones1,2-diolsepoxides
olefins
carboxylicacids
1,2-diketones
Fig. 5. Products obtainable from oxidation of olefins in phase transfer.
Si
SiO4
TiO4
OSi
OTi
OSi
OSi
SiO
SiO
SiO
SiO
Si SiO
SiO
SiO
SiO
Si
SiO
SiO
TiOOH
H
SiOO
Si
H2O2
Fig. 6. Formation andgrowth of the crystallattice of TS-1.
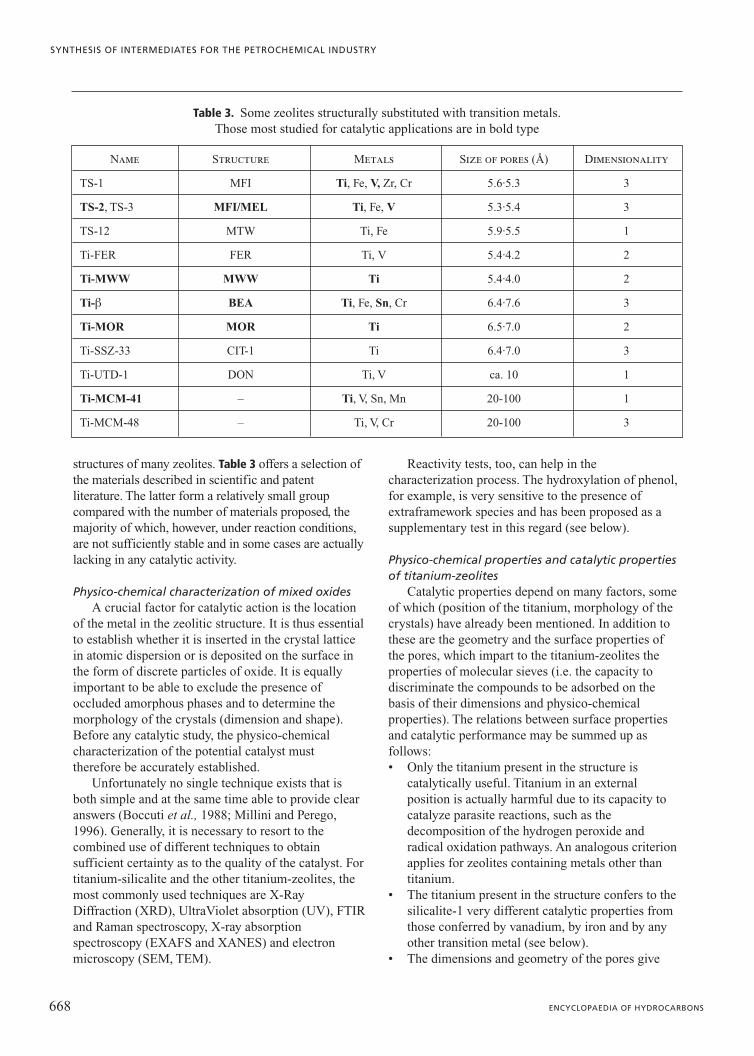
structures of many zeolites. Table 3 offers a selection ofthe materials described in scientific and patentliterature. The latter form a relatively small groupcompared with the number of materials proposed, themajority of which, however, under reaction conditions,are not sufficiently stable and in some cases are actuallylacking in any catalytic activity.
Physico-chemical characterization of mixed oxidesA crucial factor for catalytic action is the location
of the metal in the zeolitic structure. It is thus essentialto establish whether it is inserted in the crystal latticein atomic dispersion or is deposited on the surface inthe form of discrete particles of oxide. It is equallyimportant to be able to exclude the presence ofoccluded amorphous phases and to determine themorphology of the crystals (dimension and shape).Before any catalytic study, the physico-chemicalcharacterization of the potential catalyst musttherefore be accurately established.
Unfortunately no single technique exists that isboth simple and at the same time able to provide clearanswers (Boccuti et al., 1988; Millini and Perego,1996). Generally, it is necessary to resort to thecombined use of different techniques to obtainsufficient certainty as to the quality of the catalyst. Fortitanium-silicalite and the other titanium-zeolites, themost commonly used techniques are X-RayDiffraction (XRD), UltraViolet absorption (UV), FTIRand Raman spectroscopy, X-ray absorptionspectroscopy (EXAFS and XANES) and electronmicroscopy (SEM, TEM).
Reactivity tests, too, can help in thecharacterization process. The hydroxylation of phenol,for example, is very sensitive to the presence ofextraframework species and has been proposed as asupplementary test in this regard (see below).
Physico-chemical properties and catalytic propertiesof titanium-zeolites
Catalytic properties depend on many factors, someof which (position of the titanium, morphology of thecrystals) have already been mentioned. In addition tothese are the geometry and the surface properties ofthe pores, which impart to the titanium-zeolites theproperties of molecular sieves (i.e. the capacity todiscriminate the compounds to be adsorbed on thebasis of their dimensions and physico-chemicalproperties). The relations between surface propertiesand catalytic performance may be summed up asfollows:• Only the titanium present in the structure is
catalytically useful. Titanium in an externalposition is actually harmful due to its capacity tocatalyze parasite reactions, such as thedecomposition of the hydrogen peroxide andradical oxidation pathways. An analogous criterionapplies for zeolites containing metals other thantitanium.
• The titanium present in the structure confers to thesilicalite-1 very different catalytic properties fromthose conferred by vanadium, by iron and by anyother transition metal (see below).
• The dimensions and geometry of the pores give
668 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
Name Structure Metals Size of pores (Å) Dimensionality
TS-1 MFI Ti, Fe, V, Zr, Cr 5.6·5.3 3
TS-2, TS-3 MFI/MEL Ti, Fe, V 5.3·5.4 3
TS-12 MTW Ti, Fe 5.9·5.5 1
Ti-FER FER Ti, V 5.4·4.2 2
Ti-MWW MWW Ti 5.4·4.0 2
Ti-b BEA Ti, Fe, Sn, Cr 6.4·7.6 3
Ti-MOR MOR Ti 6.5·7.0 2
Ti-SSZ-33 CIT-1 Ti 6.4·7.0 3
Ti-UTD-1 DON Ti, V ca. 10 1
Ti-MCM-41 – Ti, V, Sn, Mn 20-100 1
Ti-MCM-48 – Ti, V, Cr 20-100 3
Table 3. Some zeolites structurally substituted with transition metals. Those most studied for catalytic applications are in bold type

rise to what is termed shape selectivity (i.e. thecapacity of the material to discriminate thereagents and products on the basis of their shapeand size; see below).
• Selective adsorption phenomena, governed by thenature of the surface, concentrate specificcomponents of the reaction mixture in thechannels, in the vicinity of the catalytic sites,decisively influencing the course of the reaction(activity, selectivity, deactivation). The capacity ofTS-1 to adsorb selectively non-polar compounds,even in the presence of water and other polar andprotic substances, explains the apparent differenceof the catalytic properties of titanium in TS-1 andin soluble alkoxides. The latter are more or lessinactive in an aqueous solution of hydrogenperoxide, that is under the conditions of maximumactivity of TS-1.The phenomena that take place on the catalytic site
are closely connected, on the one hand, with thetendency of framework titanium to expand its sphereof coordination from tetrahedral to octahedral,chemiadsorbing polar molecules, and, on the otherhand, with the poor resistance to hydrolysis of aTi�OSi unit (Fig. 7; Bellussi et al., 1992; Clerici andIngallina, 1993).
In accordance with this, the water and the alcoholsare coordinated on the titanium and reversiblyhydrolyse a Ti�OSi bond, producing Ti�OH andSi�OH species. The hydrogen peroxide behaves inlike manner, producing the Ti�OOH species, whichis at the origin of the oxidizing properties of the TS-1/H2O2 system. The structures illustrated in Fig. 8have been proposed for the Ti�OOH species (for thesake of clarity, the other ligands have been omitted; forthese, see again Fig. 7). The structure represented in Ais that which has the best chances of existing under theconditions suitable for catalysis (i.e. in the presence ofwater and/or of an alcohol).
TS-1 catalyzes the oxidation of many organicfunctions with hydrogen peroxide, and in particular theepoxidation of olefins and the hydroxylation ofparaffins and aromatic compounds, as well as theoxidation of alcohols, ethers and various sulphur andnitrogen compounds. On the contrary, it is not active in
oxidation processes with organic hydroperoxides. Theother metal-zeolites are active for a more limitedspectrum of reactions. Those with large pores andmesoporous materials, however, permit the use also oforganic hydroperoxides as oxidants.
TS-1, but also TS-2, Ti-b, Ti,Al-b and Ti-MWWcatalyze the epoxidation of olefins, in a dilutedsolution of hydrogen peroxide (�10%) and attemperatures below 80°C. TS-1 already shows a goodlevel of activity even below 0°C. In fact, epoxidation isthe preferred reaction when other reactive groups arepresent in the molecule. In this regard,chemoselectivity can be extremely high.
As opposed to what has been observed in the caseof soluble titanium catalysts, the solvents that favourhigh reaction rates are protic and polar: methanol(TS-1, TS-2, Ti-b, Ti,Al-b), acetone (TS-1) andacetonitrile. In a moderate quantity, water has a minoreffect on yields and selectivity. On the other hand, itspresence in the reaction environment cannot beavoided, being added with the oxidant and producedby the same reaction. The decomposition of hydrogen
669VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
SiSi
SiOH2
O
SiO
H
OH2 H2OO
OHOTi
SiSi
SiOH2
O
SiO
H
OH2O
OOHOTi
SiSi
Si Si
O
O
O
OTi
H2O/H2O2
Fig. 7. Chemical and steric properties of active sites of TS-1.
O
OO
H
H O HTi
OTi
R
Fig. 8. Structure of catalytically active speciesof TS-1.
A B
O
R
OO
H
HTi
O
R
OO
H
HTi
Fig. 9. Mechanism of epoxidation catalyzed by TS-1.

peroxide is generally slow with the catalysts indicatedor even negligible with TS-1.
Yields and selectivity are generally high, andvalues close to 100% can be reached with TS-1, inaccordance with the heterolytic mechanism (Fig. 9;Clerici and Ingallina, 1993; Neurock and Manzer,1996).
By-products of the reaction are generally productsof the hydrolysis of epoxide. Their formation may bereduced, neutralizing the residual acidity of thecatalyst, with the addition of small amounts of bases inthe reaction environment.
All zeolitic catalysts, not only oxidation ones,possess shape selectivity: only olefins capable ofbeing diffused inside the pores can be oxidized withhigh reaction rates. This explains the differentreactivity of cyclohexane on TS-1 (very poor) and onTi,Al-b (good), as its molecular size is very close tothe pore diameter of the former and significantlysmaller than that of the latter. More generally, shapeselectivity completely reverses the order of reactivityforeseen on the basis of the electronic properties of the double bond. Fig. 10 compares olefins witha different steric size in epoxidation with TS-1/H2O2,Ti,Al-b/H2O2 and with peracetic acid, which is notsignificantly affected by steric restrictions (thenumbers associated with olefins indicate their relativereactivity). The steric effects of the substituents on thedouble bond clearly prevail over the electron effectswith heterogeneous catalysts, whereas the opposite isthe case for peracetic acid.
While the activity, the selectivity and the mildreaction conditions of the TS-1/H2O2 system make it apreferable alternative to conventional oxidants, thesteric restrictions imposed by its microporous natureset evident limits to the range of its potentialapplications. Ti-b and the other titanium-zeolites withlarge pores provide a partial solution to the problem.The synthesis of mesoporous catalysts (Ti-MCM-41,Ti-MCM-48 and suchlike) could be an answer to theneed. However, the results obtained with hydrogenperoxide are not very encouraging. The conclusions ofthese studies may be summed up as follows:
• The rate of reaction diminishes rapidly in theorder: TS-1�Ti-b�Ti-MCM-41, becoming moreor less negligible on the mesoporous catalyst.
• Furthermore, Ti-b, Ti,Al-b and Ti-MCM-41 show,to an increasing extent, a structural instability inaqueous hydrogen peroxide. The drawback iscommon to other metal-zeolites, in particular thosecontaining vanadium, and takes the form of aprogressive collapse of the crystal lattice, with therelease of the metal in solution.
• The mesoporous materials available have asubstantially hydrophilic surface. Thiscircumstance favours the adsorption of water to thedetriment of the olefin or other non-polar reagent,thereby lowering the reaction rate. Unfortunately,water cannot be avoided in using hydrogenperoxide as the oxidant.
• A possible alternative is provided by the use of tert-butyl hydroperoxide (TBHP), for which theactivity increases in inverse order: Ti-b�Ti-MCM-41 while TS-1 is practicallyinactive for steric reasons.The above conclusions, therefore, do not imply any
decreasing activity of titanium sites with increasingpore size, but rather a decrease in the olefin adsorptioncapacity. It is the density of the surface Si�OHgroups and thus the hydrophilic nature of the poresthat increases in the TS-1�Ti-b�Ti-MCM-41 series,in parallel favouring the adsorption of water at theexpense of the olefin (or some other non-polarreagent). The instability, too, is to some extent aconsequence of the increase of the Si�OH groups,which may be regarded as defective sites of thesiliceous matrix and the starting point of the hydrolyticphenomena that cause the decay of the matrix.
TS-1 catalyzes the hydroxylation of paraffins, attemperatures below 100°C, in an aqueous ormethanolic solution of hydrogen peroxide (Huybrechtset al., 1990; Clerici, 1991):
It may seem surprising that commonly inertcompounds such as paraffins, even in a dilutedsolution, can be oxidized preferentially with respect tothe solvent methanol. The reason for this is probablydue to the hydrophobic nature of the catalyst, able toselectively adsorb the paraffin in the vicinity of theactive sites in accordance with the criteria alreadystated for the epoxidation of olefins. The same reason
670 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
AcO
TS-1:
Ti,Al-β:
CH3CO3H: R RR1
� � �
� �
� � � �
�
1.0
0.04 1 20-24 27 240
4.7 6.0 16.2
1 1.1 1.6 1.6
Fig. 10. Relative rate of epoxidation of some substitutedolefins with various oxidants.
TS-1
TS-1
H2O2
H2O2 TS-1 H2O2
OH OH
�
O O

likely explains the poor activity, or the lack of activity,of the large-pore titanium-zeolites and of mesoporouscatalysts, for which the order of reactivity, alreadyseen for olefins, now appears far more accentuated.
In competition with hydroxylation, the decompositionof the hydrogen peroxide also takes place, generally to anotable extent. Only secondary carbons undergooxidation, the methyl groups being inert, which producesa mix of secondary alcohols and ketones (formed byconsecutive oxidation). The vanadium-silicalites, instead,are active also for the hydroxylation of the primarycarbons, forming a mix of primary and secondaryalcohols and products of successive oxidation.
As far as the aromatic compounds are concerned,TS-1 and Ti-MOR catalyze their hydroxylation to thecorresponding phenols (Romano et al., 1990):
The reaction follows the rules of electrophilicattack: the reactivity of phenol and toluene is high,while that of nitrobenzene, chlorobenzene and benzoicacid is negligible. In alkylbenzenes, the oxidation ofthe alkyl groups competes with that of the aromaticnucleus, except for the methyl groups, which arealmost completely inert. In this case as well, thebehaviour of the vanadium-silicalites is different, asthey preferentially catalyze the hydroxylation of thealkyl side chains, including methyl groups.
The hydroxylation of paraffinic and aromaticcompounds is regulated by severe steric restrictionsimposed by the size of the pores. The kinetics of thehydroxylation of toluene is already comparable withthat of benzene, in spite of the fact that it is morenucleophilic. A second consequence of the stericeffects is that the decomposition of hydrogen peroxidecan become the predominant reaction when thesubstrates are bulky molecules.
The mechanisms of hydroxylation reactions havenot been studied in depth. However, there is strongevidence of their homolytic nature, causing them todiffer from epoxidation, which is heterolytic. Amechanism able to explain both the hydroxylation and
the competition of the decomposition of hydrogenperoxide has recently been proposed (Clerici, 2001).
The titanium-zeolites resemble other catalyticsystems in the oxidation of primary and secondaryalcohols, except for the effects of steric restrictions(Maspero and Romano, 1994). The products arealdehydes and the corresponding ketones, to which, inthe case of the primary alcohols, the carboxylic acidsformed by consecutive oxidation can also be added(Fig. 11). The primary alcohols are not as easy tooxidize as the secondary ones. It is worth noting thescanty reactivity of methyl alcohol, which makes itsuitable as a solvent for the other oxidations.
Ketones are generally stable under the oxidationconditions of the secondary alcohols. They can,however, be oxidized to lactones and the correspondingesters, selecting the appropriate catalysts. Sn-b provesto be the most selective catalyst in this context, withvalues that can reach as high as 98% (Corma et al.,2001). Contrary to the case of the titanium-zeolites, ifdouble bonds are present, they do not undergoepoxidation, as exemplified by the oxidation ofdihydrocarvone (Fig. 12). The reason for this must besought in the different mechanism, which for Sn-b isbased on the Lewis acid properties of the active site,instead of redox properties as in the case of Ti-b.
In the oxidation of amines, various products can beobtained, according to the nature of the amine and ofthe reaction conditions:
From aniline, when hydrogen peroxide is scarce,the final product obtained is azoxybenzene, while withan excess of oxidant, the main product is nitrobenzene(not shown). Oximes, alkyl-hydroxylamines and otherproducts are formed in the oxidation of the primaryand secondary aliphatic amines. Also in the case of theoxidation of amines, there is a different selectivity onthe various metal sites. In the oxidation of aniline, forexample, the vanadium-silicalites form mainlynitrobenzene even when hydrogen peroxide is scarce.
671VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
TS-1
H2O2
TS-1
H2O2
�
�
OH OH
OHHO
OH
� OHHO
O
RCH2
OO
H
HTi
OH2
H � RCHOO
Ti
O
H
OO
C
H
H
R
HTi
Fig. 11. Mechanism of alcoholoxidation catalyzed by TS-1.
NH2 NHOH
RCH2NH2 RCH�NOH RCHO
NO
O�
N�N�ArNHOH
ArNHOH

The thioethers are easily oxidized to thecorresponding sulphones and sulphoxides:
[12] R�S�R��R�SO�R��R�SO2�R
The reaction, initially hardly considered due to theease with which the thioethers oxidize, may take onimportance as a means of removing sulphur frommotor fuels. It is clear that in this application the mostsuitable catalysts are those with a mesoporousstructure, and consequently the preferred oxidant istert-butyl hydroperoxide.
Epoxidation of olefins
Industrial production of epoxidesAmong the products that can be obtained by
oxidation of olefins, epoxides have always receivedspecial attention due to their high reactivity, whichmakes them extremely useful (and used) intermediatesin organic synthesis.
As often happens, the simplest epoxide, ethyleneoxide, is produced with a method (i.e. direct oxidation ofethylene with oxygen in the gas phase) which differsfrom that used for all the other ones. The higher epoxides,instead, are prepared under milder conditions and withmore complex processes. In fact, at the relatively hightemperatures required by the use of molecular oxygen, inaddition to double-bond, also allylic C�H are oxidizedwith the consequence of obtaining a broad spectrum ofproducts. For this reason, the higher epoxides, when notobtained through oxidation of the corresponding olefinswith percarboxylic acids (a troublesome reaction, alsofrom the standpoint of safety), are generally prepared byoxidation of olefins with chlorine (through thecorresponding chlorohydrins) or with hydroperoxides. In
the future, epoxidation with hydrogen peroxide, now inthe development stage, could be added to these processes.
The manufacture of propylene oxide, which,among the higher epoxides, is by far the mostimportant commercially, will be examined below.
The production capacity for propylene oxide (PO)in the year 2000 globally amounted to 5.7·106 t, withan annual demand of around 4.7·106 t. It is estimatedthat about 7% of the propylene produced is used forthe production of this derivative.
Propylene oxide is a basic intermediate of thechemical industry, used for producing a long series ofcommercial products. For this purpose, it must first betransformed into a number of its derivatives: polyetherpolyols (65%), propylene glycols (20%), and otherintermediates such as glycolethers, alkanolamines, 1.4-butanediol, allyl alcohol and propylene carbonate.Polyether polyols, produced by the reaction of propyleneoxide with polyhydric alcohols, are used in the productionof polyurethane foams, of flexible type (applied, forexample, in the automobile industry for seats, or forhousehold or furnishing purposes for mattresses andcarpets) or of rigid type (used, for example, for heatinsulation). Propylene glycol (1.2-dihydroxypropane) is used in the production of polyester resins and, due toits biocompatibility, in the formulation of cosmetic,pharmaceutical and food products. For the same reason,dipropylene glycol and other short chain oligomers areused to produce antifreeze fluids, hydraulic fluids,cutting oils and lubricants. As opposed to analogousproducts derived form ethylene oxide, in fact, they arebiodegradable and harmless to living organisms.Mention should also be made of certain uses ofalkanolamines (detergents, anticorrosion products), ofpolyethers (antifoam agents, surface-active agents) andof esters (solvents for inks and paints).
Chlorohydrin processIntroduced about a century ago, the chlorohydrin
process is still widely applied. Propylene is reactedwith chlorine in an aqueous solution, producing amixture of two chlorohydrins, from which propyleneoxide is obtained by treatment with lime or soda(global yield around 89%):
672 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
O
O
O
OO
O
O
O
OO
Sn
O
OO
HR
H
H2O2
Ti
O
OH
OH
Fig. 12. Different oxidation mechanism of Sn-b and Ti-bcatalysts.
ula 9
CH�CH2 � H2O � Cl2CH3 n CH3 CH CH2 �
OHCl
� (1�n) CH3 CH
OH Cl
CH2 � HCl
n CH3 CH CH2�(1�n) CH3
CH3 CH
O
CH2�NaCl�H2O
CH
OH ClOHCl
CH2�NaOH
CH3 CH
O
CH2�NaCl�H2O
� (1�n) CH3 CH
OH Cl
CH2 � HCl

The chlorohydrin process is burdened by a series of problems, with environmentalimplications, such as the formation of inorganicand organic chlorinated coproducts and by-products. In fact, a large part of the chlorine used eventually forms a stoichiometric quantity ofhydrogen chloride, which is neutralized – a process taking place simultaneously with theformation of epoxide – using lime or soda. A considerable quantity of organic chlorinated by-products is also formed, whichuselessly consume propylene and complicatedownstream operations. The effluent, consisting of a diluted solution ofsodium or calcium chloride and chlorinatedorganic impurities, must be properly treated andthen disposed of. A further problem arises fromthe risks of corrosion and hence from the need touse resistant materials. In spite of this, thechlorohydrin process is economically viable, ableto satisfy approximately one-half of the worldmarket of propylene oxide.
Epoxidation with hydroperoxidesThe processes in which an organic
hydroperoxide acts as the oxidizing agent wereoriginally developed by Halcon, ARCO and Shell and were applied in the industry as ofthe 1970s. These processes produce approximatelythe other half of the propylene oxide marketed in the world. They are liquid-phase oxidationprocesses, in which the catalysts may be soluble(Halcon/ARCO) or supported (Shell).
The oxidants are TBHP (tert-butylhydroperoxide) or EBHP (ethylbenzenehydroperoxide or (1-phenyl)ethyl hydroperoxide),obtained by oxidation with air of isobutane orethylbenzene, respectively.
The alcohols coproduced, tert-butanol (TBA)or 1-phenylethanol, are used for the production ofMTBE (methyl tert-butyl ether) and styrene,respectively.
The commercial advantage of propylene oxideproduced in this way depends largely on theirvalue.
A third process also exists, introduced recently bySumitomo, in which the oxidant is cumylhydroperoxide and the alcohol coproduced is recycledinstead of being placed on the market.
Epoxidation processes with TBHPTwo versions of the process exist, as used by
Lyondell (formerly by ARCO) and by Huntsman(formerly by Texaco). The main reactions are thesame:
In the ARCO process, six stages are distinguished:oxidation of the isobutane, epoxidation of thepropylene, separation of the products, purification ofPO, purification of TBA, preparation and recovery ofthe catalyst. In the first stage, isobutane is oxidized inthe liquid phase with oxygen, at around 140°C underpressure, producing a mixture of tert-butylhydroperoxide, tert-butanol and various by-products,such as acetone and carboxylic acids. The reaction isrun in recycled tert-butanol and is a typicalautoxidation requiring no catalyst, although thepresence of initiators speeds up the initial phases. Forconversion values close to 35%, the tert-butylhydroperoxide and tert-butanol selectivities are 53%and 40%, respectively.
The stream from the autoxidation reactor issubjected to fractionation to remove the isobutane,diluted with recycled tert-butanol to bring theconcentration of TBHP to 40% and, after adding themolybdenum catalyst solution, fed to the epoxidation reactor. A large excess of propylene issupplied to maximize yields and diminish reactiontimes. The reaction is conducted at about 120°C and under pressure, with up to 98%selectivity with respect to propylene. Among the by-products are methyl formate(particularly unwelcome since its boiling point isclose to that of epoxide), carbonyl compounds,carboxylic acids and propylene glycol. Thesuccessive phases include the separation of theproducts from the excess propylene (to be recycled),the recovery of the molybdenum and the purificationof the propylene oxide and of the tert-butanol. Theprocess is characterized by the production of PO andtert-butanol in a ratio of 1:2.4.
In a second version of the process, originallydeveloped by Texaco, epoxidation is carried out in twosuccessive stages at 110°C and 135°C. This secondversion differs from the preceding one also in themethods of preparing the catalyst and the separationand purification of the products, as well as in the lowerPO/tert-butanol ratio.
673VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
�
O
CCH3 H
CH3
CH3
CCH3 OOH
CH3
CH3
CCH3 OH
CH3
CH3
�
�
CCH3 OOH
CH3
CH3
CCH3 OH
CH3
CH3
CHCH3 CH2
CHCH3 CH2

Epoxidation processes with EBHPTwo processes exist in which the oxidant is
(1-phenyl)ethyl hydroperoxide, differing in type of catalyst:
In the ARCO (now Lyondell) process, a solublecompound of molybdenum is used, whereas in theprocess developed by Shell, a silica supportedtitanium(IV), Ti/SiO2, is used. The ratio of the productsPO/styrene, instead, is similar and close to 1:2.2.
In the first stage of both processes, thehydroperoxide solution is prepared by oxidation ofethylbenzene with air at about 150°C and a pressure ofabout 3.5 bar. For safety reasons, conversion is limitedto values of less than 10%. Selectivity is 80-85% withrespect to hydroperoxide, the remainder consistingmostly of 1-phenylethanol and acetophenone. TheEBHP concentration required for the epoxidationreaction (17-19%) is reached by removing the excessof ethylbenzene by distillation.
Epoxidation is conducted at 100-115°C. In theShell process, the heterogeneous catalyst Ti/SiO2makes it possible to adopt a packed fixed-bed reactor.A notable feature of this catalyst is its stabilityregarding the release of soluble species in solution. Onthe contrary, under analogous reaction conditions,supported molybdenum, tungsten or vanadiumcatalysts release soluble species. The regeneration ofthe deactivated catalyst Ti/SiO2 takes place byoxidizing the organic deposits with air.
What characterizes the processes with EBHP is thestage dedicated to the production of monomericstyrene. The mixture of 1-phenylethanol andacetophenone, recovered from the effluent of theepoxidation reactor, is dissolved in triphenylmethaneand subjected to dehydration at a high temperature inthe presence of an acid catalyst. After the separation ofthe styrene, the residue containing acetophenone isconveyed to a hydrogenation reactor, after which theadditional 1-phenylethanol rejoins the stream sent fordehydration. Selectivity exceeds 98%.
From the standpoint of overall performance, inboth the ARCO and the Shell process the yields ofepoxide are nearly 92% and those of styrene are 94%.
Oxidation process with cumyl hydroperoxideThe process developed by Sumitomo, based on
cumyl hydroperoxide as the oxidant, is characterizedby the fact that the co-product, cumyl alcohol, isdehydrated and hydrogenated to cumene and thenrecycled. The hydroperoxide route, in this particularcase, avoids having to place another product on themarket. Furthermore, it is characterized by itstemperature and pressure conditions, significantlymilder than in other hydroperoxide processes. So far ithas been applied only in a single plant in Japan, whichhas been in operation since 2003 (200,000 t/y).
Epoxidation with hydrogen peroxide catalyzed by TS-1This process differs from the preceding ones in its
use of hydrogen peroxide as the oxidant, made possibleby adopting titanium-silicalite as the catalyst (Clerici etal., 1991). The coproduct in this process is merely water,which makes it unnecessary to market or recycle it, andoffers indisputable environmental advantages due to theabsence of chlorinated by-products.
Epoxidation is conducted in aqueous methanol at atemperature below 60°C and pressures slightly above 1atm. The reaction rate is high, with a TOF (Turn OverFrequency) of 1-2 s�1 (measured in the laboratory at40°C). Its selectivity with respect to hydrogenperoxide is nearly quantitative. Hydrogen peroxideconsumption, due to methanol oxidation and todecomposition into water and oxygen, is negligible.The by-products are propylene glycol and its twomonomethyl ethers, produced by the attack ofmethanol on the epoxide ring. The selectivity,generally high, can be made nearly quantitative(�98%), by keeping the acidity of the environmentunder control with the addition of small quantities ofbases (in the order of some ppm).
The reaction takes place by contacting, in a slurryreactor, a stream of propylene with a suspension ofTS-1 in methanol/water, into which an aqueoussolution of hydrogen peroxide is fed (Romano, 2001).The high activity of the catalyst in a diluted solutionmakes it possible to operate with oxidantconcentrations of even less than 10%. The effluent issubjected to distillation to recover the unreactedpropylene, the propylene oxide and the methanol. Theresidual aqueous solution, after possible recovery ofthe propylene glycol and the monomethyl ethers, isconveyed to an ordinary biological plant for finaltreatment.
Proper control of the operating conditions enablesthe phenomena of catalyst deactivation, caused by thedepositing of heavy organic by-products, to beminimized. These by-products can be removed bywashing with solvent at temperatures higher than100°C or, simply, by combustion with air. No
674 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
�
�
�
C H
CH3
H
C OOH
CH3
H
C OOH
CH3
H
C OH
CH3
H
C OH
CH3
HO
CH�CH2CH3
CHCH3 CH2

phenomena of deactivation due to the leakage oftitanium from the crystal lattice have been observed.
Compared with the chlorohydrin andhydroperoxide processes, the new technology ischaracterized by its low environmental impact, asimpler process layout and reduced investment costs.For example, there is five times less aqueous wasteand it is suitable for normal biological treatment. Aprototype plant of about 6 t/d capacity was set up byEniChem in 2001.
Enantioselective epoxidationsSome rather peculiar reactions, enantioselective
epoxidations, will now be addressed, which have foundvarious industrial applications, although mostly in theproduction of small quantities of compounds havingextremely high value added, typically used by thepharmaceutical industry.
The first procedure for enantioselective epoxidationwith high enantiomeric excesses was published in 1980by Barry Sharpless (then at Stanford University), whichconsists in the reaction of primary allyl alcohols with ahydroperoxide (e.g. tert-butyl hydroperoxide or cumylhydroperoxide), promoted by stoichiometric quantitiesof an alkoxide of titanium(IV) and of enantiomericallypure diethyl tartrate (Katsuki and Sharpless, 1980). Theactive species is formed in situ from the titanium thatsimultaneously binds the enantiomerically pure tartrate,the hydroperoxide and the substrate (the latter throughthe OH group). The synthetic versatility of the allylalcohols caused the reaction to rapidly become asynthetic instrument of primary importance. The roadtowards industrial applications was further eased when,in 1986, Sharpless discovered that, in the presence ofmolecular sieves, it was possible to run the oxidationsusing catalytic quantities (5-10% in mols) of thetitanium-tartrate complex.
The epoxidation of allyl alcohol was one of thefirst enantioselective epoxidation processesdeveloped at industrial scale (by ARCO; Fig. 13).Using the tartaric esters of the one form or theother, both the enantiomers of glycidol can beobtained with enantiomeric excess (ee) between 91and 95%.
The isolation of the product is greatly facilitated bytransforming in situ the glycidol (a labile, watersoluble product) into the corresponding m-nitrobenzenesulphonate, which, after crystallization,is recovered with ee�99%.
More or less at the same time as Sharpless’senantioselective epoxidation, another efficientprocedure was published, using hydrogen peroxide asthe oxidizing agent. This was the Juliá-Colonnaepoxidation, which uses polyamino-acids as chiralcatalysts, typically polyalanine (Juliá et al., 1982). Theenantiomeric excesses are very large (up to 96%), butlimited to the epoxidation of a number of a,b-unsaturated ketones.
New and important contributions followed. In1988, it was again Sharpless who introducedasymmetric dihydroxylation (which actually producesdiols and not epoxides), catalyzed by osmiumtetroxide and chiral amines; then, in 1991, a protocolwas defined for asymmetric epoxidation (catalyzed bymanganese complexes with chiral Schiff bases), whichis applied to a large number of olefins and no longeronly to allyl alcohols. In both cases, however, theoxidants used are no longer peroxides, but are amineoxides, K3[Fe(CN)6] or sodium hypochlorite.
Enantiomerically pure epoxides, prepared by theone procedure or the other, are used in the synthesis ofvarious pharmaceutical products (e.g. b-blocking orantiviral drugs) and of products for agriculture(pheromones).
675VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
H2C CH2
CH OHO
HH CH2OH
H
H
H
COOC2H5
COOC2H5
H3C OOHTi (i-PrO)4
D - (-) - DET
D - (-) - DETC� �
�
CH3
CH3
H3C OHC
CH3
CH3
HO
HO
C
C
(R)-glycidol
Fig. 13. Enantioselective epoxidation of allyl alcohol.

Oxidation of aromatics
Industrial production of phenolPhenol is one of the main chemical intermediates
with increasing world production, amounting to about8·106 t in 2004. Above all, it is used in the productionof intermediates for polycarbonates and epoxy resins,phenolic resins (used, for example, in adhesives) andcaprolactam, the raw material of Nylon 6.
At present, nearly all the global phenol productionis based upon the cumene process, which is also themain way of industrially producing acetone: for every10 t of phenol, in fact, about 6 t of acetone arecoproduced. The cumene process foresees threestages: reaction of benzene with propylene(alkylation), which gives isopropylbenzene (cumene);oxidation of the cumene to the correspondinghydroperoxide; acid decomposition of thehydroperoxide, which produces phenol and acetone:
The first reaction, the alkylation of benzene, iscatalyzed by acids: traditionally aluminium trichlorideor supported phosphoric acid. Recently, however,innovative processes have been developed that foreseethe substitution of these acids with appropriate zeoliteswhich have a very reduced environmental impact(Bellussi and Perego, 2000).
Also thanks to these improvements, the cumeneprocess is fully satisfactory in many aspects.Nevertheless, any increase in the productive capacityof phenol implies the need to find commercial outletsfor a corresponding quantity of acetone.
A first route to avoid such a need consists inrecycling the acetone, transforming it back intopropylene or, more briefly, into isopropyl alcohol,which is also able to alkylate the benzene (Girotti etal., 2003).
The processes of direct oxidation of benzene intophenol provide a more effective and definitivesolution, which would completely eliminate thecoproduction of acetone. The first of these processes isknown as the Solutia process, from the name of theAmerican company that recently finalized it incollaboration with the Boreskov Institute of Catalysisof Novosibirsk (Russia), using nitrous oxide, N2O, as
the oxidant and iron containing ZSM 5 zeolites ascatalysts:
Oxidation, run at 350°C, enables high rates ofconversion of benzene (27%) to be achieved with 98%selectivity with respect to phenol (Panov, 2000).Despite this, the use of N2O can hardly be applied on alarge scale; in fact, its supply is not adequate forphenol synthesis and its production for this purpose(e.g. by pyrolysis of ammonium nitrate or by selectiveoxidation of ammonia) would be too costly. Thus, theonly plausible scenario for the application of theSolutia process is that of foreseeing its implementationside-by-side plants for adipic acid (an importantintermediate in the nylon cycle; Bellussi and Perego,2000). N2O co-produced in the latter, instead of beingeliminated, could be used for the production of phenol,practically at zero cost.
A viable alternative to N2O is hydrogen peroxide:
The main problem encountered in the directoxidation of benzene to phenol (not only withhydrogen peroxide) is the inadequate kinetic control ofthe reaction: phenol, in fact, is oxidized more readilythan benzene and, therefore, rather than beingaccumulated, is transformed into a whole series offurther oxidized products (catechol, hydroquinone,benzoquinone, etc.), up to the complete degradation ofits cyclic structure or the formation of polymeric tars.Indeed, the ease with which phenol is oxidized withhydrogen peroxide is exploited in the production ofmixtures of catechol and hydroquinone (see below).Therefore, to obtain phenol selectively by theoxidation of benzene, strategies must be found thatenable the reactions of overoxidation to be sloweddown, allowing the desired product to be accumulated.The first step in this direction was taken by GeorgeOlah who, working with extremely concentratedhydrogen peroxide (98%) in a superacid environment(FSO3H-SbF5 1:1) at �78°C, obtained phenol with ayield of 54% based on both hydrogen peroxide and onbenzene (Olah and Ohnishi, 1978). Although theprohibitive reaction conditions meant that this workcan only be of historical value, the idea that in asuperacid medium phenol could be protonated (andhence deactivated towards subsequent oxidations) ismost definitely noteworthy.
A second approach towards reducing the rate atwhich phenol is oxidized arises from the consideration
676 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
ormula 13
Fe-zeolite� �N2O N2
OH
OH
cat.� �H2O2 H2O
CH
CH
cat.H3C
H3C CH3
CH2�
�
OH
H2SO4
C
O2
H3CH3C
OOH
O2
O
CH3C CH3

that many consecutive oxidation problems areeffectively handled and resolved by biological systems,which succeed in segregating the catalyst and thereaction product in different environments. In this way,the product substantially no longer has access to thecatalytic site and consequently can no longer betransformed, and is accumulated without any difficulty.A typical example is constituted by several enzymes ofthe oxygenase class, in which the active sites are buriedinto deep hydrophobic pockets easily accessible tolipophilic substrates, whereas the more hydrophilicproducts, once released, no longer return there.
This important feature of biological systems can bereproduced, to some extent, by adopting a reactionmedium consisting of two mutually immiscible liquidphases, one aqueous and the other one organic; thefirst one contains the catalyst and the second is veryefficient in extracting the phenol produced, conveyingit away from the catalyst and thereby minimizingfurther oxidations.
The best results have been obtained usingacetonitrile. In fact, in the presence of benzene, aheterogeneous system is formed by water andacetonitrile with two phases, one mainly aqueous andthe other mainly organic. The concentration of thebenzene in the mainly aqueous phase is more thanquadrupled. Simultaneously, a large part (85%) of thephenol formed is extracted in the organic phase and isthus protected against the overoxidation reactions.
The catalytic system consists of an acid (e.g.trifluoroacetic or sulphuric), iron sulphate and a ligandable to modulate its activity and its selectivity: 2-methylpyrazine-4-carboxylic acid N-oxide:
With this system it has been possible to convert8.4% of the benzene, obtaining phenol with aselectivity of 97% with respect to benzene and of 88%with respect to hydrogen peroxide (Bianchi et al.,2000).
The most recent developments in the oxidation ofbenzene to phenol with hydrogen peroxide regard theuse of a particular solvent (sulpholane) in combinationwith TS-1. Oxidizing benzene under these conditionsin the most commonly used solvents, the selectivityfor phenol decreases very rapidly with the increase inconversion and, typically, falls below 50% alreadywith benzene conversions of around 3%. This drop isstrongly reduced when sulpholane is used as thesolvent. In this case, the selectivity for phenol remainshigher than 80% even with benzene conversions of8%. The by-products are catechol (7%), hydroquinone(4%), 1,4-benzoquinone (1%) and tars (5%; Balducciet al., 2003). Probably sulpholane forms a complexwith phenol, by means of a hydrogen bond, once thishas been desorbed by the catalyst:
The complex is definitely larger in size thanphenol and so, although it is relatively labile, it isin any case able to delay, to some extent, a newentry of the phenol into the cavities of the zeolite.In this way the sulpholane modifies, by reducing it,the coefficient of distribution of the phenolbetween the volume of the intrazeolite cavities andthe external solvent, thereby delaying itssubsequent oxidation.
A further improvement in the performance of thecatalytic system was obtained by subjecting thetitanium-silicalite to a post-synthesis treatment withaqueous solutions of NH4HF2 and hydrogen peroxide,at 80°C. The treatment removes part of the titaniumand also substantially modifies the environment ofpart of the titanium not removed. The new catalystthus obtained (TS-1B) enables a 94% selectivity tophenol to be reached, with benzene conversions ofaround 9% (Balducci et al., 2003).
The selectivity of the whole process could befurther improved by converting the by-products intophenol by treating them with hydrogen, in water, over
677VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
CH3
HOOC
O�
N
N�
O
O
OH S
oxidation
H2O
H2O2 H2O/Na2SO4
tars
phenol
sulpholane/benzene benzenemake-up
H2SO4 H2NaOHphenol
distillation HDOsulpholanepurification
diphenolsrecovery
benzene
H2O
Fig. 14. Directoxidation of benzeneto phenol withhydrogen peroxidecatalyzed by TS-1B.HDO,hydrodeoxigenation.

commercial catalysts based on nickel andmolybdenum oxides (Bianchi et al., 2004).
A simplified block diagram of the process isshown in Fig. 14. The oxidation of benzene is carriedout continuously, between 95 and 110°C and at 6 bar,over a fixed bed containing TS-1B. At the end, theunconverted benzene, the water and the phenol areremoved through distillation, while the sulpholaneused as a solvent is purified by extracting the by-products (hydroquinone, catechol and tars) withaqueous soda and then recycled. The process permitsthe complete conversion of the hydrogen peroxide andconversion of more than 15% of the benzene, with aselectivity to phenol higher than 97% based onbenzene and 71% on hydrogen peroxide.
Conversion of the benzene obtained in directoxidation with hydrogen peroxide, although low, isfully comparable with that of the traditional process, inwhich the overall conversion after the two stages doesnot exceed 8.5% and, more often, is around 6%. Whileit is still too early to know whether the resultsdescribed will lead to a new process for the industrialproduction of phenol without the coproduction ofacetone, undoubtedly it lays the foundations for it.
Oxidation of phenol to hydroquinone and catecholIn 2002, the production capacity of hydroquinone
and of catechol in the developed countries amountedto about 50,000 and 32,000 t/y, respectively. Theproduction of the former is mainly driven by thedemand of the photographic industry. That of catecholhas undergone an appreciable development since the1970s, with the introduction on the market of syntheticvanillin and other artificial aromas. In addition, bothare used for the production of various antioxidants andinhibitors of polymerization.
In the past, diphenol synthesis processes foresaw thetransformation of pre-existing functional groups,through a succession of stoichiometric reactions. Thedemand for hydroquinone for the budding photographicindustry at the beginning of the Twentieth century, forexample, was first satisfied by reducing with sulphurdioxide the benzoquinone produced by aniline oxidationwith chromic acid. Although the environmentalcompatibility of the process was subsequently improved,by substituting chromic acid with manganese dioxide,the quantity of by-products continued to be far greaterthan that of hydroquinone. It is likely that the anilineoxidation process is still being applied in somedeveloping countries. Also an alternative method, theperoxidation of p-diisopropylbenzene, is based on aseries of stoichiometric reactions. It was not until the1970-1980s that some catalytic processes usinghydrogen peroxide as the oxidant were developed andbecame part of industrial practice. The production of
resorcinol continues to be an exception, as it is stillobtained by the alkaline fusion of m-benzendisulphonicacid or by the oxidation of m-diisopropylbenzene.
Direct hydroxylation of phenol with hydrogenperoxide
The direct hydroxylation of phenol with hydrogenperoxide was introduced during the 1970-1980s andwas a major development from the standpoint ofenvironmental safeguard.
The first commercial processes were developed byBrichima and Rhone-Poulenc. Two products are obtained,catechol and hydroquinone, in a ratio that depends bothon the process and on the working conditions. The mainby-products are water and tars, the latter being possiblyreused for the plant’s energy requirements.
In the Brichima process, no longer in operation forabout two decades, the hydroxylation of the phenoltook place with a radical-type mechanism:
This was a typical chain reaction in which iron(II)and cobalt(II) played the role of radical initiators,generating a hydroxyl radical. The propagationreaction, in which the radical species responsible forattacking the aromatic nucleus was also regenerated,consisted mainly in the oxidation of thecyclohexadienylic intermediate by the hydrogenperoxide. In this phase, the intervention of the metalions, present at the level of ppm, was overall regardedas negligible (Maggioni and Minisci, 1977).
In the Brichima process, the chief product wascatechol, obtained in a ratio of about 2.0-2.3 tohydroquinone. To minimize reactions of consecutiveoxidations, which produce tars, it was necessary to use alarge excess of phenol with respect to the oxidant,limiting its conversion to less than 25%. This entailedhaving to separate considerable quantities of phenol bydistillation from the tars and to recycle them. Selectivitywith respect to hydrogen peroxide, the more costly of thetwo reagents, slightly exceeded 60%, no longer sufficientwith the passing of time to guarantee an economicadvantage to the process as compared with others.
In the Rhone-Poulenc process (Varagnat, 1976), themechanism is heterolytic and the catalyst is a mixture ofa strong mineral acid (H2SO4, HClO4) with phosphoricacid. The latter acts as a sequestering agent of metal
678 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
OH
� .OH
OH
OHH
� H2O2
H2O2�Fe2� .OH�OH��Fe3�
� �H2O
OH
OHH
OHOH
.OH
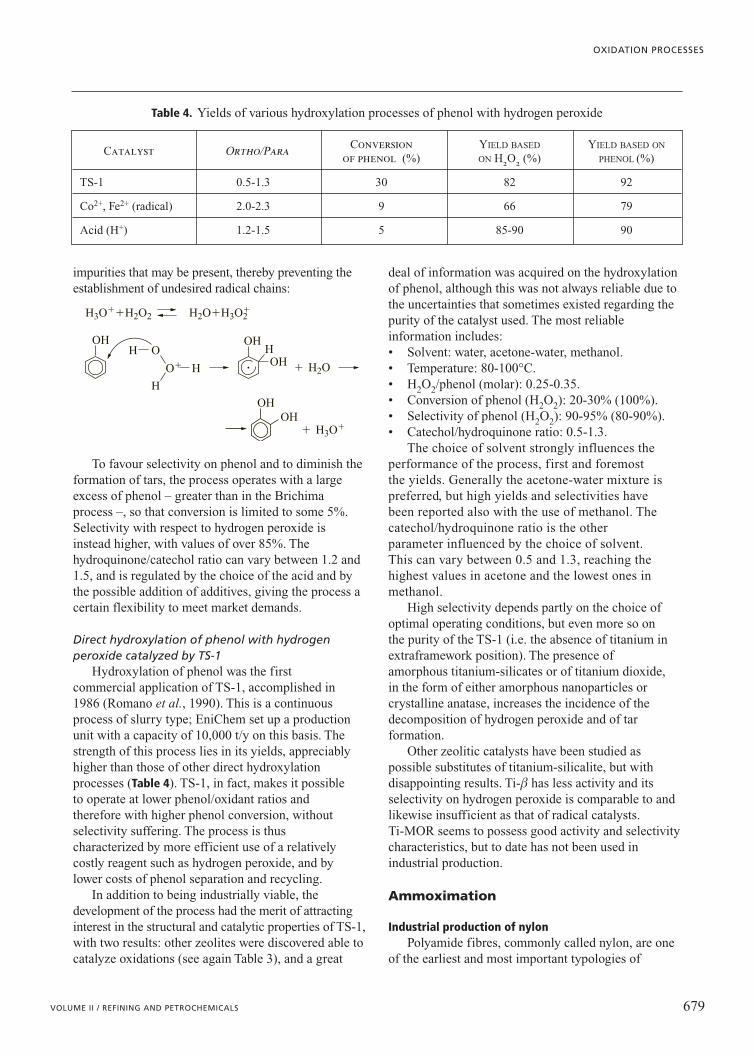
impurities that may be present, thereby preventing theestablishment of undesired radical chains:
To favour selectivity on phenol and to diminish theformation of tars, the process operates with a largeexcess of phenol – greater than in the Brichimaprocess –, so that conversion is limited to some 5%.Selectivity with respect to hydrogen peroxide isinstead higher, with values of over 85%. Thehydroquinone/catechol ratio can vary between 1.2 and1.5, and is regulated by the choice of the acid and bythe possible addition of additives, giving the process acertain flexibility to meet market demands.
Direct hydroxylation of phenol with hydrogenperoxide catalyzed by TS-1
Hydroxylation of phenol was the firstcommercial application of TS-1, accomplished in1986 (Romano et al., 1990). This is a continuousprocess of slurry type; EniChem set up a productionunit with a capacity of 10,000 t/y on this basis. Thestrength of this process lies in its yields, appreciablyhigher than those of other direct hydroxylationprocesses (Table 4). TS-1, in fact, makes it possibleto operate at lower phenol/oxidant ratios andtherefore with higher phenol conversion, withoutselectivity suffering. The process is thuscharacterized by more efficient use of a relativelycostly reagent such as hydrogen peroxide, and bylower costs of phenol separation and recycling.
In addition to being industrially viable, thedevelopment of the process had the merit of attractinginterest in the structural and catalytic properties of TS-1,with two results: other zeolites were discovered able tocatalyze oxidations (see again Table 3), and a great
deal of information was acquired on the hydroxylationof phenol, although this was not always reliable due tothe uncertainties that sometimes existed regarding thepurity of the catalyst used. The most reliableinformation includes:• Solvent: water, acetone-water, methanol.• Temperature: 80-100°C.• H2O2/phenol (molar): 0.25-0.35.• Conversion of phenol (H2O2): 20-30% (100%).• Selectivity of phenol (H2O2): 90-95% (80-90%).• Catechol/hydroquinone ratio: 0.5-1.3.
The choice of solvent strongly influences theperformance of the process, first and foremostthe yields. Generally the acetone-water mixture ispreferred, but high yields and selectivities havebeen reported also with the use of methanol. Thecatechol/hydroquinone ratio is the otherparameter influenced by the choice of solvent.This can vary between 0.5 and 1.3, reaching thehighest values in acetone and the lowest ones inmethanol.
High selectivity depends partly on the choice ofoptimal operating conditions, but even more so onthe purity of the TS-1 (i.e. the absence of titanium inextraframework position). The presence ofamorphous titanium-silicates or of titanium dioxide,in the form of either amorphous nanoparticles orcrystalline anatase, increases the incidence of thedecomposition of hydrogen peroxide and of tarformation.
Other zeolitic catalysts have been studied aspossible substitutes of titanium-silicalite, but withdisappointing results. Ti-b has less activity and itsselectivity on hydrogen peroxide is comparable to andlikewise insufficient as that of radical catalysts. Ti-MOR seems to possess good activity and selectivitycharacteristics, but to date has not been used inindustrial production.
Ammoximation
Industrial production of nylon Polyamide fibres, commonly called nylon, are one
of the earliest and most important typologies of
679VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
OH
H3O��H2O2 H2O�H3O2�
H O
H
O� H � H2O
OH
OHH
OHOH
� H3O�
OHOH
� H3O�
Catalyst Ortho/Para Conversion YIELD BASED YIELD BASED ON
of phenol (%) ON H2O2 (%) PHENOL (%)
TS-1 0.5-1.3 30 82 92
Co2+, Fe2+ (radical) 2.0-2.3 9 66 79
Acid (H+) 1.2-1.5 5 85-90 90
Table 4. Yields of various hydroxylation processes of phenol with hydrogen peroxide
synthetic fibres (Petrini et al., 1996; Bellussi andPerego, 2000). Nylon 6,6, synthesized by William H.Carothers at the DuPont company in the 1930s, wasthe first to be placed on the market. It is producedfrom adipic acid and hexamethylenediamine. Adifferent type of nylon, called Nylon 6, developed inGermany by I.G. Farben and marketed under the nameof Perlon, is obtained by ring opening polymerizationof e-caprolactam (Fig. 15).
e-Caprolactam, the world productive capacity ofwhich amounts to about 4·106 t/y, is obtainedindustrially mainly from benzene, through a complexseries of transformations. The benzene can behydrogenated to cyclohexane which can be oxidized tocyclohexanone; alternatively, cyclohexanone can beobtained by the hydrogenation of phenol, which is alsoproduced from benzene. In its turn, cyclohexanone is transformed into its oxime and the latter into e-caprolactam:
The conventional technology used in these last twostages of production raises the problem of aconsiderable coproduction of a salt, ammoniumsulphate, which is today of extremely low commercialvalue.
Moreover, the process is penalized by thecomplexity of the cycle connected with the inorganicraw materials used and with the synthesis ofhydroxylamine, as well as by the problems linked withthe emission of nitrogen oxides (NOx) and sulphuroxides (SOx).
In the Raschig process for the synthesis of e-caprolactam (Fig. 16), hydroxylamine sulphate isproduced from NH3, CO2 and SO2 through a complexseries of operations; these include the synthesis ofNOx (obtained by combustion of ammonia in air) andof ammonium carbonate (obtained by the reaction ofammonia and CO2), their combination to formammonium nitrite and the subsequent reduction of thiswith SO2.
In the synthesis of the oxime, about 2.8 kg ofammonium sulphate is coproduced for every 1 kg ofproduct. In the following conversion step of oxime tocaprolactam, by means of the Beckmannrearrangement conducted in the presence of oleum,
680 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
HO
H2NNH2
CC OH
O
O
C
O
O
N
H
N
H
n
O
C
O
[ [
N
H
n[ [NH
�
adipic acid
hexamethylenediamineNylon 6,6
Nylon 6e-caprolactam
Fig. 15. Various types of nylon and their synthesis.
preparation ofammonium
nitrite
preparation ofammoniumcarbonate
NH3oxidation
NH3
N2O3
(NH4)NO2
(NH4)2CO3
(NH4)2SO4
2.8 kg/kg
(NH4)2SO4
1.6 kg/kg
NH2OH . H2SO4
oxime
NH3
SO2
NH3
cyclohexanone
NH3
oleum
O2
NH3
CO2
CO2
preparation ofhydroxylamine
sulphate
preparation ofcyclohexanone
oxime
preparation ofcaprolactam
e-caprolactam
Fig. 16. Raschig process for production of e-caprolactam.
OOH OH
O
NH
N
NH2OH.H2SO4 2NH3
O
O
� �
(NH4)2SO4 H2O
NOH
�
� �
(NH4)2SO4H2SO4 2NH3 NH
NOH
� � �
20

another 1.6 kg is coproduced, making a total of 4.4 kgof ammonium sulphate per 1 kg of caprolactam.
This process has undergone constant improvementto reduce the coproduction of ammonium sulphate, butwith only partial results, or involving considerablecomplexity in the operative cycle. Significantexamples of this trend are the BASF and DSMprocesses, which exploit the reducing action ofhydrogen in the presence of palladium catalysts for theproduction of hydroxylamine.
The rearrangement step of the oxime into caprolactamhas also formed the object of intense studies aimed at cutting out the use of oleum and the coproduction of sulphate. Recently, Sumitomo has industrialized a vapour-phase catalytic process using a solid catalyst of a zeolitic nature instead of oleum, thus enabling thecoproduction of salt to be completely avoided.
Other forms of synthesis have tackled the problemof the production of oxime, or of caprolactam directly,in a completely different way. For example, in theToray process, the photonitrosation of the cyclohexaneto oxime takes place; the SNIA process foresees theoxidation of toluene to benzoic acid followed byhydrogenation to hexahydrobenzoic acid, and theconversion of the latter to caprolactam with nitrosylsulphuric acid. These processes have had limitedcommercial success. Other alternative routes based onbutadiene, via oxidative carbonylation (DSM) orhydrocyanation (BASF/DuPont), are at present in thefinal stage of development.
Ammoximation of cyclohexanoneIn this panorama of unresolved problems, the
ammoximation process represents a radicalinnovation (Roffia et al., 1989; Petrini et al.,1996). The term ammoximation indicates theproduction of oxime directly from ammonia. Infact, the process is based on a catalytic reactionbetween cyclohexanone, ammonia and hydrogenperoxide, and completely eliminates, on the onehand, the problems linked with the production anduse of hydroxylamine and, on the other, thecoproduction of sulphates:
The catalyst in the process is TS-1. Thecyclohexanone ammoximation reaction with ammoniaand hydrogen peroxide (or oxygen) had already beendescribed earlier, but scanty yields have been obtained.Only the use of TS-1 as the catalyst, patented for thefirst time in the 1980s by Paolo Roffia andco-workers, enabled a breakthrough to be obtained,allowing EniChem to develop the process on anindustrial basis.
In this process (see schematic diagram in Fig. 17),the reaction is carried out continuously in the liquidphase in a stirred reactor, in the presence of thecatalyst dispersed in slurry in the reaction medium at aconcentration of 2-3% in weight. The reaction istypically conducted between 80°C and 90°C, at aslight overpressure, supplying cyclohexanone, NH3and aqueous H2O2 in molar ratio 1.0:2.0:1.1 in thereaction solvent consisting of water and tert-butanol,with a residence time of around 1.5 h. The reactionproduct is separated from the catalyst, through filterssoaked in the reactor, and conveyed to a rectifyingcolumn for recovery of the unconverted excessammonia and solvent (water/tert-butanol azeotrope),which are recycled in the reaction. The aqueoussolution of crude oxime obtained from the bottom ofthe column is conveyed to the purification unit forrecovery of the cyclohexanone oxime with therequired purity, to be used in the subsequentrearrangement to caprolactam. During the process, thecatalyst must undergo periodic purging and make-upoperations since, in the presence of ammonia, thesiliceous structure of TS-1 slowly dissolves, with lossof weight of the catalyst accompanied by migration ofthe titanium to the outside surface of the solid and lossof catalytic activity.
Operating under these conditions, the conversion ofthe cyclohexanone is nearly complete (99.9%) and theselectivity to the oxime based on cyclohexanone is
681VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
NH3 � H2O2
O
� 2H2O
NOH
�cat. TS-1
catalystmake-up
NH3
H2O2
H2O
solvent make-up
vent to treatment
cyclohexanone
spent catalyst
aqueous oximeto
purification
Fig. 17. EniChem process for production of cyclohexanone oxime.
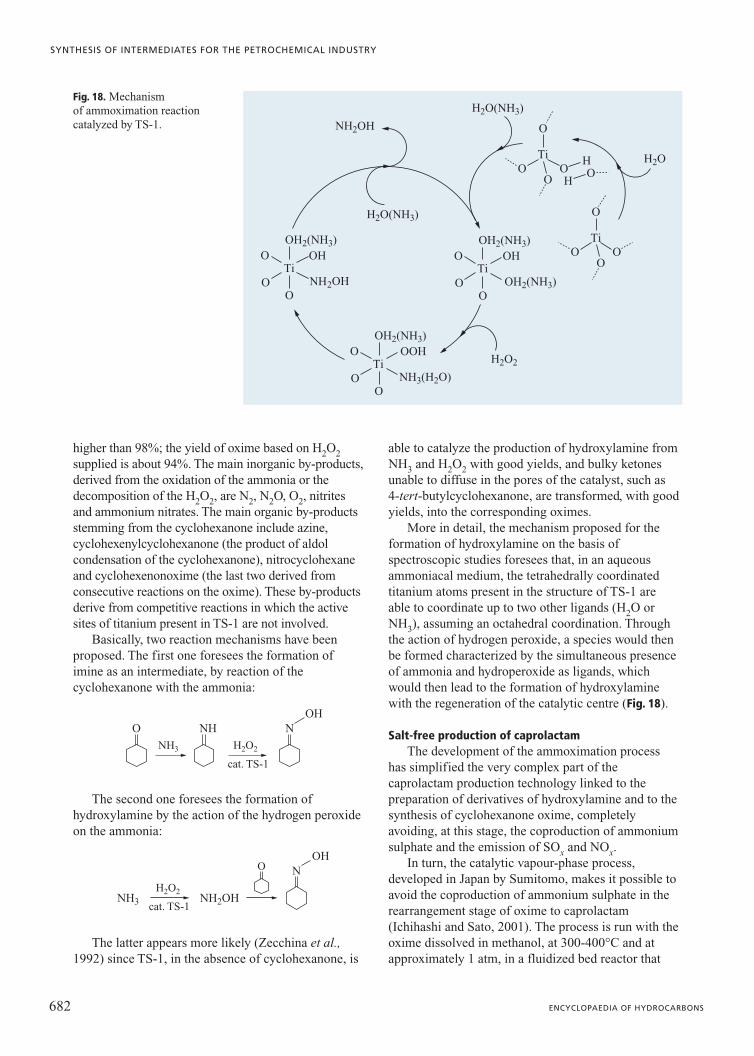
higher than 98%; the yield of oxime based on H2O2supplied is about 94%. The main inorganic by-products,derived from the oxidation of the ammonia or thedecomposition of the H2O2, are N2, N2O, O2, nitritesand ammonium nitrates. The main organic by-productsstemming from the cyclohexanone include azine,cyclohexenylcyclohexanone (the product of aldolcondensation of the cyclohexanone), nitrocyclohexaneand cyclohexenonoxime (the last two derived fromconsecutive reactions on the oxime). These by-productsderive from competitive reactions in which the activesites of titanium present in TS-1 are not involved.
Basically, two reaction mechanisms have beenproposed. The first one foresees the formation ofimine as an intermediate, by reaction of thecyclohexanone with the ammonia:
The second one foresees the formation ofhydroxylamine by the action of the hydrogen peroxideon the ammonia:
The latter appears more likely (Zecchina et al.,1992) since TS-1, in the absence of cyclohexanone, is
able to catalyze the production of hydroxylamine fromNH3 and H2O2 with good yields, and bulky ketonesunable to diffuse in the pores of the catalyst, such as 4-tert-butylcyclohexanone, are transformed, with goodyields, into the corresponding oximes.
More in detail, the mechanism proposed for theformation of hydroxylamine on the basis ofspectroscopic studies foresees that, in an aqueousammoniacal medium, the tetrahedrally coordinatedtitanium atoms present in the structure of TS-1 areable to coordinate up to two other ligands (H2O orNH3), assuming an octahedral coordination. Throughthe action of hydrogen peroxide, a species would thenbe formed characterized by the simultaneous presenceof ammonia and hydroperoxide as ligands, whichwould then lead to the formation of hydroxylaminewith the regeneration of the catalytic centre (Fig. 18).
Salt-free production of caprolactamThe development of the ammoximation process
has simplified the very complex part of thecaprolactam production technology linked to thepreparation of derivatives of hydroxylamine and to thesynthesis of cyclohexanone oxime, completelyavoiding, at this stage, the coproduction of ammoniumsulphate and the emission of SOx and NOx.
In turn, the catalytic vapour-phase process,developed in Japan by Sumitomo, makes it possible toavoid the coproduction of ammonium sulphate in therearrangement stage of oxime to caprolactam(Ichihashi and Sato, 2001). The process is run with theoxime dissolved in methanol, at 300-400°C and atapproximately 1 atm, in a fluidized bed reactor that
682 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY
OO
OOH2(NH3)
H2O(NH3)
H2O(NH3)
OH
NH2OH
NH2OH
Ti
OO
OOH2(NH3)
OH
OH2(NH3) Ti
O
OOO
H
HO
Ti
O
OOO
Ti
OO
OOH2(NH3)
OOH
NH3(H2O)
H2O2
H2O
Ti
Fig. 18. Mechanism of ammoximation reactioncatalyzed by TS-1.
cat. TS-1
O NOH
NH3
NH
H2O2
cat. TS-1NH2OHNH3
O NOH
H2O2

permits frequent regeneration of the catalyst(necessary due to its rapid deactivation caused by thedepositing of organic tars). The catalyst is a zeolite ofMFI structure with a very low aluminium (and otherhetero-elements) content, basically a silicalite-1. Itsuse, associated with that of alcohol as a component ofthe reaction medium, has enabled completeconversions of oxime and selectivities to caprolactamof more than 95% to be achieved. These performances,although slightly inferior to those of the conventionalrearrangement conducted with oleum, in which theyield exceeds 99%, have been reckoned suitable forindustrial development due to the absence of thecoproduction of any salts.
The combined process bringing together theammoximation and catalytic transpositiontechnologies results in a totally salt-free production ofcaprolactam. This process was industrialized for thefirst time in Japan by Sumitomo, which, on the basisof an agreement with EniChem, set up a plant with acapacity of 60,000 t/y. The combined process, apartfrom allowing salt-free production, eliminates allgaseous emissions of NOx and SOx and results in asignificant reduction in investment and operatingcosts. This new technology will certainly play aprimary role in caprolactam production processesbased on cyclohexanone.
Conclusions and perspectives
The prospects of using hydrogen peroxide, even forlarge-scale productions, seem encouraging. Anammoximation plant has come into production inJapan, one or more pilot plants for the epoxidation ofpropylene are operating in various chemicalcompanies, and considerable research efforts are beingmade in many industrial laboratories. Nevertheless, itcannot be denied that obstacles exist, not to beunderestimated, which are inherent in the currentmethod of producing hydrogen peroxide and whichhinder its diffusion in petrochemistry. Theanthraquinone process, in fact, is based on a complextechnology that requires considerable investments.Moreover, it is anticipated that a propylene oxideplant, based on the TS-1/H2O2 technology, will requirea hydrogen peroxide unit of a size never experimented.
Despite such considerations, however, somechemical companies have recently announced thesetting up of large-scale propylene oxide plants thatare based on this technology.
The need to exceed the limits of the anthraquinoneprocess gave fresh impetus to the studies on the directsynthesis of hydrogen peroxide, as previouslymentioned. At the same time, it led to exploring otheralternatives, such as process integration and the in situ
generation of hydrogen peroxide, made possible by themolecular sieve properties of TS-1.
On this basis, the direct use of the working solutionof the anthraquinone process for the epoxidation ofpropylene has been studied (Clerici and Ingallina, 1996):
In the process, formally, the oxidant is molecularoxygen, with all the advantages thereof with respect tothe use of hydrogen peroxide. In effect, however, thelatter is produced in the reaction environment by theoxidation in situ of the alkylanthrahydroquinone.Successively, it diffuses to the active sites within thepores, where it is immediately consumed in theepoxidation reaction, with the mechanism alreadyillustrated in Fig. 9.
The feasibility of the process is based on theimpossibility of the alkylanthraquinone and the othercomponents of the working solution to diffuse inside thepores, where they could undergo oxidative degradationprocesses or interfere with the redox mechanisms. Theirmolecular dimensions are, in fact, larger than the openingof the catalyst’s pores. However, it should be emphasizedthat the scheme envisaged does not enable the complexityof the anthraquinone process to be side-stepped, but justto eliminate the stage relative to the separation,purification and concentration of the hydrogen peroxide.
A variant foresees that the two processes, theproduction of hydrogen peroxide and epoxidation,remain substantially separate, but that the epoxidationsolvent (aqueous methanol) is used also for extractinghydrogen peroxide from the working solution (Clericiand Ingallina, 1996).
The principle foreseeing the use of aqueousmethanol as the solvent for the direct synthesis ofhydrogen peroxide is quite similar; it is targeted onobtaining a solution that can be supplied to theepoxidation reactor without further treatments, exceptfor the removal of any additives (Paparatto et al., 2003b).In general, this method (like the others mentioned above)can also be used for other TS-1-catalysed oxidations, andnot only in the case of propylene epoxidation.
Then there is a second category of studies, towhich mention has already been made, on the possibleuse of a mixture of hydrogen and oxygen directly inthe oxidation reactor. For this purpose, it is necessaryto finalize a bifunctional catalyst that possesses boththe catalytic centres necessary for synthesis of thehydrogen peroxide, and the active centres for the
683VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES
O2
H2O
� �
�
�
�
TS-1
O
O
O
ROH
OH
R
H2O��
O

oxidation reaction. TS-1, on which metallic palladiumhas been supported, is an adequate catalyst (Clericiand Bellussi, 1993; Meiers et al., 1998).
An interesting application of mixed oxides regardsthe deep desulphuration of motor fuels. The large sizeof the sulphur molecules, in this case, entails the useof mesoporous mixed oxides, instead of zeolites,which are microporous. Consequently, for the reasonsalready discussed, the oxidant cannot be hydrogenperoxide, but rather an organic hydroperoxide, asolution that, moreover, is best fitted for the actual set-up of the refinery. In principle, oxidants such astert-butyl hydroperoxide, tert-amyl hydroperoxide, (1-phenyl)ethyl hydroperoxide or other suchhydroperoxides are obtainable by oxidation with air ofstreams present in the refinery. Patents filed by variousoil companies show the interest taken in this method,as an alternative to the more traditional process ofdeep hydrotreatment.
References
Baerlocher C. et al. (2001) Atlas of zeolite framework types,Amsterdam, Elsevier.
Balducci L. et al. (2003) Direct oxidation of benzene to phenolwith hydrogen peroxide over a modified titanium silicalite,«Angewandte Chemie. International Edition», 42, 4937-4940.
Bellussi G., Perego C. (2000) Industrial catalytic aspects ofthe synthesis of monomers for nylon production, «Cattech»,4, 4-16.
Bellussi G. et al. (1992) Reactions of titanium silicate withprotic molecules and hydrogen peroxide, «Journal ofCatalysis», 133, 220-230.
Bianchi D. et al. (1999) Biphasic synthesis of hydrogen peroxidefrom carbon monoxide, water, and oxygen catalyzed bypalladium complexes with bidentate nitrogen ligands,«Angewandte Chemie. International Edition», 38, 706-708.
Bianchi D. et al. (2000) A novel iron-based catalyst for thebiphasic oxidation of benzene to phenol with hydrogenperoxide, «Angewandte Chemie. International Edition»,39, 4321-4323.
Bianchi D. et al. (2004) European Patent 1424320 to PolimeriEuropa.
Boccuti M.R. et al. (1988) Spectroscopic characterization ofsilicalite and titanium silicalite, in: Structures and reactivityof surfaces. Proceedings of a European conference, Trieste,September 13-16, Amsterdam, Elsevier, 133-144.
Clerici M.G. (1991) Oxidation of saturated hydrocarbons withhydrogen peroxide, catalysed by titanium silicalite, «AppliedCatalysis», 68, 249-261.
Clerici M.G. (2001) The role of the solvent inTS-1 chemistry:active or passive? An earlier study revisited, «Topics inCatalysis», 15, 257-263.
Clerici M.G., Bellussi G. (1993) US Patent 5235111 toEniricerche-Snamprogetti.
Clerici M.G., Ingallina P. (1993) Epoxidation of lowerolefins with hydrogen peroxide and titanium silicalite,«Journal of Catalysis», 140, 71-83.
Clerici M.G., Ingallina P. (1996) Clean oxidationtechnologies. New prospects in the epoxidation of the olefins,in: Anastas P.T., Williamson T.C. (editors) Green chemistry.Designing chemistry for the environment, Washington(D.C.), American Chemical Society, 59-68.
Clerici M.G. et al. (1991) Synthesis of propylene oxide frompropylene and hydrogen peroxide catalyzed by titaniumsilicalite, «Journal of Catalysis», 129, 159-167.
Corma A. et al. (2001) Sn-zeolite beta as a heterogeneouschemoselective catalyst for Baeyer-Villiger oxidations,«Nature», 412, 423-425.
Fenton H.J.H. (1876) On a new reaction of tartaric acid,«Chemical News», 33, 190.
Fenton H.J.H. (1894) Oxidation of tartaric acid in presenceof iron, «Journal of Chemical Society», 65, 899-910.
Girotti G. et al. (2003) Alkylation of benzene with isopropanolon β-zeolite. Influence of physical state and waterconcentration on catalyst performances, «Journal ofMolecular Catalysis A: Chemical», 204-205, 571-579.
Goor G. (1992) Hydrogen peroxide: manufacture and industrialuse for production of organic chemicals, in: Strukul G.(edited by) Catalytic oxidations with hydrogen peroxide asoxidant, Dordrecht, Kluwer Academic Publishers, 13-43.
Herrmann W.A. et al. (1991) Methyltrioxorhenium as catalystfor olefin oxidation, «Angewandte Chemie. InternationalEdition in English», 30, 1638-1641.
Hooper G.W. (1967) US Patent 3336112 to ICI.Huybrechts D.R.C. et al. (1990) Oxyfunctionalization of
alkanes with hydrogen peroxide on titanium silicalite,«Nature», 345, 240-242.
Ichihashi H., Sato H. (2001) The development of newheterogeneous catalytic processes for the productionof ε-caprolactam, «Applied Catalysis. A: General», 221,359-366.
Juliá S. et al. (1982) Synthetic enzymes. 2: Catalytic asymmetricepoxidation by means of polyamino-acids in a triphasesystem, «Journal of Chemical Society. Perkin TransactionsI», 11, 1317-1324.
Katsuki T., Sharpless K.B. (1980) The first practical methodfor asymmetric epoxidation, «Journal of the AmericanChemical Society», 102, 5974-5976.
Maggioni P., Minisci F. (1977) Catechol and hydroquinonefrom catalytic hydroxylation of phenol by hydrogen peroxide,«La Chimica e l’Industria», 59, 239-242.
Maspero F., Romano U. (1994) Oxidation of alcohols withH2O2 catalyzed by titanium silicalite-1, «Journal ofCatalysis», 146, 476-482.
Meiers R. et al. (1998) Synthesis of propylene oxide frompropylene, oxygen, and hydrogen catalyzed by palladium-platinum-containing titanium silicalite, «Journal ofCatalysis», 176, 376-386.
Milas N.A. (1937) The hydroxylation of unsaturated substances.III: The use of vanadium pentoxide and chromium trioxideas catalysts of hydroxylation, «Journal of the AmericanChemical Society», 59, 2342-2344.
Milas N.A., Sussman S. (1936) The hydroxylation of the doublebond, «Journal of the American Chemical Society», 58,1302-1305.
Millini R., Perego G. (1996) Structural evidence forisomorphous substitution in microporous materials andmethods of characterization. The case of titanium silicalite,«Gazzetta Chimica Italiana», 126, 133-140.
684 ENCYCLOPAEDIA OF HYDROCARBONS
SYNTHESIS OF INTERMEDIATES FOR THE PETROCHEMICAL INDUSTRY

Neurock M., Manzer L.E. (1996) Theoretical insights onthe mechanism of alkene epoxidation by H2O2 with titaniumsilicalite, «Chemical Communications», 1133-1134.
Olah G.A., Ohnishi R. (1978) Oxyfunctionalization ofhydrocarbons. 8: Electrophilic hydroxylation of benzene,alkylbenzenes, and halobenzenes with hydrogen peroxide,«Journal of Organic Chemistry», 43, 865-867.
Panov G.I. (2000) Advances in oxidation catalysis. Oxidationof benzene to phenol by nitrous oxide, «Cattech», 4, 18-32.
Paparatto G. et al. (2003a) European Patent 1307399 to Eni-Enichem.
Paparatto G. et al. (2003b) US Patent 6541648 to PolimeriEuropa.
Petrini G. et al. (1996) Caprolactam via ammoximation, in:Anastas P.T., Williamson T.C. (editors) Green chemistry.Designing chemistry for the environment, Washington(D.C.), American Chemical Society, 33-48.
Ricci M. (1996) Electrophilic activation of hydrogen peroxide.Some recent applications in organic synthesis, in:Proceedings of the Seminars in organic synthesis. XXISummer School A. Corbella 17-21 June, Milano, SocietàChimica Italiana, 247-266.
Roffia P. et al. (1989) Cyclohexanone ammoximation. A breakthrough in the 6-caprolactam production process, in: Newdevelopments in selective oxidation. Proceedings of aninternational symposium, Rimini (Italy), 18-22 September,43-52.
Romano U. (2001) Ossido di propilene. Nuova tecnologiaproduttiva, «La Chimica e l’Industria», 83, 30-31.
Romano U. et al. (1990) Selective oxidation with ti-silicalite,«La Chimica e l’Industria», 72, 610-616.
Taramasso M. et al. (1983) US Patent 4410501 toSnamprogetti.
Varagnat J. (1976) Hydroquinone and pyrocatechol productionby direct oxidation of phenol, «Industrial & EngineeringChemistry. Product Research and Development», 15, 212-215.
Venturello C. et al. (1983) A new, effective catalytic systemfor epoxidation of olefins by hydrogen peroxide under phase-transfer conditions, «Journal of Organic Chemistry», 48,3831-3833.
Venturello C. et al. (1985) A new peroxotungsten heteropolyanion with special oxidizing properties. Synthesis and structureof tetrahexylammonium tetra(diperoxotungsto)phosphate(3),«Journal of Molecular Catalysis», 32, 107-110.
Zecchina G. et al. (1992) Ammoximation of cyclohexanoneon titanium silicalite. Investigation of the reactionmechanism, in: Proceedings of the 10th Internationalcongress on catalysis, Budapest, 19-24 July, 719-728.
Mario G. ClericiEniTecnologie
San Donato Milanese, Milano, Italy
Marco RicciFranco Rivetti
Polimeri Europa Novara, Italy
685VOLUME II / REFINING AND PETROCHEMICALS
OXIDATION PROCESSES


















