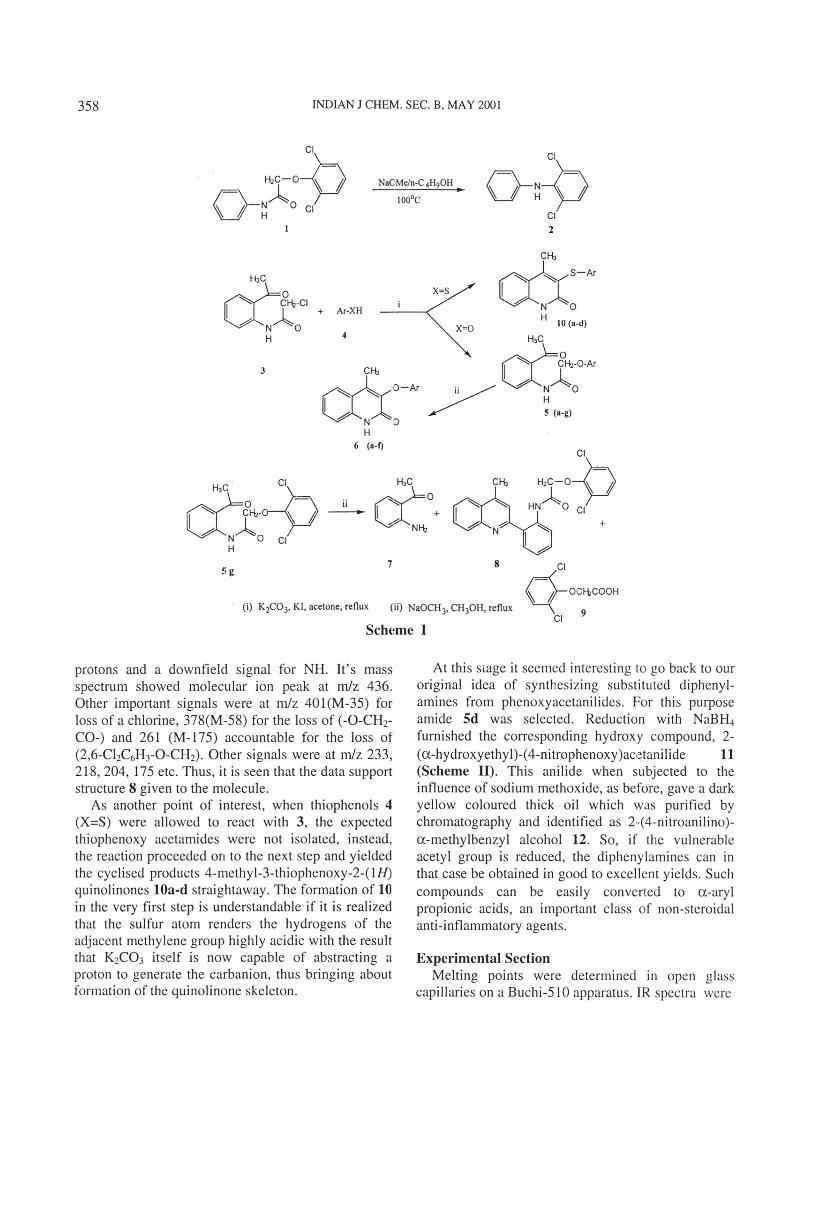
Indian Journal of Chemistry Vol. 408, May 2001 , pp. 357-360 A convenient method for the preparation of 3-phenoxy/ thiophenoxy- 2 (lH)-quinolinones t S Ria z Hashim' & P Tirupathy Reddy Organic Chemistry Division-I, Indian In stitute of Chemical Technology, Hyderabad 500 007, Indi a Received 17 August 1999; accepted (revised) 24 October 2000 A number of new 4-methyl-3-phenoxy/thiophenoxy-2 (I H) quinolinones have been sy nth es ized in excellent yields through a simple and efficient procedure from N-(2-acetylphenyl)-phenoxy/thiophenoxy acetamides under basic conditions. Recently while working on a synthetic program related to the anti-inflammatory drug diclofenac sodium, we had an occasion to synthesize 2,6-di- chlorodiphenylamine 2 from 2,6-dichlorophenoxy- acetanilide 1, by heating the alcoholic solution of the latter with sodium methoxide 1 • The reaction gave almost quantitative yield of the diphenylamine and appeared fascinating from the synthetic point of view. In particular we became interested in seeing the effect of base on amides such as 5, which are derived from a-acetyl aniline. The literature tells us that amides of this type undergo cyclization to 4-alkyl or aryl derivatives of quinolines, when treated with base 2 · 4 . Accordingly , our reaction would furnish 4-methyl-3-phenoxy/thio- phenoxy-2(11/)-quinolinones and thus provide an easy and direct access to these hitherto unreported compounds. In addition, certain quinoline derivatives have been reported to possess anti-inflammatory and diuretic activities 5 . Therefore, we thought it worthwhile to synthesizing these new chemical entities; for they might lead to compounds having useful biological properties. The sequence of reactions carried out with this end in view is indicated in Scheme I. Reaction of 2-aminoacetophenone with chloro- acetylchloride afforded N-chloroacetyl-2-acetyl aniline 3. This compound upon treatment with various phenols 4 (X=O) in presence of anhydrous potassium carbonate, dry acetone and KI , formed N-(2-acetyl- ph eny l) ph enoxyacetamides 5 a-g (Table 1), and these furnished the desired 3-(s ub stituted) phenoxy-4- t ll CT Communi cation o.: 4191 methyl carbostyrils 6a-e when treated with NaOCH 3 in methanol. The structures of the compounds were established on the basis of analytical and spectral data. Com- pound Sa showed, in it 's IR spectrum, vibrational bands at 3187 (-NH-), 1700 (CH 3 CO-) and 1663 cm· 1 (-NH-CO-). It's 1 H NMR showed signals at 82.7(s, 3H, CH 3 ), 4.65(s, 2H, CH 2 ), 7.8-8.7(m, 9H, Ar-H) and 12 .6 7(s, I H, NH) . In the case of compound 6a, the signals for CH 3 CO- and - CH 2 - were absent. The disappearance of the se signals indicated their participation in the cyclisation to give the quinolinone heterocycle. The mass spectrum of 6g showed molecular ion peak at m/z 461 and an important peak at m/z 265. Also, in this case, a series of fragme nt ions appeared at intervals of 14 mass units . However, not all anilides of the series followed this otherwise general cyclisation pattern. For example, in the case of anilide Sg an anomalous reaction was found to have taken place when subjected to the influence sodium methoxide. The reaction resulted in the formation of compounds 7-9. Isolation of o- acetylaniline 7, the quinoline derivative 8 and 2,6- dichlorophenoxy acetic acid 9, from the reaction mixture, indicated that hydrolysis of amide Sg was the main reaction occurring and as o-aminoacetophenone 7 appeared, it reacted with the unhydrolised amide to produce quinoline 8. Thus, apparently, th e steric hindrance offered by the two chlorines effectively blocked the normal course of reaction , leav in g hydrolysis as the only way open. TheIR spectrum of 8 ex hibited peak at 1660 em· ' for the -CO-NH- group. In it 's 1 H NMR spec trum , there were signals for CH 3 , CH 2 , twe lv e aromatic
Transcript
Indian Journal of Chemistry Vol. 408, May 2001 , pp. 357-360
A convenient method for the preparation of 3-phenoxy/ thiophenoxy-2 (lH)-quinolinones t
S Riaz Hashim' & P Tirupathy Reddy
Organic Chemistry Division- I, Indian Institute of Chemical Technology, Hyderabad 500 007, India
Received 17 August 1999; accepted (revised) 24 October 2000
A number of new 4-methyl-3-phenoxy/thiophenoxy-2 (I H) quinolinones have been synthes ized in excellent yields through a simple and efficient procedure from N-(2-acetylphenyl)-phenoxy/thiophenoxy acetamides under basic conditions .
Recently while working on a synthetic program related to the anti-inflammatory drug diclofenac sodium, we had an occasion to synthesize 2,6-dichlorodiphenylamine 2 from 2,6-dichlorophenoxyacetanilide 1, by heating the alcoholic solution of the latter with sodium methoxide 1
• The reaction gave almost quantitative yield of the diphenylamine and appeared fascinating from the synthetic point of view. In particular we became interested in seeing the effect of base on amides such as 5, which are derived from a-acetyl aniline.
The literature tells us that amides of this type undergo cyclization to 4-alkyl or aryl derivatives of quinolines, when treated with base2
·4
. Accordingly , our reaction would furnish 4-methyl-3-phenoxy/thiophenoxy-2(11/)-quinolinones and thus provide an easy and direct access to these hitherto unreported compounds. In addition, certain quinoline derivatives have been reported to possess anti-inflammatory and diuretic activities5
. Therefore, we thought it worthwhile to synthesizing these new chemical entities; for they might lead to compounds having useful biological properties. The sequence of reactions carried out with this end in view is indicated in Scheme I.
Reaction of 2-aminoacetophenone with chloroacetylchloride afforded N-chloroacetyl-2-acetyl aniline 3. This compound upon treatment with various phenols 4 (X=O) in presence of anhydrous potassium carbonate, dry acetone and KI , formed N-(2-acetylpheny l) phenoxyacetamides 5 a-g (Table 1), and these furnished the desired 3-(substituted) phenoxy-4-
t llCT Communication o.: 4191
methyl carbostyrils 6a-e when treated with NaOCH3
in methanol. The structures of the compounds were established
on the basis of analytical and spectral data. Compound Sa showed, in it's IR spectrum, vibrational bands at 3187 (-NH-), 1700 (CH3CO-) and 1663 cm· 1
(-NH-CO-). It's 1H NMR showed signals at 82.7(s, 3H, CH3), 4.65(s, 2H, CH2), 7.8-8.7(m, 9H, Ar-H) and 12.67(s, I H, NH) . In the case of compound 6a, the signals for CH3CO- and - CH2- were absent. The disappearance of these signals indicated their participation in the cyclisation to give the quinolinone heterocycle. The mass spectrum of 6g showed molecular ion peak at m/z 461 and an important peak at m/z 265. Also, in this case, a series of fragment ions appeared at intervals of 14 mass units .
However, not all anilides of the series followed this otherwise general cyclisation pattern. For example, in the case of anilide Sg an anomalous reaction was found to have taken place when subjected to the influence sodium methoxide. The reaction resulted in the formation of compounds 7-9. Isolation of oacetylaniline 7, the quinoline derivative 8 and 2,6-dichlorophenoxy acetic acid 9, from the reaction mixture, indicated that hydrolysis of amide Sg was the main reaction occurring and as o-aminoacetophenone 7 appeared, it reacted with the unhydrolised amide to produce quinoline 8. Thus, apparently, the steric hindrance offered by the two chlorines effectively blocked the normal course of reaction , leav ing hydrolysis as the only way open.
TheIR spectrum of 8 ex hibited peak at 1660 em· ' for the -CO-NH- group. In it 's 1H NMR spectrum, there were signals for CH 3, CH2, twelve aromatic
358 INDIAN 1 CHEM. SEC. B. MAY 200 I
Cl
· ~c-o~ o- AY
~ ° Cl
NaOMeln-C 4H90H
100°C
Cl,
o-~-o ci' 2
~S-Ar UN~O
Ar-XH - H IO(a-d)
4
3
H3C
roC~ O-N ;; ~ cCi:OA; -~ ~(a-g)
N 0 H
6 (a-f)
Sg 7 8
(i) K2C03, Kl, acetone, reflux (ii) NaOCH 3, CH30H, reflux
Cl
xo-P HN 0 Cl
!"""' .#
+
Cl
Q-oc"'coo" Cl 9
Scheme I
protons and a downfield signal for NH. It's mass spectrum showed molecular ion peak at m/z 436. Other important signals were at m/z 40l(M-35) for loss of a chlorine, 378(M-58) for the Joss of (-0-CHr CO-) and 261 (M-175) accountable for the Joss of (2,6-ChC6H3-0-CH2). Other signals were at m/z 233, 218,204, 175 etc. Thus, it is seen that the data support structure 8 given to the molecule.
As another point of interest, when thiophenols 4 (X=S) were allowed to react with 3, the expected thiophenoxy acetamides were not isolated, instead, the reaction proceeded on to the next step and yielded the cyclised products 4-methyl-3-thiophenoxy-2-(IH) quinolinones lOa-d straightaway. The formation of 10 in the very first step is understandable if it is realized that the sulfur atom renders the hydrogens of the adjacent methylene group highly acidic with the result that K2C03 itself is now capable of abstracting a proton to generate the carbanion, thus bringing about formation of the quinolinone skeleton.
At this stage it seemed interesting to go back to our original idea of synthesizing substituted diphenylamines from phenoxyacetanilides . For this purpose amide Sd was selected. Reduction with NaBH4
furnished the corresponding hydroxy compound, 2-(a-hydroxyethyl)-(4-nitrophenoxy)acetanilide 11 (Scheme II). This anilide when subjected to the influence of sodium methoxide, as before, gave a dark yellow coloured thick oil which was purified by chromatography and identified as 2-(4-nitroanilino)a-methylbenzyl alcohol 12. So, if the vulnerable acetyl group is reduced, the diphenylamines can in that case be obtained in good to excellent yields. Such compounds can be easily converted to a-aryl propionic acids, an important class of non-steroidal anti-inflammatory agents.
Experimental Section Melting points were determined in open glass
capillaries on a Buchi-51 0 apparatus. IR spectra were
HASHIM eta/.: SYNTHESIS OF QUINOLINONES 359
Table 1-Physical data of compounds Sa-g, 6a-f and lOa-d
Com pd. Ar
Sa C6Hs
Sb 4-CHJOC6H4
Sc 4-CIC6H4
Sd 4-02NC6H4
Se 4-H3COOCC6H4
Sf 3-HJ,C,sC6H4
Sg 2,6-C12C6HJ
6a C6Hs
6b 4-CHJOC6H4
6c 4-CIC6H4
6d 4-02NC6H4
6e 4-H3COOCC6H4
6f 3-HJ,C,sC6H4
lOa C6Hs
lOb 4-CIC6H4
IOc 4-HJCC6H4
IOd 2-naphthy1
m.p (OC)
75-76
85-86
128-129
186-187
146-147
71-72
156-157
285-286
264-265
297-298
143-144
311-312
136-137
268-269
227-228
224-225
263-264
i Sd-
Yield
%
85
75
80
84
86
71
84
68
74
82
77
84
70
87
82
91
92
React
period (hr)
4
6
5
3
5
4
2
2
2
0.5
2
2
6
6
8
5
· II
MS 1H NMR (8, ~~m) (M+) CH3 CH2 Ar NH Other
269 2.7 4.65 7.06-8.87 12.67
299 2.67 4.54 6.83-8.85 12.6 3.78
303 2.70 4.63 7.06-8.87 12.63
314 2.67 4.69 7.18-8.82 12.7
327 2.71 4.71 7.18-8.82 12.7 3.81
479 2.70 4.63 6.81-8.86 12.63 0.91,1 .3,2.65
337 2.67 4.6 7.07-8.87 12.7
251 2.39 6.89-8.43 11.97
281 2.36 6.68-7 .62 11.89 3.72
285 2.4 6.83-7 .67 11.97
296 2.64 6.93-8.17 10.8 1
309 2.4 7.06-7.95 12.16 3.87
461 2.4 6.63-7 .65 11.82 0.88,1.27,2.53
267 2.8 7.15-7.8 11.95
301 2.75 7.45-7.68 11.86
281 2.77 7.1-7 .8 11.95 2.54
317 2.8 7.23-7.8 11.95
Cl-\, I o.CH-OH
...? ~-o-N~ 12
(ii) Na0CH1, CH30H reflux
Scheme II
recorded on IR Nicolet 740 FfiR spectrophotometer, 1H NMR spectra on Gemini 200 MHz spectrometer and mass spectra on a VG micromass 7070 H or a Finnigan Mat 1020 B instrument.
N-(2-Acetylphenyl) phenoxyacetamide Sa: General procedure. A mixture of phenol (0.94 g; 10 mmoles), 2-acetyl-2-chloroacetanilide (2.llg; 10 mmoles), anhyd. K2C03 (2.7g; 20 mmoles) and
potassium iodide (0.15 g) in 15 mL of dry acetone was refluxed with stirring for 6 hr. The progress of the reaction was monitored by TLC. Then the reaction mixture was cooled, filtered, the inorganic residue washed with acetone and the solvent removed on a rotavapor. Addition of water (ca 10 mL) to the residue gave the title compound as a precipitate, which was collected by filtration, washed with water and crystallized from ethanol, yield 2.28 g (85 %); m.p 75-
4-Methyl-3-phenoxy-2(1H)-quinolinone 6a: General procedure. To a stirred solution of Sa (0.807g, 3 mmoles) in dry methanol (5 mL), was added dropwise a solution of sod ium meth oxide (0.27g, 5 mmoles) in methanol (5 mL). The reacti on mi xture was reflu xed fo r 2hr foll owed by removal of meth anol under reduced pressure and addition of water to the residue. The resultant prec ipitate was fi ltered, washed with water, dried and crys tall ized from ethanol, yield 0.5 1g (68%). m.p. 285-86°C; IR (KBr): 1640 cm-1 (NH-CO); 1 HNMR (DMSO-rh): 8 2.39 (s, 3H); 6.87 - 8.43 (m, 9H, Ar-H); 11 .97 (s, I H, NH); MS : mlz 251 [M t. Anal. Found : C, 76. 16; H, 4.90; N, 5.22. Calc.for C16H13 0 2: C, 76.48; H, 5.2 1; N, 5.57%.
The compounds 6b-f were simil arly prepared. Their characteri zation data are given in Table 1.
In case of Sg, a similar treatment gave a semi-solid . This was extracted from water with ether. The res idue obtained after removal of ether was subj ected to column chromatograph y (s ilica gel; pet. ether: ethyl acetate 95: 5) when two frac ti ons were coll ected. The oil obtained from the first fracti on proved to be o
am inoacetophenone 7, while the second was identified as the quinoline deri va ti ve 8. m.p. 134-35°C. IR (KBr): 3190 (NH ), 1660 (NH-CO) cm-1; 1H NMR (CDCI3): 8 2.8 (s, 3H), 4.6 (s, 2H), 7-8.76 (m, 12H, Ar-H), 13.56 (s, lH,NH). MS : mlz 436 [M t.
Acidification of the aqueous solution with dilute HCI yielded a precipitate, which was identified as 2, 6-dichlorophenoxy aceti c acid 9.
N-(4-Nitrophenyl)-2-(a-hydroxyethyl) aniline 12. To a stirred suspension of Sd (1.256 g, 4 mmoles) in methanol (10 mL) was added sodiu m borohydride (0.200 g, 5 mmoles) in small porti ons at room temperature. During addition of NaBI-14, the solution turned clear and was stirred fo r addi tional 20 min . At the end of thi s period, the solvent was removed on a rotavapor, dil. HCI added to the res idue and the contents extracted with ether (3-x 20 mL). The combined extracts were wa<;hed with water, dried over Na2S04 and concentrated. Compound 11 so obtained was crystallized from ethyl acetate-n-hexane mi xture. m.p. 161-62°C. IR (KBr): 3395 (OH), 1675 cm·1 (NH-CO); 1H NMR (DMSO-d6) : 8 1.42 (d , 3H); 4.71 8 (s, 2H); 4.875 (del , I H, CH); 5.625 (br,s, I H, OH); 6.046- 8.3 12 (m, 8H,Ar-H); 10.843 (s, I H,NH ); MS: mlz 3 14 [Mt.
To the stirred and reflu xing so lution of 11 (0.252g; 8 mmoles) in methanol (10 mL) was added a solution of NaOMe (0. 162g, 3 mmoles) in methanol (8 mL) over a peri od of 5 min . The refluxing and st irring was continued for 3 hr. The usual work up and puri ficati on of the crude material by column chromatography (s ilica gel, pet.ether: ethyl acetate) gave the title compound as ye llow oil. IR (liquid fil m) : 3385 (OH), 1470, 1330 cm·1 (N02)
1H NMR (CDCI3): 8 1.56 (d, 3H); 2.30 (brs, 1 H, OH) 4.97 (del , l H,CH ); 6.94 -8.089 (m, 8H, Ar- H), 7.75 (s, I H, !'\H) MS: mlz 257 [M-It. Anal. Found: C, 64.78 ; H, 5.15 ; N, 10.51. Calc. for C t4H14N20 3: C, 65.11 ; 1-1 , 5.46; N, 10.85 %.
Acknowledgement The au thors are thank ful to Dr KV Raghavan,
Director, Indian Institute of Chemi ca l Technology, fo r providing research fac ili ties.
References 1 G rafe I, Schi ckancdcr H & Ah re ns K H, South African
Patent, 1990, ZA8904, 667; Chem Abstr 113, 1990, 190940d. 2 Manske, R H. Chem. Rev. 30, 1942, 127. 3 Kawasc Y, Yamaguchi S, Maeda 0, Hayashi A, Hayashi 1,
Tabata K & Kondo M, J Her Chem, 16, 1979, 487 . 4 Rehwa1d M, Gewa1d K, Lankau H-J & Unvcrfc rt h K,
Heterocycles, 45, 1997, 483. 5 1-lino K, Furuk awa K, Nagai Y & Uno H, Chem Phar111 Bull