A conserved interaction between the replicative clamp loader and DNA ligase in eukaryotes; implications for Okazaki fragment joining David S. Levin 1, 6, 8 , Sangeetha Vijayakumar 2, 8 , Xiuping Liu 1, 7, 8 , Vladimir P. Bermudez 3 , Jerard Hurwitz 3 and Alan E. Tomkinson 4, 5 1 Department of Molecular Medicine, Institute of Biotechnology, The University of Texas Health Science Center at San Antonio, 15355, Lambda Drive, San Antonio, TX 78245 2 Molecular and Cellular Biology Graduate Program, University of Maryland School of Medicine, 655 West Baltimore St, Baltimore, MD21201 3 Program in Molecular Biology, Sloan Kettering Institute, Memorial Sloan-Kettering Cancer Center, New York, NY10021 4 Radiation Oncology Research Laboratory, Department of Radiation Oncology and Greenebaum Cancer Center, University of Maryland School of Medicine, 655 West Baltimore St., Baltimore, MD21201 5 Corresponding author: Radiation Oncology Research Laboratory, Department of Radiation Oncology and Greenebaum Cancer Center, University of Maryland School of Medicine, 655 West Baltimore St, Baltimore, Maryland Tel: 410-706-2375; FAX: 410-706-3000; E mail, [email protected]6 Present address: GeneTex Inc., 14785 Omicron Drive, San Antonio, TX 78245 7 Present address: Department of Molecular Genetics, M.D. Anderson Cancer Center, The University of Texas, Houston, TX 77030, USA. 8 These authors contributed equally. Running Title: DNA Ligase I interacts with the clamp loader RFC. Abbreviations footnote: BSA, bovine serum albumin; FEN-1, flap endonuclease 1; GST, glutathione S transferase; PCNA, proliferating cell nuclear antigen; RFC, replication factor C. 1 JBC Papers in Press. Published on October 23, 2004 as Manuscript M409250200 Copyright 2004 by The American Society for Biochemistry and Molecular Biology, Inc.
Transcript
A conserved interaction between the replicative clamp loader and DNA
ligase in eukaryotes; implications for Okazaki fragment joining
David S. Levin1, 6, 8, Sangeetha Vijayakumar2, 8, Xiuping Liu1, 7, 8, Vladimir P.
Bermudez3, Jerard Hurwitz3 and Alan E. Tomkinson4, 5
1Department of Molecular Medicine, Institute of Biotechnology, The University of TexasHealth Science Center at San Antonio, 15355, Lambda Drive, San Antonio, TX 782452Molecular and Cellular Biology Graduate Program, University of Maryland School ofMedicine, 655 West Baltimore St, Baltimore, MD212013Program in Molecular Biology, Sloan Kettering Institute, Memorial Sloan-KetteringCancer Center, New York, NY100214Radiation Oncology Research Laboratory, Department of Radiation Oncology andGreenebaum Cancer Center, University of Maryland School of Medicine, 655 WestBaltimore St., Baltimore, MD212015Corresponding author: Radiation Oncology Research Laboratory, Department ofRadiation Oncology and Greenebaum Cancer Center, University of Maryland School ofMedicine, 655 West Baltimore St, Baltimore, Maryland
Tel: 410-706-2375; FAX: 410-706-3000; E mail, [email protected] Present address: GeneTex Inc., 14785 Omicron Drive, San Antonio, TX 782457 Present address: Department of Molecular Genetics, M.D. Anderson Cancer Center, The University of Texas, Houston, TX 77030, USA.
8These authors contributed equally.
Running Title: DNA Ligase I interacts with the clamp loader RFC.
phenylmethanesulfonyl fluoride, 50 mM NaF, 1 mM Na3VO4). Approximately 1 mg of
the clarified extract was used for each immunoprecipitation. The extracts were pre-
cleared by incubation for 1 hour at 4°C with 50 µl of Protein A Sepharose and Protein G
Sepharose beads (Amersham Biosciences, Piscataway, NJ) equilibrated with IP buffer,
prior to the addition of antibodies against DNA ligase I, RFC p37 or Cdc25 (all from
GeneTex Inc.). After incubation at 4°C for 2 hours, 50 µl of Protein A Sepharose and
Protein G Sepharose beads were added and the incubation continued for 1 hour. The
beads were collected by centrifugation, washed three times with IP buffer lacking
ethidium bromide and then resuspended in SDS- sample buffer. After separation by
SDS-PAGE, proteins were detected by immunoblotting.
Glutathione S transferase (GST)-fusion proteins. GST fusion proteins containing either
the N-terminal 118 residues of DNA ligase I (GST-N Lig 1-118) or residues 479-919
of DNA ligase I (GST-C Lig1) were expressed and purified as described previously (9).
Sequences encoding residues 1-584 and 479-1148 of RFC p140 were amplified from
pET16a-p140 (19) by the polymerase chain reaction and subcloned in-frame with the
GST open reading frame in the pGEX vector to generate pGST-Np140 and pGST-
Cp140, respectively. Similarly, the open reading frame encoding Cdc9 DNA ligase was
amplified from S. cerevisiae genomic DNA by the polymerase chain reaction and
subcloned in-frame with the GST open reading frame in the pGSTag vector (23). After
6
expression in E. coli, GST fusion proteins were purified from cell extract affinity
chromatography using glutathione Sepharose beads.
In Vitro Transcription and Translation. Coupled in vitro transcription and translation
reactions were performed using the TNT Quick Coupled Transcription/Translation system
(Promega, Madison, WI). The plasmids for the in vitro transcription and translation of
human RFC subunits have been described previously (19). The open reading frame
encoding Rfc1 was amplified from S. cerevisiae genomic DNA by the polymerase chain
reaction and subcloned into pET-28b for coupled in vitro transcription and translation.
Labeled in vitro translated polypeptides were partially purified by ammonium sulfate
precipitation (24).
Pull down assays. To prepare GST and GST-Np140, GST-Cp140, GST-N Lig 1-118
and GST-C Lig1 beads, 5 µg of each purified protein was incubated with a 20 µl slurry of
Glutathione Sepharose beads (Amersham Biosciences) equilibrated in binding buffer for
30 min at 4°C with constant agitation. After washing with binding buffer, the beads were
resuspended in 500 µl binding buffer containing a labeled in vitro translated polypeptide
and then incubated at room temperature for 30 min with constant agitation. Next the
beads were collected by centrifugation and then washed extensively in binding buffer
prior to being resuspended in 20 µl of SDS-PAGE sample buffer. After separation by
SDS-PAGE, labeled polypeptides were visualized by phosphorImager analysis
7
(Molecular Dynamics).
Preparation of biotin-labeled linear DNA joining substrate. A 5’ biotinylated 90-mer
oligonucleotide, Bio-5-90, with a sequence corresponding to nucleotide positions 4881
and 4971 of M13mp19 single stranded DNA was purchased from Integrated DNA
Technologies, Inc. Bio15-1 5’-TGAGGCGGTCAG TAT-3’ and Bio15-2 5’-
AAGATAAAACAGAGG –3’ (Integrated Technologies, Inc) are complimentary to Bio-
5-90. Bio15-1 was 5 end labeled with 150 µCi of γ-32P ATP using T4 Polynucleotide
Kinase (New England Biolabs). After purification purified on Micro Bio-Spin 30 column
(Bio-Rad), labeled Biol5-1 and Bio15-2, were annealed to Bio-5-90 to generate a
partial duplex of 30 bp containing a single ligatable nick in the middle flanked by single-
stranded regions of 30 nucleotides.
DNA joining reaction with biotin-labeled linear substrate. Streptavidin-agarose beads
(10 µl, Pierce) were incubated with 1.6 pmol of the biotinylated linear DNA substrate in
PBS for 30 min at room temperature. After washing three times in ligation buffer (50 mM
Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol, 0.25 mg/ml BSA, 100 µM ATP,
and 100 mM NaCl), the beads were incubated with 2 pmol of RPA/pmol of DNA in the
same buffer for 15 min at room-temparature. This substrate was then incubated with 2
pmol RFC or 2 pmol of ∆NRFC in the presence and absence of PCNA (2 pmol trimer) at
30°C for 2 minutes. DNA ligase I (2 pmol), either wild type or mutant, was added and the
8
reaction incubated at room-temperature for 5 minutes. The beads were then spun down
and the reaction terminated by addition of 10 µl of stop mix (50% glycerol, 1% SDS, 20
mM EDTA and 0.05% bromophenol blue). The beads were heated at 100° C for 3 min to
denature DNA. A 2 µl aliquot was mixed with 2 µl of denaturingPAGE dye (80 %
formamide, 0.05% bromophenol and 0.05% xylene cyanol). The samples were
electrophoresed through a 12% denaturing polyacrylamide gel. After drying, the gel was
exposed to a Storage Phosphor screen and subjected to phosphorImager analysis
(Molecular Dynamics).
Preparation of circular DNA joining substrate. The oligonucleotides,
5’CGTACGGGGAAGGACGTCAA3’ and 5’
CATGAAACCAACATAAACGTTATTGCCCGG 3’ (100 pmol of each), were end-
labeled with T4 polynucleotide kinase (New England Biolabs) in the presence of 3.2 mM
ATP and 70 µCi [γ32P] ATP. After purification from free nucleotides by passage through
a 1 ml G25 spin column, both of the labeled oligonucleotides were annealed to 25 µg of
circular ΦX174 single-stranded DNA by heating at 1000C for 15 minutes in 30 mM
Tris-HCl, pH 7.5, and 300 mM NaCl (200 µl total volume) followed by slow cooling to
room temperature. The labeled partial duplex circles were purified from free
oligonucleotides by passage through a 5 ml Bio-Gel A15m (Bio-Rad) column. Peak
fractions were ethanol precipitated, resuspended in 10 mM Tris-HCl, pH 7.5 and 1 mM
9
EDTA, and then run on a 1% agarose gel with ethidium bromide to verify the presence of
the substrate DNA. When annealed to their complementary sequences in ΦX174 single-
stranded DNA, the oligonucleotides form a partial duplex region containing a single
ligatable nick.
DNA joining reaction with circular substrate. The labeled circular DNA substrate DNA
(100 fmol) was incubated with RFC (0.6 pmol) in the presence or absence of PCNA (1.2
pmol trimer) at 30°C for 1 minute in ligation buffer. Human DNA ligase I (90 fmol),
either wild type or a mutant version with a disrupted PCNA binding site (7) was then
added. Aliquots were taken after 0, 30, 90 and 300 seconds and added directly to DNA
termination dye. After boiling, samples were electrophoresed through a 6% denaturing
acrylamide gel. Labeled oligonucleotides in the dried gel were detected and quantitated
by phosphorImager analysis (Molecular Dynamics).
RESULTS
Previously we have shown that DNA ligase I forms a stable complex with PCNA
molecules that are topologically linked to a nicked DNA circle (9). Since this interaction
did not significantly increase the efficiency of DNA joining (9), we suspected that
additional DNA replication factors may be involved in promoting DNA joining by DNA
ligase I. To identify such factors, we fractionated a HeLa nuclear extract by DNA ligase
I-affinity chromatography. As reported previously, PCNA was specifically retained by
10
the DNA ligase I resin (Fig. 1A, compare lanes 3 and 4). Analysis of the same fractions
by immunoblotting with antibodies specific for the p37 subunit and p140 subunit of the
clamp loader RFC revealed that these proteins were also present in the 150 mM NaCl
eluates from the DNA ligase I column but not in the equivalent eluates from the BSA
column (Fig. 1A, compare lanes 3 and 4). PCNA and the RFC subunits, p37 and p140,
were also present in the 300 mM NaCl eluate from the DNA ligase I but not from the
BSA column (data not shown). Since RFC also binds to PCNA (25), it is possible that
DNA ligase I-bound PCNA acts as a binding site for other PCNA binding proteins.
However, FEN-1, another PCNA-binding replication protein (26), was not retained by
the DNA ligase I resin (Fig. 1A), indicating that the binding of PCNA to the DNA ligase
I beads does not result in the non-specific retention of other PCNA binding proteins. To
provide further evidence for the specific association between RFC and DNA ligase I in
HeLa cell lysates, we performed immunoprecipitations in the presence of ethidium
bromide. As shown in Figure 1B, the DNA ligase I antibody co-immunoprecipitated the
p37 subunit of RFC (Fig. 1B, lane 3) and, in reciprocal experiments, the RFC p37
antibody co-immunoprecipitated DNA ligase I (Fig. 1B, lane 4).
Since RFC p37 and DNA ligase I can be co-immunprecipitated in the presence
of ethidium bromide, it appears likely that the association between these factors is
mediated by protein-protein interactions. To determine whether there is a direct
interaction between DNA ligase I and RFC, we performed pull down assays with DNA
11
ligase I affinity beads and purified recombinant human RFC (20) (Fig. 2A, lane 2). The
binding of RFC to DNA ligase I- but not BSA-beads (Fig. 2B, compare lanes 2 and 3),
demonstrates that there is a direct interaction between RFC and DNA ligase I.
RFC is composed of a large subunit, p140 and four smaller subunits p40, p38, p37
and p36 (27,28). Recently, several different variants of RFC, in which p140 has been
replaced by different polypeptides, have been described (29,30). Since p140 is the unique
component of the replicative clamp loader, we reasoned that the specific association with
RFC is likely to involve an interaction with p140. To test this idea we expressed the N-
terminal domain of p140, which can be removed without loss of catalytic activity (19),
and the C-terminal domain of p140, which is required for complex formation with the
four small subunits (19), as GST fusion proteins. In pull down assays, in vitro translated
DNA ligase I was specifically retained on glutathione beads liganded by the non-
catalytic N- terminal domain of p140 (Fig. 3A).
If the interaction between the clamp loader and the DNA ligase plays a critical
role in lagging strand DNA synthesis then it should be conserved other eukaryotes. This
prompted us to examine whether Cdc9, the S. cerevisiae DNA ligase I homolog, interacts
with the large subunit of S. cerevisiae RFC, Rfc1. As shown in Figure 3B, in vitro
translated Rfc1 was specifically retained on glutathione beads liganded by GST-Cdc9,
indicating that the interaction between the large subunit of the replicative clamp loader
and DNA ligase is conserved in eukaryotes.
12
Since previous studies have shown that the N-terminal 118 amino acid residues
of DNA ligase I contain the binding sites for both PCNA and DNA polymerase β (11,31),
we examined whether the same region was involved in the interaction with RFC p140. As
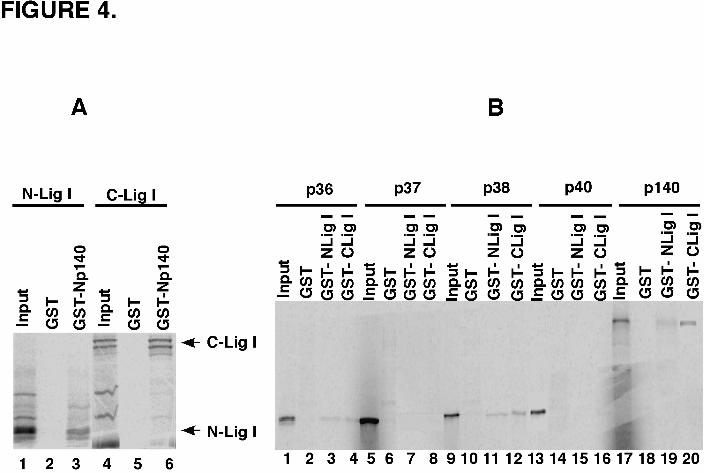
shown in Figure 4A, this fragment did bind specifically to the N-terminal domain of
RFC p140. This interaction was not disrupted by the substitution of the adjacent
phenylalanine residues by alanine residues in the PCNA binding site at the N-terminus of
DNA ligase I (data not shown). Unexpectedly, the N-terminal domain of RFC p140
interacted more efficiently with a fragment encompassing the catalytic domain of DNA
ligase I (Fig. 4A). This is the first example of a protein-protein interaction involving this
region of DNA ligase I.
The observation that more than one region of DNA ligase I binds to the N
–terminal domain of RFC p140 prompted us to examine whether DNA ligase I also
interacts with the small RFC subunits. As expected, in vitro translated RFC p140 bound
specifically to both the N- (Fig. 4B, lane 19) and C-terminal (Fig. 4B, lane 20)
fragments of DNA ligase I expressed as GST fusion proteins. Similar, albeit weaker,
interactions were observed with in vitro translated p36 (Fig. 4B, lanes 3 and 4) and p38
(Fig. 4B, lanes 11 and 12).
The results of our protein-protein interaction experiments described above
together with published studies have identified a series of pairwise interactions among
PCNA, RFC and DNA ligase I (7,9,11). To examine the effect of RFC on DNA ligase I
13
catalytic activity, we constructed a partial duplex linear substrate containing a single
ligatable nick (Fig. 5A). Pre-incubation of this substrate with RFC inhibited the extent of
DNA joining by about 50% (Fig. 5B). In accord with previous studies (9), PCNA had
essentially no effect on the DNA joining activity of DNA ligase I (Fig. 5B). Interestingly,
when PCNA was pre-incubated with RFC, the inhibitory effect of RFC on DNA joining
was alleviated (Fig. 5B). Since we have shown earlier that the N –terminal region of p140
is involved in the interaction with DNA ligase I (Fig. 3), we asked whether the
catalytically active RFC complex containing a truncated version of p140 lacking the N-
terminal region had a similar effect on DNA ligase I activity. As shown in Fig. 5C, the
RFC complex containing a truncated version of p140 inhibited DNA joining by about
50% but, as with the intact complex, this inhibition was alleviated in the presence of
PCNA. Under these reaction conditions, the RFC complex containing a truncated version
of p140 was still specifically retained by DNA ligase I beads (Fig. 5D), presumably
because of the interactions between DNA ligase I and one or more of the small RFC
subunits (Fig. 4B).
The simplest explanation for the observations described above is that the ATP-
dependent loading of PCNA by RFC induces the dissociation of RFC from the nicked
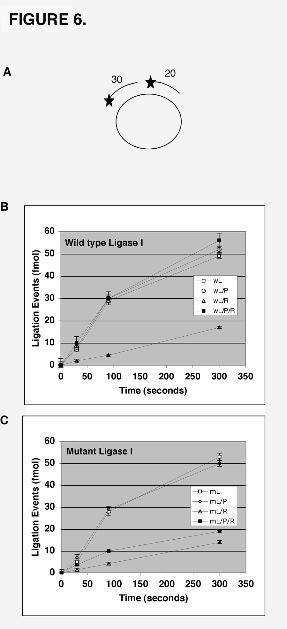
DNA, thereby making the nick accessible for ligation. Pre-incubation of a circular
ligatable substrate (Fig. 6A) with RFC also caused an inhibition of DNA ligase I activity
(Fig. 6B). In this assay, the initial rate of DNA joining was reduced by about 4-fold but
14
this inhibition was alleviated by the inclusion of PCNA (Fig. 6B). However, in similar
experiments with a mutant variant of DNA ligase I in which the PCNA binding site had
been inactivated (7), preincubation of PCNA with RFC did not alleviate the inhibitory
effect of RFC (Fig. 6C). A similar result was obtained in assay with the linear ligatable
substrate (data not shown). Thus, the ability of DNA ligase I to efficiently join DNA in
the presence of RFC and PCNA is dependent upon its ability to bind to PCNA.
DISCUSSION
There is substantial evidence supporting the notion that multiprotein DNA
transactions such as DNA replication are co-ordinated by protein-protein interactions
among the participating factors. Notably, PCNA, which is a homotrimeric sliding clamp,
interacts with multiple DNA replication factors, indicating that it plays a central role in
directing the sequential actions of these proteins (12,13). In previous studies, we have
shown that the interaction between DNA ligase I and PCNA is critical for the efficient
joining of Okazaki fragments and the completion of the repair of DNA lesions by long
patch BER (7,9). Our failure to find conditions under which PCNA stimulated the
catalytic activity of DNA ligase I (9) prompted us to look for additional protein factors
that may contribute to DNA joining events involving DNA ligase I and PCNA. In this
study, we describe a direct interaction between DNA ligase I and RFC, the clamp loader.
Unlike PCNA binding, which is dependent on a 20-amino acid sequence at the N-
15
terminus of DNA ligase I (11), the interaction with RFC involves residues from both the
non-catalytic N-terminal and the catalytic C-terminal domains of DNA ligase I.
Moreover, DNA ligase I not only interacts with the non-catalytic N-terminal region of
the large subunit of p140 but also with two of the smaller RFC subunits. Together these
results indicate that there are multiple site of contact between DNA ligase I and RFC.
Previously we had shown that PCNA tethered DNA ligase I to a nicked circular
DNA duplex but did not stimulate ligation (9). This suggested that the free-sliding DNA
ligase I-PCNA complex required an additional factor(s) to position it at a nick. In this
regard, it is intriguing that RFC binds to the 3’ hydroxyl termini of primer-template
junctions (27) and so could potentially recruit DNA ligase I to nicks. However, the recent
structures of prokaryotic and eukaryotic RFC complexes determined by electron
microscopy and x-ray crystallography suggest that the binding of RFC would prevent
DNA ligase I from gaining access to the nick (32,33). Consistent with this prediction and
a previous biochemical study (2), RFC inhibited joining by DNA ligase I. This inhibitory
effect was alleviated when PCNA was included in the reaction but only when DNA ligase
I had a functional PCNA binding site. Thus, it appears that the pairwise interactions
among RFC, PCNA and DNA ligase I co-ordinate the joining step that links Okazaki
fragments and completes certain DNA excision repair pathways. The conservation of the
interaction between the functionally homologous Saccharomyces cerevisiae proteins
supports the notion that the interaction between the replicative DNA ligase and the
16
replicative clamp loader is functionally and biologically significant.
Although PCNA is a ring-shaped molecule, the two faces of the ring are not
equivalent. RFC loads PCNA onto DNA in a particular orientation and all of the
replication proteins appear to bind to the same face of the PCNA ring (12,13). Since
PCNA is a homotrimer, it is possible that up to three replication factors can bind to the
same PCNA trimer. Alternatively, the factors may bind sequentially with the DNA
structure presumably dictating their sequential action. Only 1 molecule of DNA ligase I
was bound per PCNA trimer topologically linked to DNA (9). Moreover, we have been
unable to detect formation of a ternary complex of PCNA, DNA ligase I and FEN-1
(Varkey, J. and A.E.T., unpublished results) suggesting that the binding of one molecule
DNA ligase I to a PCNA trimer occludes the other binding sites and that the binding of
DNA ligase I and FEN-1 to a PCNA trimer are mutually exclusive. In Okazaki fragment
maturation, the removal of flaps by FEN-1 linked to PCNA generates the nicked DNA
substrate for DNA ligase I. We propose that flap removal is the signal for the dissociation
of the FEN-1/PCNA complex. Since PCNA is then free to slide away from the nick, we
suggest that RFC acts to maintain PCNA at the nick site by binding to both the nick and
PCNA. It should be noted that, based on the structure of yeast RFC (33), it appears likely
that the interaction of RFC with a 3’ OH terminus will differ depending on whether there
is an adjacent strand or not. Since there are pair wise interactions among PCNA, RFC and
DNA ligase I, we suggest that DNA ligase I initially interacts with a ternary complex of
17
RFC and PCNA bound at a DNA nick via its interaction with the N-terminal region of
the large subunit of RFC. This protein-protein interaction may induce a conformational
change in RFC, enabling DNA ligase I to contact the PCNA ring and catalyze DNA
ligation. Finally, we speculate that nick ligation is the signal for RFC to unload PCNA
and for the dissociation of the ternary complex containing DNA ligase I, PCNA and RFC.
In summary, we have identified a conserved interaction between the replicative
clamp loader and the replicative DNA ligase. Moreover, we have shown that the
interaction between DNA ligase I and PCNA is required for PCNA to overcome the
inhibitory effect of RFC on DNA ligation, suggesting that pairwise physical and
functional interactions among RFC, PCNA and DNA ligase I co-ordinate the DNA
joining step that links adjacent Okazaki fragments. Further studies are needed to delineate
the functional significance of the protein-protein interaction between RFC and DNA
ligase I, and the molecular mechanisms involved in this key reaction in lagging strand
DNA synthesis.
ACKNOWLEDGEMENTS
We are grateful to Drs. Bruce Stillman and Vladimir Podust for reagents. We thank Dr.
Sean Post for help with the immunoprecipitation experiments. This study was supported
18
by grants (GM57479 to AET and GM38559 to JH) from the National Institutes of
Health.
19
REFERENCES
1. Tomkinson, A. E., & Mackey, Z. B. (1998) Mutat. Res. 407, 1-9.2. Waga, S., Bauer, G., & Stillman, B. (1994) J. Biol. Chem. 269, 10923-10934.3. Li, C., Goodchild, J., & Baril, E. F. (1994) Nucleic Acids Res. 22, 632-638.4. Lasko, D. D., Tomkinson, A. E., & Lindahl, T. (1990) J. Biol. Chem. 265, 12618-
12622.5. Prigent, C., Satoh, M. S., Daly, G., Barnes, D. E., & Lindahl, T. (1994) Molec.
Cell. Biol. 14, 310-317.6. Mackenney, V. J., Barnes, D. E., & Lindahl, T. (1997) J. Biol. Chem. 272,
11550-11556.7. Levin, D. S., McKenna, A., Motycka, T., Matsumoto, Y., & Tomkinson, A. E.
(2000) Current Biology 10, 919-922.8. Barnes, D. E., Tomkinson, A. E., Lehmann, A. R., Webster, A. D. B., & Lindahl,
T. (1992) Cell 69, 495-503.9. Levin, D. S., Bai, W., Yao, N, and O’Donnell, M & Tomkinson, A. E. (1997)
Proc. Natl. Acad. Sci. U.S.A. 94, 12863-12868.10. Jonsson, Z., Hindges, R., & Hubscher, U. (1998) EMBO J 17, 2412-2425.11. Montecucco, A., Rossi, R., Levin, D. S., Gary, R., Park, M. S., Motycka, T. A.,
Ciarrocchi, G., Villa, A., Biamonti, G., & Tomkinson, A. E. (1998) EMBO J. 17, 3786-3795.
12. Warbrick, E. (1998) Bioessays 20, 195-199.13. Jonsson, Z., and Hubscher, U. (1997) BioEssays 19, 967-97514. Matsumoto, Y., Kim, K., Hurwitz, J., Gary, R., Levin, D. S., Tomkinson, A. E., &
Park, M. (1999) J. Biol. Chem. 274, 33703-33708.15. Mossi, R., Ferrari, E., & Hubscher, U. (1998) J. Biol. Chem. 273, 14322-14330.16. Tom, S., Henricksen, L. A., Park, M. S., & Bambara, R. A. (2001) J. Biol. Chem.
276, 24817-24825.17. Wang, Y. C., Burkhart, W. A., Mackey, Z. B., Moyer, M. B., Ramos, W., Husain,
I., Chen, J., Besterman, J. M., & Tomkinson, A. E. (1994) J. Biol. Chem. 269, 31923-31928.
18. Fien, K., & Stillman, B. (1992) Molec. Cell. Biol. 12, 155-163.19. Uhlmann, F., Cai, J., Gibbs, E., O’Donnell, M., & Hurwitz, J. (1997) J. Biol.
Chem. 272, 10058-10064.20. Podust, V. N., & Fanning, F. (1997) J. Biol. Chem. 272, 6303-6310.21. Wu, Y., Hickey, R., Lawlor, K., Wills, P., Yu, F., Ozer, H., Starr, R., Quan, J. Y.,
Lee, M., & Malkas, M. (1994) J. Cell. Biochem. 54, 32-46.22. Laemmli, U. K. (1970) Nature 227, 680-685.23. Ron, D., & Dressler, H. (1992) Biotechniques 13, 866-868.24. Bardwell, L., Cooper, A. J., & Friedberg, E. C. (1992) Mol. Cell. Biol. 12, 3041-
20
3049.25. Zhang, G., Gibbs, E., Kelman, Z., O’Donnell, M., & Hurwitz, J. (1999) Proc.
Natl. Acad. Sci. U.S.A. 96, 1869-1874. 26. Li, X., Li, J., Harrington, J., Lieber, M. R., & Burgers, P. M. J. (1995) J. Biol.
Chem 270, 22109-22112.27. Tsurimoto, T., & Stillman, B. (1991) J. Biol. Chem. 266, 1950-1960.28. Lee, S. H., Kwong, A. D., Pan, Z. Q., & Hurwitz, J. (1991) J. Biol. Chem. 266,
594-602.29. Bermudez, V. P., Maniwa, Y., Tappin, I., Ozato, K., Yokomori, K., & Hurwitz, J.
(2003) Proc. Natl. Acad. Sci. U.S.A. 100, 10237-10242.30. Lindsey-Boltz, L. A., Bermudez, V. P., Hurwitz, J., & Sancar, A. (2001) Proc.
Natl. Acad. Sci. U.S.A. 98, 11236-11241.31. Dimitriadis, E. K., Prasad, R., Vaske, M. K., Chen, L., Tomkinson, A. E., Lewis,
M. S., & Wilson, S. H. (1998) J. Biol. Chem. 32, 20540-20550. 32. Miyata, T., Oyama, Y., Mayanagi, K., Ishino, S., Ishino, Y., & Morikawa, K.
(2004) Nat. Struct. Cell Biol. 11, 632-636.33. Bowman, G. D., O’Donnell, M., & Kuriyan, J. (2004) Nature 429, 724-730.
21
FIGURE LEGENDS
Figure 1. DNA ligase I associates with RFC in human cell extracts. A. DNA ligase I
affinity chromatography. A nuclear extract from HeLa cells was incubated with Affi-gel
beads covalently linked to either DNA ligase I (Affi-Lig I) or BSA (Affi-BSA). Bound
proteins were eluted with 150 mM NaCl. The eluate from Affi-Lig I (lane 3) and Affi-
BSA beads (lane 4) were subjected to immunoblotting using the antibodies indicated on
the right. Proteins in the nuclear extract (NE, 50 µg, lane 1) and cytoplasmic extract (CE,
50 µg, lane 2) were detected by direct immunoblotting. B. DNA ligase I (Lig I, lane 3)
and RFC p37 (p37, lane 4) were immunoprecipitated from HeLa cell extracts as
described in Experimental Procedures. Immunoprecipitation with anti-Cdc25C antibody
(mock, lane 2). Proteins in the whole cell extract (WCE, 50 µg, lane 1) and the
immunopecipitates were detected by immunoblotting with the antibodies indicated on the
22
right.
Figure 2. DNA ligase I interacts directly with RFC. A. Purified recombinant proteins.
Lane 1, DNA ligase I, 2 µg; lane 2, RF-C, 1µg; lane 3, PCNA, 2 µg of PCNA. Proteins
were separated by SDS-PAGE and then stained with Coomassie blue. The positions of
molecular mass standards are indicated on the right. B. Purified RFC was incubated either
with Affi-Lig I (lane 2) or Affi-BSA (lane 3) beads as described in Experimental
Procedures. Bound proteins were detected by immunoblotting with the indicated
antibodies. Lane 1, 10% of the input RFC detected by immunoblotting.
Figure 3. The interaction between the large subunit of RFC and the replicative DNA
ligase is conserved between yeast and humans. A. In vitro translated full-length DNA
ligase I was incubated with glutathione sepharose beads liganded by; GST, lane 2; GST