1 40-500-253-AD-NEW PRODUCT INFORMATION Alcon Laboratories, Inc. STERILE UV and Blue Light Filtering Acrylic Foldable Single-Piece Posterior Chamber Lenses with the UltraSert ™ Pre-loaded Delivery System ENGLISH PRODUCT INFORMATION...................................................2 - 19 BULGARIAN ИНФОРМАЦИЯ ЗА ПРОДУКТА.....................................20 - 31 CZECH INFORMACE O VÝROBKU ......................................................32 - 43 ESTONIAN KASUTUSJUHIS.................................................................44 - 54 HUNGARIAN HASZNÁLATI ÚTMUTATÓ ..............................................55 - 66 LATVIAN INFORMĀCIJA PAR IZSTRĀDĀJUMU ..................................67 - 77 LITHUANIAN INFORMACIJA APIE GAMINĮ..........................................78 - 88 POLISH INFORMACJA O PRODUKCIE ................................................89 - 100 ROMANIAN INFORMAŢII DESPRE PRODUS......................................101 - 112 SERBIAN INFORMACIJA O PROIZVODU ............................................113 - 123 SLOVAK INFORMÁCIE O PRODUKTE ...............................................124 - 135 TURKISH ÜRÜN BİLGİLERİ ..................................................................136 - 146
Transcript
1
40-500-253-AD-NEW
PRODUCT INFORMATIONAlcon Laboratories, Inc.
STERILE UV and Blue Light Filtering Acrylic Foldable Single-PiecePosterior Chamber Lenses with the UltraSert™ Pre-loaded Delivery System
ENGLISH PRODUCT INFORMATION ...................................................2 - 19
BULGARIAN ИНФОРМАЦИЯ ЗА ПРОДУКТА .....................................20 - 31
CZECH INFORMACE O VÝROBKU ......................................................32 - 43
ESTONIAN KASUTUSJUHIS .................................................................44 - 54
LATVIAN INFORMĀCIJA PAR IZSTRĀDĀJUMU ..................................67 - 77
LITHUANIAN INFORMACIJA APIE GAMINĮ ..........................................78 - 88
POLISH INFORMACJA O PRODUKCIE ................................................89 - 100
ROMANIAN INFORMAŢII DESPRE PRODUS ......................................101 - 112
SERBIAN INFORMACIJA O PROIZVODU ............................................113 - 123
SLOVAK INFORMÁCIE O PRODUKTE ...............................................124 - 135
TURKISH ÜRÜN BİLGİLERİ ..................................................................136 - 146
2
ENGLISHPRODUCT INFORMATIONAlcon Laboratories, Inc.
STERILE UV and Blue Light Filtering Acrylic Foldable Single-PiecePosterior Chamber Lenses with the UltraSert™ Pre-loaded Delivery System
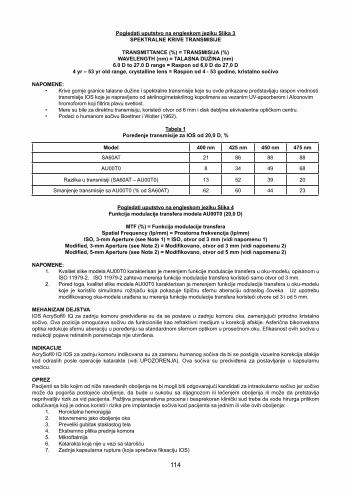
DESCRIPTIONAcrySof® IQ UV and blue light filtering acrylic foldable single-piece posterior chamber lenses are optical implants for the replacement of the human crystalline lens in the visual correction of aphakia in adult patients following cataract surgery. AcrySof® IQ IOL with Alcon’s proprietary blue light filtering chromophore filters light in a manner that approximates the human crystalline lens in the 400 – 475 nm blue light wavelength range (Boettner and Wolter, 1962). In addition to standard UV-light filtering, the AcrySof® IQ IOL reduces transmittance of blue light wavelengths from 62% at 400 nm to 23% at 475 nm (see Table 1). The optical portion consists of a high refractive index soft acrylic material. This material is capable of being folded prior to insertion. The lens gently unfolds to a full-size lens body following implantation. The lens has a biconvex optic with supporting haptics (Figure 1). The posterior aspheric surface of the AcrySof® IQ Model AU00T0 IOL is designed with negative spherical aberration to compensate for the positive spherical aberration of an average cornea. The image quality of the Model AU00T0 Single-Piece IOL (i.e., modulation transfer function) is illustrated in Figure 4. The AcrySof® IQ Model AU00T0 Single-Piece IOLs are provided in the UltraSert™ Pre-loaded Delivery System (Figure 2) for a convenient, controlled means to reliably place these lenses into the capsular bag. The physical properties of the lenses are:
Physical Characteristic DescriptionOptic Type Single-piece IOL with aspheric optic
Optics Material Ultraviolet and blue light filtering Acrylate/Methacrylate Copolymer
UV Cutoff at 10% Transmission (nm) UV (400) at 20.0 D (See Figure 3)
Index Of Refraction 1.55
Optic Powers For available power range see Alcon Product Guide
Haptic Material Ultraviolet and blue light filtering Acrylate/Methacrylate Copolymer
Optic Diameter ØB (mm) 6.0
Overall Length ØT (mm) 13.0
Haptic Angle 0º
Optic Dimensions See Figure 1
3
PHYSICAL CHARACTERISTICSAll dimensions in millimeters
Figure 1 Figure 2
Figure 3SPECTRAL TRANSMITTANCE CURVES
NOTES: • The cutoff wavelength and the spectral transmittance curves presented here represent the range of transmittance values of IOLs made from acrylate/methacrylate copolymer with bonded UV-absorber and Alcon’s proprietary blue
light filtering chromophore. • Measurements were direct transmittance using a 6mm aperture and a disc of thickness equivalent to the optic center. • Human lens data from Boettner and Wolter (1962).
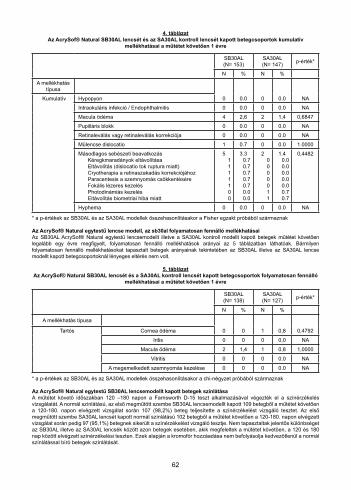
Table 1Transmittance Comparison for 20.0 D IOLs, %
Transmittance Reduction with AU00T0 (% of SA60AT) 62 60 44 23
4
Figure 4
NOTES:1. The image quality of the Model AU00T0 was characterized by measuring modulation transfer function (MTF) in a model
eye described in ISO 11979-2. The ISO 11979-2 requires MTF measurements using only a 3mm aperture. 2. In addition, the image quality of the Model AU00T0 was characterized by measuring MTF in a model eye that utilized a
simulated cornea exhibiting typical adult human spherical aberration. Using the modified model eye, MTF measurements were made using both 3mm, and 5mm apertures.
MODE OF ACTION AcrySof® IQ posterior chamber intraocular lenses are intended to be positioned in the posterior chamber of the eye, replacing the natural crystalline lens. This position allows the lens to function as a refractive medium in the correction of aphakia. The aspheric biconvex optic reduces spherical aberration as compared to a standard spherical optic in an average eye. The effectiveness of these lenses in reducing the incidence of retinal disorders has not been established.
INDICATIONS AcrySof® IQ posterior chamber intraocular lenses are indicated for the replacement of the human lens to achieve visual correction of aphakia in adult patients following cataract surgery (see WARNINGS). These lenses are intended for placement in the capsular bag.
CAUTION Patients with any of the following conditions may not be suitable candidates for an intraocular lens because the lens may exacerbate an existing condition, may interfere with diagnosis or treatment of a condition, or may pose an unreasonable risk to the patient’s eyesight. Careful preoperative evaluation and sound clinical judgment should be used by the surgeon to decide the benefit/risk ratio before implanting a lens in a patient with one or more of these conditions: 1. Choroidal hemorrhage 2. Concomitant severe eye disease 3. Excessive vitreous loss 4. Extremely shallow anterior chamber 5. Microphthalmos 6. Non-age-related cataract 7. Posterior capsular rupture (preventing fixation of IOL) 8. Severe corneal dystrophy 9. Severe optic atrophy 10. Uncontrollable positive pressure 11. Zonular separation (preventing fixation of IOL) 12. Color vision deficiencies 13. Glaucoma 14. Chronic uveitis 15. Diabetic retinopathy 16. Clinically significant macular/RPE changesStudies have shown that color vision discrimination is not adversely affected in individuals with the AcrySof® Natural IOL and normal color vision. The effect on vision of the AcrySof® Natural IOL in subjects with hereditary color vision defects and acquired color vision defects secondary to ocular disease (e.g., glaucoma, diabetic retinopathy, chronic uveitis, and other retinal or optic nerve diseases) has not been studied.
5
WARNINGS 1. As with any surgical procedure, there is risk involved. Potential complications accompanying cataract or implant
surgery may include, but are not limited to the following: corneal endothelial damage, endophthalmitis, retinal detachment, vitritis, cystoid macular edema, corneal edema, pupillary block, cyclitic membrane, iris prolapse, hypopyon, and transient or persistent glaucoma.
2. The safety and effectiveness of intraocular lens implants have not been substantiated in patients with preexisting ocular conditions (chronic drug miosis, glaucoma, amblyopia, diabetic retinopathy, previous corneal transplant, previous retinal detachment, and/or iritis, etc.). Physicians considering lens implantation in such patients should explore the use of alternative methods of aphakic correction and consider lens implantation only if alternatives are deemed unsatisfactory in meeting the needs of the patient.
3. The long-term effects of intraocular lens implantation have not been determined. Therefore, physicians should continue to monitor patients postoperatively on a regular basis.
4. Patients with preoperative problems such as corneal endothelial disease, abnormal cornea, macular degeneration, retinal degeneration, glaucoma, and chronic drug miosis may not achieve the visual acuity of patients without such problems. The physician must determine the benefits to be derived from lens implantation when such conditions exist.
5. A secondary iridectomy for pupillary block may be avoided if one or more iridectomies are performed at the time of IOL implantation (Willis, et al., 1985).
6. The safety and effectiveness of a posterior chamber lens, if placed in the anterior chamber, has not been established. Implantation of posterior chamber lenses in the anterior chamber has been shown in some cases to be unsafe (Girard, et al., 1983).
7. Some adverse reactions which have been associated with the implantation of intraocular lenses are: hypopyon, intraocular infection, acute corneal decompensation and secondary surgical intervention. Secondary surgical interventions include, but are not limited to: lens repositioning, lens replacement, vitreous aspiration or iridectomy for pupillary block, wound leak repair and retinal detachment repair.
8. Small amounts of lens decentration, occurring with an IOL having a narrow or small optic, may result in a patient experiencing glare or other visual disturbances under certain lighting conditions. Surgeons should consider this potential before implanting an IOL having a narrow or small optic. When implanting a narrow or small optic lens, it is recommended that capsulorhexis be performed.
9. Postoperative distension of the capsular bag with variable amounts of anterior chamber shallowing and induced myopia have been associated with capsulorhexis techniques and implantation of PMMA, silicone and acrylic posterior chamber lenses (Holtz, 1992).
10. Caution should be used prior to lens encapsulation to avoid lens decentrations or dislocations. Some clinical cases suggest encapsulation occurs within four weeks.
11. The clinical study of the AcrySof® Natural Single-Piece Lens (referenced in Tables 2 through 5) was conducted with the lens intended for implantation in the capsular bag only. There is no clinical data to demonstrate its safety and effectiveness for placement in the ciliary sulcus.
It is recommended that viscoelastic be removed from the eye at the close of surgery with emphasis on the space between the posterior capsule and lens. This may be accomplished by gently depressing the IOL optic posteriorly with the I/A tip and using standard irrigation/aspiration techniques to remove the viscoelastic agent from the eye. This should force any trapped viscoelastic anteriorly where it can be easily aspirated.
PRECAUTIONS 1. Do not resterilize these intraocular lenses or the UltraSert™ Pre-loaded Delivery System by any method. 2. Do not store intraocular lenses at temperatures over 45° C (113° F). 3. If required, handle lenses carefully to avoid damage to lens surfaces or haptics. 4. Do not attempt to reshape haptics in any way. 5. A high level of surgical skill is required for intraocular lens implantation. The surgeon should have observed and/
or assisted in numerous implantations and successfully completed one or more courses on intraocular lens implantation before attempting to implant intraocular lenses.
6. Contents are sterile unless package is opened or damaged. 7. The UltraSert™ Pre-loaded Delivery System is for single-use only. Discard the UltraSert™ Pre-loaded device after
use. 8. Use the UltraSert™ Pre-loaded Delivery System at Operating Room temperatures between 18° C (64° F) and 23°
C (73° F).
CALCULATION OF LENS POWERPreoperative calculation of required lens power for these posterior chamber intraocular lenses should be determined by the surgeon’s experience, preference, and intended lens placement. Lens power calculation methods are described in the following references:
6
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. et al. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. et al. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
SUGGESTED A-CONSTANT The suggested A-constant listed on the outer label is presented as a guideline and is a starting point for implant power calculations. It is recommended that you develop your own constant appropriate for you based on clinical experience with the particular lens models, surgical techniques, measuring equipment, and postoperative results.In the United States, if additional information on lens power calculation is needed, please contact Alcon Laboratories, Inc. at 1-800-TO-ALCON (1-800-862-5266). Outside the United States, contact local Alcon offices or distributors.
QUALIFIED ALCON VISCOELASTICS AND ORDER NUMBERSSee supplement included in this package. Only Alcon qualified viscoelastic(s) should be used. The use of an unqualified viscoelastic may cause damage to the lens and potential complications during the implantation process. For a full list of Alcon qualified viscoelastics for this lens, please contact your Alcon representative.
DIRECTIONS FOR USEStep 1. Use the UltraSert™ Pre-loaded Delivery System at Operating Room temperatures between 18° C (64° F) and
23° C (73° F).Step 2. Examine the label on the unopened outer box for model, power, proper configuration, and expiration date. Step 3. After opening the cardboard outer box, inspect the device package for any damage. If damage is observed, use
another AcrySof® IQ IOL and UltraSert™ Pre-loaded Delivery System. Next, verify that the lens information on the device label (e.g., model, power, and serial number) is consistent with the information on the outer box labeling.
Step 4. To remove the UltraSert™ Pre-loaded Delivery System, grip the corner of the plastic tray, peel open the TYVEK† material lid portion fully, and transfer the device to a sterile environment. If, after inspection, the device nozzle appears to have damage, particulates, or deformation, use another AcrySof® IQ IOL and UltraSert™ Pre-loaded Delivery System. If the device is not completely intact, or the plunger or plunger lock appears to have moved during shipping, use another AcrySof® IQ IOL and UltraSert™ Pre-loaded Delivery System (see RETURNED GOODS POLICY).
†Reg. TM of E.I. DuPont De Nemours & Co.
When ready to prepare the lens for delivery, perform Steps 5, 6, and 7 in sequence, with minimal delay between steps.
Step 5. Fully insert the viscoelastic cannula, perpendicular to the device, through the viscoelastic port, located in the lens stop portion of the device as shown in Figure 5. Fill the device until viscoelastic can be observed flowing to the line on the nozzle tip (see arrow in Figure 5), then retract the cannula. This will require approximately 0.2 mL of viscoelastic. Only use an Alcon viscoelastic qualified for use with the UltraSert™ Pre-loaded Delivery System, that has been allowed to come to the operating room temperature. For a list of Alcon qualified viscoelastics, refer to the Supplement included in this package.
Figure 5
7
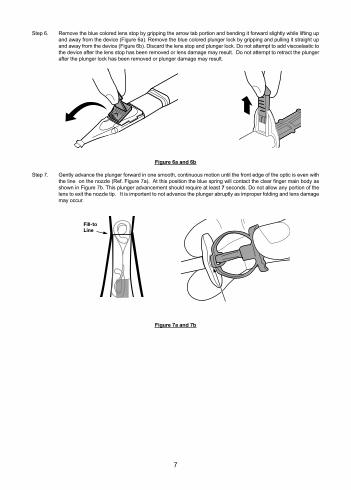
Step 6. Remove the blue colored lens stop by gripping the arrow tab portion and bending it forward slightly while lifting up and away from the device (Figure 6a). Remove the blue colored plunger lock by gripping and pulling it straight up and away from the device (Figure 6b). Discard the lens stop and plunger lock. Do not attempt to add viscoelastic to the device after the lens stop has been removed or lens damage may result. Do not attempt to retract the plunger after the plunger lock has been removed or plunger damage may result.
Figure 6a and 6b
Step 7. Gently advance the plunger forward in one smooth, continuous motion until the front edge of the optic is even with the line on the nozzle (Ref. Figure 7a). At this position the blue spring will contact the clear finger main body as shown in Figure 7b. This plunger advancement should require at least 7 seconds. Do not allow any portion of the lens to exit the nozzle tip. It is important to not advance the plunger abruptly as improper folding and lens damage may occur.
Fill-toLine
Figure 7a and 7b
8
Step 8. After the lens has been advanced to the nozzle line, the lens should be visually inspected to determine the position of the haptics. The plunger should be in contact with the trailing optic edge. No part of the lens should exit the nozzle prior to insertion through the incision (Refer to Figure 8). Once the lens is in position at the nozzle line, it should be implanted within 3 minutes.
Figure 8
Step 9. After confirming the lens is properly positioned and the haptics are folded properly, proceed with lens implantation by inserting the nozzle tip into the incision up to the depth guard (Figure 9), and aiming the nozzle tip at the anterior capsule opening.
Figure 9
9
Step 10. Gently advance the plunger in one smooth, continuous motion. Do not advance the plunger too fast or lens damage may occur. It is recommended that delivery of the lens from the visual inspection position should take at least 5 seconds. If the leading haptic is straight or looped and extended in front of the lens, rotate device to be bevel left before advancing plunger to ensure the leading haptic is correctly placed in the capsular bag. As the leading haptic begins to exit the nozzle tip, place the leading haptic into the capsular bag in its correct orientation, and continue to slowly advance the plunger to deliver the lens. As the optic exits the nozzle, rotate the device back to center or slightly bevel right if needed to ensure the lens unfolds anterior side up within the capsular bag.
Step 11. Using a suitable positioning instrument to position the lens within the capsular bag, and in a planar fashion parallel to the iris.
Step 12. Discard the entire device. Do not re-use the UltraSert™ Pre-loaded Delivery System.
PATIENT REGISTRATION AND REPORTINGThe Patient Identification Card included in the package is to be completed and given to the patient, together with instructions to keep the card as a permanent record to be shown to any eye care practitioner that the patient consults in the future.
In the United States, each patient must be registered with Alcon Laboratories, Inc., immediately following implantation of one of these lenses. Registration is accomplished by completing the Implant Registration Card that is enclosed in the lens box and mailing it to Alcon Laboratories, Inc. Patient registration is essential for the long-term patient follow up program and will assist Alcon Laboratories, Inc. in responding to reports of adverse events.Outside the United States, local laws for product registration should be followed.
Events that reasonably suggest that the lens may have caused or contributed to death or serious injury, including events occurring as a result of failure of a medical device to meet its performance specifications or otherwise perform as intended, should be reported to Alcon Laboratories, Inc. This information is being requested from all surgeons in order to document potential long-term effects of intraocular lens implantation. Surgeons should use the following address and telephone number for reporting adverse events involving these intraocular lenses:
Alcon Laboratories, Inc. Medical Safety (AB 2-6) 6201 South Freeway Fort Worth, TX 76134-2099 Call Toll free: 1-800-757-9780
For surgeons outside of the United States, contact local Alcon offices or distributors regarding reports of adverse events.
AcrySof® Foldable Posterior Chamber Lens Clinical StudiesTwo randomized, prospective well-controlled clinical studies have been performed on AcrySof® Single-Piece Foldable Posterior Chamber Lenses. The first study was conducted to demonstrate the safety and effectiveness of the AcrySof®
Natural Single-Piece Posterior Chamber Lens Model SB30AL (UV and blue light filtering) as the parent lens model. This was a randomized clinical study that included the AcrySof® Model SA30AL (UV-absorbing only) as a control lens. Only data from the first operative eye from those subjects who received either a Model SB30AL or Model SA30AL intraocular lens are included. A second randomized clinical study of the AcrySof® IQ Foldable Single-Piece IOL (with UV and blue light filtering chromophores) with an aspheric optic versus an AcrySof® Foldable Single-Piece control lens was conducted to assess the clinical/functional benefits over a traditional spherical design.
AcrySof® Natural Single-Piece Lens Model SB30AL Clinical StudyThe results achieved by the patients successfully followed for a minimum of one year postoperatively provide reasonable assurance that the AcrySof® Natural Single-Piece lens Model SB30AL is a safe and effective device for the visual correction of aphakia.
AcrySof® Natural Single-Piece Lens Model SB30AL Patient PopulationThe subject population implanted with a Model SB30AL in at least the first operative eye in this bilateral study consists of 70.6% females and 29.4% males. The subject population implanted with a Model SA30AL (control) intraocular lens consists of 60.5% females and 39.5% males. Stratifying by race for the Model SB30AL population, 95.3% are Caucasian, and 4.7% are Black. The control (SA30AL) subject population was 96.6% Caucasian, 2% Black and 1.4% other. The mean age for the total population receiving the Model SB30AL in at least the first operative eye is 72.9 years. Similarly, the mean age for the total population receiving the Model SA30AL (control) is 71.9 years.
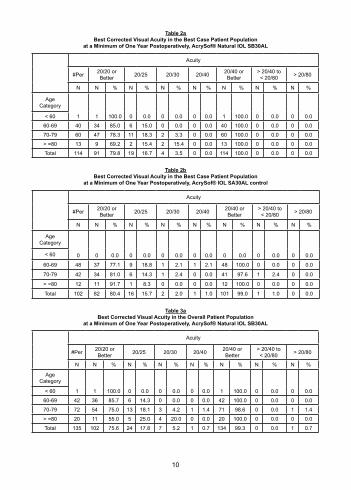
AcrySof® Natural Single-Piece Lens Model SB30AL Visual AcuityA summary of visual acuity achieved at a minimum of one year postoperatively among subjects who did not have preoperative ocular pathology, abnormal corneas, or macular degeneration at any time (Best Case) is presented in Table 2a, and visual acuity achieved by overall subject population is shown in Table 3a. Control data are found for the same data sets in Tables 2b and 3b, respectively. There was no statistically significant difference in visual acuity between Model SB30AL and the control lens, Model SA30AL, in either the best case or overall data sets.
10
Table 2aBest Corrected Visual Acuity in the Best Case Patient Population
at a Minimum of One Year Postoperatively, AcrySof® Natural IOL SB30AL
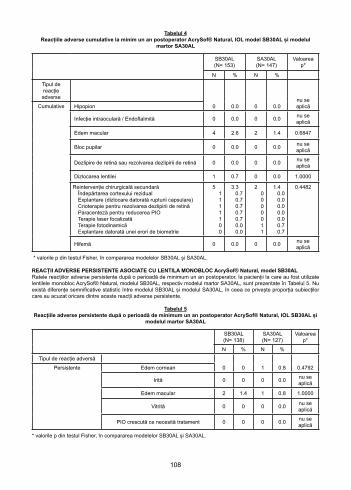
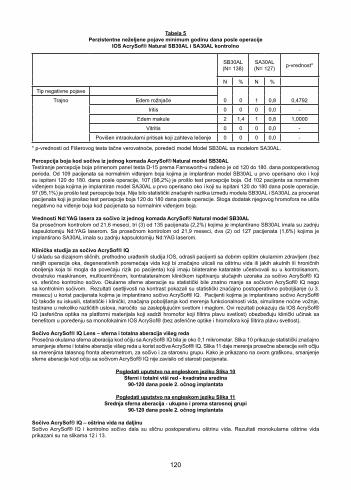
AcrySof® Natural Single-Piece Lens Model SB30AL Cumulative Adverse EventsThe cumulative rates of these adverse events up to and including a minimum of a one year postoperative period for the AcrySof® Natural Single-Piece Lens Model SB30AL and the Model SA30AL patients are shown in Table 4. There were no statistically significant differences between the Model SB30AL and the Model SA30AL for the proportion of subjects experiencing any of the cumulative adverse events.
Table 4Cumulative Adverse Events at a Minimum of One Year Postoperatively
AcrySof® Natural IOL SB30AL and SA30AL control
SB30AL (N= 153)
SA30AL(N= 147) p-value*
N % N %
Type ofAdverse Event
0 0.0 0 0.0 NACumulative Hypopyon
Intraocular Infection / Endophthalmitis 0 0.0 0 0.0 NA
Macular Edema 4 2.6 2 1.4 0.6847
Pupillary Block 0 0.0 0 0.0 NA
Retinal Detachment or Retinal Detachment Repair 0 0.0 0 0.0 NA
Lens Dislocation 1 0.7 0 0.0 1.0000
Secondary Surgical ReinterventionRemoval of Residual CortexExplant (dislocation due to capsular rupture)Cryotherapy to Repair Retinal TearParacentesis to Lower IOPFocal Laser TreatmentPhotodynamic TherapyExplant Due to Biometry Error
51 1 11100
3.3 0.7
0.70.70.70.70.00.0
20000011
1.40.00.00.00.00.00.70.7
0.4482
Hyphema 0 0.0 0 0.0 NA
*p-values from Fisher’s Exact Test comparing Model SB30AL to Model SA30AL.
AcrySof® Natural Single-Piece Lens Model SB30AL Persistent Adverse EventsThe persistent rates of these adverse events at a minimum of a one year postoperative period for the AcrySof® Natural Single-Piece Lens Model SB30AL patients and the Control Model SA30AL are shown in Table 5. There were no statistically significant differences between the Model SB30AL and the Model SA30AL for the proportion of subjects experiencing any of the persistent adverse events.
12
Table 5Persistent Adverse Events at a Minimum of One Year Postoperatively
AcrySof® Natural IOL SB30AL and SA30AL control
SB30AL (N= 138)
SA30AL(N= 127) p-value*
N % N %
Type of Adverse Event
0 0 1 0.8 0.4792Persistent Corneal Edema
Iritis 0 0 0 0.0 NA
Macular Edema 2 1.4 1 0.8 1.0000
Vitritis 0 0 0 0.0 NA
Raised IOP Requiring Treatment 0 0 0 0.0 NA
*p-values from Fisher’s Exact Test comparing Model SB30AL to Model SA30AL.
AcrySof® Natural Single-Piece Lens Model SB30AL Color PerceptionColor perception testing using the Farnsworth D-15 Panel Test was conducted at the 120 to 180 day postoperative period. Of the 109 subjects with normal color vision implanted with a Model SB30AL in the first operative eye and examined at the 120 to 180 day postoperative visit, 107 (98.2%) passed the color perception test. Of the 102 subjects with normal color vision implanted with a Model SA30AL in the first operative eye and examined at the 120 to 180 day postoperative visit, 97 (95.1%) passed the color perception test. There were no statistically significant differences between Model SB30AL and Model SA30AL for the percent of subjects that passed the color perception test at the 120 to 180 day postoperative visit. Therefore, the addition of the proprietary chromophore does not negatively affect color vision in patients with normal color vision.
AcrySof® Natural Single-Piece Lens Model SB30AL Nd:YAG Rates With a mean follow-up of 21.6 months, three (3) of the 135 subjects (2.2%) implanted with SB30AL experienced a Nd:YAG posterior capsulotomy. With a mean follow-up of 21.9 months, two (2) of the 127 subjects (1.6%) implanted with SA30AL experienced a Nd:YAG posterior capsulotomy.
AcrySof® IQ Lens Clinical StudyConsistent with the design of similar previously conducted IOL studies, adult subjects in good general ocular health (e.g. no prior ocular surgery, degenerative visual disorder which would significantly impact visual acuity, or severe acute or chronic condition that may increase patient risk) having bilateral cataracts were enrolled in a controlled, randomized, double-masked, multi-center, contralateral implant clinical investigation of the AcrySof® IQ lens versus a spherical control lens. Ocular spherical aberrations were statistically significantly less with the AcrySof® IQ lens than the control lens. Contrast sensitivity results demonstrated a statistically significant postoperative (at 3 months) improvement in favor of AcrySof® IQ implanted eyes. Eyes implanted with the AcrySof® IQ lens also experienced statistically and clinically significant improvements in a functional vision measurement, simulated night driving, under several conditions tested - especially glare and fog. These results reflect that the AcrySof® IQ IOL (an aspheric optic on a material platform containing a blue-light filtering chromophore) provides beneficial clinical performance as compared to the monofocal AcrySof® IOL (without an aspheric optic and blue-light filtering chromophore).
AcrySof® IQ Lens – Spherical and Total Higher Order AberrationsThe mean ocular spherical aberration of the AcrySof® IQ eyes was approximately 0.1 micrometers. Figure 10 represents the statistically significant reduction in spherical and total higher order aberrations observed in favor of the AcrySof® IQ lens. Figure 11 provides the mean spherical aberration measurements of all eyes with wavefront aberrometer measurements by lens and age group. As depicted in this chart, the reduction in spherical aberration of the AcrySof® IQ eyes was independent of age.
13
Figure 10Spherical and Total Higher Order RMS
90-120 Days after 2nd Eye Implant
Figure 11Mean Spherical Aberration Overall and by Age Group
90-120 Days after 2nd Eye Implant
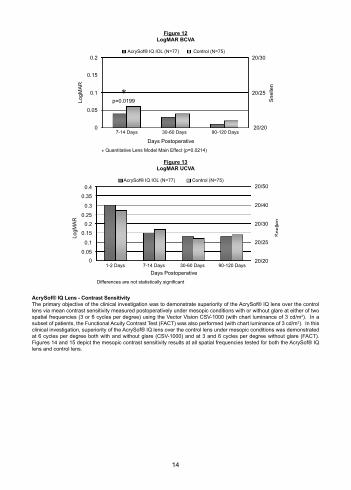
AcrySof® IQ Lens – Distance Visual AcuityThe AcrySof® IQ lens and the control lens provided clinically similar postoperative visual acuity. Monocular visual acuity results are presented in Figures 12 and 13.
14
Figure 12LogMAR BCVA
Figure 13LogMAR UCVA
AcrySof® IQ Lens - Contrast SensitivityThe primary objective of the clinical investigation was to demonstrate superiority of the AcrySof® IQ lens over the control lens via mean contrast sensitivity measured postoperatively under mesopic conditions with or without glare at either of two spatial frequencies (3 or 6 cycles per degree) using the Vector Vision CSV-1000 (with chart luminance of 3 cd/m2). In a subset of patients, the Functional Acuity Contrast Test (FACT) was also performed (with chart luminance of 3 cd/m2). In this clinical investigation, superiority of the AcrySof® IQ lens over the control lens under mesopic conditions was demonstrated at 6 cycles per degree both with and without glare (CSV-1000) and at 3 and 6 cycles per degree without glare (FACT). Figures 14 and 15 depict the mesopic contrast sensitivity results at all spatial frequencies tested for both the AcrySof® IQ lens and control lens.
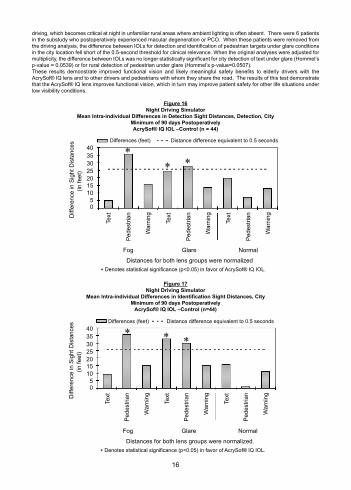
AcrySof® IQ Lens – Night Driving SimulationA subset of patients underwent testing in a validated night driving simulator. Patients were tested monocularly under conditions which simulate city and rural settings under normal, glare and fog conditions.The nighttime city driving scene employs a variety of street lights, car lights, store lights and signs to recreate the high level of ambient lighting typical under these conditions. The nighttime rural driving scene uses a minimal amount of ambient lighting. Simulated driving speeds of approximately 35 mph and 55 mph were used for the city and rural scenes, respectively.Patients were asked to detect and identify a series of targets in each scene, including white-green highway information signs, black-yellow warning signs and pedestrians. Patients were asked to respond when they saw the first target, allowing a detection distance to be recorded. Patients were then asked to respond when they could distinguish the target (e.g., what the sign says, which direction the pedestrian was walking, etc.) so that an identification distance could be recorded. Figures 16 through 19 present the average differences between the AcrySof® IQ lens and control lens in city and rural driving scenes for both detection and identification distances (e.g., the mean of the intra-individual differences).The AcrySof® IQ lens performed functionally better than the control in 34 of the 36 conditions tested, reflecting improvement in both detection and identification distances in both city and rural driving scenes under the various driving conditions tested (normal, glare, fog). Furthermore, the AcrySof® IQ lens performed statistically significantly better than the control in 12 of these conditions, with the most significant impact and greatest advantage observed in detection and identification of city pedestrians (under glare and fog conditions) and rural warning signs (under glare and fog conditions). Under reduced visibility conditions (glare, fog) in the city scene, the increased visibility distance at 35 mph provides the AcrySof® IQ lens greater than 0.5 second additional time to respond to a pedestrian target, a hazard more commonly encountered in city settings. This 0.5 second increase is functionally significant in allowing for greater time to take appropriate actions such as stopping, avoidance, etc. (Green, 2000; McBride and Matson, 2004). Under all conditions in the rural scene, the increased visibility distance at 55 mph provides the AcrySof® IQ lens more than 1 second additional time to identify warning signs, a situation frequently encountered in rural areas. A 0.5 second increase is functionally significant in allowing for greater time to take appropriate action while
16
driving, which becomes critical at night in unfamiliar rural areas where ambient lighting is often absent. There were 6 patients in the substudy who postoperatively experienced macular degeneration or PCO. When these patients were removed from the driving analysis, the difference between IOLs for detection and identification of pedestrian targets under glare conditions in the city location fell short of the 0.5-second threshold for clinical relevance. When the original analyses were adjusted for multiplicity, the difference between IOLs was no longer statistically significant for city detection of text under glare (Hommel’s p-value = 0.0539) or for rural detection of pedestrian under glare (Hommel’s p-value=0.0507).These results demonstrate improved functional vision and likely meaningful safety benefits to elderly drivers with the AcrySof® IQ lens and to other drivers and pedestrians with whom they share the road. The results of this test demonstrate that the AcrySof® IQ lens improves functional vision, which in turn may improve patient safety for other life situations under low visibility conditions.
Figure 16Night Driving Simulator
Mean Intra-individual Differences in Detection Sight Distances, Detection, CityMinimum of 90 days PostoperativelyAcrySof® IQ IOL –Control (n = 44)
Figure 17Night Driving Simulator
Mean Intra-individual Differences in Identification Sight Distances, CityMinimum of 90 days Postoperatively
AcrySof® IQ IOL –Control (n=44)
17
Figure 18Night Driving Simulator
Mean Intra-individual Differences in Detection Sight Distances, RuralMinimum of 90 days Postoperatively
AcrySof® IQ IOL –Control (n=44)
Figure 19Night Driving Simulator
Mean Intra-individual Differences in Identification Sight Distances, RuralMinimum of 90 days Postoperatively
AcrySof® IQ IOL –Control (n=44)
HOW SUPPLIEDThese posterior chamber intraocular lenses are supplied dry, in the UltraSert™ Pre-loaded Delivery System, within a primary sterilization package, and terminally sterilized with ethylene oxide. The primary sterilization package must be opened only under aseptic conditions (see DIRECTIONS FOR USE).
EXPIRATION DATESterility is guaranteed unless the primary sterilization package is damaged or opened. The expiration date is clearly indicated on the outer box label of the UltraSert™ Pre-loaded Delivery System. Any lens held after the expiration date should be returned to Alcon Laboratories, Inc. (see RETURNED GOODS POLICY).
18
RETURNED GOODS POLICYIn the United States, returned lenses will only be accepted in exchange for other products, not credit. All returns must be accompanied by an Alcon Laboratories, Inc. Returned Goods Number and be shipped via traceable means. A Returned Goods Number is obtained by contacting Alcon’s Customer Service Department. Issuance of this number does not constitute final acceptance of the returned products. For detailed policy guidelines including exchange, please contact your Sales or Customer Service Representative. Outside the United States, contact local Alcon offices or distributors regarding the applicable returned goods policy.
REFERENCES 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., et al. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., et al. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
19
SYMBOLS USED ON LABELING
SYMBOL ENGLISHIOL Intraocular lens
SP Single Piece
UV Ultraviolet
D Diopter
ØB Body diameter (Optic diameter)
ØT Overall diameter (Overall length)
2STERILIZE Do not resterilize
Do not reuse
Use by
STERILE EO Sterilized using ethylene oxide
SN Serial number
Caution
Manufacturer
EC REP Authorized Representative in the European Community
Upper Limit of Temperature
Open here
Consult instructions for use
Do not use if sterile package has been opened or damaged
Caution: Federal (USA) Law restricts this device to sale by or on the order of a physician
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 USA
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,United Kingdom
СТЕРИЛНИ филтриращи ултравиолетовата светлина и светлината в синия спектър акрилни сгъваемиеднокомпонентни заднокамерни лещи с предварително заредена система за имплантация UltraSert™
ОПИСАНИЕAcrySof® IQ филтриращи ултравиолетовата и синята светлина акрилни еднокомпонентни сгъваеми заднокамерни лещи са оптически импланти за замяна на естествената човешка кристална леща с цел корекция на афакията при възрастни пациенти при катарактна хирургия. AcrySof® IQ ВОЛ (вътреочна леща), с патентования от Alcon хромофор, филтриращ синята светлина, филтрира светлината по начин, който се приближава до естествената човешка леща за областта от 400 – 475 nm дължина на вълната, за светлината в синия спектър (Boettner и Wolter, 1962). Освен това, към стандартното UV-филтриране, AcrySof® IQ ВОЛ намалява пропускливостта на синята светлина от 62% при 400 nm до 23% при 475 nm (виж Таблица 1). Оптичната част на този тип леща се състои от мек акрилен материал с висок индекс на рефракция. Този вид материал позволява лещата да бъде сгъната, преди поставянето й. След имплантирането, лещата се разгъва леко и заема истинската си форма. Тези лещи имат биконвексна оптика с поддържаща хаптика (Figure 1). AcrySof® IQ ВОЛ модел AU00T0 е проектиран с негативна сферичната аберация, която компенсира положителната сферична аберация на средностатистическата роговица. Качеството на образа при еднокомпонентната ВОЛ модел AU00T0 (напр. модулационна предавателна функция - modulation transfer function) е показан на Figure 4. AcrySof® IQ Модел AU00T0 еднокомпонентна ВОЛ се доставя в UltraSert™ предварително заредена система за имплантация (Figure 2), предлагаща удобен и контролиран метод за сигурно поставяне на лещата в капсулния сак. Физическите свойства на тези лещи са:
Физическа характеристика Описание
Тип оптика Еднокомпонентна ВОЛ с асферична оптика
Материал на оптиката Акрилат/ Метакрилат кополимер, филтриращ ултравиолетовата и синята светлина
UV граница при 10% Пропускливост (nm) UV (400) при 20.0 D (Вижте Figure 3)
Индекс на рефракция 1.55
Диоптри на оптиката Вижте Alcon Product Guide за всички налични диоптри
Конфигурация на хаптиката STABLEFORCE® L-модифицирана хаптика
Материал на хаптиката Акрилат/ Метакрилат кополимер, филтриращ ултравиолетовата и синята светлина
Диаметър на оптиката ØB (mm) 6.0
Цялостна дължина ØT (mm) 13.0
Ъгъл на хаптиката 0º
Размери на оптиката Вижте Figure 1
BULGARIAN
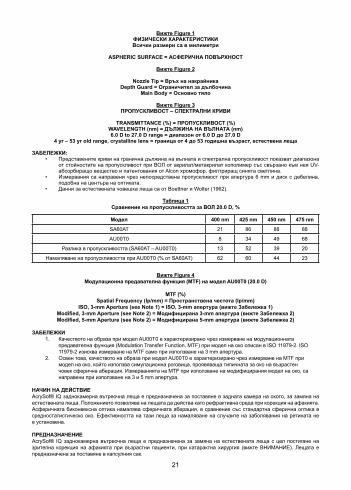
21
Вижте Figure 1ФИЗИЧЕСКИ ХАРАКТЕРИСТИКИВсички размери са в милиметри
ASPHERIC SURFACE = АСФЕРИЧНА ПОВЪРХНОСТ
Вижте Figure 2
Nozzle Tip = Връх на накрайника Depth Guard = Ограничител за дълбочина
Main Body = Основно тяло
Вижте Figure 3ПРОПУСКЛИВОСТ – СПЕКТРАЛНИ КРИВИ
TRANSMITTANCE (%) = ПРОПУСКЛИВОСТ (%)WAVELENGTH (nm) = ДЪЛЖИНА НА ВЪЛНАТА (nm)6.0 D to 27.0 D range = диапазон от 6.0 D до 27.0 D
4 yr – 53 yr old range, crystalline lens = граница от 4 до 53 годишна възраст, естествена леща
ЗАБЕЛЕЖКИ: • Представените криви на гранична дължина на вълната и спектрална пропускливост показват диапазона
от стойностите на пропускливост при ВОЛ от акрилат/метакрилат кополимер със свързано към нея UV-абсорбиращо вещество и патентования от Alcon хромофор, филтриращ синята светлина.
• Измервания са направени чрез непосредствена пропускливост при апертура 6 mm и диск с дебелина, подобна на центъра на оптиката.
• Данни за естествената човешка леща са от Boettner и Wolter (1962).
Таблица 1Сравнение на пропускливостта за ВОЛ 20.0 D, %
Модел 400 nm 425 nm 450 nm 475 nm
SA60AT 21 86 88 88
AU00T0 8 34 49 68
Разлика в пропускливостта (SA60AT – AU00T0) 13 52 39 20
Намаляване на пропускливостта при AU00T0 (% от SA60AT) 62 60 44 23
Вижте Figure 4Модулационна предавателна функция (MTF) на модел AU00T0 (20.0 D)
MTF (%) Spatial Frequency (Ip/mm) = Пространствена честота (Ip/mm)
ЗАБЕЛЕЖКИ 1. Качеството на образа при модел AU00T0 е характеризирано чрез измерване на модулационната
предавателна функция (Modulation Transfer Function, MTF) при модел на око описан в ISO 11979-2. ISO 11979-2 изисква измерване на MTF само при използване на 3 mm апертура.
2. Освен това, качеството на образа при модел AU00T0 е характеризирано чрез измерване на MTF при модел на око, който използва симулационна роговица, проявяваща типичната за око на възрастен човек сферична аберация. Измерванията на MTF при използване на модифицирания модел на око, са направени при използване на 3 и 5 mm апертура.
НАЧИН НА ДЕЙСТВИЕAcrySof® IQ заднокамерна вътреочна леща е предназначена за поставяне в задната камера на окото, за замяна на естествената леща. Положението позволява на лещата да действа като рефрактивна среда при корекция на афакията. Асферичната биконвексна оптика намалява сферичната аберация, в сравнение със стандартна сферична оптика в средностатистическо око. Ефективността на тази леща за намаляване на случаите на заболявания на ретината не е установена.
ПРЕДНАЗНАЧЕНИЕ AcrySof® IQ заднокамерна вътреочна леща е предназначена за замяна на естествената леща с цел постигане на зрителна корекция на афакията при възрастни пациенти, при катарактна хирургия (вижте ВНИМАНИЕ). Лещата е предназначена за поставяне в капсулния сак.
22
ПРЕДУПРЕЖДЕНИЯДопуска се, че вътреочните лещи не са подходящи за имплантация при пациенти с някое от следните състояния, тъй като лещата може да влоши моментното им състояние, да обърка диагнозата или хода на лечението, или може да постави неоправдан риск за зрението на пациента. Необходимо е хирургът да направи внимателна предоперативна оценка и да вземе сигурно клинично решение за съотношението полза/риск преди имплантирането на леща при пациент с едно или повече от изброените състояния: 1. Хороидални кръвоизливи 2. Съпътстващи остри очни възпаления 3. Прекалено голяма загуба на стъкловидното тяло 4. Прекалено плитка предна очна камера 5. Микрофталмия 6. Катаракта, несвързана с възрастта 7. Руптура на задната капсула (пречещи на фиксирането на ВОЛ) 8. Тежка дистрофия на роговицата 9. Тежка оптична атрофия 10. Неконтролируемо положително налягане 11. Разпадане на зонулите (пречещо на фиксирането на ВОЛ) 12. Нарушения в цветното зрение 13. Глаукома 14. Хроничен увеит 15. Диабетна ретинопатия 16. Клинично значими промени в макулата/RPE (пигментен ретинен епител)Проучванията показват, че различаването на цветовете не е засегнато при пациенти с AcrySof® Natural ВОЛ и с нормално цветно зрение. Въздействието върху зрението на AcrySof® Natural ВОЛ при пациенти с наследствени дефекти в цветното зрение и с вторично придобити дефекти в цветното зрение в следствие на очно заболяване (като глаукома, диабетна ретинопатия, хроничен увеит и други заболявания на ретината или зрителния нерв) не е изучавано.
ВНИМАНИЕ 1. Както при всяка една хирургична процедура, и при имплантацията има вероятен риск. Потенциалните
усложнения, свързани с катаракта или имплантация на вътреочна леща могат да включват, но не се ограничават само до следното: увреждане на роговичния ендотел, ендофталмити, отлепване на ретината, възпаления на стъкловидното тяло, кистоиден макулен оток, оток на роговицата, зеничен блок, цилиарна мембрана, пролапс на ириса, хипопион, временна или персистираща глаукома.
2. Безопасността и ефективността на вътреочните лещи не са доказани при пациенти с предшестващи очни проблеми (хронична лекарствена миоза, глаукома, амблиопия, диабетна ретинопатия, предишна трансплантация на роговица, предишно отлепване на ретината, и/или ирити и др.). Лекарят, който обмисля имплантация на леща при такъв пациент трябва да изследва алтернативни методи за корекция на афакията и да разглежда имплантацията като вариант само ако алтернативите бъдат преценени за неподходящи спрямо нуждите на пациента.
3. Дългосрочните ефекти от имплантацията на вътреочна леща не са определени. Ето защо лекарите трябва да продължат с постоперативно наблюдение на пациентите си през регулярен интервал от време.
4. Пациенти с преоперативни проблеми като увреждане на роговичния ендотел, анормална роговица, макулна дегенерация, дегенерация на ретината, глаукома, хронична лекарствена миоза е възможно да не достигнат такава зрителна острота, каквато се постига при пациенти без подобни проблеми. Лекарят трябва да прецени ползите от имплантирането на леща при наличие на подобни състояния.
5. Последващата иридектомия при зеничен блок може да бъде избегната ако една или повече иридектомии бъдат направени при имплантацията на ВОЛ (Willis, et al., 1985).
6. Eфективността и безопасността на заднокамерните лещи при поставяне в предната камера не са установени. Наблюденията показват, че в някои случаи имплантацията на заднокамерни лещи в предната камера не е безопасно (Girard, et al., 1983).
7. Наблюдавани са и някои нежелани реакции, които се свързват с имплантацията на вътреочни лещи като: хипопион, вътреочна инфекция, остра роговична декомпенсация и вторична хирургична интервенция. Вторичната хирургична интервенция включва, но не се ограничава само до: повторно позициониране на лещата, отстраняване на лещата, аспирация на вътреочна течност или иридектомия при зеничен блок, възстановяване на раната и отлепването на ретината
8. При леко децентриране на лещата, което може да се случи с лещи с по-малка или тясна оптика, пациентът може да усети блестене или други зрителни смущения при определени условия на осветеност. Лекарят трябва предварително да оцени тази възможност преди да имплантира ВОЛ с тясна или малка оптика. При имплантиране на леща с по-малка или тясна оптика се препоръчва да се направи капсулорексис.
9. Постоперативното разширяване на капсулния сак с индуциране на миопия и плитка предна камера се свързва с техниките на капсулорексис и имплантирането на ПММА, силиконови и акрилни заднокамерни лещи (Holtz, 1992).
10. Необходимо е да се обърне внимание преди енкапсулирането на лещата, за да се избегне децентриране или разместване. Някои клинични случаи показват енкапсулация до четири седмици след имплантацията.
23
11. Проведеното клинично проучване с AcrySof® Natural еднокомпонентни лещи (цитирано в Таблици 2 до 5) е проведено с импланитирането на лещата единствено в капсулния сак. Няма достатъчно данни, доказващи безопасността и ефективността при поставяне на лещата в цилиарния сулкус.
Препоръчва се вискоеластичното средство, използвано по време на операцията, да бъде отстранено от окото при приключването на хирургичната интервенция, с повишено внимание върху мястото между задната камера и лещата. Това може да бъде извършено като съвсем леко се притисне оптиката на лещата постериорно, с I/A накрайник и чрез използване на стандартната техника за иригация/аспирация за отстраняване на виско-субстанция от окото. Така останалото количество виско-субстанция ще бъде изтласкано навън в предната камера, откъдето лесно може да бъде аспирирано.
ПРЕДПАЗНИ МЕРКИ 1. Тези вътреочни лещи и предварително заредената система за имплантиране UltraSert™ не трябва да се
стерилизират повторно по никакъв начин. 2. Не съхранявайте тези вътреочни лещи при температура над 45° C (113° F). 3. Ако се налага, лещите трябва да се пипат много внимателно, за да се избегне увреждане на повърхността
или хаптиките. 4. Не се опитвайте да промените формата на хаптиките по никакъв начин. 5. Необходимо е високо ниво на хирургично умение за извършване на имплантация на вътреочни лещи.
Преди да се опита да имплантира леща, хирургът трябва да е наблюдавал и/или асистирал в много операции и да е завършил успешно един или повече курсове за техника на имплантиране на вътреочни лещи.
6. Съдържанието е стерилно докато опаковката не е отворена или цялостта й не е нарушена. 7. UltraSert™ предварително заредената система за имплантация е само за еднократна употреба. Изхвърлете
предварително зареденото изделие UltraSert™ след употреба. 8. Използвайте UltraSert™ предварително заредената система за имплантация при операционна стайна
температура между 18° C (64° F) и 23° C (73° F).
ИЗЧИСЛЯВАНЕ НА УВЕЛИЧИТЕЛНАТА СПОСОБНОСТ НА ЛЕЩАТА (ДИОПТЪР)Необходимата увеличителна способност на тази заднокамерна вътреочна леща трябва де бъде изчислена от хирурга преди операцията, според преценката и опита му, както и според желаното от него място за поставяне на лещата. Методите за изчисляване на увеличителната способност на лещата са описани в следните препратки:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. et al. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. et al. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
ПРЕПОРЪЧИТЕЛНА А-КОНСТАНТАПрепоръчителната А-константа, посочена на външния етикет, е представена като препоръка и е начална база за започване на изчисленията на увеличителната способност. Препоръчва се всеки оператор да разработи своя константа, базирана на клиничния опит със съответния модел лещи, хирургичните техники, измервателна техника и пост-оперативните резултати.Ако ви е необходима допълнителна информация за изчисляване на увеличителната способност на лещата, за САЩ моля свържете се с Alcon Laboratories, Inc. 1-800-TO-ALCON (1-800-852-5266).Извън САЩ, обърнете се към местния офис или дистрибутори на Алкон.
ПОДХОДЯЩИ ВИСКОЕЛАСТИЧНИ СРЕДСТВА НА ALCON И КАТАЛОЖНИ НОМЕРАМоля вижте Приложението, намиращо се в опаковката. Трябва да се използват само подходящи Alcon вискоеластични средства. Използването на неодобрени вискоеластични средства може да причини увреждане на лещата и потенциални усложнения по време на процеса на имплантация. За подробен списък с подходящите за тази леща вискоеластични средства на Alcon, моля свържете се с Вашия представител на Alcon. ИНСТРУКЦИИ ЗА УПОТРЕБАСтъпка 1. Използвайте UltraSert™ предварително заредената система за имплантация при операционна стайна
температура между 18° C (64° F) и 23° C (73° F).
Стъпка 2. Проверете внимателно етикета на неотворената опаковка за модел, диоптър, правилна конфигурация и срок на годност.
Стъпка 3. След като отворите външната картонена кутия, огледайте опаковката на изделието за наранявания. Ако забележите нарушения, използвайте други AcrySof® IQ ВОЛ и UltraSert™ предварително заредена система за имплантация. След това, отново проверете всички посочени данни (например модел, диоптър и сериен номер) и се уверете, че информацията вътре и на външната опаковка съвпада.
24
Стъпка 4. За да извадите UltraSert™ предварително заредената система за имплантация, хванете ъгъла на пластмасовата поставка, отлепете изцяло фолиото от TYVEK† материал, и преместете изделието в стерилното поле. Ако след като го огледате, накрайникът на изделието изглежда наранен, деформиран, или се забелязват частици по него, използвайте други AcrySof® IQ ВОЛ и UltraSert™ предварително заредена система за имплантация. Ако изделието не е в първоначалния си вид, или буталото или ограничителя за буталото изглежда да се е преместил напред по време на транспорт, използвайте други AcrySof® IQ ВОЛ и UltraSert™ предварително заредена система за имплантация (вижте ПОЛИТИКА ЗА ВРЪЩАНЕ НА СТОКАТА).
†Регистрирана търговска марка (TM) на E.I. DuPont De Nemours & Co.
Когато сте готови да подготвите лещата за имплантиране, изпълнете стъпки 5, 6 и 7 последователно, с минимално забавяне между тях.
Стъпка 5. Вкарайте доркай канюлата с вискоеластично средство, перпендикулярно на изделието, през порта за вискоеластично средство, който се намира в тази част на изделието, където лещата спира. Това е показано на Figure 5. Напълнете изделието докато не забележите вискоеластичното средство да преминава до преходната линия на накрайника, и след това извадете канюлата. За това се изискват около 0.2 ml вискоеластично средство. Използвайте единствено вискоеластично средство на Alcon, подходящо за употреба с UltraSert™ предварително заредената система за имплантация, което предварително сте оставили да достигне операционната стайна температура. За списък с подходящи вискоеластични средства на Alcon, моля вижте Приложението в тази опаковка.
Вижте Figure 5
Viscoelastic Port = Отвор за вискоеластично средство“Fill-To” Line = Линия „Напълнете дотук“
Стъпка 6. Отстранете синия стопер за леща, като хванете частта със стрелкичката и я извиете леко напред, докато дърпате нагоре в посока извън изделието (Figure 6a). Отстранете синия предпазител на буталото, като го хванете и издърпате право нагоре в посока извън изделието (Figure 6b). Изхвърлете стопера за лещата и предпазителя на буталото. Не се опитвайте да добавите вискоеластично средство в изделието след като стопера за леща е вече отстранен, защото в противен случай лещата може да се увреди. Не се опитвайте да приберете буталото след като предпазителя за бутало вече е премахнат. В противен случай може да увредите буталото.
Вижте Figure 6a и 6b
Стъпка 7. Плавно придвижете буталото напред с едно гладко, продължително движение, докато предната част на оптиката не се изравни с маркировката на основното тяло (вижте Figure 7a). При това положение, синята пружина ще се свърже с безцветния фланец за пръст, както е показано на Figure 7b. Това изтласкване напред на буталото изисква най-малко 7 секунди. Не позволявайте нито една част от лещата да излиза извън върха на накрайника. Много е важно буталото да не се придвижва рязко, защото е възможно да се получи неправилно сгъване или увреждане на лещата.
Вижте Figure 7a и 7b
Fill-to Line = Линия „Напълнете дотук“
Стъпка 8. След като сте придвижили лещата до линията на накрайника, трябва да огледате лещата, за да определите позицията на хаптиките. Буталото трябва да бъде в контакт с влачещия се ръб на оптика. Никаква част от лещата не трябва да се подава от накрайника преди вкарването му през направения разрез (вижте Figure 8). След като лещата е в позиция на линията на накрайника, тя трябва да бъде имплантирана в рамките на 3 минути.
Вижте Figure 8
“Proceed with full plunger advancement to fully deliver lens” = “Продължете напред, като придвижите буталото пълно напред за имплантиране на лещата“
“Proceed” = “Продължете”“Proceed with full plunger advancement to fully deliver lens” = “Продължете напред, като придвижите буталото
пълно напред за имплантиране на лещата”“Proceed with device rotations per Step 10” = “Продължете със завъртания на изделието според стъпка 10”
“Do not proceed” = “Не продължавайте нататък”
Стъпка 9. След като се уверите, че лещата е правилно позиционирана, и хаптиките са сгънати правилно, можете да пристъпите към имплантиране на лещата, като вкарате върха на накрайника в разреза, до ограничителя за дълбочина (Figure 9), и насочите върха на накрайника към отвора в предната капсула.
25
Вижте Figure 9
Depth Guard = Ограничител на дълбочина
Стъпка 10. Внимателно придвижете буталото напред с едно плавно, непрекъснато движение. Не придвижвайте буталото прекалено бързо, защото лещата може да се повреди. Препоръчителното време за имплантирането на лещата от видимата част на накрайника в окото е най-малко 5 секунди. Ако водещата хаптика е в изправено положение или оплетена и излиза пред лещата, завъртете изделието наляво с наклонената страна преди да придвижите буталото, за да сте сигурни, че водещата хаптика е правилно позиционирана в капсулния сак. При излизането на водещата хаптика през накрайника, завъртете изделието назад, за да центрирате или леко придвижете наклонената част на острието надясно, ако е необходимо, за да сте сигурни, че лещата се разгъва в капсулния сак с предната страна нагоре.
Стъпка 11. Като използвате подходящ инструмент за позициониране, ориентирайте лещата в капсулния сак така, че да е в една успоредна равнина с ириса.
Стъпка 12. Изхвърлете цялото изделие. Не използвайте повторно UltraSert™ предварително заредената система за имплантация
РЕГИСТРИРАНЕ НА ПАЦИЕНТИТЕ И ДОКЛАДВАНЕИдентификационната карта за пациента, която е включена в тази опаковка, трябва да бъде попълнена и дадена на пациента. Трябва да инструктирате пациента да пази картата постоянно, и да я показва на всеки офталмолог при бъдещи прегледи.В САЩ, всеки един пациент трябва да бе регистриран в Alcon Laboratories, Inc., непосредствено след имплантация на тази леща. Регистрацията на пациентите е изключително важна за програмата за дългосрочно проследяване на пациентите, и ще помогне на Alcon Laboratories, Inc. за реагиране при нежелани събития. Извън САЩ трябва да се спазва местното законодателство относно регистриране на продукта.Всички събития, за които има причина да се счита, че са допринесли до това лещата да е предизвикала смърт или сериозно нараняване, включително и събития, които са били резултат от неспособността на изделието да функционира според спесификациите или предназначението му, трябва да бъдат докладвани на Alcon Laboratories, Inc. Тази информация се изисква от всички хирурзи, с цел да бъдат документирани всички потенциални дългосрочни ефекти от имплантирането на вътреочната леща. Хирурзите трябва да докладват нежеланите събития, свързани с тези вътреочни лещи.
Хирурзите трябва да използват следния адрес и телефон за докладване на нежелани събития, свързани с тези вътреочни лещи:
За САЩ: Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099.Call Toll free: 1-800-757-9780 за територията на САЩ.
За хирурзи извън САЩ, свържете се с местния представител на Alcon или неговите дистрибутори относно докладването на нежелани събития.
Клинични проучвания с AcrySof® сгъваеми заднокамерни лещиПроведени са две проспективни, рандомизирани, добре контролирани клинични проучвания с AcrySof® еднокомпонентни сгъваеми заднокамерни лещи. Първото проведено проучване е проведено с цел да демонстрира безопасността и ефективността на AcrySof® Natural еднокомпонентни заднокамерни лещи, модел SB30AL (филтриращ ултравиолетовата и синята светлина) като родствен модел леща. Това е рандомизирано клинично проучване, което включва AcrySof® модел SA30AL (абсорбиращ само ултравиолетовата светлина) като контролна леща. Включени са само данните от първoто оперирано око на тези пациенти, които са получили един от двата модела SB30AL или SA30AL вътреочни лещи. Второто рандомизирано клинично проучване, включващо AcrySof® IQ сгъваеми еднокомпонентни ВОЛ (с хромофори, филтриращи ултравиолетовата и синята светлина) с асферична оптика и AcrySof® акрилни сгъваеми еднокомпонентни контролни лещи, е проведено, за да се оценят клиничните/функционалните ползи над традиционния сферичен дизайн.
Клинично проучване с AcrySof® Natural еднокомпонентна леща модел SB30AL Постигнатите резултати от успешно проследените пациенти най-малко една година след операцията показват достатъчно потвърдително, че AcrySof® Natural еднокомпонентни лещи модел SB30AL са безопасни и ефективни изделия за зрителната корекция на афакията.
26
AcrySof® Natural еднокомпонента леща модел SB30AL – Пациенти Пациентите при това двустранно проучване, на които е имплантирана леща модел SB30AL поне в първото оперирано око, се разделят на 70.6% жени и 29.4%мъже. Пациентите, при които модел SА30AL е имплантирана като контролна леща се състоят от 60.5% жени и 39.5% мъже. Разделянето им по раса за модел SB30AL е 95.3% бяла и 4.7% черна. Разделянето по раса на контролната група пациенти (с имплантиран модел SA30AL) е 96.6% бяла, 2% черна и 1.4% други. Средната възраст на проследените пациенти, на които е имплантирана леща модел SB30AL поне в първото оперирано око е 72.9 години. Съответно, средната възраст на пациентите, при които модел SА30AL е имплантирана като контролна леща е 71.9 години.
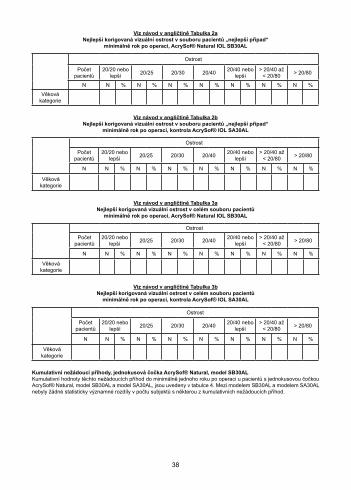
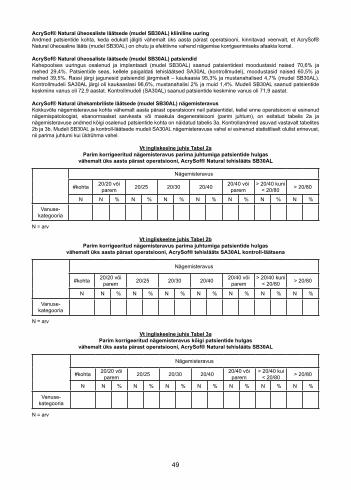
AcrySof® Natural еднокомпонента леща модел SB30AL – Зрителна остротаОбобщение на данните за зрителната острота, постигната пост-оперативно след период най-малко от една година, при пациенти без пре-оперативна очна патология, аномалии на роговицата или макулна дегенерация (най-добър случай) в нито един момент, са представени в Table 2а, а данните за зрителната острота при всички наблюдавани пациенти са представени в Table 3а. Контролните данни при същото разделяне на пациентите са дадени съответно в Таблици 2b и 3b. И в двата случая не са установени статистически значими разлики в зрителната острота между модел SB30AL и контролната леща, модел SA30AL, нито при най-добрия случай, нито при всички пациенти.
Вижте Table 2aНай-добра коригирана зрителна острота сред популацията с най-добрите случай с пациенти, най-малко
една година постоперативно, AcrySof® Natural ВОЛ SB30AL
Острота
#Per 20/20 или по-добра 20/25 20/30 20/40 20/40 или
по-добра
> 20/40 до
< 20/80> 20/80
N N % N % N % N % N % N % N %
Възрастова категория
Вижте Table 2bНай-добра коригирана зрителна острота при най-добрите случаи от пациенти, наблюдавани най-малко една
година след операцията, AcrySof® ВОЛ контролен модел SA30AL
Острота
#Per 20/20 или по-добра 20/25 20/30 20/40 20/40 или
по-добра
> 20/40 до
< 20/80> 20/80
N N % N % N % N % N % N % N %
Възрастова категория
Вижте Table 3aНай-добра коригирана зрителна острота при всички пациенти,
най-малко една година след операцията, AcrySof® Natural ВОЛ SB30AL
Острота
#Per 20/20 или по-добра 20/25 20/30 20/40 20/40 или
по-добра
> 20/40 до
< 20/80> 20/80
N N % N % N % N % N % N % N %
Възрастова категория
Вижте Table 3bНай-добре коригирана зрителна острота, постигната при всички наблюдавани пациенти, най-малко една
година след операцията, AcrySof® ВОЛ контролен модел SA30AL
Острота
#Per 20/20 или по-добра 20/25 20/30 20/40 20/40 или
по-добра
> 20/40 до
< 20/80> 20/80
N N % N % N % N % N % N % N %
Възрастова категория
27
AcrySof® Natural еднокомпонентни лещи модел SB30AL – кумулативни нежелани реакцииБроят на кумулативните нежелани събития, наблюдавани най-малко една година след операцията при пациенти с имплантирани AcrySof® Natural еднокомпонентна леща модел SB30AL и модел SA30AL са показани в Таблица 4. Няма статистически значими разлики между модел SB30AL и модел SA30AL при разделянето на пациентите, изпитали някое от кумулативните нежелани събития.
Tаблица 4 Кумулативни нежелани събития, наблюдавани най-малко една година след операцията
AcrySof® Natural ВОЛ модел SB30AL и контролен модел SA30AL
SB30AL (N= 153)
SA30AL(N= 147) p-стойност*
N % N %
Вид нежелано събитие
0 0.0 0 0.0 NAКумулативно Хипопион
Вътреочна инфекция / Ендофталмит 0 0.0 0 0.0 NA
Оток на макулата 4 2.6 2 1.4 0.6847
Зеничен блок 0 0.0 0 0.0 NA
Отлепване на ретината или възстановяване на отлепването 0 0.0 0 0.0 NA
Изместване на лещата 1 0.7 0 0.0 1.0000
Вторична хирургична интервенцияОтстраняване на остатъчен кортексЕксплантация (изместване поради руптури в капсулата)Криотерапия за възстановяване на отлепването на ретинатаПарасентезис за понижаване на ВОНФокално лазерно лечениеФотодинамична терапияЕксплантация, поради биометрична грешка
51 1
11100
3.3 0.7
0.7
0.70.70.70.00.0
200
00011
1.40.00.0
0.00.00.00.70.7
0.4482
Хифема 0 0.0 0 0.0 NA
* p-стойности от Fisher’s Exact Test, сравняващ Model SB30AL с Model SA30AL
AcrySof® Natural еднокомпонентни лещи модел SB30AL – Персистиращи нежелани събитияБроят на персистиращите нежелани събития, наблюдавани най-малко една година след операцията при пациенти с имплантирани AcrySof® Natural еднокомпонентна леща модел SB30AL и модел SA30AL, контрола, са дадени в Таблица 5. Няма статистически значими разлики между модел SB30AL и модел SA30AL при разделянето на пациентите изпитали някое от персистиращите нежелани събития.
28
Tаблица 5Персистиращи нежелани събития, наблюдавани най-малко една година след операцията
AcrySof® Natural ВОЛ SB30AL и SA30AL, контрола
SB30AL (N= 138)
SA30AL(N= 127) p-стойност*
N % N %
Тип нежелано събитие
0 0 1 0.8 0.4792Персистиращо Оток на роговицата
Ирит 0 0 0 0.0 NA
Оток на макулата 2 1.4 1 0.8 1.0000
Възпаления на стъкловидното тяло 0 0 0 0.0 NA
Повишено ВОН, изискващо лечение 0 0 0 0.0 NA
* p-стойности от Fisher’s Exact Test, сравняващ Model SB30AL с Model SA30AL.
AcrySof® Natural еднокомпонентни лещи модел SB30AL – Възприемане на цветовете Тестовете за възприемане на цветовете са направени чрез Farnsworth D-15 Panel Test в периода от 120 до 180 ден след операцията. От 109 субекта с нормално цветно зрение и имплантирана леща модел SB30AL в първото оперирано око, наблюдавани при пост-оперативна визита между 120 и 180 ден от операцията, 107 (98.2%) са преминали успешно теста за цветно зрение. От 102 субекта с нормално цветно зрение и имплантирана леща модел SА30AL в първото оперирано око, наблюдавани при пост-оперативна визита между 120 и 180 ден от операцията, 97 (95.1%) са преминали успешно изпитването за цветно зрение. Няма статистически значими разлики между модел SB30AL и модел SА30AL в процента пациенти, които са преминали успешно теста за възпремане на цветовете, проведен при пост-оперативна визита межу 120 и 180 ден. Следователно, добавеният патентован хромофор не оказва отрицателно въздейстие върху цветното зрение при пациенти с нормално цветно зрение преди операцията.
AcrySof® Natural еднокомпонентни лещи модел SB30AL Nd:YAG лазер случаи При проследяване на пациентите след средно 21.6 месеца от операцията, при три (3) от 135 субекта (2.2%), на които е била имплантирана SB30AL, е направена Nd:YAG лазер задна капсулотомия. При проследяване на пациентите след средно 21.9 месеца от операцията, при два (2) от 127 субекта (1.6%), на които е била имплантирана SА30AL, е направена Nd:YAG лазер задна капсулотомия.
Клинично проучване с AcrySof® IQ лещиПодобно на други провеждани проучвания с ВОЛ, възрастни пациенти в добро общо здраве (напр. без предишна очна операция, дегенеративни зрителни нарушения, които значително биха повлияли на зрителната острота, или остро, или хронично състояние, което може да увеличи риска за пациента) с билатерална катаракта са включени в контролирано, рандомизирано, двойно-маскирано, многоцентрово, клинично проучване с контралатерален имплант AcrySof® IQ и сферични лещи като контрола. Очните сферични аберации са статистически значително по-малко с AcrySof® IQ в сравнение с контролните лещи. Резултатите, свързани с контрастната чувствителност, показват значително пост-оперативно (след 3 месеца) подобрение при имплантация на AcrySof® IQ. При имплантация на AcrySof® IQ леща също така се установява значително статистическо и клинично подобрение при измерване на функционалното зрение, симулирано нощно шофиране при ярка светлина и мъгла. Тези резултати показват, че AcrySof® IQ ВОЛ (с асферична оптика върху материал, съдържащ хромофор, филтриращ синята светлина) има по-добри клинични показатели в сравнение с монофокална AcrySof® ВОЛ (без асферична оптика и хромофор, филтриращ синята светлина).
AcrySof® IQ лещи – сферични и други аберации от висок порядък Средната обща сферична аберация на очи с AcrySof® IQ е приблизително 0.1 микрометра. Figure 10 показва статистически значимото намаление на сферичните и другите аберации, наблюдавани в полза на лещи AcrySof® IQ. Figure 11 представя измерванията на средната сферична аберация на всички очи, направени с топограф - аберометър, по лещи и възрастова група. Както показва тази таблица, намалението на сферичната аберация при пациенти, на които е имплантирана AcrySof® IQ, е независимо от възрастта.
Вижте Figure 10Сферични и други аберации от висок порядък RMS
90-120 дни след 2ри очен имплант
Вижте Figure 11Средна сферична аберация - обща и по възрастови групи
90-120 дни след 2ри очен имплант
29
AcrySof® IQ лещи – Зрителна острота надалечЛещата AcrySof® IQ и контролната леща осигуряват клинично сходна постоперативна зрителна острота. Резултатите от монокулярно изследване на зрителната острота са представени на Figures 12 и 13.
Вижте Figure 12LogMAR BCVA
Вижте Figure 13LogMAR UCVA
AcrySof® IQ лещи – Контрастна чувствителностОсновната цел на клиничното проучване е да покаже предимството на лещите AcrySof® IQ пред контролните лещи чрез средната контрастна чувствителност, измерена пост-оперативно при мезопични условия с и без ярка светлина при всяка от двете пространстветни честоти (3 или 6 цикъла за градус), използвайки Vector Vision CSV-1000 (с осветеност на таблоцата 3 cd/m2). При част от пациентите е направен и Функционален тест за контрастна чувствителност (Functional Acuity Contrast Test - FACT) с осветеност на таблицата 3 cd/m2. При това клинично проучване, предимствата на лещите AcrySof® IQ пред контролните лещи при мезопични условия са демонстрирани при 6 цикъла за градус с и без ярка светлина (CSV-1000) и при 3 и 6 цикъла за градус без ярка светлина (FACT). Figures 14 и 15 описват резултатите на мезопична контрастна чувствителност при всички тествани пространствени честоти за лещи AcrySof® IQ и контролни лещи.
Минимум 90 дни след 2ри очен имплант AcrySof® IQ лещи – Симулиране на нощно шофиране Проведено е проучване с група пациенти за оценка на шофиране при нощни условия, като е бил използван валидиран симулатор за нощно шофиране. Пациентите са тествани монокулярно при условия, които симулират градски и извънградски условия при нормални условия, ярка светлина и мъгла. Симулирането на нощно шофиране в градски условия включва разнообразие от улични лампи, фарове на коли, светлини от магазини и знаци, за да се пресъздаде високата степен на осветеност, типична за тези условия. Симулирането на нощно шофиране в извънградски условия включва минимално количество заобикаляща светлина. Симулираната скорост на шофиране е приблизително 35 mph (=miles per hour/мили в час, или 56 km/h) и 55 mph (89 km/h) за градски и извънградски условия, респективно.Пациентите са били помолени да разграничат и идентифицират различни обекти във всяка сцена, включително бяло-зелените информационни знаци на магистралата, черно-жълтите предупредителни знаци и пешеходци. Пациентите са попитани кога са забелязали първия обект, което позволява да се определи разстояние за забелязване. След това пациентите са попитани кога са разпознали обекта (напр., какво пише на знака, в каква посока се движи пешеходецът, и др.), така че да се установи разстоянието за разпознаване. Фигури от 16 до 19 показват средните разлики между лещите AcrySof® IQ и контролни лещи, при шофиране в градски и извънградски условия, при разстоянията за забелязване и разпознаване (напр., средните интра-индивидуални разлики).
Лещите AcrySof® IQ се представят функционално по-добре от контролата при 34 от тестваните 36 условия, като се наблюдава подобрение при разстоянията за забелязване и разпознаване както в градски, така и в извънградски условия, при различните тествани условия на шофиране (нормални, ярка светлина, мъгла). Още повече, лещите AcrySof® IQ са се представили статистически значително по-добре, в сравнение с контролните, при 12 от тези условия. Най-голямото им предимство се наблюдава при забелязване и разпознаване на пешеходците в градски условия (при ярка светлина и мъгла) и предупредителните знаци извън града (при ярка светлина и мъгла). При условия на намалена видимост (ярка светлина, мъгла) в градски условия, увеличената зрителна дистанция на 35 mph (56 km/h) осигурява на пациентите с имплантирани лещи AcrySof® IQ повече от 0.5 секунди допълнително време да реагират на пешеходец - опасност, която се среща по-често в градовете. Това увеличение от 0.5 секунди е важно, защото увеличава времето за предприемане на подходящи действия като спиране, избягване и др. (Green, 2000; McBride and Matson, 2004). При всички извънградски условия, увеличаването на зрителното разстояние при 55 mph (89 km/h) осигурява на пациентите с имплантирани лещи AcrySof® IQ повече от 1 секунда допълнително време за идентифициране на предупредителните знаци, ситуация често срещана в извънградски райони. Допълнително време от 0.5 секунди е от фуинкционално значение, защото увеличава времето за предприемане на подходящи действия по време на шофиране, което е изключително важно при нощни условия в непознати извънградски райони, където уличното осветление често липсва. При 6 пациента в подизследването се наблюдава пост-оперативна макулна дегенерация или PCO. Когато тези пациенти са махнати от анализа за шофиране, разликата между ВОЛ при забелязване и разпознаване на пешеходци при условия на ярка светлина в градски условия не достига 0.5 секунди – прага на клинично значение. Когато първоначалните анализи са коригирани за множественост, разликата между ВОЛ вече не е статистически важна за забелязване на текст при ярка светлина в градски условия (Hommel‘s p-value = 0.0539) или за
30
разпознаване на пешеходци при ярка светлина в извънградски услловия (Hommel‘s p-value=0.0507).Тези резултати показват подобрение на фукнционалното зрение и възможни значителни допълнителни ползи, свързани с безопасност за възрастните шофьори с имплантирани лещи AcrySof® IQ, които са и индиректна полза за другите шофьори и пешеходци на пътя. Резултатите от този тест показват, че лещите AcrySof® IQ подобряват функционалното зрение, което може да подобри безопасността на пациента и в други ситуации с условия на ниска видимост.
Вижте Figure 16Симулиране на нощно шофиране
Средни интра-индивидуални разлики при разстояния на забелязване, градски условия минимум 90 дни след операцията
AcrySof® IQ ВОЛ – контрола (n = 44)
Вижте Figure 17Симулиране на нощно шофиране
Средни интра-индивидуални разлики при разстояния за идентификация, градски условия,минимум 90 дни след операциятаAcrySof® IQ ВОЛ – контрола (n=44)
Вижте Figure 18Симулиране на нощно шофиране
Средни интра-индивидуални разлики при разстояния за забелязване, извънградски условияминимум 90 дни след операциятаAcrySof® IQ IOL – контрола (n=44)
Вижте Figure 19Симулиране на нощно шофиране
Средни интра-индивидуални разлики при разстояния за идентификация, извънградски условия минимум 90 дни след операциятаAcrySof® IQ IOL – контрола (n=44)
ОПАКОВКАТези заднокамерни вътреочни лещи се доставят сухи, в предварително заредената система за имплантиране UltraSert™, в първична стерилизирана опаковка, финално стерилизирана с етилен оксид. Първичната стерилизирана опаковка трябва да се отваря само при асептични условия (вижте “ИНСТРУКЦИИ ЗА УПОТРЕБА”).
СРОК НА ГОДНОСТСтерилността на лещите е гарантирана, освен в случаите когато цялостта на първичната стерилизирана опаковка е нарушена. Срокът на годност на всяка леща е ясно отбелязан върху етикета на външната опаковка на UltraSert™ предварително заредената система за имплантиране. При изтичане на срока на годност, неотворените лещи трябва да се върнат обратно в Alcon Laboratories, Inc. (Вижте “ПОЛИТИКА ЗА ВРЪЩАНЕ НА СТОКАТА”).
ПОЛИТИКА ЗА ВРЪЩАНЕ НА СТОКАТАНа територията на САЩ при връщане лещите задължително се заменят с други продукти, никога срещу сума. Всички върнати лещи се съпровождат от съответен номер за върната стока на Alcon Laboratories, Inc. и трябва да бъдат изпратени по проследим начин. Номерът за върната стока се получава от Отдел Клиенти на Alcon. Издаването на този номер не представлява приемане на върнатите продукти. За повече информация относно замяна на продукти, моля свържете се с Вашия медицински или търговски представител.Извън територията на САЩ, моля обърнете се към офиса или дистрибуторите на Алкон в страната относно приложимата политика за връщане на стока.
ИЗПОЛЗВАНИ ИЗТОЧНИЦИ 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., et al. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., et al. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking
Response Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
31
СИМВОЛИ, ИЗПОЛЗВАНИ ВЪРХУ ОПАКОВКАТА
СИМВОЛ БЪЛГАРСКИIOL Вътреочна леща
SP Еднокомпонентна
UV ултравиолиетов
D диоптър
ØB Диаметър на тялото (диаметър на оптиката)
ØT Общ диаметър (обща дължина)
2STERILIZE Не стерилизирайте повторно
Не използвайте повторно
Използвайте до
STERILE EO Стерилизирано с етилен оксид
SN Сериен номер
Внимание
Производител
EC REP Упълномощен представител в Европейската общност
Горен температурен лимит
Отворете тук
Прочетете инструкцията за употреба
Не използвайте, ако стерилната опаковка е била наранена или отворена
Внимание: Федералният закон на САЩ изисква това изделие да се продава от или по препоръка на лекар
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 САЩ
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,Обединено кралство
Представител, вносител и дистрибутор за България:Алкон България ЕООДСердика Център, бул. “Ситняково” 48, ет.8, София 1505тел. 950 15 65
STERILNÍ akrylátová složitelná jednokusová čočka pro zadní oční komoru filtrující UV a modré světlo se zaváděcím systémem UltraSert™
POPISAkrylátová složitelná jednokusová čočka pro zadní oční komoru AcrySof® IQ, filtrující UV a modré světlo, je optický implantát pro nahrazení lidské krystalické čočky při vizuální korekci afakie u dospělých pacientů po operaci katarakty. Nitrooční čočky (NOČ) AcrySof® IQ s patentovaným chromoforem pro filtraci modrého světla působí tak, že napodobují lidskou krystalickou čočku v rozsahu vlnových délek modrého světla 400–475 nm (Boettner a Wolter, 1962). Kromě standardní filtrace UV světla snižuje materiál NOČ AcrySof® IQ propustnost pro vlnové délky příslušející modrému světlu od 62 % při 400 nm až po 23 % při 475 nm (viz tabulka 1). Optická část se skládá z měkkého akrylátového materiálu o vysokém indexu lomu. Tento materiál lze před vložením složit. Čočka se po implantaci jemně rozloží do svého tvaru. Tyto čočky mají bikonvexní optiku s podpůrnými haptikami (obrázek 1). Asférický povrch zadní strany nitroočních čoček AcrySof® IQ (model AU00T0) je navržen s negativní sférickou aberací, aby kompenzoval kladnou sférickou aberaci průměrné rohovky. Kvalita obrazu jednokusových NOČ, model AU00T0 (tj. modulační přenosová funkce), je uvedena na obrázku 4. Jednokusové NOČ AcrySof® IQ, model AU00T0, jsou dodávány v předplněném zaváděcím systému UltraSert™ (obrázek 2) pro pohodlné a řízené zavádění těchto čoček na správné místo do pouzdra čočky. Fyzikální vlastnosti těchto čoček jsou následující:
Fyzikální charakteristika PopisTyp optiky Jednokusové NOČ s asférickou optikou
Materiál optiky Kopolymer akrylátu/metakrylátu, filtrující ultrafialové a modré světlo
Snížení UV záření při 10% propustnosti (nm) UV (400) při 20,0 D (viz obrázek 3)
Index lomu 1,55
Optická síla Dostupný rozsah síly viz Průvodce výrobků společnosti Alcon
Uspořádání haptik STABLEFORCE® Modified-L Haptics
Materiál haptik Kopolymer akrylátu/metakrylátu, filtrující ultrafialové a modré světlo
Průměr optické části ØB (mm) 6,0
Celková délka ØT (mm) 13,0
Úhel haptik 0º
Rozměry optické části Viz obrázek 1
Viz návod v angličtině Obrázek 1FYZIKÁLNÍ CHARAKTERISTIKA
Všechny rozměry jsou v milimetrech
ASPHERIC SURFACE = ASFÉRICKÝ POVRCH
Viz návod v angličtině Obrázek 2
Nozzle Tip = Hrot nástavceDepth Guard = Chránič hloubky
Main Body = Hlavní tělo
CZECH
33
Viz návod v angličtině Obrázek 3KŘIVKY SPEKTRÁLNÍ PROPUSTNOSTI
TRANSMITTANCE (%) = PROPUSTNOST (%)WAVELENGTH (nm) = VLNOVÁ DÉLKA (nm)
6.0 D to 27.0 D range = Rozpětí 6,0 až 27,0 dioptrií4 yr – 53 yr old range, crystalline lens = Věkové rozpětí 4 až 53 let, krystalická čočka
POZNÁMKY: • Uvedené křivky spektrální propustnosti s mezní vlnovou délkou představují rozsah hodnot propustnosti
nitroočních čoček vyrobených z kopolymeru akrylátu/metakrylátu s navázaným absorbérem UV záření a chromoforem filtrujícím modré světlo patentovaným společností Alcon.
• Uvedené hodnoty byly naměřeny metodou přímé propustnosti pomocí otvoru 6 mm a destičky o tloušťce ekvivalentní optickému středu.
• Data pro lidskou čočku od autorů Boettner a Wolter (1962).
Tabulka 1Srovnání propustnosti pro nitrooční čočky 20,0 D (%)
Model 400 nm 425 nm 450 nm 475 nm
SA60AT 21 86 88 88
AU00T0 8 34 49 68
Rozdíl propustností (SA60AT – AU00T0) 13 52 39 20
Snížení propustnosti u AU00T0 (% SA60AT ) 62 60 44 23
Viz návod v angličtině Obrázek 4Modulační přenosová funkce modelu AU00T0 (20,0 D)
ISO, 3-mm Aperture (see Note 1) = ISO, 3mm otvor (viz poznámka 1)Modified, 3-mm Aperture (see Note 2) = Modifikovaný 3mm otvor (viz poznámka 2)Modified, 5-mm Aperture (see Note 2) = Modifikovaný 5mm otvor (viz poznámka 2)
POZNÁMKY: 1. Kvalita obrazu modelu AU00T0 byla charakterizována měřením modulační přenosové funkce (MTF) na modelu
oka, popsaném v ISO 11979-2. ISO 11979-2 požaduje měření MTF pouze s 3mm otvorem. 2. Kromě toho byla kvalita obrazu modelu AU00T0 charakterizována měřením MTF na modelu oka, využívajícího
simulovanou rohovku, vykazující typickou sférickou aberaci dospělého člověka. Pomocí modifikovaného modelu oka bylo provedeno měření MTF pomocí jak 3mm, tak 5mm otvoru.
ZPŮSOB ÚČINKU Nitrooční čočky pro zadní oční komoru AcrySof® IQ jsou určeny pro vložení do zadní oční komory, aby tak nahradily přírodní krystalické čočky. Tato poloha umožňuje, aby čočka fungovala jako refrakční médium při korekci afakie. Asférická bikonvexní optika snižuje sférickou aberaci v porovnání se standardní sférickou optikou lidského oka. Účinnost těchto čoček při snižování výskytu poruch sítnice nebyla stanovena.
INDIKACE Nitrooční čočky AcrySof® IQ pro zadní komoru jsou indikovány jako náhrada lidských čoček pro dosažení vizuální korekce afakie u dospělých pacientů po operaci katarakty (viz VAROVÁNÍ). Tyto čočky jsou určeny pro vložení do pouzdra.
UPOZORNĚNÍ Pacienti trpící níže uvedenými onemocněními nemusí být vhodnými kandidáty pro nitrooční čočky, protože čočky by mohly jejich současný stav zhoršovat, narušovat stanovení diagnózy nebo léčbu určitých onemocnění, nebo by mohly vytvářet neodůvodněné riziko pro zrak pacienta. Lékař by měl před implantací čočky provést pečlivé předoperační vyšetření a důkladně klinicky zhodnotit poměr mezi přínosem a rizikem, jedná-li se o pacienta s následujícími problémy: 1. krvácení do cévnatky 2. souběžné závažné oční onemocnění 3. nadměrná ztráta sklivce 4. mimořádně mělká přední komora 5. mikroftalmie 6. šedý zákal nepodmíněný věkem
34
7. ruptura zadního očního pouzdra (bránící fixaci NOČ) 8. závažná dystrofie rohovky 9. závažná atrofie optického nervu 10. nekontrolovatelný nitrooční tlak 11. zonulární separace (bránící fixaci NOČ) 12. nedostatečné barevné vidění 13. glaukom 14. chronická uveitida 15. diabetická retinopatie 16. klinicky významné změny v makulární oblasti / retinálním pigmentovém epiteluStudie prokázaly, že rozlišovací schopnost barevného vidění není u jednotlivců s nitroočními čočkami AcrySof® Natural a normálním barevným viděním negativně ovlivňována. Účinek nitroočních čoček AcrySof® Natural u jednotlivců s dědičnými poruchami barevného vidění a poruchami barevného vidění vyvolanými očními chorobami (např. glaukomem, diabetickou retinopatií, chronickou uveitidou nebo dalšími onemocněními sítnice nebo optického nervu) nebyl studován.
VAROVÁNÍ 1. Stejně jako u jiných chirurgických zákroků existuje i zde určité riziko. Potenciální komplikace doprovázející
operaci katarakty nebo zavádění implantátu mohou obnášet, aniž by byl následující výčet považován za definitivní: poškození endotelu rohovky, endoftalmitidu, odchlípení sítnice, vitritidu, cystoidní makulární edém, edém rohovky, pupilární blok, membránu řasnatého tělíska, vyhřeznutí duhovky, hypopyon a přechodný nebo přetrvávající glaukom.
2. Bezpečnost a účinnost implantátů nitroočních čoček nebyly doloženy u pacientů se stávajícími očními onemocněními (chronickou lékovou miózou, glaukomem, amblyopií, diabetickou retinopatií, dřívější transplantací rohovky, dřívějším odchlípením sítnice a/nebo iritidou atd.). Lékaři, kteří mají u těchto pacientů posuzovat implantaci čočky, by měli zvážit alternativní metody korekce afakie a o implantaci čočky by měli uvažovat pouze tehdy, jsou-li alternativní možnosti považovány za neuspokojivé při zajištění potřeb pacienta.
3. Dlouhodobé účinky implantace nitroočních čoček nebyly stanoveny. Proto by měli lékaři i nadále pacienta po operaci pravidelně sledovat.
4. Pacienti s předoperačními problémy, jako je onemocnění endotelu rohovky, abnormální rohovka, makulární degenerace, degenerace sítnice, glaukom a chronická léková mióza, nemohou docílit takové zrakové ostrosti jako pacienti, kteří těmito problémy netrpí. Pokud mají pacienti výše uvedené obtíže, měl by lékař stanovit, jaký přínos bude případná implantace čočky pro pacienta představovat.
5. Druhotné iridektomii kvůli pupilárnímu bloku lze zamezit, je-li jedna nebo více iridektomií provedena současně s implantací NOČ (Willis a spol., 1985).
6. Bezpečnost a účinnost čočky pro zadní komoru, vložené do přední komory, nebyla stanovena. Implantace čoček pro zadní komoru do přední komory se v některých případech ukázala být nespolehlivá (Girard a spol., 1983).
7. Některé nežádoucí reakce, které jsou spojeny s implantací nitroočních čoček, obnášejí: hypopyon, nitrooční infekci, akutní dekompenzaci rohovky a sekundární chirurgickou intervenci. Sekundární chirurgické intervence zahrnují, aniž by byly omezeny jenom na následující výčet: repozici čočky, výměnu čočky, aspiraci sklivce nebo iridektomii z důvodu pupilárního bloku, opravu prosakování rány a opravu odchlípnutí sítnice.
8. U malého počtu pacientů s nevycentrovanými čočkami, především u NOČ s úzkou nebo malou optikou, může dojít za určitých světelných podmínek k pocitům oslnění nebo jiným poruchám vidění. Chirurgové by tuto možnost měli uvážit před implantací NOČ s úzkou nebo malou optikou. Při implantaci úzké nebo malé optické čočky se doporučuje provedení kapsulorhexe.
9. S technikami kapsulorhexe a implantací PMMA, silikonových a akrylátových čoček pro zadní oční komoru jsou spojovány pooperační distenze pouzdra čočky s různými hodnotami změlčení přední komory a indukovanou myopií (Holtz, 1992).
10. Aby se zabránilo decentraci nebo dislokaci čoček, mělo by se před zapouzdřením čoček postupovat s nejvyšší opatrností. Některé klinické případy dokazují, že k zapouzdření dochází v průběhu čtyř týdnů.
11. Klinická studie jednokusových čoček AcrySof® Natural (viz tabulky 2 až 5) byla provedena pouze u čoček určených k implantaci v pouzdře. Neexistují žádná klinická data pro prokázání jejich bezpečnosti a účinnosti při umístění do ciliárního sulku.
Doporučuje se, aby se viskoelastický materiál odstranil na konci operačního zákroku z oka s důrazem na prostor mezi zadním pouzdrem a čočkou. Toho lze docílit lehkým stlačením optiky NOČ směrem vzad irigačním/aspiračním hrotem a použitím standardních technik irigace a aspirace pro odstranění viskoelastického materiálu z oka. To by mělo vytlačit veškerý zachycený viskoelastický materiál směrem vpřed, kde jej lze snadno odsát.
BEZPEČNOSTNÍ OPATŘENÍ 1. Tyto nitrooční čočky a předplněný zaváděcí systém UltraSert™ se nesmí podruhé sterilizovat, a to žádným
způsobem. 2. Neskladujte nitrooční čočky při teplotách přesahujících 45 °C (113 °F). 3. V případě nutnosti manipulujte s čočkami opatrně, aby nedošlo k poškození jejich povrchu nebo haptik. 4. Žádným způsobem se nepokoušejte změnit tvar haptik. 5. Pro implantaci nitrooční čočky je nezbytný vysoký stupeň chirurgické dovednosti. Chirurg by se měl nejdříve
zúčastnit pozorování velkého počtu implantací a/nebo by měl u nich asistovat a měl by úspěšně projít jedním nebo více školeními implantace nitrooční čočky, teprve potom by se měl o implantaci nitrooční čočky pokusit sám.
35
6. Obsah je sterilní, není-li balení otevřeno či poškozeno. 7. Předplněný zaváděcí systém UltraSert™ je určen pouze pro jedno použití. Po použití předplněné zařízení
UltraSert™ zlikvidujte. 8. Předplněný zaváděcí systém UltraSert™ používejte při teplotách operačního sálu mezi 18 °C (64 °F) a 23 °C
(73 °F).
VÝPOČET SÍLY ČOČKYChirurg by měl pro tyto nitrooční čočky pro zadní komoru provést předoperační výpočet požadované síly čočky na základě svých zkušeností, preferencí a zamýšleného umístění čočky. Metody výpočtu síly čočky jsou popsány v následující literatuře:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. et al. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. et al. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract. Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
DOPORUČENÁ KONSTANTA A Doporučená konstanta A, uvedená na vnějším štítku, je udávána jako vodítko a slouží jako výchozí bod pro výpočet síly implantátu. Doporučuje se, abyste si odvodili svou vlastní konstantu, která odpovídá klinické zkušenosti s určitým modelem čočky, chirurgické technice, měřicímu zařízení a pooperačním výsledkům.Potřebujete-li další informace o výpočtu síly čočky, spojte se prosím se společností Alcon Laboratories, Inc. na čísle 1-800-TO-ALCON (1-800-862-5266). Mimo USA se spojte s místními zástupci nebo distributory Alcon.
DOPORUČENÝ VISKOELASTICKÝ MATERIÁL A OBJEDNACÍ ČÍSLA SPOLEČNOSTI ALCONViz dodatek přiložený k tomuto balení. Měl by být používán pouze viskoelastický materiál schválený společností Alcon. Použití neschváleného viskoelastického materiálu může způsobit poškození čočky a případné komplikace během implantace. S žádostí o úplný seznam viskoelastického materiálu schváleného společností Alcon se prosím obraťte na svého zástupce společnosti Alcon.
NÁVOD K POUŽITÍKrok 1. Předplněný zaváděcí systém UltraSert™ používejte při teplotách operačního sálu mezi 18 °C (64 °F) a 23 °C (73 °F).
Krok 2. Prohlédněte štítek na neotevřeném balení a zkontrolujte model, sílu, odpovídající konfiguraci a datum exspirace.
Krok 3. Po otevření vnější krabice prověřte, zda je balení nepoškozené. Zjistíte-li poškození, použijte jinou NOČ AcrySof® IQ a jiný předplněný zaváděcí systém UltraSert™. Poté ověřte, zda je informace na krabičce s čočkami (např. model, síla a číslo série) shodná s informací na štítku vnějšího obalu.
Krok 4. Pro vyjmutí předplněného zaváděcího systému UltraSert™ uchopte roh plastového podnosu, úplně sloupněte materiál TYVEK† části víčka a přeneste nástavec do sterilního prostředí. Pokud se po prohlédnutí zdá, že je hubice nástavce poškozená, drolí se nebo je zdeformovaná, použijte jinou NOČ AcrySof® IQ a jiný předplněný zaváděcí systém UltraSert™. Není-li systém zcela neporušený nebo se zdá, že se při dopravě pohnul píst nebo pojistka pístu, použijte jinou NOČ AcrySof® IQ a jiný předplněný zaváděcí systém UltraSert™ (viz POSTUP PŘI VRACENÍ ZBOŽÍ).
†Registrovaná ochranná známka E.I. DuPont De Nemours & Co.
Je-li vše připraveno pro zavedení čočky, proveďte kroky 5, 6 a 7 s minimálním odstupem mezi jednotlivými kroky.
Krok 5. Úplně vložte kanylu na viskoelastikum kolmo k nástavci skrz otvor pro viskoelastikum, umístěné v části zarážky čočky nástavec tak, jak je zobrazeno na obrázku 5. Naplňte zařízení tak, aby bylo viskoelastikum vidět, jak protéká kolem rysky hubice (viz šipka na obrázku 5), poté kanylu vytáhněte. K tomu bude potřeba přibližně 0,2 ml viskoelastika. Při použití předplněného zaváděcího systému UltraSert™ používejte pouze doporučený viskoelastický materiál společnosti Alcon, který umožňuje používání při teplotě operačního sálu. Seznam doporučeného viskoelastického materiálu společnosti Alcon je uveden v dodatku přiloženém k tomuto balení.
Viz návod v angličtině Obrázek 5
Viscoelastic Port = Otvor pro viskoelastický materiál“Fill-To” Line = Ryska „Fill-To“
36
Krok 6. Vyjměte modrou zarážku čočky uchopením otvíracího kroužku se šipkou, mírně zahněte dopředu a vytáhněte rovně vzhůru ze zařízení (obrázek 6a). Vyjměte modrou pojistku pístu uchopením a vytažením rovně vzhůru ze zařízení (obrázek 6b). Zarážku čočky a pojistku pístu zlikvidujte. Nepokoušejte se přidávat do zařízení viskoelastikum po odstranění zarážky čočky, jinak může dojít k poškození čočky. Nepokoušejte se vytahovat píst po odstranění pojistky pístu, jinak může dojít k poškození pístu.
Viz návod v angličtině Obrázky 6a a 6b
Krok 7. Jemně posuňte píst dopředu hladkým a nepřetržitým pohybem, dokud se přední okraj optiky nesrovná s ryskou na hubici (viz obrázek 7a). V této poloze se bude modrá pružina dotýkat zarážky těla injektoru tak, jak je znázorněno na obrázku 7b. Tento pohyb pístu by měl trvat alespoň 7 sekund. Žádná část čočky nesmí vystupovat z hrotu hubice. Je důležité nepohybovat pístem trhavě, protože může dojít k nesprávnému složení a poškození čočky.
Viz návod v angličtině Obrázky 7a a 7b
Fill-to Line = Ryska „Fill-to“
Krok 8. Po posunu čočky k rysce na hubici je třeba čočku vizuálně prohlédnout, aby byla stanovena poloha haptiky. Píst by měl být v kontaktu s taženým koncem optiky. Žádná část čočky by neměla opustit hubici před vložením do incize (viz obrázek 8). Jakmile se čočka nachází v poloze u linky na hubici, měla by být do 3 minut implantována.
Viz návod v angličtině Obrázek 8
“Proceed with full plunger advancement to fully deliver lens” = Pokračujte v posouvání pístu, dokud se čočka zcela nezasune.
“Proceed” = Pokračujte.“Proceed with full plunger advancement to fully deliver lens” = Pokračujte v posouvání pístu, dokud se čočka
zcela nezasune.“Proceed with device rotations per Step 10” = Pokračujte s otáčením zařízení dle kroku 10.
“Do not proceed” = Nepokračujte.
Krok 9. Po potvrzení, že je čočka ve správné poloze a haptika se řádně složila, pokračujte v implantaci vložením hrotu hubice do incize až k hloubkové ochraně (obrázek 9) a nasměrujte hrot hubice k otvoru předního pouzdra.
Viz návod v angličtině Obrázek 9
Depth Guard = Chránič hloubky
Krok 10. Jemně stlačte píst jedním plynulým a nepřerušovaným pohybem. Nestlačujte píst příliš rychle, mohlo by dojít k poškození čočky. Doporučuje se, aby zavedení čočky z pozice, kde je vizuálně kontrolována, trvalo alespoň 5 sekund. Jsou-li vodicí haptiky rovné nebo zatočené a rozprostírají se před čočkou, otočte zařízením doleva před posunutím pístu, aby bylo zajištěno správné umístění vodicích haptik v pouzdře čočky. Jakmile začnou vodicí haptiky opouštět hrot hubice, umístěte vodicí haptiky do pouzdra čočky ve správném směru a pokračujte pomalým posouváním pístu pro zavedení čočky. Jakmile začne optika opouštět hrot hubice, otočte zařízením zpět pro vycentrování, nebo v případě potřeby zlehka otočte doprava, aby bylo zajištěno, že se čočka v pouzdře čočky rozvine přední stranou vzhůru.
Krok 11. Upravte polohu čočky v pouzdře čočky do roviny rovnoběžně s duhovkou za použití vhodného polohovacího nástroje.
Krok 12. Celé zařízení zlikvidujte. Předplněný zaváděcí systém UltraSert™ nepoužívejte opakovaně.
REGISTRACE PACIENTŮ A HLÁŠENÍIdentifikační kartu pacienta (Patient Identification Card), přiloženou v obalu, je třeba vyplnit a předat pacientovi společně s pokyny ohledně uložení karty jako trvalého záznamu, který je třeba předložit každému očnímu lékaři, který bude pacienta léčit v budoucnu.Ve Spojených státech se musí bezprostředně po implantaci jedné z těchto čoček každý pacient zaregistrovat u společnosti Alcon Laboratories, Inc. Registrace se provádí vyplněním karty registrace implantátu (Implant Registration Card), přiložené v krabičce s čočkami, a jejím zasláním společnosti Alcon Laboratories, Inc. Registrace pacienta je pro Alcon Laboratories, Inc. zásadní záležitostí pro dlouhodobý program péče o pacienty a pomáhá nám reagovat na případné zprávy o nežádoucích příhodách.Mimo Spojené státy by měly být dodržovány místní zákony týkající se registrace výrobku.
Nežádoucí příhody, které mohou být dávány do souvislosti s čočkami, a takové, které způsobily nebo přispěly k úmrtí nebo závažnému zranění, včetně nežádoucích příhod vyvstávajících v důsledku nesplnění výkonnostních charakteristik
37
či jiného rozporu s určeným účelem zdravotnického prostředku, by se měly nahlásit společnosti Alcon Laboratories, Inc. Tato informace je požadována od všech chirurgů, aby byly zdokumentovány potenciální dlouhodobé účinky implantace nitroočních čoček. Pro hlášení nežádoucích příhod týkajících se těchto nitroočních čoček mohou chirurgové využít následující adresu a telefonní číslo:
Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Bezplatné telefonní číslo: +1-800-757-9780
Mimo Spojené státy se spojte ohledně zpráv o nežádoucích příhodách s místním zastoupením společnosti Alcon nebo s distributory. Chirurgové by měli použít následující adresu a telefonní číslo pro nahlášení nežádoucích příhod:Alcon Pharmaceuticals (Czech Republic) s.r.o.Na Pankráci 1724/129140 00 Praha 4Tel.: +420 724 824 543 (vigilanční linka)
Klinické studie složitelných čoček AcrySof® pro zadní komoruPro jednokusové složitelné čočky AcrySof® pro zadní komoru byly provedeny dvě randomizované, prospektivní a kontrolované klinické studie. První studie byla provedena s cílem prokázat bezpečnost a účinnost jednokusové čočky AcrySof® Natural, model SB30AL (filtrující UV a modré světlo), pro zadní komoru, jako model rodičovské čočky. Jednalo se o randomizovanou klinickou studii, kde jako kontrolní čočka sloužila pouze čočka AcrySof®, model SA30AL (absorbující pouze UV světlo). Zařazena jsou pouze data z prvního operovaného oka od těch subjektů, kteří dostali nitrooční čočky – buď model SB30AL, nebo model SA30AL. Druhá randomizovaná klinická studie složitelných jednokusových čoček AcrySof® IQ (s filtrujícími chromofory pro UV a modré světlo) s asférickou optikou, provádějící srovnání s kontrolními složitelnými jednokusovými čočkami AcrySof®, byla provedena s cílem stanovit klinické a funkční výhody oproti tradičnímu sférickému návrhu.
Klinická studie jednokusových čoček AcrySof® Natural, model SB30ALVýsledky dosažené pacienty úspěšně sledovanými po dobu minimálně jednoho roku po operaci poskytují přiměřenou jistotu, že jednokusové čočky AcrySof® Natural, model SB30AL, jsou bezpečným a účinným prostředkem pro vizuální korekci při afakii.
Soubor pacientů jednokusových čoček AcrySof® Natural, model SB30ALSoubor pacientů této studie, kterým byl implantován model SB30AL do minimálně poprvé operovaného oka, obsahuje 70,6 % žen a 29,4 % mužů. Soubor pacientů, jimž byl implantován model SA30AL (kontrola), se skládá ze 60,5 % žen a 39,5 % mužů. Podle rasy u modelu SB30AL patří 95,3 % ke kavkazské rase a 4,7 % k černé rase. V kontrolním souboru pacientů (SA30AL) patřilo 96,6 % ke kavkazské rase, 2 % pacientů k černé rase a 1,4 % k jiné rase. Průměrný věk celého souboru pacientů, kterým byl implantován minimálně jednou model SB30AL, činí 72,9 roku. Stejně tak je průměrný věk pro celý soubor pacientů, kteří obdrželi model SA30AL (kontrolu), 71,9 roku.
Ostrost zraku u subjektů s jednokusovými čočkami AcrySof® Natural, model SB30ALShrnutí ostrosti zraku, které bylo dosaženo minimálně po jednom roce po operaci u subjektů, u kterých nebyl zaznamenán kdykoliv před operací žádný patologický stav, abnormální rohovka nebo makulární degenerace (tzv. Best Case, tj. „nejlepší případ“), je uvedeno v tabulce 2a. Shrnutí ostrosti zraku v celém souboru pacientů je uvedeno v tabulce 3a. Kontrolní data jsou uvedena pro stejný soubor dat v tabulkách 2b a 3b. V ostrosti zraku nebyl zaznamenán mezi modelem SB30AL a kontrolní čočkou, modelem SA30AL, žádný statisticky významný rozdíl ani v nejlepších případech nebo v celkovém souboru dat.
38
Viz návod v angličtině Tabulka 2aNejlepší korigovaná vizuální ostrost v souboru pacientů „nejlepší případ“
minimálně rok po operaci, AcrySof® Natural IOL SB30AL
Ostrost
Počet pacientů
20/20 nebo lepší 20/25 20/30 20/40 20/40 nebo
lepší> 20/40 až
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Věková kategorie
Viz návod v angličtině Tabulka 2bNejlepší korigovaná vizuální ostrost v souboru pacientů „nejlepší případ“
minimálně rok po operaci, kontrola AcrySof® IOL SA30AL
Ostrost
Počet pacientů
20/20 nebo lepší 20/25 20/30 20/40 20/40 nebo
lepší> 20/40 až
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Věková kategorie
Viz návod v angličtině Tabulka 3aNejlepší korigovaná vizuální ostrost v celém souboru pacientů
minimálně rok po operaci, AcrySof® Natural IOL SB30AL
Ostrost
Počet pacientů
20/20 nebo lepší 20/25 20/30 20/40 20/40 nebo
lepší> 20/40 až
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Věková kategorie
Viz návod v angličtině Tabulka 3bNejlepší korigovaná vizuální ostrost v celém souboru pacientů
minimálně rok po operaci, kontrola AcrySof® IOL SA30AL
Ostrost
Počet pacientů
20/20 nebo lepší 20/25 20/30 20/40 20/40 nebo
lepší> 20/40 až
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Věková kategorie
Kumulativní nežádoucí příhody, jednokusová čočka AcrySof® Natural, model SB30ALKumulativní hodnoty těchto nežádoucích příhod do minimálně jednoho roku po operaci u pacientů s jednokusovou čočkou AcrySof® Natural, model SB30AL a model SA30AL, jsou uvedeny v tabulce 4. Mezi modelem SB30AL a modelem SA30AL nebyly žádné statisticky významné rozdíly v počtu subjektů s některou z kumulativních nežádoucích příhod.
39
Tabulka 4Kumulativní nežádoucí příhody minimálně jeden rok po operaci
Sekundární chirurgický zákrokOdstranění reziduálního kortexuExplantace (dislokace kvůli roztržení pouzdra)Kryoterapie pro opravu trhliny v sítniciParacentéza pro snížení nitroočního tlakuOhnisková léčba laseremFotodynamická terapieExplantace kvůli chybě v biometrii
51 1 11100
3,30,70,70,70,70,70,00,0
20000011
1,40,00,00,00,00,00,70,7
0,4482
Hyféma 0 0,0 0 0,0 Neuvedeno
*Hodnoty p z Fisherova exaktního testu, porovnávajícího model SB30AL s modelem SA30AL.
Přetrvávající nežádoucí příhody, jednokusová čočka AcrySof® Natural, model SB30ALPřetrvávající hodnoty těchto nežádoucích příhod minimálně jeden rok po operaci u pacientů s jednokusovou čočkou AcrySof® Natural, model SB30AL a kontrolní model SA30AL, jsou uvedeny v tabulce 5. Mezi modelem SB30AL a modelem SA30AL nebyly žádné statisticky významné rozdíly v počtu subjektů s některou z přetrvávajících nežádoucích příhod.
Tabulka 5Přetrvávající nežádoucí příhody minimálně jeden rok po operaci AcrySof® Natural IOL SB30AL a kontrola SA30AL
*Hodnoty p z Fisherova exaktního testu, porovnávajícího model SB30AL s modelem SA30AL.
Barevné vnímání u jednokusových čoček AcrySof® Natural, model SB30ALTestování barevného vnímání pomocí testu s Farnsworthovým panelem D-15 bylo prováděno 120 až 180 dnů po operaci. Ze 109 subjektů s normálním barevným viděním, jimž byl implantován model SB30AL u prvního operovaného oka a kteří byli vyšetřeni na pooperační návštěvě 120 až 180 dnů po operaci, prošlo 107 (98,2 %) testem barevného vnímání. Ze 102 subjektů s normálním barevným viděním, jimž byl implantován model SA30AL u prvního operovaného oka a kteří byli vyšetřeni na pooperační návštěvě 120 až 180 dnů po operaci, prošlo testem barevného vnímání 97 pacientů (95,1 %). Mezi modelem SB30AL a SA30AL nebyly žádné statisticky významné rozdíly u procenta subjektů, kteří prošli testem barevného vnímání při pooperační návštěvě 120 až 180 dnů po operaci. Přídavek patentovaného chromoforu tedy barevné vidění u pacientů s normálním barevným viděním negativně neovlivňuje.
40
Hodnoty Nd:YAG pro jednokusové čočky AcrySof® Natural, model SB30AL Při průměrné době dalšího sledování 21,6 měsíce došlo u tří (3) subjektů ze 135 (2,2 %), jimž byla implantována čočka, model SB30AL, k Nd:YAG kapsulotomii zadního pouzdra. Při průměrné době dalšího sledování 21,9 měsíce došlo u dvou (2) subjektů ze 127 (1,6 %), jimž byla implantována čočka model SA30AL, ke Nd:YAG kapsulotomii zadního pouzdra.
Klinická studie čočky AcrySof® IQStejně, jako tomu bylo u návrhu podobných, již dříve prováděných studií s nitroočními čočkami, byly do kontrolované, randomizované, dvakrát zamaskované, multicentrické kontralaterální klinické studie s čočkami AcrySof® IQ a sférickými kontrolními čočkami zařazeny dospělé subjekty v celkovém dobrém zdravotním stavu očí (např. bez předchozí oční operace, degenerativní poruchy vidění, jež by významně ovlivňovala ostrost vidění, nebo závažným akutním či chronickým onemocněním, jež by mohlo zvyšovat riziko pro pacienta). Oční sférické aberace byly statisticky významně nižší u čoček AcrySof® IQ než u kontrolních čoček. Výsledky kontrastní citlivosti prokázaly statisticky významné zlepšení (po 3 měsících) ve prospěch očí, do nichž byly implantovány čočky AcrySof® IQ. U očí, do nichž byly implantovány čočky AcrySof® IQ, také došlo ke statisticky a klinicky významnému zlepšení při měření funkčního vidění, simulovaném nočním řízení za několika různých testovacích podmínek – především co se týče oslnění a mlhy. Tyto výsledky odrážejí skutečnost, že NOČ AcrySof® IQ (asférická optika na materiálovém základu, obsahující chromofor filtrující modré světlo) poskytují oproti monofokálním NOČ AcrySof® (bez asférické optiky a chromoforu filtrujícího modré světlo) klinický přínos.
Čočka AcrySof® IQ – Aberace sférické a celkově vyššího řáduPrůměrná oční sférická aberace očí s čočkami AcrySof® IQ byla přibližně 0,1 mikrometru. Obrázek 10 představuje statisticky významné snížení sférických aberací a aberací celkově vyššího řádu, pozorovaných ve prospěch čočky AcrySof® IQ. Obrázek 11 představuje průměrné hodnoty sférických aberací všech očí, měřených vlnovým aberometrem podle skupin dle čočky a věku. Jak je uvedeno v této tabulce, snížení sférické aberace u očí s čočkou AcrySof® IQ nebylo závislé na věku.
Viz návod v angličtině Obrázek 10Sférické a celkově vyššího řádu RMS90–120 dnů po implantaci do 2. oka
Viz návod v angličtině Obrázek 11Průměrná sférická aberace celkově a dle věkových skupin
90–120 dnů po implantaci do 2. oka
Čočka AcrySof® IQ – Vizuální ostrost na dálkuČočka AcrySof® IQ a kontrolní čočka poskytují klinicky podobnou pooperační vizuální ostrost. Výsledky monokulární vizuální ostrosti jsou uvedeny na obrázcích 12 a 13.
Viz návod v angličtině Obrázek 12LogMAR BCVA
Viz návod v angličtině Obrázek 13LogMAR UCVA
Čočka AcrySof® IQ – kontrastní citlivostPrimárním cílem klinického zkoušení bylo prokázat lepší vlastnosti čočky AcrySof® IQ oproti kontrolní čočce prostřednictvím průměrné kontrastní citlivosti, měřené po operaci za mezopických podmínek s oslněním nebo bez něj při některé ze dvou prostorových frekvencí (3 nebo 6 cyklů na stupeň) pomocí přístroje Vector Vision CSV-1000 (s navrženou svítivostí 3 cd/m2). U podmnožiny pacientů byl také proveden test kontrastu funkční ostrosti (FACT) (s navrženou svítivostí 3 cd/m2). Při tomto klinickém výzkumu byla prokázána superiorita čočky AcrySof® IQ nad kontrolní čočkou při 6 cyklech na stupeň jak s oslněním, tak bez něj (CSV-1000) a při 3 a 6 cyklech na stupeň bez oslnění (FACT). Obrázky 14 a 15 zobrazují výsledky mezopické kontrastní citlivosti při všech prostorových frekvencích, zkoušených pro čočku AcrySof® IQ i pro kontrolní čočku.
Viz návod v angličtině Obrázek 14Mezopická kontrastní citlivost (CSV-1000)
90–120 dnů po implantaci do 2. oka
Viz návod v angličtině Obrázek 15Podstudie mezopické kontrastní citlivosti (FACT)
minimálně 90 dnů po implantaci do 2. oka
Čočka AcrySof® IQ – simulace nočního řízeníPodmnožina pacientů podstoupila testování na validovaném simulátoru nočního řízení. Pacienti byli testováni monokulárně za podmínek, jež simulují podmínky ve městě i na venkově za normálních podmínek, při oslnění a při mlze.Scéna nočního řízení ve městě využívá řady pouličních světel, světel automobilů, osvětlení obchodů a značek pro vytvoření vysoké úrovně okolního osvětlení, typického pro tyto podmínky. Scéna řízení v noci na venkově využívá minimální okolní osvětlení. Pro řízení ve městě a na venkově byly použity simulované rychlosti řízení 35 mil za hodinu, resp. 55 mil za hodinu.
41
Pacienti byli požádáni, aby detekovali a identifikovali sérii cílů v každé scéně, včetně zelenobílých silničních informačních cedulí, černo-žlutých výstražných značek a chodců. Pacienti měli reagovat v okamžiku, kdy uvidí první cíl, což umožnilo zaznamenat detekční vzdálenost. Poté byli pacienti požádáni, aby reagovali, až cíl rozliší (např. co značka uvádí, kterým směrem chodec kráčí atd.), aby bylo možno zaznamenat identifikační vzdálenost. Na obrázcích 16 až 19 jsou uvedeny průměrné rozdíly mezi čočkou AcrySof® IQ a kontrolní čočkou v místech řízení ve městě a na venkově pro detekční a identifikační vzdálenosti (např. průměr intraindividuálních rozdílů).Čočka AcrySof® IQ zaznamenala u 34 ze 36 testovaných podmínek funkčně lepší výsledky než tomu bylo u kontroly, což odráží zlepšení detekčních i identifikačních vzdáleností jak ve městě, tak na venkově při různých testovaných podmínkách řízení (normálních, při oslnění a za mlhy). Kromě toho čočka AcrySof® IQ vykázala statisticky významně lepší výsledky než kontrola ve 12 z těchto podmínek, přičemž největší dopad a největší výhody byly pozorovány při detekci a identifikaci chodců ve městě (za podmínek oslnění a při mlze) a u výstražných značek na venkově (při podmínkách oslnění a za mlhy). Za snížených podmínek viditelnosti (oslnění, mlha) ve městě poskytuje zvýšená vzdálenost pro viditelnost při 35 mílích za hodinu u čočky AcrySof® IQ více než o 0,5 sekundy delší čas k reakci na chodce, což je riziko, s nímž se běžněji lze setkat za podmínek řízení ve městě. Toto zvýšení o 0,5 sekundy je funkčně významné pro získání delšího času na vhodné činnosti jako zastavení, vyhýbání atd. (Green 2000; McBride a Matson 2004). Za všech podmínek mimo město poskytuje zvýšená viditelnost při 55 mílích za hodinu u čočky AcrySof® IQ více než 1 sekundu dalšího času pro identifikaci výstražných značek, což je situace, s níž se lze za venkovských podmínek často setkat. Zvýšení o 0,5 sekundy je funkčně významné pro umožnění delší doby k přijetí vhodných opatření při řízení, což je kritické v noci v neznámých venkovských oblastech, kde často schází okolní osvětlení. V substudii bylo 6 pacientů, u nichž po operaci došlo k makulární degeneraci nebo PCO. Když byli tito pacienti vyjmuti z analýzy výsledků řízení, rozdíl mezi oběma NOČ pro detekci a identifikaci chodců za podmínek oslnění ve městě nevyhověl 0,5sekundovému prahu pro klinickou významnost. Když byly původní analýzy upraveny na multiplicitu, nebyl již rozdíl mezi NOČ pro detekci oslněného textu ve městě (Hommelova hodnota p = 0,0539) nebo pro detekci chodců na venkově při oslnění (Hommelova hodnota p = 0,0507) statisticky významný.Tyto výsledky prokazují zlepšené funkční vidění a pravděpodobně užitečný přínos k bezpečnosti u starších řidičů s čočkami AcrySof® IQ i pro ostatní řidiče a chodce, pohybující se také na vozovce. Výsledky tohoto testu prokazují, že čočka AcrySof® IQ zlepšuje funkční vidění, jež může naopak zlepšit bezpečnost pacienta pro další životní situace za podmínek snížené viditelnosti.
Viz návod v angličtině Obrázek 16Simulátor nočního řízení
Průměrné intraindividuální rozdíly detekčních vzdáleností vidění, městoMinimálně 90 dnů pooperačně
AcrySof® IQ IOL – kontrola (n = 44)
Viz návod v angličtině Obrázek 17Simulátor nočního řízení
Průměrné intraindividuální rozdíly identifikačních vzdáleností vidění, městoMinimálně 90 dnů pooperačně
AcrySof® IQ IOL – kontrola (n = 44)
Viz návod v angličtině Obrázek 18Simulátor nočního řízení
Průměrné intraindividuální rozdíly detekčních vzdáleností vidění, venkovMinimálně 90 dnů pooperačně
AcrySof® IQ IOL – kontrola (n = 44)
Viz návod v angličtině Obrázek 19Simulátor nočního řízení
Průměrné intraindividuální rozdíly identifikačních vzdáleností vidění, venkovMinimálně 90 dnů pooperačně
AcrySof® IQ IOL – kontrola (n = 44)
ZPŮSOB DODÁVKYTyto nitrooční čočky pro zadní komoru jsou dodávány v suchém stavu, v předplněném zaváděcím systému UltraSert™, v primárním sterilizačním balení a terminálně sterilizované ethylenoxidem. Primární sterilizační balení se musí otevřít pouze za aseptických podmínek (viz NÁVOD K POUŽITÍ).
DATUM EXSPIRACESterilita je zaručena, pokud není pouzdro poškozeno nebo otevřeno. Datum exspirace je jasně vyznačeno na vnějším štítku na krabičce předplněného zaváděcího systému UltraSert™. Každou čočku, u které došlo k překročení data exspirace, je třeba vrátit společnosti Alcon Laboratories, Inc. (viz POSTUP PŘI VRACENÍ ZBOŽÍ).
POSTUP PŘI VRACENÍ ZBOŽÍVe Spojených státech budou vracené čočky přijaty pouze výměnou za jiné produkty, ne za peníze. Veškeré vracené zboží musí být opatřeno číslem vraceného zboží Alcon Laboratories Inc. a musí být zasláno sledovatelnými prostředky. Číslo vraceného zboží lze získat od oddělení služeb zákazníkům Alcon. Vydání tohoto čísla neznamená, že budou vracené výrobky nakonec přijaty. Podrobné podmínky, upravující výměnu, získáte od svého obchodního zástupce nebo zástupce
42
oddělení služeb zákazníkům. Mimo Spojené státy se spojte při vracení zboží s místním zastoupením společnosti Alcon nebo s distributorem.
REFERENCE 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., a spol. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., a spol. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking
Response Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; květen 2004.
43
SYMBOLY POUŽÍVANÉ NA ŠTÍTCÍCH
SYMBOL VÝZNAMIOL Nitrooční čočka (NOČ)
SP Jednokusová
UV Ultrafialový
D Dioptrie
ØB Průměr těla čočky (průměr optiky)
ØT Celkový průměr (celková délka)
2STERILIZE Nesterilizujte znovu
Nepoužívejte opakovaně
Použijte do
STERILE EO Sterilizováno ethylenoxidem
SN Sériové číslo
Upozornění
Výrobce
EC REP Zplnomocněný zástupce v EU
Horní teplotní limit
Zde otevřete
Viz návod k použití
Nepoužívejte, je-li sterilní obal otevřen nebo poškozen
Upozornění: Federální zákony USA omezují prodej tohoto výrobku pouze pro lékaře nebo na jeho objednávku
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 USA
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,Velká Británie
Distributor/Dovozce:Alcon Pharmaceuticals (Czech Republic) s.r.o.Na Pankráci 1724/129140 00 Praha 4
Datum poslední revize textu: 04/2015
44
KASUTUSJUHISAlcon Laboratories, Inc.
STERIILSED UV ja sinist valgust filtreerivad akrüülist volditavad üheosalisedtagakambriläätsed koos UltraSert™ eellaetud sisestussüsteemiga
KIRJELDUSAcrySof® IQ UV- ja sinise valguse filtriga akrüülist volditavad üheosalised tagakambriläätsed on optilised implantaadid inimese silmaläätse asendamiseks ja nägemise parandamiseks afaakia korral täiskasvanud patsientidel pärast katarakti operatsiooni. AcrySof® IQ tehislääts on kaetud Alcon’i patenteeritud sinist valgust filtreeriva kromofooriga ning filtreerivad sinist valgust lähedaselt inimese silmaläätsele 400 – 475 nm (Boettner ja Wolter, 1962). Lisaks standardsele UV-valguse filtreerimisele vähendab AcrySof® IQ tehislääts sinise valguse lainepikkuse läbitavust alates 62% 400 nm juures kuni 23% 475 nm juures (vt tabel 1). Optiline osa koosneb kõrge refraktsiooni indeksiga pehmest akrüülmaterjalist. Sellisest materjalist valmistatud läätsi võib enne silma sisestamist kokku voltida. Pärast implantatsiooni avaneb lääts kogu suuruses. Lääts on kaksikkumer ja tal on küljes jalakesed (joonis 1). AcrySof® IQ tehisläätse mudeli AU00T0 mittesfääriline tagapind on loodud negatiivse sfäärilise aberratsiooniga, et kompenseerida keskmise sarvkesta positiivset sfäärilist aberratsiooni. Üheosalise tehisläätse mudeli AU00T0 kujutise kvaliteet (s.o modulatsiooni siirdamise funktsioon) on näidatud joonisel 4. AcrySof® IQ üheosalise tehisläätse mudel AU00T0 on tarnitud UltraSert™ eellaetud sisestussüsteemiga (joonis 2), et läätsi mugavalt ja ohutult kapslikotti paigaldada. Läätsede füüsikalised omadused on alljärgnevad:
Füüsikaline omadus KirjeldusOptika tüüp Asfäärilise optikaga üheosaline tehislääts
Optiline materjal Ultraviolett- ja sinist valgust filtreeriv akrülaat/metakrülaat kopolümeer
UV murdepunkt 10% transmissiooni juures (nm) UV (400) 20.0 D juures (vt joonis 3)
Refraktsiooniindeks 1,55
Optiline tugevus Olemasolevate tugevusvahemike kohta vt Alcon’i tootejuhendit
6.0 D to 27.0 D range = vahemik 6,0 D kuni 27,0 D4 yr – 53 yr old range, crystalline lens = vanusevahemik 4 aastat – 53 aastat, inimese silmalääts
MÄRKUSED: • Siintoodud piirlainepikkus ja spektraalläbitavuse kõverad näitavad akrülaat/metakrülaat-kopolümeerist UV-kaitsega
ja Alcon’i patenteeritud sinist valgust filtreeriva kromofooriga tehisläätsede läbitavuse väärtuste vahemikku. • Otseläbitavuse mõõtmiseks kasutati 6 mm ava ja plaati, mille paksus on sama kui läätse optilisel keskpunktil. • Inimese läätse andmed Boettner ja Wolter (1962).
ISO, 3-mm Aperture (see Note 1) = ISO, 3 mm ava (vt märkus 1)Modified, 3-mm Aperture (see Note 2) = Modifitseeritud, 3 mm ava (vt märkus 2)Modified, 5-mm Aperture (see Note 2) = Modifitseeritud, 5 mm ava (vt märkus 2)
MÄRKUSED: 1. Mudeli AU00T0 kujutise kvaliteeti kirjeldati modulatsiooni siirdamise funktsiooni mõõtmisega (MTF) silma mudelil,
mida kirjeldati standardis ISO 11979-2. Standard ISO 11979-2 nõuab MTF mõõtmiseks ainult 3 mm ava. 2. Lisaks kirjeldati mudeli AU00T0 kujutise kvaliteeti MTF mõõtmisega silmamudelil, mis kasutas järele tehtud
sarvkesta, millel oli tüüpiline täiskasvanud inimese sarvkesta sfääriline aberratsioon. Kasutades modifitseeritud silma mudelit, tehti MTF mõõtmised nii 3 mm kui ka 5 mm ava abil.
TOIMEMEHHANISM AcrySof® IQ intraokulaarseid tagakambriläätsesid kasutatakse silma loomuliku läätse asendamiseks tagakambris. Sellise paigutusega lääts toimib afaakia korrigeerimisel murdumiskeskkonnana. Võrreldes standardse sfäärilise läätsega, vähendab mittesfääriline kaksikkumer lääts sfäärilist aberratsiooni rohkem. Läätsede efektiivsust võrkkesta haiguste vähendamisel ei ole tõestatud.
NÄIDUSTUSED AcrySof® IQ tagakambri tehisläätsed on mõeldud inimese silmaläätsede asendamiseks ja nägemise parandamiseks afaakia korral täiskasvanud patsientidel pärast katarakti operatsiooni (vt HOIATUSED). Läätsed on mõeldud kapslikotti paigaldamiseks.
ETTEVAATUST Tehisläätse paigaldamine võib osutuda mittesobivaks patsientide puhul, kellel esineb alltoodud haigusseisundeid, kuna läätsed võivad selliseid seisundeid halvendada, segada nende diagnoosimist ja ravi või ohustada patsiendi nägemist. Patsientide puhul, kellel esineb üks või mitu alltoodud haigusseisundeist, peab kirurg otsuse tegemiseks hindama patsiendi seisundit enne operatsiooni ja kaaluma tõsiselt kasu/ohu vahekorda enne läätse implanteerimist: 1. Soonkesta hemorraagia 2. Kaasuv raske silmahaigus 3. Ülemäärane klaaskeha kaotus 4. Äärmiselt madal eeskamber 5. Mikroftalmia 6. Vanusega mitteseotud katarakt 7. Tagakapsli rebend (tehisläätse pole võimalik fikseerida) 8. Raske sarvkesta düstroofia
46
9. Raske nägemisnärvi atroofia 10. Kontrollimatu positiivne rõhk 11. Zonula’te rebenemine (tehisläätse pole võimalik fikseerida) 12. Värvipimedus 13. Glaukoom 14. Krooniline uveiit 15. Diabeetiline retinopaatia 16. Kliiniliselt olulised maakula/RPE muutusedUuringud on näidanud, et AcrySof® Natural läätsi omavatel ja normaalse värvusnägemisega inimestel ei teki kõrvaltoimena värvusnägemise halvenemist. AcrySof® Natural läätsede mõju päriliku ja omandatud värvusnägemise defektidega inimeste nägemisele, kellel esineb ka teisi silmahaigusi (näit glaukoom, diabeetiline retinopaatia, krooniline uveiit või muud võrkkesta või läätse haigused), pole uuritud.
HOIATUSED 1. Nagu iga kirurgilise protseduuri puhul, on ka siin olemas teatud ohud. Katarakti operatsiooni või tehisläätse
implantatsiooniga seotud potentsiaalsed ohud võivad olla, kuid mitte ainult, järgmised: sarvkesta endoteeli kahjustus, endoftalmiit, võrkkesta irdumine, vitreiit, maakula tsüstoidne ödeem, sarvkesta turse, pupillaarne blokk, tsükliitiline membraan, iirise prolaps, hüpopüon ja mööduv või püsiv glaukoom.
2. Silmasiseste tehisläätsede ohutust ja efektiivsust ei ole tõestatud varem tekkinud silma haiguslike seisundite korral (ravimitest tingitud krooniline mioos, glaukoom, amblüoopia, diabeetiline retinopaatia, varem tehtud sarvkesta transplantatsioon, varem esinenud võrkkesta irdumine ja/või iriit jm). Arstid, kes kaaluvad tehisläätse implanteerimist sellistel patsientidel, peaksid uurima afaakia korrigeerimiseks alternatiivseid meetodeid ja kaaluma läätsede implanteerimist ainult juhul, kui alternatiivsed meetodid ei vasta patsiendi vajadustele.
3. Tehisläätsede pikaajalist mõju ei ole kindlaks tehtud. Seetõttu peavad arstid patsiente pärast operatsiooni regulaarselt jälgima.
4. Patsientidel, kellel esines enne operatsiooni sarvkesta endoteeli haigus, ebanormaalne sarvkest, maakula degeneratsioon, võrkkesta degeneratsioon, glaukoom ja ravimitest põhjustatud krooniline mioos, võib nägemisteravus jääda kehvemaks võrreldes patsientidega, kellel sellised seisundid puuduvad. Arst peab selliste haigete puhul otsustama, kas läätse implantatsioon on õigustatud.
5. Pupillaarsest blokist tingitud sekundaarset iridektoomiat võib vältida, kui tehisläätse implantatsiooni ajal teha üks või mitu iridektoomiat (Willis, et al., 1985).
6. Tagakambri läätsede ohutust ja efektiivsust nende paigaldamisel eeskambrisse ei ole tõestatud. Tagakambriläätsede paigaldamine eeskambrisse on mõnel juhul osutunud ohtlikuks (Girard, et al., 1983).
7. Mõnede tehisläätsede implantatsiooniga seotud kõrvaltoimed on järgmised: hüpopüon, silmasisene infektsioon, akuutne sarvkesta dekompensatsioon, sekundaarne kirurgiline sekkumine. Sekundaarne kirurgiline sekkumine tähendab, kuid mitte ainult: läätse repositsiooni, läätse asendamist, klaaskeha aspiratsiooni või iridektoomiat pupillaarse bloki tõttu, haavalekke korrigeerimist ja võrkkesta irdumise ravi.
8. Mõningase detsentreerituse korral, eriti kui tehisläätsel on kitsas või väike optiline osa, võib patsient tunnetada teatud valgustingimuste juures peegeldust või muid nägemishäireid. Kirurg peab selliste nähtudega arvestama, kui tehisläätse optiline osa on kitsas või väike. Kitsa või väikese optikaga läätse paigaldamisel soovitatakse teha kapsuloreksis.
9. Kapslikoti venimist pärast operatsiooni ning eeskambri mõningast madaldumist ja müoopiat seostatakse kapsuloreksise tehnikatega ning tagakambri PMMA-, silikoon- ja akrüülläätsede implantatsiooniga (Holtz, 1992).
10. Enne läätsede inkapsulatsiooni tuleb olla ettevaatlik, et vältida läätsede detsentreeritust või dislokatsiooni. Mõne kliinilise juhtumi puhul soovitatakse teha inkapsulatsioon neli nädalat pärast implantatsiooni.
11. AcrySof® Natural üheosaliste läätsede kliiniline uuring (viidatud tabelites 2 kuni 5) tehti läätsedega, mis on mõeldud paigaldamiseks ainult kapsulaarkotti. Seetõttu puuduvad ka andmed nende ohutuse ja efektiivsuse kohta, kui läätsed paigaldada tsiliaarsulkusesse.
Viskoelastik soovitatakse operatsiooni lõpus silmast eemaldada, pöörates tähelepanu tagakapsli ja läätse vahelisele ruumile. Seda võib teha, vajutades I/A-otsaga õrnalt tehisläätsele ja kasutades viskoelastiku silmast eemaldamiseks standardseid irrigatsiooni-/aspiratsioonimeetodeid. Sellega surutakse liigne viskoelastik silma esiossa, kust seda on lihtne eemaldada.
ETTEVAATUSABINÕUD 1. Ärge steriliseerige tehisläätsi või UltraSert™ eellaetud sisestussüsteemi mingil viisil uuesti. 2. Tehisläätsi tuleb hoida temperatuuril alla 45° C (113° F). 3. Läätsi tuleb käsitseda ettevaatlikult, et vältida läätse pinna või jalakeste vigastusi. 4. Ärge üritage muuta jalakeste kuju. 5. Tehisläätsede implantatsioon nõuab kõrgetasemelisi kirurgilisi oskusi. Tehisläätsi võib paigaldada kirurg,
kes on osalenud ja/või assisteerinud paljudel implantatsioonidel ja lõpetanud edukalt tehisläätsede implantatsioonikursused.
6. Pakendi sisu on steriilne, kui pakend on avamata ja kahjustamata. 7. The UltraSert™ eellaetud sisestussüsteem on ainult ühekordseks kasutamiseks. UltraSert™ eellaetud seade
tuleb pärast kasutamist hävitada. 8. Kasutage UltraSert™ eellaetud sisestussüsteemi toatemperatuuridel vahemikus 18° C (64° F) kuni 23° C (73° F).
47
LÄÄTSE OPTILISE TUGEVUSE ARVESTUSSiintoodud tagakambri tehisläätsede nõutud optiline tugevus tuleb kindlaks määrata enne operatsiooni vastavalt kirurgi kogemustele, eelistustele ja läätse paigutusele. Läätse optilise tugevuse arvutusmeetodid on kirjeldatud järgmistes viidetes:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. et al. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. et al. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
SOOVITATAV A-KONSTANT Välisetiketil nimetatud A-konstant on lähtepunktiks implantaadi optilise tugevuse arvutamisele. Soovitatakse välja töötada oma konstant, mis põhineb teie kliinilisel kogemusel vastava läätse mudeliga, kirurgilistel meetoditel, mõõteseadmete iseärasustel ja operatsioonijärgsetel tulemustel.USAs võite pöörduda aadressil Alcon Laboratories, Inc., tel 1-800-TO-ALCON (1-800-862-5266), kui soovite lisainfot läätsede optilise tugevuse arvutamiseks. Väljaspool USAd võtke ühendust Alcon’i kohaliku büroo või turustajatega.
ALCON’I KVALIFITSEERITUD VISKOELASTIKUD JA TELLIMISNUMBRIDVaata pakendis olevat lisakaarti. Kasutada tohib üksnes Alcon’i kvalifitseeritud viskoelastikut (-elastikuid). Kvalifitseerimata viskoelastiku kasutamisel võite rikkuda läätse ja põhjustada komplikatsioone implantatsiooni protseduuri käigus. Selle läätsega kasutamiseks sobivate Alcon’i kvalifitseeritud viskoelastikute täieliku loetelu saamiseks pöörduge Alcon’i esindaja poole.
KASUTUSJUHISED1. Kasutage UltraSert™ eellaetud sisestussüsteemi toatemperatuuridel vahemikus 18° C (64° F) kuni 23° C (73° F).2. Kontrollige avamata välispakendi etiketilt mudeli, optilise tugevuse, õige kuju ja kõlblikkusaja andmeid. 3. Pärast välispakendi avamist kontrollige, kas seadme pakend on terve. Kui leitakse kahjustusi, kasutage teist AcrySof®
IQ tehisläätse ja UltraSert™ eellaetud sisestussüsteemi. Järgmisena kontrollige, kas seadme etiketil olev info (nt mudel, optiline tugevus ja seerianumber) vastab välispakendi etiketil olevale infole.
4. UltraSert™ eellaetud sisestussüsteemi väljavõtmiseks haarake plastaluse nurgast, tõmmake ära TYVEK† materjali kaaneosa ja paigutage seade steriilsesse keskkonda. Kui kontrollimisel selgub, et seadme otsakul leidub vigastusi, kübemeid või deformatsiooni, kasutage teist AcrySof® IQ tehisläätse ja UltraSert™ eellaetud sisestussüsteemi. Kui seade ei ole täiesti terve või kui kolb/kolvilukk on transportimise ajal liikunud, kasutage teist AcrySof® IQ tehisläätse ja UltraSert™ eellaetud sisestussüsteemi (vt TAGASTATUD KAUPADE POLIITIKA).
†Reg. TM of E.I. DuPont De Nemours & Co.
Kui olete valmis läätsi paigaldama, läbige minimaalsete viivitustega järjest punktid 5, 6 ja 7.
5. Sisestage viskoelastne kanüül perpendikulaarselt seadmesse läbi viskoelastse pordi, mis asub läätse tõkestamise osas nii, nagu joonisel 5 näidatud. Jälgige seadme täitmisel, et viskoelastik voolab läbi üleminekujoone ning täidab otsaku eesmise osa (vt nool joonisel 5), seejärel eemaldage kanüül. Vajalik viskoelastiku kogus on ligikaudu 0,2 ml. Kasutage ainult Alcon`i viskoelastikut, mis on sobiv kasutamiseks koos UltraSert™ eellaetud sisestussüsteemiga ja mis on soojenenud operatsioonisaali temperatuurini. Alcon’i kvalifitseeritud viskoelastikute loetelu nägemiseks vaadake pakendis kaasasolevat täiendavat infot.
Vt ingliskeelne juhis Joonis 5
Viscoelastic Port = Viskoelastiku port“Fill-To” Line = Otsaku üleminekujoon
6. Eemaldage sinist värvi läätsetõkestaja, haarates noolega näidatud osast ja kallutades seda veidi ettepoole, samal ajal ülespoole ja seadmest välja tõmmates (joonis 6a). Eemaldage sinist värvi kolvilukk, haarates sellest kinni ja tõmmates seda otse üles ja seadmest välja (joonis 6b). Visake läätse tõkestaja ja kolvilukk ära. Kui läätse tõkestaja on eemaldatud, ärge üritage lisada seadmesse viskoelastikut – see võib läätse kahjustada. Ärge püüdke tõmmata kolbi tagasi, kui kolvilukk on eemaldatud – see võib kolbi kahjustada.
Vt ingliskeelne juhis Joonised 6a ja 6b
7. Lükake kolbi ühtlaselt edasi kerge pideva liigutusega, kuni läätse eesmine serv jõuab otsaku üleminekujooneni (vt joonis 7a). Selles asendis puudutab sinine vedru läbipaistvat sõrmetuge nagu näidatud joonisel 7b. Kolbi edasiviimiseks kulub vähemalt 7 sekundit. Ärge laske läätsel osaliselt otsakust väljuda. Kolbi ei tohi lükata järsku, vastasel juhul võite läätse valesti kokku voltida ning seda kahjustada.
48
Vt ingliskeelne juhis Joonised 7a ja 7b
Fill-to Line = Otsaku üleminekujoon
8. Kui lääts on jõudnud otsaku üleminekujooneni, tuleb läätse visuaalselt kontrollida, et teha kindlaks jalakeste asend. Kolb peab kokku puutuma tagumise optilise servaga. Ükski läätse osa ei tohi enne otsakust välja tulla, kui otsak on viidud sisselõikesse (vt joonis 8). Kui lääts on õiges asendis otsaku üleminekujoonel, tuleb see implanteerida 3 minuti jooksul.
Vt ingliskeelne juhis Joonis 8
“Proceed with full plunger advancement to fully deliver lens” = Läätse paigaldamiseks lükake kolbi edasi“Proceed” = Jätkake
“Proceed with full plunger advancement to fully deliver lens” = Läätse paigaldamiseks lükake kolbi edasi“Proceed with device rotations per Step 10” = Jätkake seadme roteerimist nagu kirjeldatud punktis 10
“Do not proceed” = Ärge jätkake
9. Kui läätse õige asend on kontrollitud ning jalakesed õigesti kokku volditud, lükake läätse implantatsiooni, sisestades otsaku tipu läbi sisselõike kuni sügavuspiirajani (vt joonis 9) ja viies otsaku tipu eesmise kapsli avani.
Vt ingliskeelne juhis Joonis 9
Depth Guard = Sügavuspiiraja
10. Lükake kolbi ühtlase pideva liigutusega edasi. Ärge lükake kolbi edasi liiga kiiresti, muidu võib lääts viga saada. . On soovitav, et läätse toomine visuaalse kontrollimise asendist peaks kestma vähemalt 5 sekundit. Kui juhtjalg on sirge või lingus ja ulatub läätse ette, roteerige seadet vasakule, enne kui hakkate kolbi edasi lükkama, et juhtjalg paigutuks kapslikotti õigesti. Kohe kui juhtjalg hakkab väljuma otsaku tipust, paigutage juhtjalg õiges suunas kapslikotti ning jätkake kolvi aeglast edasilükkamist läätse paigaldamiseks. Kui optiline osa väljub otsakust, roteerige seadet tagasi keskele või vajadusel veidi paremale, mis tagab läätse kapslikotis lahtivoltimise eespinnaga pealpool..
11. Läätse asendi korraldamiseks kapslikotis kasutage sobivat positsioneerimisinstrumenti, et lääts oleks iirisega paralleelsel tasapinnal.
12. Visake seade minema. UltraSert™ eellaetud sisestussüsteemi ei tohi korduvalt kasutada.
PATSIENTIDE REGISTEERIMINE JA ARUANDLUSPakendisolev patsiendi identifitseerimiskaart tuleb ära täita ja anda patsiendile, paludes seda püsivalt alles hoida, et patsiendil oleks võimalik seda näidata igale silmaarstile, kellega ta tulevikus konsulteerib.Ühendriikides kehtib kohustus registreerida iga patsient Alcon Laboratories, Inc.-s otsekohe pärast ühe sellise läätse implanteerimist. Registreerimiseks tuleb täita implantatsiooni registreerimiskaart, mis on kaasa läätsekarbis ning saata see Alcon Laboratories, Inc.-le. Patsiendi registreerimine on hädavajalik pikaajalise patsiendijälgimisprogrammi toimimiseks ning võimaldab Alcon Laboratories, Inc.-l reageerida kõrvaltoime teatistele.Väljaspool Ühendriike tuleb järgida kohalikke nõudeid toote registreerimisele.
Kõrvaltoimetest, mille puhul on põhjendatud oletus, et lääts võis põhjustada või soodustada surmajuhtu või tõsist kahjustust, sh kõrvaltoimed, mis on tekkinud meditsiiniseadme mittevastavuse tõttu tehnilistele nõuetele või meditsiiniseadme muu mitteootuspärase toimimise tagajärjel, tuleb teatada Alcon Laboratories, Inc.-le. Sellist informatsiooni oodatakse kõigilt kirurgidelt, et oleks võimalik dokumenteerida intraokulaarse läätse implanteerimise võimalikke kaugmõjusid. Kirurgid peavad nende intraokulaarsete läätsedega seotud kõrvaltoimetest teatama järgmisel aadressil ja telefoninumbril:
Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Telefonikõned (tasuta): 1-800-757-9780
Kirurgid väljaspool Ühendriike: pöörduge kõrvaltoime teatiste osas Alcon’i kohaliku büroo või turustaja poole.
AcrySof® volditavate tagakambriläätsede kliinilised uuringudAcrySof® volditavate üheosaliste tagakambri läätsedega on tehtud kaks randomiseeritud prospektiivset hästi kontrollitud uuringut. Esimene uuring viidi läbi originaalse läätsemudeliga, et tõestada AcrySof® Natural üheosaliste tagakambriläätsede mudeli SB30AL (UV- ja sinise valguse filter) ohutust ja efektiivsust. Selles randomiseeritud kliinilises uuringus kasutati kontroll-läätsena AcrySof® mudelit SA30AL (ainult UV-kaitse). Esimesed andmed pärinevad patsientidelt, kellele paigaldati läätsed SB30AL või SA30AL. Teine AcrySof® IQ volditavate üheosaliste tehisläätsede randomiseeritud kliiniline uuring (koos UV ja sinist valgust filtreerivate kromofooridega) mittesfääriliste optiliste ning AcrySof® volditavate üheosaliste kontroll-läätsede osas tehti kliiniliste/funktsionaalsete eeliste hindamiseks läätsede traditsioonilise sfäärilise ehituse osas.
49
AcrySof® Natural üheosaliste läätsede (mudel SB30AL) kliiniline uuringAndmed patsientide kohta, keda edukalt jälgiti vähemalt üks aasta pärast operatsiooni, kinnitavad veenvalt, et AcrySof® Natural üheosaline lääts (mudel SB30AL) on ohutu ja efektiivne vahend nägemise korrigeerimiseks afaakia korral.
AcrySof® Natural üheosaliste läätsede (mudel SB30AL) patsiendidKahepoolses uuringus osalenud ja implantaadi (mudel SB30AL) saanud patsientidest moodustasid naised 70,6% ja mehed 29,4%. Patsientide seas, kellele paigaldati tehisläätsed SA30AL (kontrollmudel), moodustasid naised 60,5% ja mehed 39,5%. Rassi järgi jagunesid patsiendid järgmiselt – kaukaasia 95,3% ja mustanahalised 4,7% (mudel SB30AL). Kontrollmudeli SA30AL järgi oli kaukaaslasi 96,6%, mustanahalisi 2% ja muid 1,4%. Mudeli SB30AL saanud patsientide keskmine vanus oli 72,9 aastat. Kontrollmudeli (SA30AL) saanud patsientide keskmine vanus oli 71,9 aastat.
AcrySof® Natural ühekambriliste läätsede (mudel SB30AL) nägemisteravusKokkuvõte nägemisteravuse kohta vähemalt aasta pärast operatsiooni neil patsientidel, kellel enne operatsiooni ei esinenud nägemispatoloogiat, ebanormaalset sarvkesta või maakula degeneratsiooni (parim juhtum), on esitatud tabelis 2a ja nägemisteravuse andmed kõigi osalenud patsientide kohta on näidatud tabelis 3a. Kontrollandmed asuvad vastavalt tabelites 2b ja 3b. Mudeli SB30AL ja kontroll-läätsede mudeli SA30AL nägemisteravuse vahel ei esinenud statistiliselt olulist erinevust, nii parima juhtumi kui üldrühma vahel.
Vt ingliskeelne juhis Tabel 2aParim korrigeeritud nägemisteravus parima juhtumiga patsientide hulgas
vähemalt üks aasta pärast operatsiooni, AcrySof® Natural tehislääts SB30AL
Nägemisteravus
#kohta 20/20 või parem 20/25 20/30 20/40 20/40 või
parem> 20/40 kuni
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Vanuse-kategooria
N = arv
Vt ingliskeelne juhis Tabel 2bParim korrigeeritud nägemisteravus parima juhtumiga patsientide hulgas
vähemalt üks aasta pärast operatsiooni, AcrySof® tehislääts SA30AL kontroll-läätsena
Nägemisteravus
#kohta 20/20 või parem 20/25 20/30 20/40 20/40 või
parem> 20/40 kuni
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Vanuse-kategooria
N = arv
Vt ingliskeelne juhis Tabel 3aParim korrigeeritud nägemisteravus kõigi patsientide hulgas
vähemalt üks aasta pärast operatsiooni, AcrySof® Natural tehislääts SB30AL
Nägemisteravus
#kohta 20/20 või parem 20/25 20/30 20/40 20/40 või
parem> 20/40 kui
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Vanuse-kategooria
N = arv
50
Vt ingliskeelne juhis Tabel 3bParim korrigeeritud nägemisteravus kõigi patsientide hulgas
vähemalt üks aasta pärast operatsiooni, AcrySof® tehislääts SA30AL kontroll-läätsena
Nägemisteravus
#kohta 20/20 või parem 20/25 20/30 20/40 20/40 või
parem
> 20/40 kuni
< 20/80> 20/80
N N % N % N % N % N % N % N %
Vanusekategooria
N = arv
AcrySof® Natural üheosaliste läätsede (mudel SB30AL) kumulatiivsed kõrvalnähudKumulatiivsete kõrvalnähtude osakaal vähemalt ühe aasta kestel pärast AcrySof® Natural üheosaliste tehisläätsede (mudel SB30AL ja mudel SA30AL) paigaldamist on näidatud tabelis 4. Kummagi mudeli puhul ei täheldatud statistiliselt olulist erinevust kumulatiivsete kõrvalnähtudega patsientide vahel.
Tabel 4Kumulatiivsed kõrvalnähud vähemalt ühe aasta jooksul pärast operatsiooni
AcrySof® Natural tehislääts SB30AL ja kontrollmudel SA30AL
SB30AL (N= 153)
SA30AL(N= 147) p-väärtus*
N % N %
Kõrvalnähu tüüp
0 0,0 0 0,0 puudub Kumulatiivne Hüpopüon
Silmasisene infektsioon / endoftalmiit 0 0,0 0 0,0 puudub
Maakula ödeem 4 2,6 2 1,4 0,6847
Pupillaarne blokk 0 0,0 0 0,0 puudub
Võrkkesta irdumine või võrkkesta irdumise ravi 0 0,0 0 0,0 puudub
Läätse väärasetsus 1 0,7 0 0,0 1,0000
Sekundaarne kirurgiline sekkumineLäätsemassi jääkide eemaldamineEksplantaat (kapsulaarrebendist tingitud dislokatsioon)Krüoteraapia võrkkesta irdumise ravimiseksParatsentees silma siserõhu alandamiseksFokaalne laserraviFotodünaamiline raviEksplantaat biomeetriavea tõttu
51 1
11100
3,3 0,7
0,7
0,70,70,70,00,0
200
00011
1,40,00,0
0,00,00,00,70,7
0,4482
Hüfeem 0 0,0 0 0,0 puudub
*p- väärtused on saadud Fisheri testi järgi, võrreldes mudelit SB30AL mudeliga SA30AL.
AcrySof® Natural üheosaliste läätsede (mudel SB30AL) püsivad kõrvalnähudPüsivate kõrvalnähtude osakaal vähemalt ühe aasta jooksul pärast AcrySof® Natural üheosaliste tehisläätsede (mudel SB30AL ja kontrollmudel SA30AL) implantatsiooni on näidatud tabelis 5. Kummagi mudeli (SB30AL ja SA30AL) puhul ei täheldatud statistiliselt olulist erinevust püsivate kõrvaltoimetega patsientide vahel.
51
Tabel 5Püsivad kõrvalnähud vähemalt üks aasta pärast operatsiooni AcrySof® Natural tehislääts SB30AL ja kontrollmudel SA30AL
SB30AL (N= 138)
SA30AL(N= 127) p-väärtus*
N % N %
Kõrvalnähu tüüp
0 0 1 0,8 0,4792Püsiv Sarvkesta ödeem
Iriit 0 0 0 0,0 puudub
Maakula ödeem 2 1,4 1 0,8 1,0000
Vitreiit 0 0 0 0,0 puudub
Ravi nõudev silma siserõhu tõus 0 0 0 0,0 puudub
*p- väärtused on saadud Fisheri testi järgi, võrreldes mudelit SB30AL mudeliga SA30AL.
AcrySof® Natural üheosaliste läätsede (mudel SB30AL) värvitajuVärvitaju testimisel kasutati Farnsworth D-15 Panel Test´i ja see viidi läbi 120.–180. päeval pärast operatsiooni. 109 normaalse nägemisega patsiendist, kellele paigaldati mudel SB30AL läätsed esimest korda ja keda uuriti 120–180 päeval pärast operatsiooni, 107 läbisid (98,2%) värvitaju testi. 102 normaalse nägemisega patsiendist, kellele paigaldati mudel SA30AL läätsed esimest korda ja keda uuriti 120–180 päeval pärast operatsiooni, läbisid 97 (95,1%) värvitaju testi. Puudus statistiline erinevus mudelid SB30AL ja SA30AL saanud patsientide vahel, kes läbisid värvitaju testi 120–180 päeval pärast operatsiooni. Seega ei avalda patenditud kromofoor negatiivset mõju värvusnägemisele normaalse nägemisega patsientidel.
AcrySof® Natural ühekambriliste läätsede (mudel SB30AL) vahekord Nd:YAG protseduuridegaKeskmise jälgimisperioodiga 21,6 kuud, teostati kolmel (3) SB30AL läätsega patsiendil 135 hulgast (2,2%) Nd:YAG tagumine kapsulotoomia. Keskmise jälgimisperioodiga 21,9 kuud, teostati kahel (2) SA30AL läätsega patsiendil 127 hulgast (1,6%), Nd:YAG tagumine kapsulotoomia.
AcrySof® IQ läätsede kliiniline uuringSarnaselt eelnevalt läbi viidud tehisläätsede uuringutega, võeti kontrollitud, randomiseeritud, topeltpimedasse, multitsentrilisse, kontralateraalsesse implantaadi kliinilisse uuringusse hea silmade üldseisundiga täiskasvanuid (nt puudusid eelnevad silmaoperatsioonid, degeneratiivsed silmahaigused, mis võiksid märkimisväärselt nägemisteravust mõjutada või ägedad ning kroonilised seisundid, mis võivad tõsta patsiendi riski), kus võrreldi AcrySof® IQ läätsi sfääriliste kontroll-läätsedega. Silmade sfäärilised aberratsioonid olid statistiliselt vähem märgatavad AcrySof® IQ läätsedega kui kontroll-läätsedega. Kontrastitundlikkuse tulemused näitasid statistiliselt olulist operatsioonijärgset (3. kuul) paranemist AcrySof® IQ implanteeritud läätsedele. AcrySof® IQ läätsedega silmad näitasid ka statistiliselt ja kliiniliselt märkimisväärset paranemist funktsionaalse nägemise mõõtmisel, öise autojuhtimise simulatsioonil ja teistel testitud tingimustel – eeskätt sätenduse ja udu korral. Need tulemused näitavad, et AcrySof® IQ tehislääts (mittesfääriline lääts, mis koosneb sinist valgust filtreerivast kromofoorist) tagab parema kliinilise tulemuse võrreldes monofokaalsete AcrySof® tehisläätsega (ilma mittesfäärilise läätseta ja sinist valgust filtreeriva kromofoorita).
AcrySof® IQ läätsed – sfäärilised ja kõrgema astmega aberratsioonidKeskmine AcrySof® IQ silmade sfääriline aberratsioon oli ligikaudu 0,1 mikromeetrit. Joonis 10 esitab statistiliselt olulise vähenemise sfääriliste ja kõrgema astmega aberratsioonide osas, mis leiti AcrySof® IQ läätsedel. Joonis 11 näitab keskmiseid aberratsioonide mõõtmisi kõikidele silmade jaoks koos lainefrondi aberromeetria mõõtmistega läätsede ja vanusegruppide kaupa. Nagu selles tabelis kirjeldatakse, oli sfääriliste AcrySof® IQ silmade aberratsiooni vähenemine vanusest sõltuv.
Vt ingliskeelne juhis Joonis 10Sfäärilised ja kõrgema astmega ruutkeskmised
90-120 päeva pärast teise silma implantaati
Vt ingliskeelne juhis Joonis 11Keskmine sfääriline aberratsioon kokku ja vanusegrupi kaupa
90-120 päeva pärast teise silma implantaati
AcrySof® IQ läätsed – nägemisteravusThe AcrySof® IQ läätsed ja kontrollgrupi läätsed andsid kliiniliselt sarnase operatsioonijärgse nägemisteravuse. Monokulaarse nägemisteravuse tulemused on esitatud joonistel 12 ja 13.
52
Vt ingliskeelne juhis Joonis 12LogMAR BCVA
Vt ingliskeelne juhis Joonis 13LogMAR UCVA
AcrySof® IQ läätsed – kontrastitundlikkusKliinilise uuringu esmane eesmärk oli näidata AcrySof® IQ läätsede eelist kontroll-läätsede ees keskmise kontrastitundlikkuse kaudu, mida mõõdetakse operatsioonijärgselt mesoopilistel tingimustel koos või ilma peegeldusteta mõlemal sagedusel (3 või 6 tsüklit kraadi kohta), kasutades Vector Vision CSV-1000 (tabel luminestsentsiga 3 cd/m2). Osadel patsientidel tehti ka funktsionaalne nägemisteravuse kontrastsuse test (FACT) (tabel luminestsentsiga 3 cd/m2). Selles kliinilises uuringus demonstreeriti AcrySof® IQ läätsede eelist kontroll-läätsede ees mesoopilistel tingimustel 6 tsüklil kraadi kohta koos ja ilma peegelduseta (CSV-1000) ja 3 ning 6 tsüklil kraadi kohta ilma peegelduseta (FACT). Joonised 14 ja 15 kirjeldavad mesoopilist kontrastitundlikkust kõigil ruumisagedustel, mida testiti nii AcrySof® IQ läätse ja kontroll-läätsedega.
Vt ingliskeelne juhis Joonis 14Mesoopiline kontrastitundlikkus (CSV-1000)90-120 päeva pärast teise silma implantaati
Vt ingliskeelne juhis Joonis 15Mesoopilise kontrastitundlikkuse (FACT) uuringVähemalt 90 päeva pärast teise silma implantaati
AcrySof® IQ läätsed – öise autojuhtimise simulatsioonOsa patsiente testiti heakskiidetud öise autojuhtimise simulaatoril. Patsiente testiti monokulaarselt tingimustes, mis simuleerisid linna- ja maa-asulaid tavapärastel, sätenduse ja udu tingimustes.Öine linnasõidu stseen kasutas erinevaid tänavavalgusteid, autotulesid, kaupluste tulesid ja reklaame, et tekitada kõrgetasemeline ümbritsev valgus, mis on neis tingimustes tavaline. Öisel ajal maa-asulas sõitmise stseen kasutas minimaalset ümbritsevat valgust. Simuleeritavad sõidukiirused olid vastavalt ligikaudu 35 miili tunnis linna- ja 55 miili tunnis maa-asulates.Patsientidel paluti tuvastada ja identifitseerida igas stseenis sihtmärkide seeriaid, kaasa arvatud valge-rohelisi maanteeinfo liiklusmärke, kollaseid-musti hoiatusmärke ja jalakäijad. Patsientidel paluti reageerida, kui nad näevad esimest sihtmärki, võimaldades salvestamiseks tuvastamise vahemaad. Patsientidel paluti seejärel reageerida siis, kui nad suudavad eristada sihtmärke (nt mida liiklusmärk ütleb, mis suunas jalakäija kõndis jne), nii et tuvastamise vahemaa saaks salvestada.Joonised 16 kuni 19 esitavad keskmised erinevused AcrySof® IQ läätsede ja kontroll-läätsede vahel linna- ja maa-asula stseenides nii vahemaade tuvastamise kui ka identifitseerimise osas (nt inimestevaheliste erinevuste keskmine).AcrySof® IQ läätsed toimisid funktsionaalselt paremini kui kontroll-läätsed 34 tingimusel testitud 36 tingimusest, näidates paranemist tuvastamise kui ka identifitseerimise osas nii linna- kui ka maa-asulas erinevates testitud sõidutingimustes (tavapärane, sätendus, udu). Veelgi enam, AcrySof® IQ läätsed toimisid statistiliselt paremini neil 12 sõidutingimusel kui kontroll-läätsed kõige märkimisväärsema mõju ja eelisega linna jalakäijate tuvastamisel ja identifitseerimisel (sätenduse ja udu tingimustes) ning maa-asula liiklusmärkide tuvastamisel ja identifitseerimisel (sätenduse ja udu tingimustel). Vähese nähtavuse tingimustel (sätendus, udu) linna-stseenis tagasid kiirusel 35 miili tunnis AcrySof® IQ läätsed suurenenud nähtavuse üle 0,5 sekundi kiiremini jalakäijate jm linna-asulates sagedasti ette tulevatele ohtudele reageerimise osas. See 0,5 sekundit suurenenud funktsionaalsus on märkimisväärne, võimaldades rohkem aega vajalike tegevuste jaoks, nagu pidurdamine, vältimine jm (Green, 2000; McBride and Matson, 2004). Neil tingimustel maa-asula stseenis kiirusel 55 miili tunnis suurenenud nähtavus võimaldab AcrySof® IQ läätsedele rohkem kui 1 sekundit lisaaega liiklusmärkide tuvastamiseks – maa-asulates kõige sagedamini ette tulevate olukordade jaoks. 0,5 sekundiline funktsionaalsuse suurenemine on märkimisäärne, andes autoga sõites rohkem aega vastavate tegevuste ette võtmiseks, mis muutub kriitiliseks öösel tundmatutes maa-asulates sõitmisel, kus ümbritsev valgus tihti puudub. Selles alamuuringus oli 6 patsienti, kellel esines operatsioonijärgselt makulaarne degeneratsioon või tagakambri kapsli tuhmumine (PCO). Kui need patsiendid eemaldati autojuhtimise analüüsist, siis erinevus tehisläätsede osas jalakäijate tuvastamisel ja identifitseerimisel peegelduse tingimustes linnas lühenes kliiniliselt 0,5 sekundilise lävega. Kui algseid analüüse seadistati korduseks, siis erinevused tehisläätsede osas ei olnud enam statistiliselt olulised peegelduse korral linnas teksti tuvastamisel (Hommel’s p-väärtus = 0,0539) või maa-asulas peegelduse korral jalakäijate tuvastamisel (Hommel’s p-väärtus = 0,0507).Need tulemused näitavad paranenud funktsionaalset nägemist ja tõenäoliselt olulisi ohutuse eeliseid eakamate AcrySof® IQ läätsedega autojuhtide jaoks ning teiste autojuhtide ning jalakäijate jaoks, kellega nad maanteel liiklevad. Selle testi tulemused näitavad, et AcrySof® IQ läätsed parandavad funktsionaalset nägemist, mis omakorda parandab patsientide ohutust teistes halbade nähtavusega elulistes situatsioonides.
Keskmised isikutevahelised erinevused vaatekauguste tuvastamisel, maa-asulaVähemalt 90 päeva pärast operatsiooni
AcrySof® IQ lääts–kontroll (n=44)
PAKENDTagakambri tehisläätsed tarnitakse kuivas UltraSert™ eellaetud sisestussüsteemis, steriliseeritud pakendis ja eelnevalt etüleenoksiidiga steriliseeritult. Steriliseeritud pakend tuleb avada aseptilistes tingimustes (vt KASUTUSJUHEND).
KÕLBLIKKUSAEGSteriilsus on tagatud, kui pakend on kinni ja vigastamata. Kõlblikkusaeg on märgitud UltraSert™ eellaetud sisestussüsteemi välispakendi etiketil. Kõlblikkusaja ületanud läätsed tuleb tagastada Alcon’ile (Alcon Laboratories, Inc.) (vt TAGASTATUD KAUPADE POLIITIKA).
TAGASTATUD KAUPADE POLIITIKAUSAs vahetatakse tagastatud läätsed uutega, kuid raha tagasi ei maksta. Tagastatavatel läätsedel peab olema Alcon Laboratories, Inc. tagastatud kauba number ja need tuleb saata tagasi viisil, mida on võimalik jälgida. Tagastatava kauba numbri saamiseks võtke ühendust Alcon’i klienditeeninduse osakonnaga. Numbri väljaandmine ei taga tagastatavate toodete aktsepteerimist. Tagastamist puudutavates küsimustes, ka vahetamine, pöörduge oma müügiesindaja või klienditeenindaja poole. Väljaspool USAd võtke ühendust Alcon’i kohaliku büroo või tagastatavate toodetega tegeleva turustajaga.
VIITED 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., et al. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., et al. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
54
ETIKETIL KASUTATAVAD SÜMBOLID
SYMBOL EESTIIOL Silmasisene läätsSP ÜheosalineUV UltraviolettD Diopter
ØB Läätse diameeter (optiline diameeter)
ØT Ülddiameeter (üldpikkus)
2STERILIZE Mitte steriliseerida korduvalt
Mitte kasutada korduvalt
Kasutada kuni
STERILE EO Steriliseeritud etüleenoksiidiga
SN Seeria number
Hoiatus
Tootja
EC REP Volitatud esindaja Euroopa Liidus
Temperatuuri ülempiir
Ava siit
Lugege kasutusjuhendit
Mitte kasutada, kui pakend on avatud või rikutud
Ettevaatust: Föderaalseadus (USA) lubab seda seadet müüa vaid arstidel või arsti korraldusel
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 USA
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,Ühendkuningriik
Steril, ultraibolya és kék fényt szűrő, akril, hajlítható, egytestű hátsó csarnok lencsék UltraSert™ előretöltött implantáló eszközzel
LEÍRÁSAz AcrySof® IQ ultraibolya és kék fényt szűrő akril alapanyagú, hajlítható egytestű hátsó csarnok lencsék optikai implantátumok, melyek a természetes szemlencse helyettesítésével felnőtt betegeknél a szürkehályog műtétet követően az aphakia vizuális korrekciójára szolgálnak. Az AcrySof® IQ műlencse az Alcon szabadalmaztatott kék fényt szűrő kromofórral rendelkezik, amely a 400 – 475 nm hullámhosszúságú kék fény tartományában a természetes lencséhez hasonló módon szűri meg a fényt (Boettner and Wolter, 1962). A standard ultraibolya fény kiszűrésén felül az AcrySof® IQ műlencse a kék fény áteresztését 400 nm-en 62%-ról 475 nm-en 23%-ra csökkenti (lásd 1. táblázat). A lencsék optikai része egy magas törésmutatójú lágy akril alapú anyag. Ez az anyag a beültetést megelőzően összehajtható. Implantációt követően a lencse lassan eredeti méretére nyílik szét. A lencsék bikonvex optikából valamint haptikai részből állnak (lásd 1. ábra). Az AU00T0 AcrySof® IQ műlencse modell hátsó aszférikus felülete negatív szférikus aberrációval rendelkezik, hogy kompenzálja az átlagos cornea pozitív szférikus aberrációját. Az AU00T0 egytestű műlencse modell képminőségét (pl. kontrasztátviteli függvény) a 4. ábra szemlélteti. Az AU00T0 AcrySof® IQ egytestű műlencse modell az UltraSert™ előretöltött implantáló eszközben található (lásd 2. ábra), amellyel a műlencse kényelmesen és megbízhatóan a lencse tokjába helyezhető. A lencsék fizikai paraméterei:
Fizikai paraméterek LeírásAz optika típusa Egytestű műlencse aszférikus optikával
Az optika anyaga Ultraibolya és kék fényt szűrő akrilát/metakrilát kopolimer
ISO, 3-mm Aperture (see Note 1) = ISO, 3 mm-es retesz (lásd 1. Megjegyzés)Modified, 3-mm Aperture (see Note 2) = Módosított, 3 mm-es retesz (lásd 2. Megjegyzés)Modified, 5-mm Aperture (see Note 2) = Módosított, 5 mm-es retesz (lásd 2. Megjegyzés)
MEGJEGYZÉSEK: 1. Az AU00T0 modell képminőségét az 11979-2 ISO leírásban szereplő szemmodellen elvégzett kontrasztátviteli
függvény mérésével jellemezték. Az ISO 11979-2 szerint a kontrasztátviteli függvény méréseknél kizárólag 3 mm-es rekesz alkalmazható.
2. Ezen felül az AU00T0 kép minőségét olyan szemmodellen végzett kontrasztátviteli függvény mérésekkel vizsgálták, amely a tipikus, felnőttekre jellemző, szférikus aberrációt utánzó szaruhártyával rendelkezik. Módosított szemmodellt alkalmazva a kontrasztátviteli függvény méréseket 3 és 5 mm-es nyílásokkal is elvégezték.
MŰKÖDÉSI ELV:Az AcrySof® IQ hátsó csarnok lencsék a természetes lencse helyére, a szem hátulsó csarnokába helyezendők. A beültetett lencse elhelyezkedése lehetővé teszi, hogy refraktív szerepet töltsön be az aphakia korrekciójában. Az aszférikus bikonvex optika egy átlagos szem standard szférikus optikájához hasonlóan csökkenti a szferikus aberrációt. A lencsék hatékonyságát a retinális rendellenességek előfordulásának csökkentésére nem igazolták.
INDIKÁCIÓ:Az AcrySof® IQ hátsó csarnok műlencsék szürkehályogműtétet követően a humán szemlencse helyettesítésére szolgálnak felnőtt aphakiás betegek látás korrekciójához (lásd: FIGYELMEZTETÉSEK). A lencsék a lencsetokba helyezendők.
ELŐVIGYÁZAT:A műlencse használata megfontolandó mindazon betegeknél, akiknél fennáll az alábbi betegségek bármelyike, mivel a műlencse súlyosbíthatja a meglévő állapotot, zavarhatja a helyes diagnózis felállítását és a megfelelő kezelés kiválasztását, esetleg indokolatlanul veszélyeztetheti a beteg látását. A lencse beültetése előtt gondos preoperatív kivizsgálás és megfontolt
57
klinikai döntés szükséges a szemsebész részéről az előny/kockázat mérlegelésében, ha a beteg esetében az alább felsorolt állapotok közül legalább egy fennáll: 1. érhártyavérzés 2. társuló súlyos szembetegség 3. jelentős üvegtesthiány 4. extrém sekély elülső csarnok 5. microphthalmus 6. nem öregségi szürke hályog 7. hátsó tok szakadás (nem lehetséges a műlencse fixációja) 8. súlyos cornea disztrófia 9. súlyos látóideg atrófia 10. nem kontrollálható magas szemnyomás 11. zonula érintettség (nem lehetséges a lencse fixációja) 12. színlátási zavarok 13. glaukóma 14. krónikus uveitis 15. retinopathia diabetica 16. klinikailag jelentős macula/RPE elváltozásokKlinikai vizsgálatok alapján a színlátás nem változott kedvezőtlen irányba normál színlátással bíró és AcrySof® Natural műlencsét kapott betegek estében. Az AcrySof® Natural műlencséknek a látást befolyásoló hatását örökletes színlátási hibák, valamint bizonyos szembetegségekhez (például: glaukóma, retinopathia diabetica, krónikus uveitis, egyéb, retinát és a látóideget érintő betegségek) másodlagosan társuló színlátás zavarok esetében nem vizsgálták.
FIGYELMEZTETÉSEK 1. Minden sebészeti beavatkozás kockázattal jár. A szürkehályog műtéttel vagy az implantácós beavatkozással
járó lehetséges komplikációk többek között: cornea endothelium sérülés, endophthalmitis, retinaleválás, vitritis, cisztoid macula ödéma, cornea ödéma, pupilláris blokk, ciklikus membrán, iris előreesés, hypopyon, és ideiglenesen vagy folyamatosan fennálló glaukóma.
2. A beültetett intraokuláris lencse hatékonysága és biztonságossága az alábbi, már fennálló betegségekben szenvedőknél nem bizonyított: krónikus, gyógyszer okozta miózis, glaukóma, amblyopia, retinopathia diabetica, korábbi cornea transzplantáció, korábbi retinaleválás, és/vagy iritis stb. A fenti betegségeken illetve operációkon átesett betegeknél ajánlatos a szakembernek az aphakia korrekciójaként alternatív terápiás lehetőségeket is figyelembe vennie. Ha a fenti lehetőségek elégtelennek bizonyulnak, akkor célszerű csak a lencsebeültetést alkalmazni.
3. A beültetett műlencse hosszabb távú hatásai nem ismertek. Ezért a szakorvosnak ajánlatos a beteget műtét után rendszeresen ellenőrizni.
4. Ha a műtét előtt álló beteg a következő állapotok valamelyikében szenved: a cornea endotheliális elváltozásai, abnormális cornea, macula degeneráció, retina degeneráció, glaukóma, krónikus, gyógyszer okozta miózis, rosszabb műtéti kimenetelre számíthat a látásélesség tekintetében, mint az előbb felsorolt állapotoktól mentes betegek. A kezelőorvosnak ezen állapotok fennállásakor fontolóra kell vennie a műlencse beültetéséből származó előnyöket.
5 A pupilláris blokk miatt szükséges másodlagos iridektómia elkerülhető, ha egy vagy több iridektómiát hajtanak végre lencsebeültetéskor (Willis, et al., 1985.).
6. A hátsó csarnok lencse biztonságosságára és hatékonyságára vonatkozó adatok annak elülső csarnokba való beültetése esetén nem állnak rendelkezésre. Hátulsó csarnoki lencsék elülső csarnokba történő implantálása néhány esetben nem bizonyult biztonságosnak (Girard, et al., 1983.).
7. A műlencse beültetésével összefüggő mellékhatások a következők: hypopyon, intraokuláris infekció, akut cornea dekompenzáció és másodlagos sebészeti beavatkozás. Másodlagos sebészeti beavatkozások többek között: a lencse repozicionálása, lencsecsere, üvegtesti aspiráció vagy pupilláris blokk miatti iridektómia, sebszivárgás vagy retina leválás korrigálása.
8. A keskeny, vagy kisméretű optikával rendelkező lencsék enyhe decentralizációja miatt néhány beteg bizonyos fényviszonyok között káprázást, vagy más látászavart élhet meg. Az orvosnak számolnia kell ezzel a jelenséggel a keskeny vagy kisméretű optikával rendelkező lencse beültetésekor. Ilyen esetben capsulorhexis elvégzése javasolt.
9. A lencse toknak az elülső csarnok különböző mértékű elsekélyesedésével és indukált myopiával járó postoperatív kitágulása a capsulorhexis technikájával, illetve PMMA, szilikon, és akril anyagú, hátsó csarnok lencse beültetésével hozható összefüggésbe (Holz, 1992.).
10. Ügyelni kell arra, hogy a lencse betokosodása előtt ne következzen be a lencse decentralizációja illetve díszlokációja. Egyes klinikai esetek szerint a betokosodás a beültetést követő 4 héten belül jelentkezik.
11. Az AcrySof® Natural egytestű műlencsével végzett klinikai vizsgálatokban (lásd 2-5 táblázat) a lencséket kizárólag a lencsetokba ültették be. A sulcusba történő behelyezés biztonságosságával és hatékonyságával kapcsolatban nem állnak rendelkezésre klinikai adatok.
A műtéti beavatkozás végén ajánlott a viszkoelasztikus anyag eltávolítása, különös tekintettel a hátsó tok és a lencse közötti térre. Az intraokuláris lencse optikáját I/A véggel óvatosan benyomva, és standard irrigációs technikát alkalmazva lehet a viszkoelasztikus anyagot a szemből eltávolítani. Az eljárás során ugyanis az esetleges maradék viszkoelasztikus anyag a lencse mögötti térből előre mozdul, ahonnan már aspirációval könnyen eltávolítható.
58
ELŐVIGYÁZATOSSÁGI ELVEK: 1. A műlencse valamint az UltraSert™ előretöltött implantáló eszköz újrasterilizálása tilos. 2. A lencsét 45 °C alatti hőmérsékleten kell tárolni. 3. A lencsével óvatosan bánjon, hogy ne sérüljön a lencse felszíne vagy a haptikák. 4. Ne próbálja a haptika alakját megváltoztatni. 5. A műlencse beültetéséhez magas szintű sebészeti tudás szükséges. A sebésznek számos beültetést kell
végignéznie vagy leasszisztálnia és sikeresen kell egy vagy két, a műlencse beültetéséről szóló tanfolyamot elvégeznie, mielőtt megkísérelné a műlencse beültetést.
6. Ép és sértetlen csomagolás esetén a lencse steril. 7. Az UltraSert™ előretöltött implantáló eszköz egyszerhasználatos. Használatot követően dobja ki az UltraSert™
előretöltött implantáló eszközt. 8. Az UltraSert™ előretöltött implantáló eszköz használata 18oC és 23 oC közötti szobahőmérsékleten ajánlott.
A LENCSE TŐRŐEREJÉNEK MEGÁLLAPÍTÁSAA hátsó csarnok lencse szükséges törőerejének megállapítása a szakorvos tapasztalatától, preferenciájától és a beültetendő lencse kívánt elhelyezkedésétől függ. A lencse-erősség kalkulációjának módszereit a következő referenciák írják le:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. et al. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. et al. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
JAVASOLT A-KONSTANS:A külső csomagoláson feltüntetett A-konstans segítségül szolgál a megfelelő implantációs törőerő kalkulációjában. Javasoljuk a különböző lencse modellek, sebészeti technikák, mérőműszerek, és posztoperatív klinikai tapasztalatok alapján a saját A-konstans kidolgozását.Amennyiben további információra lenne szüksége a lencse erősség kiszámításában, kérjük, hívja az Amerikai Egyesült Államokban az Alcon Laboratories Inc. vállalatot, az alábbi telefonszámon: 1-800-TO-ALCON (1-800-862-5266). Az USA-n kívül lépjen kapcsolatba a helyi Alcon céggel:Alcon Hungária Kft.1114 Budapest, Bartók Bél út 43-47.Tel: 06-1-463-9080.
MINŐSÍTETT ALCON VISZKOELASZTIKUMOK ÉS RENDELÉSI SZÁMOKLásd a csomagolásban lévő mellékletet. Kizárólag Alcon minősített viszkoelasztikum(ok) használhatók. A nem minősített viszkoelasztikumok alkalmazása károsíthatja a műlencsét és lehetséges szövődményekhez vezethet a beültetés során. A műlencséhez használható Alcon minősített viszoelasztikumok teljes listáját illetően, kérjük, vegye fel a kapcsolatot az Alcon képviselőjével.
FELHASZNÁLÁSI ÚTMUTATÓ1. lépés: Az UltraSert™ előretöltött implantáló eszközt 18oC és 23 oC közötti szobahőmérsékleten használja. 2. lépés: A bontatlan doboz címkéjén ellenőrizze a modell típusát, törőerejét, alaki sajátságait és lejárati idejét.3. lépés: A külső kartondoboz felnyitása után győződjön meg az eszköz épségéről. Sérülés esetén használjon másik
AcrySof® IQ műlencsét és UltraSert™ előretöltött implantáló eszközt. Ezt követően ellenőrizze, hogy az eszköz címkéjén található információk (pl.: törőerő, modell, sorozatszám) megegyeznek a külső csomagolás címkeszövegének adataival.
4. lépés. Az UltraSert™ előretöltött implantáló eszköz kivételéhez fogja meg a műanyag tálca sarkát, és húzza le teljesen a TYVEK† fedelet, majd helyezze steril területre az eszközt. Amennyiben az eszköz vége sérültnek, deformáltnak tűnik, vagy abban szemcsék láthatók, akkor másik másik AcrySof® IQ műlencsét és UltraSert™ előretöltött implantáló eszközt használjon. Ha az eszköz nem teljesen ép, esetleg úgy tűnik, hogy a dugattyú vagy a dugattyúzár a szállítás közben elmozdult, szintén egy másik AcrySof® IQ műlencsét és UltraSert™ előretöltött implantáló eszközt vegyen elő (lásd az Áruvisszavételi szabályzatot).
†Reg. TM of E.I. DuPont De Nemours & Co.
Ha készen áll a lencse beültetésére, az 5., 6. és 7. lépést sorrendben végezze el, minimális késéssel az egyes lépések között.
5. lépés: Az eszközre merőleges irányban helyezze be teljesen az eszköz lencse elmozdulást gátló része mellett lévő viszkoelasztikus nyíláson keresztül a viszkoelasztikus kanült, az 5. ábrának megfelelően. Ezt követően legalább annyi viszkoelasztikus anyaggal töltse fel, hogy az a cső végénél lévő vonalhoz folyjon (lásd az 5. Oldalon lévő nyilat). Ehhez kb. 0,2 ml viszkoelasztikus anyagra van szükség. Kizárólag az UltraSert™ előretöltött implantáló eszközzel való együttes használathoz minősített Alcon viszkoelasztikumokat használjon, melyet használat előtt szobahőmérsékletűre kell felengedni. Az Alcon minősített viszkoelasztikumok listáját lásd ennek a csomagnak a mellékletében.
59
5. ábra
Viscoelastic Port = Viszkoelasztikus nyílás“Fill-To” Line = “Fill-To” vonal
6. lépés: Távolítsa el a kék színű, lencse elmozdulását gátló részt: ehhez fogja meg a nyíl részénél, és finoman előredöntve emelje fel, majd vegye le az eszközről (6a. ábra). Távolítsa el a kék színű dugattyú zárat: ehhez fogja meg, állítsa függőleges helyzetbe, majd vegye le az eszközről (6b. ábra). Dobja el a lencse elmozdulását gátló részt és a dugattyú zárat. A lencse elmozdulását gátló rész eltávolítását követően ne próbáljon újabb viszkoelasztikumot az eszközbe juttatni, mert megsérülhet a lencse. A dugattyú zár eltávolítását követően ne próbálja meg visszahúzni a dugattyút, mert megsérülhet a dugattyú.
6a ábra6b. ábra
7. lépés: Finom, folyamatos mozdulattal nyomja tovább a dugattyút, amíg az optika elülső széle egy vonalba nem kerül a csőn lévő vonallal (lásd 7a. ábra). Ebben a helyzetben a kék rugó hozzá fog érni az eszköz ujjperem részéhez, a 7b ábrán látható módon. A dugattyú előrehaladásának legalább 7 másodperc alatt kell végbemennie. Ne hagyja, hogy a lencse bármelyik része elhagyja a cső végét. Fontos, hogy a dugattyút ne egy hirtelen mozdulattal tolja előre, mivel így nem megfelelő hajlás és károsodás történhet a lencsében.
7a ábra7b. ábra
“Fill-To” Line = “Fill-To” vonal
8. lépés. Miután a lencse elérte a vonalat, vizuálisan ellenőrizze a lencsét, hogy meghatározhassa a haptika helyzetét. A dugattyúnak a hátsó optika széléhez kell érnie. A sebzésen keresztül történő behelyezést megelőzően figyelje meg, hogy a lencse egyik része sem hagyta el a cső végét (lásd 8. ábra). Ha a lencse már a cső vonalánál helyezkedik el, akkor azt 3 percen belül kell beültetni.
8. ábra
“Proceed with full plunger advancement to fully deliver lens” = Haladjon tovább, és a lencse belyezezéséhez tolja előre teljesen a dugattyút
“Proceed” = Haladjon tovább“Proceed with full plunger advancement to fully deliver lens” = Haladjon tovább, és a lencse belyezezéséhez tolja
előre teljesen a dugattyút “Proceed with device rotations per Step 10” = A 10. lépésben leírtak szerint fordítsa el az eszközt
“Do not proceed” = Ne haladjon tovább
9. lépés: Miután meggyőződött a lencse helyes elhelyezkedéséről és a haptikák megfelelő hajtásáról, a sebzésen keresztül a mélységjelzőig helyezze be a cső végét (9. ábra). A mélységjelző segít a cső végét az elülső tok nyílásához irányítani.
9. ábra
Depth Guard = Mélység jelző
10. lépés: Finom, folyamatos mozdulattal nyomja előre óvatosan a dugattyút. Ne nyomja túl erősen, mivel az a lencse sérüléséhez vezethet. Javasoljuk, hogy a lencse látható helyzetből való elmozdítása és behelyezése legalább 5 másodpercet vegyen igénybe. Ha az elülső haptika egyenes, vagy hurkot képez, és nyújtva van a lencse előtt, forgassa el eszköz balra, mielőtt továbbtolná a dugattyút, biztosítandó, hogy az elülső haptika megfelelően helyezkedjen el a lencsetokban. Amint az elülső haptika kezdi elhagyni a cső végét, helyezze az elülső haptikát a lencsetokba annak megfelelő pozíciójába, majd lassan tovább nyomva a dugattyút juttassa tovább a lencse többi részét is. Amint a lencse optikai része is elhagyja a cső végét, fordítsa vissza az eszközt középre vagy kissé jobbra, hogy a lencse elülső oldalával felfelé nyíljon ki a lencsetokban.
11. lépés: A megfelelő pozicionáló eszköz segítségével helyezze el a lencsét a lencsetokban, az irisszel párhuzamos síkban.
12. lépés. Az egész eszközt dobja el. Ne használja újra az UltraSert™ előretöltött implantáló eszközt.
BETEGREGISZTRÁCIÓ ÉS JELENTÉS:A dobozban lévő, kitöltendő Beteg Azonosító Kártyát át kell adni a betegnek megőrzés céljából, valamint fel kell hívni a beteg figyelmét arra, hogy az egy olyan dokumentum, amelyet a jövőben szemorvosi vizsgálat esetén meg kell mutatni az orvosnak.Az USA-ban a lencsék beültetését követően minden beteget azonnal az Alcon Laboratories Inc. cégnél kell regisztrálni. A betegek regisztrációja a dobozban található Regisztrációs Kártya kitöltéséből és annak a mellékelt borítékban az Alcon
60
Laboratories Inc. központba való visszaküldéséből áll. A betegregisztráció az Alcon Laboratories Inc. cégnek hosszú távú beteg megfigyelési programjához szükséges, valamint segít bennünket a mellékhatások megválaszolásában. Az USA-n kívül a helyi rendelkezések az irányadók.
Azokat az eseményeket, amelyek arra utalnak, hogy a műlencse okozta vagy hozzájárult az elhalálozáshoz vagy súlyos sérüléshez, ideértve azokat az eseményeket is, amelyeket az orvostechnikai eszköznek a minőségi előírások nem teljesítése vagy attól bárminemű eltérése okozott, jelenteni kell az Alcon Laboratories Inc. cégnek. A fentiek betartása minden sebészorvos számára ajánlott, így az intraokuláris lencse implantáció lehetséges hosszú távú következményei is dokumentáltak lesznek.Az USA-ban a műlencsével kapcsolatos nemkívánatos eseményeket az alábbi címen és telefonszámon kell jelenteni:
Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Call Toll free: 1-800-757-9780
Az Amerikai Egyesült Államokon kívül a nemkívánatos eseményekről szóló jelentést illetően a helyi Alconnal kell felvenni a kapcsolatot:Alcon Hungária Kft.1114 Budapest, Bartók Béla út 43-47, 4-20 tel: 36-1-463-9080, fax:+36-1-463-9081
Az AcrySof® hajlítható hátsó csarnok lencsékkel végzett klinikai vizsgálatokKét randomizált, prospektív, jól kontrollált klinikai vizsgálatot végeztek az AcrySof® egytestű, hajlítható hátsó csarnok lencsével kapcsolatban. Az első vizsgálatot az AcrySof® Natural egytestű hátsó csarnoklencse, az (UV és kék fényt szűrő) SB30AL alap lencsemodell hatékonyságának és biztonságának igazolására végezték el. A randomizált klinikai vizsgálatban a kontroll lencse az AcrySof® (csupán UV fényt abszorbeáló) SA30AL lencse modell volt. Az SB30AL vagy SA30AL intraokuláris lencsét kapott betegek esetében kizárólag az első megoperált szemre vonatkozó adatok szerepelnek. A második randomizált klinikai vizsgálat során az aszférikus optikájú AcrySof® IQ hajlítható, egytestű (UV és kék fényt szűrő kromofórt tartalmazó) intraokuláris lencsét vizsgálták az AcrySof® hajlítható, egytestű kontroll lencsével szemben, és a klinikai / funkcionális előnyeit értékelték a hagyományos szférikus modellhez képest.
Az AcrySof® Natural egytestű műlencse, az SB30AL klinikai vizsgálataA lencse beültetésétől számított legalább egy éven át megfigyelt betegektől származó adatok elfogadható bizonyítékkal szolgálnak arra vonatkozólag, hogy az AcrySof® Natural egytestű SB30AL lencsemodell biztonságos, az aphakia korrekciójára megfelelő eszköz.
SB30AL AcrySof® Natural egytestű lencsemodellt kapott betegpopulációA bilaterális vizsgálatban legalább az első megműtött szemébe SB30AL lencsemodellt kapott betegek 70,6%-a nő és 29,4 %-a férfi volt. Az SA30AL (kontroll) intraokuláris lencsemodellt kapott vizsgálati alanyok 60,5%-a nő és 39,5%-a férfi volt. Az SB30AL lencsét kapott betegek rassz szerinti megoszlása: 95,3% kaukázusi és 4,7% fekete beteg volt. A kontroll csoport (SA30AL) rassz szerinti megoszlása: 96,6% kaukázusi, 2% fekete, 1,4% egyéb rasszhoz tartozott. A vizsgálatban résztvevő, legalább az első megműtött szemébe SB30AL lencsét kapott teljes betegpopuláció átlag életkora 72,9 év volt. Hasonlóan az SA30AL lencsét kapott kontroll betegek tekintetében a teljes populáció átlagéletkora 71,9 év volt.
SB30AL AcrySof® Natural egytestű lencse modellt kapott betegek látásélességeMűtétet követően legalább egy éven át megfigyelt betegektől származó látásélesség eredményei a 2a táblázatban kerülnek bemutatásra. A betegeknek a műtétet megelőzően nem volt szembetegségük, corneájuk normális volt és nem volt macula degenerációjuk sem (legjobb esetek); a teljes betegpopulációból származó látásélesség adatai a 3a táblázatban láthatóak. Az azonos adatcsoportokhoz tartozó kontroll adatok sorrendben a 2b illetve a 3b táblázatban találhatók. A látásélesség tekintetében nem volt megfigyelhető semmilyen statisztikailag szignifikáns különbség sem az SB30AL és a kontroll SA30AL lencsét kapott betegek között, sem pedig a legjobb esetek vagy a teljes adatcsoport értékei között.
61
2a táblázatLegjobb, korrigált látásélesség az AcrySof® Natural SB30AL műlencsét kapott legjobb betegpopulációban
a beültetést követően legalább 1 évre
Élesség
Szám 20/20 vagy jobb 20/25 20/30 20/40 20/40 vagy
jobb> 20/40 - < 20/80 > 20/80
N N % N % N % N % N % N % N %
Korosztály
2b táblázatLegjobb, korrigált látásélesség az AcrySof® SA30AL műlencsét kapott legjobb betegpopulációban
a beültetést követően legalább 1 évre
Élesség
Szám 20/20 vagy jobb 20/25 20/30 20/40 20/40 vagy
jobb> 20/40 - < 20/80 > 20/80
N N % N % N % N % N % N % N %
Korosztály
3a táblázatLegjobb, korrigált látásélesség az AcrySof® Natural SB30AL műlencsét kapott teljes betegpopulációban
a beültetést követően legalább 1 évre
Élesség
Szám 20/20 vagy jobb 20/25 20/30 20/40 20/40 vagy
jobb> 20/40 - < 20/80 > 20/80
N N % N % N % N % N % N % N %
Korosztály
3b táblázatLegjobb, korrigált látásélesség az AcrySof® SA30AL műlencsét kapott teljes betegpopulációban
a beültetést követően legalább 1 évre
Élesség
Szám 20/20 vagy jobb 20/25 20/30 20/40 20/40 vagy
jobb> 20/40 - < 20/80 > 20/80
N N % N % N % N % N % N % N %
Korosztály
Az SB30AL AcrySof® Natural egytestű lencsemodell kumulatív mellékhatásaiAz AcrySof® Natural egytestű lencsemodellt, SB30AL illetve az SA30AL műlencsét kapott betegek esetében a műtétet követően legalább egy éven át megfigyelt posztoperatív időszak mellékhatásainak kumulatív arányai a 4. táblázatban táthatók. A kumulatív mellékhatásokat tapasztalt betegek arányainak tekintetében az SB30AL illetve az SA30AL lencse modellt kapott betegcsoportoknál lényeges eltérés nem volt.
62
4. táblázatAz AcrySof® Natural SB30AL lencsét és az SA30AL kontroll lencsét kapott betegcsoportok kumulatív
mellékhatásai a műtétet követően 1 évre
SB30AL (N= 153)
SA30AL(N= 147) p-érték*
N % N %
A mellékhatás típusa
0 0.0 0 0.0 NA Kumulatív Hypopyon
Intraokuláris infekció / Endophthalmitis 0 0.0 0 0.0 NA
Macula ödéma 4 2,6 2 1,4 0,6847
Pupilláris blokk 0 0.0 0 0.0 NA
Retinaleválás vagy retinaleválás korrekciója 0 0.0 0 0.0 NA
Műlencse dislocatio 1 0.7 0 0.0 1.0000
Másodlagos sebészeti beavatkozás Kéregkmaradányok eltávolítása Eltávolítás (dislocatio tok ruptura miatt) Cryotherapia a retinaszakadás korrekciójához Paracentesis a szemnyomás csökkentésére Fokális lézeres kezelés Photodinámiás kezelés Eltávolítás biometriai hiba miatt
51 1 11100
3.3 0.7
0.70.70.70.70.00.0
20000011
1.40.00.00.00.00.00.70.7
0,4482
Hyphema 0 0.0 0 0.0 NA
* a p-értékek az SB30AL és az SA30AL modellek összehasonlításakor a Fisher egzakt próbából származnak
Az AcrySof® Natural egytestű lencse modell, az sb30al folyamatosan fennálló mellékhatásai Az SB30AL AcrySof® Natural egytestű lencsemodellt illetve a SA30AL kontroll modellt kapott betegek műtétet követően legalább egy évre megfigyelt, folyamatosan fennálló mellékhatások arányai az 5 táblázatban láthatóak. Bármilyen folyamatosan fennálló mellékhatásokat tapasztalt betegek arányainak tekintetében az SB30AL illetve az SA30AL lencse modellt kapott betegcsoportoknál lényeges eltérés nem volt.
5. táblázatAz AcrySof® Natural SB30AL lencsét és a SA30AL kontroll lencsét kapott betegcsoportok folyamatosan fennálló
mellékhatásai a műtétet követően 1 évre
SB30AL (N= 138)
SA30AL(N= 127) p-érték*
N % N %
A mellékhatás típusa
0 0 1 0,8 0,4792Tartós Cornea ödéma
Iritis 0 0 0 0,0 NA
Macula ödéma 2 1,4 1 0,8 1,0000
Vitritis 0 0 0 0.0 NA
A megemelkedett szemnyomás kezelése 0 0 0 0.0 NA
* a p-értékek az SB30AL és az SA30AL modellek összehasonlításakor a chi-négyzet próbából származnak
Az AcrySof® Natural egytestű SB30AL lencsemodellt kapott betegek színlátása A műtétet követő időszakban 120 –180 napon a Farnsworth D-15 teszt alkalmazásával végezték el a színérzékelés vizsgálatát. A normál színlátású, az első megműtött szembe SB30AL lencsemodellt kapott 109 betegből a műtétet követően a 120-180. napon elvégzett vizsgálat során 107 (98,2%) beteg teljesítette a színérzékelést vizsgáló tesztet. Az első megműtött szembe SA30AL lencsét kapott normál színlátású 102 betegből a műtétet követően a 120-180. napon elvégzett vizsgálat során pedig 97 (95,1%) betegnek sikerült a színérzékelést vizsgáló tesztje. Nem tapasztaltak jelentős különbséget az SB30AL illetve az SA30AL lencsék között azon betegek esetében, akik megfeleltek a műtétet követően, a 120 és 180 nap között elvégzett színérzékelési teszten. Ezek alapján a kromofór hozzáadása nem befolyásolja kedvezőtlenül a normál színlátással bíró betegek színlátását.
63
Az AcrySof® Natural egytestű SB30AL lencsemodell Nd:YAG arányai Átlagosan 21,6 hónapos nyomonkövetés során a SB30AL lencsét kapott 135 beteg (2,2%) közül három (3) esetben végeztek el Nd:YAG hátsó kapszulotómiát. Átlagosan 21,9 hónapos nyomonkövetés során a SA30AL lencsét kapott 127 beteg (1,6%) közül két (2) esetben végeztek el Nd:YAG hátsó kapszulotómiát.
Az AcrySof® műlencse klinikai vizsgálata Hasonló, korábban elvégzett intraokuláris lencse vizsgálatok típusával megegyezően; az AcrySof® IQ és a szférikus kontroll lencse összevetése céljából végzett kontrollált, randomizált, kettős-vak, multicentrikus, kontralateriális beültetéssel járó klinikai vizsgálatába olyan, kétoldali szürkehályogban szenvedő felnőtt betegeket válogattak be, akik jó általános szemészeti egészségi állapotúak voltak (azaz például korábban nem végeztek náluk szemműtétet, illetve nem állt fenn náluk degeneratív, a látással összefüggő betegség, amely jelentősen befolyásolta volna a látásélességet, vagy súlyos akut vagy krónikus állapot, amely megnövelte volna a betegek kockázatát). A szem szférikus aberrációi jelentősen kisebbek voltak az AcrySof® IQ lencsék esetében, mint a kontroll lencsék esetében. A kontraszt érzékenységgel kapcsolatos eredmények statisztikailag jelentős posztoperatív javulást mutattak (a 3. hónapban), az AcrySof® IQ beültetett lencse javára. Az AcrySof® IQ lencse beültetésekor statisztikailag és klinikailag is jelentős javulást tapasztaltak a funkcionális látásvizsgálatok és a szimulált éjszakai vezetés során, illetve számos vizsgált helyzetben is– különösen vakító fényben és ködben. Ezek az eredmények azt tükrözik, hogy (kék fényt szűrő kromofórt tartalmazó, az anyag felületén aszférikus optikájú) AcrySof® IQ intraokuláris lencse előnyös klinikai teljesítményt biztosít, az (aszférikus optikájú és kék fényt szűrő kromofor nélküli) monofokális AcrySof® intraokuláris lencséhez képest.
AcrySof® IQ lencse – szférikus és összes, magasabb rendű aberráció Az AcrySof® IQ lencse beültetésekor az átlagos szférikus aberráció kb. 0,1 mikrométer volt. A 10. ábra az AcrySof® IQ lencse javára megfigyelt, statisztikailag jelentős szférikus és az összes, magasabb rendű aberráció csökkenését ábrázolja. A 11. ábra hullámfront aberométerrel végzett, az átlagos szférikus aberráció méréseket mutatja minden szem esetében, lencsénként és korcsoportonként egyaránt. Mint ahogy az grafikonból is kitűnik, a beültetett AcrySof® IQ lencse esetében a szférikus aberráció csökkenése kortól független volt.
10. ábraSzférikus és az összes, magasabb rendű négyzetes középérték
90-120 napoknál a 2. szembe történt beültetést követően
11. ábraÁtlagos szférikus aberráció minden esetben és korcsoportonként
90-120 napoknál a 2. szembe történt beültetést követően
AcrySof® IQ lencse - távoli látásélességAz AcrySof® IQ lencse és a kontroll lencse klinikailag hasonló posztoperatív látásélességet biztosított. A monocularis látásélességgel kapcsolatos eredmények a 12. és 13. ábrán kerülnek bemutatásra.
12. ábraLogMAR legjobban korrigált látásélesség
13. ábraLogMAR nem korrigált látásélesség
AcrySof® IQ lencse - kontraszt érzékenységA klinikai vizsgálat elsődleges célja az volt, hogy bizonyítsa az AcrySof® IQ lencse kiváló voltát a kontroll lencséhez képest a műtétet követően mért átlagos kontraszt érzékenység tekintetében, melyet mezópiás körülmények között, vakító fényben vagy a nélkül, a két térbeli frekvencia egyikén (3 és 6 ciklus/fok), Vector Vision CSV-1000 vizsgálótáblával, 3 cd/m2 megvilágítás mellett végeztek. A betegek egy alcsoportjánál elvégezték a funkcionális látásélesség kontraszt vizsgálatot (FACT) is, amely során 3 cd/m2 megvilágítást alkalmaztak. Ebben a klinikai vizsgálatban az AcrySof® IQ lencse kiváló voltát a kontroll lencséhez képest mezópiás körülmények között 6. ciklus/foknál igazolták, vakító fényben vagy a nélkül (CSV-1000), valamint 3. és 6. ciklus/foknál, vakító fény nélkül (FACT). A 14. és 15. ábrák a mezópiás kontrasztérzékenység eredményeket tartalmazzák az összes térbeli frekvencián, mind az AcrySof® IQ mind a kontroll lencse esetében.
14. ábraMezópiás kontrasztérzékenység (CSV-1000)
90-120 napoknál a 2. szembe történt beültetést követően
Min. 90. napnál, a 2. szembe történt beültetést követően AcrySof® IQ lencse– éjszakai vezetés szimulációjaA betegek egy alcsoportját validált, éjszakai vezetést szimuláló berendezésben is megvizsgálták. A betegeket monocularisan olyan körülmények között vizsgálták, amelyek a városi és vidéki környezetet utánozták, normál környezetben, vakító fényben és ködben.
64
Az éjszakai városi vezetés szimulált színhelye sokféle utcai világítást, autófényt, bolti fényeket és jelzőtáblákat tartalmaz, hogy így az ezekre a körülményekre jellemző erős környező világítást hozhassanak létre. Az éjszakai vidéki vezetés szimulált színhelye minimális mennyiségű környező világítást alkalmaz. A városi, illetve a vidéki díszletekhez kb. 65, illetve 85 km/óra szimulált vezetési sebességet alkalmaztak. Megkérték a betegeket, hogy észleljék és azonosítsák a tárgyak sorozatát mindkét díszlet esetében, ideértve a fehér-zöld színű országúti információs jelzőtáblákat, a fekete-sárga színű figyelmeztető jelzőtáblákat illetve a gyalogosokat. Továbbá megkérték a betegeket, hogy jelezzék, amikor megpillantják az első tárgyat, így az észlelés távolságát is rögzíteni tudták. Ezt követően megkérték a betegeket, hogy jelezzék, amikor már a tárgyakat is meg tudják különböztetni egymástól (pl.: mit jelez a tábla, a gyalogosok melyik irányba tartanak stb.), így pedig az azonosítás távolságát rögzítették.A 16. - 19. ábrák az AcrySof® IQ lencse és a kontroll lencse közti átlagos eltéréséket mutatják, városi és vidéki vezetési színhely esetében, mind az érzékelés és az azonosítás távolságok esetén (pl.: az egyénen belüli eltérések átlaga). Az AcrySof® IQ lencse teljesítményét tekintve a vizsgált 36 állapot közül 34-ben jobbn értéket mutatott a kontroll lencséhez képest, javulást mutatva mind az érzékelési mind az azonosítási távolságok tekintetében, mind a városi, mind a vidéki vezetési színhelyek esetében, különböző vizsgált vezetési állapotoknál (normál, vakító fény, köd). Ezen felül a vizsgált állapotok közül 12 esetben az AcrySof® IQ lencse statisztikailag is jelentősen jobb teljesítményt mutatott a kontroll lencséhez képest; amely során a legjelentősebb hatását és a legnagyobb előnyét a városi körülmények között (vakító fényben és ködben) megjelenített gyalogosok, valamint a vidéki körülmények között (vakító fényben és ködben) megjelenített figyelmeztető jelzőtáblák észlelésben, és azonosításban igazolták. Városi színhely esetében csökkent látási viszonyoknál (vakító fény, köd), a megnövekedett látási távolság 56 km/óra sebességnél az AcrySof® IQ lencse esetében több mint 0,5 másodpercnyi plusz időt biztosít ahhoz, hogy a beteg észrevegye a gyalogosokat, akik a városi körülmények között gyakorta előforduló veszélyt jelentik. A 0,5 másodperces növekedés funkcionális szempontból is jelentős, hiszen több időt biztosít a vezető számára a megfelelő intézkedések megtételére, úgymint megállás, elkerülés stb. (Green, 2000; McBride and Matson, 2004). A vidéki helyszínen minden körülmények között a megnövekedett látási távolság 85 km/óra sebességnél az AcrySof® IQ lencse esetében a több mint 1 másodpercnyi plusz időt biztosít ahhoz, hogy a beteg azonosíthassa a figyelmeztető jelzéseket, melyekkel a vidéki területeken gyakran találkozhat a vezető. A fél másodpercnyi növekedés funkcionálisan is jelentős, hiszen több időt biztosít a megfelelő intézkedés megtételére vezetés közben, amely éjszaka, ismeretlen vidéki területeken vezetve kritikus lehet, ahol a környező világítás gyakran hiányzik. Az alvizsgálatban 6 betegnél tapasztaltak a műtétet követően macula degenerációt vagy a lencse hátsó tok opacitást (PCO). Amint ezen betegeket kivették a vezetési vizsgálat elemzéséből, az intraokuláris lencsék közötti különbség a városi helyszínen, vakító fényviszonyok között a gyalogosok észlelését és azonosítását illetően hirtelen 0,5 másodperces küszöbértékre csökkent, amely már klinikai jelentőséggel bír. Abban az esetben, amikor az eredeti elemzéseket a sokszerűséghez igazították, a két intraokuláris lencse közti különbség már nem volt statisztikailag jelentős vakító fényben városi körülmények között a szöveg észlelését tekintve (Hommel’s p-érték = 0,0539) vagy vakító fényben, vidéki körülmények között a gyalogosok észlelését illetően (Hommel’s p-érték=0,0507). Az eredmények fokozott funkcionálás látást és feltehetőleg jelentőségteljes biztonsági előnyt igazolnak az AcrySof® IQ lencsét kapott idősebb beteg, valamint a szintén az utcán közlekedő többi vezető és gyalogos számára. A vizsgálat eredménye igazolja, hogy az AcrySof® IQ lencse javítja a funkcionális látást, amely pedig tovább növeli a rossz látási viszonyokkal jellemezhető élethelyzetekben a beteg biztonságát.
16. ábraÉjszakai vezetés szimulálása
Egyénen belüli átlagos különbségek a jelzőtáblák távolságának azonosításában, városi körülmények között a műtétet követően legalább 90 nap elteltével
AcrySof® IQ lencse – kontroll (n = 44)
17. ábraÉjszakai vezetés szimulálása
Egyénen belüli átlagos különbségek a jelzőtáblák távolságának azonosításában, városi körülmények között a műtétet követően legalább 90 nap elteltével
AcrySof® IQ lencse – kontroll (n = 44)
18. ábraÉjszakai vezetés szimulálása
Egyénen belüli átlagos különbségek a jelzőtáblák távolságának észlelésében, vidéki körülmények közötta műtétet követően legalább 90 nap elteltével
AcrySof® IQ – kontroll (n = 44)
19. ábraÉjszakai vezetés szimulálása
Egyénen belüli átlagos különbségek a jelzőtáblák távolságának azonosításában, vidéki körülmények között a műtétet követően legalább 90 nap elteltével
AcrySof® IQ lencse – kontroll (n = 44)
KISZERELÉSAz intraokuláris hátsó csarnok lencsék száraz állapotban, az UltraSert™ előretöltött implantáló rendszerben, egy elsődlegesen steril majd etilén-dioxiddal sterilizált csomagolásban kerül forgalomba, amely csak aszeptikus körülmények között bontható fel (lásd Felhasználási Útmutató).
65
LEJÁRAI IDŐ A sterilitás csak az elsődleges csomagolás épsége és zártsága mellett garantált. A lejárati idő az UltraSert™ előretöltött implantáló rendszer külső csomagolásán látható. A lejárt lencséket az Alcon Hungária Kft.-hez kell visszajuttatni (lásd ÁRUVISSZAVÉTELI SZABÁLYZAT).
ÁRUVISSZAVÉTELI SZABÁLYZATAz USA-ban a visszaküldött lencséket más termékre cserélik, jóváírásra nincs lehetőség. Minden visszaküldött terméket az Alcon Laboratories, Inc. cég visszaküldési számával kell ellátni és nyomon követhető módon kell visszajuttatni. A visszaküldési számot az Alcon Laboratories, Inc. ügyfélszolgálata adja meg. A visszaküldési szám kiadása nem jelenti a termék végső elfogadását. A részletes iránymutatásokért, beleértve a cserét, lépjen kapcsolatba az értékesítési vagy ügyfélszolgálati munkatársunkkal.Az Amerikai Egyesült Államokon kívül az áru visszavételi szabályzattal kapcsolatban kérjük, lépjen kapcsolatba a helyi Alcon irodával.Alcon Hungária Kft., 1114 Budapest, Bartók Béla út 43-47., tel: 36-1-463-9080, fax:+36-1-463-9081
REFERENCIÁK 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., et al. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., et al. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
66
A CSOMAGOLÁSON HASZNÁLT SZIMBÓLUMOK
SZIMBÓLUM MAGYARIOL Intraokuláris lencse
SP Egytestű
UV Ultraibolya
D Dioptria
ØB Test átmérő (optikai átmérője)
ØT Teljes átmérő (teljes hosszúság)
2STERILIZE Ne sterilizálja újra
Ne használja újra
Felhasználható
STERILE EO Etilén-dioxiddal sterilizálva
SN Sorozatszám
Figyelmeztetés
Gyártó
EC REP Meghatalmazott képviselő az Európai Közösségben
Felső hőmérsékleti korlát
Itt nyílik
Olvassa el a használati útmutatót
Ne használja, ha a steril csomagolás sérült
FIGYELEM: Az USA-ban szövetségi törvény korlátozza az eszköz orvosok részére történő árusítását, orvosok általi terjesztését vagy használatát.
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 USA
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,Egyesült Királyság
INFORMĀCIJA PAR IZSTRĀDĀJUMUAlcon Laboratories, Inc.
STERILAS UV un zilo gaismu filtrējošas akrila salokāmas viengabalamugurējās kameras lēcas ar UltraSert™ implantācijas sistēmu
APRAKSTSAcrySof® IQ UV un zilo gaismu filtrējošās akrila salokāmas viengabala mugurējās kameras lēcas ir optiski implanti, ko izmanto dabiskās lēcas aizstāšanai, lai koriģētu redzi afakijas gadījumā pieaugušiem pacientiem pēc kataraktas operācijas. AcrySof® IQ IOL ar Alcon patentēto zilo gaismu filtrējošo hromoforu filtrē gaismu līdzīgi cilvēka lēcai 400–475 nm zilās gaismas viļņa diapazonā (Boettner un Wolter, 1962). Līdztekus parastajai UV gaismas filtrēšanai AcrySof® IQ IOL samazina zilās gaismas garuma viļņu caurlaides koeficientu no 62 %, ja ir 400 nm, līdz 23 %, ja ir 475 nm (sk. 1. tabulu). Optiskā daļa sastāv no mīksta akrila materiāla ar augstu gaismas laušanas koeficientu. Pirms ievietošanas šo materiālu ir iespējams salocīt. Pēc implantēšanas lēca viegli atlokās pilnā lēcas izmērā. Lēcai ir abpusēji izliekta optiskā daļa ar haptiskajiem balstiem (skatīt 1. attēlu). AcrySof® IOL modeļa AU00T0 asfēriskā mugurējā virsma ir paredzēta negatīvas sfēriskās aberācijas iegūšanai, lai kompensētu pozitīvo radzenes sfērisko aberāciju. Modeļa AU00T0 viengabala IOL attēla kvalitāte (t. i., modulācijas pārneses funkcija) ir parādīta 4. attēlā. AcrySof® IQ modeļa AU00T0 viengabala IOL tiek piegādāta UltraSert™ implantācijas sistēmā, skatīt 2. attelu lai nodrošinātu šo lēcu ērtu un kontrolētu ievadīšanu lēcas kapsulas maisā. Lēcu fizikālās īpašības:
Fizikālās īpašības AprakstsOptikas tips Viengabala IOL ar asfērisku optiku
Optiskais materiāls Ultravioleto un zilo gaismu filtrējošs akrilāta/metakrilāta kopolimērs
UV nogriešana pie 10% transmisijas (nm) UV (400) pie 20.0D (skat. 3. attēlu)
Refrakcijas indekss 1,55
Optiskais stiprums Informāciju par pieejamā optiskā stipruma diapazonu skatīt Alcon izstrādājumu katalogā
Haptiskais materiāls Ultravioleto un zilo gaismu filtrējošs akrilāta/metakrilāta kopolimērs
Optiskais diametrs ØB (mm) 6,0
Pilnais garums ØT (mm) 13,0
Haptiskais leņķis 0º
Optiskie izmēri Skatīt 1. attēlu
Skatīt 1. attēlu tekstā angļu valodāFIZIKĀLĀS ĪPAŠĪBAS
Visi izmēri norādīti milimetros
ASPHERIC SURFACE = ASFĒRISKA VIRSMA
Skatīt 2. attēlu tekstā angļu valodā
Nozzle Tip = Uzgaļa galsDepth Guard = Dziļuma ierobežotājs
Main Body = Galvenais korpuss
LATVIAN
68
Skatīt 3. attēlu tekstā angļu valodāSPEKTRĀLĀS CAURLAIDĪBAS KOEFICIENTA LĪKNES
TRANSMITTANCE (%) = PROCENTUĀLĀ CAURLAIDĪBAWAVELENGTH (nm) = VIĻŅU GARUMS
6.0 D to 27.0 D range = 6,0–27,0 D diapazons4 yr – 53 yr old range, crystalline lens = 4–53 gadu vecuma diapazons, kristāliska lēca
PIEZĪMES • Parādītās viļņa garuma un spektrālās caurlaidības koeficienta robežvērtību līknes atspoguļo caurlaidības vērtību
diapazonu no akrilāta/metakrilāta kopolimēra izgatavotām IOL ar savienotu UV absorbētāju un Alcon patentētu zilo gaismu filtrējošu hromoforu.
• Mērījumu rezultāti atbilda tiešam caurlaidības koeficientam, izmantojot 6 mm atvērumu un optiskajam centram ekvivalentu diska biezumu.
• Dati par cilvēka lēcu ir ņemti no Boettner un Wolter (1962).
1. tabula.20,0 D IOL caurlaidības koeficienta salīdzinājums (%)
Modelis 400 nm 425 nm 450 nm 475 nm
SA60AT 21 86 88 88
AU00T0 8 34 49 68
Caurlaidības atšķirības (SA60AT pret AU00T0) 13 52 39 20
Caurlaidības koeficienta samazināšanās, izmantojot AU00T0 (% no SA60AT) 62 60 44 23
Skatīt 4. attēlu tekstā angļu valodāModeļa AU00T0 (20,0 D) modulāciju nodošanas funkcija
MTF (%) = MNF (%)Spatial Frequency (Ip/mm) = Telpiskā frekvence
ISO, 3-mm Aperture (see Note 1) = ISO, 3 mm atvērums (skatīt 1. piezīmi)Modified, 3-mm Aperture (see Note 2) = Modificēts 3 mm atvērums (skatīt 2. piezīmi)Modified, 5-mm Aperture (see Note 2) = Modificēts 5 mm atvērums (skatīt 2. piezīmi)
PIEZĪMES 1. Modeļa AU00T0 attēla kvalitāti raksturo modulāciju nodošanas funkcijas (MNF) mērījumu rezultāts modeļa
acī saskaņā ar ISO 11979-2. ISO 11979-2 standartā noteikts, ka MNF mērījumiem izmantojams tikai 3 mm atvērums.
2. Turklāt modeļa AU00T0 attēla kvalitātes raksturošanai tika izmantota MNF mērīšana modeļa acī, kas tiek izmantota, lai simulētu radzeni, kurā ir novērojama pieaugušam cilvēkam tipiska sfēriska aberācija. Izmantojot pārveidotu modeļa aci, MNF tika mērīta gan ar 3 mm, gan 5 mm atvērumu.
DARBĪBAS MEHĀNISMS AcrySof® IQ intraokulārās mugurējās kameras lēcas ir paredzētas ievietošanai acs mugurējā kamerā dabiskās kristāliskās lēcas aizstāšanai. Šāds novietojums lēcai ļauj darboties kā afakiju koriģējošai refraktīvai videi. Abpusēji izliektā optiskā daļa salīdzinājumā standarta sfērisko optisko daļu vidēji statistiskā acī samazina sfērisko aberāciju. Šo lēcu efektivitāte tīklenes patoloģiju sastopamības samazināšanā nav noteikta.
INDIKĀCIJAS AcrySof® IQ intraokulārās mugurējās kameras lēcas ir paredzētas cilvēka lēcas aizstāšanai, lai pieaugušiem pacientiem pēc kataraktas operācijas afakijas gadījumā koriģētu redzi (skatīt “BRĪDINĀJUMI”). Šīs lēcas ir paredzētas ievietošanai lēcas kapsulas maisā.
UZMANĪBU! Pacienti, kam ir kāds no turpmāk minētiem stāvokļiem, var nebūt piemēroti kandidāti intraokulārās lēcas ievietošanai, jo tā var saasināt esošo stāvokli, apgrūtināt stāvokļa diagnozi vai ārstēšanu, kā arī nepamatoti apdraudēt pacienta redzi. Ķirurgam pirms operācijas jāveic rūpīga novērtēšana un jāpieņem saprātīgs lēmums, apsverot ieguvuma un riska attiecību pirms lēcas implantēšanas pacientam, kam ir viens vai vairāki no turpmāk minētiem stāvokļiem: 1. Dzīslenes asiņošana 2. Vienlaicīga smaga acu slimība. 3. Izteikts stiklveida ķermeņa zudums. 4. Ļoti sekla priekšējā kamera. 5. Mikroftalmija.
69
6. Ar vecumu nesaistīta katarakta. 7. Mugurējās kapsulas plīsums, kas traucē IOL fiksāciju. 8. Smaga radzenes distrofija. 9. Smaga redzes nerva atrofija. 10. Nekontrolējams paaugstināts intraokulārais spiediens. 11. Zonulu dialīze, kas traucē IOL fiksāciju. 12. Krāsu redzes traucējumi. 13. Glaukoma. 14. Hronisks uveīts. 15. Diabētiska retinopātija. 16. Klīniski nozīmīgas makulas vai TPE pārmaiņas.Pētījumos ir novērots, ka personām ar normālu krāsu redzi pēc AcrySof® Natural IOL ievietošanas nav negatīvas ietekmes uz krāsu izšķirtspēju. AcrySof® Natural IOL ietekme uz redzi cilvēkiem ar pārmantotiem krāsu redzes defektiem vai pēc acu slimības (piemēram, glaukomas, diabētiskas retinopātijas, hroniska uveīta un citas tīklenes vai redzes nerva slimības) iegūtiem krāsu redzes defektiem nav pētīta.
BRĪDINĀJUMI 1. Jebkura ķirurģiska procedūra ir saistīta ar risku. Iespējamās ar kataraktas vai implantēšanas operāciju
saistītās komplikācijas ir (bet ne tikai) radzenes endotēlija bojājumi, endoftalmīts, tīklenes atslāņošanās, vitrīts, cistiska makulas tūska, radzenes tūska, zīlītes blokāde, ciklītiska membrāna, varavīksnenes dislokācija, strutu uzkrāšanās priekšējā kamerā un pārejoša vai paliekoša glaukoma.
2. Intraokulāro lēcu implantu drošums un efektivitāte nav apstiprināta pacientiem, kam jau ir acu patoloģijas (hroniska zāļu lietošanas izraisīta mioze, glaukoma, ambliopija, diabētiska retinopātija, transplantēta radzene, tīklenes atslāņošanās un/vai irīts utt.). Apsverot lēcas implantēšanu šādiem pacientiem, ārstam jāiepazīstas ar alternatīvām afakijas korekcijas metodēm, un lēmums par lēcas implantāciju jāpieņem tikai tad, ja alternatīvie varianti ir uzskatāmi par nepiemērotiem pacienta vajadzību apmierināšanai.
3. Intraokulārās lēcas implantācijas ilgtermiņa ietekme nav noteikta, tādēļ ārstiem pēc operācijas jāturpina regulāra pacientu novērošana.
4. Pacientiem, kam pirms operācijas ir kādas patoloģijas, piemēram, radzenes endotēlija slimība, radzenes anomālijas, makulas deģenerācija, tīklenes deģenerācija, glaukoma vai ilgstošas zāļu lietošanas izraisīta mioze, var netikt sasniegts tāds pats redzes asums kā pacientiem bez minētajām patoloģijām. Ārstam jānosaka lēcas implantēšanas sniegtais ieguvums pacientiem ar iepriekšminētajām slimībām.
5. No sekundāras iridektomijas zīlītes blokādes dēļ ir iespējams izvairīties, ja IOL implantēšanas laikā tiek veikta viena vai vairākas iridektomijas (Willis, et al., 1985).
6. Priekšējā kamerā ievietotas mugurējai kamerai paredzētas lēcas drošums un efektivitāte nav zināma. Dažos gadījumos mugurējās kameras lēcu implantēšana priekšējā kamerā nav bijusi droša (Girard, et al., 1983).
7. Dažas ar intraokulāro lēcu implantēšanu saistītās blakusparādības bija, piemēram, strutu uzkrāšanās priekšējā kamerā, intraokulāra infekcija, akūta radzenes dekompensācija un atkārtota ķirurģiska intervence. Sekundārai ķirurģiskai intervencei tika izmantota (bet ne tikai) lēcas repozicionēšana, lēcas nomaiņa, stiklveida ķermeņa aspirācija vai iridektomija zīlītes blokādes dēļ, brūces sulošanās novēršana un tīklenes atslāņošanās novēršana.
8. Neliela lēcas decentrēšanās, kas raksturīga intraokulārajām lēcām ar šauru vai nelielu optisko daļu, zināmos apgaismojuma apstākļos pacientiem var izraisīt apžilbšanu vai citus redzes traucējumus. Pirms implantēt IOL ar šauru vai nelielu optisko daļu, ķirurgam jāapsver šādu parādību iespējamība. Implantējot šauru vai nelielu optisko lēcu, ir ieteicams izdarīt kapsuloreksiju.
9. Ar kapsuloreksiju, kā arī PMMA, silikona un akrila mugurējās kameras lēcu implantēšanu ir bijusi saistīta lēcas kapsulas maisa izstiepšanās pēc operācijas, un tās dēļ ir notikusi priekšējās kameras dažādas pakāpes dziļuma samazināšanās un radusies tuvredzība (Holtz, 1992).
10. Lai nepieļautu lēcas decentrēšanos vai dislokāciju, pirms tās iekapsulēšanās jāievēro piesardzība. Daži klīniskie gadījumi liek uzskatīt, ka iekapsulēšanās notiek četru nedēļu laikā.
11. AcrySof® Natural viengabala lēcas klīniskais pētījums (atsauces 2.–5. tabulā) tika veikts ar lēcu, kas paredzēta implantēšanai tikai lēcas kapsulas maisā. Nav pieejami klīniski dati, kas pierāda tās drošumu un efektivitāti pēc ievietošanas sulcus ciliaris.
Operācijas beigās ieteicams izņemt no acs viskoelastīgo materiālu, īpašu uzmanību pievēršot attālumam starp mugurējās kapsulas virsmu un lēcu. Tas ir iespējams, IOL optiskajai daļai viegli piespiežot I/A uzgali un izmantojot skalošanas un aspirācijas standartmetodi viskoelastīgā materiāla izņemšanai no acs. Tā viss atlikušais viskoelastīgais materiāls tiks izspiests acs priekšējā daļā, no kurienes tas ir viegli aspirējams.
PIESARDZĪBAS PASĀKUMI 1. Šīs intraokulārās lēcas vai UltraSert™ implantācijas sistēmu nav atļauts nekādā veidā atkārtoti sterilizēt. 2. Intraokulārās lēcas jāuzglabā temperatūrā līdz 45 °C (113 °F). 3. Ar lēcām jārīkojas uzmanīgi, lai nesabojātu lēcas virsmas vai haptiskos balstus. 4. Nav atļauts jebkādā veidā mēģināt pārveidot haptiskos balstus. 5. Intraokulāro lēcu implantēšanai ir nepieciešamas augsta līmeņa ķirurga iemaņas. Pirms mēģināt implantēt
intraokulāro lēcu, ķirurgam jābūt novērojušam un/vai asistējušam vairākās implantēšanas operācijās, kā arī sekmīgi apguvušam vienu vai vairākus intraokulāro lēcu implantēšanas mācību kursus.
6. Neatvērta un nebojāta iepakojuma saturs ir sterils.
70
7. UltraSert™ implantācijas sistēma ir tikai vienreizlietojama. Pēc izmantošanas tā jāiznīcina. 8. UltraSert™ implantācijas sistēma ir izmantojama operāciju zālēs ar gaisa temperatūru 18–23 °C (64–73 °F)
LĒCAS STIPRUMA APRĒĶINĀŠANAMugurējās kameras intraokulāro lēcu vajadzīgais stiprums pirms operācijas jāaprēķina, pamatojoties uz ķirurga pieredzi, izvēli un paredzamo lēcas novietojumu. Lēcas stipruma aprēķināšanas metodes ir aprakstītas šajos atsauces materiālos:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. et al. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. et al. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
IETEIKTĀ A KONSTANTE Uz ārējās uzlīmes norādītā A konstante ir domāta kā ieteikums un implanta stipruma aprēķināšanas sākumpunkts. Ieteicams izstrādāt savu klīniskajai pieredzei atbilstošu konstanti, ņemot vērā konkrētus lēcu modeļus, operācijas metodes, pieejamās mērierīces un pēc operācijas prognozējamos rezultātus.Ja lēcas stipruma aprēķināšanai nepieciešama papildinformācija, Amerikas Savienotajās Valstīs pa tālruni 1-800-TO-ALCON (1-800-862-5266) jāsazinās ar Alcon Laboratories, Inc. Ārpus ASV jāsazinās ar Alcon vietējo biroju vai izplatītājiem.
APSTIPRINĀTIE ALCON VISKOELASTĪGIE MATERIĀLI UN PASŪTĪJUMA NUMURISkatīt iepakojuma pielikumu. Atļauts izmantot tikai Alcon apstiprinātos viskoelastīgos materiālus. Neapstiprinātu viskoelastīgo materiālu izmantošana var sabojāt lēcu un izraisīt implantācijas procesa komplikācijas. Lai saņemtu pilnu kopā ar šo lēcu izmantojamo Alcon apstiprināto viskoelastīgo materiālu sarakstu, jāsazinās ar savu Alcon pārstāvi.
NORĀDĪJUMI PAR LIETOŠANU1. solis. UltraSert™ implantācijas sistēma ir izmantojama operāciju zālēs ar gaisa temperatūru 18–23 °C
(64–73 °F).
2. solis. Pēc neatvērta ārējā iepakojuma uzlīmes pārbaudiet modeli, stiprumu, pareizo konfigurāciju un derīguma termiņu.
3. solis. Pēc ārējās kartona kārbas atvēršanas ierīces iepakojums jāpārbauda attiecībā uz bojājumiem. Ja ir atklāts bojājums, jāizmanto cita AcrySof® IQ IOL un UltraSert™ implantācijas sistēma. Pēc tam jāpārbauda, vai ierīces uzlīmē informācija par lēcu (piemēram, modelis, stiprums un sērijas numurs) atbilst informācijai uz ārējās kārbas marķējuma.
4. solis. Lai izņemtu UltraSert™ implantācijas sistēmu, jāsatver plastmasas paliktņa stūris, pilnībā jāatver TYVEK† materiāla vāciņa daļa, un ierīce jānovieto sterilā vidē. Ja pēc pārbaudes ierīces uzgalis izskatās bojāts, sadalījies vai deformējies, jāizmanto cita AcrySof® IQ IOL un UltraSert™ implantācijas sistēma. Ja ierīce nav pilnībā nebojāta vai virzulis/tā fiksators šķiet izkustējies transportēšanas laikā, jāizmanto cita AcrySof® IQ IOL un UltraSert™ implantācijas sistēma (skatīt “POLITIKA ATTIECĪBĀ UZ ATPAKAĻSŪTĀMĀM PRECĒM”).
† Reģistrēta E.I. DuPont De Nemours & Co prečzīme.
Kad lēcas ir sagatavotas implantācijai, ar minimāliem starplaikiem secīgi izpildiet 5., 6. un 7. soli.
5. solis. Viskoelastīgā kanula pa viskoelastīgo kanālu, kas atrodas ierīces kanulas virzītāja daļā, pilnībā perpendikulāri jāieliek ierīcē, kā parādīts 5. attēlā. Ierīce jāpiepilda, līdz viskoelastīgais materiāls ir redzams, ieplūstot līdz uzgaļa līnijai un (skatīt bultiņu 5. attēlā), un pēc tam jāizvelk kanula. Tam būs nepieciešami aptuveni 0,2 ml viskoelastīgā materiāla. Atļauts izmantot tikai Alcon apstiprinātu viskoelastīgu materiālu izmantošanai UltraSert™ implantācijas sistēmā, tam vispirms atļaujot sasniegt operāciju zāles temperatūru. Alcon apstiprināto viskoelastīgo materiālu sarakstu skatīt šī iepakojuma pielikumā.
Skatīt 5. attēlu tekstā angļu valodā
Viscoelastic Port = Viskoelastīgais kanāls“Fill-To” Line = Līnija “Piepildīt līdz”
6. solis. Izņemiet zilo lēcas turētāju, satverot aiz reljefās bultiņas daļas un paliecot nedaudz uz priekšu, vienlaikus ceļot uz augšu un prom no ierīces (6.a attēls). Izņemiet zilo virzuļa turētāju, to satverot un velkot taisni uz augšu un prom no ierīces (6.b attēls). Izmetiet lēcas turētāju un virzuļa turētāju. Pēc lēcas turētāja izņemšanas nemēģiniet pievienot viskoelastīgo materiālu pie ierīces, citādi iespējams sabojāt lēcu. Pēc virzuļa turētāja izņemšanas nemēģiniet izvilkt virzuli, citādi ir iespējams to sabojāt.
Skatīt 6.a un 6.b attēlu tekstā angļu valodā
71
7. solis. Ar vienmērīgu un nepārtrauktu kustību uzmanīgi bīdiet uz priekšu virzuli, līdz optiskā materiāla priekšējā mala ir pret līniju uz uzgaļa (skatīt 7.a attēlu). Šajā pozīcijā zilā atspere saskarsies ar galvenā korpusa sānu daļu, kā parādīts 7.b attēlā. Šāda virzuļa bīdīšana aizņem vismaz septiņas sekundes. Neļaujiet nevienai lēcas daļai izvirzīties ārpus uzgaļa gala. Ir svarīgi nebīdīt virzuli ar strauju kustību, jo ir iespējama nepareiza lēcas salocīšanās, un tā var tikt sabojāta.
Skatīt 7.a un 7.b attēlu tekstā angļu valodā
“Fill-To” Line = Līnija “Piepildīt līdz”
8. solis. Kad lēca ir aizbīdīta līdz līnijai uz uzgaļa, tā vizuāli jāpārbauda, lai noteiktu haptisko balstu novietojumu. Virzulim jāsaskaras ar optiskās daļas mugurējo malu. Pirms ievietošanas iegriezumā neviena lēcas nedrīkst būt izvirzījusies no uzgaļa (skatīt 8. attēlu). Kad lēca ir novietot pret uzgaļa līniju, tā jāimplantē triju minūšu laikā.
Skatīt 8. attēlu tekstā angļu valodā
“Proceed with full plunger advancement to fully deliver lens” = Turpiniet, bīdot virzuli līdz galam, lai pilnībā ievietotu lēcu
“Proceed” = Turpiniet“Proceed with full plunger advancement to fully deliver lens” = Turpiniet, bīdot virzuli līdz galam, lai pilnībā ievietotu
lēcu“Proceed with device rotations per Step 10” = Groziet ierīci, kā aprakstīts 10. solī
“Do not proceed” = Neturpiniet
9. solis. Kad ir apstiprināts pareizs lēcas novietojums un haptisko balstu izliekums, turpiniet lēcas implantēšanu, uzgaļa galu ievadot iegriezumā līdz dziļuma ierobežotājam (9. attēls), priekšējās kameras atvēruma virzienā.
Skatīt 9. attēlu tekstā angļu valodā
Depth Guard = Dziļuma ierobežotājs
10. solis. Ar vienu vienmērīgu un nepārtrauktu kustību bīdiet virzuli. Nebīdiet virzuli pārāk strauji, citādi ir iespējams sabojāt lēcu. Vēlams, lai lēcas ievadīšana, sākot ar vizuālās pārbaudes pozīciju, aizņemtu vismaz piecas sekundes. Ja priekšējais haptiskais balsts ir taisns vai salocījies cilpā un izvirzīts aiz lēcas priekšējās daļas, pirms virzuļa bīdīšanas pagrieziet ierīci slīpi pa kreisi, lai nodrošinātu, ka priekšējais haptiskais balsts ir pareizi novietots lēcas kapsulā. Tiklīdz priekšējais haptiskais balsts sāk izvirzīties no uzgaļa gala, ievietojiet to lēcas kapsulā pareizā orientācijā, un turpiniet lēni bīdīt virzuli, lai ievadītu lēcu. Kad optiskā daļa izvirzās no uzgaļa gala, ierīce pēc nepieciešamības jāgriež atpakaļ pret centru vai slīpi pa labi, lai nodrošinātu, ka lēca tās kapsulā atlokās ar priekšējo virsmu uz augšu.
11. solis. Lēca tās kapsulā ar piemērota pozicionēšanas instrumenta palīdzību pareizi jānovieto tā, lai plakne būtu paralēla varavīksnenes plaknei.
12. solis. Iznīciniet visu ierīci. UltraSert™ implantācijas sistēmu nav atļauts izmantot atkārtoti.
PACIENTU REĢISTRĀCIJA UN ZIŅOŠANAJāaizpilda iepakojumam pievienotā pacienta identifikācijas kartīte. Tā pacientam jāizsniedz kopā ar norādījumu kartīti pastāvīgi uzglabāt un uzrādīt visiem acu slimību speciālistiem, pie kuriem pacients kādreiz nākotnē varētu konsultēties.ASV visi pacienti tūlīt pēc tam, kad ir implantēta kāda no šīm lēcām, jāreģistrē Alcon Laboratories, Inc. Reģistrācija notiek, aizpildot lēcas iepakojumam pievienoto Implanta reģistrācijas kartīti, un to pa pastu nosūtot Alcon Laboratories, Inc. Pacientu reģistrācija ir obligāti nepieciešama Alcon Laboratories, Inc. ilgstošai pacientu novērošanas programmai, kā arī palīdz sniegt atbildi uz ziņojumiem par nevēlamām blakusparādībām.Ārpus ASV jāievēro vietējie tiesību akti par izstrādājumu reģistrāciju.
Par gadījumiem, kas pamatoti liecina par to, ka lēca var būt izraisījusi vai veicinājusi nāves iestāšanos vai nopietnas traumas rašanos, arī gadījumiem, kas bijuši tādēļ, ka medicīniskā ierīce nav atbildusi veiktspējas specifikācijai vai citādi nav darbojusies paredzētajā veidā, ir jāziņo Alcon Laboratories, Inc. Šāda informācija tiek pieprasīta no visiem ķirurgiem, lai dokumentētu iespējamos intraokulārās lēcas implantācijas radītos ilgtermiņa efektus. Ziņojot par intraokulāro lēcu izraisītajām nevēlamajām blakusparādībām, ķirurgiem jāizmanto šāda adrese un tālruņa numurs:
Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Bezmaksas tālruņa numurs: 1-800-757-9780
Ārpus ASV saistībā ar ziņojumiem par nevēlamām blakusparādībām ķirurgiem jāsazinās ar Alcon vietējiem birojiem vai izplatītājiem.
72
AcrySof® salokāmo mugurējās kameras lēcu klīniskie pētījumiAr AcrySof® salokāmajām viengabala mugurējās kameras lēcām ir veikti divi randomizēti prospektīvi, precīzi kontrolēti klīniskie pētījumi. Pirmais pētījums tika veikts, lai pierādītu, ka AcrySof® Natural viengabala mugurējās kameras UV starojumu un zilo gaismu filtrējošās lēcas modelis SB30AL ir tikpat drošs un efektīvs kā sākotnējais modelis. Tas bija randomizēts klīniskais pētījums, kurā kā kontroles lēca tika izmantots tikai UV starojumu absorbējošais AcrySof® modelis SA30AL. Ir iekļauti tikai dati par pirmo operēto aci tiem pacientiem, kam tika implantēta vai nu SB30AL, vai SA30AL modeļa intraokulārā lēca. Otrais ramdomizētais klīniskais pētījums tika veikts ar AcrySof® IQ salokāmām viengabala IOL ar UV starojumu un zilo gaismu filtrējošu hromoforu un asfērisku optiku, to salīdzinot ar AcrySof® F salokāmu viengabala kontroles lēcu, lai novērtētu klīniskos/funkcionālos ieguvumus salīdzinājumā ar tradicionālu sfērisku dizainu.
AcrySof® Natural viengabala lēcas modeļa SB30AL klīniskais pētījumsPar vismaz vienu gadu pēc operācijas novērotiem pacientiem iegūtie dati pamatoti pārliecina, ka AcrySof® Natural viengabala lēcas modelis SB30AL afakijas gadījumā ir drošs un efektīvs redzes korekcijas līdzeklis.
AcrySof® Natural viengabala lēcas modeli SB30AL saņēmušo pacientu populācijaDivpusējā pētījumā iekļauto pacientu grupā, kam vismaz pirmajā operējamajā acī tika implantēta SB30AL modeļa lēca, bija 70,6 % sieviešu un 29,4 % vīriešu. Pacientu grupā, kam tika implantēta SA30AL modeļa intraokulārā kontroles lēca, bija 60,5 % sieviešu un 39,5 % vīriešu. SB30AL modeļa lēcas saņēmušo pacientu populācijā bija 95,3 % baltās rases un 4,7 % melnās rases cilvēku. Kontroles (SA30AL modeļa lēcu saņēmušajā) pacientu populācijā bija 96,6 % baltās rases, 2 % melnās rases un 1,4 % citu rasu pārstāvju. Vidējais pacientu grupas, kam vismaz pirmajā operējamajā acī tika implantēta SB30AL modeļa lēca, bija 72,9 gadi. Vidējais pacientu grupas, kam tika implantēta SA30AL modeļa intraokulārā kontroles lēca, bija līdzīgs – 71,9 gadi.
AcrySof® Natural viengabala lēcas modeli SB30AL saņēmušo pacientu redzes asumsPārskats par redzes asumu, kas pacientiem, kam pirms operācijas nebija bijušas acu pataloģijas, radzenes anomālijas vai makulas distrofija (labākajā gadījumā), ir panākts vismaz vienu gadu pēc operācijas, ir parādīts 2.a tabulā, bet redzes asums, kas sasniegts kopējā populācijā, ir parādīts 3.a tabulā. Šo pašu datu kopas kontroles dati ir parādīti attiecīgi 2.b un 3.b tabulā. Statistiski nozīmīga atšķirība saistībā ar panākto redzes asumu, lietojot modeli SB30AL vai kontroles lēcu (modeli SA30AL), gan vislabākā gadījumā, gan kopējā populācijā netika konstatēta.
Skatīt 2.a tabulu tekstā angļu valodāVislabāk koriģētais redzes asums “labākā gadījuma” pacientu populācijā vismaz vienu gadu pēc operācijas, kam
izmantots AcrySof® Natural IOL modelis SB30AL
Redzes asums
#Per 20/20 vai labāks 20/25 20/30 20/40 20/40 vai
labāks> 20/40–< 20/80 > 20/80
N N % N % N % N % N % N % N %
Vecuma grupa
Skatīt 2.b tabulu tekstā angļu valodāVislabāk koriģētais redzes asums “labākā gadījuma” pacientu populācijā vismaz vienu gadu pēc operācijas, kam
izmantots AcrySof® Natural IOL modelis SA30AL (kontroles lēca)
Redzes asums
#Per 20/20 vai labāks 20/25 20/30 20/40 20/40 vai
labāks> 20/40–< 20/80 > 20/80
N N % N % N % N % N % N % N %
Vecuma grupa
73
Skatīt 3.a tabulu tekstā angļu valodāVislabāk koriģētais redzes asums kopējā pacientu populācijā vismaz vienu gadu pēc operācijas, kam izmantots
AcrySof® Natural IOL modelis SB30AL
Redzes asums
#Per 20/20 vai labāks 20/25 20/30 20/40 20/40 vai
labāks> 20/40–< 20/80 > 20/80
N N % N % N % N % N % N % N %
Vecuma grupa
Skatīt 3.b tabulu tekstā angļu valodāVislabāk koriģētais redzes asums kopējā pacientu populācijā vismaz vienu gadu pēc operācijas, kam izmantots
AcrySof® IOL modelis SA30AL (kontroles lēca)
Redzes asums
#Per 20/20 vai labāks 20/25 20/30 20/40 20/40 vai
labāks> 20/40–< 20/80 > 20/80
N N % N % N % N % N % N % N %
Vecuma grupa
AcrySof® Natural viengabala lēcas modeli SB30AL saņēmušajiem pacientiem novēroto nevēlamo blakusparādību kopskaitsPirms operācijas un vismaz vienu gadu pēc tās novēroto nevēlamo blakusparādību kopējā sastopamība AcrySof® Natural viengabala lēcas modeli SB30AL un modeli SA30AL saņēmušajiem pacientiem ir parādīta 4. tabulā. Statistiski nozīmīga ar SB30AL un SA30AL modeļa izmantošanu saistīta pacientu daļas, kam bijušas jebkuras no visām novērotajām blakusparādībām, atšķirība nav novērota.
4. tabulaVisas nevēlamās blakusparādības, kas novērotas vismaz vienu gadu pēc operācijas, izmantojot AcrySof® Natural
IOL modeli SB30AL (kontrolei tika izmantots modelis SA30AL)
SB30AL (N= 153)
SA30AL(N= 147)
p vērtība*
N % N %
Nevēlamo blakusparādību
veidi
0 0,0 0 0,0 NP Visas Strutu uzkrāšanās priekšējā kamerā
Tīklenes atslāņošanās vai tās novēršana 0 0,0 0 0,0 NP
Lēcas dislokācija 1 0,7 0 0,0 1,0000
Atkārtota sekundāra ķirurģiska intervenceLēcas masu skalošanaImplanta izņemšana (kapsulas perforācijas izraisītas dislokācijas dēļ)Krioterapija tīklenes plīsuma novēršanaiParacentēze acs iekšējā spiediena pazemināšanaiFokāla lāzerterapijaFotodinamiska terapijaImplanta izņemšana biometrijas kļūdas dēļ
51
1 11100
3,3 0,7
0,70,70,70,70,00,0
20
000011
1,40,0
0,00,00,00,00,70,7
0,4482
Hifēma 0 0,0 0 0,0 NP
* Fišera precizitātes testā iegūtas p vērtības, salīdzinot modeli SB30AL ar modeli SA30AL.
74
AcrySof® Natural viengabala lēcas modeli SB30AL saņēmušajiem pacientiem novērotās paliekošās nevēlamās blakusparādībasVismaz vienu gadu pēc operācijas novēroto paliekošo nevēlamo blakusparādību sastopamība AcrySof® Natural viengabala lēcas modeli SB30AL un kontroles modeli SA30AL saņēmušajiem pacientiem ir parādīta 5. tabulā. Statistiski nozīmīga ar SB30AL un SA30AL modeļa izmantošanu saistīta pacientu daļas, kam bijušas jebkuras novērotās paliekošās blakusparādības, atšķirība nav novērota.
5. tabulaPaliekošās nevēlamās blakusparādības, kas novērotas vismaz vienu gadu pēc operācijas, izmantojot AcrySof®
Natural IOL modeli SB30AL (kontrolei tika izmantots modelis SA30AL)
SB30AL ( N= 138)
SA30AL(N= 127)
p vērtība*
N % N %
Nevēlamo blakusparādību veidi
0 0 1 0,8 0,4792Paliekošās Radzenes tūska
Irīts 0 0 0 0,0 NP
Makulas tūska 2 1,4 1 0,8 1,0000
Vitrīts 0 0 0 0,0 NP
Paaugstināts intraokulārais spiediens, kura dēļ nepieciešama ārstēšana 0 0 0 0,0 NP
* Fišera precizitātes testā iegūtas p vērtības, salīdzinot modeli SB30AL ar modeli SA30AL.
AcrySof® Natural viengabala lēcas modeli SB30AL saņēmušo pacientu krāsu uztverePēcoperācijas perioda 120.–180. dienā ar Farnsvorta D-15 paneļa testu tika pārbaudīta krāsu uztvere. No 109 pacientiem ar normālu krāsu redzi, kuriem pirmajā operējamā acī tika implantēta SB30AL modeļa lēca un kuri tika pārbaudīti 120.–180. dienas pēcoperācijas vizītē, krāsu uztveres pārbaudi izturēja 107 pacienti (98,2 %). No 102 pacientiem ar normālu krāsu redzi, kuriem pirmajā operējamā acī tika implantēta SA30AL modeļa lēca un kuri tika pārbaudīti 120.–180. dienas pēcoperācijas vizītē, krāsu uztveres pārbaudi izturēja 97 pacienti (95,1%). Vērtējot pēc to pacientu procentuālās daļas, kas 120.–180. dienas pēcoperācijas vizītē izturēja krāsu uztveres pārbaudi, modelis SB30AL statistiski nozīmīgi neatšķīrās no modeļa SA30AL. Tas nozīmē, ka patentētā hromofora pievienošana pacientiem ar normālu krāsu redzi nerada negatīvu ietekmi uz krāsu uztveri.
Nd:YAG biežums pēc AcrySof® Natural viengabala lēcas SB30AL modeļa implantācijas Vidēji 21,6 mēnešus ilgā novērošanas periodā Nd:YAG mugurējā kapsulotomija tika veikta trijiem no 135 pacientiem (2,2 %) ar implantētu SB30AL modeļa lēcu. Vidēji 21,9 mēnešus ilgā novērošanas periodā Nd:YAG mugurējā kapsulotomija tika veikta diviem no 127 pacientiem (1,6 %) ar implantētu SA30AL modeļa lēcu.
AcrySof® IQ lēcu klīniskais pētījumsAtbilstoši jau veiktiem līdzīga dizaina IOL pētījumiem kontrolētā, randomizētā, dubultmaskētā daudzcentru kontralaterālā klīniskā implantu pētījumā, kura laikā AcrySof® IQ lēca tika salīdzināta ar sfērisku kontroles lēcu, tika iesaistīti pieauguši pacienti ar labu acu vispārējo veselību (piemēram, anamnēzē nav acu operāciju, deģeneratīvu redzes traucējumu, kas ievērojami ietekmē redzes asumu, vai smagu akūtu vai hronisku patoloģiju, kas pacientam var palielināt risku) un abpusēju kataraktu. AcrySof® IQ lēcas optiskā sfēriskā aberācija bija statistiski nozīmīgi mazāka nekā kontroles lēcai. Trīs mēnešus pēc operācijas tika novērots, ka pēc AcrySof® IQ implantācijas kontrastredzes pārbaudes rezultāti bija statistiski nozīmīgi labāki. Acīs ar implantētām AcrySof® IQ lēcām tika novērota arī statistiski un klīniski nozīmīga redzes funkcijas mērījumu un dažādos apstākļos (īpaši apžilbinošas gaismas un miglas apstākļos) simulētas transportlīdzekļa vadīšanas rezultātu uzlabošanās. Šie rezultāti apliecina, ka AcrySof® IQ IOL (ar asfērisku optiku uz zilo gaismu filtrējošu hromoforu saturoša materiāla platformas) salīdzinājumā ar monofokālo AcrySof® IOL (bez asfēriskas optikas un zilo gaismu filtrējoša hromofora) nodrošina labvēlīgāku klīnisko iedarbību.
AcrySof® IQ lēcas sfēriskā un kopējā lielākas pakāpes aberācijaAcīs ar implantētu AcrySof® IQ vidējā optiskā sfēriskā aberācija bija aptuveni 0,1 mikrometrs. 10. attēlā ir redzams, ka pēc AcrySof® IQ lēcas implantācijas statistiski nozīmīgi vairāk samazinās sfēriskā un kopējā lielākas pakāpes aberācija. 11. attēlā ir parādīti ar viļņu frontes aberometra palīdzību iegūtie acu sfēriskās aberācijas mērījumu vidējie rezultāti atkarībā no lēcas un vecuma grupas. Kā redzams šajā attēlā, acīs ar implantētu AcrySof® IQ lēcu sfēriskā aberācija samazinās neatkarīgi no pacientu vecuma.
Skatīt 10. attēlu tekstā angļu valodāSfēriskā un kopējā lielākas pakāpes PRK
90.–120. diena pēc otrā acs implanta ievietošanas
75
Skatīt 11. attēlu tekstā angļu valodāKopējā un no vecuma grupas atkarīgā vidējā sfēriskā aberācija
90.–120. diena pēc otrā acs implanta ievietošanas
Tāluma redzes asums pēc AcrySof® IQ lēcas implantācijasPēc operācijas AcrySof® IQ lēca un kontroles lēca nodrošināja klīniski līdzīgu redzes asumu. Monookulārā redzes asuma mērījumu rezultāti ir parādīti 12. un 13. attēlā.
Skatīt 12. attēlu tekstā angļu valodāLabākā redzes asuma korekcija LogMAR vienībās
Skatīt 13. attēlu tekstā angļu valodāNekoriģētas redzes asums LogMAR vienībās
Kontrastredze pēc AcrySof® IQ lēcas implantācijas Klīniskā pētījuma galvenais mērķis bija pierādīt AcrySof® IQ lēcas pārākumu salīdzinājumā kontroles lēcu, vērtējot kontrastredzes vidējos mērījumu rezultātus, kas ar Vector Vision CSV-1000 iekārtas (tabulas spilgtums bija 3 cd/m2) palīdzību pēc operācijas iegūti samazināta apgaismojuma apstākļos, izmantojot vai neizmantojot divu telpisko frekvenču gaismas zibšņus (trīs vai sešus ciklus vienai pakāpei). Pacientu apakšgrupā tika veikta arī kontrastredzes funkcijas (FACT) pārbaude (ar tabulas spilgtumu 3 cd/m2). Šajā klīniskajā pētījumā AcrySof® IQ lēcas pārākums salīdzinājumā ar kontroles lēcu samazināta apgaismojuma apstākļos tika pierādīts, izmantojot vai neizmantojot gaismas zibšņus (sešus ciklus vienai pakāpei, iekārta CSV-1000) un trīs vai sešus ciklus vienai pakāpei, neizmantojot gaismas zibšņus (FACT vērtēšanai). 14. un 15. attēlā ir parādīti pēc AcrySof® IQ lēcas un kontroles lēcas implantācijas iegūtie kontrastredzes vērtēšanas rezultāti, kas iegūti pēc visu telpisko frekvenču izmantošanas samazināta apgaismojuma apstākļos.
Skatīt 14. attēlu tekstā angļu valodāAr iekārtas CSV-1000 palīdzību iegūtie kontrastredzes mērījumu rezultāti samazināta apgaismojuma apstākļos
90.–120. diena pēc otrā acs implanta ievietošanas
Skatīt 15. attēlu tekstā angļu valodāApakšpētījums par kontrastredzi un FACT samazināta apgaismojuma apstākļos
Vismaz 90. diena pēc otrā acs implanta ievietošanas
Transportlīdzekļa vadīšanas simulācija nakts apstākļos pēc AcrySof® IQ lēcas implantācijasPacientu apakšgrupa tika pārbaudīta validētā nakts braukšanas simulatorā. Pacientu monookulārā redze tika pārbaudīta simulētā pilsētas un lauku vidē normāla apgaismojuma, apžilbinošu gaismas zibšņu un miglai raksturīga apgaismojuma apstākļos.Simulējot braukšanu naktī pa pilsētu, tiek izmantoti dažādi ielu apgaismes elementi, automašīnu gaismas, veikalu apgaismojums un reklāmas, lai radītu šādiem apstākļiem raksturīgu vides apgaismojumu. Simulējot braukšanu naktī pa lauku apvidu, tiek izmantots apkārtnes vides apgaismojums. Simulējot braukšanu pa pilsētu vai lauku apvidu, izmantotais kustības ātrums bija attiecīgi 55 un 90 km/h.Katrā simulācijā pacienti tika lūgti ieraudzīt un identificēt virkni objektu, arī zaļas informatīvās šosejas ceļazīmes ar baltiem simboliem, dzeltenmelnas brīdinājuma zīmes un gājējus. Pacienti tika lūgti reaģēt, ieraugot pirmo objektu, un tas ļāva reģistrēt attālumu, kādā pamanīts objekts. Pēc tam pacienti tika lūgti reaģēt, kad viņi spēja atšķirt objektu (piemēram, kas rakstīts uz zīmes, kādā virzienā iet gājējs utt.), un tas ļāva reģistrēt attālumu, kādā identificēts objekts. 16.–19. attēlā ir parādītas AcrySof® IQ un kontroles lēcu atšķirības, simulējot braukšanu pilsētā un lauku apvidū gan attiecībā uz objekta pamanīšanu, gan identificēšanu (piemēram, dažādiem pacientiem novērotās vidējās atšķirības).34 no 36 pārbaudītajiem apstākļiem AcrySof® IQ lēcas bija funkcionāli labākas par kontroles lēcām un dažādos pārbaudītajos simulētas braukšanas apstākļos nodrošināja labāku tāluma redzi saistībā ar objektu pamanīšanu un identificēšanu gan pilsētas, gan lauku apstākļos (normāla, apžilbinoša un miglai raksturīga apgaismojuma apstākļos). Turklāt 12 no šiem stāvokļiem AcrySof® IQ lēca bija statistiski nozīmīgi efektīvāka par kontroles lēcu, un vislielākā efektivitāte un priekšrocības tika novērotas saistībā ar gājēju pamanīšanu un identificēšanu pilsētas apstākļos un brīdinājuma zīmju pamanīšanu un identificēšanu lauku apvidū (pie gaismas uzliesmojuma un miglas apstākļos). Samazinātas redzamības apstākļos (apžilbinoša un miglai raksturīga apgaismojuma apstākļos) braucot ar ātrumu 55 km/h, lielākais ar AcrySof® IQ lēcas palīdzību iegūtais redzamības attālums nodrošina vairāk nekā 0,5 sekundes ilgu papildu laiku, reaģējot uz gājējiem, kuri pilsētas apstākļos visbiežāk rada risku. Šī 0,5 sekunžu ekonomija ir funkcionāli nozīmīga un ļauj iegūt vairāk laika atbilstošai rīcībai, piemēram, lai apturētu transportlīdzekli, izvairītos no šķēršļa utt. (Green, 2000; McBride un Matson, 2004). Lauku apvidū braucot ar ātrumu 90 km/h, ar AcrySof® IQ lēcas palīdzību iegūtais palielinātais redzamības attālums visos apstākļos nodrošina vairāk nekā 1 sekundi ilgu papildu laiku, lai identificētu brīdinājuma zīmes, un šādas situācijas lauku apvidos ir bieži sastopamas. Palielinājums par 0,5 sekundēm ir funkcionāli nozīmīgs, lai braucot iegūtu vairāk laika atbilstošai rīcībai, kas naktī nepazīstamā lauku apvidū, kur bieži vispār nav apkārtējā apgaismojuma, kļūst kritiski svarīga. Papildpētījumā piedalījās seši pacienti, kam pēc operācijas bija makulas deģenerācija vai mugurējās kapsulas apduļķojums (PCO). Kad attiecībā uz šiem pacientiem bija pabeigta braukšanas analīze, izrādījās, ka IOL lēcu efektivitāte saistībā ar gājēju pamanīšanu un identificēšanu apžilbinošā gaismā pilsētas apstākļos nesasniedza klīniskās nozīmības 0,5 sekunžu slieksni. Kad sākotnējie analīžu rezultāti bija koriģēti pēc daudzveidības, IOL lēcu efektivitātes atšķirība, vērtējot pēc teksta pamanīšanas žilbinošā apgaismojumā pilsētas apstākļos vai gājēju pamanīšanas žilbinošā apgaismojumā lauku apvidū, vairs nebija statistiski nozīmīga (Hommela p-vērtība attiecīgi 0,0539 un 0,0507).
76
Šie rezultāti pierāda, ka AcrySof® IQ lēcu implantācija gados vecākiem transportlīdzekļu vadītājiem uzlabo redzes funkciju un var nozīmīgi palielināt drošību gan viņiem pašiem, gan citiem autovadītājiem un gājējiem, kuri piedalās ceļu satiksmē. Šī testa rezultāti pierāda, ka AcrySof® IQ lēca uzlabo redzes funkciju, kas savukārt var uzlabot pacienta drošību sliktas redzamības apstākļos.
Skatīt 16. attēlu tekstā angļu valodāNakts braukšanas simulators
Dažādiem pacientiem noteiktā ar objektu pamanīšanu saistītā attāluma vidējās atšķirības pilsētas apstākļosVismaz 90 dienas pēc operācijas
AcrySof® IQ IOL salīdzinājumā ar kontroles lēcu (n = 44)
Skatīt 17. attēlu tekstā angļu valodāNakts braukšanas simulators
Dažādiem pacientiem noteiktā ar objektu identificēšanu saistītā attāluma vidējās atšķirības pilsētas apstākļosVismaz 90 dienas pēc operācijas
AcrySof® IQ IOL salīdzinājumā ar kontroles lēcu (n = 44)
Skatīt 18. attēlu tekstā angļu valodāNakts braukšanas simulators
Dažādiem pacientiem noteiktā ar objektu pamanīšanu saistītā attāluma vidējās atšķirības lauku apstākļosVismaz 90 dienas pēc operācijas
AcrySof® IQ IOL salīdzinājumā ar kontroles lēcu (n = 44)
Skatīt 19. attēlu tekstā angļu valodāNakts braukšanas simulators
Dažādiem pacientiem noteiktā ar objektu identificēšanu saistītā attāluma vidējās atšķirības lauku apstākļosVismaz 90 dienas pēc operācijas
AcrySof® IQ IOL salīdzinājumā ar kontroles lēcu (n = 44)
IEPAKOJUMSŠīs mugurējās kameras intraokulārās lēcas tiek piegādātas sausā veidā UltraSert™ implantācijas sistēmā, primārās sterilizācijas iepakojumā. Beigu sterilizācijai ir izmantots etilēna oksīds. Primārās sterilizācijas iepakojums jāatver tikai aseptiskos apstākļos (skatīt “NORĀDĪJUMI PAR LIETOŠANU”).
DERĪGUMA TERMIŅŠSterilitāte ir garantēta tikai tad, ja nav bojāts primārās sterilizācijas iepakojums. Derīguma termiņš ir nepārprotami norādīts uz UltraSert™ implantācijas sistēmas ārējās kārbas etiķetes. Visas līdz derīguma termiņam neizlietotās lēcas jānosūta atpakaļ Alcon Laboratories, Inc. (skatīt “POLITIKA ATTIECĪBĀ UZ ATPAKAĻSŪTĀMĀM PRECĒM”).
POLITIKA ATTIECĪBĀ UZ ATPAKAĻSŪTĀMĀM PRECĒMAmerikas Savienotajās Valstīs atpakaļ nosūtītās lēcas tiek pieņemtas vienīgi apmaiņai pret citiem izstrādājumiem, un naudas atmaksa nav paredzēta. Visām atpakaļ nosūtāmām precēm jābūt pievienotam Alcon Laboratories, Inc. atpakaļsūtāmo preču numuram, un tās jānosūta izsekojamā veidā. Atpakaļsūtāmo preču numuru var saņemt Alcon klientu apkalpošanas nodaļā. Šāda numura piešķiršana nenozīmē, ka atpakaļsūtāmā prece tiks pieņemta. Detalizētu informāciju par norādījumiem apmaiņas gadījumā var saņemt no tirdzniecības vai klientu apkalpošanas pārstāvja. Ārpus Amerikas Savienotajām Valstīm jāsazinās ar Alcon vietējo biroju vai izplatītājiem, lai iegūtu informāciju par piemērojamo politiku saistībā ar atpakaļsūtāmajām precēm.
ATSAUCES 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., et al. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., et al. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
77
MARĶEJUMĀ LIETOTIE SIMBOLI
SIMBOLS LATVIEŠUIOL Intraokulārā lēca
SP Viengabala
UV Ultravioletais
D Dioptrija
ØB Korpusa diametrs (optiskās daļas)
ØT Kopējais diametrs (kopējais garums)
2STERILIZE Nesterilizēt atkārtoti
Nelietot atkārtoti
Izlietot līdz
STERILE EO Sterilizēts lietojot etilēna dioksīdu
SN Sērijas numurs
Piesardzīgi
Ražotājs
EC REP Autorizētais pārstāvis Eiropas Savienībā
Augšējais temperatūras limits
Atvērt šeit
Skatīt lietošanas instrukciju
Nelietot ja sterilais iepakojums ir atvērts vai bojāts
Piesardzīgi: Federālā (ASV) likumdošana ierobežo iekārtas pārdošanu tikai ārstam vai pēc tā rīkojuma
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 USA
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,United Kingdom
STERILŪS UV ir mėlyną šviesą filtruojantys akriliniai sulankstomi vienos daliesužpakalinės kameros lęšiai su UltraSert™ parengta implantavimo sistema
APRAŠASUV ir mėlyną šviesą filtruojantys sulankstomi akriliniai vienos dalies užpakalinės kameros lęšiai AcrySof® IQ yra optiniai implantai, skirti pakeisti natūralų žmogaus akies lęšiuką koreguojant suaugusių pacientų regėjimą po kataraktos operacijos, kai yra afakija. AcrySof® IQ intraokuliniai lęšiai su Alcon patentuotu mėlyną šviesą filtruojančiu chromoforu 400–475 nm mėlynos šviesos bangos ilgių diapazone filtruoja šviesą panašiai kaip natūralus žmogaus akies lęšiukas (Boettner and Wolter, 1962). Be standartinio UV spindulių filtravimo, intraokuliniai lęšiai AcrySof® IQ sumažina skaidrumą mėlynos šviesos bangų ilgiams: nuo 62 % 400 nm ilgio bangoms iki 23 % 475 nm ilgio bangoms (žr. 1 lentelę). Optinę dalį sudaro minkšta didelio lūžio rodiklio akrilinė medžiaga. Šią medžiagą prieš implantuojant galima sulankstyti. Implantavus lęšiukas švelniai atsilenkia į pradinę formą. Šie lęšiai yra abipusiai išgaubtos formos, su atraminiais elementais (žr. 1 pav.). Intraokulinių lęšių AcrySof® IQ modelio AU00T0 užpakalinis paviršius yra su neigiama sferine aberacija, kad kompensuotų vidutinei ragenai būdingą teigiamą sferinę aberaciją. Modelio AU00T0 vienos dalies intraokulinių lęšių vaizdo kokybė (t. y. moduliacijų perdavimo funkcija) parodyta 4 pav. AcrySof® IQ modelio AU00T0 vienos dalies intraokuliniai lęšiai tiekiami parengtoje implantavimo sistemoje UltraSert™, parodyta 2 pav., kuri yra patogi ir valdoma priemonė patikimai implantuoti šiuos lęšius į kapsulės maišelį. Toliau nurodomos lęšių fizinės savybės.
Fizikinės charakteristikos AprašymasOptinis tipas Vienos dalies intraokulinis lęšis su asferine optine dalimi
Optinė medžiaga Ultravioletinius spindulius ir mėlyną šviesą filtruojantis akrilato/metakrilato kopolimeras
UV nukirtimas, prie 10% T (nm) UV (400) esant 20.0 D (Žr. 3 pav.)
Refrakcijos indeksas 1,55
Optinė galia Galimos laužiamosios gebos nurodytos Alcon gaminių žinyne
Atraminių elementų konfigūracija STABLEFORCE® modifikuoti L atraminiai elementai
Atraminių elementų medžiaga Ultravioletinius spindulius ir mėlyną šviesą filtruojantis akrilato/metakrilato kopolimeras
Optinis skersmuo ØB (mm) 6,0
Bendras ilgis ØT (mm) 13,0
Atraminio elemento kampas 0º
Optiniai skersmenys Žr. 1 pav.
Žr. anglų kalbą 1 paveiksląFIZIKINĖS SAVYBĖS
Visi išmatavimai pateikiami milimetrais
ASPHERIC SURFACE = ASFERINIS PAVIRŠIUS
Žr. anglų kalbą 2 paveiksląNozzle Tip = Antgalio galiukas Depth Guard = Gylio apsauga
Main Body = Pagrindinis korpusas
LITHUANIAN
79
Žr. anglų kalbą 3 paveiksląSPEKTRINIO LAIDUMO KREIVĖS
TRANSMITTANCE (%) = LAIDUMAS (%)WAVELENGTH (nm) = BANGOS ILGIS (nm)
6.0 D to 27.0 D range = 6,0 – 27,0 D ribos4 yr – 53 yr old range, crystalline lens = 4 metų – 53 metų amžiaus ribos, akies lęšis
PASTABOS • Čia pateikti pralaidumo ribos bangos ilgis ir spektrinio pralaidumo kreivės rodo intraokulinių lęšių, pagamintų iš
akrilato/metakrilato kopolimero su UV absorbuojančia medžiaga ir Alcon patentuotu mėlyną šviesą filtruojančiu chromoforu, pralaidumo vertes.
• Matuotas tiesioginis skaidrumas naudojant 6 mm apertūrą ir diską, kurio storis lygus lęšio storiui pagrindinėje optinėje ašyje.
• Boettner ir Wolter pateikti žmogaus lęšiuko duomenys (1962).
1 lentelė20,0 D intraokulinių lęšių pralaidumo palyginimas, %
Modelis 400 nm 425 nm 450 nm 475 nm
SA60AT 21 86 88 88
AU00T0 8 34 49 68
Pralaidumo skirtumas (SA60AT – AU00T0) 13 52 39 20
Pralaidumo sumažėjimas AU00T0 (% nuo SA60AT) 62 60 44 23
Žr. anglų kalbą 4 paveiksląModelio AU00T0 (20,0 D) moduliacijų perdavimo funkcija
ISO, 3-mm Aperture (see Note 1) = ISO, 3-mm apertūra (žr. 1 pastabą) Modified, 3-mm Aperture (see Note 2) = Modifikuota, 3-mm apertūra (žr. 2 pastabą) Modified, 5-mm Aperture (see Note 2) = Modifikuota, 5-mm apertūra (žr. 2 pastabą)
PASTABOS 1. Modelio AU00T0 vaizdo kokybė buvo nustatyta matuojant moduliacijų perdavimo funkciją (modulation transfer
function – MTF) ISO 11979-2 standarte aprašytame akies makete. ISO 11979-2 standartas reikalauja MTF matuoti naudojant tik 3 mm apertūrą.
2. Papildomai modelio AU00T0 vaizdo kokybė buvo nustatyta matuojant MTF akies makete, kuriame panaudota imitacinė ragena, turinti tipišką suaugusio žmogaus sferinę aberaciją. Naudojant šį modifikuotą akies maketą, MTF matavimai buvo atlikti naudojant tiek 3 mm, tiek 5 mm apertūras.
VEIKIMO BŪDASUžpakalinės kameros intraokuliniai lęšiai AcrySof® IQ yra skirti implantuoti į užpakalinę akies kamerą pakeičiant natūralų akies lęšiuką. Čia lęšis veikia kaip šviesą laužianti aplinka, koreguojanti regą, kai nėra natūralaus lęšiuko. Lyginant su standartine sferine optika vidutinėje akyje, abipusiai išgaubta asferinė optika mažina sferines aberacijas. Šių lęšių veiksmingumas mažinant tinklainės sutrikimų dažnį nenustatytas.
INDIKACIJOSUžpakalinės kameros lęšiai AcrySof® IQ yra skirti pakeisti natūralų žmogaus akies lęšiuką koreguojant suaugusiųjų pacientų regėjimą po kataraktos operacijos (žr. ĮSPĖJIMAI). Šie lęšiai skirti implantuoti į kapsulės maišelį.
DĖMESIOLigoniams, kuriems yra kuri nors iš toliau išvardytų būklių, intraokulinis lęšis gali netikti, nes lęšis gali paūminti esamą ligą, trukdyti šią ligą diagnozuoti ar gydyti bei sukelti nepagrįstą riziką sutrikdyti paciento regą. Prieš operaciją chirurgas turi kruopščiai ištirti pacientą ir, atsižvelgdamas į klinikinę būklė, pagrįstai nuspręsti, koks yra lęšio implantavimo naudos ir rizikos santykis, jei yra viena arba kelios iš toliau išvardytų būlių 1. Gyslainės hemoragija 2. Kita sunki akių liga, 3. Didelės stiklakūnio dalies netekimas 4. Ypač sekli priekinė kamera 5. Mikroftalmozė 6. Su amžiumi nesusijusi katarakta 7. Užpakalinės kameros įplyšimas (neleidžiantis fiksuoti intraokulinio lęšio)
80
8. Sunki ragenos distrofija 9. Sunki regos nervo atrofija 10. Nekontroliuojamas padidėjęs akispūdis 11. Zonulių atsiskyrimas (neleidžiantis fiksuoti intraokulinio lęšio) 12. Spalvų skyrimo sutrikimai 13. Glaukoma 14. Lėtinis uveitas 15. Diabetinė retinopatija 16. Kliniškai reikšmingi geltonosios dėmės ar tinklainės pigmentinio epitelio pokyčiaiTyrimai parodė, kad asmenų, anksčiau normaliai skyrusių spalvas ir kuriems implantuoti intraokuliniai lęšiai AcrySof®
Natural, gebėjimas skirti spalvas nepablogėja. Intraokulinių lęšių AcrySof® Natural įtaka pacientų, turinčių paveldėtų spalvų skyrimo sutrikimų arba spalvų skyrimo sutrikimų, atsiradusių kaip antrinė akių ligos (pvz., glaukomos, diabetinės retinopatijos, lėtinio uveito ir kitos tinklainės arba regos nervo ligos) pasekmė, regėjimui nėra tirta.
ĮSPĖJIMAI 1. Kaip ir atliekant bet kokią chirurginę procedūrą, egzistuoja tam tikra rizika. Galimos komplikacijos, susijusios su
kataraktos pažeisto lęšiuko šalinimu ir implantavimu, yra ragenos endotelio pažeidimai, endoftalmitas, tinklainės atšoka, vitritas, cistoidinė geltonosios dėmės edema, ragenos edema, vyzdžio blokas, ciklinė membrana, rainelės prolapsas, hipopionas, praeinanti ar pastovi glaukoma (tačiau galimos ir kitokios komplikacijos).
2. Intraokulinių lęšių implantų saugumas ir veiksmingumas nebuvo nustatyti pacientams, kuriems jau yra tam tikra su akimis susijusi būklė (lėtinė medikamentinė miozė, glaukoma, ambliopija, diabetine retinopatija, būklė po ragenos transplantacijos, buvusi tinklainės atšoka, iritas ir kt.). Prieš implantuodami tokiems pacientams lęšius, gydytojai turėtų išnagrinėti kitus afakijos korekcijos metodus ir pasirinkti lęšių implantavimą tik tada, kai kiti metodai laikomi neatitinkančiais paciento poreikių.
3. Ilgalaikis intraokulinių lęšių implantų poveikis nėra nustatytas. Dl šios priežasties gydytojai po operacijos pacientus turi ir toliau reguliariai stebėti.
4. Pacientų, kuriems iki operacijos buvo tokių sutrikimų kaip ragenos endotelio ligos, patologinė ragena, geltonosios dėmės degeneracija, tinklainės degeneracija, glaukoma ir lėtinė medikamentinė miozė, regos aštrumas gali netapti toks, kaip pacientų, kuriems tokių būklių nėra. Esant tokioms būklėms, gydytojas turi įvertinti lęšio implantavimo teikiamą naudą.
5. Vyzdžio bloko sukeltos iridektomijos galima išvengti, jei implantuojant intraokulinį lęšį atliekama viena ar daugiau iridektomijų (Willis et al., 1985).
6. Užpakalinės kameros lęšių implantavimo į priekinę kamerą saugumas ir veiksmingumas nenustatyti. Įrodyta, kad užpakalinės kameros lęšių implantavimas į priekinę kamerą kai kuriais atvejais yra nesaugus (Girard et al., 1983).
7. Tam tikros su intraokulinių lęšių implantavimu susijusios nepageidaujamos reakcijos yra hipopionas, intraokulinė infekcija, ūminė ragenos dekompensacija ir antrinė chirurginė intervencija. Antrinės chirurginės intervencijos metu gali būti atliekamos šios procedūros )tačiau ne vien jos): lęšio perstatymas, lęšio pakeitimas, stiklakūnio aspiracija arba vyzdžio bloko iridektomija, nesandarios žaizdos susiuvimas ir tinklainės atšokos gydymas.
8. Nedidelis lęšio nukrypimas nuo centro, kai implantuojamas siauros ar mažos optinės dalies lęšis, esant tam tikram apšvietimui gali sukelti blizgesį ar kitų regos sutrikimų. Prieš implantuodamas siauros ar mažos optinės dalies lęšį chirurgas turėtų įvertinti galimas pasekmes. Kai yra implantuojamas siauros ar mažos optinės dalies lęšis, rekomenduojama atlikti kapsuloreksiją.
9. Dėl kapsuloreksijos technikos ir implantavus PMMA, silikoninį ar akrilinį užpakalinės kameros lęšį po operacijos gali išsitempti maišelio kapsulė, priekinė kamera gali tapti sekli ir išsivystyti miopija (Holtz, 1992).
10. Iki lęšio inkapsuliacijos reikia imtis atsargumo priemonių, kad būtų išvengta lęšio nukrypimo nuo centro arba pasislinkimo. Kai kurie klinikiniai atvejai parodė, kad inkapsuliacija įvyksta per keturias savaites.
11. Klinikinis vienos dalies lęšių AcrySof® Natural tyrimas (duomenys pateikti 2–5 lentelėse) buvo atliktas su lęšiais, skirtais implantuoti tik į kapsulės maišelį. Nėra klinikinių duomenų parodyti jų saugumą ir veiksmingumą tvirtinant juos krumplyno vagelėje.
Rekomenduojama operacijos pabaigoje iš akies, ypač iš tarpelio tarp užpakalinės kameros ir lęšio, pašalinti viskoelastinę medžiagą. Tai atliekama iš pradžių plovimo/siurbimo antgaliu paspaudžiant intraokulinio lęšiuko optinę dalį atgal ir, panaudojus įprastą plovimo/siurbimo metodą, pašalinant iš akies viskoelastinę medžiagą. Taip visa užsilikusi viskoelastinė medžiaga išspaudžiama į priekį ir gali būti lengvai išsiurbta.
ATSARGUMO PRIEMONĖS 1. Šių intraokulinių lęšių ir UltraSert™ parengtos implantavimo sistemos negalima pakartotinai sterilizuoti jokiais
metodais. 2. Negalima intraokulinių lęšių laikyti aukštesnėje kaip 45 °C temperatūroje. 3. Su lęšiais reikia dirbti atsargiai, kad būtų išvengta lęšio paviršių arba atraminių elementų pažeidimų. 4. Negalima bandyti pakeisti atraminių elementų formos. 5. Intraokuliniam lęšiui implantuoti reikalingi geri chirurgo įgūdžiai. Prieš implantuodamas intraokulinį lęšį chirurgas turi
būti daug kartų stebėjęs implantavimo operacijas ir (arba) jų metu asistavęs bei sėkmingai baigęs vieną ar daugiau intraokulinių lęšių implantavimo mokymo kursų.
6. Pakuotės turinys yra sterilus, jei pakuotė nėra atidaryta ar pažeista. 7. Parengta implantavimo sistema UltraSert™ yra vienkartinė. Po naudojimo įrenginį UltraSert™ reikia išmesti. 8. Parengtą implantavimo sistemą UltraSert™ galima naudoti, kai operacinėje yra 18–23 °C temperatūra.
81
LĘŠIO LAUŽIAMOSIOS GEBOS SKAIČIAVIMASŠių užpakalinės kameros intraokulinių lęšių priešoperacinį laužiamosios gebos apskaičiavimą lemia chirurgo patirtis, pageidaujamas rezultatas ir numatomas lęšio implantavimo būdas. Lęšio laužiamosios gebos apskaičiavimo metodai aprašyti literatūroje.
Hoffer K. J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700-712, 1993.Holladay J. T. et al., A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17-24, 1988.Holladay, J. T., et al., Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations, J. Cataract Refract. Surg. 23:1356–1370, 1997.Retzlaff J. A., Sanders D. R. and Kraff M. Lens Implant Power Calculation, 3rd ed., Slack, Inc., Thorofare, N. J., 1990.
SIŪLOMA A KONSTANTA Išorinėje etiketėje nurodyta siūloma A konstanta pateikiama kaip preliminarus rodmuo, kurį naudojant turi būti atliekami implanto gebos skaičiavimai. Tinkamą konstantą rekomenduojama apskaičiuoti patiems, remiantis savo klinikine konkretaus lęšio modelio naudojimo patirtimi, taikomu chirurginiu metodu ir matavimo prietaisais bei operacijos rezultatais.Jungtinėse Valstijose, jei reikia papildomos informacijos, kaip apskaičiuoti lęšio laužiamąją gebą, kreipkitės į Alcon Laboratories Inc. biurą tel. 1-800-862-5266. Ne Jungtinėse Valstijose kreipkitės į vietinę bendrovės Alcon atstovybę ar platintojus.
TINKAMA ALCON VISKOELASTINĖ MEDŽIAGA IR UŽSAKYMO NUMERIAIŽr. su šia pakuote patiekiamą papildomą informaciją. Būtina naudoti tik Alcon patvirtintas viskoelastines medžiagas. Netinkamų viskoelastinių medžiagų naudojimas gali sukelti lęšio pažeidimą ir komplikacijas implantacijos proceso metu. Norėdami gauti šiam lęšiui Alcon patvirtintų viskoelastinių medžiagų sąrašą, kreipkitės į vietinę Alcon atstovybę.
NAUDOJIMO INSTRUKCIJA1 etapas. Parengtą implantavimo sistemą UltraSert™ galima naudoti, kai operacinėje yra 18–23 ° C temperatūra.
3 etapas. Atidarę išorinę kartoninę dėžutę, patikrinkite, ar nepažeista prietaiso pakuotė. Jei pastebima pažeidimų, naudokite kitą intraokulinį lęšį AcrySof® IQ ir parengtą implantavimo sistemą UltraSert™. Patikrinkite, ar ant prietaiso etiketės pateikti lęšio duomenys (pavyzdžiui, modelis, laužiamoji geba ir serijos numeris) atitinka išorinės dėžutės etiketėje pateiktus duomenis.
4 etapas. Išimkite implantavimo sistemą UltraSert™: paimkite plastikinio padėkliuko kampą, visiškai nulupkite TYVEK† medžiagos dangtelį ir perkelkite prietaisą į sterilią aplinką. Jei apžiūrėjus nustatoma, kad prietaiso antgalis atrodo pažeistas, ant jo yra dalelių arba jis yra deformuotas, naudokite kitą intraokulinį lęšį AcrySof® IQ ir parengtą implantavimo sistemą UltraSert™ . Jei prietaisas nėra visiškai nepažeistas arba atrodo, kad transportavimo metu buvo pajudintas stūmoklis ar jo užraktas, naudokite kitą intraokulinį lęšį AcrySof® IQ ir parengtą implantavimo sistemą UltraSert™ (žr. PREKIŲ GRĄŽINIMO TVARKA).
† Registruotas bendrovės E.I. DuPont De Nemours & Co prekės ženklas.
Kai lęšį paruošite įstatyti, atlikite 5, 6 ir 7 etapus su kuo trumpesnėmis pertraukomis tarp jų.
5 etapas. Kaip parodyta 5 pav., per viskoelastinės medžiagos jungtį, esančią prietaiso lęšio stabdymo dalyje, statmenai prietaisui iki galo įstatykite viskoelastinės medžiagos kaniulę. Pripildykite prietaisą viskoelastinės medžiagos tiek, kad matytųsi, kai ji prateka po antgalio liniją ( (žr. rodyklę 5 pav.), tada kaniulę ištraukite. Tam prireiks apie 0,2 ml viskoelastinės medžiagos. Naudokite tik Alcon viskoelastinę medžiagą, tinkamą naudoti su parengta implantavimo sistema UltraSert™, prieš tai leidę šiai medžiagai sušilti iki operacinės temperatūros. Tinkamų bendrovės Alcon tiekiamų viskoelastinių medžiagų sąrašas nurodytas šioje pakuotėje pateikiamoje papildomoje informacijoje.
Žr. anglų kalbą 5 paveikslą
Viscoelastic Port = Viskoelastinės medžiagos jungtis “Fill-To” Line = Pripildymo linija
6 etapas. Nuimkite mėlynos spalvos lęšio stabdiklį, jį suėmę už rodyklės formos rankenėlės ir šiek tiek palenkdami pirmyn bei keldami aukštyn ir tolyn nuo prietaiso (žr. 6a pav.). Nuimkite mėlynos spalvos stūmoklio užraktą, jį suėmę ir keldami tiesiai ir tolyn nuo prietaiso (žr. 6b pav.). Išmeskite lęšio stabdiklį ir stūmoklio užraktą. Nebandykite įleisti į prietaisą viskoelastinės medžiagos po to, kai jau nuimtas lęšio stabdiklis, nes gali būti pažeistas lęšis. Nebandykite atitraukti stūmoklio po to, kai jau nuimtas stūmoklio užraktas, nes gali būti pažeistas stūmoklis.
Žr. anglų kalbą 6a ir 6b paveikslus
82
7 etapas. Švelniai vienu sklandžiu nenutrūkstamu judesiu pastumkite stūmoklį į priekį, tiek, kad priekinis optinės dalies kraštas sutaptų su antgalio linija (žr. 7a pav.). Tokioje padėtyje mėlyna spyruoklė liesis su permatomu piršto antbriauniu, kaip nurodyta 7b pav. Šis stūmoklio stūmimas turi trukti mažiausiai 7 sekundes. Svarbu, kad stūmoklis nebūtų stumiamas netolygiai, nes dėl to lęšis gali neteisingai susilankstyti ir dėl to būti pažeistas.
Žr. anglų kalbą 7a ir 7b paveikslai
Fill-to Line = Pripildymo linija
8 etapas. Kai stūmoklis jau pastumtas iki antgalio linijos, reikia apžiūrėti lęšį, ar atraminiai elementai yra tinkamos padėties. Stūmoklis turi liesti optinės dalies užpakalinį kraštą. Prieš antgalio galiuką įkišant per pjūvį jokia lęšio dalis neturi būti išsikišusi iš antgalio (žr. 8 pav.). Kai lęšis yra ties antgalio linija, implantaciją reikia atlikti per 3 minutes.
Žr. anglų kalbą 8 paveikslą“Proceed with full plunger advancement to fully deliver lens” = „Stūmoklį stumkite iki galo, kol lęšis bus visiškai
išstumtas“ “Proceed” = „Tęskite“
“Proceed with full plunger advancement to fully deliver lens” = „Stūmoklį stumkite iki galo, kol lęšis bus visiškai išstumtas“
“Proceed with device rotations per Step 10” = „Sukite prietaisą, kaip nurodyta 10 etape“ “Do not proceed” = „Netęskite“
9 etapas. Įsitikinę, kad lęšis yra tinkamoje padėtyje ir atraminiai elementai yra tinkamai susilankstę, pradėkite lęšio implantavimą: įkiškite atgalio galiuką per pjūvį iki gylio apsaugos (žr. 9 pav.) ir antgalio galiuką taikykite į priekinės kapsulės angą.
Žr. anglų kalbą 9 paveikslą
Depth Guard = Gylio apsauga
10 etapas. Švelniai stumkite stūmoklį vienu sklandžiu nepertraukiamu judesiu. Nestumkite stūmoklio per greitai, nes gali būti pažeistas lęšis. Rekomenduojama iš apžiūros padėties lęšį implantuoti ne trumpiau kaip 5 sekundės. Jei priekyje esantis atraminis elementas yra tiesus arba sudaro kilpą ir išlenda į lęšio priekinę dalį, prieš stumdami stūmoklį, prietaisą šiek tiek pasukite į kairę; taip bus užtikrinta, kad priekinis atraminis elementas yra tinkamoje kapsulės maišelio vietoje. Kai priekyje esantis atraminis elementas pradės lįsti pro atgalio galiuką, priekinį atraminį elementą reikia įstatyti į kapsulės maišelį tinkamoje padėtyje ir toliau lėtai stumti stūmoklį, kad būtų išstumtas lęšis. Kai optinė dalis išstumiama pro antgalį, prietaisą reikia pasukti atgal link centro ar, jei reikia, šiek tiek į dešinę, kad būtų užtikrinta, jog lęšis kapsulės maišelyje išsilankstys priekine puse į viršų.
11 etapas. Tinkamu instrumentu perveskite lęšį kapsulės maišelyje į reikiamą padėtį, kad plokštuma būtų lygiagreti rainelei.
12 etapas. Išmeskite visą prietaisą. Nenaudokite parengtos implantavimo sistemos UltraSert™ pakartotinai.
PACIENTŲ REGISTRAVIMAS IR ATASKAITOSTuri būti užpildyta ir pacientui paduota šioje pakuotėje esanti Paciento identifikacinė kortelė, be to, pacientui turi būti nurodyta, kad jis šią kortelę saugotų ir parodytų bet kokiems akių ligų specialistams, kurie jį gydys ateityje.JAV kiekvieną pacientą būtina užregistruoti Alcon Laboratories, Inc. nedelsiant po vieno iš šių lęšių implantavimo. Registruojama užpildant Implanto registracijos kortelę, kuri būna lęšio dėžutėje, ir ją nusiunčiant Alcon Laboratories, Inc. Pacientų registravimas Alcon Laboratories, Inc. yra labai svarbus, kadangi taip vykdoma ilgalaikio stebėjimo programa ir gauti duomenys padės reaguoti į pranešimus apie nepageidaujamus reiškinius.Ne JAV reikia laikytis vietinių su produkto registravimu susijusių įstatymų.
Apie reiškinius, kurie pagrįstai gali būti laikomi susijusiais su lęšiais ir kurie sukėlė mirtį ar sunkų pažeidimą (arba prisidėjo prie jų), įskaitant reiškinius, atsiradusius dėl medicinos prietaiso neatitikimo funkcinėms specifikacijoms ar kitokio netinkamo veikimo, turi būti pranešta Alcon Laboratories, Inc. Tokios informacijos prašoma iš visų chirurgų, taip siekiant dokumentuoti galimą su ilgalaikiu intraokulinių lęšių implantavimu susijusį poveikį. Tam, kad praneštų apie su šiais intraokuliniais lęšiais susijusius nepageidaujamus reiškinius, chirurgai turi kreiptis toliau nurodytu adresu ir tel.:Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Nemokamas tel.: 1-800-757-9780
Ne JAV chirurgai dėl pranešimų apie nepageidaujamus reiškinius turi kreiptis į vietinę Alcon atstovybę ar platintoją.
83
Sulankstomų užpakalinės kameros lęšių AcrySof® klinikiniai tyrimaiSu sulankstomais vienos dalies užpakalinės kameros lęšiais AcrySof® buvo atlikti du atsitiktinių imčių, perspektyviniai, gerai kontroliuojami klinikiniai tyrimai. Pirmasis tyrimas buvo atliekamas, siekiant parodyti vienos dalies užpakalinės kameros lęšių AcrySof® Natural modelio SB30AL (filtruojančių UV ir mėlyną šviesą), kaip pirminio lęšių modelio, saugumą ir veiksmingumą. Tai buvo atsitiktinių imčių klinikinis tyrimas, kurio metu SA30AL modelio lęšiai AcrySof® (absorbuojantys tik UV) buvo naudojami kaip kontroliniai. Naudoti tik pirmosios pacientų, kuriems buvo implantuoti SB30AL arba SA30AL modelių intraokuliniai lęšiai, operuotos akies duomenys. Antrasis atsitiktinių imčių tyrimas buvo atliktas, siekiant palyginti asferinės optinės dalies sulankstomus vienos dalies intraokulinius lęšius AcrySof® IQ (su UV ir mėlyną šviesą filtruojančiais chromoforais) su sulankstomais vienos dalies kontroliniais lęšiais AcrySof® ir įvertinti klinikinius / funkcinius pranašumus lyginant su tradicine sferine forma.
Vienos dalies SB30AL modelio lęšių AcrySof® Natural klinikiniai tyrimaiPacientų, sėkmingai stebėtų ne mažiau kaip vienerius metus po operacijos, pasiekti rezultatai leidžia pagrįstai teigti, kad vienos dalies SB30AL modelio lęšiai AcrySof® Natural yra saugi ir veiksminga regos koregavimo priemonė esant afakijai.
Vienos dalies SB30AL modelio lęšiai AcrySof® Natural – demografiniai pacientų duomenysPacientų grupę, kuriai šio dvipusio tyrimo metu bent į pirmąją operuotą akį buvo implantuoti SB30AL modelio lęšiai, sudarė 70,6% moterų ir 29,4% vyrų. Pacientų grupę, kuriai buvo implantuoti SA30AL modelio lęšiai (kontroliniai), sudarė 60,5% moterų ir 39,5% vyrų. Skirstant pagal rasę, SB30AL modelio grupę sudarė 95,3% baltaodžių ir 4,7% juodaodžių. Kontrolinę grupę (SA30AL) sudarė 96,6% baltaodžių, 2% juodaodžių ir 1,4% kitų rasių. Visos tyrimo populiacijos, kuriai SB30AL modelio lęšiai buvo implantuoti bent į pirmąją operuotą akį, amžiaus vidurkis buvo 72,9 metų. Visos tyrimo populiacijos, kuriai buvo implantuoti SA30AL modelio lęšiai (kontroliniai), amžiaus vidurkis buvo 71,9 metų.
Vienos dalies SB30AL modelio lęšiai AcrySof® Natural – regėjimo aštrumasPacientų, kurie prieš operaciją niekada neturėjo akių patologijų, ragenos patologijų ar geltonosios dėmės degeneracijos, regos aštrumo(geriausias atvejis), pasiekto ne mažiau kaip po vienerių metų po operacijos, suvestinė pateikta 2a lentelėje, o visos pacientų grupės pasiektas regos aštrumas pateiktas 3a lentelėje. Tų pačių duomenų kontroliniai duomenys pateikti atitinkamai 2b ir 3b lentelėse. Tiek geriausiu atveju, tiek remiantis visos grupės duomenimis lyginant SB30AL modelio lęšius ir SA30AL modelio kontrolinius lęšius statistiškai reikšmingo regos aštrumo skirtumo nėra pastebėta.
Žr. anglų kalbą 2a lentelęGeriausio atvejo pacientų grupės pasiektas geriausias koreguotas regos aštrumas, praėjus ne mažiau kaip
vieneriems metams po lęšio AcrySof® Natural SB30AL implantavimo
Aštrumas
Iš viso 20/20 ar geresnis 20/25 20/30 20/40 20/40 ar
geresnis> 20/40 - < 20/80 > 20/80
N N % N % N % N % N % N % N %
Amžiaus kategorija
Žr. anglų kalbą 2b lentelęGeriausio atvejo pacientų grupės pasiektas geriausias koreguotas regos aštrumas, praėjus ne mažiau kaip
vieneriems metams po lęšio AcrySof® SA30AL (kontrolė) implantavimo
Aštrumas
Iš viso 20/20 ar geresnis 20/25 20/30 20/40 20/40 ar
geresnis> 20/40 - < 20/80 > 20/80
N N % N % N % N % N % N % N %
Amžiaus kategorija
Žr. anglų kalbą 3a lentelęVisų pacientų populiacijos pasiektas geriausias koreguotas regos aštrumas, praėjus ne mažiau kaip vieneriems
metams po lęšio AcrySof® Natural SB30AL implantavimo
Aštrumas
Iš viso 20/20 ar geresnis 20/25 20/30 20/40 20/40 ar
geresnis> 20/40 - < 20/80 > 20/80
N N % N % N % N % N % N % N %
Amžiaus kategorija
84
Žr. anglų kalbą 3b lentelę Visų pacientų populiacijos pasiektas geriausias koreguotas regos aštrumas, praėjus ne mažiau kaip vieneriems
metams po lęšio AcrySof® SA30AL (kontrolė) implantavimo
Aštrumas
Iš viso 20/20 ar geresnis 20/25 20/30 20/40 20/40 ar
geresnis> 20/40 - < 20/80 > 20/80
N N % N % N % N % N % N % N %
Amžiaus kategorija
AcrySof® Natural vienos dalies SB30AL modelio lęšiai: sukaupti duomenys apie nepageidaujamus reiškiniusŠių nepageiduajamų reiškinių apibendrintas dažnis iki minimalaus vienerių metų pooperacinio periodo (imtinai), pacientams implantuojant vienos dalies SB30AL ir SA30AL modelių lęšius AcrySof® Natural, pateiktas 4 lentelėje. Lyginant pacientų, patyrusių bet kokių kumuliacinių nepageidaujamų reiškinių, dalis, statistiškai reikšmingo skirtumo tarp SB30AL ir SA30AL modelio grupių nebuvo.
4 lentelėApibendrinti nepageidaujami reiškiniai per ne trumpesnį kaip vienerių metų laikotarpį po operacijos, naudojant
lęšius AcrySof® Natural SB30AL ir kontrolinius lęšius SA30AL
SB30AL (N= 153)
SA30AL(N= 147)
p-reikšmė*
N % N %
Nepageidaujamo reiškinio tipas
0 0,0 0 0,0 NėraKumuliacinis Hipopionas
Intraokulinė infekcija ar endoftalmitas 0 0,0 0 0,0 Nėra
Geltonosios dėmės edema 4 2,6 2 1,4 0,6847
Vyzdžio blokas 0 0,0 0 0,0 Nėra
Tinklainės atšoka arba tinklainės atšokos gydymas 0 0,0 0 0,0 Nėra
Lęšio nukrypimas 1 0,7 0 0,0 1,0000
Antra chirurginė intervencija Likutinės žievės šalinimas Išėmimas (nukrypimas plyšus kamerai) Krioterapija taisant tinklainės atplaišas Paracentezė, siekiant sumažinti akispūdį Kraujavimo stabdymas lazeriu Fotodinaminė terapija Išėmimas, įvykus biometrijos klaidai
51 1 11100
3,3 0,7
0,70,70,70,70,00,0
20000011
1,40,00,00,00,00,00,70,7
0,4482
Hipopionas 0 0,0 0 0,0 Nėra
*p vertės iš Fisher tikslumo testo, lyginant SB30AL ir SA30AL modelius.
Vienos dalies SB30AL modelio lęšiai AcrySof® Natural: neišnykstantys nepageidaujami reikšiniaiŠių neišnykstančių šalutinių reiškinių apibendrintas dažnis ne trumpesniu kaip vienerių metų pooperaciniu periodu, pacientams implantuojant vienos dalies SB30AL modelio lęšius AcrySof® Natural ir kontrolinį SA30AL modelio lęšį, pateiktas 5 lentelėje. Lyginant pacientų, patyrusių bet kokių kumuliacinių nepageidaujamų reiškinių, dalis, statistiškai reikšmingo skirtumo tarp SB30AL ir SA30AL modelio grupių nebuvo.
85
5 lentelėNeišnykstantys nepageidaujami reiškiniai per ne trumpesnį kaip vienerių metų laikotarpį po operacijos,
naudojant lęšius AcrySof® Natural SB30AL ir kontrolinius lęšius SA30AL
SB30AL (N= 138)
SA30AL(N= 127)
p-reikšmė*
N % N %
Nepageidaujamo reiškinio tipas
0 0 1 0,8 0,4792Neišnykstantis Ragenos edema
Iritas 0 0 0 0,0 Nėra
Geltonosios dėmės edema 2 1,4 1 0,8 1,0000
Vitritas 0 0 0 0,0 Nėra
Padidėjęs, reikalaujantis gydymo akispūdis 0 0 0 0,0 Nėra
*p vertės iš Fisher tikslumo testo, lyginant SB30AL ir SA30AL modelius.
Vienos dalies modelio SB30AL lęšiai AcrySof® Natural: spalvų skyrimas120–180 dienų laikotarpiu po operacijos buvo atlikti spalvų suvokimo tyrimai, naudojant dėžutės testą Farnsworth D-15. Iš 109 normaliai spalvas skyrusių pacientų, kuriems į pirmąją operuotą akį buvo implantuotas SB30AL modelio lęšis ir kurie buvo patikrinti po operacijos praėjus 120–180 dienų, 107 (98,2%) sėkmingai atliko spalvų skyrimo testą. Iš 102 normaliai spalvas skyrusių pacientų, kuriems į pirmąją operuotą akį buvo implantuotas SA30AL modelio lęšiukas ir kurie buvo patikrinti praėjus po operacijos 120–180 dienų, 97 (95,1%) sėkmingai atliko spalvų skyrimo testą. Vertinant pacientų, 120–180 dienų laikotarpiu po operacijos sėkmingai atlikusių spalvų skyrimo testą, procentinė dalį, statistiškai reikšmingo skirtumo tarp SB30AL ir SA30AL modelių lęšių nėra. Tai reiškia, kad pridėtas patentuotas chromoforas nedaro neigiamo poveikio normaliai spalvas skiriančių pacientų spalvų suvokimui.
Vienos dalies SB30AL modelio lęšiai AcrySof® Natural: Nd:YAG dažnis Per vidutiniškai 21,6 mėnesio stebėjimo laikotarpį trims (3) iš 135 pacientų (2,2%), kuriems buvo implantuoti SB30AL lęšiai, reikėjo atlikti Nd:YAG užpakalinės kameros kapsulotomiją. Per vidutiniškai 21,9 mėnesio stebėjimo laikotarpį dviem (2) iš 127 pacientų (1,6%), kuriems buvo implantuoti SA30AL lęšiukai, reikėjo atlikti Nd:YAG užpakalinės kameros kapsulotomiją.
Lęšių AcrySof® IQ klinikinis tyrimasLaikantis ankstesnių panašaus pobūdžio intraokulinių lęšių tyrimų struktūros, buvo atliktas kontroliuojamas, atsitiktinių imčių, dvigubai koduotas, daugiacentris, kontralateralinių implantų klinikinis tyrimas, kurio metu lęšiai AcrySof® IQ buvo lyginami su sferiniais kontroliniais lęšiais. Tyrime dalyvavo suaugusieji su sveikomis akimis (t. y. asmenys, kuriems anksčiau nebuvo atlikta akių operacija ir kurie nesirgo degeneracinėmis akių ligomis, galinčiomis ženkliai pabloginti regos aštrumą, ar sunkiomis ūminėmis ar lėtinėmis ligomis, galinčiomis padidinti pavojų pacientui) ir abiejų akių katarakta. Naudojant lęšius AcrySof® IQ, akių sferinių aberacijų atvejų skaičius buvo statistiškai reikšmingai mažesnis nei naudojant kontrolinius lęšius. Kontrastingumo jautrio rezultatai parodė, jog naudojant implantus AcrySof® IQ pasireiškia statistiškai reikšmingas pooperacinis (po 3 mėnesių) pagerėjimas. Implantavus lęšius AcrySof® IQ taip pat stebėti statistiškai ir kliniškai reikšmingi geresni regos funkcinio matavimo ir imituoto vairavimo naktį, tirto įvairiomis sąlygomis (ypač esant šviesos blyksniams ir rūkui), rezultatai. Šie rezultatai leidžia teigti, kad intraokuliniai lęšiai AcrySof® IQ (asferinė optinė dalis ant pagrindo su mėlyną šviesą filtruojančiu chromoforu) užtikrina geresnius klinikinius rezultatus nei vieno židinio intraokuliniai lęšiai AcrySof® (be asferinės optinės dalies ir mėlyną šviesą filtruojančio chromoforo).
Lęšiai AcrySof® IQ: sferinės ir bendrosios aukštesnės eilės aberacijosSu implantais AcrySof® IQ vidutinė akies sferinė aberacija buvo maždaug 0,1 mikrometro. 10 pav. vaizduojamas statistiškai reikšmingas palankesnis sferinių ir bendrųjų aukštesnės eilės aberacijų sumažėjimas, stebėtas naudojant lęšius AcrySof® IQ. 11 pav. pateikiami visų atvejų vidutinės sferinės aberacijos matavimai pagal lęšio tipą ir amžiaus grupę, atlikti bangų fronto aberometru. Kaip matyti, naudojant lęšius AcrySof® IQ sferinė aberacija mažėja neatsižvelgiant į amžių.
Žr. anglų kalbą 10 pav.Sferinė ir bendroji aukštesnės eilės RMS90–120 dienų po antrosios akies implanto
Žr. anglų kalbą 11 pav.Vidutinė sferinė aberacija – bendrieji ir amžiaus grupių duomenys
90–120 dienų po antrosios akies implanto
Lęšiai AcrySof® IQ: tolimosios regos aštrumas Naudojant AcrySof® IQ ir kontrolinius lęšius pooperaciniu laikotarpiu regos aštrumas buvo klinikiniu požiūriu panašus. Vienos akies regos aštrumo rezultatai pateikti 12 ir 13 pav.
86
Žr. anglų kalbą 12 pav.LogMAR geriausias koreguotas regos aštrumas
Žr. anglų kalbą 13 pav.LogMAR geriausias nekoreguotas regos aštrumas
Kontrastingumo jautris naudojant lęšius AcrySof® IQPagrindinis klinikinio tyrimo tikslas buvo parodyti, jog lęšiai AcrySof® IQ užtikrina didesnį vidutinį kontrastingumo jautrį nei kontroliniai lęšiai. Vidutinis kontrastingumo jautris buvo matuojamas po operacijos, mezopinėmis sąlygomis, esant arba nesant šviesos blyksnių, vienu arba dviem erdviniais dažniais (3 arba 6 ciklai laipsnyje, naudojant „Vector Vision CSV-1000“ (lentelės apšviestumas 3 cd/m2). Su pacientų poaibiu taip pat buvo atliktas funkcinio aštrumo kontrastingumo tyrimas („Functional Acuity Contrast Test“, FACT) (lentelės apšviestumas 3 cd/m2). Šio klinikinio tyrimo metu buvo nustatyta, jog mezopinėmis sąlygomis AcrySof® IQ lęšiai pranašesni už kontrolinius lęšius esant 6 ciklams laipsnyje ir esant arba nesant šviesos blyksnių (CSV-1000), ir esant 3 arba 6 ciklams laipsnyje be šviesos blyksnių (FACT). 14 ir 15 pav. pateikiami mezopinio kontrastingumo jautrio rezultatai visais erdviniais dažniais, gauti tiriant lęšius AcrySof® IQ ir kontrolinius lęšius.
Žr. anglų kalbą 14 pav.Mezopinis kontrastingumo jautris (CSV-1000)
90–120 dienų po antrosios akies implanto
Žr. anglų kalbą 15 pav.Mezopinio kontrastingumo jautrio subtyrimas (FACT)Ne mažiau kaip 90 dienų po antrosios akies implanto
Lęšiai AcrySof® IQ: vairavimo naktį imitavimas Pacientų poaibis buvo tiriamas patvirtintu naktinio vairavimo imitatoriumi. Buvo tikrinama viena pacientų akis, imituojant miesto ir kaimo vietovę įprastomis sąlygomis bei esant šviesos blyksniams ir rūkui.Siekiant atkurti naktiniam vairavimui mieste būdingą ryškų aplinkos apšviestumą, naktinio vairavimo mieste scenoje naudojami įvairūs gatvės, automobilių, parduotuvių vitrinų ir ženklų žibintai. Naktinio vairavimo kaimo vietovėje scenoje aplinkos apšviestumo beveik nėra. Miesto ir kaimo scenose buvo naudojami atitinkamai 35 ir 55 mylių per valandą imituoto važiavimo greičiai.Pacientų buvo prašoma kiekvienoje scenoje aptikti ir identifikuoti objektų grupes, įskaitant baltai žalius greitkelių ženklus, juodai geltonus įspėjamuosius ženklus ir pėsčiuosius. Kad būtų galima registruoti aptikimo atstumą, pacientų buvo prašoma reaguoti pastebėjus pirmąjį objektą. Paskui, kad būtų galima registruoti identifikavimo atstumą, pacientų buvo prašoma reaguoti atskyrus objekto informaciją (pvz., kas užrašyta ant ženklo, kuria kryptimi eina pėstieji ir t. t.).16–19 pav. pateikiami aptikimo ir identifikavimo atstumų vairuojant miesto ir kaimo vietovėse vidutiniai skirtumai naudojant AcrySof® IQ ir kontrolinius lęšius (t. y. intraindividualių skirtumų vidurkis).Iš 36 tirtų sąlygų 34 atvejais buvo nustatyta, jog naudojant AcrySof® IQ lęšius būna geresni funkciniai rezultatai nei naudojant kontrolinius lęšius: ir aptikimo, ir identifikavimo atstumai didesni vairuojant mieste ir kaime įvairiomis sąlygomis (įprastinėmis, esant šviesos blyksniams ir esant rūkui). Be to, naudojant lęšius AcrySof® IQ buvo gauti statistiškai reikšmingi geresni rezultatai tiriant 12 iš šių situacijų. Labiausiai pagerėjo pėsčiųjų mieste (esant šviesos blyksniams ir rūkui) ir įspėjamųjų ženklų kaimo vietovėje (esant šviesos blyksniams ir rūkui) aptikimo ir identifikavimo rezultatai. Prasto matomumo sąlygomis (šviesos blyksniai, rūkas) miesto scenoje, važiuojant 35 mylių per valandą greičiu naudojant AcrySof® IQ lęšius dėl padidėjusio matomumo atstumo vairuotojas turi > 0,5 sek. daugiau laiko sureaguoti į kelyje atsidūrusius pėsčiuosius; tai ypač svarbu vairuojant mieste. Šis 0,5 sek. papildomas laikas yra funkciškai reikšmingas, nes lieka daugiau laiko imtis reikiamų veiksmų, pvz., sustoti, apvažiuoti ir t. t. (Green, 2000; McBride and Matson, 2004). Kaimo vietovėje važiuojant 55 mylių per valandą greičiu (visomis sąlygomis) naudojant AcrySof® IQ lęšius dėl padidėjusio matomumo atstumo vairuotojas turi > 1 sek. daugiau laiko nustatyti įspėjamuosius ženklus (tokia situacija dažniau pasitaiko vairuojant kaimo vietovėse). 0,5 sek. papildoma trukmė yra funkciškai reikšminga, nes vairuojant lieka daugiau laiko imtis reikiamų veiksmų. Tai labai svarbu naktį važiuojant nežinomoje vietovėje, kur aplinka dažniausiai neapšviesta. Subtyrime dalyvavo 6 pacientai, kuriems po operacijos pasireiškė geltonosios dėmės degeneracija arba užpakalinės kapsulės drumstis. Šiuos pacientus pašalinus iš vairavimo analizės, skirtumas tarp tirtų lęšių miesto vietovėje esant šviesos blyksniams aptinkant ir identifikuojant pėsčiuosius tapo mažesnis už 0,5 sek. kliniškai patikimą slenkstinę reikšmę. Atlikus pradinių analizės rezultatų daugybiškumo koregavimą skirtumai tarp intraokulinių lęšių nustojo būti kliniškai reikšmingi miesto sąlygomis nustatant tekstą esant šviesos blyksniams (Hommel p reikšmė 0,0539) ir kaimo vietovėje aptinkant pėsčiuosius esant šviesos blyksniams (Hommel p reikšmė 0,0507).Šie rezultatai rodo, jog pagerėja vyresnio amžiaus vairuotojų su implantuotais lęšiais AcrySof® IQ funkcinė rega; ir patys pacientai, ir kiti vairuotojai bei pėstieji tampa saugesni. Šio tyrimo rezultatai rodo, jog naudojant lęšius AcrySof® IQ gerėja funkcinė rega, todėl pacientai gali būti saugesni ir kitomis gyvenimo situacijomis, kai būna prastos matomumo sąlygos.
Žr. anglų kalbą 16 pav.Naktinio vairavimo imitatorius
Aptikimo atstumų mieste vidutiniai intraindividualūs skirtumaiNe mažiau kaip 90 dienų po operacijoslęšiai AcrySof® IQ – kontroliniai (n = 44)
87
Žr. anglų kalbą 17 pav.Naktinio vairavimo imitatorius
Identifikavimo atstumų mieste vidutiniai intraindividualūs skirtumaiNe mažiau kaip 90 dienų po operacijoslęšiai AcrySof® IQ – kontroliniai (n = 44)
Žr. anglų kalbą 18 pav.Naktinio vairavimo imitatorius
Aptikimo atstumų kaime vidutiniai intraindividualūs skirtumaiNe mažiau kaip 90 dienų po operacijoslęšiai AcrySof® IQ – kontroliniai (n = 44)
Žr. anglų kalbą 19 pav.Naktinio vairavimo imitatorius
Identifikavimo atstumų kaime vidutiniai intraindividualūs skirtumaiNe mažiau kaip 90 dienų po operacijoslęšiai AcrySof® IQ – kontroliniai (n = 44)
PAKUOTĖŠie užpakalinės kameros intraokuliniai lęšiai tiekiami sausi parengtoje implantavimo sistemoje UltraSert™ pirminės sterilizacijos pakuotėje, po to sterilizuotoje etileno oksidu. Pirminės sterilizacijos pakuotę galima atidaryti tik steriliomis sąlygomis (žr. NAUDOJIMO INSTRUKCIJĄ).
TINKAMUMO LAIKASSterilumas yra užtikrintas tik tuo atveju, jei pirminės sterilizacijos pakuotė nėra pažeista ar atidaryta. Tinkamumo laikas yra aiškiai nurodytas ant parengtos implantavimo sistemos UltraSert™ išorinės dėžutės etiketės. Pasibaigus tinkamumo laikui visi turimi lęšiai turi būti grąžinti Alcon Laboratories, Inc. (žr. PREKIŲ GRĄŽINIMO TVARKĄ).
PREKIŲ GRĄŽINIMO TVARKAJungtinėse Valstijose lęšius galima gražinti tik juos pakeičiant į kitus produktus (pinigai negrąžinami). Visi grąžinami produktai turi turėti Alcon Laboratories, Inc. grąžinamų prekių numerį ir siunčiami tokiu būdu, kad būtų galima stebėti siuntos judėjimą. Norint gauti grąžinamų prekių numerį, reikia kreiptis į Alcon Laboratories, Inc. Klientų aptarnavimo departamentą. Šio numerio suteikimas nereiškia galutinio sutikimo priimti grąžinamą prekę. Norėdami gauti išsamesnės informacijos apie grąžinimą, įskaitant pakeitimą, kreipkitės į pardavimų ar klientų aptarnavimo departamentus. Dėl prekių grąžinimo politikos ne Junginėse Valstijose kreipkitės į vietinę Alcon atstovybę ar platintoją.
NUORODOS 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., et al. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., et al. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
88
ŽENKLINIMO SIMBOLIAI
SYMBOL LIETUVIŠKAIIOL Intraokulinis lęšis
SP Vienos dalies
UV Ultravioletiniai spinduliai
D Dioptrijos
ØB Korpuso skersmuo (optinės dalies skersmuo)
ØT Bendras skersmuo (bendras ilgis)
2STERILIZE Nesterilizuoti kartotinai
Nenaudoti kartotinai
Naudoti iki
STERILE EO Sterilizuota etileno oksidu
SN Serijos numeris
Dėmesio
Gamintojas
EC REP Įgaliotasis atstovas Europos Bendrijoje
Viršutinė temperatūros riba
Atidaryti čia
Žr. naudojimo instrukciją
Nenaudoti, jei sterili pakuotė buvo atidaryta arba yra pažeista
DĖMESIO! Pagal JAV federalinius įstatymus šis prietaisas gali būti parduodamas tik gydytojo arba jo paskyrimu.
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 JAV
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,Jungtinė Karalystė
STERYLNE akrylowe soczewki tylnokomorowe, pochłaniające promieniowanie ultrafioletowe i światło niebieskie, zwijalne, jednoczęściowe z urządzeniem UltraSert™ Pre-loaded Delivery System
OPISAkrylowe, jednoczęściowe, zwijalne soczewki do wszczepu tylnokomorowego AcrySof® IQ pochłaniające promieniowanie ultrafioletowe i światło niebieskie, są to implanty optyczne zastępujące ludzką soczewkę w korekcji bezsoczewkowości po operacyjnym usunięciu zaćmy u pacjentów dorosłych. Soczewki AcrySof® IQ z chromoforem filtrującym światło niebieskie, który jest zastrzeżonym produktem firmy Alcon, filtrują światło w sposób podobny do soczewki oka ludzkiego, w zakresie światła niebieskiego o długości fali od 400 do 475 nm (Boettner i Wolter, 1962). Oprócz standardowego filtrowania promieniowania ultrafioletowego, soczewki AcrySof® IQ zmniejszają przepuszczalność w zakresie światła niebieskiego od 62% przy długości fali 400 nm do 23% przy długości fali 475 nm (patrz Tabela 1). Część optyczna składa się z miękkiego akrylowego materiału o wysokim współczynniku refrakcji. Materiał ten można zwijać przed wszczepieniem do gałki ocznej. Soczewka delikatnie rozpręża się po implantacji do swych całkowitych rozmiarów. Soczewka ma dwuwypukłą część optyczną z pomocniczymi częściami haptycznymi (Rysunek 1). Tylna asferyczna powierzchnia soczewki wewnątrzgałkowej AcrySof® IQ Model AU00T0 IOL jest zaprojektowana z ujemną aberracją sferyczną w celu skompensowania dodatniej aberracji sferycznej przeciętnej rogówki. Jakość obrazu w jednoczęściowej soczewce wewnątrzgałkowej Model AU00T0 (tzn. funkcja przenoszenia modulacji) przedstawiona jest na Rysunku 4. Jednoczęściowe soczewki wewnątrzgałkowe AcrySof® IQ Model AU00T0 są dostarczane wraz z urządzeniem UltraSert™ Pre-loaded Delivery System (Rysunek 2). Urządzenie to umożliwia wygodną implantację soczewek do torebki soczewki, z zachowaniem odpowiedniej kontroli. Właściwości fizyczne soczewek są następujące:
Własności fizyczne OpisRodzaj części optycznej Jednoczęściowa soczewka wewnątrzgałkowa z optyką asferyczną
Materiał części optycznej Kopolimer akrylowo-metakrylowy filtrujący ultrafiolet i światło niebieskie
Pochłanianie UV przy 10% przepuszczalności (nm) UV (400) przy 20.0 D (Patrz Rysunek 3)
Współczynnik refrakcji 1,55
Moce optyczne Dostępny zakres mocy należy sprawdzić w Katalogu Produktów Alcon
Budowa części haptycznej STABLEFORCE® Modified-L Haptics
Materiał części haptycznej Kopolimer akrylowo-metakrylowy filtrujący ultrafiolet i światło niebieskie
Średnica części optycznej ØB (mm) 6,0
Długość całkowita ØT (mm) 13,0
Ukątowanie części haptycznej 0º
Wymiary części optycznej Patrz Rysunek 1
Patrz wersja angielska Rysunek 1
WŁASNOŚCI FIZYCZNEWszystkie wymiary w milimetrach
ASPHERIC SURFACE = POWIERZCHNIA ASFERYCZNA
POLISH
90
Patrz wersja angielska Rysunek 2Nozzle Tip = Końcówka wylotowa
Depth Guard = Ogranicznik głębokościMain Body = Korpus
Patrz wersja angielska Rysunek 3KRZYWE PRZEPUSZCZALNOŚCI SPEKTRALNEJ
TRANSMITTANCE (%) = PRZEPUSZCZALNOŚĆ (%)WAVELENGTH (nm) = DŁUGOŚĆ FALI (nm)
6.0 D to 27.0 D range = Zakres od 6,0 D do 27,0 D 4 yr – 53 yr old range, crystalline lens = Zakres wieku od 4 do 53 lat, soczewka naturalna
UWAGI: • Przedstawiona progowa długość fali i krzywe przepuszczalności spektralnej odpowiadają zakresowi
przepuszczalności soczewki wewnątrzgałkowej wykonanej z kopolimeru akrylowo-metakrylowego ze związanym filtrem UV oraz chromoforem filtrującym światło niebieskie, który jest zastrzeżonym produktem firmy Alcon.
• Pomiary bezpośredniej przepuszczalności wykonywano z użyciem otworu 6 mm i krążka o grubości równej grubości centrum optycznego soczewki.
• Dane dotyczące ludzkiej soczewki pochodzą od Boettner i Wolter (1962).
ISO, 3-mm Aperture (see Note 1) = ISO, otwór 3 mm (patrz Uwaga 1)Modified, 3-mm Aperture (see Note 2) = Modyfikacja, otwór 3 mm (patrz Uwaga 2)Modified, 5-mm Aperture (see Note 2) = Modyfikacja, otwór 5 mm (patrz Uwaga 2)
UWAGI: 1. Jakość obrazu w soczewce Model AU00T0 była określana przez mierzenie funkcji przenoszenia modulacji
(MTF) w modelowym oku zgodnym z ISO 11979-2. Norma ISO 11979-2 wymaga pomiarów MTF tylko przy otworze (aperturze) 3 mm.
2. Ponadto, jakość obrazu w soczewce Model AU00T0 była określana przez mierzenie MTF w modelowym oku wykorzystującym symulację rogówki wykazującą typową dla osoby dorosłej aberrację sferyczną. Przy użyciu zmodyfikowanego oka modelowego pomiary MTF wykonano przy otworze 3 mm i 5 mm.
SPOSÓB DZIAŁANIATylnokomorowe soczewki wewnątrzgałkowe AcrySof® IQ przeznaczone są do wszczepiania do komory tylnej oka w celu zastąpienia naturalnej soczewki ludzkiej. Ta pozycja umożliwia soczewce funkcjonowanie jako element refrakcyjny w korekcji afakii. Dwuwypukła optyka asferyczna zmniejsza aberrację sferyczną w porównaniu do standardowej optyki sferycznej w przeciętnym oku. Skuteczność tych soczewek w zmniejszeniu zapadalności na zaburzenia ze strony siatkówki nie jest ustalona.
WSKAZANIATylnokomorowe soczewki wewnątrzgałkowe AcrySof® IQ są wskazane do zastępowania ludzkiej soczewki w celu uzyskania korekcji bezsoczewkowości u pacjentów dorosłych po operacyjnym usunięciu zaćmy. (patrz OSTRZEŻENIA). Soczewki te przeznaczone są do wszczepiania dotorebkowego.
ŚRODKI OSTROŻNOŚCIPacjenci z którymkolwiek z niżej wymienionych stanów mogą nie być dobrymi kandydatami do wszczepienia soczewki wewnątrzgałkowej, ponieważ może ona zaostrzyć istniejący już proces, kolidować z diagnozą albo leczeniem schorzenia lub stanowić nieuzasadnione ryzyko dla zdolności widzenia pacjenta. Chirurg powinien przeprowadzić dokładną ocenę przedoperacyjną i rozważyć sytuację kliniczną, by ocenić stosunek korzyści do ryzyka przed wszczepieniem soczewki u pacjenta z jednym lub więcej z poniższych stanów:
91
1. Krwawienie naczyniówkowe 2. Współistniejąca, ciężka choroba oka 3. Nadmierna utrata ciała szklistego 4. Bardzo płytka komora przednia 5. Małoocze 6. Zaćma inna niż starcza 7. Przerwanie torebki tylnej (uniemożliwiające ufiksowanie soczewki) 8. Ciężka dystrofia rogówki 9. Ciężki zanik nerwu wzrokowego 10. Niemożliwe do wyrównania ciśnienie wypierające 11. Odłączenie obwódki rzęskowej (uniemożliwiające ufiksowanie soczewki) 12. Zaburzenia widzenia barw 13. Jaskra 14. Przewlekłe zapalenie błony naczyniowej 15. Retinopatia cukrzycowa 16. Istotne klinicznie zaburzenia plamki/nabłonka barwnikowegoBadania wykazały, że u pacjentów, którym wszczepiono soczewkę AcrySof® Natural i u których występowało prawidłowe widzenie barw, nie dochodzi do zaburzeń rozróżniania barw. Nie przeprowadzono badań nad wpływem soczewki AcrySof® Natural na widzenie u osób z wrodzonymi zaburzeniami widzenia barw albo z nabytymi zaburzeniami widzenia barw w przebiegu chorób oczu (np. jaskry, retinopatii cukrzycowej, przewlekłego zapalenia błony naczyniowej lub innych chorób siatkówki albo nerwu wzrokowego.
OSTRZEŻENIA 1. Jak w każdym przypadku interwencji chirurgicznej, istnieje pewne ryzyko. Potencjalne powikłania,
towarzyszące leczeniu operacyjnemu zaćmy i wszczepieniu soczewki wewnątrzgałkowej, mogą obejmować, lecz nie ograniczają się do następujących: uszkodzenie śródbłonka rogówki, zapalenie wnętrza gałki ocznej, odwarstwienie siatkówki, zapalenie ciała szklistego, torbielowaty obrzęk plamki, obrzęk rogówki, blok źreniczny, wysiękowe zapalenie ciała rzęskowego, wypadnięcie tęczówki, wysięk ropny w komorze przedniej oka i przejściowa lub trwała jaskra.
2. Nie określono bezpieczeństwa i skuteczności stosowania implantów wewnątrzgałkowych u pacjentów z uprzednio istniejącymi zaburzeniami oka (polekowe przewlekłe zwężenie źrenicy, jaskra, niedowidzenie, retinopatia cukrzycowa, przebyte przeszczepienie rogówki, uprzednie odwarstwienie siatkówki i/lub zapalenie tęczówki, itd.). Lekarze rozważający wszczepienie soczewki u takich pacjentów powinni zbadać ewentualne inne sposoby korekcji afakii i zadecydować o wszczepieniu soczewki jedynie wtedy, gdy owe sposoby są niezadowalające dla zaspokojenia potrzeb pacjenta.
3. Długotrwałe efekty stosowania soczewek wewnątrzgałkowych nie są określone. Dlatego należy przeprowadzać regularnie kontrole pooperacyjne u tych pacjentów.
4. Pacjenci z przedoperacyjnymi stanami, takimi jak niedomoga śródbłonka rogówki, choroba rogówki, zwyrodnienie plamki, zwyrodnienie siatkówki, jaskra, przewlekłe polekowe zwężenie źrenicy, mogą nie osiągnąć takiej ostrości widzenia, jak w przypadku pacjentów bez tych schorzeń. Lekarz musi oszacować ewentualne korzyści związane ze wszczepieniem soczewki, gdy takie stany występują.
5. Wtórnej irydektomii z powodu bloku źrenicznego można uniknąć, gdy wykona się jedną lub więcej irydektomii podczas wszczepiania soczewki wewnątrzgałkowej (Willis i wsp., 1985).
6. Nie określono bezpieczeństwa i efektywności soczewki tylnokomorowej, gdy jest stosowana jako wszczep przedniokomorowy. Implantacja soczewki tylnokomorowej do komory przedniej w niektórych przypadkach okazała się niebezpieczna (Girard i wsp., 1983).
7. Niektóre działania niepożądane, związane ze wszczepieniem soczewki wewnątrzgałkowej, to: wysięk ropny w komorze przedniej, zapalenie wewnątrzgałkowe, ostra dekompensacja rogówki i wtórna interwencja chirurgiczna. Wtórne interwencje chirurgiczne obejmują, lecz nie ograniczają się do: repozycji soczewki, wymiany soczewki, aspiracji ciała szklistego, irydektomii z powodu bloku źrenicznego, opracowania przecieku z rany pooperacyjnej, operacji odwarstwienia siatkówki.
8. Niewielka decentracja soczewki, która występuje w przypadku soczewek o wąskiej lub małej części optycznej może powodować u pacjenta objawy olśnienia lub inne zaburzenia widzenia w pewnych warunkach oświetlenia. Chirurg powinien rozważyć tę możliwość przed zastosowaniem wszczepu o wąskiej lub małej części optycznej. Kiedy wszczepiana jest soczewka o wąskiej lub małej części optycznej, zalecane jest wykonanie kapsuloreksji.
9. Pooperacyjne rozciągnięcie torebki tylnej wraz ze zróżnicowanym spłyceniem komory przedniej i wywołaną krótkowzrocznością były związane z technikami kapsuloreksji i implantacją soczewki tylnokomorowej z PMMA, silikonowej i akrylowej (Holtz, 1992).
10. Należy zachować ostrożność przed otorbieniem się soczewki, aby uniknąć decentracji lub przemieszczania soczewki. Niektóre przypadki kliniczne wskazują na to, że otorbienie się zachodzi w okresie do czterech tygodni od wszczepienia soczewki.
11. Badanie kliniczne nad stosowaniem soczewek jednoczęściowych AcrySof® Natural, (których wyniki przedstawiają Tabele od 2 do 5) przeprowadzono z użyciem soczewek przeznaczonych wyłącznie do wszczepienia do torebki soczewki. Brak danych klinicznych na temat skuteczności i bezpieczeństwa wszczepiania tych soczewek do bruzdy rzęskowej.
92
Zalecane jest, by materiał wiskoelastyczny był usunięty z gałki przed zakończeniem operacji, ze szczególnym uwzględnieniem przestrzeni między torebką tylną a soczewką. Można to osiągnąć przez delikatne uciskanie części optycznej soczewki za pomocą końcówki I/A i stosowanie klasycznych technik irygacji/aspiracji, aby usunąć materiał wiskoelastyczny z oka. To powinno przemieścić do przodu całą ilość materiału wiskoelastycznego, który mógł ewentualnie uwięznąć pod soczewką, co umożliwi jego łatwą aspirację.
ŚRODKI OSTROŻNOŚCI 1. Nie sterylizować ponownie omawianych soczewek wewnątrzgałkowych ani urządzenia UltraSert™ Pre-loaded
Delivery System żadną z metod. 2. Nie przechowywać soczewek wewnątrzgałkowych w temperaturach powyżej 45° C (113° F). 3. W razie potrzeby przytrzymywać soczewki delikatnie, by nie uszkodzić powierzchni soczewki lub części
haptycznych. 4. Nie próbować odkształcać części haptycznych w żaden sposób. 5. Wysoki poziom umiejętności chirurgicznych jest konieczny przy implantacji soczewek wewnątrzgałkowych.
Chirurg powinien obserwować i/lub asystować przy wielu implantacjach i z powodzeniem ukończyć jeden lub więcej kursów implantacji soczewek wewnątrzgałkowych przed próbą implantacji takich soczewek.
6. Zawartość jest jałowa, dopóki opakowanie nie zostanie otwarte ani uszkodzone. 7. Urządzenie UltraSert™ Pre-loaded Delivery System jest przeznaczone wyłącznie do jednorazowego
stosowania. Po zastosowaniu należy je usunąć. 8. Urządzenie UltraSert™ Pre-loaded Delivery System powinno być używane w sali operacyjnej w temperaturze
pomiędzy 18OC (64OF) a 23OC (73° F).
WYLICZENIE MOCY SOCZEWKIPrzedoperacyjne obliczenie wymaganej mocy omawianych tylnokomorowych soczewek wewnątrzgałkowych należy przeprowadzić z uwzględnieniem doświadczenia chirurga, wyboru metody oraz zamierzonego położenia soczewki. Metody obliczania mocy soczewki są przedstawione w następujących publikacjach:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. et al. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. et al. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
SUGEROWANA STAŁA ASugerowana stała A zapisana na opakowaniu zewnętrznym służy jako wskazówka i jest punktem wyjścia przy obliczaniu mocy implantu. Zaleca się, by ustalić własną wartość stałej w oparciu o doświadczenie kliniczne z poszczególnymi modelami soczewek, metodami chirurgicznymi, sprzętem pomiarowym i wynikami pooperacyjnymi.W Stanach Zjednoczonych, jeżeli potrzebna jest dodatkowa informacja o obliczaniu mocy soczewki, prosimy skontaktować się z biurem firmy Alcon Laboratories, Inc. pod nr telefonu 1-800-TO-ALCON (1-800-862-5266). Poza Stanami Zjednoczonymi prosimy o kontakt z lokalnymi biurami firmy Alcon lub dystrybutorami.
ZAKWALIFIKOWANE PREPARATY WISKOELASTYCZNE I ICH NUMERY KATALOGOWEPatrz dodatek dołączony do tego opakowania. Należy używać tylko preparaty wiskoelastyczne zakwalifikowane przez Alcon. Używanie niezakwalifikowanych preparatów wiskoelastycznych może spowodować uszkodzenie soczewki i potencjalne powikłania w czasie procesu wszczepiania. W celu uzyskania pełnego wykazu preparatów wiskoelastycznych zakwalifikowanych przez Alcon dla omawianej soczewki prosimy skontaktować się z lokalnym przedstawicielem firmy Alcon.
SPOSÓB UŻYCIAKrok 1. Używać urządzenia UltraSert™ Pre-loaded Delivery System przy temperaturze w sali operacyjnej pomiędzy 18° C
(64° F) i 23° C (73° F).
Krok 2. Obejrzeć etykietę na zamkniętym opakowaniu zewnętrznym, by sprawdzić model, moc, odpowiednią budowę i datę ważności.
Krok 3. Po otwarciu pudełka tekturowego sprawdzić opakowanie bezpośrednie wyrobu pod kątem jakiegokolwiek uszkodzenia. W przypadku stwierdzenia uszkodzenia, należy użyć innej soczewki AcrySof® IQ i urządzenia UltraSert™ Pre-loaded Delivery System. Następnie sprawdzić, czy informacje na opakowaniu bezpośrednim soczewki (np. model, moc, numer seryjny), są zgodne z informacjami na opakowaniu zewnętrznym.
Krok 4. Aby wyjąć urządzenie UltraSert™ Pre-loaded Delivery System, należy chwycić za narożnik tacki z tworzywa sztucznego, całkowicie oderwać górną folię TYVEK† i przenieść urządzenie do środowiska jałowego. Jeżeli po sprawdzeniu wydaje się, że końcówka wylotowa urządzenia jest uszkodzona, zanieczyszczona cząstkami stałymi lub zdeformowana, należy użyć innej soczewki AcrySof® IQ i urządzenia UltraSert™ Pre-loaded Delivery System. Jeżeli urządzenie nie jest całkowicie nienaruszone lub wydaje się, że blokada tłoka lub tłok przesunął się w czasie transportu, należy użyć innej soczewki AcrySof® IQ i urządzenia UltraSert™ Pre-loaded Delivery System (Patrz ZWROTY TOWARU).
† Zastrzeżony znak handlowy E.I. DuPont De Nemours & Co.
93
Po osiągnięciu gotowości do wprowadzenia soczewki, należy wykonać kolejno Kroki 5, 6 i 7 bez zwłoki.
Krok 5. Całkowicie wprowadzić kaniulę wiskoelastyku prostopadle do urządzenia przez otwór wiskoelastyku znajdujący się w części blokującej soczewkę, jak pokazano na Rysunku 5. Napełniać urządzenie co najmniej do czasu, kiedy można zaobserwować wiskoelastyk dopływający do linii na końcówce wylotowej (patrz strzałka na Rysunku 5), następnie wycofać kaniulę. Będzie to wymagało około 0,2 ml wiskoelastyku. Stosować wyłącznie preparaty wiskoelastyczne zakwalifikowane przez Alcon do stosowania z urządzeniem UltraSert™ Pre-loaded Delivery System, które osiągnęły temperaturę panującą w sali operacyjnej. Wykaz preparatów wiskoelastycznych zakwalifikowanych przez Alcon należy sprawdzić w dodatku dołączonym do tego opakowania.
Patrz wersja angielska Rysunek 5
Viscoelastic Port = Otwór wiskoelastyku“Fill-To” Line = Linia „Napełnić do tego miejsca”
Krok 6. Wyjąć niebieski ogranicznik soczewki chwytając za część oznaczoną strzałką i zginając nieco do przodu, jednocześnie wyciągając do góry z urządzenia (Rysunek 6a). Wyjąć niebieską blokadę tłoka chwytając i wyciągając na wprost do góry z urządzenia (Rysunek 6b). Wyrzucić ogranicznik soczewki i blokadę tłoka. Nie próbować dodawać wiskoelastyku do urządzenia po wyjęciu ogranicznika soczewki, ponieważ może to spowodować uszkodzenie soczewki. Nie próbować cofać tłoka po wyjęciu blokady tłoka, ponieważ może to spowodować uszkodzenie tłoka.
Patrz wersja angielska Rysunek 6a i 6b
Krok 7. Delikatnie przesunąć tłok do przodu jednym , łagodnym, ciągłym ruchem do czasu, aż przednia krawędź części optycznej zostanie ustawiona równo z linią na końcówce (Sprawdzić na Rysunkach 7a). W tej pozycji niebieska sprężyna zetknie się z przezroczystym korpusem głównym palca, jak pokazano na Rysunku 7b. Ten ruch tłoka powinien trwać co najmniej 7 sekund. Żadna z części soczewki nie powinna wystawać poza końcówkę wylotową. Ważne jest, aby nie posuwać tłoka gwałtownie, ponieważ może nastąpić nieprawidłowe zwijanie i uszkodzenie soczewki.
Patrz wersja angielska Rysunek 7a i 7b
Fill-to Line = Linia „Napełnić do tego miejsca”
Krok 8. Po przesunięciu soczewki do linii na końcówce, należy skontrolować wzrokowo soczewkę, aby upewnić się, że części haptyczne są prawidłowo ustawione. Tłok powinien stykać się tylną krawędzią części optycznej e. Żadna z części soczewki nie powinna wystawać poza końcówkę przed wprowadzeniem przez cięcie (Sprawdzić na Rysunku 8). Kiedy już soczewka znajduje się w pozycji na linii końcówki wylotowej, powinna zostać wszczepiona w ciągu 3 minut.
Patrz wersja angielska Rysunek 8
“Proceed with full plunger advancement to fully deliver lens” = Kontynuować pełne przesunięcie tłoka w celu całkowitego dostarczenia soczewki
“Proceed” = Kontynuować“Proceed with full plunger advancement to fully deliver lens” = Kontynuować pełne przesunięcie tłoka w celu
całkowitego dostarczenia soczewki“Proceed with device rotations per Step 10” = Kontynuować obroty urządzenia zgodnie z Krokiem 10
“Do not proceed” = Nie kontynuować
Krok 9. Po upewnieniu się, że soczewka jest prawidłowo ustawiona i części haptyczne są prawidłowo zwinięte, kontynuować implantację soczewki wprowadzając końcówkę wylotową przez cięcie aż do ogranicznika głębokości (Rysunek 9) i kierując ją do otworu w przedniej powierzchni torebki.
Patrz wersja angielska Rysunek 9
Depth Guard = Ogranicznik głębokości
Krok 10. Delikatnie popchnąć tłok jednym łagodnym, ciągłym ruchem. Nie popychać tłoka zbyt szybko, gdyż może nastąpić uszkodzenie soczewki. Zaleca się, aby dostarczanie soczewki od pozycji kontroli wzrokowej trwało co najmniej 5 sekund. Jeśli przednia część haptyczna jest wyprostowana lub w postaci pętli znajduje się przed soczewką, należy obrócić urządzenie na skos w lewo przed przesunięciem tłoka, aby upewnić się, że przednia część haptyczna jest prawidłowo umieszczona w torebce soczewki. Kiedy przednia część haptyczna zaczyna opuszczać końcówkę wylotową, należy umieścić ją wewnątrz torebki soczewki w odpowiednim kierunku i kontynuować powolne popychanie tłoka aby dostarczyć soczewkę. Kiedy część optyczna soczewki opuszcza końcówkę wylotową, należy obrócić urządzenie z powrotem na środek lub nieco skośnie w prawo stosownie do potrzeby, aby zapewnić, że soczewka rozwinie się przednią stroną do góry wewnątrz torebki soczewki.
94
Krok 11. Przy pomocy odpowiedniego narzędzia ustawić soczewkę wewnątrz torebki soczewki i w płaszczyźnie równoległej do tęczówki.
Krok 12. Usunąć całe urządzenie. Nie używać powtórnie urządzenia UltraSert™ Pre-loaded Delivery System.
REJESTRACJA PACJENTA I ZGŁASZANIE ZDARZEŃ NIEPOŻĄDANYCHKarta Identyfikacji Pacjenta, zawarta w opakowaniu, musi zostać wypełniona i przekazana pacjentowi razem z zaleceniem, aby przechowywał ją i pokazywał przy każdej kontroli i konsultacji okulistycznej w przyszłości.Na terytorium Stanów Zjednoczonych każdy pacjent musi być zarejestrowany w Alcon Laboratories, Inc. niezwłocznie po wszczepieniu jednej z omawianych soczewek. Rejestracji dokonuje się przez wypełnienie Karty Rejestracji Wszczepu załączonej w pudełku z soczewką i odesłanie jej do Alcon Laboratories, Inc. Rejestracja pacjenta jest istotna dla programu długotrwałego monitorowania pacjentów Alcon Laboratories, Inc. i pomoże w reagowaniu na raportowane zdarzenia niepożądane.Poza terytorium Stanów Zjednoczonych powinny być przestrzegane lokalne przepisy dotyczące rejestracji pacjenta.
Zdarzenia, które logicznie sugerują, że soczewka mogła spowodować lub przyczynić się do śmierci lub ciężkiego urazu, włącznie ze zdarzeniami następującymi w wyniku niemożności zapewnienia przez wyrób medyczny działania zgodnego ze specyfikacją lub zapewnienia zamierzonego działania pod innym względem, powinny być zgłaszane do Alcon Laboratories, Inc. Informacji takich oczekuje się od wszystkich lekarzy chirurgów w celu udokumentowania potencjalnych długoterminowych następstw mogących wystąpić po implantacji soczewek wewnątrzgałkowych. Chirurdzy w celu zgłaszania działań niepożądanych związanych z omawianymi soczewkami wewnątrzgałkowymi powinni korzystać z następującego adresu i numeru telefonu:
Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Zgłoszenia są przyjmowane pod bezpłatnym nr telefonu: 1-800-757-9780
Poza obszarem Stanów Zjednoczonych w sprawie zgłaszania zdarzeń niepożądanych prosimy o kontakt z lokalnymi biurami lub dystrybutorami firmy Alcon. W Polsce zdarzenia niepożądane należy zgłaszać korzystając z następującego adresu i numeru telefonu lub faksu: Alcon Polska Sp. z o.o., ul. Marynarska 15, 02-674 Warszawa, tel. (22) 820 34 50, faks (22) 820 34 56.
Badania kliniczne dotyczące tylnokomorowej zwijalnej soczewki AcrySof®Przeprowadzono dwa randomizowane, prospektywne, odpowiednio kontrolowane badania kliniczne dotyczące stosowania jednoczęściowych zwijanych soczewek tylnokomorowych AcrySof®. Pierwsze badanie przeprowadzono celem wykazania bezpieczeństwa stosowania i skuteczności jednoczęściowej soczewki tylnokomorowej AcrySof® Natural Model SB30AL (filtrującą promieniowanie UV i światło niebieskie) jako macierzystego modelu soczewki. Było to randomizowane badanie kliniczne, w którym stosowano soczewkę AcrySof® Model SA30AL (pochłaniającą jedynie UV) jako soczewkę kontrolną. Dane dotyczą jedynie pierwszego oka, w którym wszczepiono soczewkę u osób, u których zastosowano soczewkę wewnątrzgałkową Model SB30AL lub Model SA30AL. Drugie randomizowane badanie kliniczne dotyczące porównania jednoczęściowej zwijalnej soczewki wewnątrzgałkowej AcrySof® IQ (z chromoforem filtrującym promieniowanie UV i światło niebieskie) z optyką asferyczną w porównaniu do jednoczęściowej zwijalnej soczewki kontrolnej AcrySof® zostało przeprowadzone w celu oceny korzyści klinicznych/funkcjonalnych w stosunku do tradycyjnego projektu sferycznego.
Badanie kliniczne dotyczące jednoczęściowej soczewki AcrySof® Natural Model SB30ALWyniki uzyskane w badaniu z udziałem pacjentów obserwowanych przez okres co najmniej jednego roku po zabiegu dowodzą, że jednoczęściowe soczewki AcrySof® Natural Model SB30AL są bezpieczne i skuteczne w korygowaniu ostrości wzroku u pacjentów po usunięciu naturalnej soczewki.
Populacja pacjentów, którym wszczepiono jednoczęściowe soczewki AcrySof® Natural Model SB30ALPopulacja pacjentów, u których wszczepiono soczewkę Model SB30AL do co najmniej pierwszego operowanego oka składała się w tym dwustronnym badaniu z 70,6% kobiet i 29,4% mężczyzn. Populacja pacjentów, którym wszczepiono soczewkę Model SA30AL (grupa kontrolna) składała się z 60,5% kobiet oraz 39,5% mężczyzn. Pod względem rasy w grupie pacjentów, którym wszczepiono soczewkę model SB30AL było 95,3% osób rasy białej i 4,7% osób rasy czarnej. Populacja pacjentów, którym wszczepiono soczewkę Model SA30AL (grupa kontrolna) składała się z 96,6% osób rasy białej, 2% osób rasy czarnej oraz 1,4% osób innych ras. Średnia wieku w całej populacji osób, którym wszczepiono soczewki Model SB30AL do co najmniej pierwszego operowanego oka wynosiła 72,9 lat. Podobnie, średnia wieku w całej populacji osób, którym wszczepiono soczewki Model SA30AL (grupa kontrolna) wynosiła 71,9 lat.
Ostrość wzroku uzyskana po wszczepieniu jednoczęściowej soczewki AcrySof® Natural Model SB30ALW Tabeli 2a przedstawiono charakterystykę ostrości wzroku uzyskanej w okresie co najmniej jednego roku po zabiegu u osób, które nie miały kiedykolwiek wcześniej patologii narządu wzroku, zaburzeń rogówki lub zwyrodnienia plamki (populacja najlepszych kandydatów do zabiegu). Ostrość widzenia w ogólnej populacji pacjentów przedstawiono w Tabeli 3a. Dane dotyczące grup kontrolnych znajdują się odpowiednio w Tabelach 2b oraz 3b. Nie stwierdzono statystycznie znamiennej różnicy pod względem ostrości wzroku między grupą pacjentów, którym wszczepiono soczewkę Model SB30AL a grupą kontrolną, pacjentów, którym wszczepiono soczewkę Model SA30AL, zarówno w odniesieniu do populacji najlepszych kandydatów do zabiegu, jak i całej populacji.
95
Patrz wersja angielska Tabela 2aNajlepsza skorygowana ostrość wzroku w populacji pacjentów – najlepszych kandydatów do zabiegu – po
okresie co najmniej jednego roku po wszczepieniu soczewek AcrySof® Natural SB30AL
Ostrość
Liczba 20/20 lub lepsza 20/25 20/30 20/40 20/40 lub
lepsza> 20/40 do
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Kategoria wiekowa
Patrz wersja angielska Tabela 2bNajlepsza skorygowana ostrość wzroku w populacji pacjentów – najlepszych kandydatów do zabiegu – po
okresie co najmniej jednego roku, po wszczepieniu soczewek AcrySof® SA30AL (grupa kontrolna)
Ostrość
Liczba 20/20 lub lepsza 20/25 20/30 20/40 20/40 lub
lepsza> 20/40 do
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Kategoria wiekowa
Patrz wersja angielska Tabela 3aNajlepsza skorygowana ostrość wzroku w całej populacji pacjentów po okresie co najmniej jednego roku, po
wszczepieniu soczewek AcrySof® Natural SB30AL
Ostrość
Liczba 20/20 lub lepsza 20/25 20/30 20/40 20/40 lub
lepsza> 20/40 do
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Kategoria wiekowa
Patrz wersja angielska Tabela 3bNajlepsza skorygowana ostrość wzroku w całej populacji pacjentów po okresie co najmniej jednego roku, po
Liczba 20/20 lub lepsza 20/25 20/30 20/40 20/40 lub
lepsza> 20/40 do
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Kategoria wiekowa
Skumulowane zdarzenia niepożądane po wszczepieniu soczewki jednoczęściowej AcrySof® Natural Model SB30ALW Tabeli 4 przedstawiono skumulowaną liczbę zdarzeń niepożądanych, które obserwowano w okresie co najmniej jednego roku po wszczepieniu jednoczęściowych soczewek AcrySof® Natural Model SB30AL i Model SA30AL. Nie stwierdzono znamiennej statystycznie różnicy pomiędzy soczewkami Model SB30AL i Model SA30AL pod względem odsetka pacjentów, u których wystąpiło którekolwiek ze skumulowanych zdarzeń niepożądanych.
96
Tabela 4Skumulowane zdarzenia niepożądane w okresie co najmniej jednego roku po wszczepieniu soczewki AcrySof®
Natural SB30AL i SA30AL, grupa kontrolna
SB30AL (N= 153)
SA30AL(N= 147)
Wartość p*
N % N %
Rodzaj zdarzenia
niepożądanego
0 0,0 0 0,0 NA Skumulowane Ropostek
Zakażenie wewnątrzgałkowe / Zapalenie wnętrza gałki ocznej 0 0,0 0 0,0 NA
Obrzęk plamki 4 2,6 2 1,4 0,6847
Blok źreniczny 0 0,0 0 0,0 NA
Odwarstwienie siatkówki lub naprawa odwarstwienia siatkówki 0 0,0 0 0,0 NA
Przemieszczenie soczewki 1 0,7 0 0,0 1,0000
Wtórna interwencja chirurgicznaUsunięcie pozostałości korowychEksplantacja (przemieszczenie z powodu rozerwania torebki)Krioterapia w celu naprawy przedarcia siatkówkiNakłucie w celu obniżenia IOPOgniskowa laseroterapiaTerapia fotodynamicznaEksplantacja z powodu błędu biometrii
51 1
11100
3,3 0,7
0,7
0,70,70,70,00,0
200
00011
1,40,00,0
0,00,00,00,70,7
0,4482
Krwistek 0 0,0 0 0,0 NA
* Wartości p w teście dokładnym Fishera: porównanie między modelem SB30AL a modelem SA30AL.
Utrzymujące się zdarzenia niepożądane po wszczepieniu jednoczęściowej soczewki AcrySof® Natural Model SB30ALTabela 5 przedstawia dane na temat częstości występowania zdarzeń niepożądanych utrzymujących się przez okres jednego roku po zabiegu wszczepienia soczewki jednoczęściowej AcrySof® Natural Model SB30AL oraz Model SA30AL (grupa kontrolna). Nie stwierdzono statystycznie znamiennych różnic pod względem odsetków pacjentów, u których wystąpiły utrzymujące się zdarzenia niepożądane po wszczepieniu soczewek Model SB30AL oraz Model SA30AL.
Tabela 5Zdarzenia niepożądane utrzymujące się w okresie co najmniej jednego roku po wszczepieniu soczewki AcrySof®
Natural IOL SB30AL i SA30AL, grupa kontrolna
SB30AL (N= 138)
SA30AL(N= 127) Wartość p*
N % N %
Rodzaj zdarzenia niepożądanego
0 0 1 0,8 0,4792Utrzymujące się Obrzęk rogówki
Zapalenie tęczówki 0 0 0 0,0 NA
Obrzęk plamki 2 1,4 1 0,8 1,0000
Zapalenie ciała szklistego 0 0 0 0,0 NA
Wzrost IOP wymagający leczenia 0 0 0 0,0 NA
* Wartości p w teście dokładnym Fishera: porównanie między modelem SB30AL a modelem SA30AL.
97
Percepcja barw po wszczepieniu jednoczęściowej soczewki AcrySof® Natural Model SB30ALPo 120 do 180 dniach po zabiegu wszczepienia soczewki przeprowadzono test percepcji barw z zastosowaniem testu Farnswortha D-15. Spośród 109 pacjentów z prawidłowym widzeniem barw, którym wszczepiono soczewkę model SB30AL w pierwszym oku, których poddano testowi w okresie od 120 do 180 dni po zabiegu, u 107 (98,2%) uzyskano dobry wynik testu na percepcję barw. Spośród 102 pacjentów z prawidłowym widzeniem barw, którym wszczepiono soczewkę model SA30AL w pierwszym oku, których poddano testowi w okresie od 120 do 180 dni po zabiegu, u 97 (95,1%) uzyskano dobry wynik testu na percepcję barw. Nie stwierdzono statystycznie znamiennych różnic pomiędzy grupami pacjentów, którym wszczepiono soczewki Model SB30AL albo Model SA30AL pod względem odsetka osób, u których uzyskano dobry wynik testu na percepcję barw w okresie od 120 do 180 dni po zabiegu. Oznacza to, że zastosowanie dodatkowego opatentowanego chromoforu nie ma niekorzystnego wpływu na percepcję barw u pacjentów, u których była ona prawidłowa przed wszczepieniem soczewki wewnątrzgałkowej.
Odsetek pacjentów z wszczepioną soczewką jednoczęściową AcrySof® Natural Model SB30AL, u których przeprowadzono zabieg z użyciem lasera Nd:YAGW okresie obserwacji średnio wynoszącej 21,6 miesięcy, u trzech (3) ze 135 pacjentów (2,2%), którym wszczepiono soczewkę SB30AL wykonano kapsulotomię tylną z użyciem lasera Nd:YAG. W okresie obserwacji średnio wynoszącej 21,9 miesięcy, u dwóch (2) ze 127 pacjentów (1,6%), którym wszczepiono soczewkę SA30AL wykonano kapsulotomię tylną z użyciem lasera Nd:YAG.
Badanie kliniczne soczewki AcrySof® IQZgodnie z projektem podobnych, wcześniej przeprowadzanych badań soczewek wewnątrzgałkowych, dorośli pacjenci z obustronną zaćmą, o dobrym ogólnym stanie zdrowia narządu wzroku (np. nie mający wcześniejszego okulistycznego zabiegu chirurgicznego, zwyrodnieniowych zaburzeń wzroku, które mogłyby mieć znaczny wpływ na ostrość widzenia lub ciężkiego ostrego lub przewlekłego schorzenia, które mogłoby zwiększać ryzyko u pacjenta) zostali włączeni do kontrolowanego, randomizowanego, podwójnie maskowanego, wieloośrodkowego badania klinicznego przeciwstronnych implantów soczewki AcrySof® IQ w porównaniu do kontrolnej soczewki sferycznej. Oczne aberracje sferyczne były statystycznie znamiennie mniejsze w przypadku soczewki AcrySof® IQ niż soczewki kontrolnej. Wyniki poczucia kontrastu wykazały znamienną statystycznie poprawę po operacji (po 3 miesiącach) na korzyść oczu z implantem AcrySof® IQ. W oczach, w których wszczepiono soczewkę AcrySof® IQ wystąpiła również statystycznie znamienna poprawa w zakresie czynnościowych pomiarów wzroku, symulacji prowadzenia pojazdu w nocy, w różnych warunkach badania - szczególnie w warunkach blasku i mgły. Wyniki te świadczą, że soczewka wewnątrzgałkowa AcrySof® IQ (z optyką asferyczną wykonaną z materiału zawierającego chromofor filtrujący światło niebieskie) zapewnia korzystniejsze klinicznie działanie w porównaniu do jednoogniskowej soczewki wewnątrzgałkowej AcrySof® (bez optyki asferycznej i chromoforu filtrującego światło niebieskie).
Soczewka AcrySof® IQ – Aberracje sferyczne i całkowite aberracje wyższego rzęduŚrednia oczna aberracja sferyczna oczu z wszczepioną soczewką AcrySof® IQ wynosiła około 0,1 mikrometrów. Rysunek 10 przedstawia znamienne statystycznie zmniejszenie aberracji sferycznych i całkowitych aberracji wyższego rzędu zaobserwowane na korzyść soczewki AcrySof® IQ. Rysunek 11 przedstawia pomiary średniej aberracji sferycznej wszystkich oczu, dla których wykonano pomiary aberrometrem metodą czoła fali stosownie do rodzaju soczewki i grupy wiekowej. Jak przedstawiono na tym wykresie, zmniejszenie aberracji sferycznej w oczach z soczewką AcrySof® IQ było niezależne od wieku.
Patrz wersja angielska Rysunek 10Wartość RMS aberracji sferycznej i całkowitej aberracji wyższego rzędu
90-120 dni po wszczepieniu do drugiego oka
Patrz wersja angielska Rysunek 11Średnia aberracja sferyczna ogółem i w grupach wiekowych
90-120 dni po wszczepieniu do drugiego oka
Soczewka AcrySof® IQ - Ostrość widzenia w daliSoczewka AcrySof® IQ i soczewka kontrolna zapewniły podobną ostrość widzenia po operacji. Wyniki jednoocznego badania ostrości widzenia zostały przedstawione na Rysunkach 12 i 13.
Patrz wersja angielska Rysunek 12LogMAR BCVA
Patrz wersja angielska Rysunek 13LogMAR UCVA
Soczewka AcrySof® IQ - Poczucie kontrastuGłównym celem badania klinicznego było wykazanie przewagi soczewki AcrySof® IQ nad soczewką kontrolną za pośrednictwem pomiaru średniego poczucia kontrastu po operacji w warunkach zmierzchowych z lub bez blasku przy każdej z dwóch częstotliwości przestrzennych (3 albo 6 cykli na stopień) przy użyciu przyrządu Vector Vision CSV-1000 (z luminancją tablicy 3 cd/m2). W podgrupie pacjentów przeprowadzono także test FACT (Functional Acuity Contrast Test) (z luminancją tablicy 3 cd/m2). W tym badaniu klinicznym wykazano przewagę soczewki AcrySof® IQ nad soczewką kontrolną
98
w warunkach zmierzchowych przy 6 cyklach na stopień zarówno z, jak i bez blasku (CSV-1000) oraz przy 3 i 6 cyklach na stopień bez blasku (FACT). Rysunki 14 i 15 przedstawiają wyniki zmierzchowego poczucia kontrastu przy wszystkich częstotliwościach przestrzennych badanych zarówno dla soczewki AcrySof® IQ, jak soczewki kontrolnej.
Patrz wersja angielska Rysunek 14Zmierzchowe poczucie kontrastu (CSV-1000)90-120 dni po wszczepieniu do drugiego oka
Patrz wersja angielska Rysunek 15Zmierzchowe poczucie kontrastu (FACT) w podgrupie pacjentów
Co najmniej 90 dni po wszczepieniu do drugiego oka
Soczewka AcrySof® IQ - Symulacja prowadzenia pojazdu w nocyW podgrupie pacjentów wykonano badania na atestowanym symulatorze prowadzenia pojazdu w nocy. Pacjenci byli badani jednoocznie w warunkach symulujących otoczenie miejskie i wiejskie w warunkach normalnych, blasku i mgły.Otoczenie jazdy nocnej w mieście obejmuje różnorodne światła uliczne, światła samochodów, światła sklepów i znaki drogowe aby odtworzyć wysokie natężenie oświetlenia otaczającego, typowego w tych warunkach. Otoczenie jazdy nocnej w warunkach wiejskich stosuje minimalną ilość oświetlenia otaczającego. W warunkach miejskich i wiejskich stosowane były symulowane prędkości jazdy wynoszące odpowiednio 35 mil/godz. i 55 mil/godz.Pacjenci byli proszeni o dostrzeżenie i zidentyfikowanie szeregu celów w każdym otoczeniu, obejmujących biało-zielone drogowe znaki informacyjne, czarno-żółte znaki ostrzegawcze i pieszych. Pacjenci byli proszeni o zareagowanie po zobaczeniu pierwszego celu, co pozwoliło na zapisanie odległości dostrzeżenia. Pacjenci byli następnie proszeni o zareagowanie, kiedy mogli rozpoznać cel (np. co oznacza sygnał, w którą stronę idzie pieszy, itp.), co pozwoliło na zapisanie odległości rozpoznania. Rysunki od 16 do 19 przedstawiają średnią różnicę pomiędzy soczewką AcrySof® IQ i soczewką kontrolną podczas jazdy w otoczeniu miejskim i wiejskim zarówno w zakresie odległości spostrzeżenia jak rozpoznania (np. średnia różnic wewnątrzosobniczych).Parametry czynnościowe soczewki AcrySof® IQ były lepsze niż soczewki kontrolnej w 34 z 36 badanych warunków, co odzwierciedla poprawę zarówno w zakresie odległości dostrzegania jak rozpoznawania podczas jazdy w otoczeniu miejskim i wiejskim, badanych w różnych warunkach jazdy (warunki normalne, blask, mgła). Ponadto, soczewka AcrySof® IQ działała statystycznie znamiennie lepiej niż soczewka kontrolna w 12 z tych warunków, przy czym najsilniejszy wpływ i największą przewagę zaobserwowano w zakresie dostrzegania i rozpoznawania pieszych w otoczeniu miejskim (w warunkach blasku i mgły) oraz znaków ostrzegawczych w otoczeniu wiejskim (w warunkach blasku i mgły). W warunkach zmniejszonej widoczności (blask, mgła) w otoczeniu miejskim, zwiększenie dystansu widoczności przy prędkości 35 mil/godz. przez soczewkę AcrySof® IQ zapewnia ponad 0,5 sekundy dodatkowego czasu na reakcję na cel, jakim jest pieszy, zagrożenie częściej spotykane w warunkach miejskich. To zwiększenie o 0,5 sekundy jest istotne czynnościowo, wydłużając czas na podjęcie odpowiedniego działania, takiego jak zatrzymanie, ominięcie, itp. (Green, 2000; McBride i Matson, 2004). We wszystkich warunkach w otoczeniu wiejskim, zwiększenie dystansu widoczności przy prędkości 55 mil/godz. przez soczewkę AcrySof® IQ zapewnia ponad 1 sekundę dodatkowego czasu na rozpoznanie znaków ostrzegawczych, co jest sytuacją często spotykaną w obszarach wiejskich. Zwiększenie czasu o 0,5 sekundy jest istotne czynnościowo, pozwalając na dłuższy czas na podjęcie odpowiedniego działania podczas prowadzenia pojazdu, co nabiera krytycznego znaczenia w nocy w nieznanych obszarach wiejskich, gdzie często nie ma oświetlenia otaczającego. W podgrupie badanej było 6 pacjentów, u których po operacji wystąpiło zwyrodnienie plamki lub zmętnienie tylnej torebki soczewki (PCO). Kiedy tych pacjentów usunięto z analizy dotyczącej prowadzenia pojazdu, różnica pomiędzy soczewkami pod względem dostrzegania i rozpoznawania celu, jakim był pieszy w warunkach blasku w otoczeniu miejskim znalazła się poniżej 0,5-sekundowego progu znaczenia klinicznego. Kiedy oryginalne analizy zostały skorygowane pod względem krotności, różnica pomiędzy soczewkami wewnątrzgałkowymi przestała być statystycznie znamienna dla dostrzegania tekstu w otoczeniu miejskim w warunkach blasku (wartość p=0,0539 w teście Hommel’a) oraz dostrzegania pieszych w otoczeniu wiejskim w warunkach blasku (wartość p=0,0507 w teście Hommel’a).Wyniki te wykazują poprawę czynnościową widzenia i prawdopodobne znamienne korzyści w zakresie bezpieczeństwa dla starszych kierowców po wszczepieniu soczewki AcrySof® IQ oraz innych kierowców i pieszych, którzy znajdują się z nimi na tej samej drodze. Wyniki tego testu wykazują, że soczewka AcrySof® IQ poprawia czynnościowo widzenie, co z kolei może poprawić bezpieczeństwo pacjenta w innych sytuacjach życiowych w warunkach słabej widoczności.
Patrz wersja angielska Rysunek 16Symulator prowadzenia pojazdu w warunkach nocnych
Średnie wewnątrzosobnicze różnice dystansu dostrzegania w otoczeniu miejskimMinimum 90 dni po operacji
Soczewka AcrySof® IQ –Kontrola (n = 44)
Patrz wersja angielska Rysunek 17Symulator prowadzenia pojazdu w warunkach nocnych
Średnie wewnątrzosobnicze różnice dystansu rozpoznawania w otoczeniu miejskimMinimum 90 dni po operacji
Soczewka AcrySof® IQ –Kontrola (n=44)
99
Patrz wersja angielska Rysunek 18Symulator prowadzenia pojazdu w warunkach nocnych
Średnie wewnątrzosobnicze różnice dystansu dostrzegania w otoczeniu wiejskimMinimum 90 dni po operacji
AcrySof® IQ– Kontrola (n=44)
Patrz wersja angielska Rysunek 19Symulator prowadzenia pojazdu w warunkach nocnych
Średnie wewnątrzosobnicze różnice dystansu rozpoznawania w otoczeniu wiejskimMinimum 90 dni po operacji
AcrySof® IQ– Kontrola (n=44)
OPAKOWANIEOmawiane soczewki wewnątrzgałkowe są dostarczane w postaci suchej w urządzeniu UltraSert™ Pre-loaded Delivery System, wewnątrz sterylnego opakowania bezpośredniego i ostatecznie wysterylizowane tlenkiem etylenu. Sterylne opakowanie bezpośrednie musi być otwierane tylko w warunkach aseptycznych (patrz SPOSÓB UŻYCIA).
DATA WAŻNOŚCISterylność jest zagwarantowana, jeśli nie zostanie uszkodzone lub otwarte sterylne opakowanie bezpośrednie. Data ważności jest wyraźnie oznaczona na etykiecie opakowania zewnętrznego urządzenia UltraSert™ Pre-loaded Delivery System. Każdą soczewkę po terminie ważności należy przekazać do firmy Alcon Laboratories, Inc. (zobacz ZWROTY TOWARU).
ZWROTY TOWARUNa terytorium USA zwracane soczewki mogą być przyjęte do wymiany tylko na inne produkty, a nie za zwrotem zapłaty. Wszystkie zwroty muszą być oznaczone Numerem Zwrotu Towaru Alcon Laboratories Inc. i odesłane przesyłką poleconą. Numer Zwrotu Towaru można otrzymać po skontaktowaniu się z Działem Obsługi Klienta firmy Alcon. Wydanie tego numeru nie oznacza ostatecznej akceptacji zwrotu towaru. W celu uzyskania dodatkowych wskazówek dotyczących wymiany należy kontaktować się z lokalnymi przedstawicielami Działu Sprzedaży lub Działu Obsługi Klienta. W sprawie zwrotu towaru poza Stanami Zjednoczonymi, należy kontaktować się z lokalnymi biurami i dystrybutorami firmy Alcon.
PIŚMIENNICTWO 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., et al. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., et al. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
100
SYMBOLE UŻYWANE NA OPAKOWANIU
SYMBOL ZNACZENIE SYMBOLUIOL Soczewka wewnątrzgałkowa
SP Jednoczęściowa
UV Ultrafiolet
D Dioptrie
ØB Średnica soczewki (części optycznej)
ØT Średnica całkowita (długość całkowita)
2STERILIZE Nie sterylizować powtórnie
Nie używać ponownie
Termin ważności
STERILE EO Sterylizowane tlenkiem etylenu
SN Numer seryjny
Uwaga
Wytwórca
EC REP Autoryzowany przedstawiciel w Unii Europejskiej
Górne ograniczenie temperatury
Tu otworzyć
Sprawdzić w instrukcji użycia
Nie używać jeśli sterylne opakowanie zostało otwarte lub uszkodzone
UWAGA: Prawo federalne USA wymaga, aby wyrób ten sprzedawany był tylko na zamówienie lekarza
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 USA
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,Wielka Brytania
Lentile de cameră posterioară STERILE, ce filtrează UV și lumina albastră, acrilice, pliabile, monobloc cu sistem de administrare pre-încărcat UltraSert™
DESCRIERELentilele de cameră posterioară AcrySof® IQ ce filtrează UV și lumina albastră, acrilice, pliabile, monobloc, sunt implanturi optice pentru înlocuirea cristalinului uman în scopul corecției vizuale a afakiei la pacienți adulți după chirurgia cataractei. Lentila intraoculară AcrySof® IQ prevăzută cu agentul cromofor de filtrare a luminii brevetat de Alcon, are capacitatea de a filtra lumina într-o manieră foarte asemănătoare cristalinului uman, pentru lungimi de undă ale luminii albastre încadrate în intervalul 400 – 475 nm (Boettner and Wolter, 1962). Pe lângă filtrarea standard a razelor UV, lentila intraoculară AcrySof® IQ reduce transmiterea luminii albastre de la 62 % (la 400 nm) la 23 % (la 475 nm) (vezi Tabelul 1). Partea optică constă într-un material acrilic moale, cu indice de refracție mare. Acest material poate fi pliat înainte de inserție. După implantare, lentila se depliază lent, până la dimensiunea normală. Lentila are o parte optică biconvexă și anse de fixare (Figura 1). Suprafața posterioară asferică a lentilei intraoculare AcrySof® IQ Model AU00T0 este concepută cu o aberație sferică negativă pentru a compensa aberația sferică pozitivă a corneei umane normale. Calitatea imaginii cu lentila intraoculară monobloc Model AU00T0 (i.e. funcția de transfer a modulației) este ilustrată în Figura 4. Lentilele intraoculare monobloc AcrySof® IQ Model AU00T0 sunt furnizate în sistemul de implantare pre-încărcat UltraSert™ (Figura 2) pentru a asigura introducerea în mod convenabil și controlat a acestora în sacul capsular. Proprietățile fizice ale lentilelor sunt următoarele:
Caracteristici fizice Descriere
Tip optic IOL monobloc cu optic asferic
Materialul lentilelor Copolimer acrilat/metacrilat care filtrează radiațiile ultraviolete și lumina albastră
Blocare UV la 10% transmitanță (nm) UV (400) la 20,0 D (vezi Figura 3)
Indicele de refracție 1.55
Puterile optice Pentru puterile disponibile vezi catalogul de produse Alcon
6.0 D to 27.0 D range = Interval 6.0 D – 27.0 D 4 yr – 53 yr old range, crystalline lens = Interval de vârstă 4-53 ani, lentile cristaliniene
NOTE: • Lungimea de undă și curbele de transmitanță spectrală prezentate aici reprezintă intervalul valorilor de transmitere
ale lentilelor intraoculare fabricate din copolimer acrilat/metacrilat de care s-a fixat un absorbant de radiații UV și agentul cromofor de filtrare a luminii albastre brevetat de Alcon.
• Transmitanța a fost măsurată direct, utilizându-se o apertură/deschidere de 6 mm și un disc de grosime echivalentă cu cea a centrului părții optice.
• Informațiile despre cristalinul uman sunt citate din Boettner and Wolter (1962).
Tabelul 1Comparația transmitanței pentru lentile intraoculare de 20.0 D, %
Model 400 nm 425 nm 450 nm 475 nm
SA60AT 21 86 88 88
AU00T0 8 34 49 68
Diferența de transmitanță (SA60AT – AU00T0) 13 52 39 20
Reducerea transmitanței cu AU00T0 (% din SA60AT) 62 60 44 23
Figura 4Funcția de transfer a modulației la modelul AU00T0 (20.0 D)
MTF (%) = FTM (%) Funcția de transfer a modulațieiSpatial Frequency (Ip/mm) = Frecvența spațială (lp/mm)
ISO, 3-mm Aperture (see Note 1) = ISO, Apertură de 3 mm (vezi Nota 1)Modified, 3-mm Aperture (see Note 2) = Apertură de 3 mm modificată (vezi Nota 2)Modified, 5-mm Aperture (see Note 2) = Apertură de 5 mm modificată (vezi Nota 2)
NOTE: 1. Calitatea imaginii cu modelul AU00T0 a fost caracterizată prin măsurarea funcției de transfer a modulației (FTM)
la un model de ochi descris în ISO 11979-2. Conform cerințelor ISO 11979-2, măsurătorile FTM se realizează folosind numai o apertură de 3 mm.
2. Suplimentar, calitatea imaginii cu modelul AU00T0 a fost caracterizată prin măsurarea FTM la un model de ochi care a folosit o simulare de cornee având aberația sferică tipică unui om adult. Folosind modelul de ochi modificat, măsurătorile FTM s-au făcut folosind atât apertura de 3 mm cât și cea de 5 mm.
MOD DE ACȚIUNELentilele intraoculare de cameră posterioară AcrySof® IQ sunt concepute pentru a fi poziționate în camera posterioară a ochiului, înlocuind cristalinul natural. Această poziționare îi permite lentilei să funcționeze ca un mediu de refracție, pentru corectarea afakiei. Partea optică biconvexă asferică reduce aberația sferică în comparație cu o parte optică sferică dintr-un ochi mediu. Nu a fost stabilită eficiența acestor lentile în reducerea incidenței afecțiunilor retiniene.
INDICAȚIILentilele intraoculare de cameră posterioară AcrySof® IQ sunt indicate pentru înlocuirea cristalinului uman în scopul corecției vizuale a afakiei la pacienții adulți, după chirurgia cataractei (vezi “ATENȚIONĂRI”). Aceste lentile sunt destinate poziționării în sacul capsular.
ATENȚIEPacienții care suferă de oricare dintre următoarele afecțiuni ar putea să nu fie candidați potriviți pentru implantul de lentile intraoculare, deoarece acestea pot agrava o afecțiune existentă, pot interfera cu diagnosticul sau tratamentul unei afecțiuni sau pot determina un risc crescut pentru vederea pacientului. Chirurgul trebuie să faca o evaluare preoperatorie și un raționament clinic atent pentru aprecierea raportul beneficiu/risc, înainte de a implanta o lentilă intraoculară unui pacient cu una sau mai multe dintre următoarele afecțiuni: 1. Hemoragie coroidală 2. Afecțiune oftalmologică severă concomitentă 3. Pierdere excesivă de vitros 4. Cameră anterioară de profunzime redusă 5. Microftalmie
103
6. Cataractă non-senilă 7. Ruptură de capsulă posterioară (ce împiedică fixarea lentilei intraoculare) 8. Distrofie corneană severă 9. Atrofie optică severă 10. Presiune pozitivă incontrolabilă 11. Separare zonulară (ce împiedică fixarea lentilei intraoculare) 12. Deficiențe ale vederii colorate 13. Glaucom 14. Uveită cronică 15. Retinopatie diabetică 16. Modificări maculare/ale epiteliului pigmentar retinian, semnificative clinic Studiile au demonstrat că distincția culorilor nu este afectată în mod negativ la pacienții cu implant de lentilă intraoculară AcrySof® Natural care au o percepție normală a culorilor. La pacienții cu deficiențe de percepere a culorilor ereditare sau secundare unor afecțiuni oculare (de exemplu: glaucom, retinopatie diabetică, uveită cronică și alte boli retiniene sau ale nervului optic), efectele lentilei intraoculare AcrySof® Natural asupra vederii nu au fost studiate.
ATENȚIONĂRI 1. Ca orice procedură chirurgicală și aceasta implică anumite riscuri. Complicațiile posibile care însoțesc
intervențiile chirurgicale pentru extracția cataractei sau implantarea lentilei intraoculare pot include, fără a se limita la următoarele: lezarea endoteliului cornean, infecție (endoftalmită), dezlipire de retină, vitrită, edem macular cistoid, edem cornean, bloc pupilar, membrană ciclitică, prolaps al irisului, hipopion și glaucom tranzitoriu sau persistent.
2. Siguranța și eficiența implanturilor de lentile intraoculare nu au fost demonstrate la pacienții suferind de afecțiuni oculare pre-existente (mioză cronică iatrogenă, glaucom, ambliopie, retinopatie diabetică, antecedent de transplant cornean, antecedent de dezlipire de retină și/sau irită, etc.). Medicii care intenționează să efectueze un implant de lentilă intraoculară la astfel de pacienți trebuie să ia în considerare folosirea unor metode alternative de corectare a afakiei și să recurgă la implantul de lentilă numai dacă alternativele sunt considerate nesatisfăcătoare față de nevoile pacientului.
3. Efectele pe termen lung ale implantării de lentile intraoculare nu au fost determinate. De aceea, se recomandă urmărirea postoperatorie constantă a pacienților.
4. Pacienții cu probleme preoperatorii cum ar fi o afecțiune a endoteliului cornean, cornee anormală, degenerescență maculară, degenerescență retiniană, glaucom și mioză cronică iatrogenă s-ar putea să nu dobândească aceeași acuitate vizuală cu a celor care nu suferă de aceste afecțiuni. Atunci când aceste probleme există, medicul trebuie să determine beneficiile ce vor decurge în urma implantului de lentilă.
5. Iridectomia secundară pentru bloc pupilar poate fi evitată dacă în momentul implantării lentilei intraoculare sunt efectuate una sau mai multe iridectomii. (Willis, et al., 1985).
6. Nu au fost stabilite siguranța și eficiența poziționării în camera anterioară a unei lentile pentru camera posterioară. Implantarea în camera anterioară a lentilelor pentru camera posterioară s-a dovedit în anumite cazuri a fi nesigură (Girard, et al., 1983).
7. Unele dintre reacțiile adverse asociate cu implantarea lentilelor intraoculare sunt: hipopion, infecție intraoculară, decompensare corneană acută și intervenție chirurgicală secundară. Intervențiile chirurgicale secundare includ, dar nu se limitează la: repoziționarea lentilei, înlocuirea lentilei, aspirație de vitros sau iridectomie pentru bloc pupilar, intervenție pentru rezolvarea problemelor de etanșeitatea inciziei și rezolvarea dezlipirii de retină.
8. Descentrarea într-o mică măsură a lentilei, în cazul lentilelor intraoculare cu partea optică îngustă sau mică, poate face ca pacienții să vadă pete strălucitoare sau să sufere alte tulburări vizuale în anumite condiții de iluminare. Chirurgii trebuie să țină cont de aceste potențiale probleme înainte de implantarea unei lentile intraoculare cu partea optică îngustă sau mică. La implantarea unei lentile intraoculare cu partea optică îngustă sau mică, se recomandă efectuarea unui capsulorhexis.
9. Distensia postoperatorie a sacului capsular, însoțită de modificarea într-un grad variabil a profunzimii camerei anterioare și inducerea miopiei, au fost asociate cu tehnicile de capsulorhexis și implantarea lentilelor pentru camera posterioară din silicon, PMMA și acrilice (Holtz, 1992).
10. Înainte de poziționarea lentilei în sac trebuie acționat cu foarte multă grijă, pentru a se evita descentrarea sau dislocarea acesteia. Anumite cazuri clinice sugerează că încapsularea se produce în intervalul de 4 săptămâni de la implantare.
11. Studiul clinic cu lentila monobloc AcrySof® Natural (la care se face referință în Tabelele 2 – 5) s-a desfășurat în condițiile în care lentilele au fost implantate exclusiv în sacul capsular. Nu există date clinice care să demonstreze siguranța și eficiența plasării acestor lentile în sulcusul ciliar.
Se recomandă îndepărtarea din ochi a soluției vâscoelastice la sfârșitul intervenției chirurgicale, în special din spațiul dintre capsula posterioară și lentilă. Aceasta se realizează apăsând ușor lentila posterior cu vârful pentru irigare/aspirare și folosind tehnici standard de irigare/aspirare, pentru îndepărtarea vâscoelasticului din ochi. Această procedură ar trebui să împingă orice vâscoelastic blocat spre partea anterioară de unde poate fi aspirat cu ușurință.
104
PRECAUȚII 1. Nu resterilizați aceste lentile intraoculare sau sistemul de implantare pre-încărcat UltraSert™ prin nici o metodă. 2. Nu păstrați lentilele intraoculare la temperaturi mai mari de 45° C (113° F). 3. Dacă este nevoie, manipulați cu atenție lentilele pentru a evita deteriorarea suprafeței lor sau a anselor de fixare. 4. Nu încercați sub nici o formă să remodelați ansele de fixare. 5. Implantarea lentilelor intraoculare necesită însușirea la un nivel ridicat a tehnicilor chirurgicale. Înainte de a încerca
să implanteze lentile intraoculare, chirurgul trebuie să fi observat și/sau să fi asistat la numeroase implantări și trebuie să fi absolvit cu succes unul sau mai multe cursuri despre implantarea lentilelor intraoculare.
6. Componentele sunt sterile, exceptând cazul în care ambalajul este desfăcut sau deteriorat. 7. Sistemul de implantare pre-încărcat UltraSert™ este de unică utilizare. Aruncați sistemul de implantare pre-
încărcat UltraSert™ după utilizare. 8. Utilizați sistemul de implantare pre-încărcat UltraSert™ în sala de operații la temperaturi cuprinse între 18° C (64°
F) și 23° C (73° F).
CALCULAREA PUTERII DIOPTRICE A LENTILEIConstanta A preoperatorie a puterii dioptrice necesare lentilei intraoculare de camera posterioară este determinată de experiența și preferințele chirurgului, precum și de locul unde se intenționează poziționarea lentilei. Metodele de calculare a puterii dioptrice sunt descrise în cadrul următoarelor referințe bibliografice:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. et al. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. et al. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
CONSTANTA A SUGERATĂ Constanta A imprimată pe eticheta exterioară este prezentată cu scop orientativ și constituie un punct de pornire pentru calcularea puterii cristalinului implantat. Se recomandă să vă determinați propria constantă, adecvată pentru dumneavoastră, pe baza experienței clinice cu anumite modele de lentile, a tehnicilor chirurgicale, a echipamentului de măsurare și a rezultatelor postoperatorii. În Statele Unite, dacă sunt necesare informații suplimentare privind calcularea puterii lentilelor, vă rugăm să contactați Alcon Laboratories, Inc. la 1-800-TO-ALCON (1-800-862-5266). În afara Statelor Unite, contactați reprezentanțele Alcon locale sau distribuitorii.
SUBSTANȚE VÂSCOELASTICE RECOMANDATE DE ALCON ȘI NUMERELE DE COMANDĂ Vezi suplimentul inclus în acest pachet. Trebuie utilizate doar substanțe vâscoelastice Alcon. Utilizarea unei substanțe vâscoelastice nerecomandate poate cauza deterioararea lentilei și potențiale complicații în timpul procesului de implantare. Pentru lista completă a substanțelor vâscoelastice recomandate de Alcon, vă rugăm contactați reprezentanța Alcon.
INSTRUCȚIUNI PRIVIND UTILIZAREAPasul 1. Utilizați sistemul de implantare pre-încărcat UltraSert™ în sala de operații la temperaturi cuprinse între 18° C
(64° F) și 23° C (73° F).
Pasul 2. Examinați eticheta înainte de deschiderea ambalajului pentru a vedea modelul, puterea, configurația adecvată și data expirării.
Pasul 3. După deschiderea ambalajului din carton, verificați dacă ambalajul dispozitivului este deteriorat. Dacă observați semne de deterioare utilizați altă lentilă intraoculară AcrySof® IQ împreună cu alt sistemul de implantare pre-încărcat UltraSert™. Apoi, verificați dacă informațiile de pe cutia dispozitivului (de exemplu: modelul, puterea și numărul seriei) corespund cu cele de pe eticheta ambalajului exterior.
Pasul 4. Pentru a scoate sistemul de implantare pre-încărcat UltraSert™, prindeți colțul pungii de plastic, deschideți în totalitate porțiunea din material TYVEK† și transferați dispozitivul într-un mediu steril. Dacă în urma verificării observați că vârful dispozitivului pare să prezinte deteriorări, particule sau deformări utilizați altă lentilă intraoculară AcrySof® IQ împreună cu alt sistem de implantare pre-încărcat UltraSert™. Dacă dispozitivul nu este complet intact, sau pistonul/dispozitivul de blocare a pistonului par să fie modificate datorită trasportului, utilizați altă lentilă intraoculară AcrySof® IQ împreună cu alt sistem de implantare pre-încărcat UltraSert™ (vezi “PROCEDURA PENTRU PRODUSE RETURNATE”).
†Reg. TM of E.I. DuPont De Nemours & Co.
Când sunteți gata pentru implantarea lentilei, realizați Pașii 5, 6 și 7 în secvență, cu un interval de timp cât mai scurt între pași.
105
Pasul 5. Introduceți în totalitate canula vâscoelasticului, perpendicular pe dispozitiv, prin orificiului pentru vâscoelastic, localizat în porțiunea unde se oprește lentila în dispozitiv așa cum se poate vedea în Figura 5. Umpleți dispozitivul până în momentul în care observați prelingerea agentului vâscoelastic la linia vârfului cartușului (vezi săgeata din Figura 5), apoi retrageți canula. Această operațiune necesită aproximativ 0,2 ml de substanță vâscoelastică. Utilizați doar substanțe vâscoelastice Alcon recomandate pentru a fi utilizate cu sistemul de implantare pre-încărcat UltraSert™, care au fost aduse la temperatura din sala de operație. Pentru lista completă a substanțelor vâscoelastice recomandate de Alcon, vezi suplimentul inclus în acest pachet.
Figura 5
Viscoelastic Port = Orificiul pentru vâscoelastic“Fill-To” Line = Linia de umplere
Pasul 6. Înlăturați opritorul albastru al lentilei prin prinderea filei cu săgeata și îndoirea ușoară a acesteia în față până la înlăturarea din dispozitiv (Figura 6a). Înlăturați dispozitivul de oprire a pistonului de culoare albastră prin prinderea și tragerea ușoară a acestuia din dispozitiv (Figura 6b). Aruncați opritorul albastru al lentilei și dispozitivul de oprire a pistonului. Nu încercați să adăugați substanțe vâscoelastice în dispozitiv după ce opritorul albastru al lentilei a fost înlăturat deoarece lentila poate fi deteriorată. Nu încercați să retrageți pistonul după înlăturarea dispozitivului de oprire a pistonului deoarece poate rezulta deteriorarea pistonului.
Figura 6a și 6b
Pasul 7. Împingeți ușor pistonul înainte prin intermediul unei sigure mișcări continue și line până ce marginea superioară a lentilei ajunge la linia vârfului cartușului (vezi Figura 7a). În această poziție arcul albastru va atinge degetul așa cum apare în Figura 7b. Avansarea pistonului ar trebui să necesite cel puțin 7 secunde. Nu permiteți ieșirea oricărei părți a lentilei din vârful cartușului. Este important să nu avansați pistonul brusc deoarece poate apărea plierea necorespunzătoare și deteriorarea lentilei.
Figura 7a și 7b
Fill-to Line = Linia de umplere
Pasul 8. După avansarea lentilei până la vârful cartușului, lentila trebuie verificată vizual pentru a determina poziția anselor. Pistonul ar trebui să fie în contact cu marginea inferioară a lentilei. Nici o parte a lentilei nu trebuie să iasă prin vârful cartușului înainte de introducerea prin incizie (vezi Figura 8). Odată ce lentila este poziționată la linia vârfului cartușului, acesta ar trebui implantată în 3 minute.
Figura 8
“Proceed with full plunger advancement to fully deliver lens” = “Continuați avansarea completă a pistonului până la introducerea completă a lentilei”
“Proceed” = “Continuați”“Proceed with full plunger advancement to fully deliver lens” = “Continuați avansarea completă a pistonului până la
introducerea completă a lentilei”“Proceed with device rotations per Step 10” = “Continuați rotațiile dispozitivului ca în Pasul 10”
“Do not proceed” = “Nu continuați”
Pasul 9. După confirmarea poziționării corespunzătoare a lentilei și plierii corespunzătoare a anselor, continuați implantarea lentilei prin introducerea vârfului cartușului în incizie până la marcajul de siguranță a profunzimii (Figura 9) și îndreptarea vârfului cartușului către deschiderea capsulei anterioare.
Figura 9
Depth Guard = Marcajul de siguranță al profunzimii
Pasul 10. Împingeți ușor pistonul înainte prin intermediul unei singure mișcări continue și line. Nu avansați brusc pistonul deoarece poate apărea deteriorarea lentilei. Este recomandat ca introducerea lentilei după verificarea vizuală a poziției să dureze cel puțin 5 secunde. Dacă ansa din fața opticului este dreaptă sau buclată și extinsă în fața lentilei, răsuciți dispozitivul înspre stânga pentru a vă asigura că ansa din fața opticului este introdusă corect în sacul capsular. Pe măsură ce ansa din fața opticului iese din vârful cartușului, plasați-o în poziția sa corectă în sacul capsular și continuați să împingeți ușor pistonul până la introducerea completă a lentilei. Pe măsură ce lentila iese din vârful cartușului, răsuciți dispozitivul inapoi spre centru sau ușor într-o parte așa cum este necesar pentru a vă asigura că lentila se va deplia cu partea anterioară în sus în interiorul sacului capsular.
Pasul 11. Cu ajutorul unui instrument de poziționare corespunzător, poziționați lentila în sacul capsular, într-un plan paralel cu irisul.
Pasul 12. Aruncați întregul dispozitiv. Nu reutilizați sistemul de implantare pre-încărcat UltraSert™.
106
ÎNREGISTRAREA ȘI RAPORTAREA PACIENȚILOR“Cardul de identificare al pacientului” inclus în ambalaj trebuie completat și înmânat pacientului, care va fi instruit să păstreze cardul ca un document medical permanent și să îl prezinte oricărui medic oftalmolog/optometrist la care va apela în viitor. În Statele Unite, fiecare pacient trebuie înregistrat de către Alcon Laboratories, Inc. imediat după implantarea uneia dintre aceste lentile. Înregistrarea se efectuează prin completarea “cardului de înregistrare al implantului” care se găsește în cutia lentilei și trimiterea lui prin poștă la Alcon Laboratories, Inc. Înregistrarea pacienților este esențială pentru programul de urmărire pe termen lung al pacienților implementat de Alcon Laboratories, Inc. și ne va ajuta la oferirea unui răspuns la reacțiile adverse raportate.În afara Statelor Unite se vor respecta legile locale cu privire la înregistrarea produselor.
Reacțiile adverse ce pot fi în mod rezonabil considerate ca având legătură cu implantul (și a căror natură, gravitate sau incidență nu au fost anticipate) și care au avut ca rezultat decesul sau afectarea gravă a sănătății, inclusiv evenimente apărute ca urmare a neîndeplinirii specificațiilor de performanță sau orice alt tip de rezultat, trebuie raportate către Alcon Laboratories, Inc. (SUA). Aceste informații sunt solicitate tuturor chirurgilor pentru a documenta posibilele efecte pe termen lung ale implanturilor de lentile intraoculare. Chirurgii trebuie să folosească următoarea adresă și număr de telefon pentru a raporta reacțiile adverse legate de aceste lentile intraoculare:
Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Telefon cu taxă inversă: 1-800-757-9780
Pentru chirurgii din afara Statelor Unite, este recomandat să apeleze la reprezentanțele locale sau distribuitori pentru raportarea reacțiilor adverse.
STUDII CLINICE CU LENTILE DE CAMERĂ POSTERIOARĂ AcrySof®, ACRILICE, PLIABILE Au fost efectuate două studii clinice prospective, randomizate cu lentile de cameră posterioară AcrySof® acrilice, pliabile, monobloc. Primul studiu a fost realizat pentru a demonstra siguranța și eficacitatea lentilei monobloc de cameră posterioară AcrySof® Natural model SB30AL (ce filtrează UV și lumina albastră), ca model al lentilei de origine. Studiul clinic a fost randomizat și a inclus ca lentilă martor AcrySof® model SA30AL (care absoarbe numai UV). Sunt incluse numai datele referitoare la primul ochi operat, la pacienții cu implant de lentile intraoculare aparținând unuia dintre cele două modele SB30AL sau SA30AL. Cel de-al doilea studiu clinic randomizat, cu lentila monobloc acrilică, pliabilă AcrySof® IQ (cu cromofori de filtrare UV și lumină albastră) cu parte optică asferică, comparativ cu o lentilă martor AcrySof®, monobloc, acrilică, pliabilă a fost realizat pentru a evalua beneficiile clinice/funcționale în raport cu modelul sferic, traditional.
STUDIU CLINIC CU LENTILA MONOBLOC AcrySof® Natural, model SB30AL Rezultatele obținute de pacienții care au fost urmăriți cu succes timp de minimum un an postoperator certifică faptul că lentila monobloc AcrySof® Natural model SB30AL este un dispozitiv sigur și eficient în corectarea vizuală a afakiei.
PACIENȚII DIN STUDIUL CU LENTILA MONOBLOC AcrySof® Natural, model SB30ALDin datele disponibile până în prezent, grupul de pacienți cărora li s-au implantat lentile de tip monobloc modelul SB30AL la cel puțin primul ochi operat din acest studiu bilateral, constă în 70,6% femei și 29,4% bărbați. Grupul de pacienți din studiu cărora li s-au implantat lentile intraoculare model SA30AL (martor) constă în 60,5% femei și 39,5% bărbați. Stratificarea pe rase în cazul modelului SB30AL arată că 95.3% caucazieni și 4,7% sunt de rasă neagră. Grupul populațional martor (SA30AL) constă din 96,6% caucazieni, 2% rasă neagră și 1,4% de alte rase. Vârsta medie a pacienților ce au primit modelul SB30AL cel puțin la primul ochi operat este de 72,9 ani. În mod similar, vârsta medie a celor ce au primit modelul SA30AL (martor) este de 71,9 ani.
ACUITATEA VIZUALĂ CU LENTILA MONOBLOC AcrySof® Natural, model SB30AL În Tabelul 2a este prezentat un rezumat al rezultatelor în ceea ce privește acuitatea vizuală la minimum un an postoperator, la subiecți care nu au suferit niciodată de afecțiuni oculare preoperatorii, anomalii corneene sau degenerescență maculară (grupul de pacienți “cel mai bun caz”); acuitatea vizuală atinsă de întregul grup de pacienți este prezentată în Tabelul 3a. Datele grupului martor obținute în condiții similare sunt prezentate în Tabelele 2b, și respectiv 3b. Nu a existat o diferență semnificativă statistic între acuitatea vizuală obținută cu modelul SB30AL și modelul SA30AL (martor), nici pentru grupul “cel mai bun caz”, nici în cadrul rezultatelor globale.
107
Tabelul 2aAcuitatea vizuală maximă cu corecție la grupul de pacienți “cel mai bun caz” la cel puțin un an postoperator,
AcrySof® Natural IOL, model SB30AL
Acuitatea
#Per 20/20 sau mai bine 20/25 20/30 20/40 20/40 sau
mai bine> 20/40 to < 20/80 > 20/80
N N % N % N % N % N % N % N %
Categoria de vârstă
Tabelul 2bAcuitatea vizuală maximă cu corecție la grupul de pacienți “cel mai bun caz” la cel puțin un an postoperator,
AcrySof® IOL, model SA30AL (martor)
Acuitatea
#Per 20/20 sau mai bine 20/25 20/30 20/40 20/40 sau
mai bine> 20/40 to < 20/80 > 20/80
N N % N % N % N % N % N % N %
Categoria de vârstă
Tabelul 3aAcuitatea vizuală maximă cu corecție la întregul grup de pacienți, la cel puțin un an postoperator AcrySof®
Natural, IOL, modelul SB30AL
Acuitatea
#Per 20/20 sau mai bine 20/25 20/30 20/40 20/40 sau
mai bine> 20/40 to < 20/80 > 20/80
N N % N % N % N % N % N % N %
Categoria de vârstă
Tabelul 3bAcuitatea vizuală maximă cu corecție la întregul grup de pacienți la minimum un an postoperator, AcrySof®, IOL
model SA30AL (martor)
Acuitatea
#Per 20/20 sau mai bine 20/25 20/30 20/40 20/40 sau
mai bine> 20/40 to < 20/80 > 20/80
N N % N % N % N % N % N % N %
Categoria de vârstă
REACȚII ADVERSE CUMULATIVE ASOCIATE CU LENTILA MONOBLOC AcrySof® Natural, model SB30AL Ratele cumulative ale reacțiilor adverse apărute pe o perioadă de minimum un an postoperator, la pacienții la care au fost utilizate lentilele monobloc AcrySof® Natural modelul SB30AL, respectiv modelul SA30AL, sunt prezentate în Tabelul 4. Nu există diferențe semnificative statistic între modelul SB30AL și modelul SA30AL, în ceea ce privește proporția subiecților care au resimțit oricare dintre aceste reacții adverse cumulative.
108
Tabelul 4Reacțiile adverse cumulative la minim un an postoperator AcrySof® Natural, IOL model SB30AL și modelul
martor SA30AL
SB30AL (N= 153)
SA30AL(N= 147)
Valoarea p*
N % N %
Tipul de reacție adverse
0 0.0 0 0.0nu se aplicăCumulative Hipopion
Infecție intraoculară / Endoftalmită 0 0.0 0 0.0 nu se aplică
Edem macular 4 2.6 2 1.4 0.6847
Bloc pupilar 0 0.0 0 0.0 nu se aplică
Dezlipire de retină sau rezolvarea dezlipirii de retină 0 0.0 0 0.0 nu se aplică
Dizlocarea lentilei 1 0.7 0 0.0 1.0000
Reintervenție chirurgicală secundarăÎndepărtarea cortexului rezidualExplantare (dizlocare datorată rupturii capsulare)Crioterapie pentru rezolvarea dezlipirii de retină Paracenteză pentru reducerea PIOTerapie laser focalizatăTerapie fotodinamică Explantare datorată unei erori de biometrie
51 1 11100
3.3 0.7
0.70.70.70.70.00.0
20000011
1.40.00.00.00.00.00.70.7
0.4482
Hifemă 0 0.0 0 0.0 nu se aplică
* valorile p din testul Fisher, în compararea modelelor SB30AL și SA30AL.
REACȚII ADVERSE PERSISTENTE ASOCIATE CU LENTILA MONOBLOC AcrySof® Natural, model SB30AL Ratele reacțiilor adverse persistente după o perioadă de minimum un an postoperator, la pacienții la care au fost utilizate lentilele monobloc AcrySof® Natural, modelul SB30AL, respectiv modelul martor SA30AL, sunt prezentate în Tabelul 5. Nu există diferențe semnificative statistic între modelul SB30AL și modelul SA30AL, în ceea ce privește proporția subiecților care au acuzat oricare dintre aceste reacții adverse persistente.
Tabelul 5Reacțiile adverse persistente după o perioadă de minimum un an postoperator AcrySof® Natural, IOL SB30AL și
modelul martor SA30AL
SB30AL (N= 138)
SA30AL(N= 127)
Valoarea p*
N % N %
Tipul de reacție adversă
0 0 1 0.8 0.4792Persistente Edem cornean
Irită 0 0 0 0.0 nu se aplică
Edem macular 2 1.4 1 0.8 1.0000
Vitrită 0 0 0 0.0 nu se aplică
PIO crescută ce necesită tratament 0 0 0 0.0 nu se aplică
* valorile p din testul Fisher, în compararea modelelor SB30AL și SA30AL.
109
PERCEPȚIA CULORILOR CU LENTILELE MONOBLOC AcrySof® Natural, model SB30AL Testele de percepție a culorilor efectuate cu ajutorul panoului de testare Farnsworth D-15 au avut loc după o perioadă de 120 până la 180 de zile postoperator. Dintre cei 109 subiecți cu vederea culorilor normală, cărora li s-a implantat modelul de lentilă SB30AL la primul ochi operat și care au fost examinați la un interval curpins între 120 și 180 de zile postoperator, 107 (98,2%) au trecut testul de percepție a culorilor. Dintre cei 102 subiecți cu vederea culorilor normală, cărora li s-a implantat modelul de lentilă SA30AL la primul ochi operat și care au fost examinați la un interval cuprins între 120 și 180 de zile postoperator, 97 (95,1%) au trecut testul de percepție a culorilor. Nu au existat diferențe semnificative statistic între modelele SB30AL și SA30AL în ceea ce privește procentul de subiecți ce au trecut testul de percepție a culorilor după un interval cuprins între 120 și 180 de zile postoperator. În consecință, adăugarea agentului cromofor brevetat de Alcon nu afectează în mod negativ vederea colorată, la pacienții la care aceasta era anterior normală.
RATELE Nd:YAG PENTRU LENTILELE MONOBLOC AcrySof® Natural, model SB30AL După o perioadă de urmărire medie de 21,6 luni, trei (3) din cei 135 de subiecți (2,2%) cu implant SB30AL au necesitat capsulotomie posterioară Nd:YAG. După o perioadă de urmărire medie de 21,9 luni, doi (2) din cei 127 de subiecți (1,6%) cu implant SA30AL au necesitat capsulotomie posterioară Nd:YAG.
STUDIU CLINIC CU LENTILELE AcrySof® IQPornind de la modelul unor studii similare anterioare desfășurate cu lentilele intraoculare, subiecți adulți cu sănătate oculară generală bună (de ex: fără chirurgie oculară anterioară, tulburări vizuale degenerative care ar putea afecta semnificativ acuitatea vizuală, sau afecțiuni grave acute sau cronice care ar putea crește riscul pentru pacient), având cataractă bilaterală, au fost înrolați într-o investigație clinică controlată, randomizată dublu-orb, multicentrică privind implantul contralateral de lentile AcrySof® IQ, comparativ cu implantul de lentile sferice martor. Aberațiile oculare sferice au fost, din punct de vedere statistic, semnificativ mai reduse în cazul lentilelor AcrySof® IQ decât în cazul lentilelor martor. Rezultatele privind sensibilitatea la contrast au demonstrat o îmbunătățire postoperatorie semnificativă din punct de vedere statistic (după 3 luni), în favoarea ochilor cu implant de lentile AcrySof® IQ. În cazul ochilor cu implant de lentile AcrySof® IQ s-au constatat de asemenea îmbunătățiri semnificative din punct de vedere statistic și clinic, prin măsurarea vederii funcționale, conducerii de noapte simulate, testate în condiții severe – în special lumină orbitoare și ceață. Aceste rezultate reflectă faptul că lentila intraoculară AcrySof® IQ (cu o parte optică asferică pe un material platformă care conține un cromofor de filtrare a luminii albastre) asigură performanțe clinice benefice comparativ cu lentila intraoculară monofocală AcrySof® (fără parte optică asferică și cromofor de filtrare a luminii albastre).
LENTILELE AcrySof® IQ – ABERAȚII SFERICE ȘI TOTALE DE ORDIN SUPERIORAberația sferică oculară medie a ochilor cu lentile AcrySof® IQ a fost de aproximativ 0,1 micrometri. Figura 10 prezintă reducerea semnificativă din punct de vedere statistic în cazul aberațiilor sferice și totale de ordin superior observate, în favoarea lentilelor AcrySof® IQ. Figura 11 prezintă măsurătorile aberațiilor sferice în cazul tuturor ochilor, efectuate prin măsurători cu aberometrul cu front de undă, pe grupe de vârstă și tip de lentile. După cum se poate observa din diagramă, reducerea aberației sferice în cazul ochilor cu lentilă AcrySof® IQ a fost independentă de vârstă.
Figura 10Rădăcina medie pătrată (RMS) a aberațiilor sferice și totale de ordin superior
la 90-120 de zile după cel de-al 2-lea implant ocular
Figura 11Aberația sferică medie totală și în funcție de grupa de vârstă
la 90-120 de zile după cel de-al 2-lea implant ocular
LENTILELE AcrySof® IQ – ACUITATEA VIZUALĂ LA DISTANȚĂLentilele AcrySof® IQ și lentila martor au demonstrat clinic acuitate vizuală postoperatorie similară. Rezultatele privind acuitatea vizuală monoculară sunt prezentate în Figurile 12 și 13.
Figura 12LogMAR BCVA
Figura 13LogMAR UCVA
LENTILELE AcrySof® IQ - SENSIBILITATEA LA CONTRASTObiectivul primar al investigației clinice a fost să demonstreze superioritatea lentilei AcrySof® IQ în raport cu lentila martor, pe baza sensibilității medii la contrast măsurate postoperator în condiții mezopice, cu sau fără lumină orbitoare pentru oricare din cele două frecvențe spațiale (3 sau 6 cicluri / grad), utilizând aparatul Vector Vision CSV-1000 (cu grafic de luminanță de 3 cd/m2). La un subgrup de pacienți s-a aplicat de asemenea testul de acuitate funcțională a sensibilității la contrast (FACT) (cu grafic de luminanță de 3 cd/m2). În această investigație clinică, superioritatea lentilei AcrySof® IQ în raport cu lentila de control în condiții mezopice a fost demonstrată la 6 cicluri / grad, cu și fără lumină orbitoare (CSV-1000) și la 3 și 6 cicluri / grad, fără lumină orbitoare (FACT). Figurile 14 și 15 prezintă rezultatele privind sensibilitatea la contrast in condiții mezopice pentru toate frecvențele spațiale, testată atât pentru lentila AcrySof® IQ, cât și pentru lentila martor.
110
Figura 14Sensibilitatea la contrast în condiții mezopice (CSV-1000)
la 90-120 de zile după cel de-al 2-lea implant ocular
Figura 15Sensibilitatea la contrast în condiții mezopice (FACT) Substudiu
la minimum 90 de zile după cel de-al 2-lea implant ocular LENTILELE AcrySof® IQ – SIMULAREA CONDUCERII PE TIMP DE NOAPTEUn subgrup de pacienți a fost supus unui test într-un simulator validat pentru conducerea pe timp de noapte. Pacienții au fost testați monocular în condiții care simulează scenarii pentru conducere în mediu urban și rural, în condiții normale, de lumină orbitoare și de ceață.Cazul conducerii urbane pe timp de noapte a utilizat o mare varietate de semafoare, lumini ale vehiculelor, magazinelor, semne de circulație, pentru a crea nivelul ridicat de iluminare ambientală tipic în aceste condiții. Situația conducerii rurale pe timp de noapte a utilizat o cantitate minimă de iluminare ambientală. Au fost simulate viteze de conducere de circa 56 km/h și 88 km/h pentru mediul urban respectiv rural.Pacienților li s-a cerut să detecteze și identifice o serie de ținte în fiecare situație (urbană și rurală), inclusiv panouri luminoase de pe autostradă în culoarea alb pe verde, indicatoare de avertizare negru pe galben și pietoni. Pacienților li s-a cerut să răspundă când au văzut prima țintă, pentru a permite înregistrarea distanței de detectare. În continuare, pacienților li s-a cerut să răspundă când au putut distinge ținta (de ex: ce scria pe indicator, în ce direcție mergea pietonul, etc.), astfel încât să poată fi înregistrată viteza de identificare. Figurile 16 – 19 prezintă diferențele medii dintre lentilele AcrySof® IQ și lentilele martor în situații de conducere în mediu urban și rural, atât pentru detectarea cât și pentru identificarea distanțelor (de ex: media diferențelor intra-individuale).Lentilele AcrySof® IQ s-au comportat funcțional mai bine decât lentilele martor în 34 din cele 36 de condiții testate, reflectând îmbunătățirea atât în ceea ce privește detectarea cât și identificarea distanțelor, atât în cazul conducerii în mediu urban cât și rural, în condițiile variate de conducere testate (normale, cu lumină orbitoare, ceață). În plus, lentilele AcrySof® IQ s-au comportat semnificativ mai bine din punct de vedere statistic decât lentilele martor în 12 din aceste situații, cu cel mai mare impact și cel mai mare avantaj observate la detectarea și identificarea pietonilor în oraș (în condiții de lumină orbitoare și de ceață) și a indicatoarelor de avertizare din mediul rural (în condiții de lumină orbitoare și de ceață). În condiții de vizibilitate redusă (lumină orbitoare, ceață) în cazul condițiilor de oraș, distanța de vizibilitate mărită la 56 km/h asigură lentilelor AcrySof® IQ un timp de reacție suplimentar de peste 0,5 secunde la o țintă pieton, un pericol întâlnit mult mai des în mediile urbane. Această creștere este semnificativă din punct de vedere funcțional asigurând un timp mai lung pentru luarea măsurilor adecvate cum ar fi oprirea, evitarea pietonului/obstacolului, etc.(Green, 2000; McBride and Matson, 2004). În condițiile din mediul rural, distanța mărită de vizibilitate la viteza de 88 km/h asigură în cazul lentilelor AcrySof® IQ un timp suplimentar de peste 1 secundă pentru identificarea indicatoarelor de avertizare, o situație frecvent întâlnită în zonele rurale. O creștere de 0,5 secunde este semnificativă din punct de vedere funcțional permițând un timp mai lung de luare a măsurilor adecvate în timpul conducerii, care devine de o importanță critică pe timp de noapte în zone rurale nefamiliare unde luminatul ambiental este adesea absent. Au existat 6 pacienți din sub-studiu care prezentau postoperator degenerescență maculară sau PCO. Când acești pacienți au fost retrași din analiza capacității de conducere, diferența dintre lentilele intraoculare privind detectarea și identificarea țintelor pietonilor în condiții de lumină orbitoare în locații din oraș a scăzut sub pragul de 0,5 secunde pentru relevanță clinică. Când analizele inițiale au fost ajustate ca multiplicitate, diferența dintre lentilele intraoculare nu a mai fost semnificativă din punct de vedere statistic pentru detectarea în mediu urban a unui text în condiții de lumină orbitoare (valoarea p a lui Hommel = 0.0539) sau pentru detectarea în mediu rural a pietonilor în condiții de lumină orbitoare (valoarea p a lui Hommel = 0.0507).Aceste rezultate demonstrează vederea funcțională îmbunătățită și, în consecință, beneficii semnificative privind siguranța pentru șoferii mai în vârstă cu implant de lentile AcrySof® IQ și pentru ceilalți șoferi și pietoni participanți la trafic. Rezultatele acestui test demonstrează că lentilele AcrySof® IQ îmbunătățesc vederea funcțională, care la rândul său poate crește siguranța pacientului în cazul altor situații de viață, în condiții de vizibilitate redusă.
Figura 16Simulator pentru conducerea pe timp de noapte
Diferențe medii intra-individuale între distanțele de vizibilitate la detectarea obstacolelor, OrașLa cel puțin 90 de zile după operație
Lentilă intraoculară AcrySof® IQ – Lentilă martor (n = 44)
Figura 17Simulator pentru conducerea pe timp de noapte
Diferențe medii intra-individuale între distanțele de vizibilitate la identificarea obstacolelor, OrașLa cel puțin 90 de zile după operație
Lentilă intraoculară AcrySof® IQ – Lentilă martor (n = 44)
Figura 18Simulator pentru conducerea pe timp de noapte
Diferențe medii intra-individuale între distanțele de vizibilitate la detectarea obstacolelor, Mediu ruralLa cel puțin 90 de zile după operație
Lentilă intraoculară AcrySof® IQ – Lentilă martor (n = 44)
111
Figura 19Simulator pentru conducerea pe timp de noapte
Diferențe medii intra-individuale între distanțele de vizibilitate la identificarea obstacolelor, Mediu ruralLa cel puțin 90 de zile după operație
Lentilă intraoculară AcrySof® IQ – Lentilă martor (n = 44)
MOD DE PREZENTARELentilele intraoculare monobloc AcrySof® IQ împreună cu sistemul de implantare pre-încărcat UltraSertTM sunt livrate uscate, într-un ambalaj sterilizat cu oxid de etilenă, care trebuie desfăcut numai în condiții aseptice. (vezi “INSTRUCȚIUNI PRIVIND UTILIZAREA”).
DATA DE EXPIRARE Sterilitatea este garantată atâta timp cât ambalajul nu este deteriorat sau desfăcut. Data de expirare este clar indicată pe partea exterioară a ambalajului sistemului de implantare pre-încărcat UltraSert™. Orice lentilă păstrată după data de expirare trebuie returnată la Alcon Laboratories, Inc. (vezi “PROCEDURA PENTRU PRODUSE RETURNATE”).
PROCEDURA PENTRU PRODUSE RETURNATEÎn Statele unite, returnarea lentilelor este acceptată numai la schimb cu alte produse și nu se creditează. Toate retururile trebuie să fie însoțite de un “număr de produs returnat” acordat de Alcon Laboratories, Inc. și trebuie expediate pe căi identificabile. “Numărul de produs returnat” poate fi obținut contactând Departamentul de relații cu clienții de la Alcon. Acordarea acestui număr nu reprezintă acceptarea finală a produselor returnate. Pentru instrucțiuni detaliate referitoare la această procedură, inclusiv schimbul de produse, vă rugăm să contactați reprezentantul local de vânzări sau cel responsabil de relațiile cu clienții. În afara Statelor Unite, contactați reprezentanțele Alcon locale sau distribuitorii pentru relații privind procedura pentru produse returnate.
REFERINȚE 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., et al. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., et al. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
112
SIMBOLURI UTILIZATE PE ETICHETĂ
SIMBOL ROMÂNĂIOL Lentilă intraoculară
SP Monobloc
UV Ultraviolete
D Dioptrie
ØB Diametrul părții optice
ØT Diametrul total (lungimea totală)
2STERILIZE A nu se resteriliza
A nu se reutiliza
A se utiliza înainte de (AAAA-LL:an-lună)
STERILE EO Sterilizat cu oxid de etilenă
SN Număr de serie
Atenție: vezi instrucțiunile privind utilizarea
Producător
EC REP Reprezentant Autorizat în Uniunea Europeană
Limita superioară de temperatură
Deschideți aici
Citiți cu atenție instrucțiunile înainte de utilizare
Nu utilizați dacă ambalajul interior este deteriorat sau deschis
Atenție: În S.U.A., legislaţia federală limitează vânzarea acestui dispozitiv la comenzi emise de către sau la ordinul unui medic.
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 S.U.A.
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,Marea Britanie
Importator/Distribuitor:S.C. Alcon România S.R.L.Bd. Barbu Văcărescu nr. 301-311020276, București, RomâniaT +40 21 203 93 01; F +40 21 203 93 00
STERILNA akrilna, savitljiva sočiva iz jednog komada za zadnju komoru sa filterom za UV i plavo svetlo sa prethodno napunjenim sistemom za dopremanje UltraSert™
OPISAcrySof® IQ akrilna, savitljiva sočiva iz jednog komada za zadnju komoru sa filterom za UV i plavo svetlo su optički implantati za zamenu humanog kristalnog sočiva u vizualnoj korekciji afakije kod odraslih koja prati operaciju katarakte. AcrySof® IQ intraokularno sočivo (IOS) sa Alconovim filtrirajućim hromoforom za plavo svetlo filtrira svetlo na način koji je sličan humanom kristalnom sočivu u opsegu plave svetlosti talasne dužine od 400 – 475 nm (Boettner i Wolter, 1962). Pored standardnog filtriranja UV-svetlosti, IOS AcrySof® IQ smanjuje propuštanje plave svetlosti za 62% na 400 nm do 23% na 475 nm (vidi tabelu 1). Optički deo se sastoji od mekog akrilnog materijala sa velikim indeksom refrakcije. Ovaj materijal može da se savije pre umetanja. Sočivo se nežno ispravlja do pune veličine posle implantacije. Ima bikonveksnu optiku sa pomoćnim haptikom (slika 1). Zadnja asferična površina IOS AcrySof® IQ model AU00T0 dizajnirana je sa negativnom sferičnom aberacijom da kompenzuje pozitivnu sferičnu aberaciju prosečne rožnjače. Kvalitet slike modela AU00T0 IOS iz jednog komada (tj. funkcija modulacije transfera) ilustrovan je na slici 4. IOS AcrySof® IQ model AU00T0 iz jednog komada obezbeđena su u prethodno napunjenom sistemu za dopremanje UltraSert™ (slika 2) za pogodna, kontrolisana sredstva, da bi se ova sočiva pouzdano postavila u kapsularnu vrećicu. Fizičke karakteristike sočiva su sledeće:
Fizička karakteristika OpisOptički tip IOS iz jednog komada sa asferičnom optikom
Optički materijal Akrilni/metakrilni kopolimer koji filtrira UV i plavo svetlo
Gornja granica UV zracenja na 10% Transmisije (nm) UV (400) na 20.0 D (videti Sliku 3)
Indeks refrakcije 1.55
Optička jačina Za ponuđene opsege jačine vidi uputstvo o Alconovim proizvodima
Materijal haptika Akrilni/metakrilni kopolimer koji filtrira UV i plavo svetlo
Optički prečnik ØB (mm) 6.0
Ukupna dužina ØT (mm) 13.0
Ugao haptika 0º
Optičke dimenzije Vidi sliku 1
Pogledati uputstvo na engleskom jeziku Slika 1FIZIČKE KARAKTERISTIKESve dimenzije u milimetrima
ASPHERIC SURFACE = ASFERIČNA POVRŠINA
Pogledati uputstvo na engleskom jeziku Slika 2
Nozzle Tip = Vrh mlazniceDepth Guard = Zaštita dubine
Main Body = Glavni deo
SERBIAN
114
Pogledati uputstvo na engleskom jeziku Slika 3SPEKTRALNE KRIVE TRANSMISIJE
TRANSMITTANCE (%) = TRANSMISIJA (%)WAVELENGTH (nm) = TALASNA DUŽINA (nm)
6.0 D to 27.0 D range = Raspon od 6,0 D do 27,0 D4 yr – 53 yr old range, crystalline lens = Raspon od 4 - 53 godine, kristalno sočivo
NAPOMENE: • Krive gornje granice talasne dužine i spektralne transmisije koje su ovde prikazane predstavljaju raspon vrednosti
transmisije IOS koje je napravljeno od akrilnog/metakrilnog kopolimera sa vezanim UV-apsorberom i Alconovim hromoforom koji filtrira plavu svetlost.
• Mere su bile za direktnu transmisiju, koristeći otvor od 6 mm i disk debljine ekvivalentne optičkom centru. • Podaci o humanom sočivu Boettner i Wolter (1962).
Tabela 1Poređenje transmisije za IOS od 20,0 D, %
Model 400 nm 425 nm 450 nm 475 nm
SA60AT 21 86 88 88
AU00T0 8 34 49 68
Razlika u transmisiji (SA60AT – AU00T0) 13 52 39 20
Smanjenje transmisije sa AU00T0 (% od SA60AT) 62 60 44 23
Pogledati uputstvo na engleskom jeziku Slika 4Funkcija modulacije transfera modela AU00T0 (20,0 D)
MTF (%) = Funkcija modulacije transferaSpatial Frequency (Ip/mm) = Prostorna frekvencija (lp/mm)
ISO, 3-mm Aperture (see Note 1) = ISO, otvor od 3 mm (vidi napomenu 1)Modified, 3-mm Aperture (see Note 2) = Modifikovano, otvor od 3 mm (vidi napomenu 2)Modified, 5-mm Aperture (see Note 2) = Modifikovano, otvor od 5 mm (vidi napomenu 2)
NAPOMENE: 1. Kvalitet slike modela AU00T0 karakterisan je merenjem funkcije modulacije transfera u oku-modelu, opisanom u
ISO 11979-2. ISO 11979-2 zahteva merenja funkcije modulacije transfera koristeći samo otvor od 3 mm. 2. Pored toga, kvalitet slike modela AU00T0 karakterisan je merenjem funkcije modulacije transfera u oku-modelu
koje je koristilo simuliranu rožnjaču koja pokazuje tipičnu sfernu aberaciju odraslog čoveka. Uz upotrebu modifikovanog oka-modela urađena su merenja funkcije modulacije transfera koristeći otvore od 3 i od 5 mm.
MEHANIZAM DEJSTVAIOS AcrySof® IQ za zadnju komoru predviđena su da se postave u zadnju komoru oka, zamenjujući prirodno kristalno sočivo. Ova pozicija omogućava sočivu da funkcioniše kao refraktivni medijum u korekciji afakije. Asferična bikonveksna optika redukuje sfernu aberaciju u poređenju sa standardnom sfernom optikom u prosečnom oku. Efikasnost ovih sočiva u redukciji pojave retinalnih poremećaja nije utvrđena.
INDIKACIJEAcrySof® IQ IOS za zadnju komoru indikovana su za zamenu humanog sočiva da bi se postigla vizuelna korekcija afakije kod odraslih posle operacije katarakte (vidi UPOZORENJA). Ova sočiva su predviđena za postavljanje u kapsularnu vrećicu.
OPREZPacijenti sa bilo kojim od niže navedenih oboljenja ne bi mogli biti odgovarajući kandidati za intraokularno sočivo jer sočivo može da pogorša postojeće oboljenje, da bude u sukobu sa dijagnozom ili lečenjem oboljenja ili može da pretstavlja neprihvatljiv rizik za vid pacijenta. Pažljiva preoperativna procena i besprekoran klinički sud treba da vode hirurga prilikom odlučivanja koji je odnos koristi i rizika pre implantacije sočiva kod pacijenta sa jednim ili više ovih oboljenja: 1. Horoidalna hemoragija 2. Istovremeno jako oboljenje oka 3. Preveliki gubitak staklastog tela 4. Ekstremno plitka prednja komora 5. Mikroftalmija 6. Katarakta koja nije u vezi sa starošću 7. Zadnja kapsularna ruptura (koja sprečava fiksaciju IOS)
115
8. Jaka distrofija rožnjače 9. Jaka optička atrofija 10. Pozitivni pritisak koji ne može da se kontroliše 11. Zonularna separacija (koja sprečava fiksaciju IOS) 12. Nedostaci kod viđenja boja 13. Glaukom 14. Hronični uveitis 15. Dijabetska retinopatija 16. Klinički značajne promene makule/RPEStudije su pokazale da razlikovanje kod viđenja boja nema negativan uticaj kod osoba sa IOS AcrySof® Natural i normalnim viđenjem boja. Nije proučavan efekat na vid sa IOS AcrySof® Natural kod osoba sa nasleđenim defektima viđenja boja i sa stečenim defektima viđenja boja kao posledica okularnog oboljenja (npr. glaukom, dijabetska retinopatija, hronični uveitis i druga oboljenja retine ili očnog nerva).
UPOZORENJA 1. Kao i kod svake hirurške procedure postoji rizik. Moguće komplikacije koje prate operaciju katarakte ili hirurgiju
implantata mogu da uključe, ali nisu ograničene na sledeće: oštećenje endotela rožnjače, endoftalmitis, odvajanje retine, vitritis, cistoidni makularni edem, pupilarni blok, ciklitičnu membranu, prolaps irisa, hipopion i prolazni ili stalni glaukom.
2. Bezbednost i efektivnost implantata intraokularnog sočiva nije dokazana kod pacijenata sa prethodnim oboljenjima oka (hronični medikamentozni miozis, glaukom, ambliopija, dijabetska retinopatija, prethodna transplantacija rožnjače, prethodno odvajanje retine i/ili iritis itd.). Lekari koji razmatraju implantaciju sočiva kod takvih pacijenata treba da istraže primenu alternativnih metoda korekcije afakije i da uzmu u obzir implantaciju sočiva samo ako su alternative nezadovoljavajuće po pitanju zadovoljenja potreba pacijenta.
3. Dugoročni efekti implantacije intraokularnog sočiva nisu određeni. Stoga lekari treba redovno da prate pacijente posle operacije.
4. Pacijenti sa preoperativnim problemima kao što su oboljenje endotela rožnjače, abnormalna rožnjača, degeneracija makule, degeneracija retine, glaukom i hronični medikamentozni miozis možda neće imati oštrinu vida kao pacijenti bez takvih problema. Lekar mora da odredi benefite implantacije sočiva kada postoje takva oboljenja.
5. Sekundarna iridektomija za pupilarni blok mogla bi da se izbegne ako bi jedna ili više iridektomija bilo izvedeno u vreme implantacije IOS (Willis, et al., 1985).
6. Nije utvrđena bezbednost i efikasnost sočiva za zadnju komoru ako se ono postavi u prednju komoru. Implantacija sočiva za zadnju komoru u prednju komoru se u nekim slučajevima pokazala kao nebezbedna (Girard, et al., 1983).
7. Neke od negativnih reakcija koje su povezane sa implantacijom intraokularnih sočiva su sledeće: hipopion, intraokularna infekcija, akutna dekompenzacija rožnjače i dodatna hirurška intervencija. Dodatne hirurške intervencije uključuju, ali nisu ograničene na sledeće: repozicioniranje sočiva, zamena sočiva, aspiracija staklastog tela ili iridektomija za pupilarni blok, popravljanje rane koja vlaži i popravljanje odvajanja retine.
8. Mala decentralizacija sočiva, koja se javlja kada IOS ima usku ili malu optiku, može dovesti do toga da pacijent ima zaslepljenost ili druge poremećaje vida pod određenim uslovima svetla. Hirurzi treba da imaju u vidu ovu mogućnost pre implantacije IOS sa uskom ili malom optikom. Kada se implantira sočivo uske ili male optike preporučuje se da se izvede kapsuloreksa.
9. Postoperativna distenzija kapsularne vrećice sa varijabilnom plitkosti prednje komore i indukovane miopije vezuju se za tehnike kapsulorekse i implantaciju sočiva od PMMA, silikona i akrila u zadnju komoru (Holtz, 1992).
10. Treba biti na oprezu pre enkapsulacije sočiva da bi se izbegle decentralizacije ili dislokacije sočiva. Neki klinički slučajevi sugerišu da se enkapsulacija desi u roku od četiri nedelje.
11. Klinička studija sočiva iz jednog komada AcrySof® Natural (u tabelama 2 do 5) izvedena je sa sočivom koje je namenjeno samo za implantaciju u kapsularnu vrećicu. Ne postoje klinički podaci da prikažu njegovu bezbednost i efikasnost kod postavljanja u cilijarni sulkus.
Preporučuje se da se viskoelastika ukloni iz oka na kraju operacije sa naglaskom na prostor između zadnje kapsule i sočiva. Ovo može da se postigne nežnim pritiskanjem optike IOS na dole u zadnji deo vrhom I/A i korišćenjem standardnih tehnika irigacije/aspiracije da se ukloni viskoelastični agens iz oka. Ovo bi trebalo da protera bilo koju zaglavljenu viskoelastiku u prednji deo gde ona može lako da se usisa.
POSTUPANJE 1. Nemojte ni na koji način ponovo sterilisati ova intraokularna sočiva ili prethodno napunjen sistem za dopremanje
UltraSert™. 2. Nemojte čuvati intraokularna sočiva na temperaturama iznad 45° C (113° F). 3. Ako je potrebno, rukujte sočivima pažljivo da biste izbegli oštećenja površina sočiva ili haptika. 4. Nemojte pokušavati da preoblikujete haptik na bilo koji način. 5. Za implantaciju intraokularnog sočiva neophodna je velika veština hirurga. Hirurg treba da je posmatrao i/ili asistirao
kod brojnih implantacija i da je uspešno završio jedan ili više kurseva implantacije intraokularnog sočiva pre nego što pokuša da implantira intraokularna sočiva.
6. Sadržaj je sterilan osim ako je pakovanje otvarano ili oštećeno. 7. Prethodno napunjen sistem za dopremanje UltraSert™ je samo za jednokratnu upotrebu. Bacite prethodno
napunjen uređaj UltraSert™ posle upotrebe. 8. Koristite prethodno napunjen sistem za dopremanje UltraSert™ u operacionoj sali sa temperaturama između 18°
C (64° F) i 23° C (73° F).
116
IZRAČUNAVANJE JAČINE SOČIVAPreoperativni proračun potrebne jačine sočiva za ova intraokularna sočiva za zadnju komoru treba da bude određen iskustvom hirurga, onim, čemu on daje prednost, i nameravanim postavljanjem sočiva. Metode proračuna jačine sočiva opisane su u sledećoj literaturi:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700-7 12, 1993.Holladay, J.T. et al. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. et al. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
PREPORUČENA KONSTANTA “A”Preporučena konstanta “A” koja je data na spoljnoj etiketi predstavljena je kao smernica i ona je polazna tačka za proračun jačine implantata. Preporučuje se da razvijete svoju sopstvenu konstantu koja odgovara Vama, a bazirano na kliničkom iskustvu sa određenim modelima sočiva, hirurškim tehnikama, opremom za merenje i postoperativnim rezultatima. U Sjedinjenim državama - ako su potrebne dodatne informacije o proračunu jačine sočiva - stupite u kontakt sa Alcon Laboratories, Inc. na broju 1-800-TO-ALCON (1-800-862-5266). Van Sjedinjenih država stupite u kontakt sa lokalnim Alconovim biroima ili distributerima.
VISKOELASTIKA KOJA ODGOVARA ALCONU I BROJEVI PORUDŽBINAVidi uputstvo u ovom pakovanju. Treba da se koristi samo viskoelastika koja odgovara Alconu. Upotreba neodgovarajuće viskoelastike može izazvati oštećenje sočiva i moguće komplikacije tokom procesa implantacije. Za potpuni spisak viskoelastike za ovo sočivo koja odgovara Alconu stupite u kontakt sa svojim predstavnikom Alcona.
UPUTSTVA ZA UPOTREBUKorak 1. Koristite prethodno napunjen sistem za dopremanje UltraSert™ u operacionoj sali sa temperaturama između 18°
C (64° F) i 23° C (73° F).
Korak 2. Pročitajte etiketu na neotvorenoj kutiji da vidite model, jačinu, ispravnu konfiguraciju i rok upotrebe.
Korak 3. Posle otvaranja spoljne kartonske kutije pregledajte pakovanje uređaja da nema oštećenja. Ako se primete oštećenja upotrebite drugo IOS AcrySof® IQ i prethodno napunjen sistem za dopremanje UltraSert™. Zatim proverite da li se informacija o sočivu na etiketi uređaja (model, jačina, serijski broj) slaže sa informacijom na etiketi spoljne kutije.
Korak 4. Da izvadite prethodno napunjen sistem za dopremanje UltraSert™, uhvatite ćošak plastične posude, potpuno odvojite poklopac od materijala TYVEK† i prenesite uređaj u sterilno okruženje. Ako - posle pregledanja - izgleda da je mlaznica uređaja oštećena, ima čestice ili deformaciju, koristite drugo IOS AcrySof® IQ i prethodno napunjen sistem za dopremanje UltraSert™. Ako uređaj nije potpuno intaktan ili se vidi da su se klip ili klipna brava pomerili tokom transporta, koristite drugo IOS AcrySof® IQ i prethodno napunjen sistem za dopremanje UltraSert™ (vidi PROCEDURU VRAĆANJA ROBE).
†Reg. TM of E.I. DuPont De Nemours & Co.
Kada ste spremni da pripremite sočivo za prenos, izvedite korake 5, 6 i 7 uzastopno, sa minimalnim vremenom između koraka.
Korak 5. Potpuno uvesti viskoelastičnu kanulu, perpendikularno u odnosu na uređaj, kroz viskoelastični port, koja se nalazi u delu za zaustavljanje sočiva kao što se vidi na slici 5. Napuniti uređaj dok se ne vidi da viskoelastika teče do linije vrha mlaznice (vidi strelicu na slici 5). Ovo će zahtevati oko 0,2 ml viskoelastika. Koristiti samo Alconov viskoelastik koji odgovara za upotrebu sa prethodno napunjenim sistemom za dopremanje UltraSert™ koji je doveden na temperaturu operacione sale. Spisak viskoelastika koji su odgovarajući za Alcon nalazi se na uputstvu u ovom pakovanju.
Pogledati uputstvo na engleskom jeziku Slika 5
Viscoelastic Port = Viskoelastični port“Fill-To” Line = Linija do koje se puni
Korak 6. Ukloniti plavu kočnicu sočiva hvatanjem etikete sa strelicom i blagim savijanjem prema napred prilikom čega se ona podiže i udaljava od uređaja (slika 6a). Ukloniti plavu bravu cilindra hvatanjem i povlačenjem pravo na gore i od uređaja (slika 6b). Bacite kočnicu sočiva i bravu cilindra. Ne pokušavajte da dodate viskoelastik u uređaj kada je uklonjena kočnica sočiva da ne bi došlo do oštećenja sočiva. Ne pokušavajte da uvučete cilindar nakon što je uklonjena brava cilindra da ne bi došlo do oštećenja cilindra.
117
Pogledati uputstvo na engleskom jeziku Slike 6a i 6b
Korak 7. Pažljivo pomerati cilindar unapred nežnim, stalnim pokretom dok se prednja ivica optike ne izjednači sa linijom mlaznice (slika 7a). U ovom položaju plava opruga će dodirnuti providni deo nastavka za prste, kao što je prikazano na slici 7b. Za ovo pomeranje cilindra unapred potrebno je najmanje 7 sekundi. Ne dopustiti da neki deo sočiva izađe iz vrha mlaznice. Važno je da se cilindar ne pomera unapred odjednom jer može doći do oštećenja sočiva.
Pogledati uputstvo na engleskom jeziku Slike 7a i 7b
Fill-to Line = Linija do koje se puni
Korak 8. Kad je sočivo pomereno unapred do linije mlaznice treba ga vizuelno pregledati da bi se odredio položaj haptika. Cilindar treba da bude u kontaktu sa zadnjom ivicom optike. Nijedan deo sočiva ne treba da izađe iz mlaznice pre umetanja kroz otvor (slika 8). Kada se sočivo nađe u položaju linije mlaznice, ono treba da se implantira u roku od 3 minuta.
Pogledati uputstvo na engleskom jeziku Slika 8
“Proceed with full plunger advancement to fully deliver lens” = Nastaviti sa potpunim pomeranjem cilindra unapred da bi se potpuno dovelo sočivo.
“Proceed” = Nastaviti“Proceed with full plunger advancement to fully deliver lens” = Nastaviti sa potpunim pomeranjem cilindra unapred
da bi se potpuno dovelo sočivo.“Proceed with device rotations per Step 10” = Nastaviti sa rotacijama uređaja kao u koraku 10.
“Do not proceed” = Ne nastavljati.
Korak 9. Kada se uveri da je sočivo u ispravnom položaju i da je haptik ispravno savijen nastaviti sa implantacijom sočiva ubacivanjem vrha mlaznice u otvor do graničnika za dubinu (slika 9) i ciljati vrh mlaznice na otvor prednje kapsule.
Pogledati uputstvo na engleskom jeziku Slika 9
Depth Guard = Graničnik za dubinu
Korak 10. Pažljivo pomerati cilindar unapred nežnim, konstantnim pokretima. Ne pomerati cilindar unapred prebrzo jer može doći do oštećenja sočiva. Preporučuje se za isporučivanje sočiva da od pozicije vizuelnog pregleda treba da prođe najmanje 5 sekundi. Ako je vodeći haptik prav ili u petlji i produžen ispred sočiva, rotirati uređaj da bude nakrivljen ulevo pre pomeranja cilindra unapred da bi se osiguralo da je vodići haptik ispravno smešten u kapsularnoj vrećici. Kada vodeći haptik počne da izlazi iz vrha mlaznice postaviti vodeći haptik u kapsularnu vrećicu da bude ispravno orijentisan i nastaviti polako sa pomeranjem cilindra unapred da bi se dovelo sočivo. Kako optika izlazi iz mlaznice rotirati uređaj nazad do centra ili blago nakriviti udesno ako je potrebno da se obezbedi da se sočivo ispravi sa prednjom stranom gore u kapsularnoj vrećici.
Korak 11. Koristiti odgovarajući instrument za pozicioniranje da bi se pozicioniralo sočivo u kapsularnoj vrećici, ravno, paralelno sa irisom.
Korak 12. Baciti ceo uređaj. Ne koristiti ponovo prethodno napunjen sistem za dopremanje UltraSert™.
REGISTRACIJA PACIJENATA I IZVEŠTAVANJEIdentifikaciona karta pacijenta koja je u pakovanju treba da se popuni i dâ pacijentu, zajedno sa uputstvima da se karta čuva kao stalni zapis i da se pokaže svakom oftalmologu ili optičaru kojeg će pacijent u budućnosti konsultovati. U Sjedinjenim državama svaki pacijent mora biti registrovan kod Alcon Laboratories, Inc., odmah nakon implantacije jednog od ovih sočiva. Registracija je završena popunjavanjem registracione karte koja je priložena u kutiji i njenim slanjem na adresu kompanije Alcon Laboratories, Inc. Registracija pacijenta je od suštinske važnosti za program dugoročnog praćenja pacijenta i pomoći će Alcon Laboratories, Inc. da pruži odgovor na izveštaje o negativnim pojavama.Van Sjedinjenih država treba slediti lokalne zakone za registraciju proizvoda.Pojave, koje osnovano sugerišu da je sočivo možda izazvalo ili doprinelo smrti ili ozbiljnoj povredi, uključujući i pojave koje su rezultat nemogućnosti medicinskog uređaja da zadovolji navedeni učinak ili da na drugi način dâ rezultat kako je planirano, treba da se prijave kompaniji Alcon Laboratories, Inc. Ova informacija se zahteva od svih hirurga da bi se dokumentovali mogući dugoročni efekti implantiranja intraokularnog sočiva. Hirurzi treba da koriste sledeću adresu i broj telefona za prijavljivanje negativnih pojava koje su u vezi sa ovim intraokularnim sočivima:Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Besplatan poziv: 1-800-757-9780Hirurzi van Sjedinjenih država treba da kontaktiraju sa lokalnim Alcon-ovim biroima ili distributerima po pitanju izveštaja o negativnim pojavama.
118
Kliničke studije za savitljivo sočivo AcrySof® za zadnju komoru Izvedene su dve prospektivne, dobro kontrolisane kliničke studije slučajnih uzoraka na savitljivim sočivima AcrySof® iz jednog komada za zadnju komoru. Prva studija je vođena da demonstrira bezbednost i efikasnost sočiva za zadnju komoru iz jednog komada AcrySof® Natural model SB30AL (filtriranje UV i plave svetlosti) kao polaznog modela sočiva. Ovo je bila klinička studija slučajnih uzoraka koja je uključivala AcrySof® model SA30AL (samo UV-apsorpcija) kao kontrolno sočivo. Uključeni su samo podaci od prvog operisanog oka kod onih pacijenata koji su dobili intraokularno sočivo model SB30AL ili SA30AL. Druga klinička studija slučajnih uzoraka savitljivog IOS AcrySof® IQ iz jednog komada (sa hromoforima koji filtriraju UV i plavu svetlost) sa asferičnom optikom vs. savitljivo kontrolno sočivo AcrySof® iz jednog komada vođena je da proceni kliničke/funkcionalne benefite u odnosu na tradicionalni sferični dizajn.
Klinička studija za sočivo AcrySof® Natural iz jednog komada, model SB30AL Dobijeni rezultati kod pacijenata koji su uspešno praćeni minimum godinu dana posle operacije osnovano uveravaju da je sočivo AcrySof® Natural iz jednog komada model SB30AL bezbedan i efikasan uređaj za vizuelnu korekciju afakije.
Populacija pacijenata za sočivo AcrySof® Natural iz jednog komada, model SB30AL Populacija kod koje je implantiran model SB30AL u barem prvo operisano oko u ovoj bilateralnoj studiji sastoji se od 70,6% žena i 29,4% muškaraca. Populacija kod koje je implantiran model intraokularnog sočiva SA30AL (kontrolni) sastoji se od 60,5% žena i 39,5% muškaraca. Razlažući prema rasi populacije za model SB30AL, 95,3% su bili evropeidna rasa, a 4.7% crnci. Kontrolna populacija (SA30AL) su bili 96,6% evropeidna rasa, 2% crnci i 1,4% drugi. Prosečna starost totalne populacije, koja je dobila model SB30AL u barem prvom operisanom oku, je 72,9 godina. Slično tome, prosečna starost totalne populacije, koja je dobila model SA30AL (kontrolni) je 71,9 godina.
Oštrina vida kod sočiva iz jednog komada AcrySof® Natural, model SB30AL U tabeli 2a je dat pregled oštrine vida koja je postignuta minimum godinu dana posle operacije kod pacijenata koji nisu imali preoperativnu očnu patologiju, abnormalne rožnjače ili degeneraciju makule u bilo koje vreme (“najbolji slučaj”), a oštrina vida koja je postignuta kod ukupne populacije prikazana je u tabeli 3a. Kontrolni podaci se nalaze u istim setovima podataka u tabelama 2b odn. 3b. Nije bilo statistički značajne razlike oštrine vida između modela SB30AL i kontrolnog sočiva, model SA30AL, niti u “najboljem slučaju” niti kod setova ukupnih podataka.
Pogledati uputstvo na engleskom jeziku Tabela 2aNajbolja korigovana oštrina vida kod populacije “najboljeg slučaja” najmanje godinu dana posle operacije, IOS
AcrySof® Natural SB30AL
Oštrina
Broj osoba
20/20 ili bolje 20/25 20/30 20/40 20/40 ili
bolje> 20/40 to < 20/80 > 20/80
N N % N % N % N % N % N % N %
Starosna kategorija
Pogledati uputstvo na engleskom jeziku Tabela 2bNajbolja korigovana oštrina vida kod populacije “najboljeg slučaja” najmanje godinu dana posle operacije, IOS
AcrySof® SA30AL, kontrolno
Oštrina
Broj osoba
20/20 ili bolje 20/25 20/30 20/40 20/40 ili
bolje> 20/40 to < 20/80 > 20/80
N N % N % N % N % N % N % N %
Starosna kategorija
Pogledati uputstvo na engleskom jeziku Tabela 3aNajbolja korigovana oštrina vida kod ukupne populacije pacijenata najmanje godinu dana posle operacije, IOS
AcrySof® Natural SB30AL
Oštrina
Broj osoba
20/20 ili bolje 20/25 20/30 20/40 20/40 ili
bolje> 20/40 to < 20/80 > 20/80
N N % N % N % N % N % N % N %
Starosna kategorija
119
Pogledati uputstvo na engleskom jeziku Tabela 3bNajbolja korigovana oštrina vida kod ukupne populacije pacijenata najmanje godinu dana posle operacije, IOS
AcrySof® SA30AL, kontrolno
Oštrina
Broj osoba
20/20 ili bolje 20/25 20/30 20/40 20/40 ili
bolje> 20/40 to < 20/80 > 20/80
N N % N % N % N % N % N % N %
Starosna kategorija
Kumulativne negativne pojave kod sočiva iz jednog komada AcrySof® Natural model SB30AL Kumulativne stope ovih negativnih pojava u okviru perioda od minimum godinu dana posle operacije za pacijente sa sočivom iz jednog komada AcrySof® Natural model SB30AL i model SA30AL prikazane su u tabeli 4. Nije bilo statistički značajnih razlika između modela SB30AL i SA30AL kod dela pacijenata koji su imali bilo koju od kumulativnih negativnih pojava.
Tabela 4Kumulativne negativne pojave minimum godinu dana posle operacije
Sekundarna, ponovna hirurška intervencijaUklanjanje preostalog korteksaEksplantacija (dislokacije zbog rupture kapsule)Krioterapija zbog popravljanja poderane retineParacenteza da se smanji intraokularni pritisakLaserski tretman fokusaFotodinamička terapijaEksplantacija zbog biometrijske greške
51 1 11100
3,3 0,7
0,70,70,70,70,00,0
20000011
1,40,00,00,00,00,00,70,7
0,4482
Hifema 0 0,0 0 0,0 -
*p-vrednosti od Fišerovog testa tačne verovatnoće, poredeći model Model SB30AL sa modelom SA30AL.
Perzistentne negativne pojave kod sočiva iz jednog komada AcrySof® Natural model SB30AL Stope perzistentnosti ovih negativnih pojava u periodu od najmanje godinu dana posle operacije za pacijente sa sočivom iz jednog komada AcrySof® Natural model SB30AL i kontrolnim modelom SA30AL prikazane su u tabeli 5. Nije bilo statistički značajnih razlika između modela SB30AL i SA30AL kod dela pacijenata koji su imali bilo koju od perzistentnih negativnih pojava.
120
Tabela 5Perzistentne neželjene pojave minimum godinu dana posle operacije
IOS AcrySof® Natural SB30AL i SA30AL kontrolno
SB30AL (N= 138)
SA30AL(N= 127) p-vrednost*
N % N %
Tip negativne pojave
0 0 1 0,8 0,4792Trajno Edem rožnjače
Iritis 0 0 0 0,0 -
Edem makule 2 1,4 1 0,8 1,0000
Vitritis 0 0 0 0,0 -
Povišen intraokularni pritisak koji zahteva lečenje 0 0 0 0,0 -
* p-vrednosti od Fišerovog testa tačne verovatnoće, poredeći model Model SB30AL sa modelom SA30AL.
Percepcija boja kod sočiva iz jednog komada AcrySof® Natural model SB30AL Testiranje percepcije boja primenom panel testa D-15 prema Farnsworth-u rađeno je od 120 do 180. dana postoperativnog perioda. Od 109 pacijenata sa normalnim viđenjem boja kojima je implantiran model SB30AL u prvo operisano oko i koji su ispitani 120 do 180. dana posle operacije, 107 (98,2%) je prošlo test percepcije boja. Od 102 pacijenta sa normalnim viđenjem boja kojima je implantiran model SA30AL u prvo operisano oko i koji su ispitani 120 do 180 dana posle operacije, 97 (95,1%) je prošlo test percepcije boja. Nije bilo statistički značajnih razlika između modela SB30AL i SA30AL za procenat pacijenata koji je prošao test percepcije boja 120 do 180 dana posle operacije. Stoga dodatak njegovog hromofora ne utiče negativno na viđenje boja kod pacijenata sa normalnim viđenjem boja.
Vrednosti Nd:YAG lasera za sočivo iz jednog komada AcrySof® Natural model SB30AL Sa prosečnom kontrolom od 21,6 meseci, tri (3) od 135 pacijenata (2,2%) kojima je implantirano SB30AL imala su zadnju kapsulotomiju Nd:YAG laserom. Sa prosečnom kontrolom od 21,9 meseci, dva (2) od 127 pacijenata (1,6%) kojima je implantirano SA30AL imala su zadnju kapsulotomiju Nd:YAG laserom.
Klinička studija za sočivo AcrySof® IQ U skladu sa dizajnom sličnih, prethodno urađenih studija IOS, odrasli pacijenti sa dobrim opštim okularnim zdravljem (bez ranijih operacija oka, degenerativnih poremećaja vida koji bi značajno uticali na oštrinu vida ili jakih akutnih ili hroničnih oboljenja koja bi mogla da povećaju rizik po pacijenta) koji imaju bilateralne katarakte učestvovali su u kontrolisanom, dvostruko maskiranom, multicentričnom, kontralateralnom kliničkom ispitivanju slučajnih uzoraka za sočivo AcrySof® IQ vs. sferično kontrolno sočivo. Okularne sferne aberacije su statistički bile znatno manje sa sočivom AcrySof® IQ nego sa kontrolnim sočivom. Rezultati osetljivosti na kontrast pokazali su statistički značajno postoperativno poboljšanje (u 3. mesecu) u korist pacijenata kojima je implantirano sočivo AcrySof® IQ. Pacijenti kojima je implantirano sočivo AcrySof® IQ takođe su iskusili, statistički i klinički, značajna poboljšanja kod merenja funkcionalnosti vida, simulirane noćne vožnje, testirane u nekoliko različitih uslova, naročito sa zaslepljujućim svetlom i maglom. Ovi rezultati pokazuju da IOS AcrySof® IQ (asferična optika na platformi materijala koji sadrži hromofor koji filtrira plavu svetlost) obezbeđuju klinički učinak sa benefitom u poređenju sa monofokalnim IOS AcrySof® (bez asferične optike i hromofora koji filtrira plavu svetlost).
Sočivo AcrySof® IQ Lens – sferna i totalna aberacija višeg redaProsečna okularna sferna aberacija kod očiju sa AcrySof® IQ bila je oko 0,1 mikrometar. Slika 10 prikazuje statistički značajno smanjenje sferne i totalne aberacije višeg reda u korist sočiva AcrySof® IQ. Slika 11 daje merenja prosečne aberacije svih očiju sa merenjima talasnog fronta aberometrom, za sočivo i za starosnu grupu. Kako je prikazano na ovom grafikonu, smanjenje sferne aberacije kod očiju sa sočivom AcrySof® IQ nije zavisilo od starosti pacijenata.
Pogledati uputstvo na engleskom jeziku Slika 10Sferni i totalni viši red - kvadratna sredina
90-120 dana posle 2. očnog implantata
Pogledati uputstvo na engleskom jeziku Slika 11Srednja sferna aberacija - ukupno i prema starosnoj grupi
90-120 dana posle 2. očnog implantata
Sočivo AcrySof® IQ – oštrina vida na daljinuSočivo AcrySof® IQ i kontrolno sočivo dala su sličnu postoperativnu oštrinu vida. Rezultati monokularne oštrine vida prikazani su na slikama 12 i 13.
121
Pogledati uputstvo na engleskom jeziku Slika 12LogMAR BCVA
Pogledati uputstvo na engleskom jeziku Slika 13LogMAR UCVA
Sočivo AcrySof® IQ - osetljivost na kontrastPrimarni cilj kliničkog istraživanja bio je da se demonstrira superiornost sočiva AcrySof® IQ nad kontrolnim sočivom preko srednje osetljivosti na kontrast koja je merena posle operacije pod mezopskim uslovima sa ili bez zaslepljenja svetlom na bilo kojoj od dve prostorne frekvencije (3 ili 6 ciklusa po stepenu) koristeći Vector Vision CSV-1000 (sa dijagramom osvetljenosti od 3 cd/m2). Kod dela pacijenata je takođe urađen funkcionalni test oštrine i kontrasta (FACT; sa dijagramom osvetljenosti od 3 cd/m2). U ovom kliničkom istraživanju je superiornost sočiva AcrySof® IQ nad kontrolnim sočivom pod mezopskim uslovima demonstrirana u 6 ciklusa po stepenu i sa i bez zaslepljujuće svetlosti (CSV-1000) a u 3 i 6 ciklusa po stepenu bez zaslepljujuće svetlosti (FACT). Slike 14 i 15 prikazuju rezultate mezopske osetljivosti na kontrast na svim prostornim frekvencijama, što je testirano i kod sočiva AcrySof® IQ i kod kontrolnog sočiva.
Pogledati uputstvo na engleskom jeziku Slika 14Mezopska osetljivost na kontrast (CSV-1000)
90-120 dana posle 2. implantata
Pogledati uputstvo na engleskom jeziku Slika 15Mezopska osetljivost na kontrast (FACT) podstudija
Minimum 90 dana posle 2. implantata
Sočivo AcrySof® IQ – simulacija noćne vožnjeDeo pacijenata je podvrgnut testiranju na validiranom simulatoru noćne vožnje. Pacijenti su testirani monokularno pod uslovima koji simuliraju gradsko i ruralno okruženje i pod normalnim uslovima, zatim uslovima zaslepljenosti i magle. Noćna vožnja kroz grad uključuje razna ulična svetla, svetla automobila, izloga i znakova da bi se stvorio visok nivo ambijentalne osvetljenosti koji je tipičan za ove uslove. Ruralna noćna vožnja ima minimalno ambijentalno osvetljenje. Korišćene su simulirane brzine vožnje od oko 56 km/h odn. 88 km/h za grad odn. za ruralni predeo.Pacijenti su zamoljeni da primete i identifikuju serije meta u svakom predelu, uključujući belo-zelene znake autoputa, crno-žute znakove upozorenja i pešake. Pacijenti su zamoljeni da daju znak kada vide prvu metu, dozvoljavajući da se snimi detekcija rastojanja. Zatim su pacijenti zamoljeni da daju znak kada mogu da prepoznaju metu (šta piše na znaku, u kom je pravcu hodao pešak itd.) tako da može da se snimi detekcija rastojanja. Slike 16 do 19 prikazuju prosečne razlike između sočiva AcrySof® IQ i kontrolnog sočiva u vožnji u gradu i po ruralnom krajoliku za rastojanja primećivanja i identifikacije (npr. sredinu intraindividualnih razlika). Sočivo AcrySof® IQ je imalo bolju funkciju nego kontrolno sočivo u 34 od 36 testiranih uslova, odražavajući poboljšanje i kod rastojanja otkrivanja i kod rastojanja identifikacije, kako u gradu tako i u ruralnom krajoliku pod raznim testiranim uslovima (normalni, zaslepljenje, magla). Osim toga, sočivo AcrySof® IQ je statistički imalo značajno bolje performanse nego kontrolno sočivo u 12 ovih uslova, sa najznačajnijim uticajem i najvećom primećenom prednošću kod otkrivanja i identifikacije pešaka u gradu (u uslovima zaslepljenosti i magle) i ruralnih znakova upozorenja (u uslovima zaslepljenosti i magle). U uslovima smanjene vidljivosti (zaslepljenost, magla) u gradu povećano rastojanje vidljivosti pri brzini od 56 km/h obezbeđuje sočivu AcrySof® IQ više od 0.5 sekundi dodatnog vremena da se reaguje na pešaka - nezgoda koja je češća u gradu. Ovo povećanje od 0,5 sekundi je funkcionalno značajno zato što daje više vremena da se preduzmu odgovarajući koraci kao što je zaustavljanje, izbegavanje itd. (Green, 2000; McBride i Matson, 2004). U svim uslovima u ruralnom krajoliku povećano rastojanje viđenja pri brzini od 88 km/h obezbeđuje sočivu AcrySof® IQ više od 1 sekunde dodatnog vremena da identifikuje znake upozorenja - situacija koja je česta u ruralnim oblastima. Povećanje od 0,5 sekundi je funkcionalno značajno jer ima više vremena da se reaguje na odgovarajući način tokom vožnje, što postaje kritično noću u nepoznatim ruralnim oblastima gde često nema ambijentalne osvetljenosti. Bilo je 6 pacijenata u podstudiji koji su postoperativno imali degeneraciju makule ili opacifikaciju zadnje kapsule. Kada su ovi pacijenti sklonjeni sa analize vožnje, nije postignuta razlika između IOS za otkrivanje i identifikaciju pešaka u uslovima zaslepljenosti u gradu od 0,5 sekundi za kliničku relevantnost. Kada je originalna analiza prilagođena za brojnost, razlika između IOS nije više bila statistički značajna za otkrivanje teksta u gradu pod zaslepljenošću (Hommel-ova p-vrednost = 0,0539) ili otkrivanje pešaka u ruralnom krajoliku pod zaslepljenošću (Hommel-ova p-vrednost = 0,0507).Ovi rezultati demonstriraju poboljšani funkcionalni vid i, može se reći, značajne benefite po pitanju bezbednosti za starije vozače sa sočivom AcrySof® IQ i za druge vozače i pešake sa kojima dele ulicu. Rezultati ovog testa pokazuju da sočivo AcrySof® IQ poboljšava funkcionalni vid, što zatim može da poboljša bezbednost pacijenta u drugim životnim situacijama u uslovima slabe vidljivosti.
Pogledati uputstvo na engleskom jeziku Slika 16Simulator noćne vožnje
Srednje intraindividualne razlike kod rastojanja viđenja, otkrivanje, gradMinimum 90 dana postoperativno
IOS AcrySof® IQ – kontrola (n = 44)
122
See English language Slika 17Simulator noćne vožnje
Srednje intraindividualne razlike kod rastojanja viđenja, identifikacija, gradMinimum 90 dana postoperativno
IOS AcrySof® IQ – kontrola (n = 44)
See English language Slika 18Simulator noćne vožnje
Srednje intraindividualne razlike kod rastojanja viđenja, otkrivanje, ruralnoMinimum 90 dana postoperativno
IOS AcrySof® IQ – kontrola (n = 44)
See English language Figure 19Simulator noćne vožnje
Srednje intraindividualne razlike kod rastojanja viđenja, identifikacija, ruralno Minimum 90 dana postoperativno
IOS AcrySof® IQ – kontrola (n = 44)
KAKO SE ISPORUČUJEOva intraokularna sočiva za zadnju komoru se isporučuju suva, u prethodno napunjenom sistemu za dopremanje UltraSert™, u primarnom sterilnom pakovanje, a terminalno sterilisana etilen-oksidom. Primarno sterilno pakovanje sme da se otvori samo u aseptičnim uslovima (vidi UPUTSTVO ZA UPORTREBU).
ROK TRAJANJASterilnost je zagarantovana osim ako je primarno sterilno pakovanje oštećeno ili otvoreno. Rok trajanja je jasno naznačen na etiketi na spoljnoj kutiji prethodno napunjenog sistema za dopremanje UltraSert™. Svako sočivo, koje se čuva posle isteka roka trajanja, treba da se vrati u Alcon Laboratories, Inc. (vidi PROCEDURU VRAĆANJA ROBE).
PROCEDURA VRAĆANJA ROBEU Sjedinjenim državama se vraćena sočiva prihvataju samo za zamenu za druge proizvode, a ne povraćaj novca. Sav povraćaj mora biti praćen brojem Alcon Laboratories, Inc. za vraćenu robu i treba da se šalje tako da se pošiljka može pratiti. Broj za vraćenu robu se dobija prilikom kontaktiranja odeljenja Alcon Laboratories, Inc. za rad sa kupcima. Izdavanje ovog broja ne podrazumeva konačno prihvaćanje vraćenih proizvoda. Za detaljna uputstva u vezi sa procedurom, uključujući i zamenu, stupiti u kontakt sa distributerom ili odeljenjem za kontakt sa kupcima.Van Sjedinjenih država kontaktirati sa lokalnim Alcon-ovim biroima ili distributerima radi procedure za vraćanje robe.
LITERATURA 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., et al. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., et al. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
123
SIMBOLI NA ETIKETI
SIMBOL SRPSKIIOL Intraokularno sočivo
SP Jedan komad
UV Ultraljubičasto
D Dioptrijska jačina
ØB Prečnik tela (optički prečnik)
ØT Ukupni prečnik (ukupna dužina)
2STERILIZE Ne sterilisati ponovo
Samo za jednokratnu upotrebu.
Iskoristiti do
STERILE EO Sterilisano etilen oksidom
SN Serijski broj
Pažnja
Proizvođač
EC REP Ovlašćeni zastupnik u EZ
Gornja temperaturna granica
Ovde otvoriti
Konsultovati uputstvo za upotrebu
Ne koristiti ako je sterilno pakovanje otvoreno ili oštećeno
Upozorenje: Po saveznom zakonu (SAD), ovaj instrument sme biti prodat isključivo lekaru ili po njegovoj porudžbini.
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 USA
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,United Kingdom
Asférické vnútroočné šošovky AcrySof® IQ so zavádzacím systémom UltraSert™ Pre-loaded Delivery System
STERILNÉ akrylátové foldovateľné jednokusové zadnokomorové šošovky filtrujúce UV a modré svetlos pomôckou UltraSert™ Pre-loaded Delivery System
POPISAcrySof® IQ, akrylátové foldovateľné jednokusové zadnokomorové šošovky filtrujúce UV a modré svetlo, sú optické implantáty na nahradenie ľudskej očnej šošovky pri zrakovej korekcii afakie u dospelých pacientov po chirurgii katarakty. Vnútroočná šošovka AcrySof® IQ s chromofórom filtujúcim modré svetlo vo vlastníckom práve spoločnosti Alcon filtruje svetlo spôsobom, ktorý sa približuje ľudskej očnej šošovke v rozsahu vlnovej dĺžky modrého svetla 400 až 475 nm (Boettner a Wolter, 1962). Okrem štandardného filtrovania UV-svetla vnútroočná šošovka AcrySof® IQ znižuje priepustnosť vlnových dĺžok modrého svetla zo 62 % pri vlnovej dĺžke 400 nm na 23 % pri vlnovej dĺžke 475 nm (pozri Tabuľku 1). Optická časť pozostáva z mäkkého akrylátového materiálu s vysokým refrakčným indexom. Tento materiál je schopný sa pred vložením poskladať. Po implantácii sa šošovka rovnomerne rozvinie na svoju úplnú veľkosť. Šošovka má bikonvexnú optickú časť s podpornými haptickými časťami (Obrázok 1). Zadný asférický povrch vnútroočnej šošovky AcrySof® IQ, Modelu AU00T0 je navrhnutý s negatívnou sférickou aberáciou, aby kompenzoval pozitívnu sférickú aberáciu priemernej rohovky. Zobrazovacia kvalita vnútroočnej jednokusovej šošovky AcrySof® IQ, Modelu AU00T0 (t.j. modulačná transferová funkcia) je znázornená na Obrázku 4. Jednokusové vnútroočné šošovky AcrySof® IQ, Model AU00T0 sa dodávajú v zavádzacom systéme UltraSert™ Pre-loaded Delivery System (Obrázok 2) pre pohodlný a regulovaný spôsob spoľahlivého umiestnenia týchto šošoviek do kapsulárneho vaku. Fyzikálne vlastnosti šošovky sú nasledujúce:
Fyzikálne charakteristiky PopisOptický typ Jednokusová vnútroočná šošovka s asférickou optickou časťou
Optický materiál Kopolymér akrylátu/metakrylátu filtrujúci ultrafialové a modré svetlo
UV hranica pri 10% transmitancii (nm) UV (400) pri 20,0 D (Pozri Obrázok 3)
Refrakčný index 1,55
Optická mohutnosť Pre zistenie dostupnej škály optickej mohutnosti pozri príručku produktov Alcon
Konfigurácia haptických častí STABLEFORCE® L-modifikované haptické časti
Haptický materiál Kopolymér akrylátu/metakrylátu filtrujúci ultrafialové a modré svetlo
Optický priemer ØB (mm) 6,0
Celková dĺžka ØT (mm) 13,0
Haptický uhol 0º
Optické rozmery Pozri Obrázok 1
Pozri anglický jazyk Obrázok 1FYZIKÁLNE CHARAKTERISTIKY
Všetky rozmery v milimetroch
ASPHERIC SURFACE = ASFÉRICKÝ POVRCH
SLOVAK
125
Pozri anglický jazyk Obrázok 2Nozzle Tip = Špička nadstavca
Depth Guard = Zariadenie zabezpečujúce správnu hĺbkuMain Body = Hlavné teleso
Pozri anglický jazyk Obrázok 3KRIVKY SPEKTRÁLNEJ TRANSMITANCIE
TRANSMITTANCE (%) = TRANSMITANCIA (%)WAVELENGTH (nm) = VLNOVÁ DĹŽKA (nm)
6.0 D to 27.0 D range = Rozmedzie od 6,0 D do 27,0 D4 yr – 53 yr old range, crystalline lens = Rozmedzie 4 – 53 rokov, ľudská očná šošovka
POZNÁMKY: • Uvedené medzné vlnové dĺžky a krivky spektrálnej transmitancie predstavujú rozmedzie hodnôt transmitancie
vnútroočných šošoviek vyrobených z kopolyméru akrylátu/metakrylátu s naviazaným UV-absorbérom a chromofórom filtrujúcim modré svetlo vo vlastníckom práve spoločnosti Alcon.
• Transmitancia bola meraná priamo pomocou 6 mm clony a kotúčika s hrúbkou ekvivalentnou centrálnej optickej časti.
• Údaje o ľudskej šošovke podľa Boettner a Wolter (1962).
Tabuľka 1Porovnanie transmitancie pre vnútroočné šošovky s optickou mohutnosťou 20,0 D, %
Model 400 nm 425 nm 450 nm 475 nm
SA60AT 21 86 88 88
AU00T0 8 34 49 68
Rozdiel transmitancie (SA60AT – AU00T0) 13 52 39 20
Zníženie transmitancie s AU00T0 (% z SA60AT) 62 60 44 23
Pozri anglický jazyk Obrázok 4Modulačná transferová funkcia Modelu AU00T0 (20,0 D)
ISO, 3-mm Aperture (see Note 1) = ISO, 3 mm clona (pozri Poznámku 1)Modified, 3-mm Aperture (see Note 2) = Modifikovaná, 3 mm clona (pozri Poznámku 2)Modified, 5-mm Aperture (see Note 2) = Modifikovaná, 5 mm clona (pozri Poznámku 2)
POZNÁMKY: 1. Zobrazovacia kvalita Modelu AU00T0 bola charakterizovaná meraním modulačnej transferovej funkcie (MTF) v
modelovom oku opísanom v ISO 11979-2. ISO 11979-2 vyžaduje merania MTF s použitím len 3 mm clony. 2. Okrem toho, zobrazovacia kvalita Modelu AU00T0 bola charakterizovaná meraním MTF v modelovom oku, pri
ktorom sa využívala simulovaná rohovka vykazujúca typickú sférickú aberáciu oka dospelého človeka. Pri použití modifikovaného modelu oka sa merania MTF uskutočnili s použitím 3 mm a 5 mm clony.
MECHANIZMUS ÚČINKU Zadnokomorové vnútroočné šošovky AcrySof® IQ sú určené na umiestnenie v zadnej komore oka ako náhrada prirodzenej očnej šošovky. Toto umiestnenie umožňuje, aby šošovka fungovala ako refrakčné médium pri korekcii afakie. Asférická bikonvexná optika znižuje sférickú aberáciu v porovnaní so štandardnou sférickou optikou v priemernom oku. Schopnosť týchto šošoviek znižovať výskyt ochorení sietnice sa neskúmala.
INDIKÁCIE Zadnokomorové vnútroočné šošovky AcrySof® IQ sú určené ako náhrada ľudskej šošovky na dosiahnutie zrakovej korekcie afakie u dospelých pacientov po chirurgii katarakty (pozri UPOZORNENIA). Tieto šošovky sú určené na umiestnenie v kapsulárnom vaku.
ZVLÁŠTNE OPATRENIA Pacienti s ktorýmkoľvek z nižšie uvedených stavov nemusia byť vhodnými kandidátmi na implantáciu vnútroočnej šošovky, pretože šošovka by mohla zhoršiť existujúci stav, interferovať s diagnózou alebo liečbou stavu, alebo predstavovať neprimerané riziko pre pacientov zrak. Očný chirurg musí vykonať dôkladné predoperačné zhodnotenie a rozvážne posúdiť klinický stav za účelom zváženia pomeru prínosu a rizika pred implantáciou šošovky pacientovi s jedným alebo viacerými z nasledujúcich stavov:
126
1. Choroidálna hemorágia 2. Sprievodné závažné očné ochorenie 3. Nadmerná strata sklovca 4. Extrémne plytká predná komora 5. Mikroftalmus 6. Katarakta nesúvisiaca s vekom 7. Ruptúra zadného puzdra (zabraňujúca fixácii vnútroočnej šošovky) 8. Ťažká dystrofia rohovky 9. Ťažká atrofia očného nervu 10. Nekontrolovateľný vysoký tlak 11. Zonulárna separácia (zabraňujúca fixácii vnútroočnej šošovky) 12. Deficit farebného videnia 13. Glaukóm 14. Chronická uveitída 15. Diabetická retinopatia 16. Klinicky signifikantné makulárne/RPE zmenyŠtúdie ukázali, že rozlišovanie farebného videnia nie je nepriaznivo ovplyvnené u jednotlivcov používajúcich vnútroočnú šošovku AcrySof® Natural s normálnym farebným videním. Efekt vnútroočnej šošovky AcrySof® Natural na zrak jednotlivcov s dedičnými poruchami farebného videnia a získanými poruchami farebného videnia spôsobenými sekundárne očnými chorobami (napríklad glaukóm, diabetická retinopatia, chronická uveitída a ďalšie ochorenia sietnice alebo optického nervu) sa neskúmali.
UPOZORNENIA 1. Tak, ako pri ktoromkoľvek chirurgickom zákroku, aj tu existuje určité riziko. Možné nežiaduce udalosti spojené
s operáciou katarakty alebo s implantačným chirurgickým zákrokom môžu zahŕňať, ale nie sú obmedzené na nasledujúce: poškodenie endotelu rohovky, endoftalmitída, odlúpenie sietnice, zápal sklovca, cystoidný makulárny edém, edém rohovky, pupilárny blok, cyklitická membrána, prolaps dúhovky, hypopyón a prechodný alebo pretrvávajúci glaukóm.
2. Bezpečnosť a účinnosť vnútroočných implantovaných šošoviek nebola preukázaná u pacientov s preexistujúcimi očnými stavmi (chronická mióza vyvolaná liekmi, glaukóm, amblyopia, diabetická retinopatia, predchádzajúca transplantácia rohovky, predchádzajúce odlúpenie sietnice a/alebo iritída, atď.). Pri zvažovaní implantácie šošovky u týchto pacientov je potrebné zvážiť použitie alternatívnych metód korekcie afakie a implantácia šošovky by sa mala vykonať len v prípade, ak sú tieto alternatívne metódy z hľadiska potrieb pacienta považované za neuspokojivé.
3. Dlhodobé účinky implantácie vnútroočnej šošovky neboli stanovené. Preto je potrebné pravidelné pooperačné monitorovanie pacientov.
4. U pacientov s predoperačnými problémami, ako sú choroby endotelu rohovky, abnormality rohovky, degenerácia makuly, degenerácia sietnice, glaukóm a chronická mióza spôsobená liekmi, sa nemusí dosiahnuť rovnaká zraková ostrosť ako u pacientov, u ktorých sa takéto problémy nevyskytovali. Pri existencii takýchto stavov musí byť preto vopred stanovený prínos implantácie šošovky.
5. Je možné vyhnúť sa sekundárnej iridektómii pre pupilárny blok zrenice v prípade, že sa v čase implantácie vnútroočnej šošovky urobí jedna alebo viac iridektómií (Willis a kol., 1985).
6. Pre umiestnenie zadnokomorových šošoviek do prednej komory nebola stanovená bezpečnosť a účinnosť tohto postupu. Implantácia zadnokomorových šošoviek do prednej komory sa v niektorých prípadoch prejavila ako nebezpečná (Girard a kol., 1983).
7. Medzi niektoré nežiaduce účinky sprevádzajúce implantáciu vnútroočných šošoviek patrí: hypopyón, intraokulárna infekcia, akútna dekompenzácia rohovky a sekundárna chirurgická intervencia. Sekundárne chirurgické intervencie zahŕňajú, ale nie sú obmedzené na: repozíciu šošovky, výmenu šošovky, aspiráciu sklovca alebo iridektómiu pre pupilárny blok, uzavretie operačnej rany a nápravu odlúpenia sietnice.
8. Malá decentrácia šošovky, ktorá sa vyskytuje pri vnútroočnej šošovke s úzkou alebo malou optikou, sa u pacientov môže prejavovať pocitom oslnenia (glare) alebo inými poruchami videnia pri určitých podmienkach osvetlenia. Je potrebné, aby chirurg zvážil túto možnosť pred implantáciou vnútroočnej šošovky s úzkou alebo malou optikou. Pri implantácii očnej šošovky s úzkou alebo malou optikou sa odporúča uskutočnenie kapsulorexie.
9. Pooperačné rozšírenie kapsulárneho vaku s rozlične významným splytčením prednej komory a indukovanou myopiou sa dáva do súvislosti s technikou kapsulorexie a implantáciou PMMA, silikónových a akrylátových zadnokomorových šošoviek (Holtz, 1992).
10. Je potrebné zachovávať obozretnosť pred enkapsuláciou šošovky, aby sa zabránilo šošovkovým decentráciám alebo dislokáciám. Niektoré klinické prípady naznačujú, že enkapsulácia nastáva v priebehu štyroch týždňov.
11. Klinická štúdia jednokusovej šošovky AcrySof® Natural (údaje v Tabuľke 2 až 5) sa uskutočnila so šošovkou určenou na implantáciu výlučne do kapsulárneho vaku. Nie sú k dispozícii žiadne klinické údaje, ktoré by demonštrovali jej bezpečnosť a účinnosť pri umiestnení do sulcus ciliaris.
Na konci chirurgického zákroku sa odporúča odstrániť viskoelastický materiál z oka s dôrazom na priestor medzi zadným puzdrom a šošovkou. Toto sa môže dosiahnuť miernym stlačením vnútroočnej šošovky v zadnej časti pomocou I/A hrotu a použitím štandardných irigačných a aspiračných metód na odstránenie viskoelastického materiálu z oka. Toto by malo umožniť premiestnenie všetkého zachyteného viskoelastického materiálu do prednej časti, kde sa môže ľahko aspirovať.
127
PREVENTÍVNE OPATRENIA 1. Tieto vnútroočné šošovky alebo zavádzací systém UltraSert™ Pre-loaded Delivery System sa nesmú opätovne
sterilizovať žiadnou metódou. 2. Vnútroočné šošovky sa nesmú uchovávať pri teplotách presahujúcich 45 °C (113 °F). 3. V prípade potreby zaobchádzajte so šošovkami opatrne, aby sa nepoškodil povrch šošoviek alebo haptické časti. 4. V žiadnom prípade sa nepokúšajte zmeniť tvar haptických častí. 5. Pri implantácii vnútroočných šošoviek sa vyžaduje veľká chirurgická zručnosť. Chirurg by mal pred vlastnou
implantáciou vnútroočnej šošovky sledovať rad implantácií alebo pri nich asistovať a mal by úspešne absolvovať jeden alebo viac kurzov implantácie vnútroočných šošoviek.
6. Pomôcky sú sterilné, pokiaľ obal nie je otvorený alebo poškodený. 7. Pomôcka UltraSert™ Pre-loaded Delivery System je určená len na jednorazové použitie. Po použití UltraSert™
Pre-loaded Delivery System zlikvidujte. 8. Používajte UltraSert™ Pre-loaded Delivery System pri teplote operačnej miestnosti medzi 18° C (64° F) a 23° C
(73° F).
VÝPOČET OPTICKEJ MOHUTNOSTI ŠOŠOVKYPredoperačný výpočet požadovanej optickej mohutnosti šošovky pre tieto zadnokomorové vnútroočné šošovky by sa mal určiť na základe skúseností chirurga, jeho preferencií a zamýšľaného umiestnenia šošovky. Metódy výpočtu optickej mohutnosti šošovky sú opísané v nasledujúcich literárnych odkazoch:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. a kol. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. a kol. Standardizing constants for ultrasonic biometry, keratometry, and IOL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
NAVRHNUTÁ A-KONŠTANTA Navrhnutá A-konštanta, uvedená na vonkajšom štítku, slúži ako usmernenie a východiskový bod pre výpočty optickej mohutnosti implantátu. Odporúča sa vytvorenie vlastnej konštanty, ktorá je pre Vás vhodná na základe Vašich klinických skúseností s príslušnými modelmi šošoviek, na základe chirurgických postupov, meracích zariadení a postoperačných výsledkov. V prípade potreby ďalších informácií pre výpočet optickej mohutnosti šošoviek na území Spojených štátov amerických kontaktujte prosím Alcon Laboratories, Inc. na +1-800-TO-ALCON (+1-800-862-5266). Mimo územia Spojených štátov amerických kontaktujte miestne zastúpenie alebo distribútora spoločnosti Alcon.
SCHVÁLENÝ ALCON VISKOELASTICKÝ MATERIÁL A OBJEDNÁVACIE ČÍSLAPozri dodatok priložený k tomuto baleniu. Môže byť použitý iba viskoelastický materiál (materiály) schválené spoločnosťou Alcon. Použitie neschváleného viskoelastického materiálu môže spôsobiť poškodenie šošovky a možné nežiaduce udalosti počas procesu implantácie. Pre úplný zoznam viskoelastických materiálov odporúčaných spoločnosťou Alcon, kontaktujte prosím miestne zastúpenie spoločnosti Alcon.
NÁVOD NA POUŽITIEKrok 1. Používajte UltraSert™ Pre-loaded Delivery System System pri teplote operačnej miestnosti medzi 18° C (64° F) a
23° C (73° F).
Krok 2 Pred otvorením vonkajšieho obalu prekontrolujte štítok a údaje o modeli, optickej mohutnosti, zodpovedajúcej konfigurácii a dobe exspirácie.
Krok 3. Po otvorení vonkajšieho kartónového obalu skontrolujte, či obal pomôcky nie je poškodený. Ak zistíte poškodenie, použite inú vnútroočnú šošovku AcrySof® IQ a UltraSert™ Pre-loaded Delivery System. Ďalej si overte, či informácie o šošovke uvedené na štítku pomôcky (napríklad model, optická mohutnosť a sériové číslo) zodpovedajú informáciám uvedeným na štítku vonkajšieho balenia.
Krok 4. Aby ste vybrali UltraSert™ Pre-loaded Delivery System, uchopte roh plastovej podložky, odlúpnutím úplne otvorte vrchnú časť TYVEK† materiálu a preneste pomôcku do sterilného prostredia. Ak po prezretí nadstavec pomôcky vykazuje poškodenie, obsahuje častice alebo je deformovaný, použite inú vnútroočnú šošovku AcrySof® IQ a UltraSert™ Pre-loaded Delivery System. Ak pomôcka nie je celkom neporušená, alebo ak sa javí, že piest alebo uzáver piestu sa počas prepravy posunul, použite inú vnútroočnú šošovku AcrySof® IQ a UltraSert™ Pre-loaded Delivery System (pozri PRAVIDÁ PRE ZAOBCHÁDZANIE S VRÁTENÝMI VÝROBKAMI).
†Registrovaná ochranná známka spoločnosti E.I. DuPont De Nemours & Co.
Ak je šošovka pripravená na implantáciu, postupne uskutočnite kroky 5, 6 a 7, s minimálnym odstupom medzi jednotlivými krokmi.
128
Krok 5. Úplne zasuňte viskoelastickú kanylu kolmo k pomôcke cez otvor pre viskoelastický materiál umiestnený v časti zarážky šošovky v pomôcke tak, ako je znázornené na Obrázku 5. Napĺňajte pomôcku, až kým sa dá pozorovať, že viskoelastický materiál preteká k línii na špičke nadstavca (pozri šípku na Obrázku 5), potom kanylu odtiahnite. Toto bude vyžadovať približne 0,2 ml viskoelastického materiálu. Používajte výhradne viskoelastický materiál od spoločnosti Alcon schválený na použitie s pomôckou UltraSert™ Pre-loaded Delivery System, ktorý sa nechal zahriať na teplotu operačnej miestnosti. Zoznam viskoelastických materiálov schválených spoločnosťou Alcon je uvedený v dodatku priloženom k tomuto baleniu.
Pozri anglický jazyk Obrázok 5
Viscoelastic Port = Otvor pre viskoelastický materiál“Fill-To” Line = Línia “Fill-To”
Krok 6. Odstráňte modrú zarážku šošovky uchopením časti so šípkou a jej jemným ohnutím smerom dopredu pri súčasnom nadvihnutí smerom od pomôcky (Obrázok 6a). Odstráňte modrý uzáver piestu tak, že ho uchopíte a vytiahnete smerom nahor a preč od pomôcky (Obrázok 6b). Odstráňte zarážku šošovky a uzáver piestu. Nesnažte sa pridať viskoelastický materiál do pomôcky po odstránení zarážky šošovky, pretože by to mohlo spôsobiť poškodenie šošovky. Nesnažte sa zatiahnuť piest po odstránení uzáveru piestu, pretože by to mohlo spôsobiť poškodenie piestu.
Pozri anglický jazyk Obrázok 6a a 6b
Krok 7. Zľahka zatláčajte piest smerom dopredu plynulým, kontinuálnym pohybom, až pokým sa predná hrana optickej časti vyrovná s líniou nadstavca (Odkaz: Obrázok 7a). V tejto polohe bude modrá pružina v kontakte s bezfarebným prstovým telom, ako je znázornené na Obrázku 7b. Tento posun piestu by mal trvať minimálne 7 sekúnd. Nedovoľte, aby niektorá časť šošovky opustila špičku nadstavca. Je dôležité, aby sa piest neposúval trhane, pretože by mohlo dôjsť k nesprávnemu foldovaniu a poškodeniu šošovky.
Pozri anglický jazyk Obrázok 7a a 7b
Fill-to Line = Línia “Fill-To”
Krok 8. Po posunutí šošovky k línii nadstavca je potrebné vizuálne skontrolovať šošovku za účelom stanovenia polohy haptických častí. Piest by mal byť v kontakte s ťahaným koncom optickej časti. Žiadna časť šošovky nesmie vystúpiť z nadstavca pred vložením cez rez (Pozri Obrázok 8.) Keď sa šošovka nachádza v polohe pri línii nadstavca, musí sa implantovať do 3 minút.
Pozri anglický jazyk Obrázok 8
“Proceed with full plunger advancement to fully deliver lens” = “Pokračujte posúvaním celého piestu pre úplne vloženie šošovky”
“Proceed” = “Pokračujte”“Proceed with full plunger advancement to fully deliver lens” = “Pokračujte posúvaním celého piestu pre úplne
vloženie šošovky”“Proceed with device rotations per Step 10” = “Pokračujte otáčaním pomôcky podľa kroku 10”
“Do not proceed” = “Nepokračujte”
Krok 9. Po ubezpečení sa, že šošovka je správne umiestnená a haptické časti sú správne foldované, pokračujte v implantovaní šošovky vložením špičky nadstavca cez rez k zariadeniu zabezpečujúcemu správnu hĺbku (Obrázok 9) a namierením špičky nadstavca na predný otvor puzdra.
Pozri anglický jazyk Obrázok 9
Depth Guard = Zariadenie zabezpečujúce správnu hĺbku
Krok 10. Zľahka posúvajte piest plynulým, kontinuálnym pohybom. Neposúvajte piest príliš rýchlo, pretože by mohlo dôjsť k poškodeniu šošovky. Odporúča sa, aby vkladanie šošovky z polohy, kde je vizuálne kontrolovaná, trvalo minimálne 5 sekúnd. V prípade, že vedúca haptická časť je rovná alebo slučková a predĺžená pred šošovku, otáčajte pomôcku tak, aby bola naklonená doľava pred posúvaním piestu, aby sa zaistilo správne umiestnenie vedúcej haptickej časti v kapsulárnom vaku. Keď vedúca haptická časť začne vystupovať zo špičky nadstavca, umiestnite vedúcu haptickú časť do šošovkového puzdra so správnou orientáciou a pokračujte v pomalom posúvaní piestu, aby ste uvoľnili šošovku. Keď optická časť šošovky vystúpi zo špičky nadstavca, otáčajte pomôcku späť k centrálnej časti alebo mierne naklonenú doprava, ak je to potrebné, aby ste zaistili rozvinutie šošovky na prednej strane vo vnútri šošovkového puzdra.
Krok 11. Umiestnite šošovku vo vnútri šošovkového puzdra použitím vhodného nástroja a planárnym spôsobom paralelne s dúhovkou.
Krok 12. Zlikvidujte celú pomôcku. Pomôcka UltraSert™ Pre-loaded Delivery System sa nesmie používať opakovane.
129
REGISTRÁCIA PACIENTA A VEDENIE ZÁZNAMOV Identifikačná karta pacienta, ktorá je obsahom balenia, musí byť vyplnená a odovzdaná pacientovi spolu s pokynmi na uchovávanie karty ako trvalého záznamu, ktorý musí byť poskytnutý každému očnému lekárovi, ktorý bude pacienta v budúcnosti ošetrovať.V Spojených štátoch musí byť každý pacient registrovaný v Alcon Laboratories, Inc. bezprostredne po implantácii jednej z týchto šošoviek. Registrácia sa uskutočňuje vyplnením pripravenej registračnej karty o implantácii (Implant Registration Card), ktorá je pribalená v obale so šošovkou a jej odoslaním do Alcon Laboratories, Inc. Registrácia pacientov je nevyhnutná pre program dlhodobého sledovania spoločnosti Alcon Laboratories, Inc. a pri príprave odpovedí pre hlásenia o nežiaducich udalostiach po operácii. Mimo územia Spojených štátov amerických je potrebné dodržiavať miestne zákony v súvislosti s registráciou produktu.
Udalosti, ktoré odôvodniteľne naznačujú, že šošovka mohla spôsobiť alebo prispieť k úmrtiu alebo vážnemu zraneniu, vrátane udalostí, ktoré nastali v dôsledku zlyhania zdravotníckej pomôcky plniť svoje výkonové špecifikácie alebo fungovať inak, ako bolo zamýšľané, musia byť oznámené spoločnosti Alcon Laboratories, Inc. Táto informácia je požadovaná od všetkých chirurgov, aby sa zdokumentovali možné dlhodobé dopady implantácie vnútroočnej šošovky. Chirurgovia môžu použiť nasledujúcu adresu a telefónne číslo pre hlásenie nežiaducich udalostí v súvislosti s týmito vnútroočnými šošovkami:
Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Bezplatné volanie: +1-800-757-9780
Chirurgovia mimo územia Spojených štátov amerických musia kontaktovať miestne zastúpenie spoločnosti Alcon alebo jej obchodných zástupcov v súvislosti s nahlasovaním nežiaducich udalostí.
Klinické štúdie s foldovateľnými zadnokomorovými šošovkami AcrySof® Uskutočnili sa dve randomizované, prospektívne a kontrolované klinické štúdie s foldovateľnými jednokusovými zadnokomorovými šošovkami AcrySof®. Prvá štúdia sa uskutočnila, aby sa demonštrovala bezpečnosť a účinnosť Modelu SB30AL jednokusovej zadnokomorovej šošovky AcrySof® Natural (filtrujúcej UV a modré svetlo) ako materského modelu šošovky. Bola to randomizovaná klinická štúdia, ktorá ako kontrolnú šošovku využívala AcrySof® Model SA30AL (absorbujúcu len UV svetlo). Zahrnuli sa len údaje z prvého operovaného oka tých jedincov, ktorí dostali buď Model SB30AL alebo Model SA30AL vnútroočnej šošovky. Druhá randomizovaná klinická štúdia porovnávajúca foldovateľné jednokusové vnútroočné šošovky AcrySof® IQ (s chromofórmi filtrujúcimi UV a modré svetlo) s asférickou optickou časťou s kontrolnými foldovateľnými jednokusovými šošovkami AcrySof® bola vykonaná s cieľom stanoviť klinické a funkčné výhody v porovnaní s tradičným sférickým návrhom.
Klinická štúdia s jednokusovou šošovkou AcrySof® Natural, Model SB30AL Výsledky dosiahnuté u pacientov, ktorí boli úspešne sledovaní počas najmenej jedného roka po operácii poskytli dostatočnú istotu, že jednokusová šošovka AcrySof® Natural. Model SB30AL je bezpečnou a účinnou pomôckou pri vizuálnej korekcii afakie.
Populácia pacientov s jednokusovou šošovkou AcrySof® Natural, Model SB30AL Populácia jedincov, ktorej sa implantoval Model SB30AL aspoň do prvého operovaného oka v tejto bilaterálnej štúdii zahŕňa 70,6 % žien a 29,4 % mužov. Populácia jedincov, ktorej sa implantoval Model SA30AL (kontrola) vnútroočnej šošovky zahŕňa 60,5 % žien a 39,5 % mužov. Rozdelenie podľa rasy pre populáciu s Modelom SB30AL predstavuje 95,3 % belochov a 4,7 % černochov. Kontrolná (SA30AL) populácia jedincov predstavovala 96,6 % belochov, 2 % černochov a 1,4 % ostatných. Priemerný vek pre celkovú populáciu, ktorá dostala Model SB30AL aspoň do prvého operovaného oka predstavoval 72,9 rokov. Podobne, priemerný vek pre celkovú populáciu, ktorá dostala Model SA30AL (kontrola) predstavoval 71,9 rokov.
Zraková ostrosť pri použití jednokusovej šošovky AcrySof® Natural, Model SB30AL Súhrn zrakovej ostrosti dosiahnutej v čase minimálne jeden rok po operácii medzi jedincami, ktorí nemali predoperačný očný patologický nález, abnormálne rohovky alebo degeneráciu makuly, v ktoromkoľvek čase štúdie (najlepší prípad), je uvedený v Tabuľke 2a a zraková ostrosť dosiahnutá u celkovej populácie jedincov je uvedená v Tabuľke 3a. Kontrolné údaje pre rovnaké súbory dát sú uvedené v Tabuľkách 2b a 3b, v tomto poradí. Nezistil sa žiadny štatisticky signifikantný rozdiel v zrakovej ostrosti medzi Modelom SB30AL a kontrolnými šošovkami, Modelom SA30AL, či pre najlepší prípad alebo pre celkové súbory dát.
130
Pozri anglický jazyk Tabuľka 2aNajlepšie korigovaná pooperačná zraková ostrosť pre najlepšie prípady populácie pacientov
po minimálne jednom roku po operácii, vnútroočná šošovka AcrySof® Natural, Model SB30AL
Zraková ostrosť
#na20/20 alebo Lepšie
20/25 20/30 20/4020/40 alebo Lepšie
> 20/40 do
< 20/80> 20/80
N N % N % N % N % N % N % N %
Veková kategória
Pozri anglický jazyk Tabuľka 2bNajlepšie korigovaná pooperačná zraková ostrosť pre najlepšie prípady populácie pacientov
po minimálne jednom roku po operácii, vnútroočná šošovka AcrySof®, Model SA30AL, kontrola
Zraková ostrosť
#na20/20 alebo Lepšie
20/25 20/30 20/4020/40 alebo Lepšie
> 20/40 do
< 20/80> 20/80
N N % N % N % N % N % N % N %
Veková kategória
Pozri anglický jazyk Tabuľka 3aNajlepšie korigovaná pooperačná zraková ostrosť v celkovej populácii pacientov
po minimálne jednom roku po operácii, vnútroočná šošovka AcrySof® Natural, Model SB30AL
Zraková ostrosť
#na20/20 alebo Lepšie
20/25 20/30 20/4020/40 alebo Lepšie
> 20/40 do
< 20/80> 20/80
N N % N % N % N % N % N % N %
Veková kategória
Pozri anglický jazyk Tabuľka 3bNajlepšie korigovaná pooperačná zraková ostrosť v celkovej populácii pacientov
po minimálne jednom roku po operácii, vnútroočná šošovka AcrySof®, Model SA30AL, kontrola
Zraková ostrosť
#na20/20 alebo Lepšie
20/25 20/30 20/4020/40 alebo Lepšie
> 20/40 do
< 20/80> 20/80
N N % N % N % N % N % N % N %
Veková kategória
Kumulatívne pooperačné nežiaduce udalosti s jednokusovou šošovkou AcrySof® Natural, Model SB30AL Kumulatívny výskyt týchto pooperačných nežiaducich udalostí do a vrátane minimálne jedného roka po operácii pre jednokusovú šošovku AcrySof® Natural, Model SB30AL a Model SA30AL je uvedený v Tabuľke 4. Nezistili sa žiadne štatisticky signifikantné rozdiely medzi Modelom SB30AL a Modelom SA30AL v súvislosti s podielom jedincov, ktorí zaznamenali akékoľvek kumulatívne pooperačné nežiaduce udalosti.
131
Tabuľka 4Kumulatívne pooperačné nežiaduce udalosti po najmenej jednom roku po operácii,
vnútroočná šošovka AcrySof® Natural, Model SB30AL a SA30AL, kontrola
SB30AL(N= 153)
SA30AL(N= 147) p-hodnota*
N % N %
TypPooperačnej nežiaducej
udalosti
0 0,0 0 0,0 NA Kumulatívne Hypopyón
Vnútroočná infekcia/endoftalmitída 0 0,0 0 0,0 NA
Edém makuly 4 2,6 2 1,4 0,6847
Pupilárny blok 0 0,0 0 0,0 NA
Odlúpenie sietnice alebo náprava odlúpenej sietnice 0 0,0 0 0,0 NA
Dislokácia šošovky 1 0,7 0 0,0 1,0000
Sekundárna chirurgická opätovná intervenciaOdstránenie reziduálneho kortexuExplantácia (dislokácia z dôvodu kapsulárnej ruptúry)Kryoterapia na reparáciu natrhnutia sietniceParacentéza na zníženie vnútroočného tlakuFokálne laserové ošetrenieFotodynamická terapiaExplantácia spôsobená chybou biometrie
51 1 11100
3,30,70,70,70,70,70,00,0
20000011
1,40,00,00,00,00,00,70,7
0,4482
Hyféma 0 0,0 0 0,0 NA
*p-hodnoty podľa Fisherovho exaktného testu porovnávajúceho Model SB30AL a Model SA30AL.
Pretrvávajúce pooperačné nežiaduce udalosti v súvislosti s jednokusovou šošovkou AcrySof® Natural, Model SB30AL Pretrvávajúci výskyt týchto pooperačných nežiaducich udalostí po minimálne jednom roku u pacientov s jednokusovou šošovkou AcrySof® Natural, Modelom SB30AL a kontrolným Modelom SA30AL sú uvedené v Tabuľke 5. Nezistili sa žiadne štatisticky signifikantné rozdiely medzi Modelom SB30AL a Modelom SA30AL v súvislosti s podielom jedincov, ktorí zaznamenali akékoľvek pretrvávajúce nežiaduce udalosti.
Tabuľka 5Pretrvávajúce nežiaduce udalosti po minimálne jednom roku po operácii,
vnútroočná šošovka AcrySof® Natural, Model SB30AL a SA30AL, kontrola
SB30AL(N= 138)
SA30AL(N= 127) p-hodnota*
N % N %
Typ Nežiaducej udalosti
0 0 1 0,8 0,4792Pretrvávajúci Edém rohovky
Iritída 0 0 0 0,0 NA
Edém makuly 2 1,4 1 0,8 1,0000
Vitritída 0 0 0 0,0 NA
Zvýšený vnútroočný tlak vyžadujúci liečbu 0 0 0 0,0 NA
*p-hodnoty podľa Fisherovho exaktného testu porovnávajúceho Model SB30AL a Model SA30AL.
Vnímanie farieb pri použití jednokusovej šošovky AcrySof® Natural, Model SB30AL Testovanie vnímania farieb použitím testovacieho panelu Farnsworth D-15 sa uskutočnilo 120 až 180 dní po operácii. Zo 109 jedincov s normálnym farebným videním, ktorým sa implantovala šošovka Modelu SB30AL do prvého operovaného oka a boli vyšetrovaní 120 až 180 dní po operácii, 107 jedincov (98,2 %) úspešne prešlo testom vnímania farieb. Zo 102
132
jedincov s normálnym farebným videním, ktorým sa implantovala šošovka Model SA30AL do prvého operovaného oka a boli vyšetrovaní 120 až 180 dní po operácii, 97 jedincov (95,1 %) úspešne prešlo testom vnímania farieb. Nezistili sa žiadne štatisticky signifikantné rozdiely medzi Modelom SB30AL a Modelom SA30AL v percentuálnom počte jedincov, ktorí úspešne prešli testom vnímania farieb pri vyšetrovaní 120 až 180 dní po operácii. Pridanie chromofóru vo vlastníckom práve teda neovplyvňuje negatívne farebné videnie u pacientov s normálnym farebným videním.
Incidencia Nd:YAG pre jednokusovú šošovku AcrySof® Natural, Model SB30AL Pri priemernom sledovaní počas 21,6 mesiacov sa u troch (3) zo 135 jedincov (2,2 %), ktorým sa implantoval Model SB30AL šošovky, vyskytla zadná kapsulotómia Nd:YAG. Pri priemernom sledovaní počas 21,9 mesiacov sa u dvoch (2) zo 127 jedincov (1,6 %), ktorým sa implantoval Model SA30AL šošovky, vyskytla zadná kapsulotómia Nd:YAG.
Klinická štúdia so šošovkou AcrySof® IQKonzistentne s návrhom podobných skôr vykonaných štúdií s vnútroočnými šošovkami boli do kontrolovanej, randomizovanej, dvojito zamaskovanej, multicentrickej, kontralaterálnej klinickej štúdie so šošovkami AcrySof® IQ a sférickými kontrolnými šošovkami zaradení dospelí jedinci s celkovým dobrým zdravotným stavom očí (napr. bez predošlej očnej operácie, degeneratívnej poruchy videnia, ktoré by významne ovplyvňovali ostrosť videnia, alebo so závažným akútnym či chronickým ochorením, ktoré by mohlo zvyšovať riziko pre pacienta). Očné sférické aberácie boli štatisticky signifikantne nižšie u šošoviek AcrySof® IQ ako u kontrolných šošoviek. Výsledky kontrastnej citlivosti preukázali štatisticky signifikantné zlepšenie po operácii (po 3 mesiacoch) v prospech očí, do ktorých boli implantované šošovky AcrySof® IQ. U očí, do ktorých boli implantované šošovky AcrySof® IQ sa taktiež zistilo štatisticky a klinicky významné zlepšenie pri meraní funkčného videnia, simulovanom nočnom riadení vozidla za rôznych testovacích podmienok – predovšetkým v súvislosti s oslnením a hmlou. Tieto výsledky odrážajú skutočnosť, že vnútroočné šošovky AcrySof® IQ (asférická optika na materiálovom základe obsahujúca chromofór filtrujúci modré svetlo) poskytujú klinický prínos v porovnaní s monofokálnymi vnútroočnými šošovkami AcrySof® (bez asférickej optiky a chromofóru filtrujúceho modré svetlo).
Šošovka AcrySof® IQ – sférické aberácie a celkové aberácie vyššieho ráduPriemerná očná sférická aberácia očí so šošovkami AcrySof® IQ bola približne 0,1 mikrometrov. Obrázok 10 predstavuje štatisticky signifikantné zníženie sférických aberácií a celkových aberácií vyššieho rádu v prospech šošovky AcrySof® IQ. Obrázok 11 predstavuje priemerné namerané hodnoty sférických aberácií všetkých očí, meraných vlnoplošným aberometrom v skupinách podľa šošoviek a veku. Ako je znázornené v tomto grafe, zníženie sférickej aberácie u očí so šošovkou AcrySof® IQ nebolo závislé na veku.
Pozri anglický jazyk Obrázok 10Sférické a celkové RMS vyššieho rádu
90-120 dní po implantácii do druhého oka
Pozri anglický jazyk Obrázok 11Priemerná sférická aberácia celkovo a podľa vekových skupín
90-120 dní po implantácii do druhého oka
Šošovka AcrySof® IQ – vizuálna ostrosť na diaľkuŠošovka AcrySof® IQ a kontrolná šošovka poskytli klinicky podobnú pooperačnú vizuálnu ostrosť. Výsledky monokulárnej vizuálnej ostrosti sú uvedené na Obrázkoch 12 a 13.
Pozri anglický jazyk Obrázok 12LogMAR BCVA
Pozri anglický jazyk Obrázok 13LogMAR UCVA
Šošovka AcrySof® IQ Lens - kontrastná citlivosťPrimárnym cieľom klinického skúšania bolo preukázať lepšie vlastnosti šošovky AcrySof® IQ v porovnaní s kontrolnou šošovkou prostredníctvom priemernej kontrastnej citlivosti meranej po operácii za mezopických podmienok s oslnením alebo bez neho pri jednej z dvoch priestorových frekvencií (3 alebo 6 cyklov na stupeň) pomocou prístroja Vector Vision CSV-1000 (s navrhnutou svietivosťou 3 cd/m² ). U podskupiny pacientov bol tiež vykonaný test kontrastu funkčnej ostrosti (FACT) (s navrhnutou svietivosťou 3 cd/m²). Pri tomto klinickom výskume boli preukázané lepšie vlastnosti šošovky AcrySof® IQ v porovnaní s kontrolnou šošovkou za mezopických podmienok pri 6 cykloch na stupeň s oslnením i bez neho (CVS-1000) a pri 3 a 6 cykloch na stupeň bez oslnenia (FACT). Obrázky 14 a 15 zobrazujú výsledky mezopickej kontrastnej citlivosti pri všetkých priestorových frekvenciách testovaných pre šošovku AcrySof® IQ aj pre kontrolnú šošovku.
Pozri anglický jazyk Obrázok 14Mezopická kontrastná citlivosť (CSV-1000)90-120 dní po implantácii do druhého oka
Pozri anglický jazyk Obrázok 15Podštúdia mezopickej kontrastnej citlivosti (FACT)
minimálne 90 dní po implantácii do druhého oka
133
Šošovka AcrySof® IQ – simulácia nočného riadenia vozidlaJedna podskupina pacientov podstúpila testovanie na validovanom simulátore nočného riadenia. Pacienti boli testovaní monokulárne za podmienok, ktoré simulovali podmienky v meste a na vidieku za normálnych podmienok, pri oslnení a hmle.Scéna nočného riadenia vozidla v meste využíva rozmanité pouličné osvetlenie, svetlá automobilov, osvetlenie obchodov a značky pre vytvorenie vysokej úrovne okolitého osvetlenia, ktoré je pre tieto podmienky typické. Scéna nočného riadenia vozidla na vidieku využíva minimálne okolité osvetlenie. Pre riadenie v meste a na vidieku boli použité simulované rýchlosti riadenia 56 km/hod (35 míľ/hod), a 89 km/hod (55 míľ/hod), v tomto poradí.Pacienti boli požiadaní, aby objavili a identifikovali sériu cieľov v každej scéne, vrátane zelenobielych cestných informačných tabúľ, čierno-žltých výstražných značiek a chodcov. Pacienti mali reagovať v okamihu, keď uvideli prvý cieľ, čo umožnilo zaznamenať detekčnú vzdialenosť. Potom boli pacienti požiadaní, aby reagovali až vtedy, keď dokázali cieľ rozlíšiť (napr. čo značka uvádza, ktorým smerom chodec kráča atď.), aby bolo možné zaznamenať identifikačnú vzdialenosť. Na obrázkoch 16 až 19 sú uvedené priemerné rozdiely medzi šošovkou AcrySof® IQ a kontrolnou šošovkou v scénach riadenia vozidla v meste a na vidieku pre detekčnú aj identifikačnú vzdialenosť (napr. priemer intraindividuálnych rozdielov).Šošovka AcrySof® IQ zaznamenala v 34 z 36 testovaných podmienok funkčne lepšie výsledky, ako bolo zistené v prípade kontroly, čo naznačuje zlepšenie detekčných aj identifikačných vzdialeností v mestskom prostredí i na vidieku za rôznych testovacích podmienok riadenia (za normálnych podmienok, pri oslnení a za hmly). Okrem toho šošovka AcrySof® IQ preukázala štatisticky signifikantne lepšie výsledky ako kontrola v 12 z týchto podmienok, pričom najvýznamnejší dopad a najväčšie výhody boli pozorované pri detekcii a identifikácii chodcov v meste (za podmienok oslnenia a pri hmle) a výstražných značiek na vidieku (pri podmienkach oslnenia a za hmly). Za znížených podmienok viditeľnosti (oslnenie, hmla) v meste poskytuje zvýšená vzdialenosť pre viditeľnosť pri 56 km/hod (35 míľ/hod) u šošovky AcrySof® IQ dodatočný čas viac než o 0,5 sekundy na reakciu na chodcov, čo je riziko, s ktorým sa môžeme bežne stretnúť v mestskom prostredí. Toto zvýšenie o 0,5 sekundy je funkčne významné pre poskytnutie dlhšieho času na vhodnú reakciu, ako je napríklad zastavenie, vyhýbanie sa, atď. (Green 2000; McBride a Matson 2004). Za všetkých podmienok mimo mesta poskytuje zvýšená viditeľnosť pri 89 km/hod (55 míľ/hod) u šošovky AcrySof® IQ dodatočný čas viac než 1 sekundu na identifikáciu výstražných značiek, čo je situácia, s ktorou sa môžeme bežne stretnúť vo vidieckom prostredí. Zvýšenie o 0,5 sekundy je funkčne významné pre poskytnutie dlhšieho času na prijatie vhodných opatrení počas riadenia vozidla, čo je kritické predovšetkým v noci v neznámych vidieckych oblastiach, kde často chýba okolité osvetlenie. V podštúdii bolo 6 pacientov, u ktorých po operácii došlo k makulárnej degenerácii alebo PCO. Keď boli títo pacienti vyňatí z analýzy výsledkov riadenia, rozdiel medzi vnútroočnými šošovkami pri detekcii a identifikácii chodcov za podmienok oslnenia v meste nevyhovel 0,5-sekundovej prahovej hodnote pre klinickú významnosť. Keď boli pôvodné analýzy upravené na multiplicitu, rozdiel medzi vnútroočnými šošovkami pre detekciu oslneného textu v meste (Hommelova hodnota p = 0,0539) alebo pre detekciu chodcov pri oslnení na vidieku (Hommelova hodnota p = 0,0507) už nebol štatisticky významný.Tieto výsledky preukazujú zlepšené funkčné videnie a pravdepodobne užitočný prínos k bezpečnosti pre starších vodičov so šošovkami AcrySof® IQ a ostatných vodičov a chodcov, ktorí sa tiež pohybujú na vozovke. Výsledky tohto testu preukazujú, že šošovka AcrySof® IQ zlepšuje funkčné videnie, ktoré môže následne zlepšiť bezpečnosť pacienta pre ďalšie životné situácie za podmienok zníženej viditeľnosti.
Pozri anglický jazyk Obrázok 16Simulátor nočného riadenia
Priemerné intraindividuálne rozdiely detekčných vzdialeností videnia, mestoMinimálne 90 dní po operácii
Vnútroočná šošovka AcrySof® IQ – kontrola (n= 44)
Pozri anglický jazyk Obrázok 17Simulátor nočného riadenia
Priemerné intraindividuálne rozdiely identifikačných vzdialeností videnia, mestoMinimálne 90 dní po operácii
Vnútroočná šošovka AcrySof® IQ – kontrola (n= 44)
Pozri anglický jazyk Obrázok 18Simulátor nočného riadenia
Priemerné intraindividuálne rozdiely detekčných vzdialeností videnia, vidiekMinimálne 90 dní po operácii
Vnútroočná šošovka AcrySof® IQ – kontrola (n= 44)
Pozri anglický jazyk Obrázok 19Simulátor nočného riadenia
Priemerné intraindividuálne rozdiely identifikačných vzdialeností videnia, vidiekMinimálne 90 dní po operácii
Vnútroočná šošovka AcrySof® IQ – kontrola (n= 44)
SPÔSOB DODÁVANIATieto zadnokomorové vnútroočné šošovky sa dodávajú suché, v zavádzacom systéme UltraSert™ Pre-loaded Delivery System, v primárne sterilizovanom obale a terminálne sterilizované etylénoxidom. Primárne sterilizovaný obal sa musí otvoriť len v aseptických podmienkach (pozri NÁVOD NA POUŽITIE).
134
EXSPIRAČNÁ DOBASterilita je zaručená, ak obal nie je poškodený alebo otvorený. Exspiračná doba je zreteľne vyznačená na štítku vonkajšieho obalu pomôcky UltraSert™ Pre-loaded Delivery System. Každá šošovka, pre ktorú uplynula exspiračná doba, sa musí vrátiť spoločnosti Alcon Laboratories, Inc. (pozri PRAVIDLÁ PRE ZAOBCHÁDZANIE S VRÁTENÝMI VÝROBKAMI).
PRAVIDLÁ PRE ZAOBCHÁDZANIE S VRÁTENÝMI VÝROBKAMIV Spojených štátoch amerických sa vrátené šošovky budú prijímať len ako výmena za iné produkty, nie ako dobropis. Ku každému vrátenému tovaru musí byť priložený Returned Goods Number (Číslo vráteného tovaru) Alcon Laboratories, Inc. a je potrebné ich dopraviť sledovateľným spôsobom. Číslo vráteného tovaru sa získa pri kontaktovaní Oddelenia pre služby zákazníkom (Customer Service Department) spoločnosti Alcon. Poskytnutie tohto čísla neustanovuje konečné prijatie vrátených produktov. Pre podrobnejšie informácie o pokynoch týkajúcich sa výmeny tovaru kontaktujte Vášho zástupcu pre predaj a služby zákazníkom. Mimo územia Spojených štátov amerických vo veci vrátenia produktov kontaktujte miestne zastúpenie alebo distribútora spoločnosti Alcon.
ODKAZY 1. Boettner, E.A. a Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., a kol. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., a kol. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. a Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
135
SYMBOLY POUŽITÉ NA OBALOCH
SYMBOL VÝZNAMIOL Vnútroočná šošovka
SP Jednokusová
UV Ultrafialové svetlo
D Dioptria
ØB Priemer telesa (optický priemer)
ØT Celkový priemer (celková dĺžka)
2STERILIZE Nesmie sa opakovane sterilizovať
Nepoužívajte opakovane
Použiteľné do
STERILE EO Sterilizované etylénoxidom
SN Sériové číslo
Upozornenie
Výrobca
EC REP Splnomocnený zástupca pre Európsku Úniu
Horný teplotný limit
Tu otvoriť
Pozrite návod na použitie
Nepoužívajte, ak bol sterilný obal otvorený alebo poškodený
Upozornenie: Federálny zákon (USA) obmedzuje predaj tejto pomôcky na objednanie lekárom
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 USA
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,United Kingdom
Miestne zastúpenie v SR:Novartis Slovakia s.r.o., Alcon DivisionGalvaniho 15/ASK-821 04 Bratislava
STERİL UV ve Mavi Işık Filtreli Akrilik Katlanır Tek ParçaUltraSert™ Pre-loaded Enjektör Sistemli Arka Kamara Lensleri
AÇIKLAMAAcrySof® IQ UV ve mavi ışık filtreli akrilik katlanır tek parça arka kamara lensleri, katarakt ameliyatından sonra yetişkin hastalarda gelişen afakinin görsel iyileştirilmesinde insan göz merceğinin değişimi için kullanılan optik implantlardır. Alcon’un tescilli mavi ışık filtreleyen kromoforuna sahip AcrySof® IQ GİL, insan kristalin lensine benzer şekilde yaklaşık olarak 400 – 475 nm aralığındaki mavi ışık dalga boyunu filtreler (Boettner ve Wolter, 1962). Standart UV- ışığı filtrelemesine ek olarak AcrySof® IQ GİL, mavi ışık dalga boylarının geçirgenliğini 400 nm’de %62’den 475 nm’de %23’e kadar düşürür (bkz. Tablo 1). Optik bölüm yüksek kırılma indeksine sahip yumuşak bir akrilik maddeden oluşur. Bu madde, yerleştirme öncesinde katlanabilme özelliğine sahiptir. İmplantasyonun ardından lens tam lens gövdesi boyutunda olacak şekilde açılır. Lenste destekleyici haptik özelliklere sahip bir bikonveks optik bulunmaktadır (Şekil 1). AcrySof® IQ Model AU00T0 GİL’in arka asferik yüzeyi, ortalama bir korneanın pozitif sferik aberasyonunun kompanse edilmesi amacıyla negatif sferik aberasyon ile tasarlanmıştır. AU00T0 Model Tek Parça GİL’in görüntü kalitesi (yani modüler transfer fonksiyonu) Şekil 4’te gösterilmiştir. AcrySof® IQ AU00T0 Model Tek Parça GİL’ler, bu lensleri kapsüler keseye güvenli şekilde yerleştirmek için uygun ve kontrollü bir yöntem sağlanması amacıyla UltraSert™ Pre-loaded Enjektör Sisteminde (Şekil 2) verilmektedir. Lenslerin fiziksel özellikleri şu şekildedir:
Fiziksel Karakteristik AçıklamaOptik Türü Asferik optikli tek parça GİL
Optik Maddesi Ultraviyole ve mavi ışık filtreli Akrilat/Metakrilat Kopolimer
Haptik Materyali Ultraviyole ve mavi ışık filtreli Akrilat/Metakrilat Kopolimer
Optik Çap ØB (mm) 6.0
Toplam Uzunluk ØT (mm) 13.0
Haptik Açısı 0º
Optik Boyutlar Bkz. Şekil 1
Bkz. İngilizce dilindeki Şekil 1FİZİKSEL KARAKTERİSTİKLER
Tüm ölçüler milimetre birimindendir
ASPHERIC SURFACE = ASFERİK YÜZEY
Bkz. İngilizce dilindeki Şekil 2
Nozzle Tip = Nozul UçDepth Guard = Derinlik Koruması
Main Body = Ana Gövde
TURKISH
137
Bkz. İngilizce dilindeki Şekil 3
SPEKTRAL GEÇİRGENLİK EĞRİLERİ
TRANSMITTANCE (%) = GEÇİRGENLİK (%)WAVELENGTH (nm) = DALGA BOYU (nm)
6.0 D to 27.0 D range = 6.0 D - 27.0 D aralığı4 yr – 53 yr old range, crystalline lens = 4 yaş - 53 yaş aralığı, kristalin lens
NOTLAR: • Burada verilen dalga boyu kesimi ve spektral geçirgenlik eğrileri, UV-emici ve Alcon’un tescilli mavi ışık filtreleyen
kromoforu ile birlikte akrilat/metakrilat kopolimerden yapılmış GİL’lerin geçirgenlik değerleri aralığını yansıtmaktadır. • Ölçümlerde, 6mm’lik bir açıklık ve optik merkezine eşdeğer kalınlıkta bir disk kullanılarak doğrudan geçirgenlik
temel alınmıştır. • İnsan lens verileri Boettner ve Wolter’den (1962) alınmıştır.
Tablo 120.0 D GİL’ler için Geçirgenlik Karşılaştırması, %
Model 400 nm 425 nm 450 nm 475 nm
SA60AT 21 86 88 88
AU00T0 8 34 49 68
Geçirgenlik Farkı (SA60AT – AU00T0) 13 52 39 20
AU00T0 ile Geçirgenlik Azalması (SA60AT %’si) 62 60 44 23
Bkz. İngilizce dilindeki Şekil 4Model AU00T0’ın Modüler Transfer Fonksiyonu (20.0 D)
MTF (%) = Modüler Transfer Fonksiyonu (MTF) (%)Spatial Frequency (Ip/mm) = Uzaysal Frekans (lp/mm)
ISO, 3-mm Aperture (see Note 1) = ISO, 3-mm Açıklık (bkz. Not 1)Modified, 3-mm Aperture (see Note 2) = Modifiye, 3-mm Açıklık (bkz. Not 2)Modified, 5-mm Aperture (see Note 2) = Modifiye, 5-mm Açıklık (bkz. Not 2)
NOTLAR: 1. AU00T0 Model’in görüntü kalitesi, ISO 11979-2’de açıklanan bir model gözündeki modüler transfer fonksiyonu
(MTF) ölçülerek karakterize edilmiştir. ISO 11979-2 sadece 3mm’lik bir açıklık kullanan MTF ölçümlerine ihtiyaç duymaktadır.
2. Ayrıca AU00T0 Model’in görüntü kalitesi, tipik yetişkin insan sferik aberasyonunu ortaya koyan simüle edilmiş bir korneayı kullanan model bir gözdeki MTF ölçülerek karakterize edilmiştir. Modifiye model göz kullanımı ile MTF ölçümleri hem 3mm hem de 5mm açıklık kullanılarak yapılmıştır.
ETKİ ŞEKLİ AcrySof® IQ arka kamara göz içi lenslerinin uygulanma amacı, doğal kristalin lens yerine geçip gözün arka kamarasına konumlandırmaktır. Bu konumlandırma, lensin afakinin düzeltilmesinde bir refraktif ortam vazifesi görmesini sağlar. Asferik bikonveks optik, ortalama bir gözdeki standart sferik optikle karşılaştırıldığında sferik aberasyonu azaltır. Bu lenslerin retinal bozuklukların oluşumunun azaltılmasındaki etkinliği kanıtlanmamıştır.
ENDİKASYONLAR AcrySof® IQ arka kamara göz içi lensleri, katarakt ameliyatının ardından yetişkin hastalarda gelişen afakinin görsel olarak düzeltilmesi amacıyla insan lensinin değişimi için endikedir. Bu lensler kapsüler kese içine yerleştirmek amaçlıdır.
DİKKAT EDİLMESİ GEREKEN DURUMLAR Aşağıdaki durumlardan herhangi birinin geçerli olduğu hastalar, uygun bir göz içi lens adayı olmayabilir. Çünkü lens mevcut bir durumu kötüleştirebilir, bir durumun teşhisine veya tedavisine engel olabilir veya hastanın görme yeteneğinde aşırı bir risk teşkil edebilir. Cerrah, dikkatli preoperatif değerlendirme ve doğru klinik yargıya bağlı kalarak bir lensi aşağıdaki durumlardan bir veya daha fazlasının olduğu bir hastaya implante etmeden önce risk-yarar analizi oranını belirlemelidir: 1. Koroidal hemoraji 2. Eşlik eden ciddi göz hastalığı 3. Aşırı vitröz kaybı 4. Aşırı sığ ön kamara 5. Mikroftalmi
138
6. Yaşa bağlı olmayan katarakt 7. Arka kapsül rüptürü (GİL fiksasyonunu engelleyen) 8. Ciddi korneal distrofi 9. Ciddi optik atrofi 10. Kontrol edilemeyen pozitif basınç 11. Zonüler ayrılma (GİL fiksasyonunu engelleyen) 12. Renk körlükleri 13. Glokom 14. Kronik üveit 15. Diyabetik retinopati 16. Klinik açından önemli maküler/RPE değişimleriÇalışmalar AcrySof® Natural GİL ve normal renk görüşü olan kişilerde, renk görüşü ayrımının olumsuz etkilenmediğini göstermiştir. Kalıtsal renkli görme kusurları ve oküler hastalığa (örn. glokom, diyabetik retinopati, kronik üveit ve diğer retinal ya da optik sinir hastalıkları) bağlı sonradan edinilmiş renkli görüş kusurlarının olduğu kişilerde AcrySof® Natural GİL’in görüş üzerindeki etkisi konusunda çalışma yapılmamıştır.
UYARILAR 1. Tüm cerrahi prosedürlerde olduğu gibi bunda da risk mevcuttur. Katarakt veya implant cerrahisine eşlik eden
potansiyel komplikasyonlar -sıralananlarla sınırlı olmamak üzere- şunlardır: korneal endotel hasarı, endoftalmi, retina yırtılması, vitrit, kistoid maküler ödem, kornea ödemi, pupiler blok, siklitik membran, iris prolapsusu, hipopiyon ve geçici ya da kalıcı glokom.
2. Göz içi lens implantlarının güvenliği ve etkinliği önceden var olan durumların olduğu (kronik ilaç miyozisi, glokom, ambliyopi, diyabetik retinopati, daha önceden kornea nakli, daha önceden retina ayrılması ve/veya iritis vb.) hastalarda kanıtlanmamıştır. Bu gibi hastalarda lens implantasyonunu düşünen hekimler, afakik düzeltim için alternatif yöntemlerin kullanımını araştırmalıdır ve lens implantasyonunu sadece hastanın ihtiyaçlarının karşılanmasında tatmin edici alternatiflerin olmadığı durumlarda değerlendirmelidir.
3. Göz içi lens implantasyonunun uzun süreli etkileri belirlenmemiştir. Dolayısıyla hekimler, hastaları ameliyat sonrasında düzenli olarak izlemelidir.
4. Korneal endotelyal hastalık, anormal kornea, maküler dejenerasyon, glokom ve kronik ilaç miyozisi gibi preoperatif sorunların olduğu hastalar bu gibi sorunların olmadığı hastaların sahip olduğu görme keskinliğine erişemeyebilir. Hekim bu gibi durumlar mevcut olduğunda lens implantasyonundan ortaya çıkacak yararları belirlemelidir.
5. GİL implantasyonu sırasında bir veya daha fazla iridektomi gerçekleşirse pupiler blok için sekonder bir iridektomiden kaçınılabilir (Willis, ve ark., 1985).
6. Ön kamaraya yerleştirilen arka kamara lensinin güvenliği ve etkinliği kanıtlanmamıştır. Arka kamara lenslerinin ön kamaraya implantasyonunda bazı durumların güvenilir olmadığı görülmüştür (Girard, ve ark., 1983).
7. Göz içi lenslerin implantasyonuyla bağlantılı olan bazı advers reaksiyonlar şunlardır: hipopiyon, intraoküler enfeksiyon, akut kornea dekompozisyonu ve sekonder cerrahi müdahale. Sekonder cerrahi müdahaleler arasında, sıralananlarla sınırlı olmamak kaydıyla, şunlar bulunur: lensin yeniden konumlandırılması, lens değişimi, pupiler blok için vitröz aspirasyon veya iridektomi, yara sızıntısı onarımı ve retina ayrılmasının onarımı.
8. Dar veya küçük optikli bir GİL ile meydana gelen az miktarda lens desantralizasyonu hastanın bazı ışık koşullarında gözünün kamaşmasına veya diğer göz rahatsızlıklarına yol açabilir. Cerrahlar bu potansiyeli, dar veya küçük optikli bir GİL implantasyonunu gerçekleştirmeden önce değerlendirmelidir. Dar veya küçük bir lens implante ederken kapsuloreksis uygulanması önerilmektedir.
9. Değişken miktarda ön kamara sığlaşması ve indüklenmiş miyop ile birlikte kapsüler kesenin postoperatif distansiyonu, kapsuloreksis teknikleriyle ve PMMA, silikon ve akrilik arka kamara lenslerinin implantasyonuyla ilişkilendirilmiştir (Holtz, 1992).
10. Lens desentralizasyonlarından veya dislokasyonlardan kaçınmak için lens enkapsülasyonundan önce dikkat edilmelidir. Bazı klinik vakalar enkapsülasyonun dört hafta içinde meydana geldiğini göstermektedir.
11. AcrySof® Natural Tek Parçalı Lens (2. ve 5. tablolar arasında atıfta bulunulmuştur) klinik çalışması, sadece kapsüler kesede implantasyonu amaçlanan lensle birlikte uygulanmıştır. Siliyer sulkusa yerleştirilmesi açısından güvenliliğini ve etkililiğini gösteren bir klinik veri bulunmamaktadır.
Operasyonun sonunda arka kapsül ve lens arasındaki alana odaklanılarak viskoelastiğin gözden çıkarılması önerilmektedir. Bu işlem, I/A tip ile GİL optiğine arkadan hafifçe bastırarak ve viskoelastik maddenin gözdenin çıkarılması için standart irigasyon/aspirasyon teknikleri kullanılarak gerçekleştirilebilir. Bu işlem kolaylıkla aspire edebileceği yerdeki tüm sıkışmış viskoelastik maddeyi öne itmelidir.
ÖNLEMLER 1. Bu göz içi lensleri veya UltraSert™ Pre-loaded Enjektör Sistemini hiçbir yöntemle yeniden sterilize etmeyin. 2. Göz içi lensleri 45° C (113° F) üzerindeki sıcaklıklarda muhafaza etmeyin. 3. Lens yüzeylerine veya haptik bölümlere zarar gelmemesi için gerektiğinde lensleri dikkatli şekilde kullanın. 4. Haptik bölümleri hiçbir şekilde yeniden şekillendirmeye çalışmayın. 5. Göz için lens implantasyonunda yüksek seviyede cerrahi beceri gerekmektedir. Cerrah, göz içi lenslerin
implantasyonundan önce çeşitli implantasyonları gözlemlemiş ve/veya bu implantasyonlara yardım etmiş olmalı ve göz içi lens implantasyonuyla ilgili bir veya daha fazla kursu başarıyla tamamlamış olmalıdır.
6. Paket açılmadığı veya zarar görmediği sürece paket içeriği sterildir.
139
7. UltraSert™ Pre-loaded Enjektör Sistemi sadece tek kullanımlıktır. Kullanımdan sonra UltraSert™ Pre-loaded cihazı atın. 8. UltraSert™ Pre-loaded Enjektör Sistemini 18° C (64° F) ve 23° C (73° F) arasındaki Ameliyathane sıcaklıklarında
kullanın.
LENS GÜCÜNÜN HESAPLANMASIBu arka kamara göz içi lensleri için gereken lens gücünün preoperatif olarak hesaplanması cerrahın deneyimi, tercihi ve amaçlanan lens değişimine göre yapılmalıdır. Lens gücü hesaplama yöntemleri aşağıdaki referanslarda açıklanmıştır:
Hoffer, K.J. The Hoffer Q formula: A comparison of theoretic and regression formulas. J. Cataract Refract. Surg. 19:700- 712, 1993.Holladay, J.T. ve ark. A three-part system for refining intraocular lens power calculations. J. Cataract Refract. Surg. 14:17- 24, 1988.Holladay, J.T. ve ark. Standardizing constants for ultrasonic biometry, keratometry, and GİL power calculations. J. Cataract Refract.Surg. 23:1356-1370, 1997.Retzlaff, J.A., Sanders, D.R., and Kraff, M. Lens Implant Power Calculation, 3rd ed. Slack, Inc., Thorofare, N.J., 1990.
ÖNERİLEN A-SABİT DEĞERİ Dış etikette listelenen A-sabit değeri bir kılavuz olarak verilmiştir ve implant gücü hesaplamaları için başlangıç noktasıdır. Belli lens modelleriyle olan klinik deneyim, cerrahi teknikler, ölçme ekipmanı ve postoperatif sonuçlara bağlı olarak sizin için uygun olan sabit değeri sizin geliştirmeniz önerilmektedir.Amerika Birleşik Devletleri’nde lens gücü hesaplaması için ek bilgi gerekirse lütfen 1-800-TO-ALCON (1-800-862-5266) numaralı telefondan Alcon Laboratories, Inc. ile iletişime geçin. Amerika Birleşik Devletleri’nde değilseniz Alcon ofisleri veya bayileri ile iletişime geçin.
UYGUN ALCON VİSKOELASTİKLERİ VE SİPARİŞ NUMARALARIBu pakette bulunan eke bakın. Sadece Alcon’un uygun olarak belirlediği viskoelastik(ler) kullanılmalıdır. Uygun olmayan viskoelastik kullanımı, implantasyon işlemi esnasında lense zarar verebilir ve potansiyel komplikasyonlara yol açabilir. Alcon’un bu lens için uygun olarak belirlediği viskoelastik listesinin tamamı için lütfen Alcon temsilcinize danışın.
KULLANIM TALİMATLARI1. Adım. UltraSert™ Pre-loaded Enjektör Sistemini 18° C (64° F) ve 23° C (73° F) arasındaki Ameliyathane sıcaklıklarında
kullanın.
2. Adım. Açılmamış dış kutu üzerindeki etiketi model, güç, uygun konfigürasyon ve son kullanma tarihi yönünden kontrol edin.
3. Adım. Dış karton kutuyu açtıktan sonra cihaz paketinde herhangi bir hasar olup olmadığını kontrol edin. Hasar fark ederseniz başka bir AcrySof® IQ GİL ve UltraSert™ Pre-loaded Enjektör Sistemi kullanın. Daha sonra cihaz etiketi üzerindeki lens bilgilerinin (örn. model, güc ve seri numarası) dış kutu etiketi üzerindeki bilgilerle tutarlı olup olmadığını kontrol edin.
4. Adım. UltraSert™ Pre-loaded Enjektör Sistemini çıkarmak için, plastik tepsiyi köşesinden tutup TYVEK† madde kapağı bölümünü soyarak tamamen açın ve cihazı steril bir ortama nakledin. İncelemeden sonra cihaz nozulunde hasar, partikülat veya deformasyon görürseniz başka bir AcrySof® IQ GİL ve UltraSert™ Pre-loaded Enjektör Sistemi kullanın. Cihaz tamamen bütün değilse veya piston ya da piston kilidi sevkiyat esnasında yerinden oynamışsa, başka bir AcrySof® IQ GİL ve UltraSert™ Pre-loaded Enjektör Sistemi kullanın (bkz. İADE ÜRÜN POLİTİKASI).
†Reg. TM of E.I. DuPont De Nemours & Co.
Lens enjeksiyon için hazırlamaya uygun olduğunda sırasıyla 5., 6. ve 7. Adımları her adım arasındaki süreyi asgari düzeyde tutarak uygulayın.
5. Adım Viskoelastik kanülü, Şekil 5’te gösterildiği gibi cihazın lens stopunda bulunan viskoelastik delikten geçirerek cihaza dikey olacak şekilde iyice yerleştirin. Nozul ucundaki hatta viskoelastik akışı görülene kadar cihazı doldurun (Şekil 5’teki ok işaretine bakın), ardından kanülü çekin. Bunun için yaklaşık olarak 0,2 mL viskoelastik gerekecektir. Sadece ameliyathane sıcaklığına gelmesine izin verilen UltraSert™ Pre-loaded Enjektör Sistemi ile kullanımı uygun olan bir Alcon viskoelastiği kullanın. Alcon’un uygun olarak belirlediği viskoelastik listesi için bu pakette bulunan Eke bakın.
Bkz. İngilizce dilindeki Şekil 5
Viscoelastic Port = Viskoelastik Port“Fill-To” Line = “Doldurma Sınırı” Hattı
6. Adım. Mavi renkli lens stopunu cihazdan kaldırırken okun bulunduğu tırnak bölümünden tutup hafifçe ileriye doğru eğerek çıkarın (Şekil 6a). Mavi renkli piston kilidini tutup cihazdan yukarı doğru kaldırarak çıkarın (Şekil 6b). Lens stopunu ve piston kilidini atın. Lens stopu çıkarıldıktan sonra cihaza viskoelastik eklemeye çalışmayın, aksi halde lens zarar görebilir. Piston kilidi çıkarıldıktan sonra pistonu geri çekmeye çalışmayın, aksi halde piston zarar görebilir.
140
Bkz. İngilizce dilindeki Şekil 6a ve 6b
7. Adım. Optik bölümün ön kenarı enjektör ucu üzerindeki hatla eşit seviyede olana kadar pistonu düzgün ve devamlı bir hareketle ileri doğru hafifçe itin (Bkz. Şekil 7a) Bu noktada mavi yay Şekil 7b’de gösterildiği gibi açık parmak ana gövde ile temas edecektir. Bu piston hareketi en az 7 saniye sürmelidir. Lensin herhangi bir bölümünün enjektör ucundan çıkmasına izin vermeyin. Pistonun ani bir hareketle itilmemesi önemlidir, aksi halde uygun olmayan bükülme ve lenste hasar meydana gelebilir.
Bkz. İngilizce dilindeki Şekil 7a ve 7b
Fill-To Line = Doldurma Sınırı Hattı
8. Adım. Lens enjektör uç hattına ilerletildikten sonra haptik bölümün konumunu belirlemek için lens gözle kontrol edilmelidir. Piston arka optik kenara temas etmelidir. Lensin hiçbir kısmı, insizyon ile yerleştirme öncesinde enjekör ucundan çıkmamalıdır (Bkz. Şekil 8). Lens nozul hattındaki konumuna geldiğinde 3 dakikada implante edilmelidir.
Bkz. İngilizce dilindeki Şekil 8
“Proceed with full plunger advancement to fully deliver lens” = “Lensi tam olarak iletmek için pistonu tam olarak iterek devam edin”
“Proceed” = “Devam Edin”“Proceed with full plunger advancement to fully deliver lens” = “Lensi tam olarak iletmek için pistonu tam olarak
iterek devam edin”“Proceed with device rotations per Step 10” = “10. Adıma göre cihaz rotasyonları ile devam edin”
“Do not proceed” = “Devam etmeyin”
9. Adım. Lensin uygun şekilde konumlandığını ve haptik bölümün uygun biçimde katlandığını teyit ettikten sonra enjektör ucunu derinlik korumasına kadar insizyona yerleştirerek ve enjektör ucunu ön kapsül açıklığına yönlendirerek lens implantasyonu ile devam edin (Şekil 9).
Bkz. İngilizce dilindeki Şekil 9
Depth Guard = Derinlik Koruması
10. Adım. Pistonu düzgün ve sürekli bir hareketle tek seferde hafifçe itin. Pistonu çok hızlı itmeyin, aksi halde lenste hasar oluşabilir. Lensin iletiminin gözle takibine en az 5 saniyede devam edilmesi önerilmektedir. Öndeki haptik bölümü düz veya kıvrımlıysa ve lensin önüne çıkmışsa, öndeki haptik bölümün kapsüler kese içine doğru şekilde yerleşmesini sağlamak için pistonu itmeden önce cihazı sola doğru eğimli olacak şekilde döndürün. Öndeki haptik bölümü enjektör ucundan çıkmaya başladığında öndeki haptik bölümü kapsüler keseye doğru olacak şekilde yönlendirerek yerleştirin ve pistonu yavaşça itmeye devam ederek lensi iletin. Optik enjektör ucundan çıktığında, lensin kapsüler kese içinde ön tarafa doğru açılmasını sağlamak için gerekirse cihazı merkeze veya hafifçe sağa doğru eğimli olarak döndürün.
11. Adım. Lensi kapsüler kese içinde irise paralel olarak düzlemsel biçimde konumlandırmak için uygun bir konumlandırma aleti kullanın.
12. Adım. Cihazın tamamını atın. UltraSert™ Pre-loaded Enjektör Sistemini tekrar kullanmayın.
HASTA KAYDI VE RAPORLAMASIPakette bulunan Hasta Kimlik Kartı, hastanın ileride danışacağı herhangi bir göz hekimine kalıcı bir kayıt olarak sunması için talimatlarla birlikte doldurulup hastaya verilmelidir.Amerika Birleşik Devletleri’nde her hasta bu lenslerden herhangi birinin implante edilmesinden hemen sonra Alcon Laboratories, Inc.’ye kaydedilmelidir. Kayıt işlemi, lens kutusunda bulunan İmplant Kayıt Kartı doldurularak ve Alcon Laboratories, Inc.’ye postalanarak yapılır. Hasta kaydı uzun vadeli hasta takip programı için önemlidir ve Alcon Laboratories, Inc.’ye advers gelişme raporlarına yanıt vermesinde yardımcı olacaktır.Amerika Birleşik Devletleri dışında ürün kaydına ilişkin yerel yasalara uyulmalıdır.
Bir tıbbi cihazın performans özelliklerini yerine getirememesi veya amacın dışında kullanılması nedeniyle gerçekleşen durumlar da dahil olmak üzere lensin ölüme veya ciddi yaralanmalara neden olduğu veya bu durumların gerçekleşmesine katkıda bulunduğunu makul olarak gösteren hadiseler Alcon Laboratories, Inc.’ye bildirilmelidir. Bu bilgiler göz içi lens implantasyonunun uzun vadeli potansiyel etkilerini belgelemek amacıyla tüm cerrahlardan istenmektedir. Cerrahlar bu göz içi lenslerin dahil olduğu advers olayların bildirilmesi için aşağıdaki adresi ve telefon numarasını kullanmalıdır.
141
Alcon Laboratories, Inc.Medical Safety (AB 2-6)6201 South FreewayFort Worth, TX 76134-2099Ücretsiz telefon hattı: 1-800-757-9780Amerika Birleşik Devletleri dışındaki operatörler, advers gelişmelerin bildirilmesiyle ilgili olarak yerel Alcon ofisleri veya distribütörleri ile iletişime geçmelidir (Alcon Türkiye İrtibat Numaraları: Tel: 0 216 681 03 00, Faks: 0 216 425 68 80, e-posta: [email protected]).
AcrySof® Katlanır Arka Kamara Lensi Klinik ÇalışmalarıAcrySof® Tek Parça Katlanır Arka Kamara Lensleri üzerinde iki randomize, prospektif ve iyi kontrollü klinik çalışma yapılmıştır. İlk çalışma AcrySof® Natural Tek Parça Arka Kamara Lens Modeli SB30AL’nin (UV ve mavi ışık filtreli) hasta lens modeli olarak güvenliğini ve etkinliğini göstermek için uygulanmıştır. Bu, AcrySof® Model SA30AL’nin (sadece UV emen) bir kontrol lensi olarak yer aldığı randomize bir klinik çalışmaydı. Sadece bir Model SB30AL veya Model SA30AL göz içi lensini alan hastaların ilk operatif gözüyle ilgili veriler dahil edilmiştir. Asferik optik ve AcrySof® Katlanır Tek Parça kontrol lensli AcrySof® IQ Katlanır Tek Parça GİL’nin (UV ve mavi ışık filtreleyen kromoforlu) ikinci bir randomize klinik çalışması, geleneksel bir sferik tasarım üzerindeki klinik/fonksiyonel yararları değerlendirmek amacıyla gerçekleştirilmiştir.
AcrySof® Natural Tek Parça Lens Model SB30AL Klinik ÇalışmasıPostoperatif olarak minimum bir yıl boyunca başarıyla takip edilen hastalardan alınan sonuçlar, AcrySof® Natural Tek Parça lens Model SB30AL’nin afakinin görsel olarak düzeltilmesi için güvenli ve etkili bir cihaz olduğuna dair makul bir güvence vermektedir.
AcrySof® Natural Tek Parça Lens Model SB30AL Hasta PopülasyonuBu iki kollu çalışmada en azından ilk operatif göze Model SB30AL’nin implante edildiği hasta popülasyonunun %70,6’sı kadın, %29,4’ü ise erkektir. Model SA30AL (kontrol) göz içi lensin implante edildiği hasta popülasyonunun %60,5’i kadın, %39,5’i erkektir. Model SB30AL popülasyonundaki ırklardan %95,3’ü Beyaz, %4,7’si ise Siyahidir. Kontrol (SA30AL) hasta popülasyonunun ise %96,6’sı Beyaz, %2’si Siyahi ve %1,4’ü diğer ırklardandır. Model SB30AL’yi en azından ilk operatif gözüne alan toplam popülasyonun ortalama yaşı 72,9’dur. Benzer şekilde Model SA30AL’yi (kontrol) alan toplam popülasyonun ortalama yaşı 71,9’dur.
AcrySof® Natural Tek Parça Lens Model SB30AL Görme KeskinliğiHerhangi bir zamanda preoperatif oküler patolojisi, anormal korneası veya maküler dejenerasyonu olmayan (en iyi durum) hastalar arasında minimum bir yıl süreyle postoperatif olarak elde edilen görme keskinliğinin bir özeti Tablo 2a’da, toplam hasta popülasyonunun elde ettiği görme keskinliği ise Tablo 3a’da verilmiştir. Aynı veri setleri için kontrol verileri sırasıyla Tablo 2b ve 3b’de bulunmaktadır. Model SB30AL ve kontrol lensi Model SA30AL arasında herhangi bir en iyi durumda ve tüm veri setlerinde istatistiksel olarak önemli bir fark bulunmamaktadır.
Bkz. İngilizce dilindeki Tablo 2aPostoperatif Minimum Bir Yıllık Sürede En İyi Durumdaki Hasta
Popülasyonunun En İyi Düzeltilmiş Görme Keskinliği, AcrySof® Natural GİL SB30AL
Keskinlik
Her bir 20/20 veya Daha İyisi 20/25 20/30 20/40 20/40 veya
Daha İyisi> 20/40 ila
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Yaş Kategorisi
Bkz. İngilizce dilindeki Tablo 2bPostoperatif Minimum Bir Yıllık SüredeEn İyi Durumdaki Hasta
Popülasyonunun En İyi Düzeltilmiş Görme Keskinliği, AcrySof® GİL SA30AL kontrol
Keskinlik
Her bir 20/20 veya Daha İyisi 20/25 20/30 20/40 20/40 veya
Daha İyisi> 20/40 ila
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Yaş Kategorisi
142
Bkz. İngilizce dilindeki Tablo 3aPostoperatif Minimum Bir Yıllık SüredeTüm Hasta
Popülasyonunun En İyi Düzeltilmiş Görme Keskinliği, AcrySof® Natural GİL SB30AL
Keskinlik
Her bir 20/20 veya Daha İyisi 20/25 20/30 20/40 20/40 veya
Daha İyisi> 20/40 ila
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Yaş Kategorisi
Bkz. İngilizce dilindeki Tablo 3bPostoperatif Minimum Bir Yıllık SüredeTüm Hasta
Popülasyonunun En İyi Düzeltilen Görme Keskinliği, AcrySof® GİL SA30AL kontrol
Keskinlik
Her bir 20/20 veya Daha İyisi 20/25 20/30 20/40 20/40 veya
Daha İyisi> 20/40 ila
< 20/80 > 20/80
N N % N % N % N % N % N % N %
Yaş Kategorisi
AcrySof® Natural Tek Parça Lens Model SB30AL Kümülatif Advers Olaylar AcrySof® Natural Tek Parça Lens Model SB30AL ve Model SA30AL hastaları için -bir yıl dahil olmak üzere- minimum bir yıl postoperatif süredeki advers olaylarınkümülatif oranları Tablo 4’te gösterilmiştir. Kümülatif advers olaylardan herhangi birini yaşayan hastaların oranında, Model SB30AL ve Model SA30AL arasında istatistiksel olarak herhangi bir önemli fark görülmemiştir.
Tablo 4Minimum Bir Yıllık Postoperatif Süredeki Kümülatif Advers Olaylar
Sekonder Yeniden Cerrahi MüdahaleKalan Korteksin ÇıkarılmasıEksplant (kapsüler rüptür nedeniyle dislokasyon)Retina Yırtığının Onarılması için KriyoterapiGİB’in Düşürülmesi için ParasentezFokal Lazer TedavisiFotodinamik TerapisiBiyometri Hatası Nedeniyle Eksplant
51 1 11100
3,30,70,70,70,70,70,00,0
20000011
1,40.00.00.00.00.00,70,7
0,4482
Hifema 0 0.0 0 0.0 Uygulanamaz
*Model SB30AL ve Model SA30AL karşılaştırıldığında Fisher Kesin Testindeki p-değerleri
143
AcrySof® Natural Tek Parça Lens Model SB30AL Devam eden Advers OlaylarAcrySof® Natural Tek Parça Lens Model SB30AL hastaları ve Kontrol Modeli SA30AL için minimum bir yıl postoperatif süredeki bu advers olayların devamlılık oranları Tablo 5’te gösterilmiştir. Devam eden advers olaylardan herhangi birini yaşayan hastaların oranında Model SB30AL ve Model SA30AL arasında istatistiksel olarak herhangi bir önemli fark görülmemiştir.
Tablo 5Postoperatif Minimum Bir Yıllık Sürede Devam Eden Advers Olaylar
AcrySof® Natural GİL SB30AL ve SA30AL kontrol
SB30AL (N= 138)
SA30AL(N= 127) p-değeri*
N % N %
Advers Olay Türü
0 0 1 0,8 0,4792Sürekli Kornea Ödemi
İritis 0 0 0 0.0 Uygulanamaz
Maküler Ödem 2 1,4 1 0,8 1.0000
Vitrit 0 0 0 0.0 Uygulanamaz
Tedavi Gerektiren Artmış GİB 0 0 0 0.0 Uygulanamaz
*Model SB30AL ve Model SA30AL karşılaştırıldığında Fisher Kesin Testindeki p-değerleri
AcrySof® Natural Tek Parça Lens Model SB30AL Renk Algılama120 ila 180 günlük postoperatif sürede Farnsworth D-15 Panel Testi ile yapılan renk algılama testi uygulanmıştır. İlk postoperatif göze Model SB30AL implante edilen ve 120 ila 180 günlük postoperatif ziyarette muayene edilen ve normal renk görüşüne sahip 109 hastadan 107’si (%98,2) renk algılama testini geçmiştir. İlk postoperatif göze bir Model SA30AL implante edilen ve 120 ila 180 günlük postoperatif ziyarette muayene edilen ve normal renk görüşüne sahip 102 hastadan 97’si (%95,1) renk algılama testini geçmiştir. 120 ila 180 günlük postoperatif ziyarette renk algılama testini geçen hastaların oranı bakımından Model SB30AL ve Model SA30AL arasında istatistiksel olarak önemli oranda bir farklılık görülmemiştir. Dolayısıyla tescilli kromoforun eklenmesi normal renk görüşüne sahip hastalarda renk görüşünü olumsuz olarak etkilememektedir.
AcrySof® Natural Tek Parça Lens Model SB30AL Nd:YAG Oranları 21,6 aylık ortalama takip süresi ile SB30AL implante edilmiş 135 hastadan üçü (3) (%2,2) Nd:YAG arka kapsülotomiye maruz kalmıştır. 21,9 aylık ortalama takip süresi ile SA30AL implante edilmiş 127 hastadan ikisi (2) (%1,6) Nd:YAG arka kapsülotomiye maruz kalmıştır.
AcrySof® IQ Lens Klinik ÇalışmasıÖnceden uygulanan benzer GİL çalışmalarının tasarımıyla tutarlı olarak bilateral kataraktı olan ve genel göz sağlığı iyi durumda olan (örn.: önceden göz ameliyatı olmamış, görme keskinliğini önemli derecede etkileyecek dejeneratif görme bozukluğu veya hastanın sağlığını riske atacak ciddi akut veya kronik bir durumu olmayan) yetişkin hastalar AcrySof® IQ lens ve sferik kontrol lensinin kontrollü, randomize, çift kör, çok merkezli, kontralateral implant klinik araştırmasına dahil edilmiştir. AcrySof® IQ lensi ile oküler sferik aberasyonlar kontrol lensinden istatistiksel olarak önemli oranda daha az olmuştur. Kontrast hassasiyeti sonuçları AcrySof® IQ’nun implante edildiği gözlerin lehine istatistiksel olarak önemli bir postoperatif (3 ayda) gelişme göstermiştir. Ayrıca AcrySof® IQ lensin implante edildiği gözlerde, özellikle parlak ışık ve sis gibi test edilen zorlu koşullarda fonksiyonel görüş ölçümünde, simüle gece sürüşünde istatistiksel ve klinik olarak önemli ölçüde gelişme görülmüştür. Bu sonuçlar, monofokal AcrySof® GİL ‘e (asferik optik ve mavi ışık filtreleyen kromofor olmadan) kıyasla AcrySof® IQ GİL’in (mavi ışık filtreleyen bir kromofor içeren bir madde platformu üzerindeki bir asferik optik) yararlı klinik performans gösterdiğini yansıtmaktadır.
AcrySof® IQ Lens – Sferik ve Toplam Yüksek Dereceli AberasyonAcrySof® IQ gözlerin ortalama oküler sferik aberasyonu yaklaşık 0,1 mikrometre olmuştur. Şekil 10, AcrySof® IQ lensin lehine gözlemlenen sferik ve toplam yüksek dereceli aberasyonlarda istatistiksel olarak önemli oranda düşüşü yansıtmaktadır. Şekil 11, lens ve yaş grubuna göre wavefront aberometresi ölçümleri ile tüm gözlerin ortalama sferik aberasyon ölçümlerini vermektedir. Bu çizelgede anlatıldığı üzere AcrySof® IQ gözlerinin sferik aberasyonundaki düşüş yaştan bağımsız olmuştur.
Bkz. İngilizce dilindeki Şekil 10Sferik ve Toplam Yüksek Dereceli RMS
2. Göz İmplantından sonraki 90-120 Gün
144
Bkz. İngilizce dilindeki Şekil 11Genel ve Yaş Grubuna Göre Ortalama Sferik Aberasyon
2. Göz İmplantından sonraki 90-120 Gün
AcrySof® IQ Lens – Uzak Mesafe Görme KeskinliğiAcrySof® IQ lensi ve kontrol lensi, klinik olarak benzer postoperatif görme keskinliğini sağlamıştır. Monoküler görme keskinliği sonuçları, Şekil 12 ve 13’te gösterilmiştir.
Bkz. İngilizce dilindeki Şekil 12LogMAR BCVA
Bkz. İngilizce dilindeki Şekil 13LogMAR UCVA
AcrySof® IQ Lens - Kontrast HassasiyetiKlinik araştırmanın asıl amacı, AcrySof® IQ lensinin kontrol lensi üzerindeki üstünlüğünü Vector Vision CSV-1000 (3 cd/m2’lik çizelge parlaklığı ile) kullanılarak iki uzaysal frekanstan (derece başına 3 veya 6 devir) herhangi birinde parlama ile, veya parlama olmadan mezopik koşullar altında postoperatif olarak ölçülen ortalama kontrast hassasiyeti aracılığıyla kanıtlamaktır. Aynı zamanda hastaların bir alt kümesinde, Fonksiyonel Keskinlik Kontrast Testi (FKKT) de uygulanmıştır (3 cd/m2’lik çizelge parlaklığı ile). Bu klinik araştırmada AcrySof® IQ lensinin mezopik koşullar altında kontrol lensi üzerindeki üstünlüğü, hem parlama ile hem de parlama olmadan (CSV-1000) derece başına 6 devirde ve parlama olmadan derece başına 3 ve 6 devirde (FKKT) kanıtlanmıştır. Şekil 14 ve 15, hem AcrySof® IQ lensi ve hem de kontrol lensi için test edilen uzaysal frekanslardaki mezopik hassasiyet sonuçlarını göstermektedir.
Bkz. İngilizce dilindeki Şekil 14Mezopik Kontrast Hassasiyeti (CSV-1000)2. Göz İmplantından sonraki 90-120 Gün
Bkz. İngilizce dilindeki Şekil 15Mezopik Kontrast Hassasiyeti (FKKT) Alt Çalışması
2. Göz İmplantından sonraki 90 Gün
AcrySof® IQ Lensi – Gece Sürüş SimülasyonuHastaların bir alt kümesi onaylanmış bir gece sürüş simülatörüne tabi tutulmuştur. Hastalar, normal, parlak ve sisli atmosferde şehir ve kırsal ortamı simüle eden koşullarda monoküler olarak test edilmiştir.Gece şehirde sürüş ortamında, bu koşullarda genel olarak bulunan yüksek düzeydeki ortam ışıklandırmasını yeniden oluşturmak amacıyla çeşitli sokak ışıkları, araç ışıkları, mağaza ışıkları ve işaretler yer almaktadır. Gece kırsal sürüş ortamında ise asgari düzeyde ortam ışıklandırması kullanılmaktadır. Şehir ve kırsal ortamlar için sırasıyla yaklaşık 35 mph ve 55 mph simüle sürüş hızları kullanılmıştır.Hastalardan, yeşil-beyaz renkli karayolu bilgilendirme levhaları, siyah-sarı renkli uyarı levhaları ve yayalar dahil olmak üzere her bir ortamdaki bir dizi hedefi tespit etmeleri ve tanımlamaları istenmiştir. Hastalardan ilk hedefi gördüklerinde kayıt için bir tespit mesafesi sunarak yanıt vermeleri istenmiştir. Daha sonra bir tanımlama mesafesinin kaydedilebilmesi için hastalardan hedefi (örn.: levhada ne yazıyor, yaya hangi yöne yürüyordu gibi) ayırt edebildikleri zaman yanıt vermeleri istenmiştir. Şekil 16-19, hem tespit hem algılama mesafeleri için şehir ve kırsal sürüş ortamlarında AcrySof® IQ lensi ve kontrol lensi arasındaki ortalama farklılıkları göstermektedir (örn.: insanlar arasındaki mesafe farklarının ortalaması).AcrySof® IQ lensi, test edilen 36 durumdan 34’ünde kontrol lensinden fonksiyonel olarak daha iyi performans göstermiştir ve test edilen çeşitli sürüş koşulları altında (normal, parlak, sisli) şehir ve kırsal sürüş ortamlarında tespit ve algılama mesafelerindeki gelişmeyi yansıtmaktadır. Ayrıca AcrySof® IQ lensi istatistiksel olarak da bu koşulların 12’sinde kontrol lensinden daha iyi performans göstermiştir ve bunun en önemli etkisi ve en büyük avantajı şehirdeki yayaların (parlak ve sisli koşullarda) ve kırsal uyarı levhalarının (parlak ve sisli koşullarda) tespit edilmesi ve tanımlanmasında gözlemlenmiştir. Şehir ortamında düşük görüş koşullarında (parlak, sisli) 35 mph’deki yüksek görüş mesafesi, AcrySof® IQ lensine şehirde daha yaygın olarak karşılaşılan bir tehlike olarak bir yaya hedefine fazladan 0,5 saniye yanıt verme süresi kazanılmasını sağlamaktadır. Bu 0,5 saniyelik artış, durma, kaçma gibi uygun eylemlerin yapılması için daha fazla süre kazanılması bakımından fonksiyonel olarak önemlilik arz etmektedir (Green, 2000; McBride ve Matson, 2004). Kırsal ortamdaki tüm koşullarda 55 mph’deki yüksek görüş mesafesi AcrySof® IQ lensine, kırsal alanlarda sıklıkla karşılaşılan uyarı levhalarının algılanması için fazladan 1 saniye daha kazandırmaktadır. 0,5 saniyelik bir artış, ortam ışığının genelde olmadığı ve bilinmedik kırsal alanlarda gece saatlerinde kritik önem taşıyan sürüş esnasında uygun eylemin yapılması için daha fazla sürenin kazanılmasında fonksiyonel olarak önemlilik arz etmektedir. Alt çalışmada postoperatif olarak maküler dejenerasyon veya PCO’ya maruz kalan 6 hasta olmuştur. Bu hastalar sürüş analizinden çıkarıldığında, şehir ortamında parlak ışık koşulları altında yaya hedeflerinin tespit edilmesi ve algılanması bakımından GİL’ler arasındaki fark, klinik anlamlılık için 0,5 saniyelik eşiğin altına düşmüştür. Orijinal analizler çokluluk için ayarlandığında, parlak ışık koşulları altında (Hommel p-değeri = 0,0539) yazıların şehir ortamında tespitinde veya parlak ışık koşulları altında (Hommel p-değeri = 0,0507) yayaların kırsal ortamda tespitinde GİL’ler arasındaki fark istatistiksel olarak önemli oranda olmamıştır.Bu sonuçlar, gelişmiş fonksiyonel görüşü ve AcrySof® IQ lense sahip yaşlı sürücüler ve bu sürücülerin aynı yolu paylaştıkları diğer sürücüler ve yayalar için muhtemel olarak anlamlı güvenlik yararlarını göstermektedir. Buradaki sonuçlar AcrySof® IQ lensinin fonksiyonel görüşü geliştirdiğini ve dolayısıyla düşük görüş koşulları altında diğer yaşamsal önem arz eden durumlar bakımından hasta güvenliğini iyileştirebileceğini göstermektedir.
145
Bkz. İngilizce dilindeki Şekil 16Gece Sürüş Simülatörü
Görüş Mesafelerinin Tespitinde Kişiler Arası Farkların Ortalaması, Tespit, ŞehirPostoperatif Olarak Minimum 90 GünAcrySof® IQ GİL – Kontrol (n = 44)
Bkz. İngilizce dilindeki Şekil 17Gece Sürüş Simülatörü
Görüş Mesafelerinin Algılanmasında Kişiler Arası Farkların Ortalaması, ŞehirPostoperatif Olarak Minimum 90 GünAcrySof® IQ GİL – Kontrol (n = 44)
Bkz. İngilizce dilindeki Şekil 18Gece Sürüş Simülatörü
Görüş Mesafelerinin Tespitinde Kişiler Arası Farkların Ortalaması, KırsalPostoperatif Olarak Minimum 90 GünAcrySof® IQ GİL – Kontrol (n = 44)
Bkz. İngilizce dilindeki Şekil 19Gece Sürüş Simülatörü
Görüş Mesafelerinin Algılanmasında Kişiler Arası Farkların Ortalaması, KırsalPostoperatif Olarak Minimum 90 GünAcrySof® IQ GİL – Kontrol (n = 44)
NASIL TEDARİK EDİLMEKTEDİRBu arka kamara göz içi lensleri kuru olarak UltraSert™ Pre-loaded Enjektör Sisteminde, primer sterilizasyon paketi içinde ve nihai olarak etilen oksit ile sterilize edilmiş olarak tedarik edilmektedir. Primer sterilizasyon paketi sadece aseptik koşullarda açılmalıdır (bkz. KULLANIM TALİMATLARI).
SON KULLANMA TARİHİPrimer sterilizasyon paketi zarar görmediği veya açılmadığı sürece sterilite garanti edilmektedir. Son kullanma tarihi UltraSert™ Pre-loaded Enjektör Sisteminin dış kutu etiketinde açık biçimde gösterilmiştir. Son kullanma tarihi geçen tüm lensler Alcon Laboratories, Inc.’ye iade edilmelidir (bkz. İADE ÜRÜN POLİTİKASI)
İADE ÜRÜN POLİTİKASIAmerika Birleşik Devletleri’nde iade edilen lensler sadece diğer ürünlerin karşılığında kabul edilecektir, borç karşılığında kabul edilmeyecektir. Tüm iadelerde bir Alcon Laboratories, Inc. İade Ürün Numarası bulunmalıdır ve iade edilen ürünler takip edilebilecek şekilde sevk edilmelidir. İade Ürün Numarasını Alcon Müşteri Hizmetleri Departmanı ile iletişime geçerek alabilirsiniz. Bu numaranın verilmesi iade ürünlerin nihai olarak kabul edildiği anlamına gelmez. Takas dahil olmak üzere ayrıntılı politika ilkeleri için lütfen Satış veya Müşteri Hizmetleri Temsilcinizle iletişime geçin. Amerika Birleşik Devletleri dışındaki kişilerin geçerli iade ürün politikası için yerel Alcon ofisleri veya bayileri ile iletişime geçmesi gerekmektedir. (Alcon Türkiye İrtibat Numaraları: Tel: 0 216 681 03 00, Faks: 0 216 425 68 80, e-posta: [email protected]).
REFERANSLAR 1. Boettner, E.A. and Wolter, J.R. Transmission of the ocular media. Invest. Ophthalmol. 1:776-783, 1962. 2. Girard, L.J., ve ark. Complications of the Simcoe Flexible Loop Phacoprosthesis in the anterior chamber.
Ophthalmic Surg. 14(4):332-335, 1983. 3. Holtz, S.J. Postoperative capsular bag distension. J. Cataract Refract. Surg. 16(5):310-317, 1992. 4. Willis, D.A., ve ark. Pupillary block associated with posterior chamber lenses. Ophthalmic Surg. 16(2):108-109,
1985. 5. Green, M. “How Long Does it Take to Stop?” Methodological Analysis of Driver Perception-Brake Times.
Transportation Human Factors. 2(3):195-216, 2000. 6. McBride, D.K. and Matson, W. Assessing the Significance of Optically Produced Reduction in Braking Response
Time: Possible Impacts on Automotive Safety Among the Elderly. PIPS-50-03. Arlington, VA: Potomac Institute for Policy Studies; May 2004.
146
ETİKETTE KULLANILAN SEMBOLLER
SEMBOL TÜRKÇESİIOL Göz içi lens
SP Tek Parça
UV Ultraviyole
D Diyoptri
ØB Gövde çapı (Optik çap)
ØT Genel çap (Genel uzunluk)
2STERILIZE Yeniden sterilize etmeyin
Yeniden kullanmayın
Son kullanım tarihi
STERILE EO Etilen oksit kullanarak sterilize edilmiştir
SN Seri numarası
Dikkat
Üretici
EC REP Avrupa Topluluğundaki Yetkili Temsilci
Üst Sıcaklık Sınırı
Buradan açın
Kullanım talimatlarına bakın
Steril paket açılmış veya zarar görmüşse kullanmayın
Dikkat: ABD Federal yasaları bu cihazın satışını, sadece bir hekime veya hekimin siparişi üzerine yapılacak şekilde sınırlamaktadır.
Alcon Laboratories, Inc6201 South FreewayFort Worth, TX76134-2099 USA
EC REP
Alcon Laboratories (UK) Ltd.Frimley Business ParkFrimley, CamberleySurrey, GU16 7SR,Birleşik Krallık