8 Treating Autoimmune Bullous Skin Disorders with Biologics R. Eming, A. Niedermeier, M. Pfütze, A. Jacobi, M. Hertl 8.1 Introduction 8.1.1 Autoimmune Bullous Skin Disorders Autoimmune bullous skin disorders represent a group of severe, chronic skin diseases which are characterized by the presence of autoantibodies targeting distinct adhesion molecules of the epidermis and dermoepider- mal basement membrane zone leading to a loss of adhesive function of the target antigen(s) (Fig. 8.1). The appearance of blisters and erosions of the skin and/ or mucous membranes is the leading clinical sign of autoimmune bullous skin disorders (Fig. 8.2). While histopathology reveals the location of the blister for- mation, the detection of tissue bound autoantibodies by immunofluorescent staining of uninvolved perile- sional skin biopsies is mandatory for diagnosing auto- immune bullous skin disorders. Circulating autoanti- bodies can be visualized by indirect immunofluores- cence using tissue substrates such as monkey esopha- gus and sodium chloride-split human skin. Based on a Fig. 8.1. Schematic overview of desmosomal and hemi- desmosomal autoantigens in autoimmune bullous skin disorders. Shown are the major components of desmo- somes which have been identified as autoantigens in the different clinical variants of pemphigus (a) the specificity of the targeted antigens, several clinical- ly and immune serologically distinct bullous disorders have been defined (Fig. 8.2). 8.1.1.1 Pemphigus Desmogleins (Dsg) are transmembranous components of desmosomes, adhesion units specialized in confer- ring epidermal keratinocyte adhesion, and are linked to intercellular molecules of the desmosomal plaque which in turn interact with components of the cytoskel- eton (Fig. 8.1a). Among several forms of pemphigus, pemphigus vulgaris (PV) and pemphigus foliaceus (PF) represent the major subtypes. IgG autoantibodies against Dsg3 in PV and Dsg1 in PF lead to loss of des- mosomal adhesion of epidermal keratinocytes and intraepidermal blister formation (Fig. 8.2A–C). In PV patients suffer from flaccid blister/erosions of the mucous membranes, primarily the oral mucosa and the skin. PF is characterized by crusted painful erosions typically of the seborrheic areas such as scalp, face, Chapter 8
Transcript
8Treating Autoimmune Bullous Skin Disorderswith BiologicsR. Eming, A. Niedermeier, M. Pfütze, A. Jacobi, M. Hertl
Autoimmune bullous skin disorders represent a groupof severe, chronic skin diseases which are characterizedby the presence of autoantibodies targeting distinctadhesion molecules of the epidermis and dermoepider-mal basement membrane zone leading to a loss ofadhesive function of the target antigen(s) (Fig. 8.1).The appearance of blisters and erosions of the skin and/or mucous membranes is the leading clinical sign ofautoimmune bullous skin disorders (Fig. 8.2). Whilehistopathology reveals the location of the blister for-mation, the detection of tissue bound autoantibodiesby immunofluorescent staining of uninvolved perile-sional skin biopsies is mandatory for diagnosing auto-immune bullous skin disorders. Circulating autoanti-bodies can be visualized by indirect immunofluores-cence using tissue substrates such as monkey esopha-gus and sodium chloride-split human skin. Based on
a
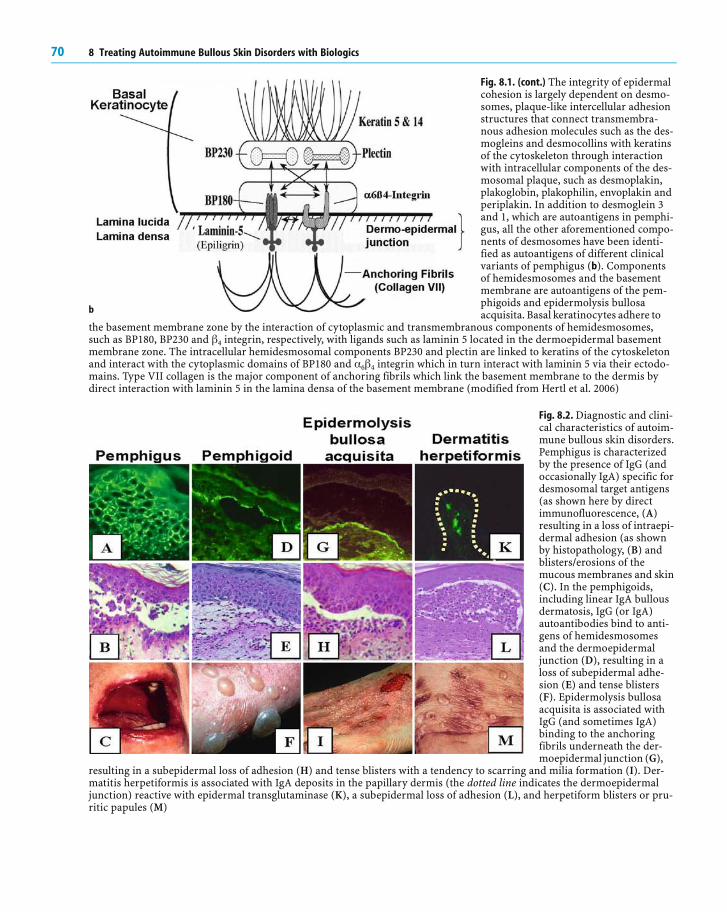
Fig. 8.1. Schematic overview of desmosomal and hemi-desmosomal autoantigens in autoimmune bullous skindisorders. Shown are the major components of desmo-somes which have been identified as autoantigens inthe different clinical variants of pemphigus (a)
the specificity of the targeted antigens, several clinical-ly and immune serologically distinct bullous disordershave been defined (Fig. 8.2).
8.1.1.1Pemphigus
Desmogleins (Dsg) are transmembranous componentsof desmosomes, adhesion units specialized in confer-ring epidermal keratinocyte adhesion, and are linkedto intercellular molecules of the desmosomal plaquewhich in turn interact with components of the cytoskel-eton (Fig. 8.1a). Among several forms of pemphigus,pemphigus vulgaris (PV) and pemphigus foliaceus(PF) represent the major subtypes. IgG autoantibodiesagainst Dsg3 in PV and Dsg1 in PF lead to loss of des-mosomal adhesion of epidermal keratinocytes andintraepidermal blister formation (Fig. 8.2A–C). In PVpatients suffer from flaccid blister/erosions of themucous membranes, primarily the oral mucosa and theskin. PF is characterized by crusted painful erosionstypically of the seborrheic areas such as scalp, face,
Chapter 8
b
Fig. 8.1. (cont.) The integrity of epidermalcohesion is largely dependent on desmo-somes, plaque-like intercellular adhesionstructures that connect transmembra-nous adhesion molecules such as the des-mogleins and desmocollins with keratinsof the cytoskeleton through interactionwith intracellular components of the des-mosomal plaque, such as desmoplakin,plakoglobin, plakophilin, envoplakin andperiplakin. In addition to desmoglein 3and 1, which are autoantigens in pemphi-gus, all the other aforementioned compo-nents of desmosomes have been identi-fied as autoantigens of different clinicalvariants of pemphigus (b). Componentsof hemidesmosomes and the basementmembrane are autoantigens of the pem-phigoids and epidermolysis bullosaacquisita. Basal keratinocytes adhere to
the basement membrane zone by the interaction of cytoplasmic and transmembranous components of hemidesmosomes,such as BP180, BP230 and q 4 integrin, respectively, with ligands such as laminin 5 located in the dermoepidermal basementmembrane zone. The intracellular hemidesmosomal components BP230 and plectin are linked to keratins of the cytoskeletonand interact with the cytoplasmic domains of BP180 and [ 6 q 4 integrin which in turn interact with laminin 5 via their ectodo-mains. Type VII collagen is the major component of anchoring fibrils which link the basement membrane to the dermis bydirect interaction with laminin 5 in the lamina densa of the basement membrane (modified from Hertl et al. 2006)
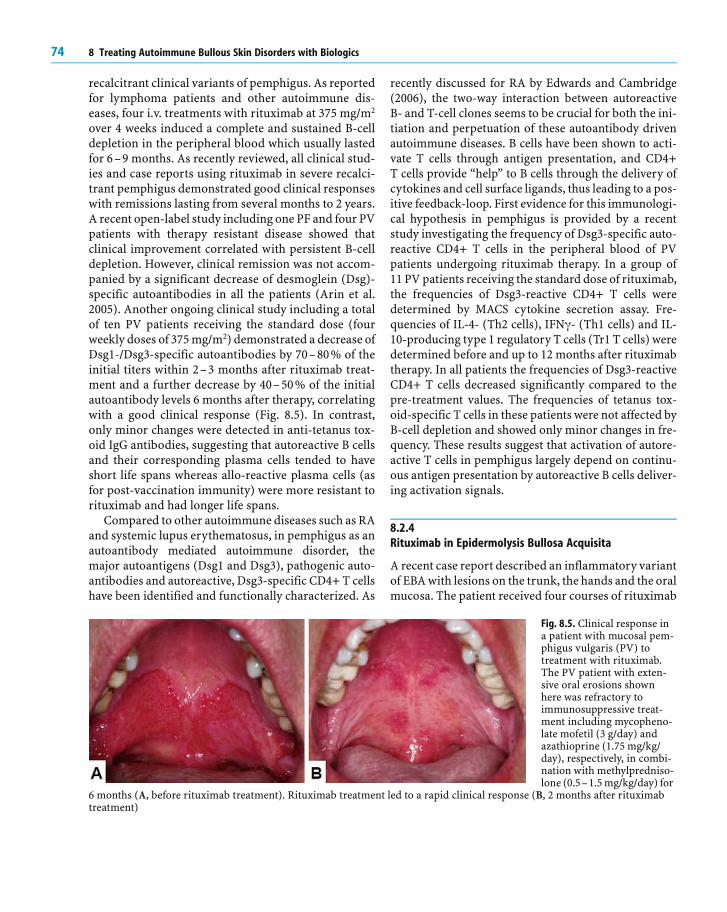
Fig. 8.2. Diagnostic and clini-cal characteristics of autoim-mune bullous skin disorders.Pemphigus is characterizedby the presence of IgG (andoccasionally IgA) specific fordesmosomal target antigens(as shown here by directimmunofluorescence, (A)resulting in a loss of intraepi-dermal adhesion (as shownby histopathology, (B) andblisters/erosions of themucous membranes and skin(C). In the pemphigoids,including linear IgA bullousdermatosis, IgG (or IgA)autoantibodies bind to anti-gens of hemidesmosomesand the dermoepidermaljunction (D), resulting in aloss of subepidermal adhe-sion (E) and tense blisters(F). Epidermolysis bullosaacquisita is associated withIgG (and sometimes IgA)binding to the anchoringfibrils underneath the der-moepidermal junction (G),
resulting in a subepidermal loss of adhesion (H) and tense blisters with a tendency to scarring and milia formation (I). Der-matitis herpetiformis is associated with IgA deposits in the papillary dermis (the dotted line indicates the dermoepidermaljunction) reactive with epidermal transglutaminase (K), a subepidermal loss of adhesion (L), and herpetiform blisters or pru-ritic papules (M)
70 8 Treating Autoimmune Bullous Skin Disorders with Biologics
chest and upper back whereas mucosal lesions areabsent in PF. In general, the treatment of pemphigus isbased on immunosuppressive treatment regimentsconsisting of high dose glucocorticoids and immuno-suppressive “steroid-sparing” adjuvants, including aza-thioprine or mycophenolate mofetil.
8.1.1.2Bullous Pemphigoid
In the pemphigoids, IgG autoantibodies against com-ponents of the dermoepidermal basement membrane,such as bullous antigen 180 (BP180/BP antigen 2/typeXVII collagen), BP antigen 230 (BP230/BP antigen 1),and laminin 5 interfere with the adhesion of basal epi-dermal keratinocytes to the dermoepidermal basementmembrane zone (Fig. 8.1b). BP180 and BP230 aretransmembranous and intracellular components,respectively, of hemidesmosomes of basal epidermalkeratinocytes while laminin 5 is a ligand of BP180located in the lamina lucida of the basement membranezone (Fig. 8.1b). Bullous pemphigoid (BP) is the mostcommon autoimmune blistering disease in adults. Pro-dromal, non-bullous periods of BP may occur in whichpatients demonstrate pruritic eczematiform papules orurticarial plaques. The bullous manifestations of BPpresent as tense, fluid-filled vesicles and bullae on nor-mal or erythematous skin in combination with urticat-ed plaques (Fig. 8.2D–F). Involvement of the oralmucosa is rarely observed in about 10–30% ofpatients. In contrast to pemphigus, the use of potenttopical corticosteroids such as clobetasol propionatehas been shown to be effective even in the treatment ofgeneralized BP. Nevertheless, BP patients with exten-sive disease are usually treated with systemic predni-sone at doses of 0.5–1.0 mg/kg/day, which is then pro-gressively tapered.
8.1.1.3Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare chronicsubepidermal bullous disease of the skin and mucousmembranes characterized by the presence of autoanti-bodies against type VII collagen, a major component ofanchoring fibrils. There is a great diversity in the clini-cal presentation of the disease. Generally, it can be dif-ferentiated into the mechanobullous or classic form ofEBA and inflammatory variants, resembling BP, Brun-
sting-Perry pemphigoid, mucous membrane pemphi-goid or linear IgA bullous dermatosis. MechanobullousEBA presents as a non-inflammatory disease with anacral distribution and skin fragility over trauma pronesurfaces. The blisters and erosions heal with scarringand milia formation (Fig. 8.2G–I). Especially themechanobullous form of EBA often reveals itself refrac-tory to high doses of systemic glucocorticoids com-bined with immunosuppressive adjuvants. Other treat-ment options include the use of colchicines, high doseimmunoglobulins or immunoadsorption.
8.1.1.4Dermatitis Herpetiformis Duhring
Dermatitis herpetiformis (DH) represents a bullous orpruritic autoimmune disorder with subepidermal blis-ter formation which is considered to be a specific cuta-neous manifestation of celiac disease, although mostDH patients do not present gastrointestinal symptoms.The autoantigen of dermatitis herpetiformis, epider-mal transglutaminase, is targeted by autoantibodies ofthe IgA class. Immunofluorescent staining of unin-volved perilesional skin biopsies reveals granular IgAdeposition in the papillary dermis (Fig. 8.2K–M). DHpatients show an intense pruritus with eruption of ery-thematous papules and herpetiform vesicles distribut-ed symmetrically on the extensor surfaces. In additionto a gluten-free diet, the sulphonamide diamino-diphe-nyl sulfone (dapsone) is most commonly used in thetreatment of DH.
8.1.2Immune Pathogenesis of Bullous Autoimmune Disorders
Pemphigus and pemphigoid are considered to be pro-totypic bullous disorders based on their well-charac-terized immune pathogenesis. Apart from pemphigusand BP, there is only circumstantial evidence that auto-reactive T cells are present and involved in the patho-genesis of the autoimmune bullous disorders epider-molysis bullosa acquisita and dermatitis herpetiformis.In PV and BP, autoreactive CD4+ T lymphocytes thatare presumably crucial in initiating the autoimmuneresponse recognize distinct epitopes of the extracellu-lar portions of Dsg3 and BP180, components of desmo-somal and hemidesmosomal adhesion complexes ofhuman skin, respectively. Dsg3- and BP180-reactive Tcells produce T-helper 2 (Th2) cytokines, such as IL-4,
8.1 Introduction 71
Fig. 8.3. Schematic overviewof the immune pathogenesisof pemphigus vulgaris. Pem-phigus vulgaris (PV) is theprototype of an autoanti-body-mediated immunobul-lous skin disorder and ischaracterized by a loss ofintraepidermal adhesion pri-marily caused by IgG auto-antibodies specific for des-moglein (Dsg)3 and Dsg1,components of the desmo-somal adhesion complex ofepidermal keratinocytes thatare connected to the keratincytoskeleton through inter-action with the intracellularplaque proteins plakoglobin(PG) and desmoplakin (DP)(inset). IgG production bythe autoreactive B cells ispresumably regulated byDsg3- and Dsg1-reactive Th1and Th2 cells
IL-5 and IL-13, and presumably foster the productionof autoantibodies of the Th2-dependent IgG4 subtypewhich are preferentially seen in active stages of thesedisorders (Fig. 8.3).
8.2Rituximab (Anti-CD20 Monoclonal Antibody)in the Treatment of Autoimmune BullousSkin Disorders8.2.1Biological Activity of Rituximab
Rituximab is a chimeric human/mouse IgG1 monoclo-nal antibody with human constant regions and variablemurine regions derived from a murine anti-CD20 anti-body (IDEC 2B8). Rituximab is directed toward CD20,a pan B-cell glycoprotein which is expressed on B lym-phocytes from the pre-B-cell stage to the pre-plasma-cell stage. CD20 is a four-transmembrane glycoproteinthat is involved in B-cell differentiation and activation,although its exact physiological function is not yet fullyunderstood. Several characteristics make the CD20antigen attractive for immunotherapy; since CD20does not circulate in the plasma, it is neither internal-ized nor downregulated and there is no evidence thatantibody binding leads to shedding from the cell sur-
face of the CD20+ target B cells. Among several mecha-nisms involved in B-cell killing by rituximab, its B-cellcytolytic activity mainly depends on antibody-depen-dent cell-mediated cytotoxicity (ADCC). Polymor-phisms of the Fc * RIII receptor which mediates ADCCseem to influence the response to rituximab. Othermechanisms such as complement-mediated lysis andinduction of apoptosis may also contribute to the B-cell-depleting activity of rituximab. Interestingly, thereis recent evidence suggesting that the mechanism of B-cell killing by rituximab depends on the tissue micro-environment (Fig. 8.4).
In vivo studies using hCD20 BAC (bacterial artificialchromosome) transgenic mice demonstrate that deple-tion of B lymphocytes varies among different B-cellcompartments. In these mice, over 90% of the circulat-ing B cells in the peripheral blood are depleted withinminutes. In lymph nodes and the spleen, B-cell deple-tion occurs within 24 h after application of rituximaband within 7 days in the peritoneal cavity where signifi-cant B-cell depletion is delayed. Even though more than90% of follicular B cells in the spleen are depleted with-in 2 days after rituximab application, germinal center Bcells, particularly marginal zone B cells, appear to bemore resistant to killing. These differences in resistanceto rituximab are neither due to lack of CD20 expressionnor due to differences in drug bioavailability. Thus, the
72 8 Treating Autoimmune Bullous Skin Disorders with Biologics
Fig. 8.4. Mode of action of rituximab. B-cell killing is largelymediated by antibody-dependent cell mediated cytotoxicity(ADCC). Recent data indicates that the patient’s Fc * RIII allo-type seems to influence the response to rituximab. In vitrostudies have shown that rituximab effectively induces comple-ment-dependent cytotoxicity (CDC) against B-cell lines andlymphoma cells. Induction of apoptosis may also contribute toB-cell killing
differences in kinetics and sensitivity to the B-cell cyto-lytic activity of rituximab seem to depend on protectivemicroenvironmental factors. The expression of theCD20 antigen is not restricted to B cells. A small num-ber of T cells and NK cells also express low levels ofCD20. A recent study with 24 rheumatoid arthritis(RA) patients showed that these cell populations disap-pear for a mean of 5 months after rituximab therapy.The authors did not find significant changes in the totalnumber or frequency of other T-cell subpopulations inthe peripheral blood.
8.2.2Clinical Experience with Rituximab Therapy
In December 1997 the US Food and Drug Administra-tion (FDA) approved rituximab for the treatment ofrelapsing or refractory indolent CD20+ B-cell non-Hodgkin’s lymphoma (NHL). More recently, rituximabwas also approved for the treatment of aggressive NHLin combination with standard chemotherapy in the USand in Europe. The use of rituximab in the treatment ofRA was endorsed by the FDA approval in March 2006.
The current application of rituximab in autoantibodymediated autoimmune diseases was catalyzed by find-ings that treatment of B-cell NHL improved symptomsof lymphoma-associated autoimmune phenomena,since malignant B-cell clones are capable of secretinglow-affinity self-reactive antibodies, for example auto-antibodies specific for self antigens on red blood cellsleading to autoimmune hemolytic anemia (Boye et al.2003). Moreover, the rationale for applying rituximabin primary autoimmune disorders is the long-termdepletion of pathogenic, autoantibody-secreting B-cellclones, thus restoring tolerance. Follow-up of rituxi-mab-induced B-cell depletion in 24 patients with activeRA showed that B-cell repopulation in the peripheralblood occurred around 8 months after rituximab treat-ment. Increased numbers of naıve and immature Bcells (CD19+, IgD+, CD38high, CD10low, CD24high) wereidentified during repopulation in the peripheral bloodsimilar to the B-cell populations found following bone-marrow transplantation. In contrast, patients whoexperienced a relapse of RA tended to show highernumbers of memory B cells during repopulation(Edwards et al. 2006). Another report by Rouziere et al.(2005) demonstrated changes in the immunoglobulinheavy-chain repertoire after rituximab treatment, sug-gesting that new clones of B cells emerged from thebone-marrow dominating the repopulation process. Inmost clinical studies treating autoimmune disorders,the oncological regimen consisting of four weekly i.v.doses of 375 mg/m2 rituximab is adopted. However, dif-ferent treatment protocols are reported with two singleinfusions, multiple courses including four weekly infu-sions or single doses with up to 1 g rituximab. Besidesits use in RA, rituximab is being investigated in numer-ous autoimmune disorders including idiopathic throm-bocytopenic purpura, autoimmune hemolytic anemia,lupus erythematosus, myasthenia gravis, Sjögren’s syn-drome and, finally, autoimmune bullous disorders.Within the latter group of disorders, mainly severecases of paraneoplastic pemphigus, pemphigus vulga-ris, pemphigus foliaceus and, to a lesser extent, epider-molysis bullosa acquisita have been successfully treat-ed with rituximab.
8.2.3Rituximab in Pemphigus
There is a body of evidence demonstrating that rituxi-mab treatment is highly beneficial in the treatment of
8.2 Rituximab (Anti-CD20 Monoclonal Antibody) in the Treatment of Autoimmune Bullous Skin Disorders 73
recalcitrant clinical variants of pemphigus. As reportedfor lymphoma patients and other autoimmune dis-eases, four i.v. treatments with rituximab at 375 mg/m2
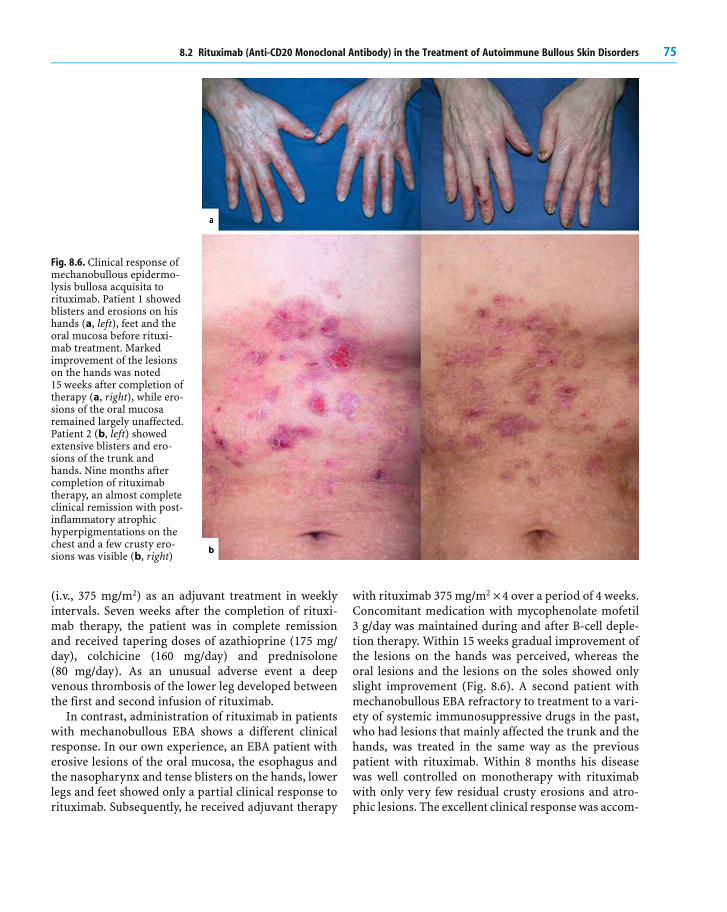
over 4 weeks induced a complete and sustained B-celldepletion in the peripheral blood which usually lastedfor 6–9 months. As recently reviewed, all clinical stud-ies and case reports using rituximab in severe recalci-trant pemphigus demonstrated good clinical responseswith remissions lasting from several months to 2 years.A recent open-label study including one PF and four PVpatients with therapy resistant disease showed thatclinical improvement correlated with persistent B-celldepletion. However, clinical remission was not accom-panied by a significant decrease of desmoglein (Dsg)-specific autoantibodies in all the patients (Arin et al.2005). Another ongoing clinical study including a totalof ten PV patients receiving the standard dose (fourweekly doses of 375 mg/m2) demonstrated a decrease ofDsg1-/Dsg3-specific autoantibodies by 70–80% of theinitial titers within 2–3 months after rituximab treat-ment and a further decrease by 40–50% of the initialautoantibody levels 6 months after therapy, correlatingwith a good clinical response (Fig. 8.5). In contrast,only minor changes were detected in anti-tetanus tox-oid IgG antibodies, suggesting that autoreactive B cellsand their corresponding plasma cells tended to haveshort life spans whereas allo-reactive plasma cells (asfor post-vaccination immunity) were more resistant torituximab and had longer life spans.
Compared to other autoimmune diseases such as RAand systemic lupus erythematosus, in pemphigus as anautoantibody mediated autoimmune disorder, themajor autoantigens (Dsg1 and Dsg3), pathogenic auto-antibodies and autoreactive, Dsg3-specific CD4+ T cellshave been identified and functionally characterized. As
Fig. 8.5. Clinical response ina patient with mucosal pem-phigus vulgaris (PV) totreatment with rituximab.The PV patient with exten-sive oral erosions shownhere was refractory toimmunosuppressive treat-ment including mycopheno-late mofetil (3 g/day) andazathioprine (1.75 mg/kg/day), respectively, in combi-nation with methylpredniso-lone (0.5–1.5 mg/kg/day) for
6 months (A, before rituximab treatment). Rituximab treatment led to a rapid clinical response (B, 2 months after rituximabtreatment)
recently discussed for RA by Edwards and Cambridge(2006), the two-way interaction between autoreactiveB- and T-cell clones seems to be crucial for both the ini-tiation and perpetuation of these autoantibody drivenautoimmune diseases. B cells have been shown to acti-vate T cells through antigen presentation, and CD4+T cells provide “help” to B cells through the delivery ofcytokines and cell surface ligands, thus leading to a pos-itive feedback-loop. First evidence for this immunologi-cal hypothesis in pemphigus is provided by a recentstudy investigating the frequency of Dsg3-specific auto-reactive CD4+ T cells in the peripheral blood of PVpatients undergoing rituximab therapy. In a group of11 PV patients receiving the standard dose of rituximab,the frequencies of Dsg3-reactive CD4+ T cells weredetermined by MACS cytokine secretion assay. Fre-quencies of IL-4- (Th2 cells), IFN * - (Th1 cells) and IL-10-producing type 1 regulatory T cells (Tr1 T cells) weredetermined before and up to 12 months after rituximabtherapy. In all patients the frequencies of Dsg3-reactiveCD4+ T cells decreased significantly compared to thepre-treatment values. The frequencies of tetanus tox-oid-specific T cells in these patients were not affected byB-cell depletion and showed only minor changes in fre-quency. These results suggest that activation of autore-active T cells in pemphigus largely depend on continu-ous antigen presentation by autoreactive B cells deliver-ing activation signals.
8.2.4Rituximab in Epidermolysis Bullosa Acquisita
A recent case report described an inflammatory variantof EBA with lesions on the trunk, the hands and the oralmucosa. The patient received four courses of rituximab
74 8 Treating Autoimmune Bullous Skin Disorders with Biologics
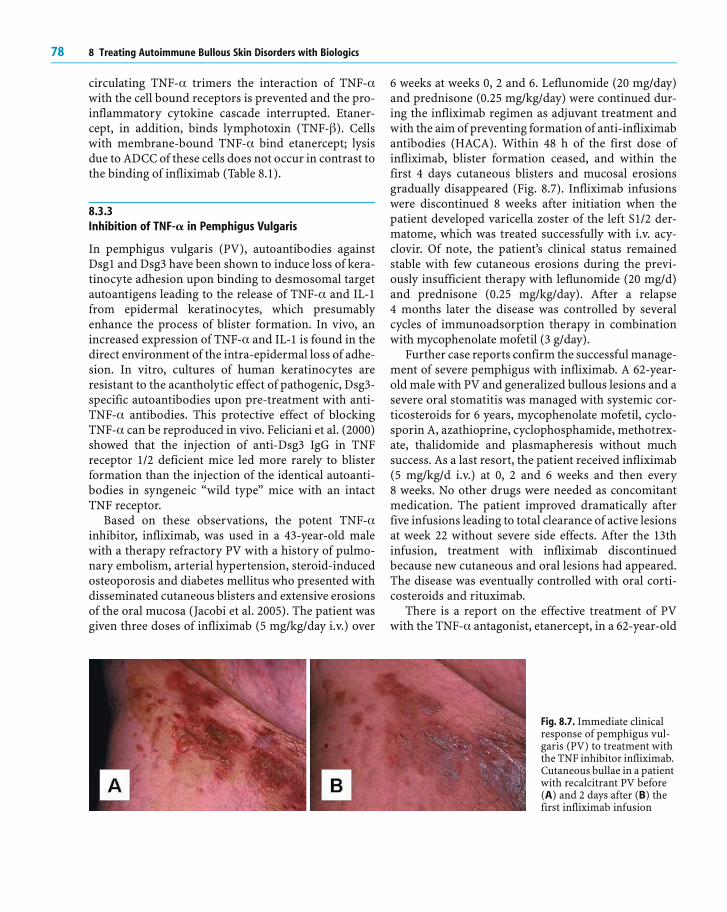
Fig. 8.6. Clinical response ofmechanobullous epidermo-lysis bullosa acquisita torituximab. Patient 1 showedblisters and erosions on hishands (a, left), feet and theoral mucosa before rituxi-mab treatment. Markedimprovement of the lesionson the hands was noted15 weeks after completion oftherapy (a, right), while ero-sions of the oral mucosaremained largely unaffected.Patient 2 (b, left) showedextensive blisters and ero-sions of the trunk andhands. Nine months aftercompletion of rituximabtherapy, an almost completeclinical remission with post-inflammatory atrophichyperpigmentations on thechest and a few crusty ero-sions was visible (b, right)
(i.v., 375 mg/m2) as an adjuvant treatment in weeklyintervals. Seven weeks after the completion of rituxi-mab therapy, the patient was in complete remissionand received tapering doses of azathioprine (175 mg/day), colchicine (160 mg/day) and prednisolone(80 mg/day). As an unusual adverse event a deepvenous thrombosis of the lower leg developed betweenthe first and second infusion of rituximab.
In contrast, administration of rituximab in patientswith mechanobullous EBA shows a different clinicalresponse. In our own experience, an EBA patient witherosive lesions of the oral mucosa, the esophagus andthe nasopharynx and tense blisters on the hands, lowerlegs and feet showed only a partial clinical response torituximab. Subsequently, he received adjuvant therapy
with rituximab 375 mg/m2 ×4 over a period of 4 weeks.Concomitant medication with mycophenolate mofetil3 g/day was maintained during and after B-cell deple-tion therapy. Within 15 weeks gradual improvement ofthe lesions on the hands was perceived, whereas theoral lesions and the lesions on the soles showed onlyslight improvement (Fig. 8.6). A second patient withmechanobullous EBA refractory to treatment to a vari-ety of systemic immunosuppressive drugs in the past,who had lesions that mainly affected the trunk and thehands, was treated in the same way as the previouspatient with rituximab. Within 8 months his diseasewas well controlled on monotherapy with rituximabwith only very few residual crusty erosions and atro-phic lesions. The excellent clinical response was accom-
8.2 Rituximab (Anti-CD20 Monoclonal Antibody) in the Treatment of Autoimmune Bullous Skin Disorders 75
panied by a decline in titres of circulating autoanti-bodies, which were undetectable & 9 months after com-pletion of rituximab therapy. Complete B-cell depletionpersisted for & 9 months. In both patients rituximabwas well tolerated with no side effects.
8.2.5Toxicity of Rituximab Treatment and Adverse Effects
In general, rituximab is well tolerated in both patientswith malignancies and those with autoimmune bullousdisorders. Except for two reports of serious infectionsincluding septic arthritis, Pneumocystis carinii pneu-monia and pneumonia after treatment of pemphiguspatients with rituximab, the most common adverseevents are infusion-related events occurring during orshortly after the first infusion. Larger studies with lym-phoma patients demonstrated that about 95% of theinfusion-related symptoms such as chills, fever, head-ache, rhinitis, pruritus or vasodilatation were mild tomoderate. Most of these symptoms could be avoided bypre-treatment with antihistamines, antipyretics andsteroids. Another report showed that a severe cytokinerelease syndrome more frequently occurred in lym-phoma patients with a higher tumor burden in theirperipheral blood which might be correlated withincreased serum levels of tumor necrosis factor-alpha(TNF- [ ) and interleukin 6 (IL-6) after infusion of ritu-ximab. At present, severe cytokine release syndromeshave not yet been reported in patients with bullousautoimmune disorders. Hematological toxicity hasbeen reported in about 10% of patients including atemporary reduction of platelets or neutrophils.Despite the depletion of B cells for several months therisk for infectious diseases does not seem to be signifi-cantly higher in patients treated with rituximab. Arecent study in RA patients demonstrated a significantdecrease of serum rheumatoid factor (IgM, IgG and IgAisotypes) over several months, whereas total immuno-globulin levels decreased by only 25% for IgG and IgAand by 43% for IgM and the titers for pneumococcalpolysaccharide-reactive antibodies remained un-changed. Another recent report showed unaffected lev-els of anti-tetanus toxoid antibodies in patients treatedwith rituximab. These observations help to explainwhy, despite complete depletion of B cells for severalmonths, the risk for secondary infections does notseem to be significantly higher in patients treated withrituximab. As rituximab is a chimeric antibody with
reduced immunogenicity, the presence of human anti-chimeric antibodies (HACA) is a rare event which hasbeen reported in less than 1% of treated patients withNHL. In patients with HACA impairing the function ofrituximab, a newly designed humanized monoclonalanti-CD20 antibody (hCD20) might be effective.
8.2.6Contraindications for Treatment with Rituximab
Ongoing or recurrent infectious disorders includingclinically apparent bacterial or viral infections excludethe application of rituximab. Patients with a history ofmalignancies or immunodeficiency syndromes, as wellas pregnant or lactating women, should not undergorituximab therapy. Patients with cardiac disordersshould be closely monitored during therapy with ritu-ximab. A history of anaphylactic hypersensitivity reac-tions against mouse proteins also excludes treatmentwith rituximab.
8.3Inhibitors of TNF-� in the Treatment ofAutoimmune Bullous Skin Disorders8.3.1Central Role of TNF-� in Inflammation
Tumor necrosis factor- [ is a proinflammatory cytokinewhich plays a key role in most of the inflammatory pro-cesses as well as in immune responses to infections andtumor antigens. TNF- [ is largely produced andreleased by macrophages and monocytes. It exists as asoluble 17-kDa sized protein made of three subunits.The cytokine stands at the beginning of a cascade ofpro-inflammatory cytokines and triggers off pro-inflammatory signals at the target cell by binding tomembrane-based TNF receptors, e.g., on T cells andmacrophages. Furthermore, TNF- [ causes a rise inbody temperature as well as the induction of the acutephase proteins, an increased migration of dendriticcells from the periphery to regional lymph nodes andan activation of neutrophils. In inflammatory diseases,TNF- [ possesses pleiotropic effects and leads, amongother factors, through the activation of a pro-inflam-matory cytokine cascade, to an induction of adhesionmolecules on endothelial cells (E-selectin, ICAM-1),which leads to an increased migration of leukocytes.Furthermore, TNF- [ induces the secretion of metallo-
76 8 Treating Autoimmune Bullous Skin Disorders with Biologics
proteinases and the release of pro-inflammatory cyto-kines (IL-1, IL-6, IL-8, GM-CSF). TNF- [ -conveyedinduction of pro-inflammatory cytokines, leukocytechemotaxis and angiogenesis possibly play a funda-mental role in autoimmune diseases of the skin, pre-sumably diseases which are characterized by elevatedTNF- [ serum concentrations, fever and an increase ofacute phase proteins. Elevated serum levels of TNF- [are detectable in many autoimmune diseases includingRA, psoriasis and Crohn’s disease.
8.3.2Inhibition of TNF-� by Biologics
A new class of TNF- [ inhibitors are the so-called “bio-logics,” which are either recombinant monoclonal anti-bodies or soluble TNF receptor fusion proteins. Theseproteins can be either isolated from animal tissues orcommonly synthesized by biotechnological methods.The more defined understanding of the pathophysiolo-gy of autoimmune diseases has led to the therapeuticuse of biologics in chronic inflammatory disorders(Scheinfeld 2004). At present, a few uncontrolled casereports suggest that the therapeutic blockade of TNF- [may be a novel option for the short-term control of oth-erwise recalcitrant autoimmune bullous skin disorders(Table 8.1).
Infliximab is a chimeric monoclonal antibody con-sisting of a murine anti-TNF- [ Fab fragment and the
Table 8.1. TNF- [ inhibitors and application in autoimmune bullous dermatoses
Pemphigus vulgaris IgA pemphigus Pemphigus vulgarisJacobi et al. 2005 Howell et al. 2005 Berookhim et al. 2004Pardo et al. 2005 Lin et al. 2005
Mucous membrane pemphigoidSacher et al. 2002Bullous PemphigoidYamauchi et al. 2006
constant region (Fc) of human IgG1. Infliximab bindswith high specificity and affinity to free and mem-brane-bound TNF- [ , which is expressed at the surfaceby activated T cells and macrophages. Besides theblocking of TNF- [ , the lysis of the target cell may alsooccur through activation of complement. This proba-bly accounts for the antibody conveying a cytotoxiceffect of infliximab. Inhibition of the pro-inflammatoryeffects of TNF- [ results through the formation of stablecomplexes. In addition, the migration of leukocytesand the release of TNF- [ -dependent proinflammatorycytokines like IL-1 and IL-6 is inhibited.
Adalimumab is a human monoclonal IgG1 antibodycontaining only human peptide sequences. It bindswith high specificity and affinity to soluble and mem-brane-bound TNF- [ and blocks its interaction with thep55 and p75 cell surface TNF receptors, thereby neu-tralizing the biological activities of this cytokine. Ada-limumab also modulates TNF-induced or regulatedbiological responses, such as changes in the levels ofadhesion molecules responsible for leukocyte migra-tion.
Etanercept is a recombinant human fusion proteinwhich consists of two soluble p75 TNF receptors andthe Fc portion of human IgG1. Etanercept possesses adimeric structure with high affinity to TNF- [ , and thelinkage to the Fc portion of human IgG produces a lon-ger half-life. Etanercept neutralizes TNF- [ better thanthe monomeric soluble p75 receptor. By blocking the
8.3 Inhibitors of TNF-� in the Treatment of Autoimmune Bullous Skin Disorders 77
circulating TNF- [ trimers the interaction of TNF- [with the cell bound receptors is prevented and the pro-inflammatory cytokine cascade interrupted. Etaner-cept, in addition, binds lymphotoxin (TNF- q ). Cellswith membrane-bound TNF- [ bind etanercept; lysisdue to ADCC of these cells does not occur in contrast tothe binding of infliximab (Table 8.1).
8.3.3Inhibition of TNF-� in Pemphigus Vulgaris
In pemphigus vulgaris (PV), autoantibodies againstDsg1 and Dsg3 have been shown to induce loss of kera-tinocyte adhesion upon binding to desmosomal targetautoantigens leading to the release of TNF- [ and IL-1from epidermal keratinocytes, which presumablyenhance the process of blister formation. In vivo, anincreased expression of TNF- [ and IL-1 is found in thedirect environment of the intra-epidermal loss of adhe-sion. In vitro, cultures of human keratinocytes areresistant to the acantholytic effect of pathogenic, Dsg3-specific autoantibodies upon pre-treatment with anti-TNF- [ antibodies. This protective effect of blockingTNF- [ can be reproduced in vivo. Feliciani et al. (2000)showed that the injection of anti-Dsg3 IgG in TNFreceptor 1/2 deficient mice led more rarely to blisterformation than the injection of the identical autoanti-bodies in syngeneic “wild type” mice with an intactTNF receptor.
Based on these observations, the potent TNF- [inhibitor, infliximab, was used in a 43-year-old malewith a therapy refractory PV with a history of pulmo-nary embolism, arterial hypertension, steroid-inducedosteoporosis and diabetes mellitus who presented withdisseminated cutaneous blisters and extensive erosionsof the oral mucosa (Jacobi et al. 2005). The patient wasgiven three doses of infliximab (5 mg/kg/day i.v.) over
Fig. 8.7. Immediate clinicalresponse of pemphigus vul-garis (PV) to treatment withthe TNF inhibitor infliximab.Cutaneous bullae in a patientwith recalcitrant PV before(A) and 2 days after (B) thefirst infliximab infusion
6 weeks at weeks 0, 2 and 6. Leflunomide (20 mg/day)and prednisone (0.25 mg/kg/day) were continued dur-ing the infliximab regimen as adjuvant treatment andwith the aim of preventing formation of anti-infliximabantibodies (HACA). Within 48 h of the first dose ofinfliximab, blister formation ceased, and within thefirst 4 days cutaneous blisters and mucosal erosionsgradually disappeared (Fig. 8.7). Infliximab infusionswere discontinued 8 weeks after initiation when thepatient developed varicella zoster of the left S1/2 der-matome, which was treated successfully with i.v. acy-clovir. Of note, the patient’s clinical status remainedstable with few cutaneous erosions during the previ-ously insufficient therapy with leflunomide (20 mg/d)and prednisone (0.25 mg/kg/day). After a relapse4 months later the disease was controlled by severalcycles of immunoadsorption therapy in combinationwith mycophenolate mofetil (3 g/day).
Further case reports confirm the successful manage-ment of severe pemphigus with infliximab. A 62-year-old male with PV and generalized bullous lesions and asevere oral stomatitis was managed with systemic cor-ticosteroids for 6 years, mycophenolate mofetil, cyclo-sporin A, azathioprine, cyclophosphamide, methotrex-ate, thalidomide and plasmapheresis without muchsuccess. As a last resort, the patient received infliximab(5 mg/kg/d i.v.) at 0, 2 and 6 weeks and then every8 weeks. No other drugs were needed as concomitantmedication. The patient improved dramatically afterfive infusions leading to total clearance of active lesionsat week 22 without severe side effects. After the 13thinfusion, treatment with infliximab discontinuedbecause new cutaneous and oral lesions had appeared.The disease was eventually controlled with oral corti-costeroids and rituximab.
There is a report on the effective treatment of PVwith the TNF- [ antagonist, etanercept, in a 62-year-old
78 8 Treating Autoimmune Bullous Skin Disorders with Biologics
woman with long-standing cutaneous PV and concom-itant seronegative arthritis. Upon initiation of treat-ment with etanercept for arthritis, the cutaneous blis-ters gradually disappeared. Lin et al. (2005) reportedthe successful treatment of recalcitrant PV and pem-phigus vegetans with etanercept and carbon dioxidelaser.
The human anti-TNF- [ monoclonal antibody, adali-mumab, has in combination with mycophenolate mofe-til been successfully employed in the subcorneal pustu-lar dermatosis subtype of IgA pemphigus. A 41-year-old male with IgA pemphigus failed multiple treat-ments, including acitretin, broadband ultraviolet Btherapy, dapsone, methotrexate and oral steroids. Heshowed improvement to alefacept, but treatment wasdiscontinued because of a low peripheral CD4+ T-cellcount. Cyclosporine was also effective, but was discon-tinued secondary to elevated creatinine levels. Adali-mumab (40 mg s.c. every other week) was started, andafter the third dose his skin completely cleared. Myco-phenolate mofetil (1 g/day) was used as concomitantmedication. The patient has been symptom-free for5 months, besides the occasional occurrence of a fewpustules.
8.3.4Inhibition of TNF-� in Bullous Pemphigoid
There are a few reports on the effect of TNF- [ antago-nists in clinical variants of BP, an autoimmune bullousskin disorder associated with IgG autoantibodiesagainst components of the dermo-epidermal basementmembrane zone, such as BP180 and BP230. In the clini-cal variant mucous membrane pemphigoid, a disorderwhich is characterized by chronic blistering of themucous membranes with secondary scarring, the TNF-[ blocker etanercept has been applied with great suc-
cess. A 72-year-old woman with long-standing mucousmembrane pemphigoid and acute exacerbation of orallesions had already been treated with prednisone(1 mg/kg/day) in combination with azathioprine(100 mg/day) and mycophenolate mofetil (2×1 g/day)over a year, leading to a moderate clinical response.Etanercept (25 mg s.c. ×2/week) in combination withprednisone (initially 60 mg/day) led after the thirdcycle to the disappearance of newly developed blisters.Even though the corticosteroids were graduallytapered, the patient was symptom-free after a total ofsix etanercept injections and clinical remission was
maintained for more than 8 months with low-doseprednisone treatment (1 mg/day).
There is also a report about the treatment of coexist-ing BP and psoriasis with the TNF- [ antagonist, eta-nercept. A 64-year-old man with a long history ofplaque-type psoriasis developed acute symptoms of BP.Initial therapy consisted of mycophenolate mofetil (2 g/day), which had no effect on the clinical activity of BP.Mycophenolate mofetil was discontinued, and he wasstarted on prednisone (60 mg/day). The cutaneous bul-lae disappeared almost completely by the 10th day andalso the plaque psoriasis improved. To prevent reboundof psoriasis and BP during the tapering phase of pred-nisone, etanercept (50 mg s.c. weekly) was added to thetreatment regimen. At a dose of 20 mg/day prednisone,new cutaneous blisters developed. Etanercept wasincreased to 50 mg twice weekly and the blisters subse-quently resolved. When prednisone treatment waseventually discontinued, the clinical activity of BPremained silent; no adverse events occurred.
8.4Summary
Rituximab has clearly evolved as a novel therapeuticoption in refractory autoimmune bullous skin disor-ders such as severe pemphigus vulgaris and epidermo-lysis bullosa acquisita. The mode of action suggeststhat not only the production of autoantibodies is inhib-ited by depletion of autoreactive B cells but also thecritical interaction of autoreactive T and B cells, whichis essential for the perpetuation of the ongoing autoim-mune response. Treatment of pemphigus with rituxi-mab has been shown to be highly effective in refractorymucocutaneous pemphigus with a prolonged biologi-cal activity due to the long-term depletion of autoreac-tive B cells and a lack of major side effects. In addition,single case reports suggest that TNF- [ blockers may bea therapeutic option in pemphigus and mucous mem-brane pemphigoid with life-threatening or therapyrefractory disease course, particularly as a short-termintervention therapy. However, potential side effectsdue to the immunosuppressive potential of TNF- [antagonists such as severe bacterial infections, viralinfections and tuberculosis should be carefully consid-ered and constitute a serious hazard in patients onchronic immunosuppressive therapy.
8.4 Summary 79
References
Arin MJ, Engert A, Krieg T, Hunzelmann N (2005) Anti-CD20monoclonal antibody (rituximab) in the treatment of pem-phigus. Br J Dermatol 153: 620–625
Berookhim B, Fischer HD, Weinberg JM (2004) Treatment ofrecalcitrant pemphigus vulgaris with the tumor necrosisfactor alpha antagonist etanercept. Cutis 74(4):245–247
Boye J, Elter T, Engert A (2003) An overview of the current clin-ical use of the anti-CD20 monoclonal antibody rituximab.Ann Oncol 14:520–533
Edwards JCW, Cambridge G (2006) B-cell targeting in rheuma-toid arthritis and other autoimmune diseases. Nature RevImmunol 6:394–403
Feliciani C, Toto P, Amerio P, Pour SM, Coscione G, Shivji G,Wang B, Sauder DN (2000) In vitro and in vivo expression ofinterleukin-1alpha and tumor necrosis factor-alpha mRNAin pemphigus vulgaris: interleukin-1alpha and tumornecrosis factor-alpha are involved in acantholysis. J InvestDermatol 114(1):71–77
Hertl M, Eming R, Veldman C (2006) T cell control of autoim-mune bullous skin disorders. J Clin Invest 116:1159–1166
Howell SM, Bessinger GT, Altman CE, Belnap CM (2005) Rapidresponse of IgA pemphigus of the subcorneal pustular der-matosis subtype to treatment with adalimumab and myco-phenolate mofetil. J Am Acad Dermatol 53(3):541–543
Jacobi A, Manger B, Schuler G, Hertl M (2005) Rapid control oftherapy-refractory pemphigus vulgaris by treatment withthe tumour necrosis factor-alpha inhibitor infliximab. Br JDermatol. 153(2):448–449
Lin MH, Hsu CK, Lee JY (2005) Successful treatment of recalci-trant pemphigus vulgaris and pemphigus vegetans with eta-nercept and carbon dioxide laser. Arch Dermatol 141(6):680–682
Pardo J, Mercader P, Mahiques L, Sanchez-Carazo JL, Oliver V,Fortea JM (2005) Infliximab in the management of severepemphigus vulgaris. Br J Dermatol 153(1):222–223
Rouziere AS, Kneitz C, Palanichamy A, Dorner T, Tony HP(2005) Regeneration of the immunoglobulin heavy-chainrepertoire after transient B-cell depletion with an anti-CD20antibody. Arthritis Res Ther 7(4):R714–724
Sacher C, Rubbert A, Konig C, Scharffetter-Kochanek K, KriegT, Hunzelmann N (2002) Treatment of recalcitrant cicatri-cial pemphigoid with the tumor necrosis factor alpha antag-onist etanercept. J Am Acad Dermatol 46(1):113–115
Scheinfeld NA (2004) Comprehensive review and evaluation ofthe side effects of the tumor necrosis factor alpha blockersetanercept, infliximab and adalimumab. J Dermatol Treat15:280–294
Yamauchi PS, Lowe NJ, Gindi V (2006) Treatment of coexistingbullous pemphigoid and psoriasis with the tumor necrosisfactor antagonist etanercept. J Am Acad Dermatol 54(3Suppl 2):S121–122
80 8 Treating Autoimmune Bullous Skin Disorders with Biologics