53
5 th VISEGRAD SYMPOSIUM ON STRUCTURAL SYSTEMS BIOLOGY PROGRAM & ABSTRACTS 17 th - 20 th June 2015 Szeged Hungary
| Date post: | 13-Jun-2018 |
| Category: |
Documents |
| Upload: | nguyenthuy |
| View: | 216 times |
| Download: | 0 times |

5th VISEGRAD SYMPOSIUM ON
STRUCTURAL SYSTEMS
BIOLOGY
PROGRAM & ABSTRACTS
17th- 20th June 2015
Szeged
Hungary

1

2
Organization
Department of Chemical Informatics, University of Szeged
Partners
Institute of Nanobiology and Structural Biology Global Change Research Centre Academy of Sciences of the Czech Republic Comenius University Bratislava University of Warsaw Jagellonian University in Krakow Medical University of Vienna Academy and University Center of Nove Hrady Infrastructure for System Biology Europe
Scientific Organizing Committee Babak Minofar
Rüdiger Ettrich
David Řeha,
Ján Urban
Béla Viskolcz
Imre G. Csizmadia
Thomas Stockner
Marta Pasenkiewicz-Gierula
Joanna Sułkowska
Local Organizing Team Béla Viskolcz
Imre G. Csizmadia
Csaba Hatvani
Anita Rágyanszki
János J. Szórád
Balázs Jójárt
Milán Szőri

3
5th Visegrad Symposium on Structural System Biology program Wednesday June 17 10.00-18.00 Registration
13:30-13:45 Béla Viskolcz: Conference opening
13:45-14:00 Imre G. Csizmadia: Past, present and future of V4 Opening session Chairperson: Richard Buchner 14:00-14:45 Maxim Fedorov (Glasgow): Modelling solvation properties of bio-active
molecules by molecular theory: achieving chemical accuracy 14.45-15:30 Pál Jedlovszky (Budapest): Effect of anaesthetics on the properties of a lipid
membrane in the biologically relevant phase. A computer simulation study 15:30-16:15 Joanna Sułkowska (Warsaw): Free energy landscape of protein with
complex topology, Tadpoles: new entangled motifs in proteins
16:15-16:30 Coffee break
1st Session Chairperson: Babak Minofar 16:30-17:00 Christian Schröder (Vienna): Computational dielectric spectroscopy of
proteins in various solvents 17:00-17:30 Richard Buchner (Regensburg): Dielectric Spectroscopy of Hydration and
Ion Binding 17:30-18:00 Svend Knak Jensen (Aarhus): The quest for methane on Mars – Methane as
a biomarker for past or present life
19:00 - Welcome party, buffet dinner (Jósika Pince)
Thursday June 18 2nd Session Chairperson: Jost Ludwig 09:00-09:30 Michelle A. Sahai (London): Exploring a Common Thread between
Addiction and Disease: The Dopamine Transporter 09:30-10:00 Michael C. Owen (Jülich): The Study of Oxidative Stress in
Alzheimer's Disease: A Theoretical Approach 10:00-10:30 Pavel Grinkevich (Nové Hrady): Structure and function of C-terminal
helical domain of the motor subunit HsdR from the type I restriction-modification system EcoR124I
10:30-11:00 Coffee break
3rd Session Chairperson: Joanna Sułkowska 11:00-11:30 Giorgia Brancolini (Oristano): Computational Strategies for
Amyoloidogenic Proteins interacting with Gold Nanoparticles 11:30-12:00 Jost Ludwig (Nové Hrady): The roles of conserved residues in the
Saccharomyces cerevisiae K+-translocation protein Trk1 analyzed by theoretical and experimental approaches
12:00-12:30 Ben Luisi (Cambridge): The structure and mechanism of a molecular

4
machine of drug transport 12:30-12:45 Anita Rágyanszki (Szeged): Analytical functional representation of quantum
chemical potential energy curves and surfaces
12:45-14:00 Lunch
4th Session Chairperson: Ján Urban
14:00-14:30 James J. Valdés (České Budějovice): Ticks as venomous animals: a
pharmacological perspective
14:30-15:00 Szilárd Fejér (Szeged): Cooperative rearrangements in clusters of anisotropic
particles
15:00:15.30 Nacer Idrissi (Oran): Luteolin- organic solvent interactions. A
moleculardynamics simulation analysis
15:30-16:00 Coffee break 5th Session Chairperson: Giorgia Brancolini 16:00-16:30 Milan Melicherčí: Changes induced by ligand binding to arginine repressor
from Bacillus subtilis
16:30-17:00 Stefan Balint (Timisoara): Prediction of the pollution due to a pesticide
17:00-17:30 Imre Jákli (Budapest): Extraordinary thermal stability of Trp-cage
miniproteins observed by chiroptical and NMR spectroscopy
18:00 - Dinner, Freetime Activities, Walk around Town, Poster Session
Friday June 19 6th Session Chairperson: Ben Luisi
09:00-09:15 Balázs Fábián (Budapest): Floating Patches of HCN at the Surface of Their
Aqueous Solutions – Can They Make “HCN World” Plausible?
09:15-09:30 Zsófia Borbála Rózsa (Szeged): Mapping out the 366 shades of the C4H8O2
isomers
09:30-10:00 Caroline Lynn Kamerlin (Uppsala):
10:00-10:30 Coffee break
7th Session Chairperson: Caroline Lynn Kamerlin
10:30-11:00 Ján Urban (Bratislava): Computational study concerning RNA or DNA
aptamers
11:00-11:30 Sebastian Kmiecik (Warsaw): Protein-peptide docking with significant
conformational changes and without prior knowledge of the binding site using
the CABS-dock web server
11:30-12:00 Balázs Jójárt (Szeged): MODYDB – just an other database?

5
12:30-13:30 Lunch
8th Session Chairperson: Nacer Idrissi 13:30-13:45 Lily Hunnisett (Szeged): The Cannabinoids: Does Cooperative Interplay
Underlie Their Therapeutic Nature? 13:45-14:15 Piotr Setny (München): Hydration in discrete water - from bulk free energies
to localised water molecules 14:00-14:30 Giancarlo Franzese (Barcelona): How water contributes to pressure and
cold denaturation of proteins
15:30-18:30 Trip around Szeged finished at the Monastery
20:00 - Conference dinner at the Monastery
Saturday June 20
Morning Departure

6
Abstracts of oral presentations
The authors of the abstracts bear the full responsibility for the scientific and
linguistic content.

7
Modelling solvation properties of bio-active molecules by molecular
theory: achieving chemical accuracy
Maksim Misin1, David S. Palmer2, Maxim V. Fedorov1* 1Department of Physics, SUPA, University of Strathclyde, 107 Rottenrow, Glasgow, G4 0NG, UK
2Department of Pure and Applied Chemistry, University of Strathclyde, 16 Richmond Street, Glasgow, G4 0NG,
UK
Corresponding author: [email protected]
Keywords: Free energy, solvents, integral equations, chemical potential, correlation functions.
Molecular integral equations theories (IETs) provide an alternative way to molecular simulations
to calculate properties of biomolecular solvation [1,2]. In this report we will overview recent
developments in IETs with particular focus on IET-based methods for predicting thermodynamic
properties of bioactive molecules.
We recently developed a new model for computing hydration free energies based on the 3D
Reference Interaction Site Model (3D-RISM) [3]. The new adjustment to 3D-RISM theory
significantly improves hydration free energy predictions for various classes of organic molecules
at both ambient and non-ambient temperatures. An extensive benchmarking against experimental
data shows that, at least for uncharged compounds, the accuracy of the model is comparable to
(much more computationally expensive) molecular dynamics simulations.
Accuracy of predictions of other properties (aqueous solubility, octanol-water partition
coefficients, caco-2 cell permeability) by IET-based methods will be also discussed.
References
[1] E.L. Ratkova, D.S. Palmer and M.V. Fedorov, Chem. Rev., 2015, available online, DOI:
10.1021/cr5000283.
[2] D.S. Palmer, A.I. Frolov, E.L. Ratkova and M.V. Fedorov, Molecular Pharmaceuticals, 2011,
8, 1423-1429.
[3] M. Misin, M.V. Fedorov and D.S. Palmer, J. Chem. Phys., 2015, 142, 091105.

8
Effect of anaesthetics on the properties of a lipid membrane in the
biologically relevant phase. A computer simulation study
Balázs Fábián1, Mária Darvas2, Sylvain Picaud3, Marcello Sega4, and Pál
Jedlovszky1,5,6
1Institute of Chemistry, ELTE University, Budapest, Hungary 2SISSA, Sector of Molecular and Statistical Biophysics, Trieste, Italy
3Institut UTINAM Université de Franche-Comté, Besançon, France 4Institut für Computergestützte Biologische Chemie, University of Vienna, Austria
5MTA-BME Research Group of Technical Analytical Chemistry, Budapest, Hungary 6EKF Department of Chemistry, Eger, Hungary
Molecular dynamics simulations of the fully hydrated neat dipalmitoylphosphatidylcholine
(DPPC) membrane as well as DPPC membranes containing four different general anaesthetic
molecules, namely chloroform, halothane, diethyl ether and enflurane have been simulated at two
different pressures, i.e., at 1 bar and 1000 bar, at the temperature of 310 K. At this temperature
the model used in this study is known to be in the biologically most relevant liquid crystalline
(L) phase. To find out which properties of the membrane might possibly be related to the molecular
mechanism of anaesthesia, we have been looking for properties that change in the same way in
the presence of any general anaesthetic molecule, and change in the opposite way by the increase
of the pressure. This way, we have ruled out the density distribution of various groups along the
membrane normal axis, orientation of the lipid heads and tails, self-association of the
anaesthetics, as well as the local order of the lipid tails as possible molecular reasons of
anaesthesia. On the other hand, we have found that the molecular surface area, and hence also the
molecular volume of the membrane is increased by the presence of any anaesthetic molecule, and
decreased by the pressure, in accordance with the more than half a century old critical volume
hypothesis. We have also found that anaesthetic molecules prefer two different positions along
the membrane normal axis, namely the middle of the membrane and the outer edge of the
hydrocarbon region, close to the polar headgroups. This dual preference is explained by the
interplay of steric and electrostatic effects. The increase of the pressure is found to decrease the
former, and increase the latter preference, and hence it might also be related to the pressure
reversal of anaesthesia.

9
Free energy landscape of protein with complex topology, Tadpoles:
new entangled motifs in proteins
Joanna Sułkowska1
1 Chemistry Department and Center of New Technology, University of Warsaw
Pasteura 1, Warsaw, Poland
Corresponding author: [email protected]
We identify new entangled motifs in proteins that we call tadpoles. Tadpoles arise in proteins
with disulphide bridges (or in proteins with amide linkages), when termini of a protein backbone
pierces through an auxiliary surface of minimal area, spanned on a covalent loop. We find that
as much as 18\% of all proteins with disulphide bridges in a non-redunant subset of PDB form
tadpoles, and classify them into five distinct geometric classes. Based on biological classification
of proteins we find that tadpoles are much more common in viruses, plants and fungi than in
other kingdoms of life. During the talk I will also discuss possible functions of tadpoles. Tadpoles
and associated surfaces of minimal area provide new, interesting geometric characteristics not
only of proteins, but also of othery biomolecules, with many potential applications.

10
Computational dielectric spectroscopy of proteins in various
solvents
Christian Schröder, Michael Haberler, Gregor Neumayr, Tibor Rudas, Othmar
Steinhauser
Department of Computational Biological Chemistry, Währingerstrasse 17, A-1090 Vienna (Austria)
Dielectric spectroscopy is a wide-spread used experimental technique to study relaxation in
amorphous media, in particular liquid systems. As compared to nuclear magnetic resonance,
inelastic neutron scattering and similar techniques, dielectric experiments probe the complete
sample, i.e. they measure collective translational and rotational motion in systems governed by
electrostatic forces.
-helices are characteristic for a parallel alignment of peptide dipoles. Therefore, in this case the
collective dipole reaches its maximum value. In contrast, -sheets are typical for dipole
compensation whereas coil structures exhibit a certain residual correlation of dipoles [1-2]. In
addition, the interaction with ions, solvation water and bulk water complicates the dielectric
spectrum of these systems [1-4]. However, in computational dielectric spectroscopy, the overall
frequency-dielectric spectrum can be decomposed into its major contributions which can be
analyzed separately.
In here, we would like to present our results concerning the interaction of ubiquitin with water at
a molecular [1] and mesoscopic resolution [2] as well as solvation shell resolved [3]. Particular
interactions with ionic liquid/water mixtures and their effects on the protein structure and
dynamics are also discussed [4]. In a recent study we combined experimental and dielectric
spectroscopy to investigate the oligomerization of insulin monomers in aqueous solution [5]
showing the importance of pre-aggregated states.
References
[1] C. Schröder, T. Rudas, S. Boresch, and O. Steinhauser „Simulations studies of the protein-
water interface. I. Properties at the molecular resolution”, J. Chem. Phys. 124 (2006), 234907
[2] T. Rudas, C. Schröder, S. Boresch, and O. Steinhauser „Simulations studies of the protein-
water interface. II. Properties at the mesoscopic resolution”, J. Chem. Phys. 124 (2006), 234908
[3] G. Neumayr, T. Rudas, and O. Steinhauser “Global and local Voronoi analysis of solvation
shells of proteins”, J. Chem. Phys. 133 (2010), 084108
[4] M. Haberler, C. Schröder, and O. Steinhauser „Solvation studies of a zinc finger protein in
hydrated ionic liquids“, Phys. Chem. Chem. Phys. 13 (2011), 6924
[5] C. Schröder, O. Steinhauser, P. Sasisanker and H. Weingärtner „Orientational alignment of
amyloidogenic proteins in pre-aggregated solutions“, Phys. Rev. Lett. 114 (2015), 128101

11
Dielectric Spectroscopy of Hydration and Ion Binding
Richard Buchner1
1Institute of Physical and Theoretical Chemistry, University of Regensburg, Regensburg, Germany
Corresponding author: [email protected]
Our understanding of the physiological action of many small organic solutes, like urea or TMAO,
in water is still rather patchy. For instance, in the case of urea it is still debated whether its
denaturating effect on proteins arises from direct interaction or through its disturbing effect on
protein hydration [1,2]. Clearly, the balance of hydrophilic and hydrophobic moieties on the
molecule and their respective hydration is of major importance for the behavior of these
substances. Of at least similar relevance is the interaction of biomolecules with ions since the
latter are always present under physiological conditions.
Dielectric relaxation spectroscopy in the microwave region is a convenient tool to study hydration
as well as ion-binding phenomena [3,4]. In particular, this technique can detect various types of
ion pairs and distinguishes between bulk-like, moderately-bound (slow) and “frozen”
(irrotationally bound) water molecules. After a short introduction into the basics of DRS, its
advantages and pitfalls, we will discuss what kind of information can be deduced from dielectric
spectra and how this is linked to results from other techniques. In particular, the connections to
computer simulations and statistical mechanics will be highlighted, focusing on our recent
investigations of aqueous solutions of methylated ureas and of the osmolyte ectoine as examples.
References
[1] Funkner, S.; Havenith, M.; Schwaab, G.; Urea, a Structure Breaker? Answers from THz
Absorption Spectroscopy, J. Phys. Chem. B 116, 13374-13380 (2012).
[2] Hunger, J.; Ottosson, N.; Mazur, K.; Bonn, M.; Bakker, H.J.; Water-mediated interactions
between trimethylamine-N-oxide and urea, Phys. Chem. Chem. Phys. 17, 298-306 (2015).
[3] Buchner, R.; Hefter, G.; Interactions and dynamics in electrolyte solutions by dielectric
spectroscopy, Phys. Chem. Chem. Phys. 11, 8984-8999 (2009).
[4] Rahman, H.M.A; Hefter, G.; Buchner, R.; Hydrophilic and Hydrophobic Hydration of
Sodium Propanoate and Sodium Butanoate in Aqueous Solution, J. Phys. Chem. B 117, 2142-
2152 (2013).

12
The quest for methane on Mars. Methane as a biomarker for past
or present life.
Ebbe N. Bak1, Kai Finster1,2, Per Nørnberg1, and Svend J. Knak Jensen3 1Department of Bioscience, Aarhus University, Denmark
2Stellar Astrophysics Center, Department of Physics and Astronomy, Aarhus University, Denmark 3Department of Chemistry, Aarhus University, Denmark
Corresponding author: [email protected]
The observation of methane on Mars is of much interest as it may indicate past or present life on
the planet. The concentration of methane shows a substantial spatial and temporal variation. The
source of methane on Mars is unknown; likewise, a fast destruction mechanism is needed to
understand the observations [1, 2]. Here we present experiments that mimic the wind mediated
erosion of surface material. The erosion creates activated sites that can react with methane and
account for the fast disappearance of methane from the Martian atmosphere [3]. A preliminary
quantum mechanical based mechanism is presented for the reaction of activated material with
methane.
References
[1] C. R. Webster et al., Low Upper Limit to Methane Abundance on Mars. Science 342 355-356
(2013)
[2] C. R. Webster et al., Mars methane detection and variability at Gale crater. Science 347 415-
417 (2015)
[3] S. J. K. Jensen et al., A sink for methane on Mars? The answer is blowing in the wind. Icarus
236 24-27 2014

13
Exploring a Common Thread between Addiction and Disease: The
Dopamine Transporter
Dr. Michelle A. Sahai1
1 University of Roehampton, Whitelands College, Holybourne Avenue, London, SW15 4JD, United Kingdom
Corresponding author: [email protected]
Dopamine neurotransmission has been the subject of research for nearly half a century. However,
it was not until the late eighties, when it was implicated with the reinforcing effects of cocaine,
that the dopamine transporter (DAT) was identified as a potential target for pharmacological
treatments of cocaine abuse. Despite intensive pharmacological investigations, no effective
pharmacotherapy for cocaine abuse has demonstrated efficacy for long-term use, but tremendous
scientific advances have been made toward understanding DAT and its role in a variety of
neurological disease states and disorders. This includes Parkinson’s disease, schizophrenia,
attention-deficit/hyperactivity disorder and drug abuse [1, 2]. With the aid of sophisticated
mathematical/molecular modelling methods such as Quantitative Structure-Activity Relationship
models (QSAR) and molecular dynamics simulations we aim to assess novel psychoactive
substances (NPS) as potential treatments for the stimulant effects of cocaine in DAT. The results
that characterise the stimulant potential of the screened NPS will be compared with those derived
from the so-far used experimental techniques, such as autoradiography and voltammetry [3].
References
[1] F. H. Hansen, T. Skjørringe, S. Yasmeen, N. V. Arends, M. A. Sahai , K. Erreger, T. F.
Andreassen, V. Neergheen, M. Karlsborg, A. H. Newman, S. Pope, S. Heales, L. Friberg, L. H.
Pinborg, C. Loland, L. Shi, H.Weinstein, A. Galli, L. E. Hjermind, L. B. Møller and U. Gether.
Missense dopamine transporter mutations associate with adult parkinsonism and ADHD, J. Clin.
Invest . 124(7); 3107-3120. (2014)
[2] P. J. Hamilton, N. G. Campbell, S. Sharma, K. Erreger, F. H. Hansen, C. Saunders, A. N.
Belovich, NIH Autism Seq. Consort., M. A. Sahai , E. H. Cook, U. Gether, H. S. Mchaourab, H.
J. G. Matthies, J. S. Sutcliffe and A. Galli. De novo mutation in the dopamine transporter gene
associates dopamine dysfunction with autism spectrum disorder, Mol. Psychiatry. 18(12); 1315-
1323. (2013)
[3] M. Sahai, V. Barrese, N. Dutta, J. Opacka-Juffry and C. Davidson. The benzofuran 5-MAPB
(1-(benzofuran-5-yl)-n-methylpropan-2-amine) binds to the dopamine transporter and prolongs
dopamine efflux in rat nucleus accumbens. (in preparation)

14
The Role of Oxidative Stress in Alzheimer's Disease: A Theoretical
Approach
Michael C. Owen1, Waldemar Kulig2, Ilpo Vattulainen2, and Birgit Strodel.1,3
1 Institute of Complex Systems: Structural Biochemistry (ICS-6), Forschungszentrum Jülich GmbH, Jülich,
Germany
2 Department of Physics, Tampere University of Technology, Tampere, Finland
3 Institute of Theoretical and Computational Chemistry, Heinrich Heine University Düsseldorf, Düsseldorf,
Germany
Corresponding author: [email protected]
Alzheimer’s disease (AD) is the most common form of dementia in the elderly. In AD, the
amyloid-β peptide (Aβ) has been identified as a clinical hallmark in its disease pathology. A
conformational transition of the native Aβ peptide conformation into a β-sheet-rich state results
in the formation of toxic oligomers, which is believed to be a crucial event in the initiation of
AD. Aβ has been shown to induce pore formation in neuronal membranes, which subsequently
disrupts neuronal Ca2+ ion homeostasis [1]. However, neither the relationship between the
conformational change and oligomerization nor that between Aβ-membrane interactions and pore
formation are properly understood. Free radicals, produced either by A, respiration, or the
Fenton reaction, can oxidize the lipids that form the plasma membrane, a result which has shown
to promote A-membrane interactions. Meanwhile, free radicals have also been shown to abstract
hydrogen from the C of Gly and Ala residues, which initiated the to unfolding of oxidized
model peptides [2,3].
In an effort to dilineate the role of oxidation AD pathology we monitored the changes to the
structure of oxidized A peptides in different solvents and a bilyer comprised of oxidized 1-
palmitoyl-2-oleoylphosphatidylcholine (POPC) and cholesterol using MD simulations. An
oxidized Gly25 caused A to form -sheets in a solvent-dependent manner, whereas oxidized
POPC affected the membrane curvature, bilayer thickness, and the area per lipid in a dose-
dependent manner. These ongoing studies are an attempt to dilineate the role of oxidative stress
in the pathology of AD as it relates to A conformational change and A-membrane interactions.
References
[1] J. Nasica-Labouze, et al. Amyloid protein and Alzheimer's disease: When computer
simulations complement experimental studies. Chem. Rev. 115, 3518-3563 (2015).
[2] M. Owen, B. Viskolcz, I. G. Csizmadia. Quantum chemical analysis of the unfolding of a
penta-alanyl 310-helix initiated by HO, HO2 and O2-. J. Phys. Chem B 115, 8014-8023 (2011).
[3] M. C. Owen, M. Szori, I. G. Csizmadia, B. Viskolcz Conformation-dependent OH/H202
hydrogen abstraction reaction cycles of Gly and Ala residues: A comparative theoretical study.
J. Phys. Chem. B 116, 1143-1154 (2012).

15
Structure and function of C-terminal helical domain of the motor
subunit HsdR from the type I restriction-modification system
EcoR124I
Pavel Grinkevich1,2, Amanda Li3, Tatsiana Baikova1,2, Mikalai Lapkouski1, Alena
Guzanova4, Eva Csefalvay1,2, Marie Weiserova4 and Rüdiger Ettrich1,2 1 Institute of Nanobiology and Structural Biology of GCRC, Academy of Sciences of the Czech Republic, Nove
Hrady, Czech Republic 2 Faculty of Sciences, University of South Bohemia, Czech Republic
3 Chemistry Department, Princeton University, Princeton, New Jersey, USA 4 Institute of Microbiology, Academy of Sciences of the Czech Republic, Prague, Czech Republic
The structure of the HsdR subunit of EcoR124I [1] suggests that the motor subunit is a planar
array of four functionally integrated domains, with the fourth, C-terminal domain being all helical
and implied to play a role in complex assembly and/or DNA binding. However, the last 150
amino acids of this domain are unresolved in the crystal structure.
A single point mutation lead to a new crystal structure that allowed to trace the backbone of the
unresolved C-terminal residues, and homology and energetic modeling was applied to generate
an all-atom 3-D model of wild-type HsdR, complemented by in vivo and in vitro studies to
establish the function of the helical domain.
In vitro DNA cleavage assays, gel mobility shift assays and in vivo restriction tests were
performed on the wild-type and mutant HsdRs with selectively deleted parts of the helical
domain. In addition, further techniques, such as screening for stably expressing HsdRs with
Exoluclease III mediated random-length deletions of N-terminus, and the fusion of HsdR’s C-
terminus with green fluorescent protein, are being developed in effort to obtain a higher-
resolution crystal structure.
Our results strongly support the role of the C-terminal domain of HsdR in subunit interaction and
demonstrate the importance of the C-terminus in complex assembly.
Reference
[1] M. Lapkouski, S. Panjikar, P. Janscak, I. Kuta Smatanova, J. Carey, R. Ettrich, E. Csefalvay,
Structure of the motor subunit of type I restriction-modification complex EcoR124I, Nat Struct
Mol Biol. 16, 94-95 (2009).

16
Computational Strategies for Amyoloidogenic Proteins interactingwith
Gold Nanoparticles
Stefano Corni1 and Giorgia Brancolini1
1CNR-NANO S3, Institute of Nanoscience, via Campi 213/A, 41100 Modena, Italy Corresponding author: [email protected]
Nanoparticles (NPs) are recognized to exhibit distinct physical and chemical properties
compared with the same materials in the bulk form. [1] NPs have been repeatedly reported to interact with proteins, and this interaction can be exploited to affect processes undergone by
proteins, such as fibrillogenesis. Fibrillation is common to many proteins, and in living organisms, it causes tissue-specific or systemic amyloid diseases.
I will review our recent computational modeling advances which were pursued in the quest for a
theoretical framework elucidating the association mechanisms and the ability to design and
control the recognition events of interfaces between amyloidogenic protein and nanoparticles.
The effects of two different systems on 2-microglobulin, namely gold nanoparticles covered with
hydrophilic surfactants and gold nanoparticles functionalized with hydrophobic ligands, are
presented. Recent simulations at multiple levels (enhanced sampling molecular dynamics,
Brownian dynamics, and Poisson_Boltzmann electrostatics) and NMR measurements have
explained the origin of the observed protein perturbations, showing that in the presence of
citrate-capped gold NPs at physiological-like conditions, the interaction is weak and it is not able
to induce protein fibrillation (Fig.1). [2] On the contrary, the interactions with hydrophobic
ligands can block active sites of the protein domain from binding to another protein, thus
potentially inhibiting the fibrillation activity.[3]
The interaction of the gold-nanoparticles with two amyloidogenic variants of 2-microglobulin is
further discussed, namely (a) the truncated N6 and (b) the mutated D76N, and a fibrillization pathway
is proposed. The results offer possible strategies for controlling the desired effect of NPs on the
conformational changes of amyloidogenic proteins, which have crucial roles in the fibrillation
process.[4]
Fig. 1 Monomeric protein interacting with citrate-capped gold nanoparticle: (left) 20 ns of T-REMD
simulations (right) NMR protein signal intensities [1] Kumar et al., 2013. Manual on Critical Issues in Nanotechnology R&D Management: An Asia-
Pacific Perspective. APCTT-ESCAP
[2] Brancolini, G. et al. Nanoscale, 2014, 6, 7903-7911
[3] (i) Brancolini, G. et al. ACS NANO, 2015, 9, 2600-2613 (ii) Brancolini, G. et al. ACS Nano,
2012, 6, 9863-9878
[4] (i) Mangione, P. P. et al., J. Biol. Chem. 2013, 288, 30917-30930 (ii) G. Esposito et al. Subcell
Biochem. 2012, 65, 165-83.

17
The roles of conserved residues in the Saccharomyces cerevisiae
K+-translocation protein Trk1 analyzed by theoretical and
experimental approaches.
Vasilina Zayats, Thomas Stockner, Saurabh Kumar Pandey, Katharina Wörz,
Rüdiger Ettrich, Jost Ludwig
Institute of Nanobiology & Structural Biology, GCRC, v.v i., Nove Hrady, Czech Republic and IZMB / Molecular
Bioenergetics, University of Bonn, Bonn, Germany
Potassium ion (K+) uptake in yeast is mediated mainly by the Trk1/2 proteins that enable cells to
survive on external K+ concentration as low as a few µM. Yeast (and other fungal) Trks are
related to prokaryotic TRK and Ktr and plant HKT K+ transport systems. Based on the (weak)
similarity of these proteins to prokaryotic K-channels, homology models were developed (Durell
& Guy, 1999) after the first K-channel structure was available. Crystal structures of prokaryotic
TrkH and KtrB that are more closely related to yeast Trks were published recently (Cao et al.,
2011, Vieira-Pires et al., 2013) and allowed to develop more precise homology models. Overall
sequence similarity is however very low, thus requiring experimental verification of homology
models. Here a refined structural model of the Saccharomyces cerevisiae Trk1 is presented that
was obtained by combining homology modeling, molecular dynamics simulation and
experimental verification through functional analysis of mutants. The structural models and the
experimental results showed that glycines within the selectivity filter, conserved amongst the K-
translocation protein family are not only important for protein function, but are also required for
correct folding/ membrane targeting. Furthermore, the roles of several conserved charged
residues were analyzed. An earlier proposed interaction was verified and another -yet not
considered- interaction identified that might enhance folding and stability of yeast Trk1. The
model could provide the structural basis for addressing the long standing question if Trk1 is a
passive or active ion-translocation system.
References
[1] S.R. Durell, H.R. Guy, Structural models of the KtrB, TrkH, and Trk1,2 symporters based on
the structure of the KcsA K+ channel, Biophys. J. 77 (1999) 789-807.
[2] Y. Cao, X. Jin, H. Huang, M. G. Derebe, E.J. Levin, V. Kabaleeswaran, Y. Pan, M. Punta, J.
Love, J. Weng, M. Quick, S. Ye, B. Kloss, R. Bruni, E. Martinez-Hackert, W.A. Hendrickson,
B. Rost, J.A. Javitch, K.R. Rajashankar, Y. Jiang, M. Zhou, Crystal structure of a potassium ion
transporter TrkH, Nature 471 (2011) 336-40.
[3] R.S. Vieira-Pires, A. Szolloressi, J.H. Morais-Cabral, The structure of the KtrAB potassium
transporter, Nature 496 (2013) 323–328.

18
Structure and mechanism of a bacterial multi-drug efflux pump
Dijun Du1 , Zhao Wang2, Nathan R. James1 , Jarrod E. Voss1 , Ewa Klimont1 ,
Thelma Ohene-Agyei1 , Henrietta Venter1 , Wah Chiu2 , and Ben Luisi1
1 Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge CB2 1GA, UK
2Department of Bichemistry and Molecular Biology, Baylor College of Medicine, Houston, Texas 77030, USA
Corresponding author: [email protected]
Microorganisms encode several classes of transmembrane molecular pumps that can expel a wide
range of chemically distinct toxic substances. These machines contribute to the capacity of the
organisms to withstand harsh environments, and they help to confer resistance against clinical
antimicrobial agents. In Gram-negative bacteria, the pumps comprise tripartite assemblies that
actively transport drugs and other harmful compounds across the cell envelope. We describe
recent structural and functional data that have provided insights into the architecture and transport
mechanism of the AcrA-AcrB-TolC pump of Escherichia coli. This multi-drug efflux pump is
powered by AcrB, a member of the resistance/nodulation/cell division (RND) family of
transporters, which are energised by proton electrochemical gradients. Crystallographic data
reveal how a small protein AcrZ is engaged in a concave surface in the transmembrane domain
of AcrB, and we discuss how this interaction may affect the efflux activities of AcrB and other
RND family members.
References
[1] Du, D., Wang, Z., James, N.R., Voss, J.E., Klimont, E., Ohene-Agyei, T., Venter, H. Chiu,
W. and Luisi, B.F. (2014). Structure of the AcrAB-TolC multidrug efflux pump. Nature. 509,
512-515. Doi:10.1038/nature13205
[2] Du, D., van Veen, H.W. and Luisi, B.F. (2015) Assembly and operation of bacterial
tripartite multidrug efflux pumps. Trends in Microbiology . doi:10.1016/j.tim.2015.01.010.

19
Analytical functional representation of quantum chemical potential
energy curves and surfaces
Anita Rágyanszki1, Svend J. Knak Jensen2, Imre G. Csizmadia1, Béla Viskolcz1, 1 Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary
2 Department of Chemistry, Aarhus University, Langelandsgade 140, DK-8000 Aarhus C, Denmark
Corresponding author: [email protected]
The long term aims for the present research is to find mathematical functions which describe the
folding of the peptide residues. In order to find eventually the solution to solve the protein folding
problem and to build up the conformational network for the folding but it is reasonable to start
with the description of small compounds (I-V), and aim for a bottom up solution.
C
CC
HH
HH
Propane
I
HHH H
C
CC
CC
H
HH
H
H H
HH
HH HH
n-Pentane
II
C
CN
HH
H
NC
O
CC
HH
HH HH O
HIII IV
N-Acetyl-Glycine
N-methylamide
C
CN
H
NC
O
CC
HH
HH HH O
H
N-Acetyl-Alanine
N-methylamide
HH3C
V
C
CN
H
NC
O
CC
HH
HH HH O
H
N-Acetyl-Valine
N-methylamide
CH
CC
H H
H
H H
HH
The aims for the present research:
to achieve a reasonable accuracy of the fitted functions for the folding of the internal rotation
of typical organic functional groups which have only one independent variable [1],
to extend the one dimensional mathematical functions to describe the folding of the molecules
which have two independent torsional angles and they are include peptide bonds [2],
the fitted functions, which are describe the conformational spaces, can be analyzed to obtain
the critical points, minima, transition states and maxima.
References
[1] A. Rágyanszki, A. Surányi, I.G. Csizmadia, A. Kelemen, S. J. K.k Jensen, S. Y. Uysal, B.
Viskolcz. Fourier type potential energy function for conformational change of selected organic
functional groups. Chem. Phys. Lett. 599 (2014) 169–174
[2] A. Rágyanszki, K. Z. Gerlei, A. Surányi, I. G. Csizmadia, A. Kelemen, S. J. K. Jensen, B.
Viskolcz. Big Data reduction by fitting mathematical functions. A search for appropriate
functions to fit Ramachandran surfaces. Chem. Phys. Lett. 625 (2015) 91-97

20
Ticks as venomous animals: a pharmacological perspective James J. Valdés1
1Institute of Parasitology, Biology Centre of ASCR, Branišovská 1160/31 České Budějovice, Czech Republic
37005
Ticks are considered ectoparasites, but, according to the criteria revised by Fry et al. [1], ticks
are venomous animals [2]. Tick saliva contains an arsenal of macromolecules that target host
defense mechanisms to obtain a blood meal. Kunitz peptides are expressed by venomous animals
and are one of the major families encoded in tick salivary glands. Tick salivary Kunitz are diverse
in their primary sequence, but maintain a conserved tertiary structural motif. Some tick salivary
Kunitz modulate an array of targets. We were able to hypothesize on this target promiscuity by
analyzing the molecular dynamics of two Kunitz salivary peptides from two geographically
distinct tick species [3]. Understanding the promiscuity of tick salivary macromolecules may
benefit the drug industry.
Ticks also secrete lipocalins that sequester histamine during blood feeding. By sequestering
histamine, these lipocalins prevent host inflammatory responses by competing with the host’s
native receptor for histamine. Competition for ligand/drug binding between two proteins (or
pharmacological promiscuity) is extensively studied experimentally, but there are no studies on
the all-atom exploration of this competition. A novel method was developed to simulate, visualize
and analyze this competition at the host-tick interface [4] using an algorithm developed at the
Barcelona Supercomputing Center [5]. Pharmacological promiscuity is an obstacle in the drug
industry and simulating the host-tick interface may aid in rational drug design.
References:
[1] Fry, B.G. et al. (2009). The Toxicogenomic Multiverse: Convergent Recruitment of
Proteins Into Animal Venoms. Annu. Rev. Genomics Hum. Genet. 10:483.
[2] Cabezas-Cruz, A., Valdés, J.J. (2014) Are ticks venomous animals? Frontiers in Zoology.
11:47.
[3] Valdés, J.J., et al. (2013). Tryptogalinin is a tick Kunitz serine protease inhibitor with a
unique intrinsic disorder. PLoS ONE. 8(5):e62562.
[4] Valdés, J.J. (2014). Antihistamine response: a dynamically refined function at the host-
tick interface. Parasites & Vectors. 7:491.
[5] Borrelli, K.W., Vitalis, A., Alcantara, R., Guallar, V. (2005) PELE: Protein energy
landscape exploration. A novel Monte Carlo based technique. J Chem Theory Comput.
1(6):1304-1311

21
Cooperative rearrangements in clusters of anisotropic particles
Szilard Fejer1, Dwaipayan Chakrabarti2 and David J. Wales3
1University of Szeged, Faculty of Education, Department of Chemical Informatics, H-6725 Szeged,
Boldogasszony sgt. 6, Hungary 2School of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, United Kingdom
3University Chemical Laboratory, Lensfield Road, Cambridge CB2 1EW, United Kingdom
Corresponding author: [email protected]
The shape and interaction anisotropy of building blocks defines not only the way such particles
aggregate, but their dynamical properties as well. The building block anisotropies are therefore
of particular importance in the functioning of biological systems and the design of mesoscale
structures with well-defined properties. Low-energy aggregates of simple anisotropic building
blocks usually interconvert via highly cooperative motions that are specific to the system itself.
I will present different mechanisms specific to complex structures such as sliding rearrangements
in open tubes, hinge-moves in Bernal spirals, string bends in clusters of triblock Janus particles
etc. Among these, some can be seen experimentally, but in nanoscale structures such mechanisms
are yet to be confirmed.

22
Luteolin- organic solvent interactions. A molecular dynamics
simulation analysis
Khadidja Smail1, Noureddine Tchouar2, B. Marekha3, A. Sietsonen4, M. Barj,3
A. Idrissi3 1 Département de Biotechnologie, Faculté SNV, Université des Sciences et de la Technologie Université des Sciences
et de la Technologie d'Oran-Mohamed Boudiaf (USTO-MB), BP 1505 El M'naouer, Oran 31000, Algeria. 2 Laboratoire de Modélisation et Optimisation des Systèmes Industriels (LAMOSI), Université des
Sciences et de la Technologie d'Oran- Mohamed Boudiaf (USTO-MB), BP 1505 El M'naouer, Oran 31000,
Algeria. 4Ecole Normale Supérieure, UMR CNRS-ENS-UPMC n 8640, 24 rue Lhomond, F-75005 Paris, France
3 Université Lille 1 Sciences et Technologies, UMR 8516, LASIR, Villeneuev d'Ascq, France 59650.
Flavonoid biological activity is correlated with the structural elements of these molecules,
including the number and position of hydroxyl groups, It is also affected by the molecular
environment (polar or non-polar solvent, cellular environment).1 For this reason the current
theoretical investigation is carried out using molecular dynamics (MD) simulation on a
flavonoid molecule (Luteolin: Lut) in various solvents. The idea is to characterize the local
structure around the C=O and the various OH groups of Lut in these environments. Indeed,
previous works have shown the correlation between the different patterns of the local structure
around the OH moieties of quercetine and its solubility in various solvent such as chloroform,
water, acetone and ter-butanol.2 Furthermore, one of the important issues in the study of the
photochemistry of flavonoids and particularly Lut is to rationalize to what extent the nature of the
solvent determines their photochemistry, and more precisely how the nature of the solvent may
induce a special local organization around the OH and C=O moieties. As a consequence, the MD
simulations of Lut in four alcohols (methanol, 1-propanol, 2- propanol, 1-butanol), in
dimethylsulfoxide, acetone and hexane have been performed with a dual purpose: first, to
characterize the local structure around the OH and C=O moieties of Lut and particularly to
investigate the effect of these solvents on the intra molecular H-bonding between the OH and
the C=O groups of Lut, and, second, to correlate these data with the experimental properties of
Lut in these solvents.3 To this end the behavior of radial distributions (RDF), nearest neighbor
RDF, Voronoi polyhedra as well as the behavior of the number of H-bonding as a function of the
type the solvent, have been analyzed.
References
[1] Gould, K. S.; Lister, C., Flavonoid Functions in Plants. CRC Press, Taylor and Francis: Boca Raton,
2006.
[2]. Chebil, L. et al. J. Phys.Chem. B 2010, 114 (38), 12308-12313.
[3] Peng, B. et al. J. Chem. Eng. Data 2006, 51 (6), 2038-2040.

23
Changes induced by ligand binding in arginine repressor from Bacillus
subtilis
Milan Melicherčík,1,2 Saurabh Pandey2,3, Thomas Stockner4, Jannette Carey5,
Rüdiger Ettrich2,3
1 Department of Nuclear Physics and Biophysics, Comenius University in Bratislava, Mlynská dolina, SK-842 48,
Bratislava, Slovak Republic
2 Institute of Nanobiology and Structural Biology, Global Change Research Center, Academy of Sciences of the
Czech Republic, Zamek 136, CZ-373 33, Nove Hrady, Czech Republic
3 Faculty of Sciences, University of South Bohemia in Ceske Budejovice, Zamek 136, CZ-373 33, Nove Hrady,
Czech Republic
4 Medical University of Vienna, Vienna, Austria
5 Chemistry Department, Princeton University, Princeton, NJ, 08544-1009, USA
Corresponding author: [email protected]
The arginine repressor protein is main regulon of whole arginine metabolism in procaryotic cells.
The whole protein is functional as hexamer (as two trimers) composed of six identical monomers
(each has approx. 150 aminoacids). Each monomer has two domains connected thru short linker.
Domain at the C end (C domain) is responsible for hexamer forming and ligand binding. The
domain at the N end (N domain) binds to the DNA.
The most studied protein is from Escherichia coli. Previously we simulated this system and
shown the ligand binding induces the allosteric changes in domain positions – the trimer stop
rotation around the axis, which goes through their centers of mass. [1, 2] Unfortunately for E.
coli protein there are no crystal structures of whole protein (only independent N domain and
hexamer of C domains). Because of this missing structure, we can’t study the DNA-protein
binding. We used structures from Bacillus subtilis species, whose are available in PDB database
with codes 1F9N, 2P5L and 2P5M. The primary structure is very similar, although there are some
important differences. B. subtilis doesn’t have arginine residue in position to compete with ligand
(in E. coli the ligand competes with Arg110 for creating a salt bridge with Asp125 and the ligand-
Asp salt bridges stop the trimers rotation), has one helix (H4) inserted just after linker and has
arginine residue in linker instead E. coli, which it has on the very beginning of N domain.
We have simulated above mentioned structures to find out what are the differences between B.
subtilis and E. coli proteins. In apo state of protein the H4 has similar function as the salt bridge
between E. coli's Arg110-Asp125 – keeping together the trimers. Also the missing arginine
results in different rotation of trimers after ligand binding. In simulations with the whole protein
the biggest movements were done by N domains. They were able to switch their beta-turn-beta
structures, which allow binding to the DNA molecule.
References
[1] Rebecca Strawn, Milan Melichercik, Michael Green, Thomas Stockner, Jannette Carey,
Rudiger Ettrich (2010) Symmetric allosteric mechanism of hexameric Escherichia coli arginine
repressor exploits competition between L-arginine ligands and resident arginine residues PLOS
Computational Biology 6:6.e1000801.
[2] S K Pandey, D Reha, V Zayats, M Melichercik, J Carey, R Ettrich (2014) Binding-competent
states for L-arginine in E. coli arginine repressor apoprotein. Journal of Molecular Modeling 20
(7):2330.

24
Prediction of the pollution due to a pesticide
Agneta M. Balint1 and Stefan Balint1
1West University of Timisoara
Corresponding author: [email protected]
In this paper the cause of the pollution is a pesticide. The degradation of the pesticide as well the
spatial concentration distribution in soil of its toxic by-products is investigated . A mathematical
model is presented in which the degradation of the pesticide and the migration in soil of the toxic
by-products is described. This model facilitates the understanding and prediction of the pollution
caused by a pesticide. Based on the model, numerical simulations are presented.
25
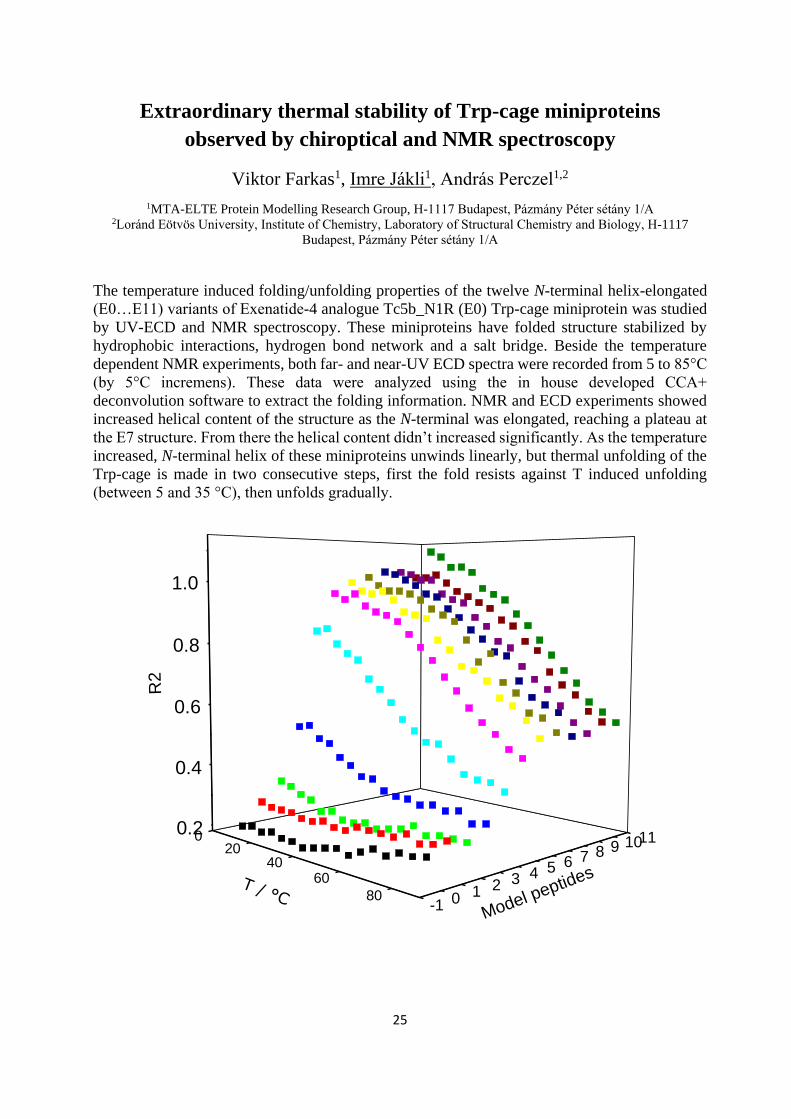
Extraordinary thermal stability of Trp-cage miniproteins
observed by chiroptical and NMR spectroscopy
Viktor Farkas1, Imre Jákli1, András Perczel1,2
1MTA-ELTE Protein Modelling Research Group, H-1117 Budapest, Pázmány Péter sétány 1/A 2Loránd Eötvös University, Institute of Chemistry, Laboratory of Structural Chemistry and Biology, H-1117
Budapest, Pázmány Péter sétány 1/A
The temperature induced folding/unfolding properties of the twelve N-terminal helix-elongated
(E0…E11) variants of Exenatide-4 analogue Tc5b_N1R (E0) Trp-cage miniprotein was studied
by UV-ECD and NMR spectroscopy. These miniproteins have folded structure stabilized by
hydrophobic interactions, hydrogen bond network and a salt bridge. Beside the temperature
dependent NMR experiments, both far- and near-UV ECD spectra were recorded from 5 to 85°C
(by 5°C incremens). These data were analyzed using the in house developed CCA+
deconvolution software to extract the folding information. NMR and ECD experiments showed
increased helical content of the structure as the N-terminal was elongated, reaching a plateau at
the E7 structure. From there the helical content didn’t increased significantly. As the temperature
increased, N-terminal helix of these miniproteins unwinds linearly, but thermal unfolding of the
Trp-cage is made in two consecutive steps, first the fold resists against T induced unfolding
(between 5 and 35 °C), then unfolds gradually.
020
4060
80-1 0 1 2 3 4 5 6 7 8 9 1011
0.2
0.4
0.6
0.8
1.0
Model peptides
R2
T / °C

26
Floating Patches of HCN at the Surface of Their Aqueous Solutions
– Can They Make “HCN World” Plausible?
Balázs Fábián1,2, Milán Szőri3, Pál Jedlovszky1,4,5
1 Laboratory of Interfaces and Nanosize Systems, Institute of Chemistry, Eötvös Loránd University, Pázmány P.
Stny 1/A, H-1117, Budapest, Hungary 2 Department of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szt.
Gellért tér 4, H-1111 Budapest, Hungary 3 Department of Chemical Informatics, Faculty of Education, University of Szeged, Boldogasszony sgt. 6, H-6725
Szeged, Hungary 4 MTA-BME Research Group of Technical Analytical Chemistry, Szt. Gellert tér 4, H-1111 Budapest, Hungary
5 EKF Department of Chemistry, Leényka utca 6, H-3300 Eger, Hungary
Corresponding author: [email protected], [email protected]
The liquid/vapor interface of the aqueous solutions of HCN of different concentrations has been
investigated using molecular dynamics simulation and intrinsic surface analysis. Although HCN
is fully miscible with water, strong interfacial adsorption of HCN is observed at the surface of its
aqueous solutions, and, at the liquid surface, the HCN molecules tend to be located even at the
outer edge of the surface layer. It turns out that in dilute systems the HCN concentration can be
about an order of magnitude larger in the surface layer than in the bulk liquid phase. Furthermore,
HCN molecules show a strong lateral self-association behavior at the liquid surface, forming thus
floating HCN patches at the surface of their aqueous solutions. Moreover, HCN molecules are
staying, on average, an order of magnitude longer at the liquid surface than water molecules, and
this behavior is more pronounced at smaller HCN concentrations. Because of this enhanced
dynamical stability, the floating HCN patches can provide excellent spots for polymerization of
HCN, which can be the key step in the prebiotic synthesis of partially water-soluble adenine. All
of these findings make the hypothesis of “HCN world” more plausible.
Reference
[1] B. Fábián, M. Szőri, P. Jedlovszky, Floating Patches of HCN at the Surface of Their
Aqueous Solutions – Can They Make “HCN World” Plausible? J. Phys. Chem. C 118, 21469-
21482 (2014).

27
Mapping out the 366 shades of the C4H8O2 isomers
Zsófia Borbála Rózsa1, Milán Szőri1
1 Department of Chemical Informatics, University of Szeged, H-6725, Szeged, Boldogasszony sgt 6.
Corresponding author: [email protected]
Constitutional isomers of the oxygen containing organic compounds can make a huge variety of
molecules and complexes. In this study all possible 366 singlet state constitutional isomers of the
C4H8O2 formula had been examined, including all molecules, bi-, tri- and tetramolecular
complexes, by comparing their thermodynamic properties, trying to find correlation between
their stability and structural features. This specific chemical formula includes molecules such as
butyric acid, which is a preferred substrate for colonocytes [1], or dioxanes which are used as
industrial solvents and may cause xenobiotic contamination of waters [2]. By using graph theory
all these molecules and multi-molecular complexes can be made, where atoms make the nodes
of the graph, and bonds make the edges. With this technique we can generate all the isomers of
a given formula by taking into consideration the binding specificities of the atoms using valence
shell electron pair repulsion coded by general MM2 force field parameters. After the creation of
the isomers the structures were optimized and the thermodynamic values were computed using
the G3MP2B3 ab initio computational methods. In order to validate our method, atomization
scheme was used to calculate heat of formation to compare the results with literature values. The
chemical species were ranked according to their Gibbs free energy and entropy which resulted a
thermodynamic map of molecules. This map can be divided into domains where great difference
can be found based on the types of the isomers, like their number of fragments. The Guinness
molecules of the system [3] were also described in details as well as the effect of structural
elements such as the size and the oxygen content of the ring.
[1] D. L. Topping and P. M. Clifton, “Short-Chain Fatty Acids and Human Colonic Function: Roles
of Resistant Starch and Nonstarch Polysaccharides,” Physiol Rev, vol. 81, no. 3, pp. 1031–1064, Jul.
2001.
[2] H. Barndõk, D. Hermosilla, L. Cortijo, E. Torres, and a Blanco, “Electrooxidation of industrial
wastewater containing 1,4-dioxane in the presence of different salts.,” Environ. Sci. Pollut. Res. Int., vol.
21, no. 8, pp. 5701–12, Apr. 2014.
[3] M. A. Suhm, “Guinness molecules: identifying lowest-energy structures.,” Angew. Chem. Int.
Ed. Engl., vol. 53, no. 7, pp. 1714–5, Feb. 2014.

28
The Effect of High Density Lipoprotein (HDL) on the Catalytic
Activity of Serum Paraoxonase-1 (PON1)
Klaudia Szeler,1 Moshe Ben-David,2,3 Joel L. Sussman,3 Dan S. Tawfik2, Shina
Caroline Lynn Kamerlin1
1Science for Life Laboratory, Department of Cell and Molecular Biology, Uppsala University, Sweden 2Departments of Structural Biology
3Biological Chemistry, Weizmann Institute of Science, Israel
Corresponding author: [email protected]
Serum paraoxonase 1 (PON1) is a calcium-dependent lipo-lactonase that promiscuously
catalyzed the hydrolysis of various organophosphate pesticides and nerve agents [1]. PON1
interacts with high-density lipoprotein (HDL) affecting both PON1's stability and activity [2,3]
(Fig. 1). However, how PON1-HDL interactions regulate PON1's activity remains unclear. We
present here a combination of kinetic, crystallographic and computational work (using the
empirical valence bond approach) of the PON1 catalyzed hydrolysis of phosphotriester and
lactone substrates. We provide a detailed structural and mutational analysis for the native
lactonase activity of the enzyme, and for the promiscuous phosphatase activity using paraoxon as
a model substrate. Our results provide a detailed model for the activation of PON1 by HDL and
demonstrate the detrimental role too much flexibility can play on an enzyme's catalytic activity. Figure 1: Proposed model for the anchoring of PON1 to HDL based on the substrate-free crystal
structure of rePON1 (PDB ID:3SRE).
References
[1] M. I. Mackness, S. Arrol, C. Abbot and P. N. Durrington, Protection of high-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis 104 129-135 (1993). [2] L. Gaidukov L and D. S. Tawfik, High affinity, stability and lactonase activity of serum paraoxonase PON1 anchored on HDL with ApoA-I. Biochemistry 44, 11843-11854 (2005). [3] L. Gaidukov, R. I. Viji, S. Rosenblat, M. Aviram and D. S. Tawfik, ApoE induces serum paraoxonase PON1 activity and stability similar to ApoA-I. Biochemistry 49, 532-538 (2010). [4] M. Ben-David, J. L. Sussman, K. Szeler, C. I. Maxwell, S. C. L. Kamerlin and D. Tawfik,
Catalytic stimulation by restrained active-site floppiness - the case of high-density lipoprotein-bound serum paraoxonase 1. J. Mol. Biol. 427, 1359-1374 (2015)

29
Computational study concerning RNA or DNA aptamers
Katarína Skúpa1, Milan Melicherčík1 and Ján Urban1
1Department of nuclear physics and biophysics
Faculty of Mathematics, Physics and Informatics
Comenius University in Bratislava, Slovakia
Aptamers are short nucleic acid segments, which can bind with high affinity and specifity to a wide
range of molecules. The RNA/DNA aptamers form an attractive class of molecules which are widely
applicable. They can be used in medicine, pharmaceutics, environmental and food analytics as
diagnostic and therapeutic agents and as recognition elements. Our contribution is oriented to the
application of theoretical methods for the study of properties of dopamine and RNA/DNA
nucleosides molecular systems.
For the description of these systems molecular dynamics simulations (MD) and quantum
chemical calculations have been applied. The obtained results are discussed.
Acknowledgements: Support from the VEGA 1/0878/15 grant is acknowledged.

30
Protein-peptide docking with significant conformational changes
using the CABS-dock web server
Mateusz Kurcinski, Michal Jamroz, Maciej Blaszczyk, Andrzej Kolinski and
Sebastian Kmiecik*
University of Warsaw, Warsaw, Poland
*Corresponding author: [email protected]
Protein-peptide interactions play a key role in cell functions. Their structural characterization,
although very challenging, is important for discovery of new drugs. Based on our methodology
for highly efficient simulation of proteins [1, 2], we developed the CABS-dock web server for
protein-peptide molecular docking [3]. While other docking algorithms require pre-defined
localization of the binding site, CABS-dock doesn’t require such knowledge. Given a protein
receptor structure and a peptide sequence (and starting from random conformations and positions
of the peptide), CABS-dock performs simulation search for the binding site allowing for full
flexibility of the peptide and small fluctuations of the receptor backbone [3-5]. This protocol was
extensively tested over the largest dataset of non-redundant protein-peptide interactions available
to date (including bound and unbound docking cases) [3]. For over 80% of the dataset cases, we
obtained models with high or medium accuracy (sufficient for practical applications). CABS-
dock method for coupled binding site search and protein-peptide docking can be easily
complemented by other computational tools (e.g. high-resolution docking refinement protocols)
or experimental data to improve the results of the docking experiment. CABS-dock web server
is freely available at http://biocomp.chem.uw.edu.pl/CABSdock
References
[1] Jamroz M, Kolinski A, Kmiecik S. (2013) CABS-flex: Server for fast simulation of protein
structure fluctuations. Nucleic Acids Res. 41, W427-31.
[2] Blaszczyk M, Jamroz M, Kmiecik S, Kolinski A. (2013) CABS-fold: Server for the de novo
and consensus-based prediction of protein structure. Nucleic Acids Res. 41, W406-11.
[3] Kurcinski M, Jamroz M, Blaszczyk M, Kolinski A, Kmiecik S. (2015) CABS-dock web
server for the flexible docking of peptides to proteins without prior knowledge of the binding
site. Nucleic Acids Res. doi: 10.1093/nar/gkv456.
[4] Kurcinski M, Kolinski A, Kmiecik S. (2014) Mechanism of Folding and Binding of an
Intrinsically Disordered Protein As Revealed by ab Initio Simulations. Journal of Chemical
Theory and Computation. 10, 2224-2231.
[5] Blaszczyk M, Kurcinski M, Kouza M, Wieteska L, Debinski A, Michal J, Andrzej K,
Kmiecik S. (2015) Modeling of protein-peptide interactions using the CABS-dock web server
for binding site search and flexible docking. Methods (submitted), preprint at
arXiv:1505.01138.

31
MoDyDB – just an other database?
Béla Fiser1, László Müller2, Béla Viskolcz2, Balázs Jójárt2
1Department of Organic Chemistry I, University of the Basque Country/UPV-EHU, Manuel de Lardizabal 3,
Donostia-San Sebastián, Spain-20018
2Department of Chemical Informatics, Faculty of Education, University of Szeged, Boldogasszony sgt. 6, Szeged,
Hungary-6725
Corresponding author: [email protected]
In this lecture we introduce a new database – called Molecular Dynamics and Docking DataBase,
will be freely available at modydb.org - containing parameter and coordinate files for molecular
dynamics and docking calculations of receptor – ligand complexes. AMBER related parameters
were assigned for the complexes, and for docking calculations files are in AutoDock and
AutoDock Vina format. The database provides a coherent, standardized set of structures which
is useful in different fields of computational chemistry.

32
Cannabinoids. Does Cooperative Interplay Underlie Their
Therapeutic Nature?
Lily M. Hunnisett1,2 , Kun V. Tian3 , Bela Viskolcz1 , Imre G. Csizmadia1,4,
Gregory A. Chass2
1 Department of Chemical Informatics, University of Szeged, Szeged, Hungary
2 School of Biological and Chemical Sciences, Queen Mary University of London, London, UK 3 Materials Science Research Institute, Semmelweis University, Budapest Hungary
4 Department of Chemistry, University of Toronto, Toronto, Ontario, Canada
The regulation of cannabinoid ratios existing in cannabis samples, and therefore the
accompanying psychological side effects, is a major concern when considering its use as a
therapeutic or recreational drug. It is therefore of great interest to understand, on a molecular
level, their interconverting-abilities, and molecular behaviour in biologically-relevant media.
This work presents exploratory structural and energetic analyses of neutral models of Δ8 and Δ9
isomers of Tetrahydrocannabinol (THC) and Cannabidiol (CBD), completed at the B3LYP/6-
311++G(d,p) level of theory in H2O solvent.
Free-energy results showed Δ8-THC to be ~9 and ~22 kJ.mol-1 more stable than Δ9-THC and
CBD respectively, a result in agreement with previous experimental data. High sensitivity to
cannabinoid conformation was observed, in particular for the alkyl side chain, suggesting a
potentially significant influence over their pharmacological activities. Overlap of relative free-
energies of the three cannabinoids, complemented by energetically-accessible barriers to inter-
conversion supports the hypothesis that they co-exist, and that their dynamic switching plays an
important role in their bio-activites. Complementary methodological development allowed for
effective general dissemination of the multi-dimensional analyses carried out.

33
Hydration in discrete water – from bulk free energies to localised
water molecules
Piotr Setny1
1Centre of New Technologies, University of Warsaw
Banacha 2c, 02-097 Warsaw, Poland
Corresponding author: [email protected]
Biomolecules typically exist and function in water. The influence of aqueous environment needs
to be taken into account in all theoretical and computational studies of living matter at atomistic
level. Approaches, such as computer simulations, involving water in explicit, all-atom
representation are often prohibitively expensive, while the existing simplified, so called implicit
solvent models are only moderately successful.
Here, we will present a new method for modeling of biomolecular hydration. It is based on
discrete solvent representation and mean field description of solute-water and water-water
interactions. While maintaining computational efficiency of typical implicit solvent models, it
avoids many of their important deficiencies. The proposed model correctly reproduces
experimental hydration free energies for an extensive set of roughly 700 diverse organic
compounds, including atomic and molecular ions. At the same time it accurately predicts the
distribution and binding free energies of isolated water molecules buried within protein
structures. The combination of such unique capabilities, makes it a useful tool for receptor-ligand
docking as well as a valuable aid for experimental methods aimed at the prediction of
macromolecular structures such as X-ray crystallography or NMR.

34
How water contributes to pressure and cold denaturation of
proteins
Giancarlo Franzese1 1Universitat de Barcelona, Spain
Corresponding author: [email protected]
The mechanisms of cold- and pressure-denaturation of proteins are matter of debate and are
commonly understood as due to water-mediated interactions. Here we study several cases of
proteins, with or without a unique native state, with or without hydrophilic residues, by means of
a coarse-grain protein model in explicit solvent. We show, using Monte Carlo simulations, that
taking into account how water at the protein interface changes its hydrogen bond properties and
its density fluctuations is enough to predict protein stability regions with elliptic shapes in the
temperature-pressure plane, consistent with previous theories. Our results clearly identify the
different mechanisms with which water participates to denaturation and open the perspective to
develop advanced computational design tools for protein engineering [1-5].
References
[1] K. Stokely, M. G. Mazza, H. E. Stanley, and G. Franzese, Proc. Natl. Acad. Sci. 107, 1301 (2010).
[2] V. Bianco, S. Iskrov, and G. Franzese, J. Biol. Phys. 38, 27 (2012). [3] G.
Franzese and V. Bianco, Food Biophys. 8, 153 (2013). [4] V. Bianco and G.
Franzese, Sci. Rep. 4, 4440 (2014).
[5] V. Bianco and G. Franzese, "How water contributes to pressure and cold denaturation of
proteins", arXiv:1505.07594 (2015)

35
Abstracts of poster presentations
The authors of the abstracts bear the full responsibility for the scientific and
linguistic content.

36
Computational study of hydrophilic ephedrine derivatives
(C10H16NO2) and interactions with β2-adregeneric receptor
Joshua Campbell1, János J. Szórád3, Lily M. Hunnisett3, Béla Fiser3,4, Anita
Rágyanszki3, Balázs Jójárt3, Milán Szőri3, Imre G. Csizmadia2,3, Béla Viskolcz4
1-Department of Chemical &Physical Sciences, University of Toronto Mississauga, L5L6A2, Ontario Canada
2-Department of Chemistry, University of Toronto, M5S 3H6 Toronto, Ontario, Canada 3-Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary
4-Department of Organic Chemistry I, University of the Basque Country, Manuel Lardizábal 3, 20018 Donostia-
San Sebastian, Gipuzkoa, Spain
As a preliminary step to drug design, the interactions that promote the formation of stable
complex should be considered. Activation of the G-coupled protein, specifically the β2-
adregeneric receptor (β2AR) is used to treat asthma and preterm labour as these receptors exist at
higher densities in smooth muscles.[1,2] One such known agonist is ephedrine, however it is
unselective in binding to this G-coupled protein.[3,4]
As such, this study explored the binding affinity of the isomers with stoichiometry C10H16NO2
and the human β2AR. Generation of the molecular library constrained the isomers to include a
phenyl ring and protonated amine nitrogen. Probing the depth of the library within the constraints,
the various protonation states and stereoisomers were included. Simulation of physiological pH
(Protein preparation wizard in Schrödinger suites) determined the protonation stated of the
various residues.
After which they were sequentially docked (with Glide apart of the Schrödinger suites) with
increasing precision. From the docking scores and interaction energies (Eint) it was observed that
the Asp113 accounts for approximately 60% of the total Eint.
References:
[1] Solanki, P.; Yadav, P.; Kantharia, N. Ephedrine: Direct, Indirect or Mixed Acting
Sympathomimetic? Int. J. Basic Clin. Pharmacol. 2014, 3 (3), 431
[2] Barnes, P. J. Beta-Adrenoceptors on Smooth Muscle, Nerves and Inflammatory Cells. Life
Sci. 1993, 52 (26), 2101–2109.
[3] Butz, P.; Kroemer, R. T.; Macleod, N. a.; Simons, J. P. Conformational Preferences of
Neurotransmitters: Ephedrine and Its Diastereoisomer, Pseudoephedrine. J. Phys. Chem. A
2001, 105 (3), 544–551.
[4] Ahlquist, R. . A Study of Adrenotopic Receptors. Am. J. Physiol 1948, 153, 586–600.

37
Molecular recognition of constitutional isomers of epinephrine by
GPCR Balázs Fábián1, János J. Szórád2, Lily M. Hunnisett2, Béla Fiser2,3, Anita
Rágyanszki2, Balázs Jójárt2, Milán Szőri2, Imre G. Csizmadia2,4, Béla Viskolcz2
1Department of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics,
Műegyetem rkp. 3-9 H-1111 Budapest, Hungary 2Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary
3Department of Organic Chemistry I, University of the Basque Country, Manuel Lardizábal 3, 20018 Donostia-San
Sebastian, Gipuzkoa, Spain 4Department of Chemistry, University of Toronto, M5S 3H6 Toronto, Ontario, Canada
Corresponding author: [email protected]
Epinephrine (C9H13NO3), also known as adrenaline, is a hormone1 and neurotransmitter2
belonging to the group of catecholamines, that is, it is a monoamine having a benzene ring with
two hydroxyl side groups (catechol group). Epinephrine, along with other catecholamines like
norepinephrine and dopamine is produced in the adrenal medulla and is released into the blood-
stream as a part of the fight-or-flight response3. Epinephrine works as a nonselective agonist of
α and β adrenergic receptors, which are responsible the majority of cellular responses.
Our aim was to study molecules based on the same formula as the protonated epinephrine
(C9H14NO3) in order to evaluate their pharmacological potency as a stimulant of adrenergic
receptors. In the current work, extensive docking calculations were performed using the
Schrödinger program package to gain insight into properties of the possible ligands. The
molecules were suitably prepared and docked into the R chain of the 3SN6 β adrenergic receptor4
of the Protein Data Bank. Out of all possible isomers containing the pharmacophore benzene
ring, the top 10 scoring were analyzed in detail.
To the best of our knowledge, these structures are novel and also have better docking score than
the parent molecule epinephrine. The differences in their scores are within the accuracy of the
method. The molecules always interact strongly with the ASP 113, the SER and ASN residues
located at the binding site of the receptor. The stability of each molecule was assessed based on
their relative free energies. The comparison was made using Density Functional Theory (DFT)
with the B3LYP functional in water solvent. Based on this, most of the analyzed structures show
greater stability than the parent molecule epinephrine. Finally, the plot of the docking scores
against the DFT energies indicates 3 families of molecules. Although there were no obvious
structural similarities in these groups, this question needs to be investigated in detail.
References
[1] Babisch, W.; "Stress hormones in the research on cardiovascular effects of noise"; Noise &
Health; 2003, 5(18), 1-11
[2] Berecek, K. B. M.; Brody, M. J.; "Evidence for a neurotransmitter role for epinephrine
derived from the adrenal medulla"; American Journal of Physiology; 1982, 242 (4), H593–
H601
[3] "Catecholamines"; Health Library. San Diego: University of California.
[4] Rasmussen, S. G. F.; DeVree, B. T.; Zou, Y. et al.; "Crytal structure of the β2 adrenergic
receptor – Gs protein complex"; Nature, 2011, 477, 549-555

38
Development of Fluctuating Finite Element Analysis - Dynamics
and Kinetics of Mesoscale Biomolecules
Ben Hanson, Daniel Read, Oliver Harlen, Sarah Harris
University of Leeds
The increasing resolution capability of x-ray crystallography, and more recently cryo-em [1], is
leading to more interest in the structure and dynamics of larger and more complex proteins.
Proteins of this nature, such as molecular motors, arguably exist at the mesoscale in both the
spatial and temporal regimes. Fluctuating Finite Element Analysis (FFEA), a continuum
mechanical representation of biomaterial [2], has been previously reported to give insights into
the dynamical range of motors such as rotary ATPases [3]; i.e. the spatial mesoscale. Here we
present preliminary results of a kinetic scheme integrated with FFEA, implemented in order to
take FFEA fully into the temporal mesoscale. The molecule of study is cytoplasmic dynein, a
molecular motor powered by an ATP cycle which transports cargo along microtubules [4], and
for which a purely kinetic model currently exist [5].
References
[1] Kühlbrandt, W., Science 343, 1443-44 (2014)
[2] Oliver, R., Read, D. J., Harlen, O. G. and Harris, S. A., J. Comp. Phys. 239, 147-65 (2013)
[3] Richardson, R. A. et al., Proteins 82(12), 3298-311 (2014)
[4] Roberts, A. J., Kon, T., Knight, P. J., Sutoh, K. and Burgess, S. A., Nature Reviews Mol. Cell Biol. 14,
713-726 (2013)
[5] Sarlah, A. and Vilfan, A., Biophys. J. 107, 662-671 (2014)

39
Dynamics of protein ligand interactions – impact on drug discovery
Sarah Harris1 , Anastasia Zhuravleva1 and Outi Kamarainen1
1University of Leeds, Leeds, United Kingdom
Corresponding author: [email protected]
Introducing a new drug to market is a lengthy and expensive process (10-15 years and
$1.2billion). Better understanding of how and why a drug molecule binds to a target and what
changes in the structure and chemistry could improve the binding affinity may help to shorten
the drug design process and in recent years the role of molecular motions in binding selectivity
and efficiency attracts an increasing attention from drug design research. Whilst calorimetric
methods can quantify the total free energy and entropy change, it is difficult to estimate
contributions from the different components of entropy, one of the largest unknowns being the
magnitude of the configurational entropy change.
Molecular dynamics simulations of the drug and target protein can provide more details of the
different atomistic movements contributing to the total entropy change, thus potentially providing
valuable clues for lead optimisation. However, the relatively short time scale of simulations (100
ns to few µs) and potential issues with obtaining suitable parameters for ligands remain a
challenge. For this study, we use the well-characterized N-terminal domain of the chaperone
protein Hsp90 as a model system to study the vibrational entropy of Hsp90 binding to different
small-molecule inhibitors. We employ the NMR spectroscopy to validate the data from molecular
dynamics simulations and improve our in silico methods. The aim of this project is to develop a
generic computational approach that can be used to predict dynamic features for different protein-
ligand/drug systems and thus, to improve drug design and lead optimisations processes in
pharmaceutical sector.

40
Synthesis and characterization of NGR peptides to design
radiopharmaceuticals
Z. Lovrity1, G. Mező2, D. Szikra3, I. Kertész3 1Department of Nanobiotechnology, University of Miskolc, Hungary
2Research Group of Peptide Chemistry, Hungarian Academy of Sciences, Eötvös L. University, Hungary 3Department of Nuclear Medicine, University of Debrecen, Hungary
Angiogenesis is the formation of new blood vessels from preexisting vasculature. This process might be
triggered and enhanced by many human diseases including cancer, atherosclerotic plaque and peripheral
artery disease. Aminopeptidase N (APN, also known CD13) has been associated with the growth of the
new vasculatures and suggested as a suitable target for anti-cancerous therapy. An asparaginyl-glycinyl-
arginine (NGR) sequence containing peptides have been identified as a specific ligands of CD13.
Conjugation of NGR peptides with chemotherapeutic drugs might improve the tumor therapy.
Labeled NGR derivatives with 64Cu, 68Ga and 99mTc are useful radiotracers for the in vivo imaging of
CD13-expression of tumors and neovasculature by binding to APN.
The aim of this study was to design, synthetize different, chemically stable NGR derivatives for using them
radiotracers.
Cyclic NGR peptides with amide- and thioether bond were synthetized by development of precursor linear
peptides on Rink-Amide MBHE resin with standard Fmoc/tBu strategy. Cyclization was carried out in
solution. The linear semi-protected H-Lys(CIZ)-Asn-Gly-Arg-Glu-NH2 peptide was cleaved from the
resin using TFA mixture, which was cyclized: the amide bond was formed between the N-terminus
and side chain of Glu. The crude cyclic peptide was purified by RP-HPLC. The CIZ protecting group from
the side chain of Lys residue was removed by HF-p-cresol mixture. The cyclic peptides were dissolved
in sodium-carbonate buffer. p-SCN-Bn-NOTA (or p-NCS-benzyl NODAGA) was dissolved in
DMSO and was introduced to the cyclic peptide solution. The chelator-conjugated NGR derivate was
purified by semi-preparative RP-HPLC.
The pure products were identified by LC-MSMS (LCMS-IT-TOF, Shimadzu Co.). External mass
calibration, resolution and sensitivity of the ion trap and TOF analyzer were adjusted using a standard
TFANa solution. Mobile phase A was composed 5% acetonitrile in water, 0,1% TFA and B 5% water in
acetonitrile, 0,1% TFA. Isocratic elution was performed at a flow rate of 0,2 ml/min. During a 3-min
run, independent infusion analysis was carried out in the positive ionization mode. The MS operating
conditions were as follows: electrospray voltage 4,5 kV; CDL and heat block temperatures 200°C;
nebulizing gas flow 1,5 L/min, drying gas pressure 100 kPa. Mass spectra were acquired in the range of
m/z 100-1500 Da for MS1, 50-1000 for MS2 and MS3. The collision energy was set 50% both for MS2,
MS3. Shimadzu's Composition Formula Predictor software was used to provide chemical formula of
NGR peptides and their fragments.

41
Constitutional Isomers of Norepinephrine (C8H12NO3) Interactions
with β2-Adrenergic Receptor
Min-Yen Lu1, János J. Szórád2, Lily M. Hunnisett2, Béla Fiser23, Anita
Rágyanszki2, Balázs Jójárt2, Milán Szőri2, Imre G. Csizmadia12, Béla Viskolcz2
1: Department of Chemistry, University of Toronto, M5S 3H6 Toronto, Ontario, Canada
2: Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary
3: Department of Organic Chemistry I, University of the Basque Country, Manuel Lardizábal 3, 20018 Donostia-San Sebastian, Gipuzkoa,
Spain
Norepinephrine is a catecholamine with multiple roles often acting as a hormone or
neurotransmitter. It often plays a role in adrenergic agonism as a drug through the activation by
ligand binding of GPCRs. Most of the known noradrenaline effects such as regulation of vascular
tone and sympathetic system are carried out via common bindings to α1 and α2-adrenergic
receptors instead of β subclass. Therefore, our investigation of Norepinephrine (C8H12NO3)
structure and its isomers is a key element to discover a new binding endogenous ligand. Due to
the difference of ligand specificity and signalling process, this structural-based approach will
identify the potential ligands through examination of docking score and interaction energy.
Approximately 7000 isomeric structures of C8H12NO3 are generated by Schrödinger program
including Molgen, Stereoizoer and Epik. By performing molecular docking against β2-adrenergic
receptors with Glide utilizing levels of HTVS, SP and XP precison , the top 10 XP high-rankig
molecules display binding affinities with targeted receptor. Of the 10 structures, 5 molecules
were unknown, displaying low docking scores and favorable interaction energy values. Among
the best docking hits, interactions of the neibouring amino acids residues are examined to
determine the most favorable structure. The XP results of the new discovered ligand structures
can be further synthesized for potential pharmaceutical research.
This computational data induced through experimentation may create new opportunities for
therapeutic drug development.
References:
[1] S. Ferguson, R. Feldman; “Beta-Adrenoceptors as Molecular Targets in the treatment of
Hypertension” Canadian Journal of Cardiology, 2014, vol 30, pps3-s8
[2] S. Rasmussen, B. Devree, Y. Zou, A. Kruse, K. Chung, T. Kobilka, F. Thian, P. Chae, E. Pardon,
D. Calinski, J. Mathiesen, S. Shah, J. Lyons, M. Caffrey, S. Gellman, J. Steyaerr, G. Skinitotis, W. Weis,
R, Sunahara; Cyrstal structure of the beta 2 adenergic receptor-Gs protein complex; Nature, 2011, vol477
pp549-555
[3] P. Kolb, D. Rosenbaum, J. Irwin, J. Fung, B. Kolbllka, B. Schoichet, “Structure-based discovery
of beta 2-adrenergic receptor ligands”, PNAS 2009, vol 16, pp6843-6848
[4] D. Vassilatis, J. Hohmann, H. Zeng, F Li, J Rachalis, M. Mortrud, S Rodriquez, J weller, A
Wright, J Bergmann, G Gaitanaris; “The G protein-coupled receptor repertoires of human and mouse”
PNAS, 2003, vol 100, pp4903-4908

42
Binding-Competent States for L-arginine in E. coli Arginine
Repressor Apoprotein
Saurabh K. Pandey1,2, David Reha 1,2, Vasilina Zayats 1,2, Milan Melichercik 1,3,
Jannette Carey 4,
Rudiger Ettrich 1,2 1Academy of Sciences of the Czech Republic, Nove Hrady, Czech Republic
2 University of South Bohemia, Nove Hrady, Czech Republic
3 Comenius University, Bratislava, Slovakia
4 Princeton University, Princeton, NJ, USA.
Arginine repressor of E. coli is a multifunctional hexameric protein that provides feedback
regulation of arginine metabolism upon activation by the negatively cooperative binding of L-
arginine. A molecular mechanism of allostery has been described earlier in which
conservedarginine and aspartate residues in each ligand-binding pocket promote rotational
oscillation of the trimers within the hexameric domain that binds L-arginine by engagement and
release of hydrogen-bonded salt bridges. Binding of exogenous L-arginine displaces resident
arginine residues and arrests oscillation, shifting the equilibrium quaternary ensemble and
promoting motions that maintain the configurational entropy of the system [1]. Interpretation of
this complex system requires an understanding of the protein's conformational landscape. In this
work the ~50 kDa hexameric C-terminal domain was studied by 100 ns molecular dynamics
simulations in presence and absence of the six L-arg ligands that bind at the trimer-trimer
interface. A rotational shift between trimers followed by rotational oscillation occurs in the
production phase of the simulations only when L-arg is absent. Analysis of the system reveals
that the degree of rotation is correlated with the number of hydrogen bonds across the trimer
interface. The trajectory presents frames with one or more apparently open binding sites into
which one L-arg could be docked successfully in three different instances, indicating that a
binding-competent state of the system is occasionally sampled. Simulations of the resulting
singly-liganded systems reveal for the first time that the binding of one L-arg results in a
holoprotein-like conformational distribution.
References
[1] R Strawn, M Melichercik, M Green, T Stockner, J Carey, R Ettrich (2010) Symmetric
vallosteric mechanism of hexameric Escherichia coli arginine repressor exploits competition
between L-arginine ligands and resident arginine residues. PLOS Computational Biology 6: 6.
e1000801

43
Molecular Recognition Involving Combinatorial Chemistry: A
Case Study of Norepinephrine Derivative, C8H12NO2
Seung Gwan Ryoo1, János J. Szórád2, Lily M. Hunnisett2, Béla Fiser2,3, Anita
Rágyanszki2, Balázs Jójárt2, Milán Szőri2, Imre G. Csizmadia1,2, Béla Viskolcz2
1Department of Chemistry, University of Toronto, M5S 3H6 Toronto, Ontario, Canada
2Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary 3Department of Organic Chemistry I, University of the Basque Country, Manuel Lardizábal 3, 20018
Norepinephrine is a neurotransmitter responsible for regulating stress responses and autonomic
activity within the body[1]. It strongly binds α1, α2, and β1 adrenergic receptors (ARs) and weakly
binds β2AR[2]. For this study, receptor-ligand interactions between β2AR and norepinephrine
derivatives corresponding to the molecular formula, C8H12NO2, are investigated to discover
ligand structures with tight binding interactions with β2AR, and to see if they have relevance in
drug design. Constitutional isomers containing a phenyl ring and a protonated nitrogen atom are
generated using Molgen[3], and prepared using Schrödinger LigPrep module[4]. The β2AR
structure is retrieved from the PDB database (PDB ID: 3SN6) and prepared via Schrödinger
Protein Preparation Wizard[5]. Molecular docking is then performed to predict the orientation
and conformation of the receptor-ligand complex using Glide[6]. Three docking methods –
HTVS, SP, and XP – are used to yield the 10 most stable ligand structures with XP docking
scores ranging from -10.141 to -8.821. The average docking score of these 10 structures (-9.3414)
is higher than that of norepinephrine derivatives containing one less oxygen atom (-8.5742), and
lower than that of norepinephrine, which contains one more oxygen atom (-10.3224). This trend
suggests that the number of oxygen atoms is proportional to both the docking score and ligand
stability. Six residues, namely ASP113, PHE193, SER203, PHE289, PHE290, and ASN312,
interact with the ligand structures. ASP113 contributes most significantly by forming hydrogen
bonds with -OH groups.
References
[1] A. P. Kohm, and V. M. Sanders, Norepinephrine and beta 2-adrenergic receptor stimulation
regulate CD4+ T and B lymphocyte function in vitro and in vivo, Pharmacol. Rev. 4, 487-525
(2001)
[2] K. Mori, E. Ozaki, B. Zhang, L. Yang, A. Yokoyama, I. Takeda, N. Maeda, M. Sakanaka,
and J. Tanaka, Effects of norepinephrine on rat cultured microglial cells that express α1, α2, β1
and β2 adrenergic receptors, Neuropharmacology 43, 1026-1034 (2002)
[3] R. Gugisch, A. Kerber, A. Kohnert, R. Laue, M. Meringer, C. Rücker and A. Wassermann,
MOLGEN 5.0, a Molecular Structure Generator, Bentham Science Publishers Ltd., Bayreuth,
Germany (2012)
[4] LigPrep, version 3.4, Schrödinger, LLC, New York, NY (2015)
[5] G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, Protein and ligand
preparation: Parameters, protocols, and influence on virtual screening enrichments, J. Comput.
Aid. Mol. Des. 27, 221-234 (2013)
[6] R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P.
C. Sanschagrin and D. T. Mainz, Extra Precision Glide: Docking and Scoring Incorporating a
Model of Hydrophobic Enclosure for Protein-Ligand Complexes, J. Med. Chem. 49, 6177-6196
(2006)

44
The role of motif III and its extended region in positioning the two
helicase domains in the motor subunit of the restriction-
modification system EcoR124I
Sinha D.1, Bialevich V.1, Shamayeva K.1, Řeha D.1, Ettrich R.1
1INSB, Academy of Sciences of the Czech Republic
The type I restriction-modification enzymes differ significantly from the type II enzymes
commonly used as molecular biology reagents. On hemi-methylated DNAs type I enzymes act
as conventional adenine methylases at their specific target sequences, but unmethylated targets
induce them to translocate thousands of basepairs through the enzyme before cleaving distant
sites nonspecifically. EcoR124I belongs to the same superfamily as yeast dsDNA translocase
Rad54 and two RecA-like helicase domains are involved in translocation and contain the seven
characteristic motifs for DEAD box helicases. In Rad54 an extended region of motif III is
involved ATPase activity. In EcoR124I this extended region, although it bears sequence and
structural similarities with Rad54, does not influence ATPase activity nor restriction activity.
However, the conserved glycin residue in motif III plays a role in the positioning of the two
helicase domains towards each other and its mutation alters ATPase activity and cleavage
activity.
References
[1] Mikalai Lapkouski, et al.[2009]Structure of the motor subunit of type I restriction-
modification complex EcoR124I.,Nat Struct Mol Biol 16:1 94-95 Jan
[2] D Sinha, et al.[2014]Interdomain communication in the endonuclease/motor subunit of Type
I restriction-modification enzyme EcoR124I,Journal of Molecular Modeling20 (7):2334
[3] Eva Csefalvay, et al.[2015]Functional coupling of duplex translocation to DNA cleavage in
a type I restriction enzyme PLOS One accepted with minor revisions 21.1.2015.
[4] Bialevich V, et al.The role of motif III and its extended region in positioning the two helicase
domains in the motor subunit of the restriction-modification system EcoR124I [manuscript in
preparation]
Acknowledgements: We gratefully acknowledge support from the Czech Science Foundation
(project number GACR P207/12/2323), and the Grant Agency of the University of South
Bohemia (grant no. 170/2010/P). Some computations were performed in MetaCentrum
SuperComputer facility.

45
A computational study of the interaction between dopamine and
DNA/RNA nucleosides
Katarína Skúpa, Milan Melicherčík, Ján Urban
Faculty of Mathematics, Physics and Informatics, Comenius University in Bratislava
The interaction between protonated dopamine and neutral RNA and DNA nucleosides was
studied by means of density functional theory calculations in vacuum and in implicit water. On
the most stable complexes formed with each of the nucleosides, the vertical absorption excitation
energies were evaluated and compared with the values of separated dopamine and corresponding
nucleoside. The most stable complex was formed with guanosine and the spectral changes in this
complex resulted in a significant reduction of the oscillator strength of the first dopamine’s
transition. In the first guanosine’s transition, a redshift of 0.2 eV was found combined with a
reduction of the oscillator strength.

46
Molecular Recognition of Norepinephrine Derivatives C8H12NO by
GPCR
Cheng Min Sung,1 János J. Szórád,2 Lily M. Hunnisett,2 Béla Fiser,2,3 Anita
Rágyanszki,2 Balázs Jójárt,2 Milán Szőri,2 Imre G. Csizmadia,2,4 Béla Viskolcz.2
1Department of Immunology and Human Biology, University of Toronto, M5S 3H6 Toronto, Ontario, Canada 2Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary
3Department of Organic Chemistry I, University of the Basque Country, Manuel Lardizábal 3, 20018 Donostia-San
Sebastian, Gipuzkoa, Spain 4Department of Chemistry, University of Toronto, M5S 3H6 Toronto, Ontario, Canada
Norepinephrine with a protonated amino group, C8H12NO3, is an essential monoamine
neurotransmitter in human body that exerts its effect on several brain regions, resulting in fight-
or-flight response.[1] As a vasopressor, norepinephrine is used as an immediate treatment for
vasodilatory states such as septic shocks.[2] Norepinephrine has high affinity to α1 and α2
adrenergic receptors (AR); it also binds to β1 and β3, but only weakly binds to β2AR.[3]
In this study, molecular recognition of norepinephrine derivatives (C8H12NO) by β2AR was
performed by Maestro 10.1 software package. All of the possible isomers of C8H12NO were
generated, and β2AR structure (PDB name: 3sn6) was prepared.[4] After step-by-step precision
state calculations from HTVS to SP to XP, 10 conformations with the lowest docking scores were
analyzed. Optimizations of the 10 conformations were performed in solvation of water by
Gaussian 09, and free energies were calculated.
Figure1. The most stable norepinephrine derivative (C8H12NO) found to bind with β2AR
with the lowest docking score.
With computational method, the top 10 ligands for β2AR with molecular formula of C8H12NO
were found. Further synthesis and investigation can be used to visualize their drug potential.
References
[1] Katzung, BG, Trevor, AJ. (2014). Basic and clinical pharmacology 13th edition. McGraw-
Hill Education. ISBN: 978-0-07-182505-4.
[2] De Backer, D, Biston, P, Devriendt, J, Madl, C, Chochrad, D, Aldecoa, C, Brasseur, A,
Defrance, P, Gottignies, P, Vincent, J. (2010). Comparison of dopamine and norepinephrine in
the treatment of shock. New Engl. J. Med. 362(9):779-789
[3] Piascik, M.T., Perez, D.M. (2001). Alpha1-adrenergic receptors: new insights and
directions. J. Pharmacol. Exp. Ther. 298 (2):403–410.
[4] Rasmussen, S.G.F., DeVree, B.T., Zou, Y., Kruse, A.C., Chung, K.Y., Kobilka, T.S., Thian,
F.S., Chae, P.S., Pardon, E., Calinski, D.,Mathiesen, J.M., Shah, S.T.A., et al. (2011). Crystal
structure of the beta-2 adrenergic receptor-Gs protein complex. Nature. 477:549-557

47
Where are the isopeptides?
Veronika Szentirmai1, Milán Szőri1
1 Department of Chemical Informatics, University of Szeged, H-6725, Szeged, Boldogasszony sgt 6.
Corresponding author: [email protected], [email protected]
Conventional peptide (amide) bond is covalent linkage of NH2 and COOH groups of α-carbons
of two essential amino acid residues, which can form proteins (e.g hormones, enzymes and
structural proteins) in the living cells. However, most of the essential amino acid have other
functional groups (e.g. COOH, NH2 groups in the cases of Asp and Lys, respectively) which is
suitable to form alternative linkage between amino acid residues in principles. These species are
called as isopeptides in general and the best known existing example for such is the disulfide
bond between two cysteine (Cys) residues. Several other types of isopeptides can be also found
in Nature. In cyanobacteria and a few heterotrophic bacteria, other multiple isopeptide bonds
(multi-Arg-poly-γAsp) can be found with function of temporary nitrogen and reserve.1 It also has
been found that Streptococcus pyogenes, like many other Gram-positive bacteria, contains
extracellular proteins stabilized by spontaneous formation of the intramolecular isopeptide bonds.
The reason of the relatively rare presence of the isopeptides in living cell is not quite understood,
since some of them can have higher thermodynamic stability than the conventional peptide
linkage between the amino acid residues (ΔrG0 (Gly-Ala) = 17.3 kJ/mol), as it is the case for
gluthatione (γ-Glu-Cys-Gly) in our previous work.2 In order to shed some more light on the
thermodynamic background of the isopeptide formations, they are described quantitatively by the
change of thermochemical functions (ΔrH0, ΔrS0 and ΔrG0). These calculations were carried out
using the combination of the G3MP2B33 composite model and SMD implicit solvent model.4
According to our preliminary results serine-γ-asparagine (Ser_Asn) and Nε-(γ-glutamyl)lysine
(Glu_Lys) show relative large thermodynamical stability (ΔrG0 < -30.5 kJ/mol, above the Gibbs
free energy of hydrolysis of 1 mol ATP). Amongst these isopeptides, Glu_Lys motif had also
been detected experimentally in the spore coat proteins of Bacillus Subtilis5 and during the action
of transglutaminases.6
References
(1) Krehenbrink, M.; Oppermann-Sanio, F. B.; Steinbüchel, A. Arch. Microbiol. 2002, 177
(5), 371–380.
(2) Fiser, B.; Jojart, B.; Szőri, M.; Lendvay, G.; Csizmadia, I. G.; Viskolcz, B. J. Phys. Chem.
B 2015, 119.
(3) Baboul, A. G.; Curtiss, L. a.; Redfern, P. C.; Raghavachari, K. J. Chem. Phys. 1999, 110
(16), 7650.
(4) Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Chem. Theory Comput. 2009, 5 (9),
2447–2464.
(5) Kobayashi, K.; Kumazawa, Y.; Miwa, K.; Yamanaka, S. FEMS Microbiol. Lett. 1996, 144
(2-3), 157–160.
(6) Steinert, P. M.; Marekov, L. N. Mol. Biol. Cell 1999, 10 (12), 4247–4261.

48
Binding Affinities of Epinephrine Derivatives (C9H14NO2) to ß2
Adrenergic Receptors; A Computational Study
Wendy Wang1, János J. Szórád3, Lily M. Hunnisett3, Béla Fiser3,4, Anita
Rágyanszki3, Balázs Jójárt3, Milán Szöri3, Imre G. Csizmadia2,3, Béla Viskolcz2
1Department of Pharmacology and Toxicology, University of Toronto, M5S 3H6 Toronto, Ontario, Canada 2Department of Chemistry, University of Toronto, M5S 3H6 Toronto, Ontario, Canada
3Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary 4Department of Organic Chemistry I, University of the Basque Country, Manuel Lardizábal 3, 20018 Donostia-San
Sebastian, Gipuzkoa, Spain
Over the past decades, ß2 adrenergic receptors have garnered attention as drug targets for many
pharmacological developments. Combined with a number of G-protein coupled receptors, a well-
known ligand that binds to the cavity of this protein is epinephrine [1]. The depolarization of this
receptor plays a crucial role in the fight-or-flight response, smooth muscle contractions as well
as bradycardia [1]. Therefore, the investigation of the relationship between the epinephrine ligand
and its receptor has great implications upon the development of novel compounds, as epinephrine
today has known applications in medicine, particularly in the treatment of select cardiovascular
conditions—such as heart arrhythmia—as well as roles in inducing smooth muscle relaxation in
the bronchioles when coupled with ß2 adrenergic receptors [2].
The aim of this investigation is to generate all the possible isomeric forms given the molecular
formula: C9H14NO2. The relative lack of one oxygen atom compared to the epinephrine molecular
formula makes this compound an epinephrine derivative. Noting that the binding pocket of the
3SN6 receptor is hydrophobic, the aim is to investigate the elements that increase the affinity of
the ligand with its active site. Computational software along with elements from the Schrödinger
Suite 2015 will be used to generate the library of possible isomers of the epinephrine derivative
(C9H14NO2). Density functional theory techniques will be carried out in the end to finalize the
best structure that dock to this protein.
References
[1] Marki, Arpad, et al. "Phytoecdysteriods and anabolic-androgenic steroids-structure and
effects on humans." Curr. Med. Chem 15.1 2008 75-91.
[2] Packer, Milton. "Current role of beta-adrenergic blockers in the management of chronic
heart failure." Am. J. Med 110.7 2001 81-94.

49
Computational Molecular Modeling of Ephedrine and its Isomers
(C10H16NO) and Molecular Recognition by GPCR protein
Vivian Xie1, János J. Szórád2, Lily M. Hunnisett2, Béla Fiser2, 3, Anita
Rágyanszki2, Balázs Jójárt2, Milán Szőri2, Imre G. Csizmadia1, 2, Béla Viskolcz2
1Department of Chemistry, University of Toronto, M5S 3H6 Toronto, Ontario, Canada 2Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary
3Department of Organic Chemistry I, University of the Basque Country, Manuel Lardizábal 3, 20018 Donostia-San Sebastian,
Gipuzkoa, Spain
A computational chemistry approach was used to create a molecular library of isomers with the
molecular formula C10H16NO (ephedrine). These isomers were studied using computational
chemistry principles to find their respective binding affinities with the β2 adrenergic receptor
protein. Programs such as the Schrodinger Maestro suite [1] were used to prepare and dock the
isomers. The stability of the isomers were calculated by density functional theory using Gaussian
09 [2]. Ephedrine plays an important role as a sympathomimetic drug, mimicking transmitters such as
adrenaline [3]. This makes it an interesting molecule to study for the purpose of drug synthesis
which target the receptors of adrenaline. The target receptor, β2 adrenergic receptor, is linked to
cardiovascular and pulmonary functions [4]. The isomer with the highest docking score suggests
the highest binding affinity with this receptor.
Figure 1 The most stable isomer of C10H16NO
Results showed that Figure 1 had the highest binding affinity with the receptor due to
intermolecular hydrogen bonding with surrounding amino acids of the receptor. The stability of
the isomer in solvation was had a fair correlation with the docking score, highlighting why
some isomers docked with the receptor better than others.
References
[1] Schrödinger Release 2015-2: Maestro, version 10.2, Schrödinger, LLC, New York, NY, 2015.
[2] Gaussian 09, Revision D.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et. al. Gaussian,
Inc., Wallingford CT, 2009.
[3] Casella, M., Russo, A.D., Izzo, G., Pieroni, M., Andreini, D., Russo, E., Colombo, D.,
Bologna, F., Bolognese, L., Zeppilli, P., Tondo, C. (2014). Ventricular arrhythmias induced by
long-term use of ephedrine in two competitive athletes. Heart Vessels. 30(10). 280-283
[4] Ramussen, S. G. F., Choi, H., Rosenbaum, D. M., Kobilka, T. S., Thian, F. S., Edwards, P.
C., Burghammer, M., Ratnala, V. R. P., Sanishvili, R., Fischetti, R. F., Schertler, G. F. X., Weis,
W. I., Kobilka, B. K. (2007). Crystal Structure of the human β2 adreneric G-protein-coupled
receptor. Nature. 10(1038). 1-6.

50
Interactions of epinephrine derivative (C9H14NO) with GPCR
protein
Rui Yue1, János J. Szórád2, Lily M. Hunnisett2, Béla Fiser2,3, Anita Rágyanszki2,
Balázs Jójárt2, Milán Szőri2, Imre G. Csizmadia1,2, Béla Viskolcz2 and the
presenting author Rui Yue1
1 Department of Chemistry, University of Toronto, M5S 3H6 Toronto, Ontario, Canada 2 Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary
3 Department of Organic Chemistry I, University of the Basque Country, Manuel Lardizábal 3, 20018 Donostia-
San Sebastian, Gipuzkoa, Spain
Epinephrine (C9H13NO3) is a common primary treatment of anaphylaxis and cardiac arrest [1, 2].
When epinephrine binds to a β2 GPCR receptor in the human liver, heart and smooth muscle to
form a complex, it triggers second messages to increase glycogenosis, increase heart rate and
relax the smooth muscle [3]. In the interest of understanding the strength of biological activity of
a protein-ligand complex, determination of the docking score of the most stable complex of β2
G-protein coupled receptor and epinephrine derivative C9H14NO and analysis the interaction
energy are the purposes of the project. All possible benzene- ring containing constitutional
isomers were generated using Molgen 5.0, then all of them was docked into β2 GPCR by Maestro
Suits 2015-1 program package. The top 10 ranked structures were selected for further analysis.
Figure 1. Docking score comparison for top three most stable structures in epinephrine family.
Results suggest the non-covalent interactions between H atoms of the ligand and the oxygen atom
of the ASP113 residue form the most stable binding interaction. The increasing trend of the
stability of epinephrine family: epinephrine derivative (C9H14NO), another epinephrine
derivative (C9H14NO2) and epinephrine (C9H14NO3) suggests that increasing number of oxygen
in the formula increases chances of non-covalent interactions and the complex stability.
References
[1] M. C. Bhalla, et al, Predictors of epinephrine autoinjector needle length inadequacy.
American Journal of Emergency Medicine 31, 1671-1676 (2013)
[2] P. A. Greenberger, Epinephrine for anaphylaxis. Annals of Allergy, Asthma &
Immunology. 94 (5). 515-516 (2005)
[3] J. Pearce, Links between nerves and glands: the story of adrenalin. Advances in Clinical
Neuroscience & Rehabilitation 9: 22–28. (2009)

51
List of Participants
1 Agneta Balint [email protected] West University of Timisoara
2 Stefan Balint [email protected] West University of Timisoara
3 Paul Bauer [email protected] Department of Cell and Molecular Biology,
Uppsala University
4 Giorgia Brancolini [email protected] Center S3, CNR-NANO Institute Nanocience
5 Richard Buchner richard.buchner@
chemie.uni-regensburg.de
Institute of Physical and Theoretical Chemistry,
University of Regensburg
6 Jaroslav Burda [email protected] Charles University in Prague
7 Joshua Campbell [email protected] University of Szeged
8 Imre G. Csizmadia [email protected] University of Szeged
9 Balázs Fábián [email protected] University of Szeged
10 Maxim Fedorov [email protected] University of Strathclyde
11 Jacob Fondriest [email protected] Princeton University
12 Pavel Grinkevich [email protected] The University of South Bohemia in Ceske
Budejovice
13 Ben Hanson [email protected] University of Leeds
14 Pradeep Hiriyur
Nagaraj
[email protected] INSB, Czech Academcy of Sciences
15 Brianna Hnath [email protected] Princeton University
16 Lily Hunnisett [email protected] University of Szeged
17 Abdenacer Idrissi [email protected] University Lille1 Sciences and Technologies
18 Imre Jákli [email protected] MTA-ELTE Protein Modelling Research Group
19 Pál Jedlovszky [email protected] ELTE University
20 Balázs Jójárt [email protected] University of Szeged
21 Deepika Kale [email protected] Institute of Nanobiology and Structural Biology,
Global Change Research Center, Academy of
Sciences of the Czech Republic, Nove Hrady,
Czech Republic
22 Outi Kamarainen [email protected] University of Leeds
23 Lynn Kamerlin [email protected] Uppsala University, Department of Cell and
Molecular Biology
24 Sebastian Kmiecik [email protected] University of Warsaw
25 Sudhir Kumar Pal [email protected] Institute of Nanobiology and structural biology of
GCRC
26 Michal Kutý [email protected] The Institute of Nanobiology and Structural
Biology
27 Virginia Lane [email protected] Princeton University
28 Zita Lovrity [email protected] University of Miskolc, Department of
Nanobiotechnology
29 Min Yen Lu [email protected] University of Szeged
30 Jost Ludwig [email protected] INSB of GCRC, Academy of Sciences of the
Czech Republic, Nove Hrady, Czech Republic
31 Nicholas Luedtke [email protected] Princeton University
32 Ben Luisi [email protected] University of Cambridge

52
33 Mikael Lund [email protected] Lund University
34 John Martin [email protected] University of Cincinnati
35 Milan Melicherčík [email protected] Comenius University, Faculty of Mathematics,
Physics and Informatics
36 Babak Minofar [email protected] INSB
37 Wojciech Mrozik [email protected] Newcastle University
38 Judit Némethné
Sóvágó
[email protected] University of Miskolc
39 Martin Novák [email protected] Centrum výzkumu globání změny AV ČR, v.v.i.
40 Michael Owen [email protected] Forschungszentrum Jülich
41 Anna Pabis [email protected] Uppsala University, Department of Cell and
Molecular Biology
42 Saurabh
Kumar
Pandey [email protected] Institute of Nanobiology and Structural Biology
43 Miha Purg [email protected] Department of Cell and Molecular Biology,
Uppsala University
44 Anita Rágyanszki [email protected] University of Szeged
45 David Reha [email protected] INSB, Academy of Sciences of the Czech
Republic
46 Zsófia
Borbála
Rózsa [email protected] University of Szeged
47 Seung Gwan Ryoo [email protected] University of Szeged
48 Michelle Sahai [email protected] University of Roehampton
49 Christian Schröder [email protected] Department of Computational Biological
Chemistry
50 Dhiraj Sinha [email protected] INSB, Academy of Sciences of the Czech
Republic
51 Katarína Skúpa [email protected] Comenius University in Bratislava, Faculty of
Mathematics, Physics and Informatics
52 Emma Stavropoulos [email protected] Princeton University
53 Cheng Min Sung [email protected] University of Szeged
54 Veronika Szentirmai [email protected] University of Szeged
55 János Szórád [email protected] University of Szeged
56 Milán Szőri [email protected] University of Szeged
57 Laura Tociu [email protected] Princeton University
58 Jan Urban [email protected] Faculty of Mathematics, Physics and Informatics,
Comenius University in Bratislava
59 Andrea Vachová [email protected] Centrum výzkumu globání změny AV ČR, v.v.i.
60 James Valdés [email protected] Institute of Parasitology, Biology Centre of ASCR
61 Béla Viskolcz [email protected] University o Szeged
62 Jiawen Wang [email protected] University of Szeged
63 Daniel Wood [email protected] Princeton University
64 Vivian Xie [email protected] University of Szeged
65 Rui Yue [email protected] University of Szeged

















