Page 1
5
In recent years, much attention has been focused on lipid-based formulations (Humberstone
et al., 1997) to enhance the solubility of poorly water soluble drugs and improving
bioavailability to administer them through oral route resulting in increasing their clinical
efficacy, cost effectiveness and ease of preparation.
2.1. Lipid based drug delivery systems (Pouton, 2000)
Lipid based drug delivery systems (LBDDS) are one of the most popular approaches to
improve the oral bioavailability of poorly water soluble drugs (Charman, 2000; Gershanik et
al., 2000). Lipid vehicles are known to enhance the absorption of lipophilic drugs and there
are various mechanisms by which LBDDS enhance the absorption of lipophilic drugs:
Enhanced dissolution/solubilization
The presence of lipids in the GI tract stimulates gall bladder contractions, biliary and
pancreatic secretions, including bile salts (BS), phospholipids (PL) and cholesterol (Ch)
(Fleisher et al., 1999). These products, along with the gastric shear movement form a crude
emulsion and promote the solubilization of the co-administered lipophilic drug (Tso, 1985).
Surfactants present in the delivery system may also improve the solubilization of the
lipophilic compound.
Affecting intestinal permeability
A variety of lipids have been shown to change the physical barrier function of the gut wall
and hence, to enhance permeability. For BCS class II compounds, permeability through the
GI wall is not a limiting step towards absorption and hence, this mechanism is not thought to
be a major contributor for the absorption enhancement of lipophilic drugs. However, this may
be helpful for BCS class IV drugs.
Reduced metabolism and efflux activity
Certain lipids and surfactants also reduce the activity of efflux transporters in the GI wall and
hence, to increase the fraction of drug absorbed (Dintaman et al., 1999; Nerurkar et al.,
1996). Because of the interplay between P-gp and CYP3A4 activity this mechanism may
reduce intra-enterocyte metabolism as well.
Page 2
6
Stimulation of lymphatic transport
Bioavailability of lipophilic drugs may also be improved by the stimulation of the intestinal
lymphatic transport pathway (Trevaskis et al., 2008; Charman et al., 1996).
Prolongation of gastric residence time
Gastric transit time is prolonged with the presence of lipids in GI tract (Van Citters et al.,
1999). As a result, the residence time of the lipophilic drug in the small intestine increases.
This improves dissolution of the drug and thereby improves absorption.
Classification of lipid delivery systems (Pouton, 2000)
The simplest lipid products are those in which the drug is dissolved in digestible oil, usually a
vegetable oil or medium chain triglyceride (fractionated coconut oil). These are safe food
substances and do not present a toxicological risk to formulators. Oil solution has been the
standard way of administering oil-soluble vitamins (A and D) for many years. Bioavailability
from oil solutions is likely to be very good because the triglycerides are rapidly digested to
free fatty acids and 2-mono-glycerides, and these products are solubilised to form a colloidal
dispersion within bile salt-lecithin mixed micelles. A hydrophobic drug is also likely to be
solubilised in mixed micelles resulting in a reservoir of drug in colloidal solution from which
it can partition, allowing efficient, passive (transcellular) absorption. The low solvent
capacity of triglycerides often prevents formulation in oil, but oil solutions may be a realistic
option for potent drugs. Solvent capacity for less hydrophobic drugs can be improved by
blending triglycerides with other oily excipients which include mixed monoglycerides and
diglycerides. Since these are similar to the natural degradation products of triglycerides (the
difference being that their monoglyceride content is mostly 1 monoglyceride rather than 2-
monoglyceride) they do not prevent efficient digestion. As excipients with GRAS status they
have the same advantages as triglycerides, and are useful for blending with triglycerides or
for use as an alternative. Formulations which comprise drug in solution in triglycerides and/or
mixed glycerides are classified here as „Type I‟ (Table 2.1).When an appropriate dose of the
drug can be dissolved, a Type I formulation may well be the system of choice, in view of its
simplicity and biocompatibility.
The inclusion of a lipophilic surfactant (HLB 12) may improve the solvent capacity of the
formulation, but this approach has mainly been used to promote emulsification. Under
Page 3
7
optimum conditions it is possible to formulate a self-emulsifying drug delivery system
(SEDDS) which emulsifies in aqueous solutions under very gentle conditions of agitation, to
result in a dispersion of colloidal dimensions. If the surfactant is insufficiently hydrophilic to
dissolve and form micelles in aqueous solution, then it exists itself as a dispersed phase,
either with or separated from the oily components. This type of formulation is likely to retain
its solvent capacity for the drug after dispersion and is referred to as Type II (Table 2.3). The
distinguishing features of Type II systems are (a) efficient self-emulsification and (b) absence
of water-soluble components. Formulation of Type II SEDDS has been described in detail
elsewhere (Pouton, 1982, 1984, 1985a, b; Wakerley et al., 1986a,b, 1987; Charman et al.,
1992) and reviewed recently (Pouton, 1997). Hydrophilic surfactants (water soluble with
HLB> 12) and/ or water-soluble cosolvents have also been blended with oils to produce self-
emulsifying systems. When the surfactant content is high (for example 40%, w/w, or more)
or co-solvents are included in addition to surfactants, it is possible to produce very fine
dispersions <100 nm in diameter) under conditions of gentle agitation (Constantinides, 1995;
Constantinides and Scalart, 1997). This approach was used for the reformulation of
cyclosporin A as 'Neoral' (Vonderscher and Meinzer, 1994). Formulations which include
water-soluble components are referred to in Table 2.1 as Type III formulations, and have
been referred to as 'self microemulsifying' systems, due to the optical clarity which can be
achieved with Type III systems.
Table 2.1. Classification of lipid delivery system. (Pouton, 2000)
Components Type I Type II Type III Type IV
Triglycerides or
mixed glycerides
100 40-80 40-80 <20
Surfactants - 20-60 (HLB>11) 20-40 (HLB>11) 20-50 (HLB>11)
Hydrophilic
cosolvents
- - 0-40 20-50
Particle size of
dispersion (nm)
Coarse 100-250 100-250 50-100
Significance of
aqueous dilution
Limited
importance
Solvent capacity
unaffected
Some loss of
solvent capacity
Significant phase
changes
Significance of
digestibility
Cruical Not crucial but
likely to occur
Not crucial but
may be inhibited
Not likely to occur
Advantages GRAS status;
simple;excellent
capsule
compatibility
Unlikely to lose
solvent capacity
on dispersion
Clear or almost
clear dispersion
Formulation has
good solvent
capacity for many
drugs
Disadvantages Poor solvent
capacity unless
drug is highly
lipophilic
Turbid o/w
dispersion
Possible loss of
solvent capacity
on dispersion;
less digested
Likely loss of
solvent capacity
on dispersion;may
not be digestible
Page 4
8
2.2. Self-emulsifying drug delivery systems (SEDDS)
Self-emulsifying drug delivery systems (SEDDS) are defined as isotropic mixtures of natural
or synthetic oils, solid or liquid surfactants, or alternatively, one or more hydrophilic solvents
and co-solvents/surfactants (Wakerly et al., 1986; Craig et al., 1993; Constantinides, 1985;
Craig, 1993). Upon mild agitation followed by dilution in aqueous media, such as GI fluids,
these systems can form fine oil in water (o/w) emulsions or microemulsions (SMEDDS).
Self-emulsifying formulations spread readily in the gastro intestinal tract and the digestive
motility of the stomach and the intestine provide the agitation necessary for self-
emulsification (Shah et al., 1994; Kommuru et al., 2001).
SEDDS typically produce emulsions with a droplet size between 100 and 300 nm while
SMEDDS form transparent microemulsions with a droplet size of less than 50 nm. An
additional advantage of SEDDS over simple oily solutions is that they provide a large
interfacial area for partitioning of the drug between oil and water. Thus, for lipophilic drug
compounds that exhibit dissolution rate limited absorption, these systems may offer an
improvement in the rate and extent of absorption and result in more reproducible blood time
profiles (Charman et al., 1992) and have been shown to enhance the oral bioavailability of
lipophilic drugs (Humberstone et al., 1997; Pouton et al., 1997) such as cyclosporine (Klauser
et al., 1997), halofantrine (Khoo et al., 1998), ontazolast (Jayaraj et al., 1998), and
progesterone (Solomon et al., 1998). The ease of dispersion and the very small particle size of
the resultant colloidal microemulsion have been viewed as the principal reasons for their
utility in the delivery of lipophilic drugs. Consequently, most of the commercially available
lipid formulations are complex mixtures of lipids,surfactants, and cosolvents/cosurfactants
constructed to improve drug solubility in the formulation (and therefore increase drug pay
load) and also to maximize dispersion of the dose form on exposure of the capsule fill to the
GI contents (Porter et al., 2004).
When compared with emulsions, which are sensitive and metastable dispersed forms, SEDDS
are physically stable formulations that are easy to manufacture. Microemulsions are readily
distinguished from normal emulsions by their transperancy, their low viscosity, and more
fundamentally their thermodynamic stability. The dividing line, however, between the size of
a swollen micelle (approx. 10-140 nm) and a fine emulsion droplet (approx. 100-600 nm) is
not well defined, although microemulsions are very labile systems and a microemulsion
Page 5
9
droplet may disappear within a fraction of a second whilst another droplet forms
spontaneously elsewhere in the system. In contrast, ordinary emulsion droplets, however
small, exist as individual entities until coalescence or Ostwald ripening occurs.
2.2.1. Advantages of SEDDS
Increased stability (Gursay, et al., 2003)
SMEDDS present small particle sizes, thus avoiding the problems of large particle size in
conventional composition.
Efficient transport
The particle sizes in the aqueous dispersions of the SMEDDS are much smaller (l00 nm-250
nm) than the larger particle characteristics of vesicular and emulsion phases. This reduced
particle size enables more efficient drug transport through the intestinal aqueous boundary
layer, and through the absorption brush border membrane.
Less dependence on lipolysis (Chris de Smidt, et al., 2004)
The smaller particle size triglyceride components make SMEDDS less dependent upon
lipolysis and other factors, which affect the rate and extent of lipolysis. The reduced lipolysis
dependence further provides transport, which is less prone to suffer from any lag time
between administration and absorption caused by the lipolysis process, enabling a more rapid
onset of therapeutic action and better bioperformance characteristics.
Non-dependence on biles and meal food contents (Charman & Pouton, 1997)
Food is a major factor affecting the therapeutic performance of the drug in the body. Due to
the higher solubilization potential over bile salt micelles, the SMEDDS are less dependent on
endogenous bile and bile related patient disease states and meal fat contents. These
advantages overcome meal dependence with meal-dosage restriction.
Superior solubilization
SMEDDS enables efficient loading capacity over conventional formulations. In addition, the
particular combination of surfactant used can be optimized for a specific therapeutic agent to
more closely match the polarity distribution of the therapeutic agent, resulting in still further
enhanced solubilization.
Page 6
10
Faster dissolution and release
Dissolution being the rate limiting step for absorption of drugs, it is a major factor that limits
the bioavailability of numerous poorly water soluble drugs. Due to the robustness of
SMEDDS to dilution, the therapeutic agent remains solubilized and thus does not suffer
problem of precipitation of the therapeutic agent in the time frame relevant for absorption. In
addition, the therapeutic agent is presented in small particle carriers and is not limited in
dilution rate by entrapment in the emulsion carriers. These factors avoid liabilities associated
with the poor partitioning of lipid solubilized drug to the aqueous phase, such as large
emulsion droplet surface area, and high interfacial transfer resistance and enable rapid
completion of the critical partitioning stage.
Consistent performance
Aqueous dispersions of the SMEDDS are thermodynamically stable for the time period
relevant for absorption and can be predictably reproduced, thereby limiting variability in
bioavailability a particularly important advantage for therapeutic agents with a narrow
therapeutic index.
Efficient release
The composition of the SMEDDS are designed using components that help to keep the
therapeutic agent or absorption promoter, such as permeation enhancer, an enzyme inhibitor,
etc., solubilized for transport to the absorption sites, but readily available for absorption, thus
providing a more efficient transport and release.
Less prone to gastric emptying delays
Unlike conventional triglycerides containing formulations, which form bigger droplets on
dispersion, the SMEDDS are less prone to gastric emptying delays, resulting in faster
absorption and avoid unwanted retention in the gastro-intestinal tract.
Small size
Because of small size in aqueous dispersion, the SMEDDS allow for the faster transport of
the therapeutic agent through the aqueous boundary layer.
Page 7
11
Ease of manufacture and scale up (Patraval, et al., 2003)
Ease of manufacture and scale up are the most important advantages that make SMEDDS
unique when compared to other drug delivery systems like solid dispersions, liposomes,
nanoparticles, etc., for improving bioavailability. SMEDDS require very simple and
economical manufacturing facilities like simple mixer with agitator and volumetric liquid
filling equipment for large scale manufacturing. This explains the interest of industry in the
SMEDDS.
Self-emulsification is a phenomenon, which has been exploited commercially for many years
in formulations of emulsifiable concentrates of herbicides and pesticides. These formulations
are used to produce concentrates of crop sprays which are diluted by users, such as farmer or
household gardner, allowing very hydrophobic compounds to be transported efficiently. In
contrast SMEDDS, using excipients acceptable for oral administration to humans have not
been widely exploited. There are number of reasons why self-microemulsifying drug delivery
system (SMEDDS) is not in common use. Some relate to custom and habit of pharmaceutical
development laboratories and some to genuine reasons (Pouton, C. W., 1997);
1. Most pharmaceutical companies have a commitment to tabletting machinery, which
leads them to favor tablet formulation at early stage often before sufficient data are
available on the bioavailability of the drug. Once the path to formulation of a solid
dosage form is selected, it is difficult to revert to oily formulation at a later stage.
2. Encapsulation of an oily formulation may have to be outsourced.
3. The solvent capacity of oily formulation is not high unless the drug is very
hydrophobic/lipophilic (log P>4), so that formulation in oils is usually limited to
potent compounds.
4. There may be some suspicion that chemically the drug will be unstable in an oily
formulation than in crystalline form, and this may be so, but at present there is
insufficient data to predict chemical stability in self-micro emulsifying drug delivery
system (SMEDDS).
5. The use of high concentration of surfactant is a legitimate concern from a
toxicological standpoint.
Page 8
12
Additionally, formulators favor utilizing tried and tested materials to prevent problems
surfacing at a later stage of development of new chemical entity. The above are the issues
which will tend to direct a formulator away from SMEDDS, though it is now accepted that
there are a group of compounds for which SMEDDS, is the delivery system of choice,
exemplified by cyclosporin A. The development of this compound by Sandoz has confirmed
the potential of se1f-microemulsifying drug delivery system (SMEDDS) in bringing
hydrophobic compounds to the market place. There are several drugs which are poorly
bioavailable from marketed conventional formulation which could have been formulated in
oily systems, the potential of SMEDDS will not be realized until more human bioavailability
studies have been published and until more information is available on the chronic toxicology
of self-micro emulsifying drug delivery system (SMEDDS).
2.2.2. Applications of SEDDS (Tang et al., 2007)
Supersaturable SEDDS (S-SEDDS)
The high surfactant level typically present in SEDDS formulations can lead to GI side effects
and a new class of supersaturable formulations, including supersaturable SEDDS (S-SEDDS)
formulations, have been designed and developed to reduce the surfactant side-effects and
achieve rapid absorption of poorly soluble drugs (Poelma et al., 1991; Gao et al., 2003; Gao
et al., 2004). The S-SEDDS approach is to generate a protracted supersaturated solution of
the drug when the formulation is released from an appropriate dosage form into an aqueous
medium. Surpersaturation is intended to increase the thermodynamic activity to the drug
beyond its solubility limit and, therefore, to result in an increased driving force for transit into
and across the biological barrier (Gao et al., 2004). The S-SEDDS formulations contain a
reduced level of surfactant and a polymeric precipitation inhibitor to yield and stabilize a
drug in a temporarily supersaturated state. Hydroxypropyl methylcellulose (HPMC) and
related cellulose polymers are well recognized for their propensity to inhibit crystallization
and, thereby, generate and maintain the supersaturated state for prolonged time periods
(Pellet et al., 1994; Pellet et al., 1997; Usui et al., 1997; Yamada et al., 1999; Raghavan et al.,
2000; Lervolino et al., 2000). A supersaturable self-emulsifying drug delivery system (S-
SEDDS) of paclitaxel was developed employing HPMC as a precipitation inhibitor with a
conventional SEDDS formulation. In vitro dilution of the S-SEDDS formulation results in
formation of a microemulsion, followed by slow crystallization of paclitaxel on standing.
Page 9
13
This result indicates that the system is supersaturated with respect to crystalline paclitaxel,
and the supersaturated state is prolonged by HPMC in the formulation. In the absence of
HPMC, the SEDDS formulation undergoes rapid precipitation, yielding a low paclitaxel
solution concentration. A pharmacokinetic study showed that the paclitaxel S-SEDDS
formulation produces approximately a 10-fold higher maximum concentration (Cmax) and a 5-
fold higher oral bioavailability (F = 9.5%) compared with that of the orally administered
Taxol formulation (F = 2.0%) and the SEDDS formulation without HPMC (F =1%) (Gao et
al., 2003). It is worth emphasizing that the significantly reduced amount of surfactant used in
the S-SEDDS formulation approach provides a better toxicity/safety profile than the
conventional SEDDS formulations. However, the underlying mechanism of the inhibited
crystal growth and stabilized supersaturation by means of these polymers is poorly
understood even although several studies have been carried out to investigate this (Gao et al.,
2003; Raghavan et al., 2001; Pellet et al., 1997; Hasegawa et al., 1988).
Solid SEDDS
SEDDS are normally prepared as liquid dosage forms that can be administrated in gelatin
capsules. An alternative method is the incorporation of liquid self-emulsifying ingredients
into a powder in order to create a solid dosage form (tablets, capsules). A pellet formulation
of progesterone in SEDDS has been prepared by the process of extrusion/spheronization to
provide a good in vitro drug release (100% within 30 min, T50% at 13 min). The same dose
of progesterone (16 mg) in pellets and in the SEDDS liquid formulation resulted in similar
AUC, Cmax and Tmax values (Tuleu et al., 2004). Attama et al. used goat fat and Tween 65
admixtures to formulate self-emulsifying tablets containing diclofenac by pour moulding
using a plastic mould. The tablets showed good release profiles, as well as acceptable tablet
properties. Under mild agitation, such as occurs under gastrointestinal conditions, the release
rates are comparable with those of conventional tablets (Attama et al., 2003). Encapsulating
the emulsion lipid droplets in HPMC by spray-drying has been demonstrated to produce an
improved bioavailability following release of the lipid droplets from the powder in vivo. Tue
et al. (Hansen et al., 2005) have investigated the oral bioavailability of a directly
compressible dry emulsion as a tablet and compared it with an HPMC dry emulsion powder
and a simple lipid solution. Four female beagle dogs received a single dose of each
formulation containing the same amount of MCT and model drug, Lu 28-179. Cyclodextrin
solutions administered orally and intravenously were used as references. The absolute
Page 10
14
bioavailability decreased in the order: cyclodextrin solution (0.14) > HPMC dry emulsion
(0.11) > technically improved dry emulsion (0.10) > MCT solution (0.06). The directly
compressible dry emulsion tablets were concluded to be comparable with the HPMC dry
emulsion powder in terms of bioavailability (Woo et al., 1961).
SEDDS for TCM
Silybin, the principal component of a Carduus marianus extract, is known to be very
effective in protecting liver cells from harmful effects caused by smoking, drinking,
overworking, environmental contaminants, stress or liver damaging drugs. However, the
bioavailability of orally administered silybin is very low due to its low solubility in water.
Woo et al. discloses an oral microemulsion consisting of a Carduus marianus extract
containing a major amount of silybin, or a silybin derivative as an active ingredient. The
composition of the invention consists of miglyol 812 and ethyl linoleate as oils, HCO 50 and
Tween 20 as surfactant, dimethyl isosorbide as co-surfactant and D-tocopherol as an anti-
oxidant. The formulation provides a greatly increased level of in vivo bioavailability of
silybin, the level being atleast 4-fold higher than that achievable by conventional
formulations (You et al., 2005) Curcuma zedoaria (Berg.) Rose. (Zingiberaceae), also called
„er-zhu‟ in Chinese, has long been used as a folk medicine. The essential oil, zedoary
turmeric oil (ZTO), was extracted from the dry rhizome of C. zedoaria. A series of studies on
ZTO indicated that it exhibits potent pharmacological actions including the suppression of
tumors, antibacterial and antithrombotic activity, increased white blood cell count, and
increased gastric motility (You et al., 2005). To increase the in vivo absorption of zedoary
turmeric oil (ZTO) and develop new formulations of a water insoluble oily drug, Li
formulated SEDDS using ZTO as the oil (Li et al., 2002). Pueraria lobata is a traditional
Chinese medicinal herb. In China, its extract has been used for the treatment of hypertension,
senile ischemic cerebrovascular disease and anginapectoris. Studies of its pharmacology and
clinical application shave shown that the active constituents in the extractare isoflavones,
mainly puerarin. It is known to dilate coronary arteries, reduce myocardial oxygen
consumption and improve microcirculation in both animals and humans suffering from
cardiovascular disease (Li et al., 2002). Yufengningxin tablets are a formulation of total
isoflavones obtained from Pueraria lobata, and are available commercially in China. The
dissolution rate of Yufengningxin tablets is very low and, therefore, a SMEDDS formulation
of Pueraria lobata isoflavone was developed to improve the oral bioavailability. An optimized
Page 11
15
formulation consisted ethyl oleate, Tween 80 and transcutol P as cosurfactant. The
dissolution of SMEDDS after 10 min was more than 90%, and the dissolution of
Yufengningxin tablets at 60 min was less than 30%. The absorption of puerarin from the
SMEDDS of Puerarialobata isoflavone resulted in a 2.2-fold increase in bioavailability
compared with Yufengningxin tablets Ginkgo biloba L., the last surviving member of a
family of trees (Ginkgoaceae) that appeared more than 250 million years ago, has been
mentioned in the Chinese Materia Medica for more than 2500 years. A standardized Ginkgo
biloba extract (GBE) contains 5-7% terpene lactones (ginkgolides and bilobalide) and 22-
27% ginkgo flavonol glycosides (eg., the flavones quercetin, kaempferol, and isorhamnetin)
(Kleijnen et al., 1992). Many pharmacological and clinical studies have demonstrated that the
extracts of Ginkgo biloba possess antioxidant, antiischemic, neuro protective, cardiovascular
and cerebrovascular activities, and have beneficial effects oncognitive deficits, including
Alzheimer‟s type and multi infarct dementia, as well as peripheral vascular disease
(Kressmann et al., 2002). The dissolution and bioavailability of the active components from
the oral solid preparations of different Ginkgo biloba brands were obviously different and
irreproducible, due to the lower solubility of the active components (Kressmann et al., 2002).
The SEDDS formulation of GBE was accordingly developed to increase the dissolution rate
and thus improve oral absorption and acquire the reproducible blood-time profiles of the
active components of GBE. The prepared SEDDS was compared with the conventional GBE
tablets following administration to fasted dogs. The active components of GBE, terpene
lactones, were determined using liquid chromatography with electrospray ionization mass
spectrometric detection (Tang et al., 2006). The relative bioavailability of SEDDS for
ginkgolide was 154%, compared with the reference tablets.
2.2.3. Formulation of SEDDS (Patel et al., 2010)
SEDDS formulation containing following components
1) Oil phase
2) Surfactant
3) Co-surfactant/Co-Solvent
Self-emulsification has been shown to be specific to the nature of the oil/surfactant pair, the
surfactant concentration and oil/surfactant ratio and the temperature at which self-
Page 12
16
emulsification occurs (Wakerly et al., 1986; Wakerly et al., 1987; Pouton et al., 1985). In
support of these facts, it has also been demonstrated that only very specific pharmaceutical
excipient combinations could lead to efficient self-emulsifying systems (Charman et al.,
1992; Shah et al., 1994; Chanana et al., 1995; Kimura et al., 1994; Hauss et al., 1998; Karim
et al., 1994).
Oils
The oil represents one of the most important excipients in the SEDDS formulation not only
because it can solubilise marked amounts of the lipophilic drug or facilitate self-
emulsification but also and mainly because it can increase the fraction of lipophilic drug
transported via the intestinal lymphatic system, thereby increasing absorption from the GI
tract depending on the molecular nature of the triglyceride (Gershanik et al., 2000; Lindmark
et al., 1995; Charman et al., 1991; Holm et al., 2002). Both long and medium chain
triglyceride oils with different degrees of saturation have been used for the design of self-
emulsifying formulations. Furthermore, edible oils which could represent the logical and
preferred lipid excipient choice for the development of SEDDS are not frequently selected
due to their poor ability to dissolve large amounts of lipophilic drugs. Modified or hydrolyzed
vegetable oils havebeen widely used since these excipients form good emulsification systems
with a large number of surfactants approved for oral administration and exhibit better drug
solubility properties (Constantinides et al., 1995; Kimura et al., 1994; Hauss et al., 1998).
They offer formulative and physiological advantages and their degradation products resemble
the natural end products of intestinal digestion. Novel semisynthetic medium chain
derivatives, which can be defined as amphiphilic compounds with surfactant properties, are
progressively and effectively replacing the regular medium chain triglyceride oils in the
SEOFs (Constantinides et al., 1995; Karim et al., 1994).
Surfactants
Several compounds exhibiting surfactant properties may be employed for the design of self-
emulsifying systems, the most widely recommended ones being the non-ionic surfactants
with a relatively high hydrophilic−lipophilic balance (HLB). The commonly used emulsifiers
are various solid or liquid ethoxylated polyglycolyzed glycerides and polyoxyethylene 20
oleate (Tween 80). Safety is a major determining factor in choosing a surfactant. Emulsifiers
of natural origin are preferred since they are considered to be safer than the synthetic
Page 13
17
surfactants (Constantinides et al., 1995; Hauss et al., 199; Yuasa et al., 1994; Georgakopoulos
et al., 1992). However, these excipients have a limited self-emulsification capacity. Non-ionic
surfactants are less toxic than ionic surfactants but they may lead to reversible changes in the
permeability of the intestinal lumen (Swenson et al., 1994; Wakerly et al., 1986). Usually the
surfactant concentration ranges between 30 and 60% w/w in order to form stable SEDDS. It
is very important to determine the surfactant concentration properly as large amounts of
surfactants may cause GI irritation.The surfactant involved in the formulation of SEDDS
should have a relatively high HLB and hydrophilicity so that immediate formation of o/w
droplets and/or rapid spreading of the formulation in the aqueous media (good self-
emulsifying performance) can be achieved. For an effective absorption, the precipitation of
the drug compound within the GI lumen should be prevented and the drug should be kept
solubilized for a prolonged period of time at the site of absorption (Serajuddin et al., 1988;
Shah et al., 1994). Surfactants are amphiphilic in nature and they can dissolve or solubilize
relatively high amounts of hydrophobic drug compounds. The lipid mixtures with higher
surfactant and co-surfactant/oil ratios lead to the formation of SMEDDS (Constantinides et
al., 1995; Karim et al., 1994; Meinzer et al., 1995; Vondercher et al., 1994). There is a
relationship between the droplet size and the concentration of the surfactant being used. In
some cases, increasing the surfactant concentration could lead to droplets with smaller mean
droplet size such as in the case of a mixture of saturated C8-C10 polyglycolized glycerides
(Labrafac CM-10). This could be explained by the stabilization of the oil droplets as a result
of the localization of the surfactant molecules at the oil-water interface (Levy et al., 1990).
On the other hand, in some cases the mean droplet size may increase with increasing
surfactant concentrations (Wakerly et al., 1987; Kommuru et al., 2001; Craig et al., 1995).
This phenomenon could be attributed to the interfacial disruption elicited by enhanced water
penetration into the oil droplets mediated by the increased surfactant concentration and
leading to ejection of oil droplets into the aqueous phase (Pouton et al., 1997).
Co-surfactants
Role of co-surfactant together with surfactant is to lower the interfacial tension to a very
small, even transient, negative value. At this value the interface would expand to form fine
dispersed droplets, and subsequently adsorb more surfactant and surfactant/co-surfactant until
their bulk condition is depleted enough to make interfacial tension positive again. This
process is known as “spontaneous emulsification” forms the microemulsion. However, the
Page 14
18
use of co-surfactant is crucial not only to formation of microemulsion, but also to
solubilisation in microemulsion. Other variables such as the chemical nature of oil, salinity,
and temperature are also expected to influence the curvature of the interfacial film.
Co-solvents
The production of an optimum SEDDS requires relatively high concentrations (generally
more than 30% w/w) of surfactants. Organic solvents such as, ethanol, propyleneglycol (PG),
and polyethylene glycol (PEG) are suitable for oral delivery, and they enable the dissolution
of large quantities of either the hydrophilic surfactant or the drug in the lipid base. These
solvents can even act as co-surfactants in microemulsion systems. On the other hand,
alcohols and other volatile co-solvents have the disadvantage of evaporating into the shells of
the soft gelatin, or hard, sealed gelatin capsules in conventional SEDDS leading to drug
precipitation. Thus, alcohol free formulations have been designed (Constantinides et al.,
1995), but their lipophilic drug dissolution ability may be limited.
2.2.4. Mechanism of self-emulsification (Gursoy et al., 2004)
Self-emulsification takes place when the entropy change favouring dispersion is greater than
the energy required to increase the surface area of the dispersion (Reiss et al., 1975). The free
energy of a conventional emulsion formulation is a direct function of the energy required to
create a new surface between the oil and water phases. The two phases of the emulsion tend
to separate with time to reduce the interfacial area and thus the free energy of the systems.
The conventional emulsifying agents stabilize emulsions resulting from aqueous dilution by
forming a monolayer around the emulsion droplets, reducing the interfacial energy and
forming a barrier to coalescence. On the other hand, emulsification occurs spontaneously
with SEDDS because the free energy required to form the emulsion is both low and positive
or negative (Constantinides et al., 1995). It is necessary for the interfacial structure to show
no resistance against surface shearing in order for emulsification to take place (Dabros et al.,
1999). The ease of emulsification was suggested to be related to the ease of water penetration
into the various LC or gel phases formed on the surface of the droplet (Wakerly et al., 1986;
Groves et al., 1974; Rang et al., 1999). The interface between the oil and aqueous continuous
phases is formed upon addition of a binary mixture (oil/non-ionic surfactant) to water
(Wakerly et al., 1986). This is followed by the solubilisation of water within the oil phase as a
result of aqueous penetration through the interface. This will occur until the solubilisation
Page 15
19
limit is reached close to the interphase. Further aqueous penetration will lead to the formation
of the dispersed LC phase. In the end, everything that is in close proximity with the interface
will be LC, the actual amount of which depends on the surfactant concentration in the binary
mixture. Thus, following gentle agitation of the self-emulsifying system, water will rapidly
penetrate into the aqueous cores and lead to interface disruption and droplet formation. As a
consequence of the LC interface formation surrounding the oil droplets, SEDDS become very
stable to coalescence. Detailed studies have been carried out to determine the involvement of
the LC phase in the emulsion formation process (Wakerly et al., 1986; Rang et al., 1999;
Pouton et al., 1987; Wakerly et al., 1987). Also, particle size analysis and low frequency
dielectric spectroscopy (LFDS) were utilized to examine the self-emulsifying properties of a
series of Imwitor 742 (a mixture of mono and diglycerides of capric and caprylic
acids)/Tween 80 systems (Craig et al., 1993; Craig et al. 1995). The results suggested that
there might be a complex relationship between LC formation and emulsion formation (Craig
et al. 1995). Moreover, the presence of the drug compound may alter the emulsion
characteristics, probably by interacting with the LC phase (Craig et al., 1993). Nevertheless,
the correlation between the LC formation and spontaneous emulsification has still not been
established (Craig et al., 1993; Gursoy et al., 2003).
2.2.5. Biopharmaceutical aspects (Tang et al., 2007)
The ability of lipids and/or food to enhance the bioavailability of poorly water soluble drugs
has been comprehensively reviewed and the interested reader is directed to these references
for further details (Humberstone et al., 1997; Charman et al., 1997). Although incompletely
understood, the currently accepted view is that lipids may enhance bioavailability via a
number of potential mechanisms, including (Porter et al., 2001).
a) Alterations (reduction) in gastric transit, thereby slowing delivery to the absorption
site and increasing the time available for dissolution (Porter et al., 2001).
b) Increases in effective luminal drug solubility. The presence of lipids in the GI tract
stimulates an increase in the secretion of bile salts (BS) and endogenous biliary lipids
including phospholipid (PL) and cholesterol (CH), leading to the formation of
BS/PL/CH intestinal mixed micelles and an increase in the solubilisation capacity of
the GI tract. However, intercalation of administered (exogenous) lipids into these BS
structures either directly (if sufficiently polar), or secondary to digestion, leads to
Page 16
20
swelling of the micellar structures and a further increase in solubilisation capacity
(Porter et al., 2001).
c) Stimulation of intestinal lymphatic transport. For highly lipophilic drugs, lipids may
enhance the extent of lymphatic transport and increase bioavailability directly, or
indirectly via reduction in first pass metabolism (Porter et al., 1997; Porter et al.,
2001; Muranishi et al., 1991).
d) Changes in the biochemical barrier function of the GI tract. It is clear that certain
lipids and surfactants may attenuate the activity of intestinal efflux transporters, as
indicated by the p-glycoprotein efflux pump, and may also reduce the extent of
enterocyte based metabolism (Benet at al., 2001; Dintaman et al., 1999; Nerurkar et
al., 1996).
e) Changes in the physical barrier function of the GI tract. Various combinations of
lipids, lipid digestion products and surfactants have been shown to have permeability
enhancing properties (Aungst et al., 2000; Muranishi et al., 1990). For the most part,
however, passive intestinal permeability is not thought to be a major barrier to the
bioavailability of the majority of poorly water soluble, and in particular, lipophilic
drugs.
Enhanced drug absorption by lymphatic delivery
Charman et al. proposed that drug candidates for lymphatic transport should have a log P >5
and, in addition, a triglyceride solubility >50 mg/ml. The importance of lipid solubility was
illustrated by comparing the lymphatic transport of DDT (log P 6.19) with
hexachlorobenzene (HCB, log P 6.53).
Khoo et al. showed significant lymphatic transport of the poorly lipid soluble (1 mg/ml) HCl
salt of halofantrine (Hf-HCl), following oral post prandial administration to dogs. The authors
suggest that the high level of lymphatic transport of Hf-HCl (43.7% of dose), which was
similar to that of the lipid soluble Hf base, was due to conversion of Hf-HCl in the intestinal
lumen, during lipolysis, to the more lipophilic free base, which then becomes associated with
chylomicron production (Khoo et al., 1999)
Page 17
21
Although enhanced lymphatic transport has been suggested as a potential mechanism of
enhanced bioavailability, few studies have investigated the lymphotropic potential of
SEDDS.
Haus et al. investigated the effects of a range of lipid based formulations on the
bioavailability and lymphatic transport of ontazolast, following oral administration to
conscious rats. This drug undergoes extensive hepatic first pass metabolism and it has
solubility in soybean oil of 55 mg/ml, and a log P of 4. The formulations of ontazolast
investigated included a suspension (lipid free control), a 20% soyabean o/w emulsion, two
SEDDS containing Gelucire 44/14 and Peceol in the ratios 50:50 and 80:20, respectively, and
a solution of the drug in Peceol alone. All the lipid formulations increased the bioavailability
of ontazolast relative to the control suspension, while the SEDDS promoted more rapid
absorption.
The Effect of excipients on efflux transport
Drug efflux mediated by broad specificity xenobiotic transporters present in the intestinal
epithelium may be an important factor in the poor or variable absorption of orally
administered drugs (Makhey et al., 1998).
Lo et al. have shown that bile salts, fatty acids, phospholipids, and surfactants were potent
absorption enhancers and efflux reducing agents in Caco-2 cells and the rat intestine (Lo et
al., 1998; Lo et al., 2000; Lo et al., 2000).
Other researchers also investigated the non-ionic surfactants, such as Tween 80, Pluronic
P85, and Cremophor EL in vitro and in vivo in animals and in humans for their potential
ability to reverse MDR caused by p-glycoprotein (P-gp) and multidrug resistance-associated
proteins (MRP) (Batrakova et al., 1999; Yamazaki et al., 2000).
Recently, Cremophor, Tween 80, and Solutol HS-15 have been proven to reverse the MDR
phenotype in cultured cells at concentrations likely to be achieved clinically (Yamazaki et al.,
2000; Dudeja et al., 1995)
TPGS (d-alpha-tocopheryl polyethylene glycol 1000 succinate) has been shown to be an
effective inhibitor of P-gp mediated drug resistance and has been used to enhance the
bioavailability of CsA in liver transplant patients as well as significantly improving
Page 18
22
absorption and reducing the daily drug cost (Dintaman et al., 1999). Inhibition of MDR-
related pumps by various excipients has been proposed to occur due to binding competition,
ATP depletion, and membrane perturbation (Yamazaki et al., 2000; Friche et al., 1990). For
example, Tween 80 has been shown to modulate anthracycline and vinca alkaloid resistance
in MDR cells by inhibiting the binding of these drugs to P-gp (Yamazaki et al., 2000; Friche
et al., 1990). The ability of Pluronic copolymer, one poly (ethylene oxide) block copolymer,
to antagonize P-gp and sensitize MDR cells appears to be a result of ATP depletion, and
inhibition of P-gp and MRP drug efflux proteins (Kabanov et al., 2002). Studies with MDR
modifiers such as bile salts indicated that perturbations of the cell membrane structure may
influence P-gp-mediated drug transport (Lo et al., 1998; Schuldes et al., 2001; Dolderer et al.,
2000). These modifiers may influence cytotoxic drug action by producing structural changes
to the lipid domains in the plasma membrane. The membrane perturbation caused by
pharmaceutical excipients, such as Tween 20, Tween 80, Brij 30, and Myrj 52, may result in
a change in the fluidity of Caco-2 cell membranes, and thus inhibit the activity of membrane
spanning proteins, such as P-gp and MRPs which substantially reduce the basolateral to
apical efflux of epirubicin across Caco-2 monolayers (Lo et al., 2003). Tween 20, Tween 80,
Brij 30, and Myrj 52 may also inhibit protein kinase C (PKC) activity, reduce
phosphorylation of P-gp, and modulate P-gp mediated drug efflux (Komarov et al., 1996).
Inhibition of the efflux and/or enterocyte based metabolism will increase the concentration
and residence time of the intact drug in the cell. This may result in increased drug available
for partitioning into the lymphatics (O‟Driscoll et al., 2002).
Role of lipolysis
Digestion of dietary triglyceride in the small intestine is very rapid, and many other non-ionic
esters, such as mixed glycerides and surfactants, will be substrates for pancreatic lipase
(Embleton et al., 1997). Digestion of formulations will inevitably have a profound effect on
the state of dispersion of the lipid formulation, and the fate of the drug (Macgregor et al.,
1997). Fortunately, the liberation of free fatty acid during lipolysis can be titrated using
NaOH in a pH stat, allowing quantitative data about the kinetics of digestion to be obtained.
The location of the drug can be assayed in various fractions after ultracentrifugation of the
products of digestion, which allows investigation of the likely fate of the drug after lipolysis
(Pouton et al., 2000). The inclusion of highly lipophilic compounds in SEDDS is often
reported to result in strongly enhanced oral absorption although it is still controversial
Page 19
23
whether further lipolysis of the dispersed lipid material is required for final transfer to the
enterocyte membranes.
In order to assess the relative roles of lipid vehicle dispersion and vehicle digestibility in the
oral absorption of penclomedine (Pcm), a series of formulations of Pcm in medium chain
triglyceride (MCT)/TPGS was developed having three sizes (160 nm, 720 nm, and mm sized
(crude oil); with or without the inclusion of tetrahydrolipstatin (THL), a known lipase
inhibitor. Oral absorption of Pcm was studied after administration of small volumes of these
formulations to conscious rats. Formulations with a particle size of 160 nm had the highest
relative bioavailability (set at F = 1), whereas administration in particle 720 nm in size
resulted in a slightly lower bioavailability (F = 0.79). Co-inclusion of THL yielded similar
bioavailability for these two SEDDS. Crude oil formulations had an F= 0.62 (without THL)
and 0.25 (with THL).
Positively charged SEDDS
Many physiological studies have proved that the apical potential of absorptive cells, as well
as that of all other cells in the body, is negatively charged with respect to the mucosal
solution in the lumen (Corbo et al., 1990; Rojanasakul et al., 1992). A novel SEDDS, which
results in positively charged dispersed oil droplets upon dilution with an aqueous phase,
showed an increase in the oral bioavailability of progesterone in young female rats
(Gershanik et al., 1996). More recently, it has been shown that the enhanced electrostatic
interactions of positively charged droplets with the mucosal surface of the everted rat
intestine are mainly responsible for the preferential uptake of the model drug cyclosporine A
(CsA) from positively charged droplets (Gershanik et al., 1998). The Caco-2 cell model was
used for the investigation of the charge dependent interactions of the SEDDS with human
intestinal epithelial cells. The positively charged emulsions affected the barrier properties of
the cell monolayer at high concentrations and reduced the cell viability. However, at the
dilution with aqueous phase used in the study (1:2000), the positively charged SEDDS did
not produce any detectable cytotoxic effect. The binding of the fluorescent dye DiIC18(3)
was much higher from the positively charged SEDDS, compared with the negatively charged
formulation, suggesting increased adhesion of the droplets to the cell surface due to
electrostatic attraction (Gershanik et al., 2000).
Page 20
24
Table 2.2. List of SEDDS of various drugs.
Compound Formulation Excipients Observations
Simvastatin (Kang et
al., 2004)
SMEDDS 37% Capryol 90, 28%
Cremophor EL, 28% Carbitol
BA 1.5 fold higher
from SMEDDS
Indomethacin (Kim
et al., 2000)
SEDDS 70% ethyl oleate, 30%
Tween 85
BA significantly
increased from
SEDDS
Progesterone (Tuleu
et al., 2004)
SEDDS Mono-di-glycerides:
polysorbate 80, 50/50 w/w
BA 9 fold higher
from SEDDS
Danazol (Porter et
al., 2004)
SMEDDS Medium chain lipids,
Cremophor EL, ethanol
BA 6 fold higher
from SMEDDS
Carvedilol (Wei et
al., 2005)
SEDDS Labrafil M1944CS, Tween
80, Transcutol
BA 4 fold higher
from SEDDS
Cyclosporin (Trull et
al., 1994)
SEDDS Corn oil, ethanol Increased BA and
Cmax from SEDDS
Vitamin E (Julianto
et al., 2000)
SEDDS Tween 80: Span 80: Palm oil
in 4:2:4
BA 3 fold higher
from SEDDS
Coenzyme Q10
(Kommuru et al.,
2001)
SMEDDS 40% Myvacet, 90%
Labrasol, 10% Lauroglycol
BA 2 fold higher
from SEDDS
Silymarin (Wu et al.,
2006)
SMEDDS Tween 80,ethyl alcohol,
ethyl linoleate, PEG 400
BA 2 fold higher
from SMEDDS
Win 54954 (Charman
et al., 1992)
SEDDS 40% Neobee M5, 25% Tagat
To
Increased Cmax, No
difference in BA
Halofantrine (Khoo
et al., 1998)
SEDDS 47% Captex 355, 23%
Capmul MCM, 15%
Cremophor, Ethanol
High BA
Itraconazole (Hong et
al., 2006)
SEDDS Transcutol, pluronic L64,
tocopherol acetate
Increased BA and
reduced Food effect
Atorvastatin (Shen et
al., 2006)
SMEDDS Cremophor RH 40,
propylene glycol, Labrafil,
Labrafac
BA increased
significantly
Atovaquone (Sek et
al., 2006)
SMEDDS LC lipids, ethanol,
cremophor EL
BA 3 fold higher
from SMEDDS
PNU 91325 (Gao et
al., 2004)
SEDDS 6% LC lipids, 30%
Cremophor, 18% Pluronic
L44, 9% PEG, 5% DMA,
20% HPMC
BA 5-6 fold increase
from SEDDS
Page 21
25
2.2.6. Characterization of SEDDS (Patel et al., 2010)
Differential scanning calorimetry
Differential scanning calorimetry for SMEDDS can be determined using DSC 60. Liquid
sample and Solid sample should be placed in the aluminum pan and result can be recorded.
Any type of chemical interaction should be determined using DSC (Taha et al., 2004)
Fourier transform-infrared spectroscopy
Fourier transform-infrared for SMEDDS can be determined using FT-IR. Liquid sample
should be placed in the liquid sample holder and result can be recorded. Any type of chemical
interaction should be determined using FT-IR (Nazzal et al., 2002).
Macroscopic evaluation
Macroscopic analysis is carried out in order to observe the homogeneity of microemulsion
formulations. Any change in color and transparency or phase separation occurred during
normal storage condition (37±2 ºC) was observed in optimized microemulsion formulation.
Visual assessment
To assess the self-emulsification properties, formulation is introduced into 100 ml of water in
a glass Erlenmeyer flask at 25 °C and the contents are gently stirred manually. The tendency
to spontaneously form a transparent emulsion is judged as good and it is judged bad when
there was poor or no emulsion formation. Phase diagrams are constructed identifying the
good self-emulsifying region (Shen et al., 2006).
Determination of self-emulsification time
The emulsification time of SMEDDS is determined according to USP XXII dissolution
apparatus. Each formulation is added drop wise to purified water at 37 ºC. Gentle agitation
can be provided by a standard stainless steel dissolution paddle rotating at 50 rpm.
Emulsification time is assessed visually (Wei et al., 2005).
Solubility studies
Page 22
26
Unknown amount of selected vehicles are added to each cap vial containing an excess of
drug. After sealing, the mixtures are heated at 40 ºC in a water bath to facilitate the
solubilization. Mixing of the systems is performed using a vortex mixer. Formed suspensions
are then shaken with a shaker at 25 ºC for 48 h. After reaching equilibrium, each vial is
centrifuged at 3000 rpm for 5 minutes, and excess insoluble LOV is discarded by filtration
using a membrane filter (0.45 μm, 13 mm, Whatman, India). The concentration of drug is
then quantified (Shen et al., 2006).
Transmittance test
Stability of optimized microemulsion formulation with respect to dilution is checked by
measuring Transmittance through U.V. Spectrophotometer. Transmittances of samples are
measured at 650 nm and for each sample three replicate assays are performed (Shen et al.,
2006).
Droplet size determination
It is a precise method for evaluation of stability. Size of droplet is measured by photon-
correlation spectroscopy (PSC) with Zetasizer. All measurements are carried out at scattering
angle of 90 ° and 25 °C temperatures. Prior to measurement, microemulsion is diluted in two
steps with pure water then it is filtered through a 0.22 µm filter just before it is added to
cuvette. In first step it is diluted with equal amount of water. In second step the mixture is
further diluted to appropriate concentration for the measurement. That depends on droplet
size (Usually diluted 100-200 times) (Zhang et al., 2007).
Zeta potential measurement
Zeta potential for microemulsion is determined using Zetasizer. Samples are placed in clear
disposable zeta cells and results are recorded. Before putting the fresh sample, cuvettes are
washed with the methanol and rinsed using the sample to be measured before each
experiment (Wei et al., 2004).
Page 23
27
Stability
Temperature
Shelf life as a function of time and storage temperature is evaluated by visual inspection of
the SMEDDS system at different time period. SMEDDS are diluted with purified distilled
water and to check the temperature stability of samples, they are kept at three different
temperature range 2-8 °C (refrigerator), room temperature and observed for any evidences of
phase separation, flocculation or precipitation.
Centrifugation
In order to estimate metastable systems, the optimized SMEDDS formulations are diluted
with purified distilled water. Then microemulsion is centrifuged at 1000 rpm for 15 minute at
0 °C and is observed for any change in homogeneity of microemulsions.
In vitro release
The quantitative in vitro release test is performed in purified distilled water/dissolution
medium, which is based on USP 24 method. SMEDDS are placed in dialysis bag during the
release period to compare the release profile with conventional dosage form. Sample
solutions are withdrawn at predetermined time intervals, filtered through a 0.45 μ membrane
filter, dilute suitably and analyzed spectrophotometrically. Equal amount of fresh dissolution
medium is replaced immediately after withdrawal of the test sample. Percent drug dissolved
at different time intervals is calculated using the Beer Lambert‟s equation (Kang et al., 2004)
2.3. Diabetes mellitus
Diabetes mellitus is a chronic metabolic disorder characterized by a high blood glucose
concentration (hyperglycemia) caused by insulin deficiency, often combined with insulin
resistance. Hyperglycemia occurs because of uncontrolled hepatic glucose output and
reduced uptake of glucose by skeletal muscle with reduced glycogen synthesis. When the
renal threshold for glucose reabsorption is exceeded, glucose spills over into the urine
(glycosurea) and causes an osmosis diuresis (polyurea), which in tum results in dehydration,
thirst and increased drinking (polydipsia). Depending on the etiology of the diabetes mellitus,
factors contributing to hyperglycemia may include reduced insulin secretion, decreased
Page 24
28
glucose usage and increased glucose production (Davis and Granner, 1996; Nolte and Karam,
2001; Rang et al., 2003).
2.3.1. Complications of diabetes mellitus
Diabetes mellitus leads to various acute and chronic complications. Diabetic ketoacidosis and
nonketotic hyperosmolar state are acute complications of diabetes. Both are associated with
potentially serious complications if not promptly diagnosed and treated. Chronic
complications can be divided into vascular and nonvascular complications. The vascular
complications are further subdivided into microvascular that include eye disease (retinopathy,
macular edema, cataracts and glaucoma), neuropathy (sensory and motor and autonomic) and
nephropathy and macrovascular that include coronary artery disease, peripheral vascular
disease, and cerebrovascular disease and dennatologic complications. Nonvascular
complications include problems such as gastroparesis, sexual dysfunction and skin changes
(Powers et al., 2001).
2.3.2. Classification of diabetes mellitus
Several distinct types of diabetes mellitus exist and are caused by a complex interaction of
genetics, environmental factors and lifestyle choices (Table 2.3). However, there are two
major types of diabetes mellitus. Type 1 diabetes (Insulin Dependent Diabetes Mellitus;
IDDM) is a severe form where there is an absolute deficiency of insulin resulting from
autoimmune destruction of P cells. If untreated, it will ultimately lead to ketosis and death.
The incidence of each type of diabetes mellitus varies widely throughout the world. In the
United States, about 10% of all diabetic patients have IDDM with an incidence of 18 per
100,000 inhabitants per year (Davis and Granner, 1996; Powers, 2001). Type 2 (Non Insulin
Dependent Diabetes Mellitus; NIDDM) diabetes is accompanied both by insulin resistance
and by impaired insulin secretion. Such patients are often obese and usually present in adult
life; the incidence rising progressively with age as P cell function declines. In this case,
insulin secretion may appear normal or even excessive but it is insufficient to compensate for
insulin resistance. Circulatory endogenous insulin is sufficient to prevent ketoacidosis but is
often subnormal or relatively inadequate because of tissue insensitivity. NIDDM is far more
common than IDDM, accounts for 80-90% of all cases of diabetes. The incidence rates of
NIDDM increase with age, with a mean rate of about 440 per 100,000 per year by the sixth
decade in males in the United States. A few patients, perhaps 5 percent, who appear to have
Page 25
29
NIDDM may actually have a slowly progressive form of IDDM, and eventually become
dependent on insulin. NIDDM further subdivided into non-obese and obese types; the latter is
more common (incidence, approximately 80% of NIDDM patients) (Davis and Granner,
1996; Powers, 2001).
Table 2.3. Different forms of diabetes mellitus (Davis and Granner, 1996)
General
Insulin dependent diabetes mellitus (IDDM, type I)
Non Insulin Dependent Diabetes Mellitus (NIDDM, type II)
Gestational diabetes mellitus
Specific Maturity onset diabetes of youth (MODY, glucokinase gene mutations)
Mutations of the insulin receptor (including leprechunism)
Insulin gene mutations
Tropical diabetes Diabetes secondary to pancreatic disease or surgery
Diabetes associated with genetic syndromes, e.g., Prader-Willi syndrome
Diabetes secondary to endocrinopathies
2.3.3. Drugs used for the treatment of diabetes mellitus
a. Insulin: Insulin is the mainstay for treatment of virtually all IDDM and many NIDDM
patients. Different insulin preparations available depending on their duration of action are:
Ultra-short acting (Insulin Lispro), Short acting (Regular insulin and Prompt insulin zinc
suspension i.e., Semilente), Intermediate acting (Isophane insulin suspension and Insulin zinc
suspension i.e., Lente) and Long acting (Protamine zinc insulin suspension and Extended
insulin zinc suspension i.e., Ultralente) (Davis and Granner, 1996; Nichols, 2000).
b. Oral hypoglycemic agents: Most patients with NIDDM can be treated without insulin
(Gerich, 1989; Davis and Granner, 1996). The oral antidiabetic agents have been proved
efficacious in NIDDM. They can be classified according to their predominant mechanism of
action (Reynolds, 1997; Nichols, 2000; Satoskar et a1., 2001).
1. Stimulators of beta cells, e.g., sulfonylureas, repaglinide
2. Inhibitors of gluconeogenesis, e.g., biguanides
3. Inhibitors of intestinal a gIucosidases, e.g., acarbose, meglitol
4. Drugs which reduce insulin resistance, e.g., glitazones
5. Aldose reductase inhibitors, e.g., epalrestat, sorbinil
Page 26
30
Sulfonyl ureas
Among the oral hypoglycemic agents listed above, sulfonylureas are an important class
currently available for treating hyperglycemia in NIDDM. They are sulfonamide derivatives
but do not possess antibacterial activity. All members of this class are substituted
arylsulfonylureas. They differ at the para position on the benzene ring and at one nitrogen
residue of the urea moiety. They increase the release of endogenous insulin as well as its
peripheral effectiveness (Davis and Granner, 1996; Nolte and Karam, 2001). The
sulfonylureas are divided traditionally into first and second groups or generations of agents
(Table 2.4) (Reynolds, 1997).
Table 2.4. Different sulfonylurea drugs.
First-generation sulfonylureas Second-generation sulfonylureas
Clorpropamide, tolbutamide, tolazamide,
acetohexamide, carbutamide
Glibenclamide (glyburide), glimipiride,
gliclazide, glipizide , glibunuride,
gliquidone, glisentide, glisolamide,
glisoxepide, glyclopyramide,
glycyclamide
The hypoglycemic activity of synthetic sulphur containing compounds had been known for
some time when researchers found during studies of the antibacterial potency of p-amino-
sulfonamide-isopropyl-thiodiazole in patients with typhoid fever that it could cause severe
hypoglycemia (Gerich, 1989). By the mid-1960s, four compounds- tolbutamide,
acetohexamide, tolazamide and chlorpropamide were in widespread clinical use for the
treatment of NIDDM. In the late 1970s the use of sulfonylureas revived and in 1984 both
glibenclamide and glipizide were approved for use in United States. Both the number and
percentage of the patients whose diabetes is treated with sulfonylureas is increasing. Three
agents, chlorpropamide, glibenclamide and glipizide, account for about 75% of the market
(Gerich, 1989). Some patients who do not respond initially to tolbutamide, acetohexamide or
tolazamide may respond to glibenclamide, glipizide or chlorpropamide. Glibenclamide also
offer a wider dose range as compared to other sulfonylureas. It also offers the theoretical
advantage of fewer pharmacological interactions because of their nonionic binding to plasma
proteins (Gerich, 1989). In comparison to Glibenclamide, gliclazide is comparatively newer
sulfonylurea and has good efficacy, appears of particular benefit in patients previously
untreated with oral antidiabetic drugs and is generally well tolerated. In the long term, it
reduces hepatic gluconeogenesis, and increases insulin effects by acting at receptor or post
Page 27
31
receptor sites. It also inhibits platelet aggregation and increases fibrinolysis. Because of these
therapeutic effects it has emerged as one of the important and promising drug substances for
diabetes mellitus, especially due to the noticeable improvement of survival rates in patients
with chronic increase in glucose level.
The principal action of glibenclamide and gliclazide is on beta cells, stimulating insulin
secretion from pancreatic cells and thus reducing plasma glucose. High affinity receptors for
sulfonylureas are present on the KATP channels in beta cell plasma membranes and the
binding of various sulfonylureas parallels their potency in stimulating insulin release. The
drugs reduce the K+ permeability of the beta cells by blocking KATP channels causing
depolarization, Ca2+ entry and insulin secretion. These drugs may also inhibit the release of
glucagon in vitro. Reduced plasma glucagon levels have been found in patients with NIDDM
after the long term administration of sulfonylureas in many but not all. They also may further
increase insulin levels by reducing hepatic clearance of the hormone (Gerich, 1989; Davis
and Granner, 1996; Rang et al., 2003). These agents have the potential to cause profound and
persistent hypoglycemia. Hypoglycemia is the major risk that must be weighed against any
benefits of these drugs. It is usually related to delayed meals, increased physical activity,
alcohol intake or renal insufficiency. Most sulfonylureas are metabolized in the liver to
compounds that are cleared by the kidney. Thus their use in individuals with significant
hepatic or renal dysfunction is not advisable. Weight gain, a common side effect of
sulfonylurea therapy, results from the increased insulin levels and improvement in glycemic
control. Gastrointestinal (GI) disturbances such as nausea, vomiting, heartburn, anorexia,
diarrhea and a metallic taste may occur. Skin rashes and pruritus may take place and
photosensitivity has been reported. Other severe side effects may be manifestations of a
hypersensitivity reaction. They include leucopenia, thrombocytopenia, agranulocytosis,
hemolytic anemia and cholestatic jaundice (Gerich, 1989; Davis and Granner, 1996; Satoskar
et al., 200 1). Since glibenclamide and glipizide are usually intended to be taken for a longer
period, patient compliance is also very important (Takahshi et al., 1997). In addition, they
require more than once a day dosing because of the shorter half-life, which adversely, affects
the patient compliance (Gerich, 1989).
Diabetes mellitus, long considered a disease of minor significance to world health, is now
taking its place as one of the main threats to human health in the 21st century (Zimmet et al.,
2000). The past two decades have seen an explosive increase in the number of people
Page 28
32
diagnosed with diabetes worldwide (Amos et al., 1997; King et al., 1998). Diabetes mellitus
has reached epidemic proportions and affects more than 170 million individuals worldwide.
Global estimates for the year 2010 predict a further growth of almost 50%, with the greatest
increases in the developing countries of Africa, Asia, and South America (Zimmet et al.,
2001). In more developed societies, the prevalence of diabetes mellitus has reached about 6%
(King et al., 1998) and, even more alarmingly, among obese white adolescents 4% had
diabetes and 25% had abnormal glucose tolerance (Sinha et al., 2002). Some 90% of diabetic
individuals have type 2 (non-insulin-dependent) diabetes mellitus, and within this category no
more than 10% can be accounted for by monogenic forms such as maturity onset diabetes of
the young (Fajans et al., 2001) and mitochondrial diabetes (Maassen et al., 2004) or late onset
autoimmune diabetes of the adult, which is actually a late-onset type 1 diabetes (Pozzilli et
al., 2001). Thus, most diabetes in the world is accounted for by “common” type 2 diabetes,
which has a multifactorial pathogenesis caused by alterations in several gene products. The
medical and socioeconomic burden of the disease is caused by the associated complications
(Wei et al., 1998; UKPDS., 1998) which impose enormous strains on healthcare systems. The
incremental costs of patients with type 2 diabetes arise not only when the diagnosis is
established but at least 8 years earlier (Nichols et al., 2000). The devastating complications of
diabetes mellitus are mostly macrovascular and microvascular diseases as a consequence of
accelerated atherogenesis. Cardiovascular morbidity in patients with type 2 diabetes is two to
four times greater than that of non-diabetic people (Zimmet et al., 2001). Over the past 30
years, the status of diabetes has changed from being considered as a mild disorder of the
elderly to one of the major causes of morbidity and mortality affecting the youth and middle
aged people (Huizinga et al., 2006). It is important to note that the rise in prevalence is seen
in all six inhabited continents of the globe (Wild et al., 2004). According to the Diabetes
Atlas 2006 published by the International Diabetes Federation, the number of people with
diabetes in India currently around 40.9 million is expected to rise to 69.9 million by 2025
unless urgent preventive steps are taken. Besides significant mortality, diabetes related
morbidities such as diabetic retinopathy, neuropathy and cardiovascular disease have also
placed a heavy financial burden on society (Henriksson et al., 1998; Warner et al., 1996;
Simell et al., 1996). For example, in the United States alone, the total annual economic cost
of diabetes in 1997 was estimated to be US$98 billion. This included US$44 billion in direct
medical and treatment costs and US$54 billion for indirect costs attributed to disability and
mortality. Healthcare professionals as well as public policy makers are well aware of the
Page 29
33
public health impact of diabetes. Diabetes is a silent disease – many sufferers became aware
that they have diabetes only when they develop one of its life-threatening complications.
Knowledge of diabetes mellitus can assist in early detection of the disease and reduce the
incidence of complications. Thus, considerable efforts had been put in to inform the public
about diabetes.
2.4. Drugs profile
2.4.1. Gliclazide (BP 2008; Martindale 2006)
Structure:
Chemical formula C15H21N3O3S
Molecular weight 323.41
Category In NIDDM (Sulfonylurea derivative)
Chemical name 1-(3-azabicyclo(3.3.0)oct-3-yl)-3-p-tolylsulphonylurea
pKa 5.8
Log p (Oct/Water) 2.1
Melting point
Appearance White or almost white powder
Solubility Practically insoluble in water, freely soluble in methylene
chloride, sparingly soluble in acetone, slightly soluble in alcohol
Loss on drying Not more than 0.25%, determined on 1.000 g by drying in an
oven at 100 oC to 105
oC for 2h
Content NLT 99.0% and NMT 101.0%
Dosage form Tablet
Strength 20 mg, 30 mg, 40 mg, 80 mg
160 mg daily are given in 2 divided doses
Route of administration Oral
Mechanism Inhibition of ATP-dependent potassium channels(sulfonylurea);
treatment of diabetes mellitus
Indications Noninsulin-Dependent Diabetes Mellitus
T1/2 10-12 h
Tmax (h) 4-6 h
Bioavailability Variable (approx. 60-110%)
Page 30
34
2.4.2. Glibenclamide (Glyburide) (IP 2007; BP 2008; Martindale)
Structure:
Chemical formula C23H28ClN3O5S
Molecular weight 494.0
Category In NIDDM (Sulfonylurea derivative)
Chemical name 1-{4-[2-(5-chloro-2-
methoxybenzmido)ethyl}benzenesulphonyl}-3-cyclohexylurea
pKa 5.3
Log p (Oct/Water) 4.8
Melting point 172 to 174 °C
Appearance A white or almost white, crystalline powder
Solubility Practically insoluble in water, sparingly soluble in methylene
chloride, slightly soluble in alcohol and in methanol
Loss on drying Not more than 1.0%, determined on 1.0 g by drying in an oven
at 105 oC
Content 99.0% to 101.0% (dried substance)
Dosage form Tablet
Strength 2.5 mg-5 mg daily with breakfast
Increments of 2.5 – 5 mg daily up to 15 mg daily
Route of administration Oral
Mechanism Inhibition of ATP-dependent potassium channels(sulfonylurea);
treatment of diabetes mellitus
Indications Noninsulin-Dependent Diabetes Mellitus
Tmax (h) 2-4 h
Bioavailability Variable (approx. 60-90%)
Page 31
35
2.5. Excipients profile
2.5.1. Sunflower oil
Chemical Name Sunflower oil
Structural Formula Sunflower oil is classified as anoleic–linoleic acid oil. Its
composition includes linoleic acid (66%), oleic acid
(21.3%), palmitic acid (6.4%), arachidic acid (4.0%), stearic
acid (1.3%), and behenic acid (0.8%). The USP32–NF-27
describes sunflower oil as a refined fixed oil obtained from
the seeds of Helianthus annus Linne (Family: Asteraceae).
The PhEur 6.2 describes sunflower oil as the refined fatty oil
obtained from the seeds of Helianthus annus C. by
mechanical expression or by extraction. A suitable
antioxidant may be added.
Description Sunflower oil occurs as a clear, light yellow-colored liquid
with a bland, agreeable taste
Boiling point
40–60 oC
Density (g/cm3)
0.915–0.919
Hydroxyl value 14–16
Iodine number 125–140
Melting point -18 oC
Refractive index
1.472–1.474
Specific gravity 0.914–0.924
Saponification value 180–200
Functional Category Diluent; emollient; emulsifying agent; solvent; tablet binder.
Stability and Storage
Conditions
Sunflower oil should be stored in an airtight, well-filled
container, protected from light. Stability may be improved
by the addition of an antioxidant such as butylated
hydroxytoluene
Page 32
36
2.5.2. Palm Oil
Source Kernels of fruits of Elaeis guineensis
Chemical Name Palm Oil
Appearance Liquid
Description Orange-red liquid or solid with a pleasant odor. Insoluble in water
and less dense than water. Hence floats on water. Contains
principally glycerides of palmitic, oleic, and linoleic acids
Melting point
30-40 °C (lit.)
Water
0.3% max
Acid value 251 – 261
Iodine number 16-22
Solubility Soluble in water, 1.141e-011 mg/L @ 25 °C
Saponification value 252 – 262
Stability Stable. Combustible. Incompatible with strong oxidising agents
2.5.3. Polyethylene Glycol 600 (PEG 600)
Chemical Name Structural formula
Emperical formula HOCH2(CH2OCH2)mCH2OH where m represents the average
number of oxyethylene groups.m= 13.2
Molecular weight 570–613
Density
1.11–1.14 g/cm3 at 25
oC
Viscosity (dynamic)
[mPa s (cP)]
15–20
Moisture content Hygroscopic,although hygroscopicity decreases with increasing
molecular weight.
Freezing point
15–25 oC
Refractive index
1.467
Description
Liquid grades (PEG 200–600) occur as clear, colorless or
slightlyyellow-colored, viscous liquids. They have a slight but
characteristic odor and a bitter, slightly burning taste. PEG 600 can
occur as a solid at ambient temperatures
Category Ointment base; plasticizer; solvent; suppository base; tablet
Stability and Polyethylene glycols are chemically stable in air and in solution,
Page 33
37
Storage Conditions
although grades with a molecular weight less than 2000 are
hygroscopic. Polyethylene glycols do not support microbial growth,
and they do not become rancid.
2.5.4. Tween 20
Chemical Name Polyoxyethylene 20 sorbitan monolaurate
Structural formula
Emperical formula C58H114O26
Molecular weight 1128
Viscosity (mPa s): 400
Moisture content 3.0
Color and Form at
25 oC
Yellow oily liquid
Specific gravity at
25 oC
1.1
HLB value 16.7
Acidity/alkalinity pH 6.0–8.0 for a 5% w/v aqueous solution
Flash point 149 oC
Acid value (%) 2.0
Hydroxyl value 96–108
Saponification
Value
40–50
Description
Polysorbates have a characteristic odor and a warm, somewhat
bitter taste. The absolute color intensity of the products may vary
from batch to batch and from manufacturer to manufacturer
Functional Category
Dispersing agent; emulsifying agent; nonionic surfactant;
solubilizing agent; suspending agent; wetting agent.
Page 34
38
2.5.5. Tween 80
Chemical Name Polyoxyethylene 20 sorbitan Monooleate
Structural formula
Emperical formula C58H114O26
Molecular weight 1310
Viscosity (mPas): 425
Moisture content 3.0
Color and Form at 25 oC Yellow oily liquid
Specific gravity at 25 oC 1.1
HLB value 15
Acidity/alkalinity pH 6.0–8.0 for a 5% w/v aqueous solution
Flash point 149 oC
Acid value (%) 2.0
Hydroxyl value 65-80
Saponification value 45–55
Description
Polysorbates have a characteristic odor and a warm,
somewhat bitter taste. Absolute color intensity of the
products may vary from batch to batch and from
manufacturer to manufacturer
Functional Category
Dispersing agent; emulsifying agent; nonionic surfactant;
solubilizing agent; suspending agent; wetting agent.



















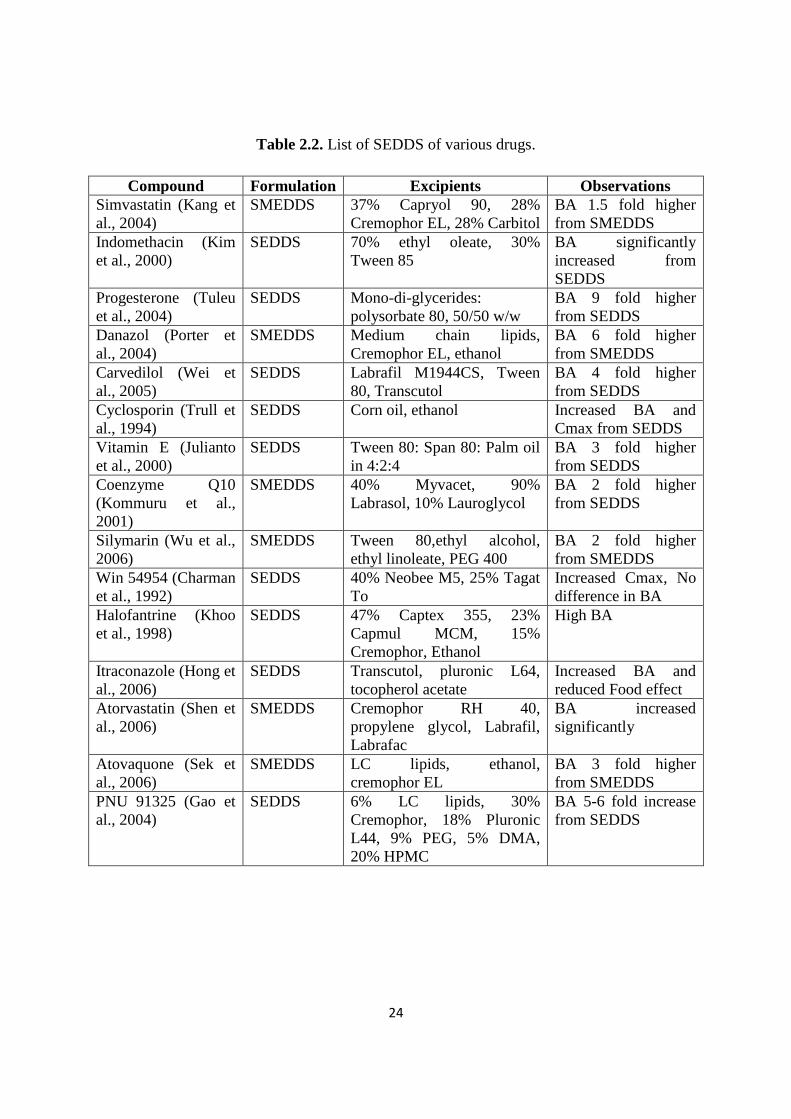





























![REVIEW ON SOLUBILITY ENHANCEMENT TECHNIQUES...absorption of poorly water-soluble drugs. [8,9] In solid dispersion a poorly soluble drug is dispersed in a highly soluble solid hydrophilic](https://static.documents.pub/doc/80x56/5fab6e219a0b2d27a86e8e9a/review-on-solubility-enhancement-techniques-absorption-of-poorly-water-soluble.jpg)

