Page 1
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 1/12
DOI: 10.1002/cssc.200900134
Towards the Sustainable Production of Acrolein byGlycerol Dehydration
Benjamin Katryniok,[a] Sbastien Paul,[b] Mickal Capron,[a] and Franck Dumeignil*[a]
1. Introduction
The exhaustion of nonrenewable fossil fuels is foreseen in the
next several decades. During the last years, the production of
alternative gasolines by making use of vegetable oils and fats
has attracted the attention of many academic and industrial re-
searchers. In addition to renewability, the resulting biodiesels
have the advantage of providing a neutral CO2 balance with a
much lower environmental impact than petrodiesels. The raw
materials for biodiesel production are vegetable oils and fats—
from canola, soy, corn, or others—and a mono-alcohol (usually
methanol), which is used to cleave the fatty acids from theglycerol backbone to finally yield the fatty-acid-esters
(Scheme 1). These acid-esters can be used as biodiesels either
directly or after blending with fossil-fuel-derived diesels.
The use of biodiesels as substitutes or additives for petrol-
based diesels can be seen as a political choice, rather than a
decision driven socio-economic reasons. Recently, this choice
has even been criticized owing to the concurrent use of the
same raw materials for food production. Nevertheless, the Eu-
ropean Union has planned to progressively increase the bio-
diesel proportion in commercial fuels to reach a target of 10%
in 2015.[1] This is a general trend, and the capacity for biodiesel
production is continuously increasing all over the world. For
example, in 2007 the USA and the EU produced 1.2 and 5.7
million tonnes (Figure 1) of biodiesel, respectively, and these
quantities are expected to double by 2012.[2]
This growth is accompanied by a significant increase in glyc-
erol production, as this compound is a major byproduct of bio-
diesel production (ca. 10 wt%; Scheme 1). A projection of the
global production forecasts that world-wide 1.2 million tonnes
of glycerol will be generated in 2010,[3] which has to be pro-
cessed to achieve a sustainable industry. Crude glycerol ob-
tained from the biodiesel process contains 80 wt% of glycerol
but, as a drawback, also contains water, methanol, traces of
fatty acids, and various inorganic and organic compounds (re-
[a] B. Katryniok, Dr. M. Capron, Prof. F. Dumeignil
Universit des Sciences et Technologies de Lille
Unit de Catalyse et de Chimie du Solide (UMR, CNRS 8181)
Cit Scientifique, 59655 Villeneuve d’Ascq (France)
Fax: ( +33)320-436561
E-mail: [email protected]
[b] Dr. S. Paul
Univ. Lille Nord de France, Ecole Centrale de Lille
Unit de Catalyse et de Chimie du Solide (UMR, CNRS 8181)
Cit Scientifique, BP 48, 59651 Villeneuve d’Ascq (France)
The massive increase in biodiesel production by transesterifica-
tion of vegatable oils goes hand-in-hand with the availability
of a large volume of glycerol, which must be valorized. Glycer-
ol dehydration to acrolein over acid catalysts is one of the
most promising ways of valorization, because this compound
is an important chemical intermediate used in, for example,
the DL-methionine synthesis. In this Minireview, we give a de-
tailed critical view of the state-of-the-art of this dehydration re-
action. The processes developed in both the liquid and the gas
phases are detailed and the best catalytic results obtained so
far are reported as a benchmark for future developments. The
advances on the understanding of the reaction mechanism are
also discussed and we further focus particularly on the main
obstacles for an immediate industrial application of this tech-
nology, namely catalyst coking and crude glycerol direct-use
issues.
Scheme 1. The transesterification of vegetable oils to yield the so-called ‘bio-
diesels.’
Figure 1. European biodiesel production (reproduced with permission from
the European Biodiesel Board).
ChemSusChem 2009 , 2, 719 – 730 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 719
Page 2
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 2/12
ferred to as MONG: matter organic non-glycerol). As a conse-
quence, crude glycerol must in most cases be purified by an
expensive distillation step prior to further use. The proportion
of glycerol that is actually refined is now decreasing owing to
the high cost of the distillation step accompanied by a too-
rapid growth of the quantity of produced crude glycerol, but
mostly because of the absence of any market able to absorb
the overproduction (Figure 2). In contrast, a model based on
the production and sale of crude glycerol predicts an inverse
linear relationship between the net production costs of biodie-sel and the price of glycerol. [4] New and economical ways of
using glycerol must therefore be developed to greatly increase
the demand and price of crude glycerol, thereby ensuring the
sustainability of the biodiesel sector; [5] an issue addressed by a
consortium of academic and industrial researchers recently set
up in the UK.[6]
More than 1500 direct applications of glycerol are already
known, especially in the cosmetic, pharmaceutical, and food in-
dustries.[8] The large versatility of glycerol is based on both its
chemical and physical properties. Because of its three hydroxyl
groups, glycerol is completely soluble in water and alcohols
whereas it is completely insoluble in hydrocarbons. It is a very
hydrophilic species and is employed as such when the amount
of water has to be controlled, that is, in glue or other adhe-
sives. Furthermore, the presence of hydroxyl groups leads to
the formation of intra- and intermolecular hydrogen networks,
which explains its high boiling point (563 K at atmospheric
pressure) and high viscosity. This latter rheological property
leads to the use of glycerol as a softener in resins and plastics
but also as a lubricant, for example in pharmaceutical applica-
tions. In addition, glycerol is nontoxic and has a sweet taste. It
can be incorporated into foods, medicines, and cosmetics but,
as mentioned above, the crude glycerol from biodiesel pro-
cesses contains impurities and is therefore not suitable for
these applications without a prior purification stage. In addi-
tion, as noted earlier the size of the existing markets is not suf-
ficient to absorb the huge amounts of glycerol currently being
produced, and the gap between the absorption capacity of
the market and the glycerol production will steadily increase in
the near future. Today, crude glycerol that is not refined is gen-
erally burned (Figure 2), which must be considered as a dra-
matic waste of a potentially very interesting organic raw mate-
rial. Glycerol is a molecule with a large potential for functionali-
zation that offers many opportunities of chemical or biochemi-
cal conversions to produce value-added chemicals. A selection
of these possibilities is shown in Scheme 2 and briefly further
discussed below. More detailed information is available in
recent Review papers.[5,9–11] The reforming of glycerol on Pt–Rh
catalysts to yield syngas, which can be used for either the syn-
thesis of alkanes in the Fischer–Tropsch process or the synthe-
sis of methanol, has been recently reported.[12,13] A potential
application derived from this technology is the production of
hydrogen by the water-gas-shift process. Another interesting
possibility for valorizing glycerol is its selective reduction,
which leads to either propylene glycol (MPG) or to 1,3-pro-
panediol (PD), which are used in the polymer industry. It is also
possible to halogenate glycerol to epichlorhydrin, an important
intermediate for epoxy resins, by using hydrochloric acid in the
presence of organic acids, such as caprylic acid (Solvay) or
acetic acid (Dow),[14] as catalysts working in the gaseous phase
at 353–393 K under 1–5 bar.[15] This technology is now mature,
as Solvay has run this process commercially in France since
2007, using an existing production facility where formerly glyc-
erol was produced from epichlorhydrin.
Another way of valorizing glycerol is etherification to glycer-
ol-tert -butylether (GTBE), which is an excellent additive for
Figure 2. Global crude glycerol production and distillation (the difference
corresponds to the quantity of glycerol that is not upgraded and usually
burned).[7]
Scheme 2. A selection of glycerol valorization pathways.
720 www.chemsuschem.org 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemSusChem 2009 , 2, 719 – 730
F. Dumeignil et al.
Page 3
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 3/12
diesel blending. Furthermore, esterification of glycerol to mon-
oacylglycerol (MAG) or diacylglycerol (DAG), used as emulsifiers
in for example foods (margarines and sauces) and cosmetics, is
also a possibility. The latter process can be catalyzed either by
a conventional alkaline catalyst or by (lipase-type) enzymes. [16]
Glycerol partial oxidation leads to a rich chemistry with many
possible products, such as glyceric, tartronic and ketomalonic
(or mesoxalic) acids, glyceraldehyde, or dihydroxyacetone
(DHA). The challenge is to find catalysts that are selective to
the target molecule among many possible products. For exam-
ple, Bi/Pt catalysts were proven efficient for DHA production
with a yield of 37% at a glycerol conversion of 70%.[17] Oxida-
tion can also be performed by using modified bacteria.[18] Re-
cently, the anodic oxidation of glycerol gave a DHA yield of
25%, which is comparable to that obtained with the aforemen-
tioned biotechnological process.[19] However, one of the most
promising ways of glycerol valorization lies in its catalytic de-
hydration to acrolein, which is an important intermediate for
the chemical and agricultural industries. Because of its toxicity,
acrolein is usually directly converted into derivatives such asacrylic acid. The most important use of isolated acrolein is the
synthesis of DL-methionine via 3-methylthiopropionaldehyde
as an intermediate (Scheme 3).[20] DL-methionine is an essential
amino acid that cannot be synthesized by living organisms. It
is widely used in meat production to accelerate animal growth.
The annual world-wide production capacity of DL-methionine
is about 500000 tonnes.[21] Because the amounts offered by
natural methionine sources such as plants and micro-organ-
isms are too low, it has to be industrially synthesized at an ex-
tensive scale to meet the demand. It is estimated that the
global demand will increase by 3–7% in the near future, irre-
spective of the region.[22]
Currently, the synthesis of acrolein is based on the selective
oxidation of propene over complex multicomponent BiMoO x -
based catalysts. The selectivity obtained in this process is close
to 85% at 95% conversion.[23] New approaches with propane
as a starting material are currently explored on a laboratory
scale, but they still suffer from insufficient yields that are in-
compatible with commercialization.[24] Considering the deple-
tion issues associated with petrochemical feedstocks, sustaina-
ble resources will become more and more competitive, not to
mention the positive effect in terms of impact on the climate.
In this context, obtaining acrolein from bioresources in sub-
stantial yields is an important challenge. An economical study
has shown that a competitive production of acrolein from glyc-
erol would be possible at a glycerol price lower than ca.
US$ 300 t1.[25] Currently (June 2009), refined glycerol still costs
US$ 400–500 t1 but crude glycerin is around US$ 100–200 t1,
which makes it a potentially very competitive raw material for
acrolein production.[26]
In this Minireview, we draw the state-of-the-art of the sus-
tainable production of acrolein by dehydration of glycerol. Pro-
cesses developed in the liquid and the gas phase are detailed,
and the best catalytic results obtained up to now are reported
and commented. The fundamentals of the reaction are also
discussed in a section devoted to its mechanism. Finally, the
main obstacles to an immediate industrial development of the
process are discussed in order to outline new insights for
future research efforts.
2. Historical Context
It has been well-known for a long time that heating glycerol
induces its decomposition into acrolein and water, with the
production of byproducts. However, an acid catalyst is needed
to have better control over the reaction and to obtain a signifi-
cant yield of the unsaturated aldehyde at a moderate tempera-
ture. The first patent on this subject was published in France
in 1930.[27] The reaction was carried out in the gaseous phase
over a supported lithium phosphate catalyst, which already
provided an acrolein yield close to 75%. Later, in 1934, Groll
and Hearne claimed a patent for the Shell company for the de-
hydration of an aqueous glycerol solution in the presence of
sulfuric acid at 463 K, which was supposedly the boiling pointof the solution.[28] The resulting acrolein stream was a con-
densed vapor with a yield of nearly 50%. Hoyt and Manninen,
17 years later, patented the dehydration of glycerol using sup-
ported H3PO4.[29] This heterogeneous catalyst was prepared by
impregnation of a clay with 25 wt% of acid. The authors chose
petroleum oil with a boiling point higher than 573 K as a reac-
tion medium, which enabled the use of higher reaction tem-
peratures compared to those used by Groll and Hearne,[28] who
worked in the aqueous phase. Even when the glycerol was
used at a larger concentration (95 wt%) a high yield in acrolein
(72.3%) could be reached. However, these early works, in both
gaseous and liquid phases, remained without further follow-up
until the end of the 20th century and the upcoming of the
massive production of cheap glycerol resulting from biodiesel
production processes.
3. Dehydration of Glycerol in the Liquid Phase
Encouraged by the increased availability of cheap glycerol,
Ramayya et al. studied the dehydration of glycerol in a solution
of sulfuric acid, and under conditions close to the critical point
of water (i.e., P =22.1 MPa and T =647 K).[30] (Near-)supercritical
water offers the advantage that its physical properties, such as
its dielectric constant or its ion product, can be adjusted by
varying the temperature and pressure. The mixture was heated
Scheme 3. Chemical pathways for the industrial manufacturing of DL-me-
thionine.
ChemSusChem 2009 , 2, 719 – 730 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemsuschem.org 721
Towards the Sustainable Production of Acrolein by Glycerol Dehydration
Page 4
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 4/12
in a batch reactor at 623 K, pressurized to 34.5 MPa, and acidi-
fied with 5 mm of sulfuric acid. In these conditions, and at a
residence time of 25 s, the conversion of glycerol and the se-
lectivity towards acrolein were 55% and 86%, respectively.
Considering the fact that sulfuric acid is soluble in supercritical
water, this process can be classified as homogeneous catalysis
with, as a consequence, catalyst/reaction mixture separation
issues. Nevertheless, the idea of using supercritical water as a
reaction medium was a new aspect, and Bhler et al. decided
to deepen the subject in 2001.[31] Instead of working in a batch
reactor, they built a flow-type reactor and used supercritical
water as both solvent and catalyst. They observed various de-composition products, such as acetaldehyde, formaldehyde, al-
lylic alcohol, propionaldehyde, and, of course, acrolein. The
acrolein yield varied between 10 and 23% depending on the
reaction conditions, with the best result (23%) obtained at a
reaction temperature of 623 K at a pressure of 45MPa. As
aforementioned, the tests were in the absence of any catalyst
or acid component, which explains the poor yields in acrolein.
Recently, a combination of the ideas of Hoyt and Manni-
nen[29] and Ramaya et al.[30], that is, using a supported catalyst
and using supercritical water as a reaction medium, respective-
ly, was proposed by Ott and co-workers.[3] These authors inves-
tigated the dehydration of glycerol in supercritical water with
zinc sulfate as a catalyst, which is surprising because this cata-
lyst is supposedly not acidic enough to efficiently promote the
reaction. However, they justified their choice considering mate-
rial stress issues: water in its supercritical form is a highly cor-
rosive agent and therefore requires special and expensive steel
grades for reactors; this corrosive power would become even
stronger if acidic compounds were subsequently added in the
medium, leading to unacceptable reactor material stress. In
these conditions, the authors showed that supercritical condi-
tions are not necessarily the optimal ones. Actually, near-critical
water (633 K at 25 MPa) showed better conversion and selec-
tivity than supercritical water, that is, 75% selectivity at 50%
conversion. This might be explained by the degradation of
acrolein following its formation when it is subjected to too
high temperatures. In 2007, Lehr et al. also published on the
dehydration of biomass-derived polyols in sub- and supercriti-
cal water. As far as glycerol is concerned, they reported 59%
glycerol conversion with almost
60% acrolein selectivity, using
ZnSO4 as a catalyst in subcritical
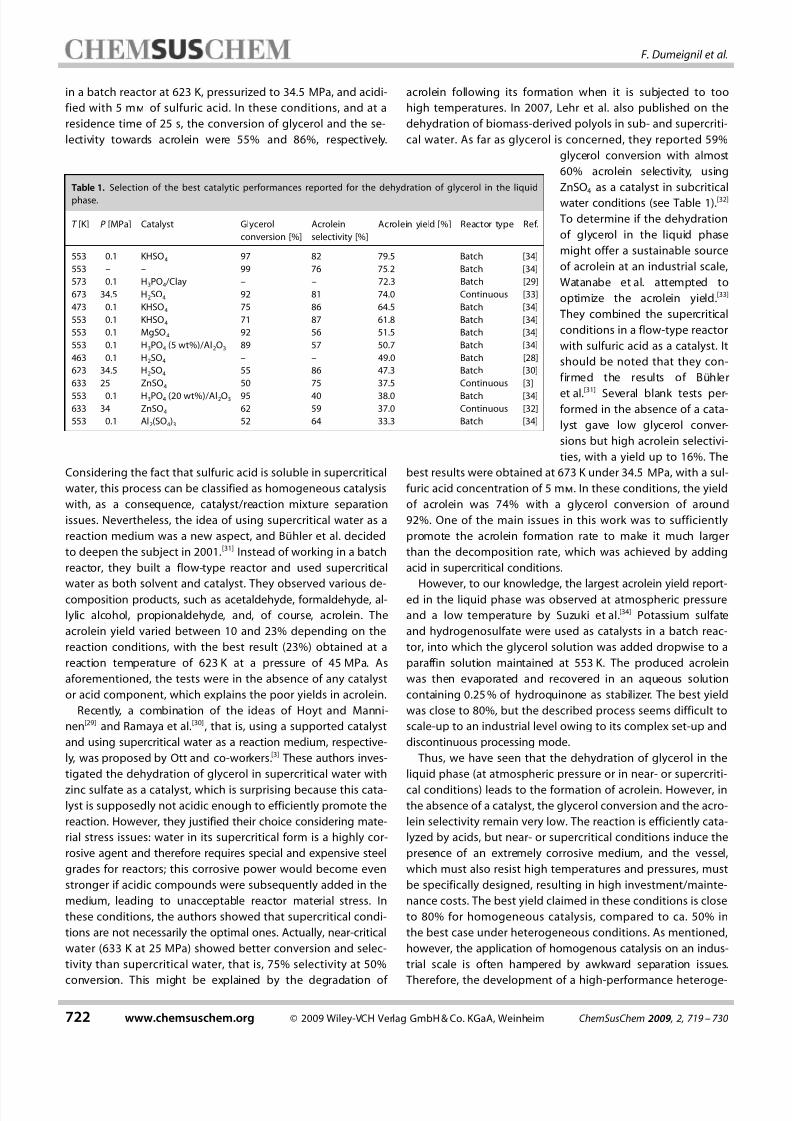
water conditions (see Table 1).[32]
To determine if the dehydration
of glycerol in the liquid phase
might offer a sustainable source
of acrolein at an industrial scale,
Watanabe et al. attempted to
optimize the acrolein yield.[33]
They combined the supercritical
conditions in a flow-type reactor
with sulfuric acid as a catalyst. It
should be noted that they con-
firmed the results of Bhler
et al.[31] Several blank tests per-formed in the absence of a cata-
lyst gave low glycerol conver-
sions but high acrolein selectivi-
ties, with a yield up to 16%. The
best results were obtained at 673 K under 34.5 MPa, with a sul-
furic acid concentration of 5 mm. In these conditions, the yield
of acrolein was 74% with a glycerol conversion of around
92%. One of the main issues in this work was to sufficiently
promote the acrolein formation rate to make it much larger
than the decomposition rate, which was achieved by adding
acid in supercritical conditions.
However, to our knowledge, the largest acrolein yield report-ed in the liquid phase was observed at atmospheric pressure
and a low temperature by Suzuki et al.[34] Potassium sulfate
and hydrogenosulfate were used as catalysts in a batch reac-
tor, into which the glycerol solution was added dropwise to a
paraffin solution maintained at 553 K. The produced acrolein
was then evaporated and recovered in an aqueous solution
containing 0.25 % of hydroquinone as stabilizer. The best yield
was close to 80%, but the described process seems difficult to
scale-up to an industrial level owing to its complex set-up and
discontinuous processing mode.
Thus, we have seen that the dehydration of glycerol in the
liquid phase (at atmospheric pressure or in near- or supercriti-
cal conditions) leads to the formation of acrolein. However, in
the absence of a catalyst, the glycerol conversion and the acro-
lein selectivity remain very low. The reaction is efficiently cata-
lyzed by acids, but near- or supercritical conditions induce the
presence of an extremely corrosive medium, and the vessel,
which must also resist high temperatures and pressures, must
be specifically designed, resulting in high investment/mainte-
nance costs. The best yield claimed in these conditions is close
to 80% for homogeneous catalysis, compared to ca. 50% in
the best case under heterogeneous conditions. As mentioned,
however, the application of homogenous catalysis on an indus-
trial scale is often hampered by awkward separation issues.
Therefore, the development of a high-performance heteroge-
Table 1. Selection of the best catalytic performances reported for the dehydration of glycerol in the liquid
phase.
T [K] P [MPa] Catalyst Glycerol
conversion [%]
Acrolein
selectivity [%]
Acrolein yield [%] Reactor type Ref.
553 0.1 KHSO4 97 82 79.5 Batch [34]
553 – – 99 76 75.2 Batch [34]
573 0.1 H3PO4/Clay – – 72.3 Batch [29]
673 34.5 H2SO4 92 81 74.0 Continuous [33]
473 0.1 KHSO4 75 86 64.5 Batch [34]
553 0.1 KHSO4 71 87 61.8 Batch [34]
553 0.1 MgSO4 92 56 51.5 Batch [34]
553 0.1 H3PO4 (5 wt%)/Al2O3 89 57 50.7 Batch [34]
463 0.1 H2SO4 – – 49.0 Batch [28]
623 34.5 H2SO4 55 86 47.3 Batch [30]
633 25 ZnSO4 50 75 37.5 Continuous [3]
553 0.1 H3PO4 (20 wt%)/Al2O3 95 40 38.0 Batch [34]633 34 ZnSO4 62 59 37.0 Continuous [32]
553 0.1 Al2(SO4)3 52 64 33.3 Batch [34]
722 www.chemsuschem.org 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemSusChem 2009 , 2, 719 – 730
F. Dumeignil et al.
Page 5
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 5/12
neous catalyst and/or a practical process remains a topical
issue before commercial applications of liquid-phase dehydra-
tion of glycerol to acrolein can be envisaged.
4. Dehydration of Glycerol in the Gas Phase
4.1. Early work
Similar to the case of the liquid phase reaction, research on de-
hydration in the gaseous phase was abandoned until the
1990s, when Neher et al. published two patents[35,36] as a
follow-up on the old work of Schering–Kahlbaum.[27] They re-
produced the tests over the lithium phosphate catalyst and
compared its performance to those of a set of acid catalysts
with well-defined Hammet acidities (HAs) ranging from +3 to
8.2. The authors claimed that aqueous solutions of 10–
40 wt% glycerol could be converted at 573 K over alumina-
supported phosphorous acid to yield acrolein with selectivities
of up to 75% at full glycerol conversion (see Table 2). The main
byproduct was hydroxyacetone (selectivity close to 10 %). Haaset al. also continued the research on alumina-supported H 3PO4,
but with a view towards producing 1,2- and 1,3-propanediol. [37]
The yield of the intermediately formed acrolein was, however,
a bit lower (70.5%).
4.2. Catalyst acidity as an important parameter
Obviously, the acidity of the active phase is a crucial parameter
influencing the catalytic performance and stability. Its effect
has been thoroughly studied by several groups. In 2006,
Dubois et al. published two patents on the dehydration of
glycerol over different types of catalysts.[38,39] They carried out
several tests with zeolites, Nafion, heteropolyacids, and also
with different types of acid-impregnated metal oxides. All of
these catalysts have a well-defined HA, ranging between 9
and 18. The selectivity towards acrolein depended on the
catalyst. For Nafion catalysts (HA=12) and for tungsten
oxide on zirconium oxide (HA=14.5), the selectivity was
close to 70% at full conversion, while the selectivity over zeo-
lite catalysts (HA<2) did not exceed 60%. The authors con-
cluded that catalysts with a HA within the range of 10 to
16 are the best candidates.
The second research group that has focused its activities on
the influence of the catalysts’ acidity is headed by Chai. In
2007, they first published two papers on sustainable produc-
tion of acrolein from glycerol. Whereas the first paper focuses
on niobium oxide as a catalyst,[40] the second compares the
catalytic performance of different types of acid catalysts.[41] Be-
cause the acidity of niobium oxide strongly depends on the
calcination temperature (the higher the calcination tempera-ture of the hydrous niobium oxide precursor, the lower the
acidity, as previously described by Chen et al.[42] and Lizuka
et al.[43]), this compound is particularly suitable as a model cata-
lyst for studying the influence of acidity on a given reaction.
The best results were obviously obtained for catalysts calcined
at a low temperature (523–573 K). In addition to their high
acidity, these catalysts are also advantageous as they show
higher Brunauer–Emmett–Teller (BET) surface areas than the
catalysts calcined at higher temperatures. Nevertheless, the se-
lectivity to acrolein barely exceeded 50% at 90% glycerol con-
version. In addition, the catalysts
calcined at low temperaturesshowed high carbon deposition
levels and, therefore, quick deac-
tivation. The second article of
Chai’s group reported the
screening of two dozen catalysts,
which they classified into four
different groups according to
their acidity.[41] Group 1 included
basic catalysts with HA values
higher than 7, such as magnesi-
um oxide. These catalysts
showed no selectivity to acrolein
at all. Group 2 contained cata-
lysts with a HA value ranging be-
tween 3 and 7, such as zirconi-
um oxide. According to the work
of Neher,[35] these catalysts
should give high acrolein selec-
tivities but, in practice, the selec-
tivity did not exceed 30% al-
though the performances re-
mained rather stable for 10 h
on-stream. Group 3, which was
more promising, comprised cata-
lysts with a HA value between
Table 2. Selection of catalytic performances reported for the dehydration of glycerol in the gas phase.[a]
Active phase Support T [K] Glycerol
conversion [%]
Acrolein
yield [%]
STY [mmol h1
gcat1]
Time on
stream [h]
Ref.
H4SiW12O40 SiO2 548 100.0 87.0 5.3 5 [48]
H4SiW12O40 SiO2 523 100.0 85.9 1.6 5 [49]
H4SiW12O40 SiO2 548 98.3 84.7 1.6 5 [47]
H-MC M49 – 633 100.0 82.8 n.a. n.a. [54]
H-MFI 1 % Au – 633 100.0 82.8 6.3 1 [66]
ZSM-11 – 633 99.0 81.3 n.a. n.a. [54]
H-MFI 0.1 % Pt – 633 100.0 80.7 6.2 3 [66]
b-zeolite – 633 100.0 80.3 n.a. n.a. [54]
MCM-22 – 633 100.0 80.1 n.a. n.a. [54]
Fe2 (PO4)3 Pumice stone 673–693 n.a. 80.0 n.a. n.a. [27]
Nd4
(P2O7)3 – 593 96.4 79.7 n.a. 7 [57]MFI+Ba – 633 100.0 79.0 6.2 2.5 [51]
H2WO4 – 533 100.0 79.0 1.2 5 [60]
WO3 ZrO2/montmorillonite 533 100.0 79.0 1.2 5 [63]
H2WO4 1 wt % Pd – 533 100.0 77.0 1.2 5 [62]
MCM-56 – 633 100.0 76.8 n.a. n.a. [54]
Nafion SiO 573 100.0 76.0 19.8 7 [38]
H3PO4 a-Al2O3 573 n.a. 75.0 0.3 n.a. [35]
Li3PO4 Pumice stone 673–693 n.a. 75.0 n.a. n.a. [27]
Gd4 (P2O7)3 – 593 98.2 74.3 n.a. 1 [57]
Sm4 (P2O7)3 – 593 99.6 73.9 n.a. 1 [57]
ZSM-5 – 588 98.3 73.6 n.a. n.a. [56]
WO3 ZrO2 573 100.0 73.5 19.2 7 [38]
H3PW12O40 ZrO2 588 100.0 70.0 12.6 4 [55]
[a] n.a.=data not available in the publication; STY =Space-time yield of acrolein.
ChemSusChem 2009 , 2, 719 – 730 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemsuschem.org 723
Towards the Sustainable Production of Acrolein by Glycerol Dehydration
Page 6
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 6/12
8 and 3. In this group, alumina-supported phosphorous
acid could be found, and also alumina-supported heteropolya-
cids and niobium oxide (calcined at temperatures between
673 K and 773 K). The group also contained a HZSM zeolite
and pure alumina. In good agreement with previous re-
sults,[38,39] the selectivity was generally higher than that ob-
served for Group 2 catalysts, with the exception of pure alumi-
na and niobium oxide calcined at 773 K. Interesting results
were observed with the alumina-supported phosphotungstic
heteropolyacid and a mixed phase of tungsten oxide/zirconi-
um oxide with approximately 70% selectivity at a conversion
of 70%, in both cases after 2 h on-stream. Unfortunately, these
catalysts showed poor stability, and their performance signifi-
cantly decreased during time-on-stream. Actually, for both cat-
alysts, the glycerol conversion dropped from 68–69% to 23–
25% between 1 and 10 h under reactant flow; the selectivity
being rather constant at roughly 66–70%. The other catalysts
were overall less efficient, and gave selectivities between 35%
and 55% at conversions between 55% and 100% after 10 h
on-stream. Group 4 contained catalysts with a HA value of lessthan 8, such as Hb-zeolite, niobium oxide calcined at 623 K,
and also alumina-silicate as well as sulfonated zirconium oxide.
These catalysts were less selective to acrolein than those of
Group 3 but they showed increasing performances during
time-on-stream. For example, the glycerol conversion on an
alumina-silicate dropped from 94 to 75% between 1 to 10 h
under reactant flow, the selectivity being rather unchanged at
roughly 43–46 %. Nevertheless, similar to niobium oxide,
Group 4 catalysts were also subjected to a detrimental carboni-
zation, with a carbon deposit that represented between
100 mg and 400 mg of carbon per gram of catalyst after 10 h
on-stream.The same team further published two other papers in 2008
and 2009 dealing with zirconia- and silica-supported 12-tung-
stophosphoric acid catalysts.[44,45] These studies showed that
the nature of the catalyst support has a significant effect on
the thermal stability and on the dispersion of the Keggin-type
active phase. The use of ZrO2 as a support led to better results
as far as the deactivation of the catalyst is concerned. The
Keggin-anion density at the surface of the support was identi-
fied as a key parameter for tuning the activity and the selectivi-
ty of the HPA for acrolein production, which is in good agree-
ment with the conclusions of Ning et al. who used activated
carbon-supported silicotungstic acid catalysts.[46] The latter
claimed that a 10 wt% H4SiW12O40 supported catalyst gives the
best acrolein space-time yield ever reported in the literature
(i.e., 68.5103 molacrolein h1 gactivephase1). This promising perfor-
mance was attributed to the good dispersion of the HPA on
the support surface and also to the relative quantities of
strong acid sites.
In this section we have seen that the tuning of the acidity of
the active phase is of prime importance for getting a good cat-
alytic performance. However, this characteristic alone is not
sufficient to also get a sufficient stability of the active phase
and a good selectivity to acrolein. It seems that the properties
of the support and the way the active phase is dispersed on
can also have large consequences. This point is discussed in
section.
4.3. Influence of the textural properties: A strong effect
This very interesting aspect has been recently discussed by
Tsukuda and co-workers.[47] This group works with silica-sup-
ported heteropolyacids and focuses on the influence of the
textural properties of the porous support on the selectivity.
They used three silicas with pore sizes of 3, 6, and 10 nm. The
active phase was either a heteropolyacid or a conventional in-
organic acid, such as phosphoros or boric acid. Good results
were obtained when using Keggin-type heteropolyacids such
as phosphotungstic acid (H3PW12O40) or silicotungstic acid
(H4SiW12O40) on the 6 nm silica support. At full conversion, the
selectivities towards acrolein were 65% and 75%, respectively,
whereas the molybdenum-based homologous heteropolyacid
(i.e., H3PMo12O40) did not yield a high selectivity (34%). These
values could be increased even further by changing the sup-
port to a silica with a larger pore diameter. The maximum se-lectivity measured was then 86% for the silicotungstic hetero-
polyacid deposited on a 10 nm pore diameter silica with a
30 wt% loading (at full glycerol conversion). For very small
pore diameters (i.e., 3 nm), the selectivity remained nearly un-
changed with time-on-stream whereas the conversion de-
creased by a factor of two, probably because of coking related
to steric limitations. The influence of temperature was also
studied and the optimum, in terms of acrolein yield, was
548 K.
These results show that the size of the pores has a direct in-
fluence on the stability of the catalyst. Actually, if steric limita-
tions do not enable the rapid desorption and diffusion of theproducts in the porous network of the catalyst, then coking is
more likely to occur, which results in blocking of the access to
the active phase and, thus, leads to the deactivation of the cat-
alyst.
4.3. The combined effect of acidity and porous structure
The above-mentioned studies point out two important param-
eters that govern the catalytic performances: the acidity and
the textural properties of the catalyst. These two parameters
were concomitantly investigated by Chai and Tsukuda, who
gathered their efforts, resulting in the publication of two
common patents.[48,49] These patents actually focus on support-
ed heteropolyacids as catalysts and on their catalytic perform-
ances in relation to the textural properties of the porous sup-
port. They carried out a large number of tests, varying the
active catalyst phase, loading amounts, and supports. As previ-
ously reported, phosphomolybdic acid was unselective com-
pared to phosphotungstic acid and silicotungstic acid.[47] They
obtained the largest—to our knowledge—acrolein selectivity
ever reported in the literature using a catalyst comprising 20–
30 wt% silicotungstic acid deposited onto porous silica with a
pore diameter of 10 nm (i.e., 87% at 548 K). Smaller pore diam-
eters (3 nm) or higher reaction temperatures (573 K) always led
to a decrease of the catalytic performance indicators (selectivi-
724 www.chemsuschem.org 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemSusChem 2009 , 2, 719 – 730
F. Dumeignil et al.
Page 7
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 7/12
ty, conversion) by a factor two. The influence of the BET specif-
ic surface area (SSA) of the support was also studied but
proved to have little effect. They compared two silicas with
identical pore diameter but with different SSAs. A twofold in-
crease of the surface area did not result in any significant
effect on the catalytic performance.
Apparently, the use of heteropolycompounds as active
phase offers an easy way for controlling the acidity, and there-
by the catalytic performance, by carefully tuning the composi-
tion of the Keggin HPA. Encouraged by the work of Tsukuda
et al. on heteropolyacids,[47] Atia et al. published a comparison
of the performances of silicotungstic acid and phosphomolyb-
dic acid supported on alumina, silica, or aluminosilicates.[50]
The samples, prepared by incipient wetness impregnation,
were calcined at 673 K for 4 h prior to use and their acidity
was determined by ammonia temperature-programmed de-
sorption. The silica-supported heteropolycompound was more
acidic than the alumina-supported one. Moreover, the acidity
of the alumina-supported species was similar to that of pure
alumina, which was explained by the substitution of aluminaBrønsted sites by HPA ones. The reaction conditions for the
catalytic tests were chosen similar to those used by Tsukuda, [47]
for the sake of easier direct comparison. Irrespective of the
type of alumina-supported catalyst, total glycerol conversion
was observed for a temperature higher than 548 K but signifi-
cant differences in selectivity were observed (i.e., between 30
and 60%), underlining the strong influence of the support. The
best results were obtained for silicotungstic acid supported on
alumina and aluminosilicates with acrolein yields of 63% and
67%, respectively, but the authors preferred to rule out any
conclusion about silica-supported silicotungstic acid because
the deactivation was too fast and reliable data could not thusbe acquired. They did indicate the instantaneous initial selec-
tivity over silicotungstic acid supported on a silica with a mean
pore diameter of 11 nm, which was 73%. The contact time was
further varied by flow adjustment, and full conversion was ach-
ieved for values larger than 0.2 s, which illustrates the high re-
activity of glycerol. Although acrolein is subjected to degrada-
tion by consecutive reactions, the selectivity did not drastically
decrease with the increase in contact time from 0.15 to 0.7 s.
4.5. Recent progress: Other classes of active catalysts
The use of supported heteropolycompounds is not the only
way to reach good catalytic performance, as demonstrated by
Dubois et al. in recent work. [38] Actually, zeolitic structures are
also promising and have been well-studied for three years.
In 2007, Okuno and coworkers patented metallosilicate cata-
lysts with an MFI structure; a typical structure of zeolites with
a 3D pore network.[51] These catalysts are prepared in a four-
step synthesis, starting from the impregnation of a silica sup-
port with a solution containing a metal component such as
aluminum hydroxide, tetraalkylammonium components such
as tetramethyl- or tetraethylammonium, and a certain amount
of an alkaline compound. This leads to a precursor with the
formula Si1T x L y (SDA) z , where T is the metal atom, L is the alka-
line, and SDA the tetraalkyl component. In the second step,
this precursor is crystallized by hydrothermal treatment to give
a metallosilicate of MFI structure. In the third step, the intro-
duced alkali atoms are substituted by ammonia in an ion-ex-
change reaction before the final catalyst is obtained in the
fourth step, which is called the burning step and means calci-
nation at a temperature of up to 873 K.
The catalytic performance tests revealed good results for
aluminosilicates and gallosilicates whereas ferrosilicates were
less selective. The aluminosilicates offered even stable perfor-
mance without addition of any alkali atom or ion-exchange,
with a selectivity for acrolein of around 65%. This selectivity
did not change significantly with the aforementioned addition/
exchange, which was also observed over the gallosilicates. In
this patent, the results claimed over zeolitic catalysts are quite
similar to those reported in the work by Dubois et al. (i.e., 70%
acrolein selectivity at glycerol full conversion).[38] In further
publications, the same group reported tuning of the acidity by
modifying the Si/Al ratio of the zeolite, and the best results
were achieved using a ratio of 28 with a 71.2% yield of acrole-
in.[52,53]
A second group, headed by Li, specifically studied aluminosi-
licates catalysts.[54] The authors reported about protonated zeo-
lites, for example, MCM-49, which was synthesized under hy-
drothermal conditions and further protonated by treatment
with ammonium nitrate before calcination. This procedure
seems very similar to those reported by Okuno et al.[51] and
Dubois et al.,[38] but the catalytic performances observed by Li
et al. are better. The selectivity to acrolein reached a value
close to 83% at full glycerol conversion, which is the best
result ever published for a zeolitic catalyst (see Table 2). This
might be due to the optimization of the reaction parameters
such as temperature and contact time.Zhou and co-workers proposed a synthesis procedure using
dual templates of micro- and mesoporous ZSM-5 composites
used as catalysts in the dehydration of glycerol to acrolein.[56]
The best result achieved with these catalysts was 73.6% acrole-
in selectivity at 98.3 % glycerol conversion, which is a good
performance. Rare-earth pyrophosphates were also claimed by
Liu and co-workers to be efficient catalysts for the production
of acrolein by vapor-phase dehydration of glycerol[57] with an
acrolein yield as high as 80% achieved over Nd4(P2O7)3. Matsu-
nami et al. also reported results on silica-supported phosphates
doped by alkali and metal salts but in this case, the acrolein
yield did not exceed 67 %.[58,59] More recently, Redlingshofer
et al. proposed tungstates as efficient catalysts for this reac-
tion.[60,61] The reaction was carried out at a relatively low tem-
perature (533 K) and exhibited good performances with an
acrolein yield between 77 and 79%. Nevertheless, the catalysts
tended to deactivate under the stream, whereby the acrolein
yield decreased at a rate of 5% per 10 h. Additionally, the au-
thors described the possibility to regenerate the catalyst by
oxygen treatment at 623 K for 5 h and claimed to recover the
initial catalytic performance after this treatment.[62,63]
As discussed above and further summarized in Table 2, a
great variety of active phases can be used to selectively dehy-
drate glycerol to acrolein in the gas phase. One key parameter
is the tuning of the solid acidity, but the physical properties of
ChemSusChem 2009 , 2, 719 – 730 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemsuschem.org 725
Towards the Sustainable Production of Acrolein by Glycerol Dehydration
Page 8
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 8/12
the catalyst are also of prime importance to achieve good per-
formances. Here, the supports can play a major role through
modifying the textural properties of the catalyst, which has a
dramatic effect on the selectivity probably because of steric
and diffusion issues. The best results published so far were ob-
tained at a relatively low temperature (523–548 K) using silica-
supported HPAs. These catalysts, however, tend to deactivate
quite rapidly with time-on-stream. The zeolite family is also
promising but needs higher working temperatures (more than
623 K) to achieve high yields.
When the necessary data were available in the papers, we
calculated the acrolein space-time yield (STY) taking the total
catalyst mass as a reference. The obtained values are reported
in Table 2. Accordingly, the best result obtained up to now
were achieved by Dubois et al. on a SiO2-supported Nafion cat-
alyst.[38] The STY was 20103 molacrolein h1 gcatalyst1. However,
another method to calculate the STY takes into account only
the active phase mass, and when using this method the best
result was achieved by Ning et al. with an activated carbon-
supported silicotungstic acid catalyst (10 wt% H4SiW12O40).[64]
The STY was 68.5103 molacrolein h1 gactive phase1.
5. Reaction Mechanism
Whereas the research for an efficient catalyst can follow a
more or less purely applied approach, an understanding of the
reaction mechanism is a more fundamental aspect. This implies
the identification of the intermediate steps and the explana-
tion of formation of the byproducts. A first proposal for the
glycerol activation in near- or supercritical conditions was
made by Bhler et al.[31] Two pathways were claimed, via either
an ionic or a radical mechanism. The ionic reaction(Scheme 4a) begins with the protonation of glycerol on either
one primary hydroxyl group or the secondary hydroxyl group.
Afterwards, the elimination of a water molecule leads to the
formation of a carbocation. In the case of the secondary carbo-
cation, the only possible product after cleavage of H3O+ is
acrolein, whereas either acrolein or acetaldehyde and formal-
dehyde can be formed starting from the primary carbocation.
The radical pathway (Scheme 4b) starts with the abstraction of
a hydrogen from a primary carbon by an OH C radical. The re-
sulting radical species further loses an OHC, which leads to the
formation of acrolein.
Tsukuda et al. subsequently proposed a more formal reac-
tion framework (Scheme 5).[47] The first step, glycerol dehydra-
tion, leads to the formation of two enols, which are in tauto-
meric equilibrium with the corresponding ketone (hydroxyace-
tone) and aldehyde (3-hydroxypropionaldehyde). The latter
subsequently reacts in a second step either via dehydration to
acrolein or via a retroaldol reaction to give formaldehyde and
acetaldehyde, which may be easily oxidized to acetic acid (in
the presence of O2). As an important conclusion, Tsukuda, in
agreement with Bhler’s previous proposal,[31] pointed out that
the key to obtaining a high selectivity for acrolein lies in the
control of the first dehydration step to favor the formation of
3-hydroxypropionaldehyde while suppressing the formation of
hydroxyacetone, which is identified as the main byproduct of
the process.
The same mechanism has been further completed by Chai
et al. (Scheme 5),[41] who added the two hydrogenation steps
that can occur starting from acrolein and hydroxyacetone to
form allylic alcohol and 1,2-propanediol, respectively, which are
sometimes actually observed. The hydrogenation of the form-
aldehyde to methanol or its decomposition into CO H2 is also
mentioned.
These schemes are interesting for a first approach, but an
even deeper analysis is needed to explain the formation of all
the observed byproducts. Corma postulated a more complex
Scheme 4. Reaction pathways to the formation of acrolein considering (a) an
ionic and (b) a radical pathway.[31]
Scheme 5. Mechanism proposed by Tsukuda et al. and by Chai et al. [41,47]
726 www.chemsuschem.org 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemSusChem 2009 , 2, 719 – 730
F. Dumeignil et al.
Page 9
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 9/12
reaction framework (Scheme 6).[25] The first dehydration step is
obviously the same as that previously described by Tsukuda
and Chai, and leads to hydroxyacetone or 3-hydroxypropional-
dehyde, which further reacts in a second dehydration step to
yield acrolein.[47] To go further and investigate the secondary
reactions, the authors directly used hydroxyacetone as a reac-
tant at 623 K and classified the main reaction products in three
groups: acids (9.4 %), other aldehydes (52.0 %), and coke
(27.7%). They also observed the formation of low amounts of acetone (4.0 %), acetaldehyde (1.4%), and carbon monoxide
(1.2 %). Nevertheless, the conversion of hydroxyacetone did
not exceed 25%, which means that this compound is relatively
stable and is thus usually identified as the major byproduct of
the reaction of dehydration of glycerol to acrolein.
To investigate the consecutive reactions of acetone, they re-
peated this method by reacting acetone at 623 K. The resulting
product distribution is dominated by unsaturated hydrocar-
bons such as butene (27.9%), propylene (1.7%), C5 C6–8 aro-
matics (6.6 %), and coke (31.9 %; note that the latter is the
known consecutive product of unsaturated hydrocarbons). The
other identified products were various acids (20.2%) and other
aldehydes (7.3%) at an overall conversion that did not exceed
14%. With their findings, they thus completed the reaction
scheme proposed by Chai and Tsukuda by adding further side-
reactions originating from hydroxyacetone. Thereby, they ex-
plained the formation of oligomers and coke by consecutive
reactions starting from hydroxyacetone via acetone and acetal-
dehyde.
Suprun et al. chose a similar approach to investigate the
consecutive reactions of 3-hydroxypropionaldehyde and hy-
droxyacetone.[65] They could confirm the retroaldol reaction
previously postulated by Tsukuda,[47] which leads to the forma-
tion of formaldehyde and acetaldehyde issued from 3-hydroxy-
propionaldehyde. In addition, significant amounts of coke
were found, which is explained by the formation of cyclic C 6-
compounds identified by GC-MS analysis (Scheme 7a). The au-
thors further identified various furan derivates (Scheme 7 b),
which must be thus taken into account when one specifically
seeks for fine information on the minor reaction byproducts.
6. Catalyst Deactivation: A Key Issue
It has been reported above that very efficient catalysts for the
dehydration of glycerol into acrolein can be prepared. Unfortu-
nately, these catalysts are also very unstable under reaction
conditions and prove to deactivate very quickly, most probably
because of the formation of coke, which makes their straight-
forward use in an industrial plant difficult at this time. There-
fore, solutions have to be found to avoid or limit the deposi-
tion of coke onto the catalyst surface or at least to optimize
the regeneration of the catalysts.
At least three kinds of solutions have been proposed to con-
tinuously regenerate the catalyst: (1) co-injection of oxygen
with the gas feed to yield in situ regeneration (co-feeding),[39]
(2) cyclic regeneration of the used catalyst by injection of a
flow/pulses of air or oxygen,[67] and (3) circulation of the cata-
lyst in a moving bed reactor with regeneration in a parallel
vessel (as for the FCC process).[25] Whereas the first option is as-
sociated with the risk of generating explosive conditions and/
Scheme 6. Reaction network proposed by Corma et al.[25]
Scheme 7. Products derived from (a) 3-hydroxypropionaldehyde and (b) hy-
droaxyacetone. [65]
ChemSusChem 2009 , 2, 719 – 730 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemsuschem.org 727
Towards the Sustainable Production of Acrolein by Glycerol Dehydration
Page 10
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 10/12
or oxidizing the various reaction products, the second alterna-
tive is accompanied by the disadvantage of a loss of productiv-
ity. The third option does not suffer from these drawbacks but
the construction and the operation of a circulating bed reactor
implies serious technological difficulties that can only be over-
taken by skilled personnel and fine process control.
The idea of working with air in the reaction gas was first
proposed by Dubois.[38] To stay out of the explosivity range,
the oxygen fraction must never exceed 7 vol%. Therefore, the
composition of the reaction feed was adjusted to 6 % of
oxygen for 4.5 % of glycerol, the remaining 89.5% being
steam. Thereby, the author claimed to reduce catalyst deactiva-
tion and even to inhibit the formation of byproducts such as
hydroxyacetone in these conditions. Kasuga et al. injected air
into the gas feed to have a feed ratio of 7% of oxygen for
27% of glycerol, the remaining 66% being steam and nitrogen
from air. Nevertheless, the selectivity towards acrolein was
then rather low and did not exceed 45% after 24 h under reac-
tant flow.[67] To optimize the regeneration, Kasuga et al. also
proposed to modify their MFI protonated-zeolite with a smallamount of metal (Pt, Pd, Ru, Cu, Ir, or Au). By this doping, they
claimed to accelerate the split of the dioxygen used for the re-
generation.[66] The best results were obtained with 0.1 wt% Pt
and 1 wt% Au (80.7 and 79.7% of acrolein yield, respectively,
at full glycerol conversion after 150 min under reaction condi-
tions).
The possibility of periodic regeneration was studied by Arita
et al., who regenerated a used H-ZSM5 catalyst under air flow
and showed the possibility of recovering the initial perfor-
mance. However, the hot-spot temperature during regenera-
tion surpassed the regeneration temperature by more than
100 K.
[67]
Atia et al. carried out long-term runs (up to 300 h)and observed that the selectivity to acrolein remained stable
whereas glycerol conversion decreased more or less linearly.
This effect could be well explained by the deposit of coke
onto the catalyst surface and the concomitant decrease of the
number of accessible active sites. To verify this hypothesis,
coked catalysts were regenerated under a flow of 1% oxygen
in nitrogen at 598 K during 24 h. After this treatment, the cata-
lyst performed identical to the fresh catalyst. To quantify this
effect, the used catalyst was also analyzed by thermogravime-
try, which confirmed the carbon deposit hypothesis. [50]
Meanwhile, Corma et al. adopted the idea of a circulating
bed reactor.[25,68] The authors studied the opportunity of inject-
ing crude glycerol directly into FCC plants. An advantage
would be the use of existing facilities and, therefore, no need
for investments for building specific infrastructures. It is worth
noting that this idea has already been proposed in a different
form by Dubois, who pointed out the opportunity of injecting
glycerol into propylene oxidation plants to concomitantly yield
acrolein from both sources.[69] In the FCC plants, the heat re-
covered by the burning of coke could be used to provide the
energy necessary for the evaporation of glycerol. Corma et al.
concluded that an autothermal process is possible in that case.
Nevertheless, the authors did not limit their view on the dehy-
dration but also investigated the possibilities for reforming the
glycerol at 773 K to 873 K to produce ethylene and propylene.
Thereby, the glycerol feedstock may be an alterative or a com-
plement to naphta cracking.
An original approach was followed by Kasuga et al., who
used some pretreatment of the catalysts (protonated MFI zeo-
lite or Alox alumina).[70] Three types of pretreatments were
tested: (1) under a flow of hydroxyacetone, water, and nitro-
gen; (2) under a flow of acrolein, water, and nitrogen; and
(3) under a flow of hydroxyacetone, water, and air. Irrespective
of the pretreatment method, the Alox catalyst always led to
very poor acrolein yields while over the MFI catalyst the pre-
treatments enabled higher selectivities in the first minutes
under the reactant flow. However, after 150 min on-stream, the
catalytic performances recovered values similar to those ob-
tained without preliminary pretreatment, which means that
this effect is only effective in the early stage of the reaction.
Nevertheless, in this work the authors underlined the possibili-
ty of recovering the initial performance by regenerating the
catalysts at 773 K under air flow.
7. Direct Use of Crude Glycerol as Reactant:An Important Economic Issue
The price of glycerol drastically depends on the required tech-
nical grade. Whereas refined glycerol currently costs between
US$ 500 and 600 t1 (June 2009), crude glycerol can be ob-
tained for only US$ 100–200 t1.[26] Nevertheless, the glycerol
market has been highly volatile in 2007–2008, with glycerol
costs of up to US$ 1800 t1 for refined glycerol and up to
US$ 500 t1 for crude glycerol.[26] It should be mentioned that
the availability of glycerol—and therefore its price—also de-
pends on political decisions concerning biodiesel production
and agricultural subsidies. Therefore, enabling the use of crudeglycerol is a crucial issue for achieving a sustainable and eco-
nomically viable production of acrolein. However, as men-
tioned earlier crude glycerol is generally contaminated with by-
products issued from biodiesel process (inorganic traces such
as water, sodium and potassium carbonate, and organic traces
such as esters, fatty acids, and alcohols). The use of crude glyc-
erol as a feedstock may therefore cause problems either by
poisoning the catalyst or by causing plugs because of the dep-
osition of high-boiling-point organic materials or inorganic
salts. From our knowledge, at this date no catalytic test using
this type of feedstock has been reported in the literature. All
the disclosed works were carried out using diluted aqueous
solutions prepared from refined glycerol. Furthermore, it has
been found that the acrolein yield strongly depends on the
glycerol concentration in the reactor feed, with a decrease of
the yield with increasing concentration.[35,36] However, the use
of crude glycerol is of prime economical importance and differ-
ent concepts have already been proposed to avoid the costly
distillation of crude glycerol and to overtake the obstacles
raised by its use as a feedstock. Kijenski et al. proposed a
modified evaporation system in which the crude glycerol is
brought into contact with an inert liquid at high tempera-
ture.[71] The glycerol is evaporated and fed into the reactor by
an inert carrier gas while the impurities remain in the inert
liquid. The author describes a process example where a solu-
728 www.chemsuschem.org 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemSusChem 2009 , 2, 719 – 730
F. Dumeignil et al.
Page 11
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 11/12
tion of 75 wt% of glycerol with up to 3 wt% impurities was
added to a silicon-oil heated at 603 K. The glycerol vapors
were then carried by a nitrogen flow to feed the reactor.
To facilitate the use of crude glycerol, Dubois rather suggest-
ed a fluidized inert solid heated at high temperature.[72] This
leads to the evaporation of glycerol vapors while the impuri-
ties remain inside the fluidized bed, which can be continuously
regenerated. The regeneration may consist in a flushing step
with water to dissolve inorganic salts followed by a thermal
treatment to burn organic deposits. The author describes an
example process in which a solution of 18 wt% of glycerol
with 2 wt% of sodium chloride is fed into a fluidized bed of
silica particles. The fluidization is performed using nitrogen at
583 K. As a result, the author claimed a recovery of 99.9% of
the introduced sodium chloride.
8. Conclusions
The finite nature of the fossil fuel supply and the global warm-
ing problem have led to a change in energy policies. In thiscontext, biofuels have received increased attention as CO2-neu-
tral alternatives to crude oils. Beside bioethanol as a substitute
for gasoline, the use of biodiesel has drastically progressed—
especially in the European Union. The increase in biodiesel
production has also led to an oversupply of glycerol as a co-
product. Because glycerol is a highly functionalizable molecule,
many processes for its valorization have been recently pub-
lished. In particular the catalytic dehydration to acrolein—a
precursor of DL-methionine used for cattle feeding—is of eco-
nomic interest.
This reaction can proceed in either the gas or the liquid
phase. The reaction conditions in the liquid phase include thepossibility to use supercritical solvents. In terms of economical
interest, the use of heterogeneous catalysis is usually preferred
rather than the use of homogenous catalysis, as the separation
of the catalyst from the reaction medium is simplified. There-
fore, the possibility of using supported acids instead of dis-
solved ones is preferable, even if it has been demonstrated
that both technologies show comparable catalytic performan-
ces. A second important step on the road to commercialization
is demonstrating the feasibility of working in continuous
mode. Experimental results have already been published, and
show only slightly decreased performances when compared
with discontinuous experiments.[33] At last, the use of supercrit-
ical water as a reaction medium has been proven to be adapt-
able to the reaction. Nevertheless, the combination of super-
critical and acid conditions leads to high demands on the ma-
terials used for the reaction vessel and all peripherals. For all
these reasons, and even if an acrolein yield of 80% has already
been achieved in the liquid phase, the possibility of a commer-
cial application seems questionable at this date.
Concerning the gas-phase reaction, this Minireview has out-
lined a great variety of efficient catalysts. We can classify them
according to three different categories: (1) supported inorganic
acids, with a subcategory for supported heteropolyacids;
(2) protonated zeolites; and (3) mixed-oxide type catalysts. The
best acrolein yields are, up to now, achieved with supported
inorganic acids and are in the range of 75 to 87%. On zeolitic
and mixed-oxide catalysts, the acrolein yield is generally slight-
ly lower (around 70 to 80%) and the temperature needed to
reach high conversions is higher (around 573–623 K against
523–573 K for the inorganic acids). For supported heteropolya-
cids, the published results already give a clear image of the
properties required to design efficient catalysts. These are a
controlled proper acidity of the active phase, and appropriate
textural properties (mainly porosity) brought by the support to
avoid steric and diffusion limitations effects. Applying these
two concepts, an acrolein yield of 87% has already been ach-
ieved.
Nevertheless, the main problem remains the rapid catalyst
deactivation owing to carbon deposition. Up to now, no cata-
lyst exceeds a half-life time of a few days, which is necessary
for commercial applications. Different approaches have been
published to regenerate the coked catalyst. These include the
use of a fluidized circulating catalyst that is continuously re-
generated in a parallel reactor, the introduction of air or
oxygen into the reaction feed for a continuous regeneration of the catalyst directly inside the reactor, and the discontinuous
regeneration by alternating the glycerol feed and the oxygen
feed. Modifying the catalyst formulations to facilitate the split-
ting of the dioxygen used for regeneration has also been pro-
posed. Currently, none of these three has proven technologi-
cally superior to the others.
Finally, we have shown the stringent need for using crude
glycerol as a feedstock with regard to economical aspects. We
did not find any attempt of directly using crude glycerol over
catalytic formulations but two promising processes have been
presented, which intend to eliminate the impurities of the
crude glycerol during evaporation and avoid costly crude glyc-erol distillation. As in the case of the catalyst regeneration,
none of these processes have proven superior to the others at
this moment.
Acknowledgements
The authors would like to thank the French Research Network
No. 1 (RDR1) of the CPDD program as well as CE (through con-
tract number MIRG-CT-2007-046383) for their financial support
for realizing this bibliographic study.
Keywords: acrolein · biomass · catalysis · dehydration ·
glycerol
[1] Directive 2003/30/EC of the European Parliament and of the council of
05/08/2003 on the promotion of the use of biofuels or other renewable
fuels for transport.
[2] Eurostat European Commission, http://epp.eurostat.ec.europa.eusion.
[3] L. Ott, M. Bicker, H. Vogel, Green Chem. 2006, 8, 214.
[4] M. J. Haas, A. J. Mc Aloon, W. C. Yee, T. A. Foglia, Bioresour. Technol.
2006, 97 , 671.
[5] M. Pagliaro, M. Rossi, The Future of Glycerol: New Uses of a Versatile Raw
Material , RSC Green Chemistry Book Series, 2008.
[6] http:/ /theglycerolchallenge.org/index.htm.
[7] Data sources: European Biodiesel Board and HB I.
ChemSusChem 2009 , 2, 719 – 730 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemsuschem.org 729
Towards the Sustainable Production of Acrolein by Glycerol Dehydration
Page 12
8/15/2019 2009 Towards the Sustainable Production of Acrolein By
http://slidepdf.com/reader/full/2009-towards-the-sustainable-production-of-acrolein-by 12/12
[8] C. S. Callam, S. J. Singer, T. L. Lowary, C. M. Hadad. , J. Am. Chem. Soc.
2001, 123, 11743.
[9] G. Paulo da Silva, M. Mack, J. Contiero, Biotechnol. Adv. 2009, 27 , 30.
[10] Y. Zheng, X. Chen an d Y. Shen, Chem. Rev. 2008, 108, 5253.
[11] M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi, C. Della Pina, Angew.
Chem. 2007, 119, 4516; Angew. Chem. Int. Ed. 2007, 46, 4434.
[12] D. A. Simonetti, J. Ross-Hansen, E. L. Kunkes, R. R. Soares, J. A. Dumesic,
Green Chem. 2007, 9, 1073.
[13] R. R. Soares, D. A. Simonetti, J. A. Dumesic, Angew. Chem. 2006, 118,4086; Angew. Chem. Int. Ed. 2006, 45, 3982.
[14] D. Siano, E. Santacesaria, V. Fiandra, R. Tesser, G. Di Nuzzi, M. Di Serio, M.
Nastasi (ASER), WO2006111810, 2006.
[15] D. Schreck, W. Kruper, F. Varjian, M. Jones, R. Campbell, K. Kearns, B.
Hook, J. Briggs, J. Hippler (DOW Chemicals), WO2006020234, 2006.
[16] N. O. V. Sonntag, J. Am. Ceram. Soc. 1992, 75, 795.
[17] H. Kimura, Appl. Catal. A: Gen. 1993, 105, 147; H. Kimura, K. Tsuto, T. Wa-
kisaka, Y. Kazumi, Y. Inaya, Appl. Catal. A 1993, 96, 217.
[18] K. Nabe, N. Izuo, S. Yamada, I. Chibata, Appl. Environ. Microbiol. 1979,
38, 1056.
[19] R. Ciriminna, G. Palmisano, C. Della Pina, M. Rossi, M. Pagliaro, Tetrahe-
dron Lett. 2006, 47 , 6993.
[20] A. Yamamoto, Encyclopedia of Chemical Technology 3rd ed. , 1978, Vol. 2,
403.
[21] M. P. Malveda, H. Janshek ar, K. Yokose, CEH Marketing Research Report—
SRI Consulting, Major Amino Acids, June 2006.[22] M. P. Malveda, H. Janshekar, K. Yokose, Chemical Economics Handbook-
SRI Consulting, June 2006, 502.5000 B.
[23] G. W. Keulks, L. D. Krenzke, T. M. Notermann, Adv. Catal. 1978, 27 , 183.
[24] M. M. Lin, Appl. Catal. A: Gen. 2001, 207 , 1 .
[25] A. Corma, G. W. Huber, L. Sauvanaud, P. O’Connor, J. Catal. 2008, 257 ,
163.
[26] Petrosil Glycerine Report, June 12, 2009, www.glycerinereport.com.
[27] Schering-Kahlbaum AG, FR 695931, 1930.
[28] H. Groll, G. Hearne (Shell), US 2042224, 1936.
[29] H. Hoyt, T. Manninen (US Ind. Chemicals. Inc.), US 2558520, 1951.
[30] S. Ramayya, A. Brittain, C. DeAlmeida, W. Mok, M. J. Antal, Fuel 1987, 66,
1364.
[31] W. Bhler, E. Dinjus, H. J. Ederer, A. Kruse, C. Mas, J. Supercrit. Fluids
2002, 22, 37.
[32] V. Lehr, M. Sarlea, L. Ott, H. Vogel, Catal. Today 2007, 121, 121.[33] M. Watanabe, T. Iida, Y. Aizawa, T. M. Aida, H. Inomata, Bioresour. Technol.
2007, 98, 1285.
[34] N. Suzuki, M. Takahashi (KAO Corp.), JP 2006290815, 2006.
[35] A. Neher, T. Haas, A. Dietrich, H. Klenk, W. Girke (Degussa), DE 4238493,
1994.
[36] A. Neher, T. Haas, A. Dietrich, H. Klenk, W. Girke (Degussa), US 5387720,
1995.
[37] A. Neher, T. Haas (Degussa), US 5426249, 1995.
[38] J.-L. Dubois, C. Duquenne, W. Hoelderich, J. Kervennal (Arkema), WO
2006087084, 2006.
[39] J.-L. Dubois, C. Duquenne (Arkema), WO 2006087083, 2006.
[40] S.-H. Chai, H.-P. Wang, Y. Liang, B.-Q. Xu, J. Catal. 2007, 250, 342.
[41] S.-H. Chai, H.-P. Wang, Y. Liang, B.-Q. Xu, Green Chem. 2007, 9, 1130.
[42] Z. H. Chen, T. Lizuka, K. Tanabe, Chem. Lett. 1984, 13, 1085.
[43] T. Lizuka, K. Ogasawara, K. Tanabe, Bull. Chem. Soc. Jpn. 1983, 56 , 2927.
[44] S-H. Chai, H-P. Wang, Y. Liang, B-Q. Xu, Green Chem. 2008, 10, 1087.
[45] S.-H. Chai, H.-P. Wang, Y. Liang, B.-Q. Xu, Appl. Catal. A 2009, 353, 213.
[46] L. Ning, Y. Ding, W. Chen, L. Gong, R. Lin, Y. L, Q. Xin, Chin. J. Catal.
2008, 29, 212.
[47] E. Tsukuda, S. Sato, R. Takahashi, T. Sodesawa, Catal. Commun. 2007, 8 ,
1349.
[48] B.-Q. Xu, S-H. Chai, T. Takahashi, M. Shima, S. Sato, R. Takahashi (Nippon
Catalytic Chem. Ind.), WO 2007058221, 2007.[49] T. Sato, R. Takahashi (Nippon Catalytic Chem. Ind.), JP 2008088149,
2008.
[50] H. Atia, U. Armbruster, A. Martin, J. Catal. 2008, 258, 71.
[51] M. Okuno, E. Matsunami, T. Takahashi, H. Kasuga, M. Okada, M. Kirishik
(Nippon Catalytic Chem. Ind.), WO 2007132926, 2007.
[52] M. Okuno, E. Matsunami, T. Takahashi, H. Kasuga (Nippon Catalytic
Chem. Ind.), JP 2007301505, 2007.
[53] M. Okuno, E. Matsunami, T. Takahashi, H. Kasuga (Nippon Catalytic
Chem. Ind.), JP 2007301506, 2007.
[54] X.-Z. Li (Shanghai Huayi Acrylic Acid Co), CN 101070276, 2007.
[55] H. Jo, S.-H. Chai, T. Takahashi, M. Shima (Nippon Catalytic Chem. Ind.),
JP 2007137785, 2007.
[56] C.-J. Zhou, C.-J. Huang, W.-G. Zhang, H.-S. Zhai, H.-L. Wu, Z. S. Chao,
Stud. Surf. Sci. Catal. 2007, 165, 527.
[57] Q. Liu, Z. Zhang, Y. Du, J. Li, X. Yang, Catal. Lett. 2009, 127 , 419.
[58] E. Matsunami, T. Takahashi, H. Kasuga (Nippon Catalytic Chem. Ind.), JP2007268363, 2007.
[59] E. Matsunami, T. Takahashi, H. Kasuga (Nippon Catalytic Chem. Ind.), JP
2007268364, 2007.
[60] H. Redlingshoefer, C. Weckbecker, K. Huthmacher, A. Doerflein (Evonik
Degussa), WO 2008092533, 2008.
[61] H. Redlingshoefer, C. Weckbecker, K. Huthmacher, A. Doerflein (Evonik
Degussa), DE 102007004351, 2007.
[62] H. Redlingshoefer, C. Weckbecker, K. Huthmacher, A. Doerflein (Evonik
Degussa), WO 2008092534, 2008.
[63] H. Redlingshoefer, C. Weckbecker, K. Huthmacher, A. Doerflein (Evonik
Degussa), DE 102007004350, 2007.
[64] L. Ning, Y. Ding, W. Chen, L. Gong, R. Lin, Y. L, Q. Xin, Chin. J. Catal.
2008, 29, 212.
[65] W. Suprun, M. Lutecki, T. Haber, H. Papp, J. Mol. Cat. A: Chem. 2009,
309, 71.[66] H. Kasuga, M. Okada (Nippon Catalytic Chem. Ind.), JP 2008137950,
2008.
[67] Y. Arita, H. Kasuga, M. Kirishiki (Nippon Catalytic Chem. Ind.), JP
2008110298, 2008.
[68] P. O’Connor, A. Corma, G. W. Huber, L. Savanaud (Bioecon. Internat.
Holding), WO 2008052993, 2008.
[69] J.-L. Dubois (Arkema), FR 2897058, 2007.
[70] H. Kasuga (Nippon Catalytic Chem. Ind.), JP 2008137952, 2008.
[71] J. Kijenski, A. Migdal, O. Osawaru, E. Smigiera (Inst. Chemii Przemy-
slowe), EP 1860090, 2007.
[72] J.-L. Dubois (Arkema), WO 2008129208, 2008.
Received: May 12, 2009
730 www.chemsuschem.org 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemSusChem 2009 , 2, 719 – 730
F. Dumeignil et al.