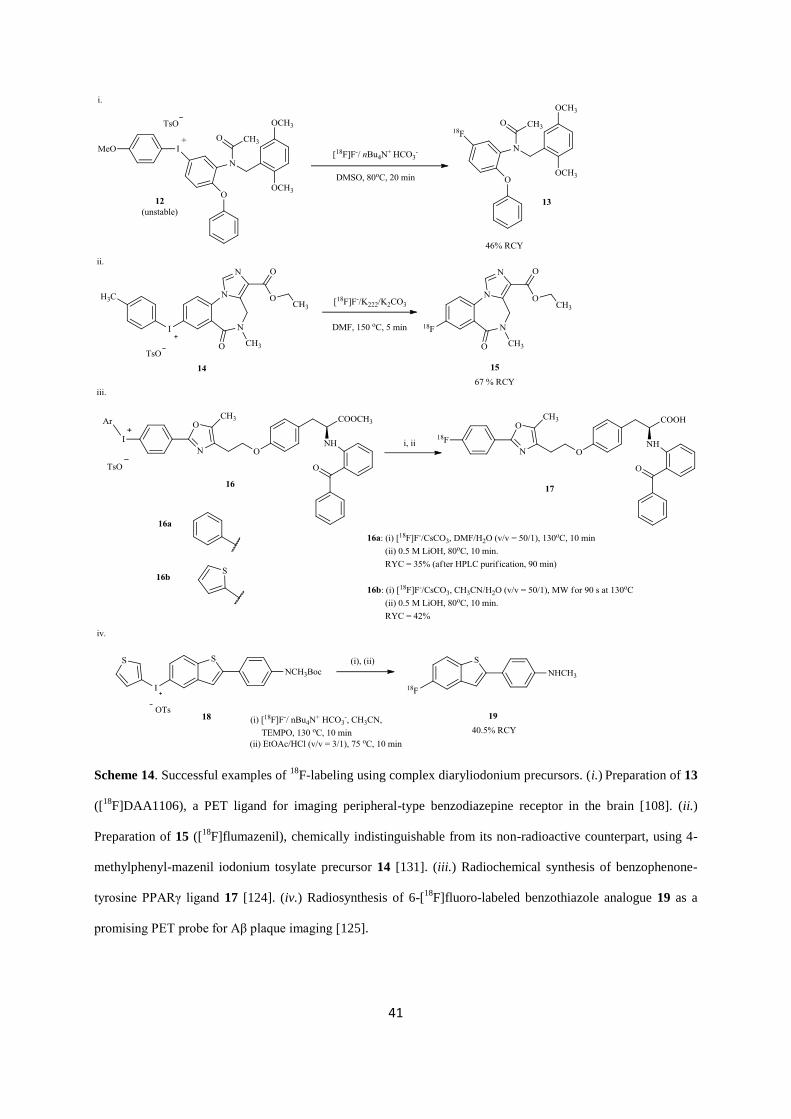
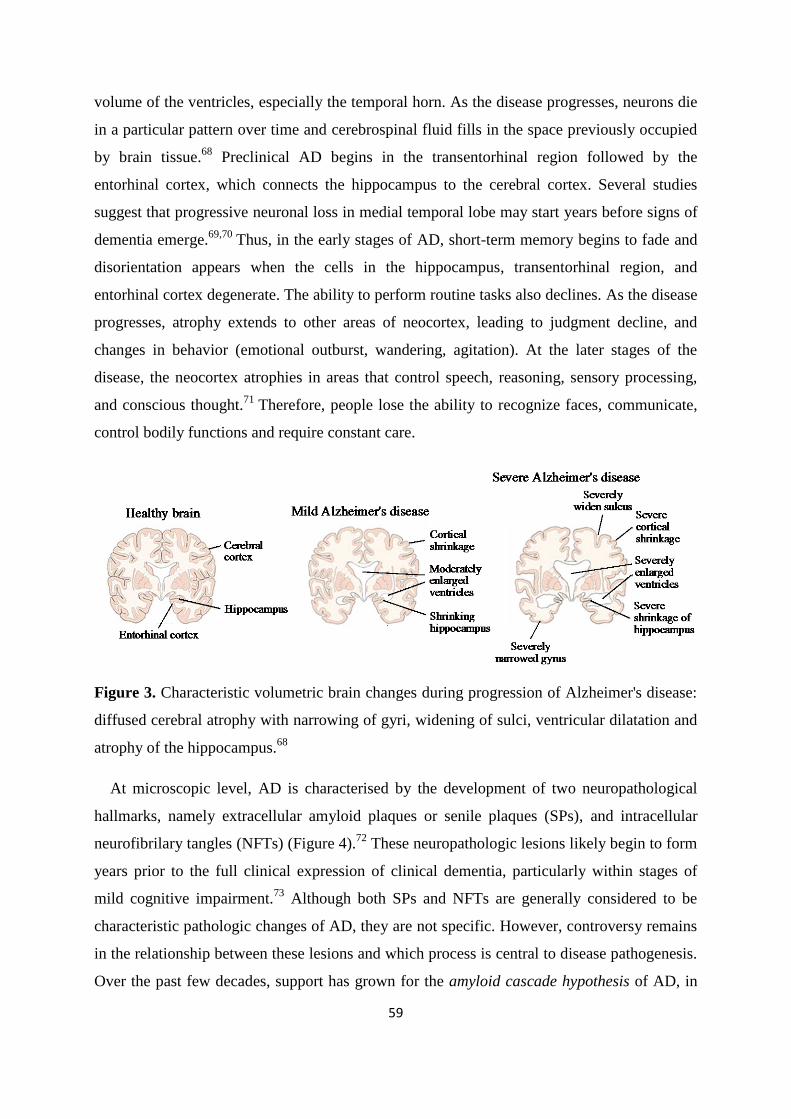
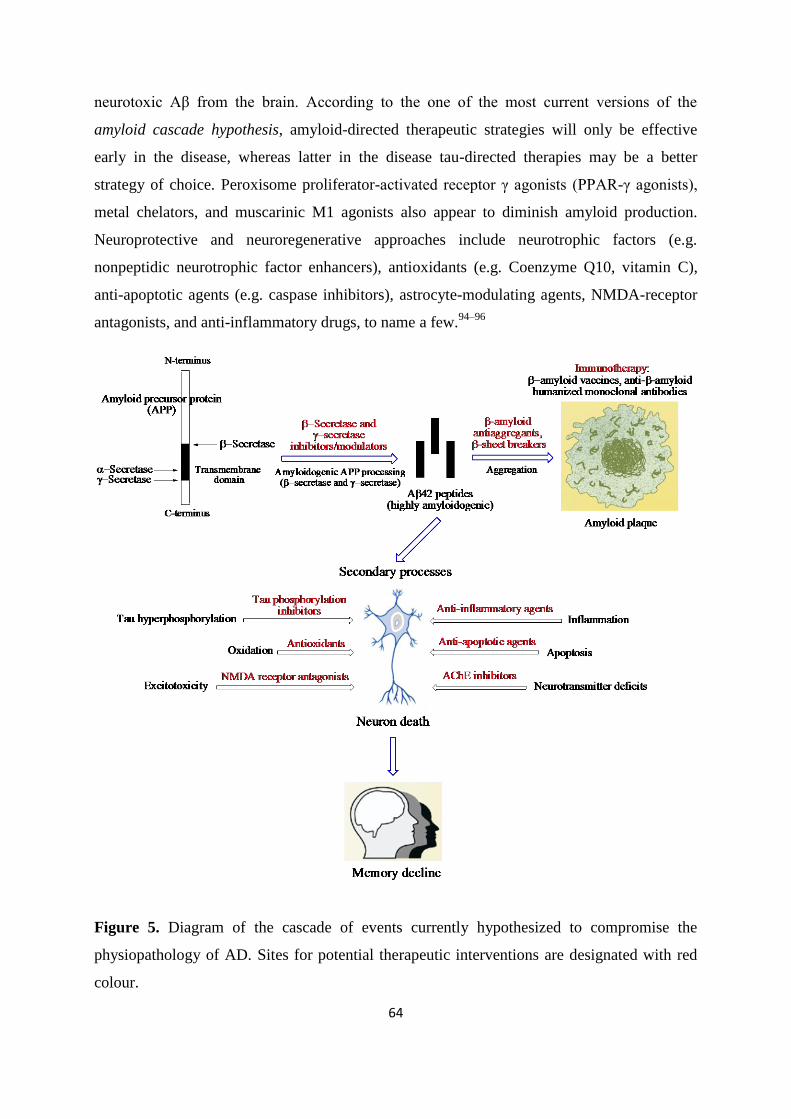
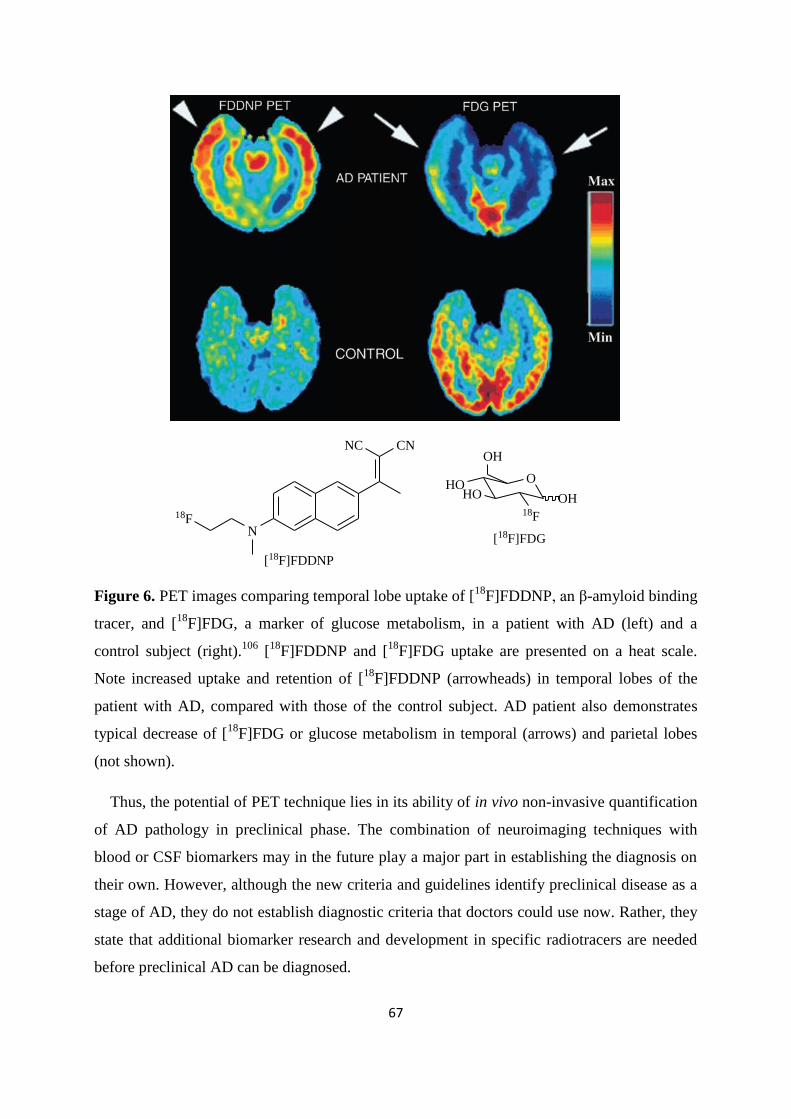
Page 1
UNIVERSITÉ FRANÇOIS – RABELAIS DE TOURS
École Doctorale « Santé - Sciences Biologiques - Chimie du Vivant »
and
UNIVERSITY OF LJUBLJANA, FACULTY OF PHARMACY
«Department of Pharmaceutical Chemistry»
A cotutelle thesis submitted in fulfillment of the requirements for
the degree of «Docteur» at the University François Rabelais of
Tours (France) and Doctor of Pharmacy at the University of
Ljubljana (Slovenia) In
Pharmaceutical Chemistry
Publicly defended on the 1st of March 2013 by
Mitja KOVAČ in Ljubljana
FLUORATION DE DERIVES DU BENZOVESAMICOL POUR L'OBTENTION
DE RADIOLIGANDS POTENTIELS DU TRANSPORTEUR VESICULAIRE DE
L'ACETYLCHOLINE
Under the co-direction of:
Associate Professor Sylvie Mavel (MCU, Tours) and Associate Professor Marko Anderluh
(Ljubljana)
----------------- JURY for Oral Defense:
Ms MAVEL Sylvie – Associate Professor, University François-Rabelais, Tours, France
Mr ANDERLUH Marko – Associate Professor, University of Ljubljana, Slovenia
Mr DOLLÉ Frédéric – Service Hospitalier Frédéric Joliot, Institut d'Imagerie
BioMédicale - CEA, Orsay, France (Reviewer)
Mr EMOND Patrick – Professor, University François-Rabelais, Tours, France
Ms GMEINER STOPAR Tanja – Assistant Professor, University of Ljubljana, Slovenia
(Reviewer)
Mr GOBEC Stanislav – Professor, University of Ljubljana, Slovenia (Chairman)
Page 2
This cotutelle PhD was carried out with the collaboration between the University of Tours
(Laboratoire de Biophysique Médicale et Pharmaceutique, Unité INSERM U930 - FRANCE)
and the University of Ljubljana (Faculty of Pharmacy, Department of Pharmacutical
Chemistry - SLOVENIA).
The work was supported by a grant from the Slovene Human Resources Development and
Scholarship Fund, by a grant from the University of Ljubljana (Inovativna shema za
sofinanciranje doktorskega študija za spodbujanje sodelovanja z gospodarstvom in reševanja
aktualnih družbenih izzivov - generacija 2010 Univerza v Ljubljani), and by a Slovenia-
French bilateral collaboration project (project n° BI-FR/12-13-PROTEUS-007).
Page 3
AKNOWLEDGEMENTS
I would like to extend my most sincere gratitude to my supervisors Dr. Sylvie Mavel and
Dr. Marko Anderluh for their continued direction, training, and encouragement. Their
guidance has been inspirational and instructive throughout my journey along this path, and I
will continue to draw on the wisdom they have imparted as I move forward.
Special thanks go to Dr. Johnny Vercouille and his team for their training and supervision
in the radiopharmaceutical laboratory CERRP (Centre d'Etude et de Recherche sur les
Radiopharmaceutiques).
I would like to extend my thanks to Dr. Patrick Emond and Dr. Frédérick Dollé for their
valuable advices and reading the thesis.
Thanks are given to Dr. Tanja Gmeiner Stopar and Dr. Stanislav Gobec for reading the
thesis.
Thanks also to Dr. Sylvie Chalon, and Dr. Mohamed Abarbri.
Finally, I would like to thank my mother and all the family who have always been loving,
supportive, and a guiding light throughout my life.
Page 4
RÉSUMÉ
La maladie d’Alzheimer (MA) est une maladie neurodégénérative progressive et l’une des
principales démences. Les plaques amyloïdes extracellulaires, les dégénérescences
neurofibrillaires intracellulaires, et la dégénérescence synaptique sont des marqueurs
neurophysiopathologiques de la MA. Il a été montré que la diminution en transporteur
vésiculaire de l’acétylcholine (VAChT) est un paramètre neurologique précoce, précédant les
signes cliniques de la maladie, et fortement corrélée avec la démence associée à la maladie.
L’utilisation de radiotraceur sélectif et spécifique pouvant être utilisé en tomographie par
émission de positrons (TEP) ou par tomographie d'émission monophotonique (TEMP) offre la
possibilité d’identifier de subtils changements neurologiques aux stades précoces de la
maladie, et ainsi aider au diagnostic différentiel de la MA avec d’autres démences corticales
ou sous-corticales.
Le (2R,3R)-5-IBVM, dérivé du benzovésamicol, est un ligand de haute affinité et sélectivité
du VAChT, et est le seul radiotraceur utilisé en imagerie humaine par TEMP pour le
diagnostic de la MA. Or, la TEP présente des avantages par rapport à la TEMP telle qu'une
meilleure détection, meilleure résolution de l'image et possibilité de quantification. Sachant
que l’analogue fluoré du 5-IBVM devrait présenter une affinité et sélectivité pour le VAChT
du même ordre, nous avons donc synthétisé les énantiomères du 5-FBVM et nous avons
développé des méthodes d'introduction régiosélective d'ion fluorure en position 5 du
benzovésamicol, qui est une position non activée. Pour cela, nous avons choisi comme
précurseur, la fonction triazène (Ar-N=N-NR2) en tant que «groupe partant » pour l'obtention
d'aryles fluorés.
En partant d'études théoriques de fluoro-dediazénation, nous avons synthétisé les
énantiomères du 5-FBVM, dans un rendement de 25%, en utilisant le t-butyle de nitrite
comme agent de diazotation et l'éthérate de trifluorure de bore en tant qu'agent de fluoration.
Des études de modélisation moléculaire (QSAR), faites sur 32 composés de type vésamicol,
ont été réalisées en tenant compte de la stéréospécificité du site de fixation du VAChT.
L'évaluation in vitro a montré la très bonne affinité pour le VAChT des 2 énantiomères, du
même ordre que le 5-IBVM, comme le prédisaient les études QSAR. Le (2S,3S)-5-FBVM
présente une meilleure sélectivité vis-à-vis des récepteurs 1 et pourrait être un traceur
potentiel pour l'imagerie in vivo des neurones cholinergiques.
Page 5
Actuellement un des facteurs limitants du développement de la technologie TEP est
l'introduction d'un ion fluorure sur un système aromatique non activé. Nous nous sommes
donc focalisés dans un deuxième temps sur la fluoration d'aryles non activés possédant un
triazène. Nous avons recherché un acide n'interférant pas lors d'un potentiel radiomarquage.
Nous avons étudié différentes conditions expérimentales (acide, solvant, et agent de
fluoration) pour la fluoration du 3,3-diéthyl-1-naphtyltriazène choisi comme modèle d'étude.
À partir des résultats obtenus en chimie froide, l'acide polyphosphorique (PPA) dans un
solvant chloré est le plus prometteur et de plus innovant dans ce type de réaction. De plus, en
se basant sur la chimie de coordination des triazènes avec le trifluorure de bore, nous
proposons que la fluoro-detriazénation pourrait être obtenue, avec uniquement de l'éthérate de
trifluorure de bore, sans addition d'acide protique, à partir d'une température suffisante. Nous
avons confirmé cette hypothèse sur le naphtyltriazène ainsi que sur des phényltriazènes para-
substitués en comparant chauffage traditionnel et chauffage par micro-ondes. Nous avons
validé notre méthode en fluorant un système plus complexe, à savoir le 5-TBV qui a conduit
au 5-FBVM dans un rendement de 72%, sous micro-onde, dans le tétrachlorure de carbone.
Des tests préliminaires de radiomarquage du 5-TBV, en utilisant le PPA dans le
chloroforme sous micro-onde ont donné des résultats prometteurs.
Mots Clés: fluorination, triazène, benzovésamicol, VAChT, TEP
Page 6
ABSTRACT
Alzheimer's disease (AD), a progressive neurodegenerative and terminal disorder, is the
most common cause of dementia in the elderly. Extracellular amyloid plaques, intracellular
neurofibrillary tangles, and degeneration of the synaptic terminals are the most characteristic
neuropathophysiological hallmarks of AD. It has been shown that deficiencies in vesicular
acetylcholine transporter (VAChT) are among the earliest neuronal changes, preceding
clinical symptoms of the disease, and showing a strong correlation with the severity of
dementia. Thus, the use of selective and specific radiotracer functional imaging modalities,
such as positron emission tomography (PET) and single photon emission computed
tomography (SPECT), offers non-invasive in vivo identification of subtle neurological
changes in the early stages of AD and, therefore, offers value in the differential diagnosis of
AD from other cortical and subcortical dementias.
Benzovesamicol-related ligand (2R,3R)-5-IBVM has a high affinity and enough selectivity
for the VAChT, and is the only SPECT VAChT radiotracer used in human to obtain an early
diagnosis of AD. Regarding physico-chemical properties of fluorine-19 and fluorine-18, and
PET advantages over SPECT in terms of higher detection efficiency, better spatial resolution
and possibility for quantification, it is expected that the fluoro analog of 5-IBVM should be of
the same order of affinity and selectivity for the VAChT. Thus, we have prepared pure
enantiomers of 5-FBVM and developed method for regioselective introduction of fluorine
into the 5-position of non-activated benzovesamicol scaffold. We have chosen as a precursor
system triazene function (Ar-N=N-NR2) as a leaving group for arylfluorination.
Firstly, we built theoretical model by studying characteristics of fluoro-de-diazoniation
process. Accordingly, we have synthesized pure enantiomers of 5-FBVM from amino analog
5-ABV in around 25% yield by using t-butyl nitrite as diazonating agent and boron trifluoride
etherate as fluorinating agent. Furthermore, to demonstrate the suitability of a triazene as a
leaving group, the fluoro-de-triazenation of the corresponding triazene precursor (5-TBV),
using triflic acid to trigger triazene moiety decomposition and boron trifluoride etherate,
afforded 5-FBVM in reasonable yield (25%). QSAR studies based on 32 vesamicol
derivatives taking into account the stereoselectivity of the VAChT binding site were
performed. Both enantiomers exhibited high in vitro VAChT binding affinities determined by
radioligand displacement studies and were in the same range as 5-IBVM as predicted by 3D
Page 7
QSAR studies. (2S,3S)-5-FBVM was more selective over σ1 receptors and could be a
potential PET radioligand for in vivo mapping of cholinergic terminals.
Actually, one of the main limitations in aromatic nucleophilic fluorination is that
arylfluorides are only satisfactory obtained on “activated system”. As new techniques to
incorporate fluoride are needed for PET technology, we focused our research in the second
step on fluorination of non-activated aryl skeleton from triazene precursor. We sought for the
appropriate acid to trigger triazene decomposition but with no interference in the
radiofluorination step. We studied different conditions (acid, solvent, and fluorinating agent)
for the fluorination of 3,3-diethyl-1-naphthyltriazene (1-NT), chosen as precursor model.
According to the results in the non-radioactive attempts, polyphosphoric acid (PPA) proved to
be the most suitable one in chlorinated solvents, although had never been used in this type of
reaction. Furthermore, from coordination chemistry of triazene derivatives with boron
trifluoride, we proposed that fluoro-de-triazenation can be successfully accomplished by the
only presence of boron trifluoride without any protic acid at elevated temperature. Our
hypothesis was first confirmed on 1-NT. This methodology was also extended on several
para-substituted 3,3-diethyl-1-aryltriazenes by conventional and microwave heating. To
prove that the method is applicable to obtain more complex arylfluorides too, 5-FBVM was
accomplished in high yield (72%) with microwave heating in tetrachloromethane.
Preliminary tests were transposed to F-18 radiolabelling. Encouraging results were obtained
by radiofluorination of 5-TBV using PPA in chloroform with microwave heating.
Keywords: fluorination, triazene, benzovesamicol, VAChT, PET
Page 8
ABBREVIATIONS
A- conjugate base
ACh acetylcholine
AChE acetylcholine esterase
AChEI acetylcholinesterase inhibitor
AD Alzheimer's disease
Aβ amyloid beta peptide
APP amyloid precursor protein
ATP adenosine triphosphate
ChAT choline acetyltransferase
ChT choline transporter
CSF cerebrospinal fluid
CT computerized tomography
d deuteron
D.c. decay corrected
DIPEA diisopropylethylamine
DLB dementia with Lewy body
EWG electron-withdrawing group
FTD frontotemporal dementia
GC gas chromatography
HPLC high-pressure liquid chromatography
LG leaving group
MCI mild cognitive impairment
MRI magnetic resonance imaging
n neutron
N.d.c. non-decay corrected
NFT neurofibrilary tangles
NMR nuclear magnetic resonance
p proton
PA phosphoric anhydride
PDD Parkinson's disease dementia
Page 9
PP phosphatase
PET positron emission tomography
PPA polyphosphoric acid
PPAR peroxisome proliferator-activated receptor
PS presenilin
RCY radiochemical yield
SA specific activity
sMRI structural magnetic resonance imaging
SNAP synaptosome-associated proteins
SP senile plaque
SPECT single photon emission computed tomography
tR retention time
TEA triethylamine
TLC thin-layer chromatography
VAChT vesicular acetylcholine transporter
VaD vascular dementia
VAMP vesicle-associated membrane proteins
ν neutrino
Page 10
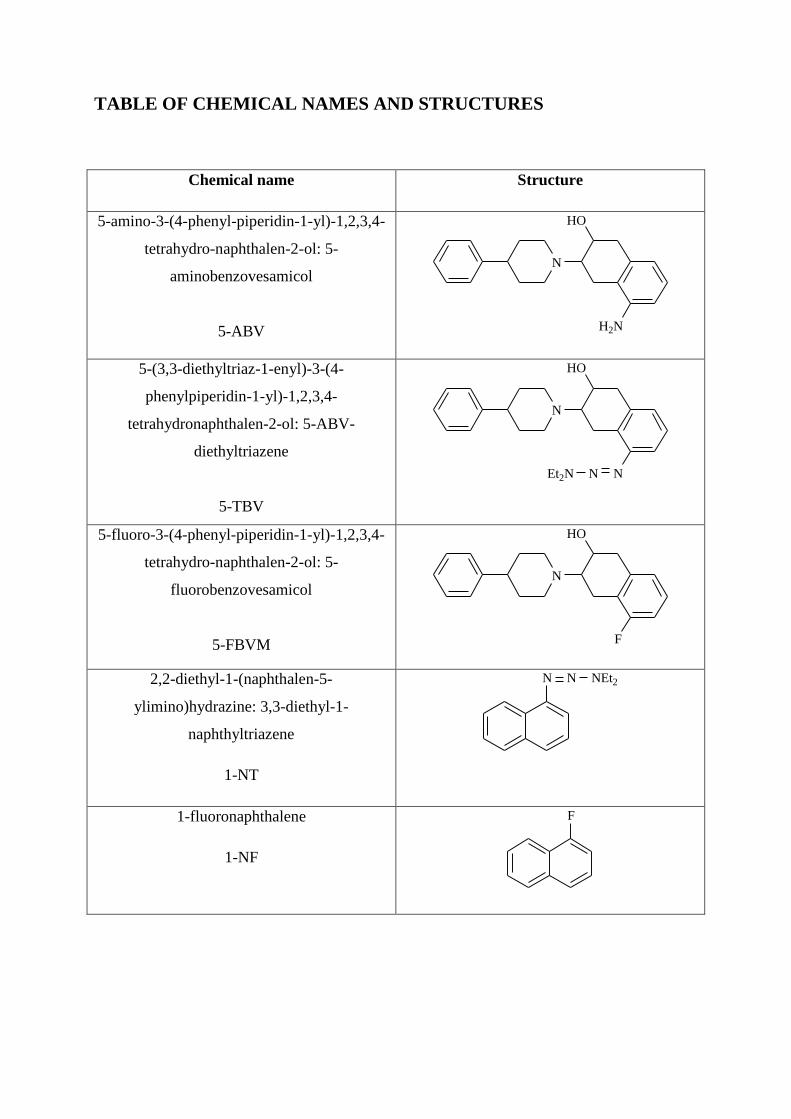
TABLE OF CHEMICAL NAMES AND STRUCTURES
Chemical name Structure
5-amino-3-(4-phenyl-piperidin-1-yl)-1,2,3,4-
tetrahydro-naphthalen-2-ol: 5-
aminobenzovesamicol
5-ABV
N
HO
H2N
5-(3,3-diethyltriaz-1-enyl)-3-(4-
phenylpiperidin-1-yl)-1,2,3,4-
tetrahydronaphthalen-2-ol: 5-ABV-
diethyltriazene
5-TBV
N
HO
NNEt2N
5-fluoro-3-(4-phenyl-piperidin-1-yl)-1,2,3,4-
tetrahydro-naphthalen-2-ol: 5-
fluorobenzovesamicol
5-FBVM
N
HO
F
2,2-diethyl-1-(naphthalen-5-
ylimino)hydrazine: 3,3-diethyl-1-
naphthyltriazene
1-NT
N N NEt2
1-fluoronaphthalene
1-NF
F
Page 11
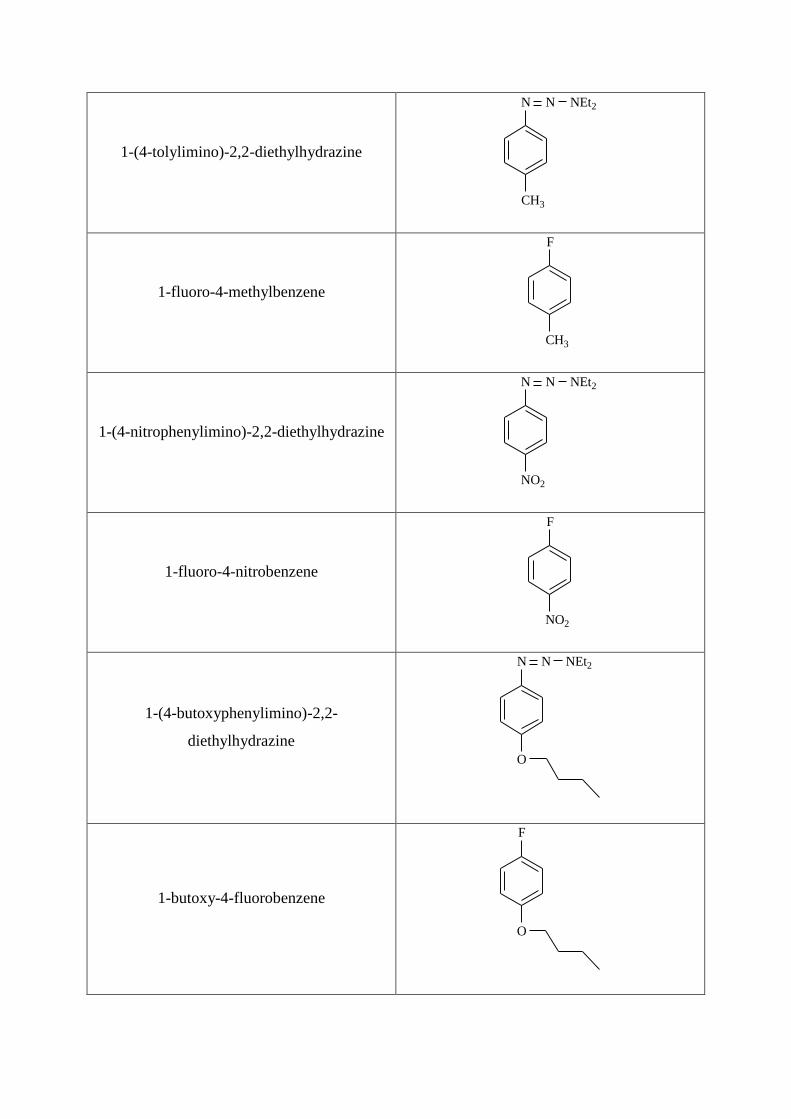
1-(4-tolylimino)-2,2-diethylhydrazine
N N NEt2
CH3
1-fluoro-4-methylbenzene
F
CH3
1-(4-nitrophenylimino)-2,2-diethylhydrazine
N N NEt2
NO2
1-fluoro-4-nitrobenzene
F
NO2
1-(4-butoxyphenylimino)-2,2-
diethylhydrazine
N N NEt2
O
1-butoxy-4-fluorobenzene
F
O
Page 12
1-(4-iodophenylimino)-2,2-diethylhydrazine
N N NEt2
I
1-fluoro-4-iodobenzene
F
I
1-(4-cyanophenylimino)-2,2-diethylhydrazine
N N NEt2
CN
4-fluorobenzonitrile
F
CN
Page 13
TABLE OF CONTENTS
1. INTRODUCTION ................................................................................................................ 1
1.1. Positron Emision Tomography ........................................................................................ 2
1.2. Strategies for 18
F-labelling ............................................................................................... 7
1.2.1. Direct electrophilic 18
F-fluorination.......................................................................... 8
1.2.2. Direct nucleophilic 18
F-fluorination .......................................................................... 9
1.2.2.1. Direct aliphatic 18
F-nucleophilic substitution reactions ....................................... 11
1.2.2.2. Direct aromatic 18
F-nucleophilic substitution reactions....................................... 12
1.2.3. Indirect 18
F-labelling reactions................................................................................ 16
1.3. 18
F-labelled Aryl-Tracers through Direct Introduction of [18
F]fluoride into
Electron-Rich Arenes .................................................................................................... 18
1.4. Alzheimer's disease ........................................................................................................ 57
1.4.1. Epidemiology and risk factors of Alzheimer’s disease ........................................... 57
1.4.2. Neurophysiology and pathology of Alzheimer’s disease ....................................... 58
1.4.3. Pharmaceutical management and research directions ............................................. 62
1.4.4. The diagnosis of Alzheimer’s disease by PET ....................................................... 65
1.5. Vesicular acetylcholine transporter (VAChT) and the most promising
PET imaging tracers ...................................................................................................... 68
1.6. Synthesis of 5-aminobenzovesamicol (5-ABV) and its enantiomers ............................ 71
2. AIMS AND SCOPE ........................................................................................................... 73
3. RESULTS AND DISCUSSION ......................................................................................... 76
3.1. Theoretical model for efficient one-pot fluoro-de-diazoniation .................................... 77
3.2. Synthesis of 5-FBVM and its enantiomers via fluoro-de-diazoniation ......................... 82
3.3. Theoretical model for efficient fluoro-de-triazenation and synthesis of
5-FBVM from the corresponding triazene precursor .................................................... 86
3.4. 3D QSAR study, synthesis, and in vitro evaluation of (+)-5-FBVM as potential
PET radioligand for the vesicular acetylcholine transporter (VAChT) ......................... 88
3.5. Aromatic fluoro-de-triazenation with boron trifluoride diethyl etherate under non
protic acid conditions ..................................................................................................... 99
3.6. Examination of reaction parameters for radio-fluoro-de-triazenation of 5-TBV ........ 106
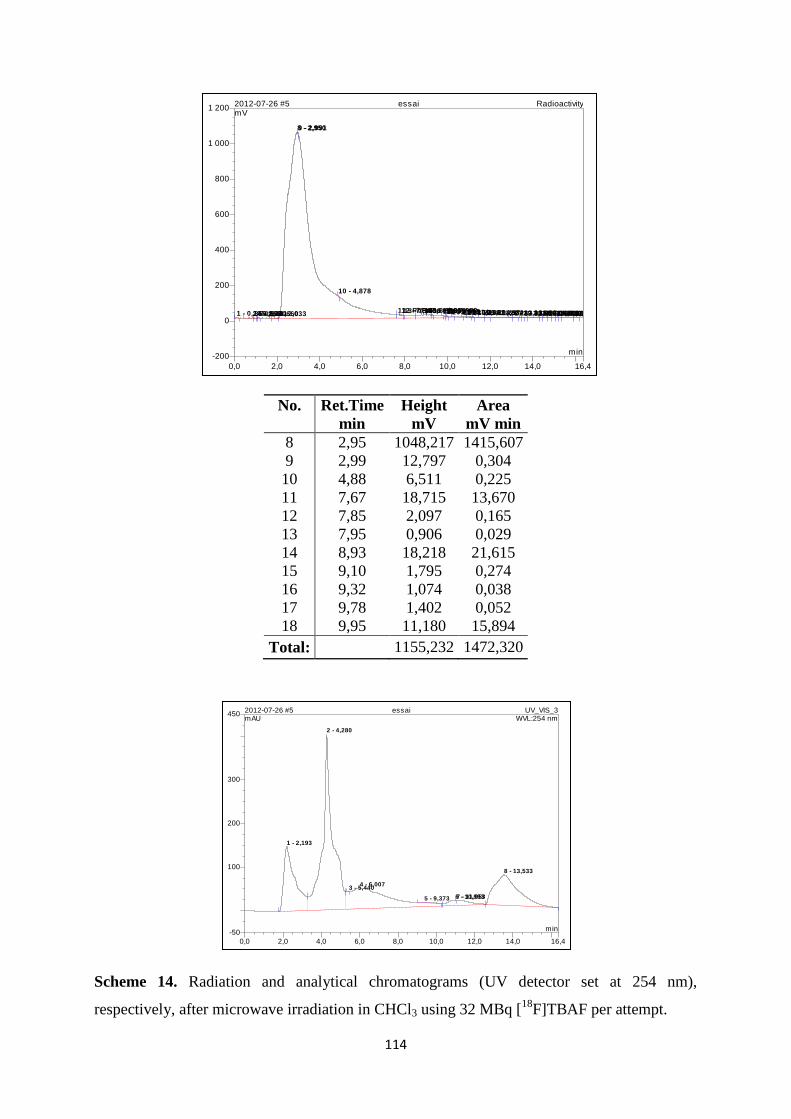
3.7. Radiochemistry ............................................................................................................ 110
Page 14
3.7.1. Preparation of [18
F]TBAF using TRACERlab™
FX F-N synthesizer ................... 110
3.7.2. N.c.a. 18
F-radiofluoro-de-triazenation of 5-TBV .................................................. 112
4. EXPERIMENTAL ........................................................................................................... 116
4.1. General information ..................................................................................................... 117
4.2. Chemistry ..................................................................................................................... 117
4.2.1. One-pot fluoro-de-diazoniation of 5-aminobenzovesamicol (5-ABV) in
1,2-dichlorobenzene .............................................................................................. 117
4.2.2. One-pot fluoro-de-diazoniation of 5-aminobenzovesamicol (5-ABV)
in ionic liquid ........................................................................................................ 118
4.2.3. Fluoro-de-triazenation of 3,3-diethyl-1-naphthyltriazene (1-NT) using
KF/Kryptofix®
complex and triflic acid (TfOH) in dichloromethane ................... 118
4.2.4. Fluoro-de-triazenation of 3,3-diethyl-1-naphthyltriazene (1-NT) using tetra-n-
butylammonium fluoride (TBAF) and triflic acid (TfOH) in chloroform ........... 119
4.2.5. Fluoro-de-triazenation of 3,3-diethyl-1-naphthyltriazene (1-NT) using tetra-n-
butylammonium fluoride (TBAF) and polyphosphoric acid (PPA)
in chloroform ........................................................................................................ 119
4.2.6. General procedure for the fluoro-de-triazenation of
3,3-diethyl-1-naphthyltriazene (1-NT) with different amounts of
polyphosphoric acid (PPA) ................................................................................... 120
4.2.7. General procedure for the fluoro-de-triazenation of
3,3-diethyl-1-naphthyltriazene (1-NT) with different fluoride sources ................ 120
4.2.8. General procedure for the de-triazenation of 5-TBV
with polyphosphoric acid (PPA) .......................................................................... 121
4.3. Radiochemistry. ........................................................................................................... 121
4.3.1. Preparation of [18
F]TBAF using TRACERlab™
FX F-N synthesizer .................. 121
4.3.2. General procedure for the thermal n.c.a. 18
F-radiofluoro-de-triazenation
of 5-TBV ............................................................................................................... 121
4.3.3. General procedure for the microwave-assisted n.c.a.
18
F-radiofluoro-de-triazenation of 5-TBV ............................................................ 122
5. CONCLUSION ................................................................................................................. 123
6. REFERENCES ................................................................................................................. 126
Page 15
1
1. INTRODUCTION
Page 16
2
1.1. Positron Emision Tomography
Positron emission tomography (PET) is a very powerful non-invasive in vivo quantitative
molecular and functional imaging technique that is used to study and visualize human and
animal physiology and biochemical events by monitoring the distribution and concentration of
positron-emitting radiopharmaceuticals in the body over time.1 Information about metabolism,
receptor or enzyme function, and biochemical mechanisms in living tissue can be obtained
directly from PET experiments in a quantitative manner. Unlike conventional anatomic
imaging techniques (e.i. computerized tomography/CT), which mainly provide detailed
anatomical images, PET can provide an early diagnosis (chemical changes that occur before
macroscopic signs of a disease are observed), more accurate staging, monitoring response to
therapy, and assessment of recurrence of disease. For all these reasons PET is increasingly
used in oncology,2 cardiology,
3,4 neurology,
5 drug development
6,7 and therapy.
8 It is
anticipated that PET studies will improve the selection of potential drug candidates in early
stages of development, give a greater understanding of drug's mechanism of action, and aid in
guiding dose selection.
One of the main challenges for radiochemists is the development of rapid synthetic
methods for introducing short-lived positron-emitting isotopes into the molecule of interest.
The radiolabelled probe has to be synthesized, purified, analyzed, and formulated roughly
within three isotope half-lives to ensure there is enough radiolabelled material to be
administered to a subject (animal or human) undergoing the PET scan. Except for fluorine-18,
the extremely short-lives of the isotopes shown in Table 1 necessitate that the labelled probes
be prepared in proximity to where the isotopes are produced and used almost immediately
after their synthesis. A number of modern PET facilities house cyclotrons for radioisotope
production, radiosynthetic laboratories, and PET scanners are under one roof to allow
efficient production and transport of short-lived PET probes from the laboratory to the PET
scanner.1
Page 17
3
Table 1. The most important and commonly used short-lived positron-emitting radionuclides
for PET imaging.
Radionuclide
Half-life, t1/2
(min)
Nuclear
reaction Target Product
Decay
product
18F 109.8
20Ne(d,α)
18F
18O(p,n)
18F
Ne(+F2)
[18
O]H2O
[18
F]F2
[18
F]F-
18O
11C 20.4
14N(p,α)
11C
N2(+O2)
N2(+H2)
[11
C]CO2
[11
C]CH4 11
B
13N 9.97
16O(p,α)
13N
H2O
H2O + EtOH
[13
N]NOx
[13
N]NH3
13C
15O 2.07
15N(d,n)
15O N2(+O2) [
15O]O2
15N
Radioisotope production begins in a cyclotron. This is a (compact) particle accelerator,
which is capable of producing proton (p) or deuteron (d) beams of the required energy range
to generate 18
F, 11
C, 13
N, and 15
O species.9
The beam is directed onto a target system at the
exit of the cyclotron, which contains the target material suitable for the production of the
required radioisotope (Table 1).
Dealing with high-energy short lived radioactive compounds safely and effectively is a
priority, and traditional bench-top synthetic chemistry is clearly not an option. State-of-the-art
PET radiosynthesis laboratories use “hot cells”, which are basically enclosed lead-lined
versions of a fumehood with lead glass windows many inches thick, to carry out
radiolabelling procedures. The radioactive isotope is transferred to the hot cell (usually under
an inert gas stream) where it is converted by a series of chemical steps into the final
radiolabelled product. Typically, computer-controlled robotic or automated systems are used
for such labelling to restrict, as much as possible, exposure of the user to radiation.1
Page 18
4
Figure 1. The process of PET radiotracer production begins at a cyclotron and end at the PET
scanner. The whole process typically takes few hours. From left to right: (A.) commercially
available biomedical cyclotron; (B.) “Hot-Cell” - automated radiolabelling system controlled
from outside the hot cell; (C.) combined PET/CT scanner and processed PET image (bottom
right).
An important advance in scanner technology and evolution in imaging technology has been
the physical integration of PET and CT within the same device (hardware fusion approach).
The combined PET/CT scanners allow to acquire co-registered anatomic and functional
images in a single scan session and therefore allows accurate localization of functional
abnormalities.10
Since PET labelling reactions are performed with nanomolar amounts of radioisotopes,
there is normally a vast stoichiometric excess of “cold” reagents which results in pseudo-first-
order reaction kinetics with respect to the radioisotope concentration. Advantageously,
reactions, which may normally need hours or days to reach completion on a macroscopic
scale, can often be performed in minutes using PET radioisotopes. It is also worth noting that
even minor impurities found in precursors, solvents and reagents can become significant when
performing such small-scale reactions.
Before a PET radiotracer can be administered to a patient, usually by intravenous injection
in the form of saline solution, it must be suitable and rapidly characterized, and sterilized.
PET radiotracers are often characterized by using a combination of high-pressure liquid
chromatography (HPLC), thin-layer chromatography (TLC), and gas chromatography (GC) in
conjuction with suitable radioactivity and mass detectors. Quality control procedures for
radiotracers and radiopharmaceuticals are similar to those applied to non-radioactive
pharmaceuticals. There are two categories for quality control tests: physiochemical test and
biological test. Physiochemical tests give the level of radioisotope and radiochemical
Page 19
5
impurities, chemical impurities, pH value, ionic strength, osmolality, and physical state of the
sample, while the biological tests determine the sterility, apyrogenicity, and toxicity of the
sample.1
The radiochemist has to consider the radiochemical yield (RCY) of the radiosynthesis and
the specific activity (SA) of the final radiolabelled compound. The RCY is a function of both
the chemical yield and half-life of the radioisotope, and is expressed as a fraction of the
radioactivity originally present in the sample following a radiochemical separation. The value
is often quoted as being either non-decay corrected (n.d.c.) or decay corrected (d.c.). Decay
corrected figures are mathematically adjusted measurements that take into account radioactive
decay that has occurred between two different times to give a single value (decay equation:
A(t2) = A(t1)e-0.693Δt/t1/2). It is desirable, but not always essential, to have a high RCY as it is a
useful gauge to measure the efficiency of the radiolabelling procedure. The specific activity is
a measure of the radioactivity per unit of mass of the labelled compound, and is commonly
expressed as giga-Becquerel (SI unit) per micromol (GBq/μmol) or Curies per micromol
(Ci/μmol). The theoretical maximum values of the specific activity (e.g. for fluorine-18 is
63344 GBq/μmol or 1712 Ci/μmol) are never reached for radiolabelled compounds, because
of unavoidable isotopic dilution by the naturally occurring stable isotope. This effect is
particularly apparent when a “cold” fluorine-19 gas is added to the target to allow recovery of
molecular 18
F[F2] for electrophilic fluorinations. Typical specific activities of PET-labelled
products are in the order of 50-500 GBq/μmol (ca. 1-15 Ci/μmol). Since a small amount of
radioactivity can lead to a good quality PET image, only very low amounts (tracer dose) of
compound need to be administered - typically in the sub-micro-gram level. This implies that
the fate of labelled molecules can be studied in vivo without perturbing the biological system
being measured and that very potent or toxic compounds can be studied in human at
subpharmacological and subtoxicological doses.1,11
Due to the characteristic physico-chemical properties, fluorine-18 as artificial radionuclide
is among the available PET radionuclides the most favoured and widely used radiolabel for in
vivo imaging. Fluorine-18 emits quite low energy positron (β+ particle; max. 0.635 MeV; 97%
positron emission and 3% electron capture), which has short path in vivo (~0.5 mm in water,
max. 2.3 mm in tissue) before it annihilates with an electron (e-) giving rise to two opposed
and coincidentally detected by PET camera 511 keV γ-rays. This is the physical basis to
reconstruct the highest resoluted PET images of all the available positron emitters (Figure
Page 20
6
2).1,11
The decay of fluorine-18 gives innocuous 18
O as the product atom (Table 1, Figure 2).
It can be produced as the single-atom species [18
F]fluoride ion (no-carrier-added) in very high
specific radioactivities (~37-370 GBq/μmol or ~1-10 Ci/μmol ) by irradiation of [18
O]-
enriched water (available from commercial vendors) with 11-19 MeV energy proton (p) beam
from small compact cyclotrons according to the 18
O(p,n)18
F reaction.1,11
The ease of
production of high amounts of fluorine-18 coupled with its almost 2 hour half-life (t1/2 = 109.8
min) allows either more time consuming multistep labelling reactions or longer lasting in vivo
investigation or commercial distribution of a tracer over reasonable distances from remote
cyclotron to the clinical PET centers that lack radiochemistry facilities. Furthermore, fluorine
may to some extent mimic a hydrogen atom or hydroxy group in an organic molecule and in
parallel can lead to favourable conformational and physico-chemical changes (such as pKa,
logD) with improved pharmacokinetic (e.g. bioavailability), pharmacodynamic (e.g. enhanced
target binding affinity and selectivity) and toxicologic profiles of organofluorine
compounds.12–15
Figure 2. The physical principles of PET imaging shown schematically. After intravenous
administration to the patient or animal, 18
F-labelled tracer distributes to target tissue(s) and
selectively binds to particular biochemical target (e.g. neuroreceptor). Fluorine-18 is neutron-
Page 21
7
deficient isotope that achieves stability through the nuclear transmutation of a proton (p) into
a neutron (n). This process involves emission of positron (β+) and neutrino (ν). Positron
quickly collides with a free or loosely bound electron (e-) in surrounding tissue, and both are
annihilated to form two 180 degrees separated 511 keV gamma-ray photons (γ-rays). PET
camera/scanner (circular configuration of scintillation detectors around the subject) detects a
large number of pairs of annihilation photons in coincidence (“annihilation coincidence
detection”) in all the lines by opposing detectors as the basis for the approximate
determination of location of the fluorine-18 nuclei/PET probe in the patient. Millions of
individual annihilation events are required to give enough data to reconstruct a high-
resolution PET image.
In comparison with SPECT that utilizes the single photons emitted by gamma-emitting
radionuclides, PET has some essential advantages in comparison to SPECT: higher detection
efficiency, better three-dimensional resolution of a studied image, possibility to quantify the
studied target, and after all, minimization of radiation dose to the patient due to low positron
energy of fluorine-18.
1.2. Strategies for 18
F-labelling
The main synthetic strategies behind 18
F-labelling can be generally divided into two main
distinct areas: (1) direct fluorination and (2) indirect fluorination. In direct fluorination
fluorine-18 is introduced directly into the target molecule of interest in one step, and in
indirect fluorination fluorine-18 is introduced via so-called prosthetic groups and requires a
multistep synthetic approach. These prosthetic groups are typically small 18
F-labelled alkyl or
aryl compounds that have reactive functional groups. They are used to react with more
complex biological molecules which may not be suitable or stable enough to tolerate direct
fluorination methods. The direct 18
F-labelling strategies can be subdivided into two
categories: electrophilic and nucleophilic. Of these two methods, nucleophilic 18
F-
fluorination has dominated in importance because of its greater selectivity and capability to
give highly specific radioactive compounds suitable for PET imaging.
Page 22
8
Synthetic methods for the introduction of fluorine-18 into organic molecules need to be
convenient, rapid and of reasonable high RCY and SA. High SA also enables radiotracers to
be administered to subjects in low mass doses (1-10 nmol or sub-microgram level) to avoid
any toxic or pharmacological effects and perturbation of the biological target or process.
1.2.1. Direct electrophilic 18
F-fluorination
Electrophilic 18
F-fluorinations are less favoured nowadays for two reasons: (A.) they
generally give labelled products with low specific activity, because of the carrier-added
method of [18
F]F2 production, and (B.) labelling with electrophilic reagents such as very
reactive [18
F]F2 is generally non-regioselective and can result in mixtures of 18
F-labelled
products. However, electrophilic fluorination has played an important and historical role in
the development of 18
F-labelled molecules for PET imaging. For example, the first synthesis
of the hugely important PET tracer 2-deoxy-2-[18
F]fluoro-D-glucose ([18
F]FDG) was carried
out by electrophilic fluorination.16
Other more recent examples of important PET tracers,
which still rely on electrophilic 18
F-fluorination methods of synthesis, include [18
F]fluoro-L-
DOPA and 2-L-[18
F]fluorotyrosine.
The most common reagent for electrophilic fluorination is [18
F]F2, which is obtained from
the nuclear reactions of 20
Ne(d,α)18
F or 18
O(p,n)18
F. It can be used as it is or converted into
the less reactive but more selective derivative, such as acetyl hypofluorite ([18
F]CH3COOF)
(Scheme 1, Example A.).1,11
Electrophilic 18
F-reagents can be used to fluorinate electron-rich
substrates by either direct electrophilic substitution or by more regioselective direct
demetallation reactions using organometallic intermediates precursors, such as organomercury
and organotin preursors (Scheme 1, Example B.).
OAcOAcO
OAc
1. [18F]CH3COOF
2. HCl
OHO
HO
OH
18F
OH
[18F]FDG
A.
Page 23
9
BocO
Me3Sn
OH
NHBoc
1. CCl3F/AcOH,
[18F]F2
2. HBr
HO
18F
OH
NH2
B.
4-[18F]FMR
Scheme 1. (A.) Electrophilic 18
F-fluorination using [18
F]CH3COOF was the early synthetic
method for the production of [18
F]FDG, a powerful imaging agent for determining cerebral
and myocardial glucose metabolism, tumour localization and determining its respons to
therapy.1,16
(B.) Synthesis of (1R,2S)-4-[18
F]fluorometaraminol (4-[18
F]FMR), a radiotracer
for in vivo mapping of adrenergic nerves in the heart, from Boc-protected organotin precursor
by electrophilic aromatic substitution with [18
F]F2.17
1.2.2. Direct nucleophilic 18
F-fluorination
Nucleophilic 18
F-fluorination reactions are routinely used to efficiently produce some of the
most important 18
F PET radiotracers: [18
F]FDG widely used in oncology investigations,
[18
F]fallypride,18
[18
F]haloperidol,19
and [18
F]N-methylspiperone20
used in dopamine receptor
studies, and [18
F]fluoroazomycin arabinoside ([18
F]FAZA)21
and [18
F]fluoromisonidazole
([18
F]FMISO)22
for non-invasive tumour hypoxia imaging in vivo with PET.
As [18
F]fluoride ion ([18
F]F-) is in most cases produced by the proton irradiation of highly
enriched [18
O]water target (> 95%) which renders the anion poorly nucleophilic due to its
high degree and strength of hydration in aqueous solution (ΔHhydr = 506 kJ/mol),23
the bulk
[18
O]water is removed and can be recovered for reuse by adsorption of [18
F]fluoride onto an
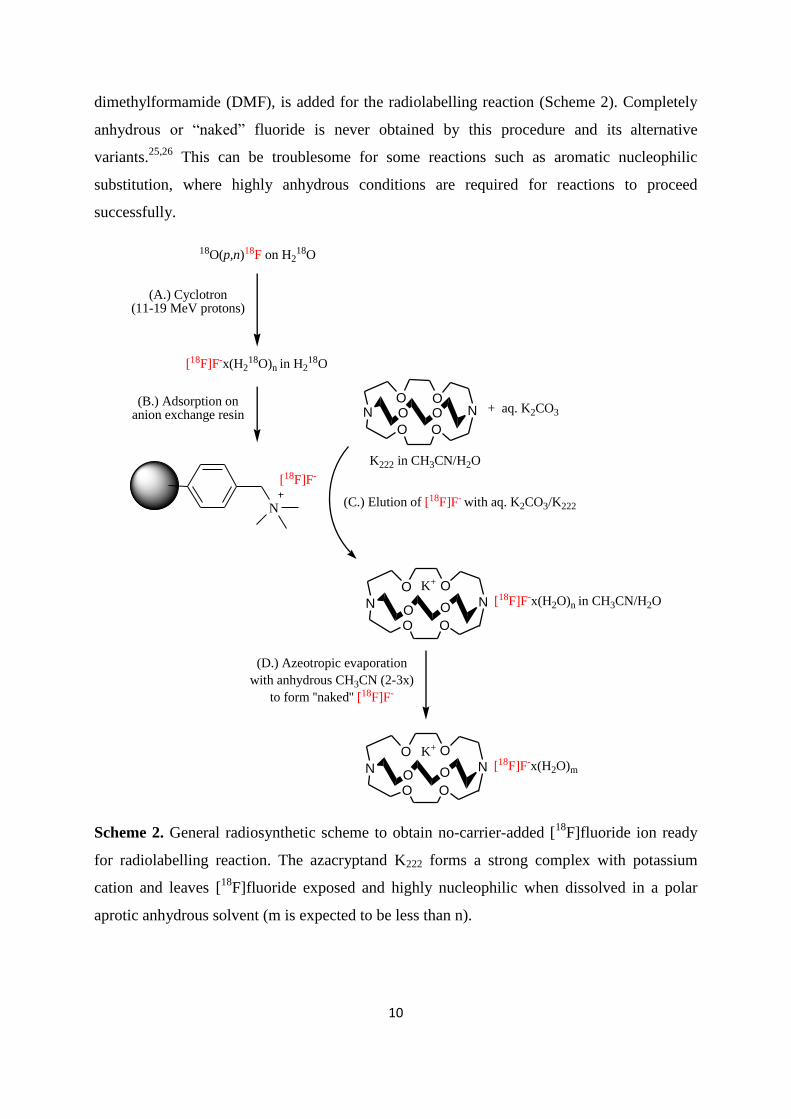
anion exchange resin.24
[18
F]Fluoride is eluted from the anion-exchange resin using either an
alkali carbonate, hydrogencarbonate and phase transfer catalyst in a water/acetonitrile solution
or using dilute aqueous solution of tetra-n-butylammonium bicarbonate (n-TBAHCO3). The
most commonly used cryptand in combination with potasium carbonate (K2CO3) in order to
activate [18
F]fluoride ion is Kryptofix 2.2.2. (K222). Water is then removed via repeated cycles
of azeotropic evaporation with anhydrous acetonitrile (CH3CN) to form the reactive (“naked”)
fluoride.1,25
Finally, a solution of precursor mostly dissolved in polar aprotic anhydrous
organic solvent, such as acetonitrile (CH3CN), dimethyl sulfoxide (DMSO) and
Page 24
10
dimethylformamide (DMF), is added for the radiolabelling reaction (Scheme 2). Completely
anhydrous or “naked” fluoride is never obtained by this procedure and its
alternative
variants.25,26
This can be troublesome for some reactions such as aromatic nucleophilic
substitution, where highly anhydrous conditions are required for reactions to proceed
successfully.
18O(p,n)18F on H218O
(A.) Cyclotron(11-19 MeV protons)
[18F]F-x(H218O)n in H2
18O
N
(B.) Adsorption on anion exchange resin
N
O O
N
O OO O
+ aq. K2CO3NO O
N
O O
O O
K222 in CH3CN/H2O
(C.) Elution of [18F]F- with aq. K2CO3/K222
K+
[18F]F-x(H2O)n in CH3CN/H2O
(D.) Azeotropic evaporation
with anhydrous CH3CN (2-3x)
to form ''naked'' [18F]F-
N
O O
N
O OO O
K+
[18F]F-x(H2O)m
[18F]F-
Scheme 2. General radiosynthetic scheme to obtain no-carrier-added [18
F]fluoride ion ready
for radiolabelling reaction. The azacryptand K222 forms a strong complex with potassium
cation and leaves [18
F]fluoride exposed and highly nucleophilic when dissolved in a polar
aprotic anhydrous solvent (m is expected to be less than n).
Page 25
11
Direct 18
F nucleophilic labelling can be subdivided into aliphatic and aromatic 18
F-labelling
strategies.
1.2.2.1. Direct aliphatic 18
F-nucleophilic substitution reactions
Direct aliphatic nucleophilic 18
F reactions are generally straightforward. Unlike aromatic
substitution reactions, activating groups are not required. The only requirement for aliphatic
nucleophilic 18
F reactions is a good leaving group, such as triflate (TfO) (Scheme 3), tosylate
(TsO), mesylate, iodo, or bromo group. Labelling at aliphatic carbon atoms using sulfonate
ester (mesylate, tosylate) or halide leaving groups can be accomplished very efficiently even
in the presence of trace water27
or in sterically hindered alcohols as a protic reaction medium
(e.g. tert-butyl alcohol, t-BuOH).28,29
The main drawback of aliphatic method is the need to
protect any potentially competing sites of nucleophilic attack in the molecule (principally
acid, alcohol, or amine groups). Additionally, fluorine-18 bound to an aliphatic carbon atom
can be prone to de-fluorination in vivo, giving rise to [18
F]F-. The latter binds avidly to bone,
including skull, and compromise PET measurements with failure to image specific target in
vivo.30–32
OAcOAcO
OAc
1. [18F]KF/K222
2. HCl or NaOH
OHOHO
OH
18F[18F]FDG
O
SO2CF3
OHOAc
Scheme 3. Direct nucleophilic n.c.a. 18
F-fluorination and subsequent deprotection (acidic or
basic hydrolysis) of acetyl-protected mannose triflate for the preparation of [18
F]FDG.33
Direct aliphatic nucleophilic 18
F reactions have been used to effectively label a number of
complex organic molecules in either one step where no protecting groups are necessary
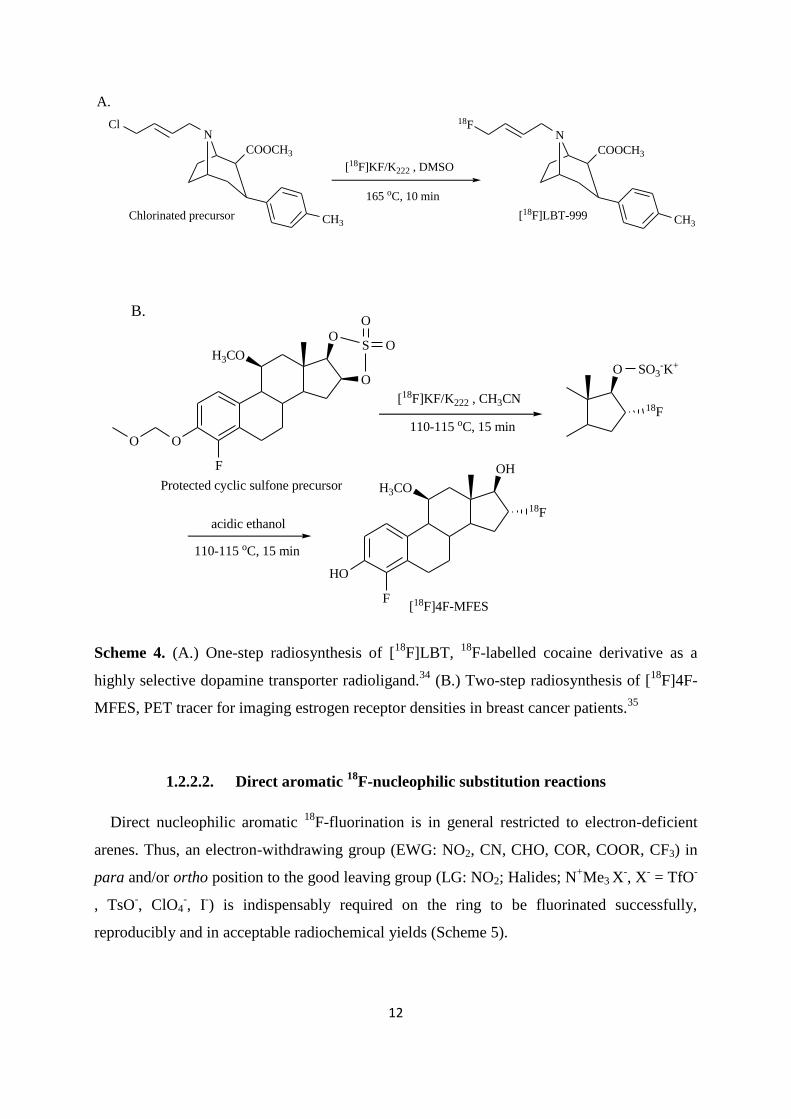
(Scheme 4, Example A.) or two synthetic steps which involves a deprotection (Scheme 4,
Example B.).
Page 26
12
N
COOCH3
CH3
18F
[18F]LBT-999
A.
N
COOCH3
CH3
Cl
[18F]KF/K222 , DMSO
165 oC, 10 min
Chlorinated precursor
OH
H3CO
F
HO
18F
[18F]4F-MFES
B.
O
H3CO
F
O
O
S
O
O
O110-115 oC, 15 min
[18F]KF/K222 , CH3CN
O
18F
SO3-K+
acidic ethanol
110-115 oC, 15 min
Protected cyclic sulfone precursor
Scheme 4. (A.) One-step radiosynthesis of [18
F]LBT, 18
F-labelled cocaine derivative as a
highly selective dopamine transporter radioligand.34
(B.) Two-step radiosynthesis of [18
F]4F-
MFES, PET tracer for imaging estrogen receptor densities in breast cancer patients.35
1.2.2.2. Direct aromatic 18
F-nucleophilic substitution reactions
Direct nucleophilic aromatic 18
F-fluorination is in general restricted to electron-deficient
arenes. Thus, an electron-withdrawing group (EWG: NO2, CN, CHO, COR, COOR, CF3) in
para and/or ortho position to the good leaving group (LG: NO2; Halides; N+Me3
X
-, X
- = TfO
-
, TsO-, ClO4
-, I
-) is indispensably required on the ring to be fluorinated successfully,
reproducibly and in acceptable radiochemical yields (Scheme 5).
Page 27
13
N
O
NO2 or -Cl
[18F]KF/K222
[18F]haloperidol
Nitro- or chloro-precursor
HO
Cl
N
O
18F
HO
Cl
Scheme 5. Direct synthesis of [18
F]haloperidol (butyrophenone neuroleptic) from the
corresponding nitro-36
or chloro-precursor.19
In spite of the presence of EWG quite high reaction temperatures are still required for n.c.a.
18F-labelling.
Synthesis of [18
F]fluoropyridine derivatives proceeds in a similar fashion since these
compounds are reactive toward nucleophilic substitution at the C(2) and C(4) positions to the
ring nitrogen (Scheme 6).37
Thus, only a good leaving group (a halogen, NO2 or N+Me3) is
required, except if fluorination on C(3) to the ring nitrogen is desired. As direct nucleophilic
aromatic 18
F-fluorination is restricted to electron-deficient arenes, extra steps after labelling
have to be performed occasionally to modify or eliminate the activating group completely on
account of a certain loss of overall RCY and specific activity (SA).
NBoc
N
X
[18F]norchlorofluoroepibatidine
[18F]KF/K222, DMSO
150-180 oC, 10 min or
MW (100 W), 1-2.5 min
NBoc
N
18F
NH
N
18F
X = Br, NO2, N+Me3 I-
CF3COOH
CH2Cl2, rt, 2-5 min
Scheme 6. Synthesis of [18
F]norchlorofluoroepibatidine as a selective central nicotinic
cholinergic α4β2 PET radioligand. The high radiochemical yield for the first step (up to 70%)
and the quantitative conversion in the deprotection with trifluoroacetic acid afforded overall
d.c. radiochemical yields of up to 65%.38,39
Page 28
14
There are only a few available methods, namely, Balz-Schiemann reaction,40
Wallach
reaction,41
and more recently diaryliodonium salt method,42
to allow direct introduction of
n.c.a. [18
F]fluoride into arenes without the need for further activating groups (Scheme 7).
18F
R
N2 BF4
R
Balz-Schiemann reaction
N
R
N NWallach reaction
LG
EWG
Classical aromatic nucleophilic
substitution (SNAr)
Diaryliodonium salt method
I
R R'
A
LG = N+Me3, NO2, halogen
EWG = o- or p-NO2, -CN, -CF3, CHO, RCO, COOR
Alkyl
Alkyl
[18F]F
[18F]F
[18F]F-
[18F]F
Scheme 7. Direct regioselective one-step n.c.a. methods to the radiosyntheses of
[18
F]fluoroarenes using [18
F]fluoride anion.
Since only one of the four fluorine atoms is retained in the final product after thermal
decomposition of diazonium tetrafluoroborate salt (ArN2+ BF4
-), Balz-Schiemann reaction is
very inefficient from a radiochemical point. Therefore, the yield of [18
F]fluoride labelling is
theoretically limited to only 25%, and more importantly, the dilution of SA by the non-
radioactive fluorine in the counterion [18
F]BF4- (carrier added [
18F]F
- in the form of
18F-
labelled tetrafluoroborate anion) is high with the consequential great possibility of failure to
localize the biochemical target in the desired tissue(s). More precisely, low specific
radioactivity would saturate targets with the co-administered non-radioactive tracer, and so
Page 29
15
annul any signal from radiotracer binding. In spite of described limitations, Balz-Schiemann
reaction was the first method used in nucleophilic 18
F-labelling of arenes.43
Aryl-3,3-dialkyltriazenes (Ar-N=N-NR'R'') are regarded as protected form of anilines and
stable surrogates of aryldiazonium ions.44
Consequently, the decomposition of aryltriazenes
proceeds via a diazonium ion or corresponding radical, and cause the same yield and
reproducibility problems as Balz-Schiemann method or its modifications. For this reason
there are very few examples of the successful application of the Balz-Schiemann and fluoro-
de-triazenation reaction (Wallach reaction) to the preparation of complex [18
F]fluoroarenes
usually with low RCYs.45–47
Because of limited RCYs, [18
F]fluoro-de-triazenation is rarely
applied for the production of 18
F-labelled tracers nowadays.
On the other hand, diaryliodonium salts have been proven to be useful precursors for the
introduction of n.c.a. [18
F]fluoride into simple as well as more complex arenes (Scheme 8)
and heterocycles. Thus, they gain more and more interest for direct radiofluorination of
otherwise unfavourable electron-rich arenes.25,48
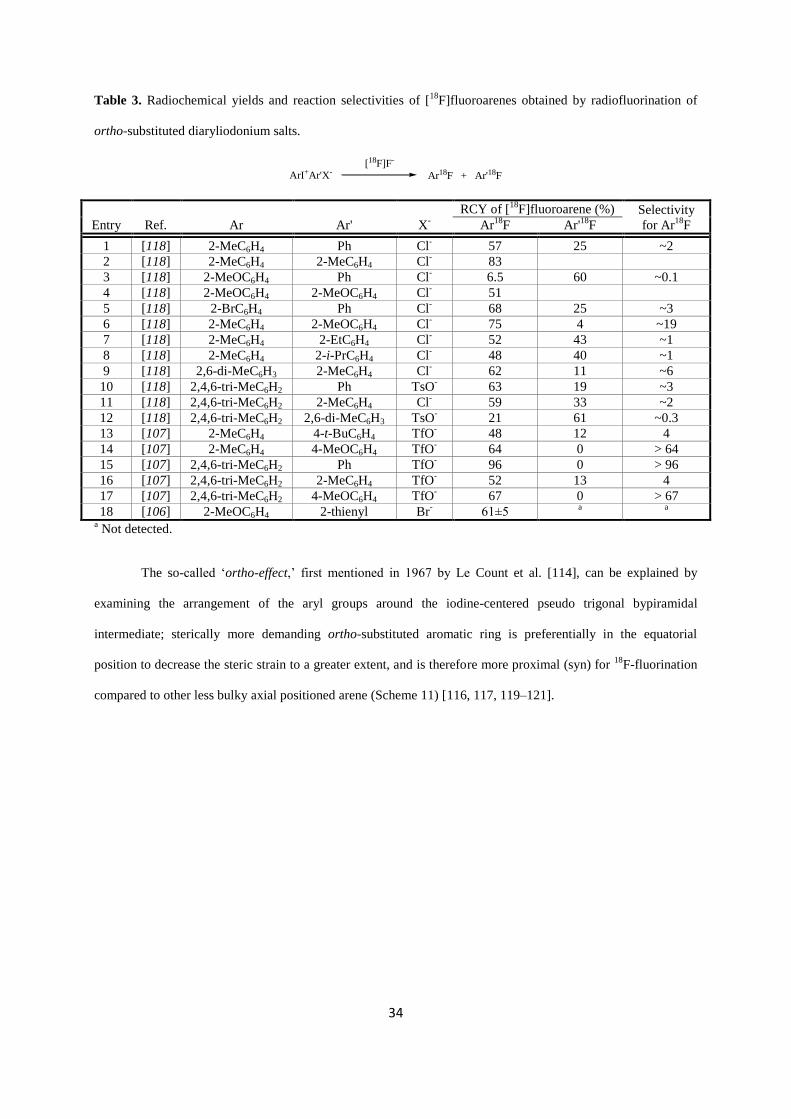
The prominent effect of diaryliodonium
precursors in radiofluorination is the so-called ortho-effect.49–52
Generally higher Ar18
F yields
compared to Balz-Schiemann and Wallach reactions probably arise from the direct vicinity of
[18
F]fluoride to the proximal equatorial aryl ring due to [18
F]fluoride “fixationˮ to the
hypervalent iodine, and because of short-lived nature of the subsequently formed transition
state at elevated temperature all together limit the formation of reactive intermediates (e.g.
aryl cations, aryl radicals).51,53
The principal limitation of the method is the preparation of
highly pure complex precursors suitable for radiolabelling.
N
N
N O
OCH3
I
H3C
O CH3
N
N
N O
OCH3
O CH3
[18F]KF/K222
DMF, 150 oC, 5 min
TsO
67% RCY
[18F]flumazenil
18F
Scheme 8. Preparation of [18
F]flumazenil, chemically indistinguishable from its non-
radioactive counterpart, using 4-methylphenyl-mazenil iodonium tosylate precursor.54
Page 30
16
For more information of n.c.a. nucleophilic 18
F-fluorination of electron-rich arenes see
Chapter 1.3 (pages 18-56): 18
F-labelled Aryl-Tracers through Direct Introduction of
[18
F]fluoride into Electron-Rich Arenes.
1.2.3. Indirect 18
F-labelling reactions
Direct 18
F-fluorination methods are not always suitable or possible for the synthesis of 18
F-
target compound, because either the compound is not sufficiently activated or it can't tolerate
harsh reaction conditions (high reaction temperatures, basic conditions, polar organic
solvents). In these cases indirect introduction of the 18
F radioisotope by reaction with small
18F-labelled reactive precursors or suitable
18F-labelled prosthetic groups under milder
reaction conditions (e.g. room temperature) is used.
Simple mono[18
F]fluoroalkyl halide or sulfonate derivatives with methyl, ethyl, propyl, and
butyl carbon backbones are important synthetic precursors for the introduction of
[18
F]fluoroalkyl groups into complex target molecules, and thus provide alternative synthetic
path to label target biological compounds. These derivatives are prepared by the reaction of
[18
F]fluoride with dihalo or disulfonate alkyl starting materials where the vast excess of the
alkyl starting material compared to [18
F]fluoride
allows exclusive formation of the
mono[18
F]fluoroalkyl halide or sulfonate reagent (Scheme 9).
XX
n = 0-3
[18F]KF/K22218F
X
n = 0-3
Nu18F
Nu
n = 0-3
X = tosylate, Br, I
A.-
B.
N
COOCH3
18F
[18F]LBT-999
HN
COOCH3
CH3
OTs
KI, DMF, 90 oC, 20 min
CH3
18F
Scheme 9. (A.) Synthesis and general reaction of simple [18
F]fluoroalkyl halide/sulfonate
derivatives. (B.) Alternative two-step synthesis of [18
F]LBT-999 using the reactive (E)-
[18
F]fluoro-4-tosyloxybut-2-ene group and the corresponding secondary amine precursor.55
Page 31
17
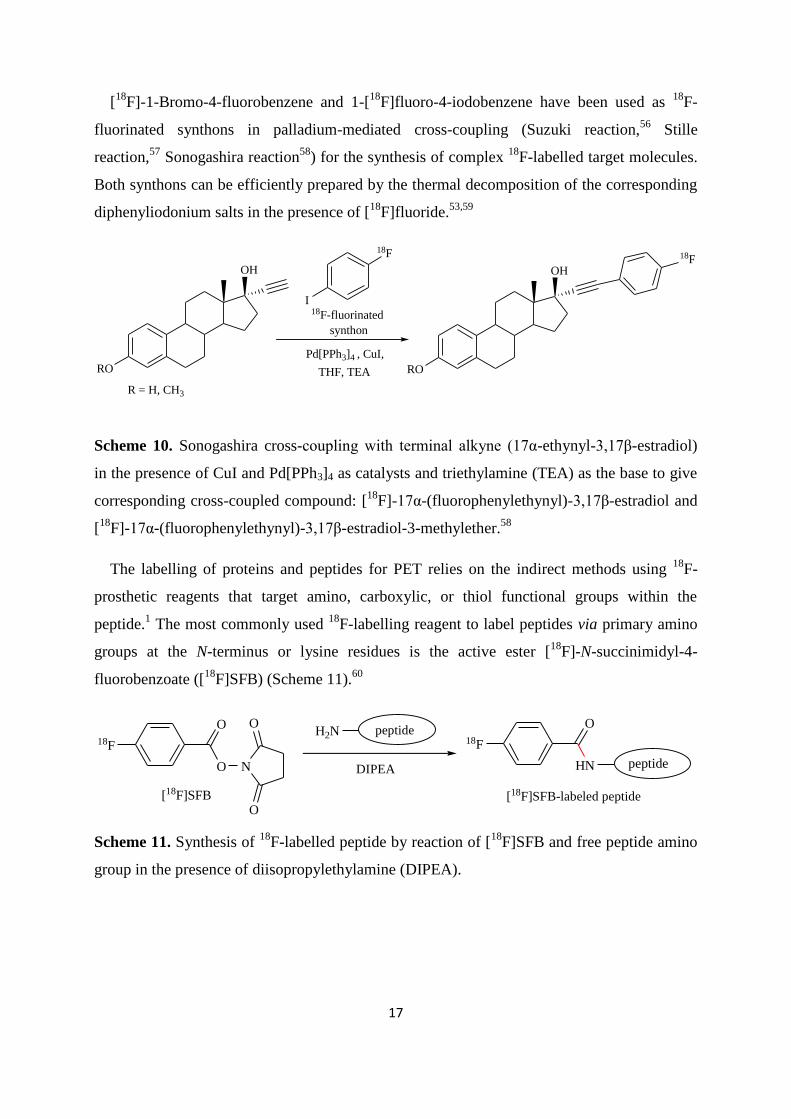
[18
F]-1-Bromo-4-fluorobenzene and 1-[18
F]fluoro-4-iodobenzene have been used as 18
F-
fluorinated synthons in palladium-mediated cross-coupling (Suzuki reaction,56
Stille
reaction,57
Sonogashira reaction58
) for the synthesis of complex 18
F-labelled target molecules.
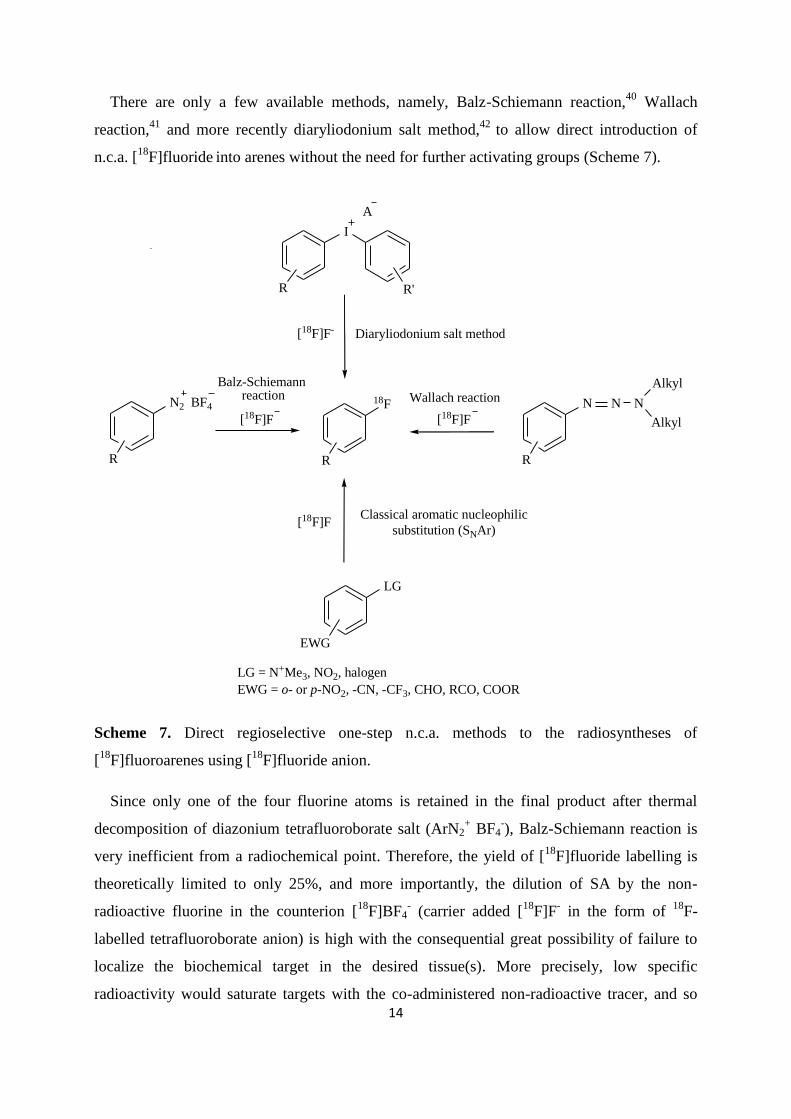
Both synthons can be efficiently prepared by the thermal decomposition of the corresponding
diphenyliodonium salts in the presence of [18
F]fluoride.53,59
18F
I
THF, TEA RO
OH
18F
RO
OH
R = H, CH3
Pd[PPh3]4 , CuI,
18F-fluorinated
synthon
Scheme 10. Sonogashira cross-coupling with terminal alkyne (17α-ethynyl-3,17β-estradiol)
in the presence of CuI and Pd[PPh3]4 as catalysts and triethylamine (TEA) as the base to give
corresponding cross-coupled compound: [18
F]-17α-(fluorophenylethynyl)-3,17β-estradiol and
[18
F]-17α-(fluorophenylethynyl)-3,17β-estradiol-3-methylether.58
The labelling of proteins and peptides for PET relies on the indirect methods using 18
F-
prosthetic reagents that target amino, carboxylic, or thiol functional groups within the
peptide.1 The most commonly used
18F-labelling reagent to label peptides via primary amino
groups at the N-terminus or lysine residues is the active ester [18
F]-N-succinimidyl-4-
fluorobenzoate ([18
F]SFB) (Scheme 11).60
18F
O
O N
O
O[18F]SFB
18F
O
HNDIPEA peptide
[18F]SFB-labeled peptide
H2N peptide
Scheme 11. Synthesis of 18
F-labelled peptide by reaction of [18
F]SFB and free peptide amino
group in the presence of diisopropylethylamine (DIPEA).
Page 32
18
1.3. 18F-labeled Aryl-Tracers through Direct Introduction of
[18
F]fluoride into Electron-Rich Arenes
Mitja Kovača,b
, Sylvie Mavela, Marko Anderluh
b,*
a Université François-Rabelais de Tours, INSERM U930, 37000 Tours, France
b University of Ljubljana, Faculty of Pharmacy, Department of Pharmaceutical Chemistry,
Aškerčeva 7, 1000 Ljubljana, Slovenia
Accepted in: Current Organic Chemistry
Page 33
19
SUMMARY:
Radiolabelling of electron-rich arenes directly with no-carried-added [18
F]fluoride is a
challenging problem for radiochemists specialized in labelling of organic molecules with
fluorine-18. In this review we described critically chemical methods, namely Balz-Schiemann
reaction with its modifications, Wallach reaction, and more recently developed
diaryliodonium salt methodology that are used to label non-activated arenes with no-carried-
added [18
F]fluoride anion regioselectively. The review is focused on diaryliodonium salts,
because they have been shown to be the most suitable precursors for direct single-step
nucleophilic [18
F]fluorination of simple as well as more complex electron-rich arenes
recently.
STATEMENT: I declare, that nobody of co-authors has used the article 18
F-labelled Aryl-
Tracers through Direct Introduction of [18
F]fluoride into Electron-Rich Arenes, which was
submitted in the Current Organic Chemistry, for his/her own thesis.
Page 34
20
ABSTRACT
Rapid and efficient methods using no-carried-added [18
F]fluoride as the source of fluorine-18 for nucleophilic
aromatic fluorination play an important role in the development of new radiopharmaceuticals for positron
emission tomography (PET). Molecules that bear electron-rich aromatic moieties are especially difficult to label
by direct single-step nucleophilic no-carrier-added radiofluorination. Classical Balz-Schiemann reaction with its
modifications, Wallach reaction and diaryliodonium salts methodology are a few methods to enable this. The
present review provides a critical overview of these chemical methods with the emphasis on diaryliodonium salt
as precursors for the direct introduction of [18
F]fluoride into electron-rich arenes in synthesis of 18
F-labeled
molecules for PET scanning.
GRAPHICAL ABSTRACT
18F
R
N2 BF4
R
Balz-Schiemann reaction
N
R
N N
Wallach reaction
Diaryliodonium salt method
I
R R'
A
Alkyl
Alkyl[18F]F-
[18F]F-
[18F]F-
Keywords: aromatic fluorination, arylfluoride, Balz-Schiemann reaction, diaryliodonium salts, 18
F-labeled
molecules, PET, triazene.
Page 35
21
1. INTRODUCTION
Positron emission tomography (PET) is a very powerful non-invasive in vivo molecular imaging
technique that allows visualization, characterization, and quantification of biochemical target function and
physiopathological processes at the cellular or molecular levels even before macroscopic anatomical and clinical
signs of a disease are observed in animal and human subjects [1, 2, 3]. Thus, it is increasingly applied in clinical
research and diagnosis, as well as in drug discovery, development, and therapy [1, 4–6]. Expansion of PET
utility depends on the development and availability of selective and specific positron-emitting radionuclide-
labeled molecular probes for particular biochemical targets or pathways that enable their non-invasive imaging
and quantification in vivo [4, 7, 8]. The development of receptor-specific probes is far from trivial and represents
an important challenge for synthetic and medicinal chemists. Fluorine-18 is the most attractive and favored
radiolabel for in vivo imaging among the available PET radionuclides due to its characteristic physical and
chemical properties [1-3, 9–15]. No-carrier-added (n.c.a.) [18
F]fluoride ([18
F]F-) is nowadays mostly produced
via the proton irradiation of an [18
O]-enriched cyclotron water target, and according to the 18
O(p,n)18
F reaction
[9, 11, 12] which renders the anion, due to its high degree and strength of hydration in aqueous solution, poorly
nucleophilic [16]. Still, a variety of rapid phase-transfer-type protocols have been developed based on trapping
and subsequent elution of [18
F]F- from the anion-exchange resin [17], with the addition of either bulky counter-
cations (e.g. of Bu4N+ HCO3
-) or cryptands (e.g. diazacryptand Kryptofix 2.2.2., K222) with alkali metal salts
(e.g. K2CO3) in order to obtain (after azeotropic drying step(s)) a highly nucleophilic [18
F]F-
system, such as
Bu4N+ [
18F]F
- ([
18F]TBAF) and [
18F]KF/K222 complex [9, 18]. Although completely anhydrous or “naked” [
18F]F
-
reagents are never obtained by these procedures, their degree of dryness are high enough to perform difficult
reactions, such as aromatic nucleophilic substitution in polar aprotic anhydrous organic solvents (e.g. DMSO,
CH3CN, DMF).
A major challenge in PET radiotracer development is to find an efficient and rapid method for n.c.a.
incorporation of cyclotron-produced [18
F]F- into an organic molecule. This may be achieved at aliphatic and
aromatic sites by substitution reactions [9-12]. Labeling at aliphatic carbon atoms using sulfonate ester
(mesylate, tosylate) or halide leaving groups can be accomplished very efficiently, even in the presence of trace
water [19] or in sterically hindered alcohols such as tert-butyl alcohol (t-BuOH) as a protic reaction medium [9,
18, 20]. However, fluorine-18 bound to an aliphatic carbon atom is often prone to de-fluorination in vivo, giving
rise to [18
F]F-, which binds avidly to bone, including the skull, and compromises PET measurements with the
Page 36
22
failure to image specific target in vivo [21–23]. However, attachment of fluorine-18 to an aromatic carbon atom
through a stronger C-F bond compared to fluoroalkyl bond greatly reduces the tendency for radio-de-
fluorination. Consequently, methods for the introduction of fluorine-18 into aromatic ring systems play an
important role in the development of new radiopharmaceutical for PET.
There are principally two common strategies for the direct 18
F-labeling of the arenes: (1) electrophilic;
and (2) nucleophilic 18
F-substitution, among which the latter dominates in importance of researches as will be
discussed in the following sections. Several nucleophilic 18
F-substitution methods to obtain 18
F-labeled aryl
fluorides have been established, evaluated, and applied [2, 3, 9, 11, 12]. As nucleophilic aromatic substitution is
an energetically demanding reaction and not all biological molecules or drug candidates contain a suitably
activated aryl ring for fluorination by the addition-elimination mechanism, the direct incorporation of [18
F]F- into
electron-rich arenes represents a significant challenge in the synthesis of PET tracers. In order to perform
radiofluorination successfully, reproducibly, and in acceptable to high radiochemical yields (RCYs), radiotracers
are usually designed so that the electron-withdrawing group (EWG), such as NO2, CN, CHO, COR, COOR, and
CF3, is easily incorporated in the para and/or ortho position to the good leaving group (LG) (NO2, Halides,
Me3N+
X-; X
- = TfO
-, TsO
-, ClO4
-, I
-) [9, 24–29]. In spite of the presence of EWG, quite high reaction
temperatures are still required for n.c.a. [18
F]-labeling. Synthesis of [18
F]fluoropyridine derivatives proceeds in a
similar fashion since these compounds are reactive toward nucleophilic substitution at the C(2) and C(4)
positions [30]. In some cases, extra steps after labeling have to be performed occasionally to modify or
completely eliminate the activating group on account of a certain loss of overall RCY and specific activity (SA)
[31, 32]. Electron-rich aromatic rings can in principle be more conveniently directly radiolabeled by electrophilic
18F-substitution using [
18F]fluorine gas ([
18F]F2 =
18F-
19F) or less reactive but more selective electrophilic
18F-
fluorination reagents derived from it, such as acetyl [18
F]hypofluorite (CH3COO[18
F]F) [1, 2, 33, 34]. Fluoro-
demetalation reactions using organomercuric or preferably less toxic organostannane precursors afford more
regioselective aromatic 18
F-fluorination with [18
F]F2 and [18
F]CH3COOF as electrophilic radiofluorinating
agents. In this manner some important radiopharmaceuticals such as 6-[18
F]fluoro-L-3,4-dihydroxyphenylalanine
(6-[18
F]fluoro-L-DOPA) [35–37], 2-[18
F]fluoro-L-tyrosine [38] (1R,2S)-4-[18
F]fluorometaraminol [39] have been
prepared. Important consideration of using an organometallic approach is to ensure that there are no residual
amounts of the metals in the final product which would complicate the quality control analysis. However,
electrophilic radiofluorination of organic compounds has several significant shortcomings [1, 2, 11, 12]. Firstly,
Page 37
23
the theoretical maximum achievable RCY can be only 50% because only half the radioactivity of [18
F]F2 can be
utilized for mono-radiofluorination of an organic compound (only one of the fluorine atoms in molecular
[18
F]fluorine carries the 18
F label, the other 50% of the input activity is lost in the form of fluoride) which has not
been realized in practice. Secondly, electrophilic radiofluorination is not applicable to n.c.a. labeling, because
[18
F]F2 is produced along with non-radioactive fluorine gas (19
F2) as a carrier in order to increase the recovering
efficiency of [18
F]F2 from the cyclotron target after its production. Thus, the addition of 19
F2 significantly lowers
(100-1000x) the specific radioactivity (SA) of [18
F]F2 compared to the SA of [18
F]F- even when [
18F]F2 is
generated via 18
O(p,n)18
F reaction [40]. Consequently, the SA of radiotracers prepared by the electrophilic
approach are typically less than 0.4 GBq/μmol (~ 0.011 Ci/μmol) and usually too low for PET investigations of
low density in vivo imaging. High SA also enables radiotracers to be administered to subjects in low mass doses
(1-10 nmol or sub-microgram level) to avoid any toxic or pharmacological effects and perturbation of the
biological target or process [9].
Scheme 1. Direct regioselective n.c.a. methods for the radiosyntheses of [18
F]fluoroarenes using [18
F]fluoride
anion.
Given the above reasons, the ultimate goal is to perform direct regioselective n.c.a. [18
F]F- incorporation
into complex electron-rich arenes as late as possible in the synthetic sequence to obtain a radiotracer of high SA.
Page 38
24
This is a particular challenge in arenes of high electron density, where an electrophilic aromatic carbon or
intermediate should be generated. Only a limited number of available methods proceed via generation of
mentioned electrophilic species, namely, Balz-Schiemann reaction [41], Wallach reaction [42], and more
recently with the use of diaryliodonium salt precursors (Scheme 1) [43, 44]. This review focuses on
radiolabeling strategies applied in the synthesis of 18
F-labeled aryl-tracers from electron rich aryl precursors.
Furthermore, pros and cons of each method are highlighted, and an overview of the successful and most recent
examples with an emphasis on diaryliodonium salt precursors is provided.
2. RADIOFLUORINATION OF ELECTRON-RICH ARNES VIA A BALZ-SCHIEMANN REACTION
Although known for almost a century, the Balz-Schiemann reaction [41] is still the broadest substrate
scope method for the regioselective nucleophilic introduction of fluorine into aromatic ring. This is a
deaminative fluorination type of reaction composed of three sequential steps: (1) diazonitation of primary
aromatic amine in aqueous medium with sodium nitrite (NaNO2) and fluoroboric acid (HBF4) at 0-5oC to
produce arenediazonium tetrafluoroborate (ArN2+BF4
-); (2) isolation and drying of ArN2
+BF4
- to avoid side
formations of phenols and biaryl ethers [45]; and (3) thermal fluorinated decomposition of ArN2+BF4
- (fluoro-de-
diazoniation) [46, 47]. However, this method suffers from yield reproducibility problems because isolation and
complete drying can be tedious and unsafe, and controlled thermal decomposition of ArN2+BF4
- is problematic
[45, 48]. To overcome reproducibility problems, simplify the procedure, broaden substrate tolerance, improve
safety, and to increase the yields, alternative approaches based mostly on one-pot methodology (in situ fluoro-
de-diazoniation) in non-aqueous solvents have been developed during the last few decades [48–56].
Decomposition of aryldiazonium cations can occur by an ionic (heterolytic) pathway via aryl cation
intermediates or by a homolytic pathway that generates aryl radical intermediates which quickly react with
fluoride or any other nucleophile due to their high reactivity and non-selectivity via a SNAr1 type of reaction
mechanism [57–59]. The delicate decomposition pathway balance is crucially dependent on the substituents in
the aromatic ring and reaction conditions. More precisely, substituents and their substitution pattern affect
stability of the aryldiazonium ion, its redox potential, and consequently its decomposition temperature and
pathway [47, 59, 60]. For successful fluoro-de-diazonitation, conditions should be carefully chosen to promote
aryl cation formation. The solvent, pH of the medium, the nature of the counterion, and the presence of reducing
Page 39
25
agents and/or radical sources decisively influence arylfluoride yields. The choice of the solvent is one of the
most important parameters [59, 60] and so to facilitate fluoro-de-diazonitation it should possess the following
properties: (1) it should dissolve all the reagents with minimal solvation of fluoride anion; (2) it should be non-
nucleophilic; (3) it should have suitably high redox potential to avoid reduction of the aryldiazonium ion and
consequently suppress the homolytic decomposition pathway; (4) it should be aprotic; and (5) it should have a
high enough boiling point as radical decomposition pathway is kinetically and thermodynamically favored to an
ionic pathway. Chlorinated solvents (e.g. CCl4) have been reported to have a beneficial effect on arylfluoride
yields via probable enhancement of the ionic decomposition pathway [60]. Selection of the suitable counter-
anion with non-nucleophilic and non-reducing properties is also an important consideration to avoid its
interference with fluorination. Since only one of the four fluorine atoms is retained in the final product after
thermal decomposition of diazonium tetrafluoroborate salt, the Balz-Schiemann reaction is from a radiochemical
point very inefficient (Scheme 2). Therefore, the yield of [18
F]F- labeling is theoretically limited to only 25%,
and more importantly, the dilution of SA by the non-radioactive fluorine in the counterion [18
F]BF4-/18
F-BF3-/
(carrier added 18
F- in the form of
18F-labeled tetrafluoroborate anion) is high with the consequential great
possibility of failure to localize the biochemical target in the desired tissue(s) [61]. Namely, co-administered
non-radioactive tracer would saturate targets by competitive displacement of radiotracer and thereby remove
most of the localized signal from the radiotracer binding. In spite of the described limitations, the Balz-
Schiemann reaction was the first method used in nucleophilic 18
F-labeling of arenes [62] and adapted to prepare
some of the electron-rich 18
F-labeled amino acids, such as 18
F]fluorophenylalanine isomers [63–65], 5- and 6-
[18
F]fluorotryptophan [66] as potential pancreas scanning agents, and 3,4-dihydroxy-5-[18
F]fluorophenylalanine
(5-[18
F]fluoro-DOPA) as a potential brain capillaries scanner [67–71].
Scheme 2. The Balz-Schiemann reaction for the preparation of [18
F]fluoroarenes; 18
F is introduced as 18
F-labeled
tetrafluoroborate anion (carrier-added) via exchanged reaction.
Page 40
26
The syntheses of typical butyrophenone antipsychotic [18
F]haloperidol (1) [70] and antifungal 4-
[18
F]fluconazole (1-2% decay non-corrected yield) [71] were performed by the modified Balz-Schiemann
reaction. Also noteworthy, a 35.5% incorporation of 18
F into the [18
F]haloperidol product from the corresponding
diazonium fluoroborate precursor was reported (Scheme 3) [70]. A higher RCY than the maximal theoretical
yield of 25% might be explained by the fact that 18
F introduced into the arene can come from [18
F]F-
which
exchanges into the fluoroborate salt (carrier-added) as well as coming from unexchanged [18
F]F-
(non-carrier-
added) [66]. Nevertheless, [18
F]haloperidol can be more conveniently prepared with higher SA using a single-
step 18
F-for-LG exchange reaction [72] because the butyrophenone system is moderately activated toward a
direct aromatic nucleophilic substitution.
Scheme 3. Synthesis of [18
F]haloperidol (1) by Balz-Schiemann reaction [70].
Knöchel and Zwernemann investigated several reaction parameters (solvents, reaction temperature and
time, pH of the reaction medium, counter-anions, phase transfer catalysts, and the fluoride sources) influencing
the [18
F]F- fluorination RCYs of p-tolyldiazonium ion used as a model precursor in a modified Balz-Schiemann
reaction [73, 74]. The best result was obtained with p-tolyldiazonium 2,4,6-tri-isopropylbenzenesulfonate in p-
chlorotoluene [74]. Under optimized conditions n.c.a. labeling with [18
F]fluoride gave a decay corrected (EOB)
radiochemical yield of 39% (60 minutes) 4-[18
F]fluorotoluene (2) with a calculated specific radioactivity around
1GBq/μmol in a total synthesis time of 48 minutes (Scheme 4) [74]. In comparison, this method gave
approximately 104
times higher specific radioactivity of 4-[18
F]fluorotoluene than fluoro-de-diazonitation using
[18
F]-BF3 as counter-anion in preparation of [18
F]haloperidol [70]. It should be noted that during optimization
much lower or no fluorination yields of non-radioactive 4-fluorotoluene were obtained using K222/KF and/or 18-
Crown-6/KF complexes of the solubilized fluoride ion. The authors believe that the poor fluorination yields and
slow reaction rates were related to ability of K222 and 18-Crown-6 to complex the aryldiazonium ions [75–77]. In
this regard solubilization of the fluoride was reduced, since parts of the cryptand reacted with aryldiazonium ion
Page 41
27
instead of mobilizing the potassium fluoride, thus enhancing the thermal stability of the complexed
aryldiazonium ion and giving much lower fluorination yields.
N2
CH3
X18F
CH3
110 oC, 30 min
15-Crown-5 / Na18F
4-chlorotoluene
X = SO3
total synthesis time 48 min
39% decay corrected RCY(60 minutes)
SA ~ 1 GBq/mol
dryed2
Scheme 4. No-carrier-added 18
F-labeling of p-tolyldiazonium 2,4,6-triisopropylbenzenesulfonate by a modified
Balz-Schiemann reaction [74].
3. RADIOFLUORINATION OF ELECTRON-RICH ARENES BY WALLACH REACTION
1-Aryl-3,3-dialkyltriazenes (Ar-N=N-NR'R''), compounds having a diazoamino group are regarded as a
protected form of anilines and stable surrogates of aryldiazonium ions [78]. Aryltriazenes are safely and mostly
readily prepared by the coupling of diazotized aniline with amine, or by the action of Grignard reagents on aryl
azides [79], and can be stored for a long period of time in cold temperatures protected from light. They are
adaptable to numerous synthetic transformations with wide applicability in the chemical, medical, and
technological fields [80, 81]. An essential advantage over aryldiazonium ions (Balz-Schiemann reaction) is their
solubility in a number of (anhydrous) organic and ionic solvents that allows in situ generation of the
corresponding aryldiazonium ion upon reacting with acids, and therefore enabling a one-pot fluoro-de-
triazenation reaction [60, 82]. In this respect laborious isolation, drying, and the accumulation of the potential
hazardous and thermally unstable diazonium intermediates is avoided. Moreover, aryltriazenes can be easily
isolated, chromatographically purified, introduced in the early stages of the synthesis, and functionalized and
thermally decomposed in the presence of protic acid in the latest stages if the preceding reactions have been
performed under non-acidic conditions [83, 84]. Thus, fluoro-de-triazenation can be an attractive means of
forming 18
F-labeled fluoroarenes by direct nucleophilic substitution with [18
F]F- due to both the one-pot
Page 42
28
methodology and rapid nature of triazene transformation to fluoroarenes in order to obtain the tracers with good
SA. Nevertheless, the decomposition of aryltriazenes proceeds via diazonium ions and consequently leads to the
same yield and reproducibility problems as (modified) Balz-Schiemann method (Scheme 5).
Pages and Langlois [84] investigated the acidic decomposition of simple, non-activated 1-aryl-3,3-
dialkyltriazenes in the presence of non-radioactive fluoride anions (19
F-) by examining different parameters such
as solvent, acid, fluoride source, stoichiometry, reaction temperature, time, and introduction order of the reaction
components, in order to build a good model for radiofluorination so the radical decomposition pathway could be
suppressed and the ionic pathway maximized. According to their findings, they have successfully
radiofluorinated protected (S)-[18
F]-3-fluoro-α-methylphenylalanine (1) under optimized conditions through
acidic decomposition of the corresponding 1-aryl-3,3-dimethyltriazene precursor with 15 % RCY (decay
corrected (d.c.) RCY , Scheme 6) [85].
Scheme 5. General competitive processes during n.c.a. [18
F]fluoro-de-triazenation and [18
F]fluoro-de-
diazonitation in the protic acid mediated decomposition of 1-aryl-3,3-dialkyltriazenes [84].
Scheme 6. Radiofluorination of protected form (S)-[18
F]-3-fluoro-α-methylphenylalanine (3) through acidic
decomposition of the corresponding 1-aryl-3,3-dimethyltriazene precursor [85].
The choice of the suitable protic acid is an important consideration to transform triazeno moiety via
heterolytic decomposition into a diazonium group in order to obtain arylfluorides in reasonable yields. As the
Page 43
29
phenyl cation is highly reactive and a non-selective species, fluoro-de-triazenation is often accompanied by the
formation of a substantial amount of the acid counterion-substituted byproduct (Ar-A). This is especially true
when the protic acid is used in excess, even if its conjugate base (A-) is considered non-nucleophilic (e.g. triflate
anion) [84–87]. Since PET labeling reactions are performed with nanomolar amounts of [18
F]F-, there is a vast
stoichiometric excess, typically about 103-10
4-fold, of unlabeled precursor and acid [85]. Thus, the use of an acid
constitutes a severe limitation to obtain considerable yields of Ar18
F by fluoro-de-triazenation. The competitive
reactions during fluoro-de-triazenation are the main reason for very few examples of the successful application
of the Wallach reaction in the preparation of complex [18
F]fluoroarenes with RCYs not exceeding 2%: [18
F]-1-
methyl-4-(2-fluorophenyl)-1,2,3,6-tetrahydropyrine (2'-18
F-MPTP) [88], [18
F]haloperidol, and [
18F]spiroperidol
[89].
Recently, Riss et al. [90] reported successful solid phase supported [18
F]fluoro-de-triazenation of 2-
phenoxy-1-(aryldiazenyl)piperazine to afford 1-[18
F]fluoro-2-phenoxybenzene (4) in up to 14% RCYs using
reaction conditions similar to that of Pages et al. (Scheme 7) [85]. It is however questionable whether the solid
phase synthesis offers a significant advantage over the synthesis in solution, since the last method yielded the
18F-labeled product under essentially the same conditions in a higher RCY of 23%.
Scheme 7. (i.) N.c.a. [18
F]fluoro-de-triazenation of solid phase supported 1-(aryldiazenyl)piperazine and (ii.)
''soluble'' n.c.a. [18
F]fluoro-de-triazenation of 1-(aryldiazenyl)piperazine to obtain 1-[18
F]fluoro-2-
phenoxybenzene (4) [90].
Page 44
30
Because of limited RCYs, [18
F]fluoro-de-triazenation is rarely applied for the production of 18
F-labeled tracers
nowadays.
4. RADIOFLUORINATION OF ELECTRON-RICH ARENES BY DIARYLIODONIUM SALTS
Hypervalent iodine compounds have more than eight electrons in their valence shell [91]. The
fundamental feature of these compounds is the highly polarized three-centered-four-electron (3c-4e) bond, in
which the central iodine atom is electron-deficient or bears a positive charge, and monovalent ligands (L) share
the corresponding negative charge. Thus, hypervalent iodine compounds react as electrophiles or/and oxidants at
the iodine center. They belong to two general structural types: (1) iodine (III) compounds A and B, also named
λ3-iodanes; and (2) iodine (V) compounds C and D, termed λ
5-iodanes according to IUPAC nomenclature
(Scheme 8) [91, 92]. In 10-I-3 species the interchange of axial and equatorial ligands via Berry pseudorotation as
well as turnstile rotation is rapid, while such fluxional processes in 12-I-5 species is slower [93–95]. Hypervalent
iodine compounds are used as mild, non-toxic (compare to heavy metals) and selective reagents due to
exploitation of their electrophilic and excellent leaving-group character in a wide range of applications [91, 92,
96–99].
Scheme 8. General oxidation of iodine compounds. Polyvalent iodine compounds differ in the number of
valence electrons surrounding the central iodine atom, the number of ligands and their chemical structure. In
terms of the Martin-Arduengo N-X-L designation/notation [100], 8-I-2 (A) and 10-I-3 (B) species are derivatives
of trivalent iodine and are termed according to IUPAC λ3-iodanes. 10-I-4 (C) and 12-I-5 (D) species are
derivatives of pentavalent iodine and are termed λ5-iodanes (periodanes). According to the hypervalent model
Page 45
31
[101], the apical hypervalent 3c-4e bond in 10-I-3 species is close to linear, longer, and weaker compared to a
regular covalent and equatorial bond and is responsible for high electrophilic reactivity of λ3-iodanes [96].
Scheme 9. Structure and configurations of diaryliodonium salts in solution and in the solid state.
The most common, stable, and well established class among polyvalent iodine compounds are
diaryliodonium salts (ArI+Ar' X
-). Their chemistry, preparative methods, and synthetic applications have been
covered in previous reviews [43, 91, 92, 96]. According to conventional classification, they are defined as diaryl-
λ3-iodanes or as positively charged 8-I-2 species with two aryl ligands and a closely associated negatively
charged counterion. In a solid state, the overall X-ray experimentally determined geometry is pseudo-trigonal
bipyramidal or approximately T-shaped with characteristic 3c-4e linear hypervalent bond (Schemes 8 and 9) [91,
92, 102, 103]. The least electronegative aromatic carbon or the most sterically demanding aryl group and both
electron pairs reside in equatorial positions. On the other hand, the configuration of λ3-iodanes in solution is still
debated, but a certain amount of dissociation is expected depending on the aryl substituents, anion of the salt,
and the type of the solvent used [103]. Diaryliodonium salts are air- and moisture-stable, mild, nontoxic, and
versatile, selective arylating agents delivering one of the aryl moieties to the nucleophile under polar, catalytic,
or photo-chemical conditions. The salt is referred to as a symmetrical salt if R1 = R
2, and as an unsymmetrical
salt if R1 ≠ R
2 (Scheme 9).
When utilizing diaryliodonium salts in reactions with nucleophiles, the nucleophile has a choice of
displacing either of the two aryl groups due to the exceptional leaving group ability of the -IAr fragment, which
has been estimated to be roughly six orders of magnitude greater than that of triflate [104]. In this instance, one
of the more recent application areas of diaryliodonium salts, firstly introduced in 18
F-radiochemistry by Pike and
Page 46
32
Aigbirhio in 1995 [105], is their use as suitable precursors for the preparation of n.c.a. 18
F-labeled arenes as
potential radiotracers for PET imaging. Diaryliodonium salts allow direct introduction of [18
F]fluoride ion in a
single step into aromatic systems without the need for further activating groups and with little or no restriction
on the nature of the functionality present [9]. Thus, they gain more and more interest for direct radiofluorination
of otherwise unfavorable electron-rich arenes. At this, symmetric diaryliodonium salts are generally preferred
over unsymmetrical salts as no regioselectivity problems arise (Scheme 10). However, the use of unsymmetrical
salts is in some situations desirable and necessary, such as when the starting materials are expensive or if one of
the aryl substituent is very complex.
Scheme 10. Regioselectivity problems of direct n.c.a. [18
F]-fluorination of unsymmetrical diarylidonium salts.
Table 1. Synthesis of simple n.c.a. [18
F]fluoroarenes via nucleophilic displacement of corresponding 4-
substituted diaryliodonium salt by n.c.a. [18
F]fluoride. Selectivity for the product 6 increases with relative
increase of electron density on the partner para-substituted phenyl ring.
Compound Ref. R R' X-
RCY 5 (%) RCY 6 (%) Selectivity for 6
5a/6a [105]
CH3 H TfO-
~13a
~26a
2
5b/6b [105]
OCH3 H Br
- b ~62
a > 62
5c/6c [105] OCH3 4-OCH3 CF3CO2-
~55a
5d/6d [107] OCH3 H TfO- 0 96 > 96
5e/6e [107] t-BuO H TfO- 0 95 > 95
5f/6f [107] OCH3 3-CH3 TfO- 0 66 > 66
5g/6g [28] OCH3 3-Br TfO-
3±1 43±5 ~ 14
5h/6h [108] CH3 H TsO-
10 43 ~ 4
5i/6i [108] OCH3 H TsO-
~3a ~77
a ~ 26
5j/6j [108] OCH3 4-CH3 TsO- 9 36 4
5k/6k [108] OCH3 4-Cl TsO- 6 80 ~ 13
5l/6l [108] OCH3 4-OCH3 TsO
- 90
5m/6m [109]
OCH3 3-CN TsO
- 11 82 ~ 7
5n/6n [109]
OCH3 3-NO2 TsO
- 7 58 ~ 8
5o/6o [109]
OCH3 3-CF3 TsO
- < 1 53 > 53
a Average RCY,
b Not detected
Page 47
33
The outcome and regioselectivity of the fluorination has been shown to be strongly dependent on three
parameters: (1) the electronic density (inductive and resonance effects of substituent groups on the aryl ring); (2)
the substitution pattern; and (3) the steric structure of the diaryliodonium precursor [106]. Following the trend of
SNAr, the n.c.a. [18
F]fluoride attack occurs preferably on the most electron-deficient arene in the absence of an
ortho effect [9]. The strategy is therefore to make one of the aryl rings more electron-rich. Iodonium salts
containing p-methoxyphenyl (p-anisyl) (Table 1) [28, 105, 107–109] and 2-thienyl (Table 2) [106, 109–112] as
highly electron rich arenes were found to lead to highly regioselective nucleophilic 18
F-fluorination of the partner
aryl ring.
Table 2. Radiochemical yields of n.c.a. 18
F-fluorination of 4-phenyl(2-thienyl)iodonium bromides to obtain
simple electron-rich and electron-poor 4-substituted 18
F-fluoroarenes [106].
R H CH3 OCH3 OBn Cl Br I
RCY(%) 64 ± 4 32 ± 2 29 ± 3 36 ± 3 62 ± 4 70 ± 5 60 ± 8
Carroll et al. [113] have performed fluorination of simple 2-thienyliodonium salts and found that the
process is not selective, and the 2-thienyl group, although highly electron-rich, may not be the ideal non-
participating ring for production of fluoroarenes by the fluorination of diaryliodonium salts as had been
described the same year by Ross et al. [106]. They concluded that it is the analysis, characterization, and
isolation of 2-fluorothiophene that may be (extremely) problematic. Iodonium compounds with bulky aryl rings
tend to undergo nucleophilic substitution on the bulky ring. However, the bulkiness alone is not an exclusive
factor for stereoselective fluorination. The decisive factor is one (hydrophobic) group (e.g. methyl) at the ortho-
position that shows a directing steric effect and induces an attack in the ortho-substituted aromatic ring [114–
117], even though it is more electron-rich than the partner ring (Table 3, Entries 1, 3, 9-11, 15-16) [107, 118].
This preference increases further along with RCYs for doubly ortho- and/or more alkyl/methyl-substituted aryl
rings in spite of increasing steric hindrance and electronic deactivation (Table 3, Entries 9-11, 15-17) [107, 118–
120].
Page 48
34
Table 3. Radiochemical yields and reaction selectivities of [18
F]fluoroarenes obtained by radiofluorination of
ortho-substituted diaryliodonium salts.
ArI+Ar'X- [18F]F-
Ar18F + Ar'18F
RCY of [18
F]fluoroarene (%) Selectivity
for Ar18
F Entry Ref. Ar Ar' X- Ar
18F Ar'
18F
1 [118] 2-MeC6H4 Ph Cl-
57 25 ~2
2 [118] 2-MeC6H4 2-MeC6H4 Cl- 83
3 [118] 2-MeOC6H4 Ph Cl- 6.5 60 ~0.1
4 [118] 2-MeOC6H4 2-MeOC6H4 Cl- 51
5 [118] 2-BrC6H4 Ph Cl- 68 25 ~3
6 [118] 2-MeC6H4 2-MeOC6H4 Cl-
75 4 ~19
7 [118] 2-MeC6H4 2-EtC6H4 Cl- 52 43 ~1
8 [118] 2-MeC6H4 2-i-PrC6H4 Cl- 48 40 ~1
9 [118] 2,6-di-MeC6H3 2-MeC6H4 Cl- 62 11 ~6
10 [118] 2,4,6-tri-MeC6H2 Ph TsO- 63 19 ~3
11 [118] 2,4,6-tri-MeC6H2 2-MeC6H4 Cl- 59 33 ~2
12 [118] 2,4,6-tri-MeC6H2 2,6-di-MeC6H3 TsO- 21 61 ~0.3
13 [107] 2-MeC6H4 4-t-BuC6H4 TfO-
48 12 4
14 [107] 2-MeC6H4 4-MeOC6H4 TfO- 64 0 > 64
15 [107] 2,4,6-tri-MeC6H2 Ph TfO-
96 0 > 96
16 [107] 2,4,6-tri-MeC6H2 2-MeC6H4 TfO-
52 13 4
17 [107] 2,4,6-tri-MeC6H2 4-MeOC6H4 TfO- 67 0 > 67
18 [106] 2-MeOC6H4 2-thienyl Br-
61±5 a a
a Not detected.
The so-called ‘ortho-effect,’ first mentioned in 1967 by Le Count et al. [114], can be explained by
examining the arrangement of the aryl groups around the iodine-centered pseudo trigonal bypiramidal
intermediate; sterically more demanding ortho-substituted aromatic ring is preferentially in the equatorial
position to decrease the steric strain to a greater extent, and is therefore more proximal (syn) for 18
F-fluorination
compared to other less bulky axial positioned arene (Scheme 11) [116, 117, 119–121].
Page 49
35
Scheme 11. Suggested outline mechanism for the radiofluorination of an unsymmetrical substituted
diaryliodonium salt through transition states TS1 and TS2 to give 18
F-labeled products P1 and P2, respectively
[118].
As studied and rationalized by Chun et al. [118] the electronic features of the ortho-substituent and transition
state stabilities are important in determining product selectivity, because the ortho-effect is not purely related to
substituent bulkiness (Table 3, Entries 3, 6-8). Opposing (Table 3, Entry 3) and reinforcing (Table 3, Entry 5)
electronic properties of ortho substituent appeared to be important in determining product selectivity. Thus,
when both of the ortho substituents are of similar effective bulkiness, [18
F]fluoride together with the more
electron-deficient ligand would eliminate in order to decrease the positive charge on the iodine atom (Table 3,
Entries 6-8). They suggested that ortho-hydrophobic groups create lipophilic micro-environment in which the
incoming [18
F]fluoride can act as a powerful nucleophile (even in moderately hydrated state), by first loosely
binding to the hypervalent iodine atom and then attacking the locally lipophilic ortho-substituted ring (Scheme
11). They also found evidence that the reactions of [18
F]fluoride with unsymmetrical diaryliodonium salt (2-
Page 50
36
methylphenyl)(phenyl)iodonium chloride) comply with the Curtin-Hammett principle [118, 122]. Accordingly,
they proposed that radiofluorination of an unsymmetrical diaryliodonium salt involves an attack of [18
F]fluoride
onto either of the two rapidly interconverting conformers [94, 95] relative to the rate of product formation,
yielding two transition states (TS1 and TS2) that each give a single radiofluorinated product (P1, P2), whereas
the products do not undergo interconversion (Scheme 11). Consequently, the ratio of radiofluorinated products
(P1/P2) was not in direct proportion to the relative concentrations of the conformational isomers in the
substrate, but is dependent only on the difference in standard free energies of the respective transition states
(P1/P2=e-(GTs1-GTs2)/RT
), which is relatively small compared to the activation energies for the same reactions. In
other words, P1/P2 will increase with increasing difference between free energies of the respective transition
states. As depicted in the Scheme 11, generally higher Ar18
F yields compared to Balz-Schiemann and Wallach
reactions probably arise from a [18
F]fluoride preferential attack on the proximal equatorial aryl ring, and
subsequent decomposition of “trigonal” transition state which limits the formation of reactive intermediates, e.g.
aryl cations or aryl radicals. This might explain why even the addition of water is not detrimental for successful
radiofluorination [118, 123–125]. Other mechanisms for reactions of diaryliodonium salts with [18
F]fluoride
have also been proposed, such as ʽturnstileʼ mechanism by Grushin [117] and SNAr by Ross et al. [106] on the
basis of a reasonable good linear fit between reaction rates of para-substituted aryl(2-thienyl)iodonium bromides
and Hammett substituent constants. The application of the Hammett constants failed for the ortho-OMe
substituted precursor, as claimed by Authors, due to the strong ortho-effect which could not be taken into
account by that approach. Meta-derivatives are known to be electron-rich in comparison to corresponding para-
and ortho-derivatives, and are therefore more problematic for nucleophilic fluorination. Unsymmetrical
diaryliodonium salts have also been shown to be effective precursors to obtain simple 3-[18
F]fluoroarenes
bearing meta electron-withdrawing or meta electron-donating substituents [109, 112, 126] 3-
[18
F]fluoroheteroarenes [127] in moderate to high RCYs. Also noteworthy is that pyridyliodonium salts are the
most appropriate precursors for the preparation of fluorine-18 labeled 3-fluoropyridine that is more stable in
vivo, but less easily available via conventional SNAr reaction than 2-fluoro and 4-fluoropyridines. The use of 4-
methoxyphenyl moiety as a partner aromatic ring in 4-methoxyphenyl(3-pyridine) iodonium salt gave 3-
[18
F]fluoropyridine in radiochemical yields of about 60%. The same approach has been employed to yield 3-
[18
F]fluoroquinoline in about 25% RCY via a 4-methoxyphenyl(3-quinoline) iodonium salt precursor [127].
Page 51
37
Besides the substitution pattern and electronic properties, parameters such as solvent, counter-anion(s),
[18
F]fluoride source, stoichiometry, reaction temperature, and time, strongly affect the outcome (product
distribution) and RCYs of nucleophilic 18
F-labeling reaction with diaryliodonium salts. An appropriate solvent
should be non-nucleophilic with weak power of cation and anion solvation, and with suitable redox potential to
exclude or limit any redox processes between iodine (III) and solvent molecules [28, 106, 128]. Organic aprotic
solvents like CH3CN and DMF have appeared to be the most beneficial, according to the RCYs in contrast to
DMSO which is very useful for direct SNAr. To avoid the homolytic aryl-iodine bond fission [117], and
consequently to improve RCYs and reproducibility, addition of radical scavengers (e.g. TEMPO) has been
shown to be advantageous [125, 129–131]. According counter-anionʼs influence on RCYs, stability, reaction
rates, and selectivity, inorganic and organic counter-anions such as bromide [105, 106], tosylate (TsO-) [108,
109, 124, 125, 131, 132], and triflate (TfO-) [105, 107] have appeared attractive considering their low
nucleophilicity and good leaving group ability. Lee et al. determined X-ray structure of a representative
unsymmetrical iodonium salt, 2-methylphenyl(2ʼ-methoxy-phenyl)iodonium chloride (7), in order to achieve a
deeper understanding of its structure, and to assist in understanding radiofluorination mechanism and similar
reactions of diaryliodonium salts with nucleophiles in organic media [133]. Their X-ray study unveiled that the
hypervalent iodine in 7 acts as a previously unrecognized stereogenic center within a dimeric structure as the unit
cell in a centrosymmetric crystal, composed of conformational M and P enantiomers (Scheme 12). They
investigated racemization process of 7 in CH3CN solution with the ab initio replica path method, thereby
revealing two additional pairs of conformational enantiomers. All identified six conformers of 7 were calculated
to be comparable in energy and thus, all are likely to exist in CH3CN at room temperature due to fast
interconversion via two essentially isoenergetic transition states (calculate energy barrier of 9,1 kcal/mol in
CH3CN). In addition, their quantum chemical and dimerization energy calculations together with LC-MS
observations of clusters of 7 suggested that it predominantly exists as dimers (dimeric anion-bridge clusters) in
CH3CN due to the secondary bonding interaction between I and the Cl atoms within an enantiomeric pair. The
evidence of the existence of dimeric solution clusters of 7 further indicates that the reactions of diaryliodonium
salts similar to 7 with nucleophiles (e.g. [18
F]F-) in organic solvents may require dissociation of dimers or
possibly even higher-ordered clusters (e.g. tetramers), preceding replacement of chloride ion with [18
F]F- and
subsequent attack of the bound fluoride onto an aryl carbon atom to give either of the two possible
[18
F]fluorarene products (Scheme 12).
Page 52
38
I
Cl
O
I
Cl
O
M
[18F]F-
disssociation
I
Ar
Ar'
Cl
I
Cl
Ar
Ar'
Cl-bridged dimer in organic solvent(square planar configuration)
ArI+Ar'Cl-
Ar18F/Ar'18F + Ar'I/Ar
P
i ii
7
Scheme 12. (i.) M and P are conformational enantiomers of 2-methylphenyl(2ʼ-methoxy-phenyl)iodonium
chloride (7). (ii.) Favorable dimerization energy calculation and LC-MS observations of clusters suggest that
dissociation of dimeric Cl-bridge cluster, held together by iodine-chloride ionic bonds, of diaryliodonium salt
similar to 7 is preceding necessary step to allow formation of the two possible [18
F]fluorarene products [133].
So far, diaryliodonium salts have mainly been proven to be useful precursors for the introduction of
[18
F]F- onto simple aromatic rings via straightforwardly prepared precursors where comparison with the Wallach
reaction has shown much greater efficiency for the former methodology [28, 134]. It was assumed that the 18
F-
labeling via iodonium precursors is somehow limited by the molecule size and complexity of the structure [120].
With further mechanistic, stability, permutational, and intramolecular interference studies, and also with an
improvement of existent and development of new synthetic methods to obtain stable and highly pure complex
diaryliodonium precursors, this approach will likely find wider application in preparing more complex 18
F-
labeled tracers for PET imaging. More complex 18
F-labeled radiopharmaceuticals from corresponding
diaryliodonium salts are represented in Schemes 13 and 14.
Page 53
39
Scheme 13. Successful examples of 18
F-labeling using complex diaryliodonium precursors. (i.) Radiosynthesis
of metabolically stable 4-[18
F]fluorophenyl pyrazolo steroid (9) as high affinity ligand for brain glucocorticoid
receptors [132]. (ii.) Radiosynthesis of 11 ([18
F]F-ADTQ), classified as non-competitive AMPA receptor
antagonist [120].
Radiosynthesis of metabolically stable 4-[18
F]fluorophenyl pyrazolo steroid 9 as high affinity ligand for brain
glucocorticoid receptors was accomplished by Wüst et al. [132]. Since the aromatic ring is not sufficiently
activated by a strong electron-withdrawing group, diaryliodonium salts 8a and 8b were used as precursors for the
incorporation of n.c.a. [18
F]F-
to obtain 9 in low decay-corrected radiochemical yields of 0.2 and 2.0%,
respectively. The use of the more electron-donating tolyl-functionalized iodonium salt 8b favored the formation
of corticosteroid 9 which is in accordance of the para-substituted electronic effects of the counter rings. Authors
Page 54
40
also detected by radio-TLC analysis the formation of large amounts of [18
F]fluorobenzene and [18
F]fluorotoluene
as by-products. Ross reported radiosynthesis of 11 ([18
F]F-ADTQ), classified as a putative, non-competitive
AMPA receptor antagonist, from iodonium precursors 10a, 10b, and 10c [120]. Comparing the precursors 10a
and 10b, the RCYs showed an increase from the phenyliodonium group (1.2 %) to the 2-thienyliodonium group
(2.9 %), as expected for the electronic differences between the iodonium precursors and the corresponding aryl
iodides as leaving groups. It should be noted that the synthesis starting from iodonium precursor 10c with a
bromide as a counter-anion showed the best RCY of about 3.6%, and was obtained in only 3% yield.
Consequently, 11 was not isolated or prepared for pharmacological evaluation studies because of a very low
RCY.
Preparation of 13 ([18
F]DAA1106) by Zhang et al., a PET ligand for imaging peripheral-type
benzodiazepine receptor in the brain, was accomplished in much higher d.c. RCY of 46 % than in previous noted
attempts and was therefore the first report of a complex and electron rich practical PET ligand synthesized by the
reaction of diphenyliodonium salt with n.c.a. [18
F]F- in high RCY [108]. Based on consideration that the
[18
F]fluoride attacks the diphenyliodonium salt preferably at the electron-deficient benzene ring, p-iodoanisole as
a leaving group was designed to increase the regioselectivity of 18
F into a desired ring. Since 12 was unstable, it
was used for radiosynthesis after the respective coupling reaction without further purification. Another
successful radiofluorination in high RCY via iodonium salts is the preparation of [18
F]flumazenil (15) using 4-
methylphenyl-mazenil iodonium tosylate precursor 14 without any structural modifications of the parent
molecule [131]. Interestingly, 14 was superior to other precursors, in spite of the fact that 2-thienyl-, 3- thienyl-,
and 4-methoxyphenyl-mazenil iodonium tosylate have relatively high electron densities. The authors observed
that the more electron-rich diaryliodonium tosylate precursors have lower stability and selectivity for the desired
product formation and that these correspond with the tendency of the [18
F]fluoride incorporation yield. Thus, the
best result was obtained for the reaction of 14 with n.c.a. [18
F]fluoride and K222/K2CO3 (0.6 equiv. of K2CO3
relative to the precursor) in the presence of TEMPO in DMF at 150 °C for 5 min. Under these conditions, d.c.
RYC was 67 ± 2.7 % with more than 99% radiochemical purity after HPLC purification. The total synthesis time
for 15 was about 55 min, including HPLC purification and the specific activity was in the range of 370-450
GBq/μmol (10-12 Ci/μmol). Further studies showed that the optimized reaction conditions were well adapted to
the reproducible (n = 26 with no failure) high-scale automatic production of [18
F]flumazenil (RCY 63.5 ± 3.2 %
in total synthesis time 60 ± 1.1 min) in a commercial automated device (Scheme 14).
Page 55
41
Scheme 14. Successful examples of 18
F-labeling using complex diaryliodonium precursors. (i.) Preparation of 13
([18
F]DAA1106), a PET ligand for imaging peripheral-type benzodiazepine receptor in the brain [108]. (ii.)
Preparation of 15 ([18
F]flumazenil), chemically indistinguishable from its non-radioactive counterpart, using 4-
methylphenyl-mazenil iodonium tosylate precursor 14 [131]. (iii.) Radiochemical synthesis of benzophenone-
tyrosine PPARγ ligand 17 [124]. (iv.) Radiosynthesis of 6-[18
F]fluoro-labeled benzothiazole analogue 19 as a
promising PET probe for Aβ plaque imaging [125].
Page 56
42
Lee et al. developed radiochemical synthesis of 18
F-labeled analog of the potent and selective PPARγ
agonist farglitazar (17), a tyrosyne-benzophenone class of PPARγ regulators) by radiofluorination of a
diaryliodonium tosylate precursors 16a and 16b [124]. The radiosynthesis of 17 was accomplished in
approximately 90 minutes with a good d.c. RCY of up to 42% and the SA after decay correction of
approximately 37 GBq/μmol (1 Ci/μmol). Authors concluded that although the compound had high and selective
PPARγ binding affinities and also good metabolic stability, its nonselective target-tissue (brown and/or white
fat) biodistribution uptake versus non-target tissues in rats indicated that it was likely to be unspecific for
effective imaging of breast cancer or vascular disease in humans. It should be noted that the addition of some
water significantly increased RCYs as suggested due to increased solubility of Cs[18
F]F- salt in the studied
solvents (DMF and CH3CN). It is also interesting to note that they were unable to obtain radiolabeled product 17
from the p-methoxyphenyl-based iodonium salt precursor, even though this precursor worked quite well to
produce the unlabeled fluoroproduct [124, 135]. Similarly, the same authors (Lee et al.) have recently
synthesized various diaryliodonium tosylate precursors (containing the more electron-rich p-methoxyphenyl, p-
methylphenyl, 2- and 3-thienyl compare to phenyl ring on 6-position of benzothiazole ring) to allow aromatic
18F-labeling at 6-position of benzothiazole ring [125]. The highest d.c. RYC 40.5% with SA 110 GBq/μmol
(2.97 Ci/μmol) of one of the most promising Aß plaque-specific PET imaging probes 19 was obtained via one-
pot radiofluorination and deprotection of benzothiazole iodonium tosylate precursor 18 in the presence of
TEMPO using nBu4N+[
18F]fluoride as the fluoride source.
5. CONCLUSIONS
A direct, one-step, no-carrier-added synthesis of inactivated or electron-rich [18
F]fluoroaromatic
compounds with high specific activity represent an important challenge for radiochemists. This is limited to only
a few methods in preparative organic chemistry. The traditional Balz-Schiemann reaction with its modification
and Wallach reaction are not particularly efficient due to the competing reactions of the highly reactive and non-
selective intermediates leading to low radiochemical yields and specific activities of 18
F-labeled arenes. On the
other hand, diaryliodonium salts have been shown to be the most suitable precursors for direct single-step
nucleophilic [18
F]fluorination of simple arenes with little or no restriction on the nature of the functionality
present. The regioselectivity of this reaction has been found to be controlled electronically as well as by the
steric bulkiness of the substituents. The so-called ortho-effect is the most prominent feature of this methodology
Page 57
43
and almost quantitatively radiochemical yields of small electron-rich arenes have been reported due to this
effect. Higher radiochemical yields compared to Balz-Schiemann and Wallach reactions have also been
explained by the synchronous reductive elimination of the transition state which limits formation of reactive
intermediates. The principal drawback of this method is that radiochemical yield is limited by the additional
formation of undesirable counter [18
F]fluoroarene. Thus, to improve the radiochemical yield, symmetrically
substituted diaryliodonium salts as precursors are preferred, which is not the case for complex arenes. So far, this
approach has been limited for simple readily prepared diaryliodonium salts. However, very promising for future
investigation are a few recent successful examples of direct aromatic nucleophilic 18
F-labeling of complex non-
activated radiopharmaceuticals using corresponding diaryliodonium salts as precursors. With further mechanistic
studies and with an improvement of existent synthetic methods and development of new to obtain stable and
highly pure complex diaryliodonium precursors, this approach will likely find wider application in
radiofluorination.
List of abbreviations: AMPA - 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid, EWG - electron-
withdrawing group, LG - leaving group, N.C.A. - no-carrier-added, PET - positron emission tomography, PPAR
- peroxisome proliferator-activated receptor, RCY - radiochemical yield, SA - specific activity, TEMPO -
2,2,6,6-tetramethylpiperidin-1-yl)oxyl.
ACKNOWLEDGEMENTS
This work was supported by Egide for graduate grant (PHC PROTEUS, 2012, n°26502QF). The authors would
also like to acknowledge Slovenian Research Agency for financial support of Slovenian-French bilateral
collaboration (project n° BI-FR/12-13-PROTEUS-007).
REFERENCES
[1] Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular Imaging with PET. Chem. Rev., 2008, 108(5), 1501-
1516.
[2] Antoni, G.; Långström, B. Radiopharmaceuticals: molecular imaging using positron emission tomography.
Handb. Exp. Pharmacol., 2008, 185, 177-201.
Page 58
44
[3] Dollé, F.; Roeda, D.; Kuhnast, B.; Lasne, M.-C. In: Fluorine and Health, Molecular Imaging, Biomedical
Materials and Pharmaceuticals; Alain Tressaud and Günter Haufe Eds.; Elsevier Science B. V: Amsterdam,
2008; pp. 3-65.
[4] Paans, A.M.J.; Vaalburg, W. Positron Emission Tomography in Drug Development and Drug Evaluation.
Curr. Pharm. Des., 2000, 6(16), 1583-1591.
[5] Lin, M.; Shon, I.H.; Lin, P. Positron Emission Tomography: Current Status and Future Challenges. Intern.
Med. J., 2010, 40(1), 19-29.
[6] Zimmer, L.; Luxen, A. PET Radiotracers for Molecular Imaging in the Brain: Past, Present and Future.
NeuroImage, 2012, 61(2), 363-370.
[7] Lee, C.-M.; Farde, L. Using Positron Emission Tomography to Facilitate CNS Drug Development. Trends
Pharmacol. Sci., 2006, 27(6), 310-316.
[8] Vallabhajosula, S. 18F-Labeled Positron Emission Tomographic Radiopharmaceuticals in Oncology: An
Overview of Radiochemistry and Mechanisms of Tumor Localization. Semin. Nuc. Med., 2007, 37(6), 400-419.
[9] Cai, L.; Lu, S.; Pike, V.W. Chemistry with [18
F]Fluoride Ion. Eur. J. Org. Chem., 2008, 2008(17), 2853-
2873.
[10] Cai, L.; Lu, S.; Pike, V.W. Chemistry with [18
F]Fluoride Ion. Eur. J. Org. Chem., 2008, 2008(17), 2843-
2848.
[11] Coenen, H.H. Fluorine-18 labelling methods: features and possibility of basic reactions. In: PET
Chemistry, The Driving Force in Molecular Imaging; P. A. Schubiger, L. Lehmann, M. Friebe, Eds.; Springer-
Verlag: Berlin Heidelberg, 2007; pp. 15-50.
[12] Kilburn, M.R.; Huizenga J.R. Fluorine-18 labelling of radiopharmarmaceuticals; Nuclear Science Series,
Natural Academic Press, Washington, DC, 1990.
[13] O’Hagan, D. Understanding Organofluorine Chemistry. An Introduction to the C-F Bond. Chem. Soc. Rev.,
2008, 37, 308-319.
Page 59
45
[14] Zhou, P.; Zou, J.; Tian, F.; Shang, Z. Fluorine Bonding - How Does it Work in Protein-Ligand Interactions?
J. Chem. Inf. Model., 2009, 49(10), 2344-2355.
[15] Shah, P.; Westwell, A.D. The Role of Fluorine in Medicinal Chemistry. J. Enzyme Inhib. Med. Chem.,
2007, 22(5), 527-540.
[16] Kemp, D.D.; Gordon, M.S. Theoretical Study of the Solvation of Fluorine and Chlorine Anions by Water. J.
Phys. Chem. A, 2005, 109(34), 7688-7699.
[17] Schlyer, D.J.; Bastos, M.A.V.; Alexoff, D.; Wolf, A.P. Separation of [18F]Fluoride from [18O]Water Using
Anion Exchange Resin. Int. J. Rad. Appl. Instrum. A., 1990, 41(6), 531-533.
[18] Kim, D.W.; Jeong, H.J.; Lim, S.T.; Sohn, M.-H. Recent Trends in the Nucleophilic [18F]-radiolabeling
Method with No-carrier-added [18F]fluoride. Nucl. Med. Mol. Imaging, 2010, 44(1), 25-32.
[19] Briard, E.; Pike, V.W. Substitution-reduction: an Alternative Process for the [18F]N-(2-fluoroethylation) of
Anilines. J. Labelled Compd. Radiopharm., 2004, 47(4), 217-232.
[20] Kim, D.W.; Ahn, D.-S.; Oh, Y.-H.; Lee, S.; Kil, H.S.; Oh, S.J.; Lee, S.J.; Kim, J.S.; Ryu, J.S.; Moon, D.H.;
Chi, D.Y. A New Class of SN2 Reactions Catalyzed by Protic Solvents: Facile Fluorination for Isotopic
Labeling of Diagnostic Molecules. J. Am. Chem. Soc., 2006, 128(50), 16394-16397.
[21] Pike, V.W. PET Radiotracers: Crossing the Blood-Brain Barrier and Surviving Metabolism. Trends
Pharmacol. Sci., 2009, 30(8), 431-440.
[22] Baumann, C.A.; Mu, L.; Wertli, N.; Krämer, S.D.; Honer, M.; Schubiger, P.A.; Ametamey, S.M. Syntheses
and Pharmacological Characterization of Novel Thiazole Derivatives as Potential mGluR5 PET Ligands. Bioorg.
Med. Chem., 2010, 18(16), 6044-6054.
[23] Al Hussainy, R.; Verbeek, J.; van der Born, D.; Molthoff, C.; Booij, J.; Herscheid, J.D.M. Synthesis,
Biodistribution and PET Studies in Rats of 18F-Labeled Bridgehead Fluoromethyl Analogues of WAY-100635.
Nucl. Med. Biol., 2012, 39(7), 1068-1076.
[24] Attiná, M.; Cacace, F.; Wolf, A.P. Displacement of a Nitro-Group by [18F] fluoride ion. A New Route to
Aryl Flourides of High Specific Activity. J. Chem. Soc., Chem. Commun., 1983, 108-109.
Page 60
46
[25] Angelini, G.; Speranza, M.; Wolf, A.P.; Shiue, C.-Y. Nucleophilic Aromatic Substitution of Activated
Cationic Groups by 18F-labeled Fluoride. A Useful Route to No-Carrier-Added (NCA) 18F-labeled Aryl
Fluorides. J. Fluorine Chem., 1985, 27(2), 177-191.
[26] Haka, M.S.; Kilbourn, M.R.; Watkins, G.L.; Toorongian, S.A. Aryltrimethylammonium
Trifluoromethanesulfonates as Precursors to Aryl [18F]Fluorides: Improved Synthesis of [18F]GBR-13119. J.
Labelled Compd. Radiopharm., 1989, 27(7), 823-833.
[27] Karramkam, M.; Hinnen, F.; Bramoullé, Y.; Jubeau, S.; Dollé, F. Ortho-[18F]Fluoronitrobenzenes by No-
Carrier-Added Nucleophilic Aromatic Substitution with K[18F]F-K222-A Comparative Study. J. Labelled
Compd. Radiopharm., 2002, 45(13), 1103-1113.
[28] Ermert, J.; Hocke, C.; Ludwig, T.; Gail, R.; Coenen, H.H. Comparison of Pathways to the Versatile Synthon
of No-Carrier-Added 1-bromo-4-[18F]fluorobenzene. J. Labelled Compd. Radiopharm., 2004, 47(7), 429-441.
[29] Sun, H.; DiMagno, S.G. Competitive Demethylation and Substitution in N,N,N-trimethylanilinium
fluorides. J. Fluorine Chem., 2007, 128(7), 806-812.
[30] Dollé, F. Fluorine-18-Labelled Fluoropyridines: Advances in Radiopharmaceutical Design. Curr. Pharm.
Des., 2005, 11(25), 3221-3235.
[31] Sobrio, F.; Amokhtari, M.; Gourand, F.; Dhilly, M.; Dauphin, F.; Barré, L. Radiosynthesis of [18F]Lu29-
024: A Potential PET Ligand for Brain Imaging of the Serotonergic 5-HT2 Receptor. Bioorg. Med. Chem., 2000,
8(10), 2511-2518.
[32] Langer, O.; Dollé, F.; Valette, H.; Halldin, C.; Vaufrey, F.; Fuseau, C.; Coulon, C.; Ottaviani, M.; Någren,
K.; Bottlaender, M.; Maziére, B.; Crouzel, C. Synthesis of High-Specific-Radioactivity 4- and 6-
[18F]Fluorometaraminol- PET Tracers for the Adrenergic Nervous System of the Heart. Bioorg. Med. Chem.,
2001, 9(3), 677-694.
[33] Ishiwata, K.; Ishii, S.-I.; Senda, M.; Tsuchiya, Y.; Tomimoto, K. Electrophilic Synthesis of 6-[18F]fluoro-l-
dopa: Use of 4-O-pivaloyl-l-dopa as a Suitable Precursor for Routine Production. Appl. Radiat. Isot., 1993,
44(4), 755-759.
Page 61
47
[34] Hatano, K.; Ishiwata, K.; Yanagisawa, T. Co production of 2,6-[18F] difluoroDOPA during Electrophilic
Synthesis of 6-[18F]fluoro-l-DOPA. Nucl. Med. Biol., 1996, 23(1), 101-103.
[35] Luxen, A.; Perlmutter, M.; Bida, G.T.; Van Moffaert, G.; Cook, J.S.; Satyamurthy, N.; Phelps, M.E.; Barrio,
J.R. Remote, Semiautomated Production of 6-[18F]fluoro-l-dopa for Human Studies with PET. Int. J. Rad. Appl.
Instrum. A., 1990, 41(3), 275-281.
[36] Namavari, M.; Bishop, A.; Satyamurthy, N.; Bida, G.; Barrio, J.R. Regioselective radiofluorodestannylation
with [18F]F2 and [18F]CH3COOF: A High Yield Synthesis of 6-[18F]fluoro-l-dopa. Int. J. Rad. Appl. Instrum.
A., 1992, 43(8), 989-996.
[37] Luxen, A.; Guillaume, M.; Melega, W.P.; Pike, V.W.; Solin, O.; Wagner, R. Production of 6-[18F]fluoro-l-
DOPA and its Metabolism in vivo-A Critical Review. Int. J. Rad. Appl. Instrum. B., 1992, 19(2), 149-158.
[38] Hess, E.; Sichler, S.; Kluge, A.; Coenen, H.H. Synthesis of 2-[18F]Fluoro-L-Tyrosine via Regiospecific
Fluoro-De-Stannylation. Appl. Radiat. Isot., 2002, 57(2), 185-191.
[39] Eskola, O.; Grönroos, T.; Bergman, J.; Haaparanta, M.; Marjamäki, P.; Lehikoinen, P.; Forsback, S.;
Langer, O.; Hinnen, F.; Dollé, F.; Halldin, C.; Solin, O. A Novel Electrophilic Synthesis and Evaluation of
Medium Specific Radioactivity (1R,2S)-4-[18F]Fluorometaraminol: A Tracer For The Assessment Of Cardiac
Sympathetic Nerve Integrity with PET. Nucl. Med. Biol., 2004, 31(1), 103-110.
[40] Bergman, J.; Solin, O. Fluorine-18-labeled fluorine gas for synthesis of tracer molecules. Nucl. Med. Biol.,
1997, 24(7), 677-683.
[41] Balz, G.; Schiemann, G. Über aromatische Fluorverbindungen, I.: Ein neues Verfahren zu ihrer Darstellung.
Ber. Dtsch. Chem. Ges., 1927, 60(5), 1186-1190.
[42] Wallach, O. Ueber das Verhalten einiger Diazo- und Diazoamidoverbindungen. Justus Liebigs Ann. Chem.,
1886, 235(3), 233-255.
[43] Merritt, E.A.; Olofsson, B. Diaryliodonium Salts: A Journey from Obscurity to Fame. Angew. Chem. Int.
Ed., 2009, 48(48), 9052-9070.
Page 62
48
[44] Coenen, H.H.; Ermert, J. Direct Nucleophilic 18F-Fluorination of Electron Rich Arenes: Present Limits of
No-Carrier-Added Reactions. Curr. Radiopharm., 2010, 3(3), 163-173.
[45] Roe, A. In: Organic Reactions; Roger Adams, Ed.; John Wiley & Sons Inc.: New York, 1949; Vol. 5, pp.
193-228.
[46] Swain, C.G.; Rogers, R.J. Mechanism of Formation of Aryl Fluorides from Arenediazonium Fluoborates. J.
Am. Chem. Soc., 1975, 97(4), 799-800.
[47] Koval’chuk, E.P.; Reshetnyak, O.V.; Kozlovs’ka, Z.Y.; Błażejowski, J.; Gladyshevs’kyj, R.Y.; Obushak,
M.D. Mechanism of the Benzenediazonium Tetrafluoroborate Thermolysis in the Solid State. Thermochim. Acta,
2006, 444(1), 1-5.
[48] Yoneda, N.; Fukuhara, T. Facile Preparation of Aromatic Fluorides by Deaminative Fluorination of
Aminoarenes using Hydrogen Fluoride Combined with Bases. Tetrahedron, 1996, 52(1), 23-36.
[49] Doyle, M.P.; Bryker, W.J. Alkyl Nitrite-Metal Halide Deamination Reactions. 6. Direct Synthesis of
Arenediazonium Tetrafluoroborate Salts from Aromatic Amines, tert-butyl Nitrite, and Boron Trifluoride
Etherate in Anhydrous Media. J. Org. Chem., 1979, 44(9), 1572-1574.
[50] Yong, H.K.; Chun, H.L.; Ki, Y.C. Facile Direct Conversion of Amino-Heterocycles to Fluoro-Heterocycles
using t-butylthionitrite or t-butylthionitrate with Sodium Tetrafluoroborate. Tetrahedron Lett., 1990, 31(21),
3019-3022.
[51] Yoneda, N. The Combination of Hydrogen Fluoride with Organic Bases as Fluorination Agents.
Tetrahedron, 1991, 47(29), 5329-5365.
[52] Milner, D.J. Fluoroaromatics from Arylamines, a Convenient One-Pot Conversion Using Nitrosonium
Tetrafluoroborate. Synth. Commun., 1992, 22(1), 73-82.
[53] Shinhama, K.; Aki, S.; Furuta, T.; Minamikawa, J.-I. Facile Conversion of Arenediazonium Salts to the
Corresponding Fluoroarenes Using Boron Trifluoride Diethyl Ether Complex. Synth. Commun., 1993, 23(11),
1577-1582.
Page 63
49
[54] Laali, K.K.; Gettwert, V.J. Fluorodediazoniation in ionic liquid solvents: new life for the Balz-Schiemann
reaction. J. Fluorine Chem., 2001, 107(1), 31-34.
[55] Tamura, M.; Shibakami, M.; Sekiya, A. Deaminative Fluorination of Anilines with Silicon Tetrafluoride:
Utility of Silicon Tetrafluoride as a Fluorine Source. Eur. J. Org. Chem., 1998, 1998(4), 725-727.
[56] Garel, L.; Saint-Jalmes, L. One-pot fluoro-de-diazoniation of anilines in Organic Medium. Tetrahedron
Lett., 2006, 47(32), 5705-5708.
[57] Swain, C.G.; Sheats, J.E.; Harbison, K.G. Evidence for Phenyl Cation as an Intermediated in Reactions of
Benzenediazonium Salts in Solution. J. Am. Chem. Soc., 1975, 97(4), 783-790.
[58] Galli, C. Radical Reactions of Arenediazonium Ions: An Easy Entry into the Chemistry of the Aryl Radical.
Chem. Rev., 1988, 88(5), 765-792.
[59] Canning, P.S.J.; McCrudden, K.; Maskill, H.; Sexton, B. Rates and Mechanisms of the Thermal Solvolytic
Decomposition of Arenediazonium Ions. J. Chem. Soc., Perkin Trans. 2, 1999, 2735-2740.
[60] Satyamurthy, N.; Barrio, J.R.; Schmidt, D.G.; Kammerer, C.; Bida, G.T.; Phelps, M.E. Acid-catalyzed
Thermal Decomposition of 1-aryl-3,3-Dialkyltriazenes in the Presence of Nucleophiles. J. Org. Chem., 1990,
55(15), 4560-4564.
[61] Eakins, M.N.; Palmer, A.J.; Waters, S.L. Studies in the Rat with 18F-4-Fluoro-Oestradiol and 18F-4-
Fluoro-Oestrone as Potential Prostate Canning Agents: Comparison with 125I-2-iodo-oestradiol and 125I-2,4-
Di-iodo-oestradiol. Int. J. Appl. Radiat. Isot., 1979, 30(11), 695-700.
[62] Nozaki, T.; Tanaka, Y. The preparation of F18-labelled aryl fluorides. Int. J. Appl. Radiat. Isot., 1967,
18(2), 111-119.
[63] Hoyte, R.M.; Lin, S.S.; Christman, D.R.; Atkins, H.L.; Hauser, W.; Wolf, A.P. Organic
Radiopharmaceuticals Labeled with Short-Lived Nuclides. III. 18F-Labeled Phenylalanines. J. Nucl. Med., 1971,
12(6), 280-286.
[64] Goulding, R.W.; Palmer, A.J. The Preparation of Fluorine-18 labelled p-Fluorophenylalanine for Clinical
Use. Int. J. Appl. Radiat. Isot., 1972, 23(3), 133-137.
Page 64
50
[65] Goulding, R.W.; Gunasekera, S.W. Fluorine-18 labelled L-p-Fluorophenylalanine. Int. J. Appl. Radiat. Isot.,
1975, 26(9), 561-564.
[66] Atkins, H.L.; Christman, D.R.; Fowler, J.S.; Hauser, W.; Hoyte, R.M.; Klopper, J.F.; Lin, S.S.; Wolf, A.P.
Organic Radiopharmaceuticals Labeled with Isotopes of Short Half-Life. V. 18F-Labeled 5- and 6-
Fluorotryptophan. J. Nucl. Med., 1972, 13(10), 713-719.
[67] Firnau, G.; Nahmias, C.; Garnett, S. Synthesis of 3,4-dihydroxy-5-fluoro-DL-Phenylalanine and 3,4-
Dihydroxy-5-[fluorine-18]fluoro-DL-phenylalanine. J. Med. Chem., 1973, 16(4), 416-418.
[68] Firnau, G.; Nahmias, C.; Garnett, S. The Preparation of [18F]5-fluoro-DOPA with Reactor-Produced
Fluorine-18. Int. J. Appl. Radiat. Isot., 1973, 24(3), 182-184.
[69] Argentini, M.; Wiese, C.; Weinreicht, R. Syntheses of 5-fluoro-d/l-dopa and [18F]5-fluoro-l-dopa. J.
Fluorine Chem., 1994, 68(2), 141-144.
[70] Kook, C.S.; Reed, M.F.; Digenis, G.A. Preparation of Fluorine-18-labeled Haloperidol. J. Med. Chem.,
1975, 18(5), 533-535.
[71] Livni, E.; Fischman, A.J.; Ray, S.; Sinclair, I.; Elmaleh, D.R.; Alpert, N.M.; Weiss, S.; Correia, J.A.; Webb,
D.; Dahl, R. Synthesis of 18F-labeled Fluconazole and Positron Emission Tomography Studies in Rabbits. Int. J.
Rad. Appl. Instrum. B, 1992, 19(2), 191-199.
[72] Hashizume, K.; Tamakawa, H.; Hashimoto, N.; Miyake, Y. Single-step Synthesis of [18F]Haloperidol from
the Chloro-precursor and its Applications in PET Imaging of a Cat’s Brain. Appl. Radiat. Isot., 1997, 48(9),
1179-1185.
[73] Knöchel, A.; Zwernemann, O. Aromatic n.c.a. labelling with 18F- by modified Balz-Schiemann-
decomposition. Int. J. Radiat. Appl. Instrum. A., 1991, 42(11), 1077-1080.
[74] Knöchel, A.; Zwernemann, O. Development of a No-Carrier-Added Method for 18F-Labelling of Aromatic
Compounds by Fluorodediazonation. J. Labelled Compd. Radiopharm., 1996, 38(4), 325-336.
[75] Bartsch, R.A.; Chen, H.; Haddock, N.F.; Juri, P.N. Stabilization of Aryldiazonium Ions by Crown Ether
Complexation. J. Am. Chem. Soc., 1976, 98(21), 6753-6754.
Page 65
51
[76] Krane, J.; Skjetne, T. Studies of Crown Ether Complexes Aryldiazonium Ion Complexes. Tetrahedron Lett.,
1980, 21(18), 1775-1778.
[77] Nakazumi, H.; Kitao, T.; Zollinger, H. Dediazoniation of Arenediazonium Ions. 24. Dual and Triple
Substituent Parameter Evaluation of Competitive Heterolytic and Homolytic Dediazoniations of Diazonium Ions
Complexed with 18-Crown-6 Ether. J. Org. Chem., 1987, 52(13), 2825-2830.
[78] Gross, M.L.; Blank, D.H.; Welch, W.M. The Triazene Moiety as a Protecting Group for Aromatic Amines.
J. Org. Chem., 1993, 58(8), 2104-2109.
[79] Nifontov, V.I.; Bel’skaya, N.P.; Shtokareva, E.A. Manufacture Methods of Synthesizing Triazenes
(Review). Pharm. Chem. J., 1993, 27(9), 652-665.
[80] Nifontov, V.I.; Bel’skaya, N.P.; Shtokareva, E.A. The Reactivity and Mechanism of Action of Triazenes
(Review). Pharm. Chem. J., 1994, 28(10), 687-706.
[81] Kimball, D.B.; Haley, M.M. Triazenes: A Versatile Tool in Organic Synthesis. Angew. Chem. Int. Ed.,
2002, 41(18), 3338-3351.
[82] Chu, C.-K.; Kim, J.-H.; Kim, D.W.; Chung, K.-H.; Katzenellenbogen, J.A.; Chi, D.Y. Aromatic
Fluorination by Decomposition of Triazenes in Ionic Liquids. Bull. Korean Chem. Soc., 2005, 26(4), 599-602.
[83] Threadgill, M.D.; Stevens, M.F.G. Selective Reactions in the Triazene Series; I. Reduction of 1-(4-
Acetylphenyl)-triazenes. Synthesis, 1983, 1983(4), 289-291.
[84] Pages, T.; Langlois, B.R. Fluorination of Aromatic Compounds from 1-aryl-3,3-Dimethyltriazenes and
Fluoride Anions in Acidic Medium: 1. A model for 18F labelling. J. Fluorine Chem., 2001, 107(2), 321-327.
[85] Pages, T.; Langlois, B.R.; Le Bars, D.; Landais, P. Fluorination of aromatic compounds from 1-aryl-3,3-
dimethyltriazenes and fluoride anions in acidic medium: 2. Synthesis of (S)-[18F]-3-fluoro-α-
methylphenylalanine. J. Fluorine Chem., 2001, 107(2), 329-335.
[86] Yoneda, N.; Fukuhara, T.; Mizokami, T.; Suzuki, A. A Facile Preparation of Aryl Triflates. Decomposition
of Arenediazonium Tetrafluoroborate Salts in Trifluoromethanesulfonic Acid. Chem. Lett., 1991, 20(3), 459-
460.
Page 66
52
[87] Picherit, C.; Wagner, F.; Uguen, D. The Sequel to a Carbocyclic Nucleoside Synthesis: A Divergent Access
to Both Arenediazonium Ions and Aryl Triflates. Tetrahedron Lett., 2004, 45(12), 2579-2583.
[88] Berridge, M.S.; Sayre, L.M.; Arora, P.K.; Terris, A.H.; Riachi, N.J.; Harik, S.I. Design and Synthesis of
18F-Labeled Neurotoxic Analogs of MPTP. J. Med. Chem., 1993, 36(9), 1284-1290.
[89] Kilbourn, M.R.; Welch, M.J.; Dence, C.S.; Tewson, T.J.; Saji, H.; Maeda, M. Carrier-Added and No-
Carrier-Added Syntheses of [18F]Spiroperidol and [18F]Haloperidol. Int. J. Appl. Radiat. Isot., 1984, 35(7),
591-598.
[90] Riss, P.J.; Kuschel, S.; Aigbirhio, F.I. No Carrier-Added Nucleophilic Aromatic Radiofluorination using
Solid Phase Supported Arenediazonium Sulfonates and 1-(aryldiazenyl)Piperazines. Tetrahedron Lett., 2012,
53(14), 1717-1719.
[91] Stang, P.J.; Zhdankin, V.V. Organic Polyvalent Iodine Compounds. Chem. Rev., 1996, 96(3), 1123-1178.
[92] Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev., 2008, 108(12), 5299-5358.
[93] Ugi, I.; Marquarding, D.; Klusacek, H.; Gillespie, P.; Ramirez, F. Berry pseudorotation and turnstile
rotation. Acc. Chem. Res., 1971, 4(8), 288-296.
[94] Cass, M.E.; Hii, K.K.; Rzepa, H.S. Mechanisms That Interchange Axial and Equatorial Atoms in Fluxional
Processes: Illustration of the Berry Pseudorotation, the Turnstile, and the Lever Mechanisms via Animation of
Transition State Normal Vibrational Modes. J. Chem. Educ., 2006, 83(2), 336.
[95] Couzijn, E.P.A.; Slootweg, J.C.; Ehlers, A.W.; Lammertsma, K. Stereomutation of Pentavalent Compounds:
Validating the Berry Pseudorotation, Redressing Ugi’s Turnstile Rotation, and Revealing the Two- and Three-
Arm Turnstiles. J. Am. Chem. Soc., 2010, 132(51), 18127-18140.
[96] Zhdankin, V.V.; Stang, P.J. Recent Developments in the Chemistry of Polyvalent Iodine Compounds.
Chem. Rev., 2002, 102(7), 2523-2584.
[97] Wirth, T. Hypervalent Iodine Chemistry in Synthesis: Scope and New Directions. Angew. Chem. Int. Ed.,
2005, 44(24), 3656-3665.
[98] Zhdankin, V.V. Hypervalent iodine(III) reagents in organic synthesis. ARKIVOC, 2009, 2009, 1-62.
Page 67
53
[99] Zhdankin, V.V. Organoiodine(V) Reagents in Organic Synthesis. J. Org. Chem., 2011, 76(5), 1185-1197.
[100] Perkins, C.W.; Martin, J.C.; Arduengo, A.J.; Lau, W.; Alegria, A.; Kochi, J.K. An Electrically Neutral
.Sigma.-Sulfuranyl Radical from the Homolysis of a Perester with neighboring Sulfenyl Sulfur: 9-S-3 species. J.
Am. Chem. Soc., 1980, 102(26), 7753-7759.
[101] Musher, J.I. The Chemistry of Hypervalent Molecules. Angew. Chem. Int. Ed., 1969, 8(1), 54-68.
[102] Alcock, N.W.; Countryman, R.M. Secondary bonding. Part 1. Crystal and molecular structures of (C6H5)2
IX (X = Cl, Br, or I). J. Chem. Soc., Dalton Trans., 1977, 217-219.
[103] Ochiai, M. In: Topics in Current Chemistry; Thomas Wirth, Ed.; Springer-Verlag: Berlin Heidelberg New
York, 2003; Vol. 224, pp. 5-68.
[104] Okuyama, T.; Takino, T.; Sueda, T.; Ochiai, M. Solvolysis of Cyclohexenyliodonium Salt, a New
Precursor for the Vinyl Cation: Remarkable Nucleofugality of the Phenyliodonio Group and Evidence for
Internal Return from an Intimate Ion-Molecule Pair. J. Am. Chem. Soc., 1995, 117(12), 3360-3367.
[105] Pike, V.W.; Aigbirhio, F.I. Reactions of Cyclotron-produced [18F]Fluoride with Diaryliodonium Salts-A
Novel Single-Step Route to No-Carrier-Added [18]Fluoroarenes. J. Chem. Soc., Chem. Commun., 1995, 2215-
2216.
[106] Ross, T.L.; Ermert, J.; Hocke, C.; Coenen, H.H. Nucleophilic 18F-Fluorination of Heteroaromatic
Iodonium Salts with No-Carrier-Added [18F]Fluoride. J. Am. Chem. Soc., 2007, 129(25), 8018-8025.
[107] Shah, A.; Pike, V.W.; Widdowson, D.A. The Synthesis of [18F]Fluoroarenes from the Reaction of
Cyclotron-Produced [18F]Fluoride Ion with Diaryliodonium Salts. J. Chem. Soc., Perkin Trans. 1, 1998, 2043-
2046.
[108] Zhang, M.-R.; Kumata, K.; Suzuki, K. A Practical Route for Synthesizing a PET ligand Containing
[18F]Fluorobenzene Using Reaction of Diphenyliodonium Salt with [18F]F-. Tetrahedron Lett., 2007, 48(49),
8632-8635.
Page 68
54
[109] Chun, J.-H.; Lu, S.; Pike, V.W. Rapid and Efficient Radiosyntheses of meta-Substituted [18F]Fluoroarenes
from [18F]Fluoride Ion and Diaryliodonium Tosylates within a Microreactor. Eur. J. Org. Chem., 2011,
2011(23), 4439-4447.
[110] Ross, T.L.; Ermert, J.; Coenen, H.H. N.C.A. 18F-Fluorination of Various Arenes via Aryl(2-
thieny)iodonium Salts. J. Label. Compd. Radiopharm., 2005, 48(S1), S153.
[111] Yan, R;, Brichard, L.; Soloviev, D.; Aigbirhio, F.I.; Carroll, M.A. The First Single-Step-Single-Post
Synthesis of 4-[18
F]SFB. J. Label. Compd. Radiopharm., 2009, 52(6), 216.
[112] Yan, R;, Brichard, L.; Soloviev, D.; Aigbirhio, F.I.; Carroll, M.A. The First Synthesis of Ethyl 3-
[18
F]fluorobenzoate Using [18
F]fluoride. J. Label. Compd. Radiopharm., 2009, 52(6), 217.
[113] Carroll, M.A.; Jones, C.; Tang, S.-L. Fluoridation of 2-Thienyliodonium Salts. J. Label. Compd.
Radiopharm., 2007, 50(5-6), 450-451.
[114] Le Count, D.J.; Reid, J.A.W. Iodonium Salt Synthesis. Evidence for the Formation of Isomers in the
Synthesis of Diaryliodonium Salts. J. Chem. Soc. C, 1967, 1298-1301.
[115] Yamada, Y.; Okawara, M. Steric Effect in the Nucleophilic Attack of Bromide Anion on Diaryl- and Aryl-
2-thienyliodonium Ions. Bull. Chem. Soc. Jpn., 1972, 45(6), 1860-1863.
[116] Lancer, K.M.; Wiegand, G.H. The Ortho Effect in the Pyrolysis of Iodonium Halides. A Case for a
Sterically Controlled Nucleophilic Aromatic (SN) Substitution Reaction. J. Org. Chem., 1976, 41(21), 3360-
3364.
[117] Grushin, V.V. Carboranylhalonium ions: from Striking Reactivity to a Unified Mechanistic Analysis of
Polar Reactions of Diarylhalonium Compounds. Acc. Chem. Res., 1992, 25(11), 529-536.
[118] Chun, J.-H.; Lu, S.; Lee, Y.-S.; Pike, V.W. Fast and High-Yield Microreactor Syntheses of ortho-
Substituted [18F]Fluoroarenes from Reactions of [18F]Fluoride Ion with Diaryliodonium Salts. J. Org. Chem.,
2010, 75(10), 3332-3338.
Page 69
55
[119] Gail R., Hocke C., Coenen H.H. Direct N.C.A. 18F-fluorination of Halo- and Alkylarenes via
Corresponding Diphenyliodonium Salts. J. Label. Compd. Radiopharm., 1997, 40(1), 50-52.
[120] Ross, T.L. Direct no-carrier-added 18F-labelling of arenes via nucleophilic substitution on aryl(2-
thienyl)iodonium salts. PhD Thesis, The German National University: Köln, December 2005.
[121] Martín-Santamaría, S.; Carroll, M.A.; Carroll, C.M.; Carter, C.D.; Pike, V.W.; Rzepa, H.S.; Widdowson,
D.A. Fluoridation of Heteroaromatic Iodonium Salts-Experimental Evidence Supporting Theoretical Prediction
of the Selectivity of the Process. Chem. Commun., 2000, 649-650.
[122] Seeman, J.I. Effect of Conformational Change on Reactivity in Organic Chemistry. Evaluations,
Applications, and Extensions of Curtin-Hammett Winstein-Holness Kinetics. Chem. Rev., 1983, 83(2), 83-134.
[123] Wadsworth, H.J.; Devenish, T. Fluoridation Method. International Patent WO2005097713, October 20,
2005.
[124] Lee, B.C.; Dence, C.S.; Zhou, H.; Parent, E.E.; Welch, M.J.; Katzenellenbogen, J.A. Fluorine-18 Labeling
and Biodistribution Studies on Peroxisome Proliferator-Activated Receptor-Γ Ligands: Potential Positron
Emission Tomography Imaging Agents. Nucl. Med. Biol., 2009, 36(2), 147-153.
[125] Lee, B.C.; Kim, J.S.; Kim, B.S.; Son, J.Y.; Hong, S.K.; Park, H.S.; Moon, B.S.; Jung, J.H.; Jeong, J.M.;
Kim, S.E. Aromatic Radiofluorination and Biological Evaluation of 2-aryl-6-[18F]Fluorobenzothiazoles as a
Potential Positron Emission Tomography Imaging Probe for β-amyloid Plaques. Bioorg. Med. Chem., 2011,
19(9), 2980-2990.
[126] Basuli, F.; Wu, H.; Griffiths, G.L. Syntheses of Meta-[18F]Fluorobenzaldehyde and Meta-
[18F]Fluorobenzylbromide from Phenyl(3-Formylphenyl) Iodonium Salt Precursors. J. Labelled Compd.
Radiopharm., 2011, 54(4), 224-228.
[127] Carroll, M.A.; Nairne, J.; Woodcraft, J.L. Diaryliodonium salts: a solution to 3-[18
F]fluoropyridine. J.
Label. Compd. Radiopharm., 2007, 50(5-6), 452-454.
[128] Wüst, F.R.; Kniess, T. Synthesis of 4-[18F]Fluoroiodobenzene and its Application in Sonogashira Cross-
Coupling Reactions. J. Labelled Compd. Radiopharm., 2003, 46(8), 699-713.
Page 70
56
[129] Wadsworth, H.J.; Widdowson, D.A.; Wilson, E.; Carroll, M.A. Radical Trap in Fluoridation of Iodonium
Salt. International Patent WO2005061415, July 07, 2005.
[130] Carroll, M.A.; Nairne, J.; Smith, G.; Widdowson, D.A. Radical Scavengers: A Practical Solution to the
Reproducibility Issue in the Fluoridation of Diaryliodonium Salts. J. Fluorine Chem., 2007, 128(2), 127-132.
[131] Moon, B.S.; Kil, H.S.; Park, J.H.; Kim, J.S.; Park, J.; Chi, D.Y.; Lee, B.C.; Kim, S.E. Facile Aromatic
Radiofluorination of [18F]Flumazenil from Diaryliodonium Salts with Evaluation of their Stability and
Selectivity. Org. Biomol. Chem., 2011, 9(24), 8346-8355.
[132] Wüst, F.; Carlson, K.E.; Katzenellenbogen, J.A. Synthesis of novel Arylpyrazolo Corticosteroids as
Potential Ligands for Imaging Brain Glucocorticoid Receptors. Steroids, 2003, 68(2), 177-191.
[133] Lee, Y.-S.; Hodošček, M.; Chun, J.-H.; Pike, V.W. Conformational Structure and Energetics of 2-
Methylphenyl(2′-methoxyphenyl)iodonium Chloride: Evidence for Solution Clusters. Chem. Eur. J., 2010,
16(34), 10418-10423.
[134] Huiban, M.; Gee, A.D.; Martarello, L.; Passchier, J. Labelling Methods and Radiosynthesis–Organic
Positron Emitters. J. Label. Compd. Radiopharm. 2007, 50(S1), S130.
[135] Lee, B.C.; Lee, K.C.; Lee, H.; Mach, R.H.; Katzenellenbogen, J.A. Strategies for the Labeling of Halogen-
Substituted Peroxisome Proliferator-Activated Receptor γ Ligands: Potential Positron Emission Tomography
and Single Photon Emission Computed Tomography Imaging Agents. Bioconjugate Chem., 2007, 18(2), 514-
523.
Page 71
57
1.4. Alzheimer's disease
1.4.1. Epidemiology and risk factors of Alzheimer's disease
Deterioration of intellectual ability and cognitive decline was and is considered an
inevitable and natural consequence of aging that was recognized as early as the 7th
century
BC.61,62
Alzheimer’s disease (AD) has become one of the leading health concerns as aging of
the population has now become a worldwide phenomenom and is no longer limited to the
developed Western societies. The number of people surviving into their 80s and 90s and
beyond is expected to grow dramatically due to advances in medical treatment and medical
technology, as well as socio-economic conditions, further contributing to the increase in
prevalence of AD.63,64
The increasing number of people with AD have a marked impact on
health care systems, not to mention families and caregivers.
Alzheimer’s disease (AD) is a neurodegenerative and terminal disease characterised with
insidious onset, gradual memory loss and progressive decline in other cognitive functions
over time. The term AD is today use for mental dementia (without antecedent causes as
stroke, head trauma, alcohol, …) irrespective of the age of onset. It is the most common type
of dementia in the elderly and affects approximately half of all patients with dementia
worldwide. An estimated 5.4 million Americans of all ages have AD in 2012 and one in eight
people age 65 and older (13 percent) has AD in the United States.63
As demonstrated by the
number of incidence and prevalence studies, advancing age is the greatest and most consistent
risk factor for the disease, where incidence and prevalence for AD show approximately
exponential increase with increasing age.63,64
Most of the prevalence and incidence studies are
consistent with the hypothesis that genetic factors predispose a person to develop AD, while
other factors modulate the age of onset of clinical dementia.
Once AD has been diagnosed, the average life expectancy is approximately 4 to 8 years,
while very small percent of the patients live as long as 15 years. As the disease progresses, the
individual’s cognitive and functional abilities decline. In advanced AD, people need help with
basic activities of daily living and those in the final stages of the disease become bed-bound
and reliant on caregivers.
Analytic epidemiologic studies are important for identification of risk factors, which can be
in general divided into non-modifiable and modifiable ones. Modifiable factors are obvious
Page 72
58
targets for preventive treatment, while non-modifiable ones (gender, level of education,
genetic factors and family history) are important in understanding the pathogenesis of the
disease. Although in numerous studies women were found to be at greater risk for AD than
men, they are not more likely than men to develop dementia at any given age. The larger
proportion of women with AD or other dementias is primarily explained by the fact that
women live longer on average than men. Data from a number of studies show that a low
education level is associated with a greater risk of developing and greater likelihood of having
dementia and AD.63,64
These findings led to the hypothesis that lifetime cognitive experience
and engagement in leisure activities of intellectual and social nature may influence the
number of neurons and synapses that survive into adult life (cognitive reserve hypothesis) -
the greater the number of synapses formed between neurons, the longer the time needed for
the synapses to degenerate, and later the onset of dementia.65
However, other believe that the
increased risk of dementia among those with lower educational level may be explained by
other factors common to people in lower socioeconomic groups, such as increased risk for
disease in general and less access to medical care.63
Many studies have also shown that family
history is another risk factor for AD, especially for occurrence in a first-degree relative
(parent or sibling). Currently four genes have been found to be definitely associated with AD.
Mutations in the amyloid precursor protein (APP), presenilin 1 (PS1), and presenilin 2 (PS2)
are known to cause early onset of AD, with PS1 mutations being the most frequent.66
Molecular studies of these three mutations are all associated with an increased production of
the Aβ42 amyloid peptide, and provide support for the amyloid cascade hypothesis of AD
pathogenesis. By contrast, individuals who inherit one or two genes of the apolipoprotein-ε4
gene (ApoE-ε4 gene) are at increased risk to develop AD and to develop it at an earlier age
than those who inherit the ε2 or/and ε3 forms of the ApoE gene.67
Nevertheless, ApoE-ε4 is a
susceptibility risk factor, as it is neither necessary nor sufficient that an individual will
develop AD.63,64
1.4.2. Neurophysiology and pathology of Alzheimer’s disease
AD pathology can be characterized on a macro level as the progressive loss of brain tissue
due to localised degeneration of neurons and synapses, which roughly correlates with the
severity of cognitive decline. Diffused cerebral atrophy is manifested by narrowing of the
cerebral gyri, widening of sulci, thinning of the cortical ribbon, and by enlargement of the
Page 73
59
volume of the ventricles, especially the temporal horn. As the disease progresses, neurons die
in a particular pattern over time and cerebrospinal fluid fills in the space previously occupied
by brain tissue.68
Preclinical AD begins in the transentorhinal region followed by the
entorhinal cortex, which connects the hippocampus to the cerebral cortex. Several studies
suggest that progressive neuronal loss in medial temporal lobe may start years before signs of
dementia emerge.69,70
Thus, in the early stages of AD, short-term memory begins to fade and
disorientation appears when the cells in the hippocampus, transentorhinal region, and
entorhinal cortex degenerate. The ability to perform routine tasks also declines. As the disease
progresses, atrophy extends to other areas of neocortex, leading to judgment decline, and
changes in behavior (emotional outburst, wandering, agitation). At the later stages of the
disease, the neocortex atrophies in areas that control speech, reasoning, sensory processing,
and conscious thought.71
Therefore, people lose the ability to recognize faces, communicate,
control bodily functions and require constant care.
Figure 3. Characteristic volumetric brain changes during progression of Alzheimer's disease:
diffused cerebral atrophy with narrowing of gyri, widening of sulci, ventricular dilatation and
atrophy of the hippocampus.68
At microscopic level, AD is characterised by the development of two neuropathological
hallmarks, namely extracellular amyloid plaques or senile plaques (SPs), and intracellular
neurofibrilary tangles (NFTs) (Figure 4).72
These neuropathologic lesions likely begin to form
years prior to the full clinical expression of clinical dementia, particularly within stages of
mild cognitive impairment.73
Although both SPs and NFTs are generally considered to be
characteristic pathologic changes of AD, they are not specific. However, controversy remains
in the relationship between these lesions and which process is central to disease pathogenesis.
Over the past few decades, support has grown for the amyloid cascade hypothesis of AD, in
Page 74
60
which accumulation of neurotoxic amyloid beta (Aβ) peptide in brain tissue is believed to be
an early and necessary step. This triggers a series of events including inflammatory response,
free radical formation, oxidative stress, lipid peroxidation, excessive excitotoxicity of
glutaminergic neurons, and formation of neurofibrillary tangles that lead to
neurodegeneration, neurotransmitter dysfunction and dementia (Figure 5 on page 64).74,75
While there is substantial evidence supporting the amyloid cascade hypothesis, there are also
limitations because SPs and NFTs may develop independently, and they may be a protective
response against inflammatory cascade rather than the cause of neurodegeneration in AD.
Furthermore, plaque pathology and spread do not always correlate with clinical findings in
AD and can exist in normal individuals.76,77
Thus, the amount of defficient nerve cells and
synaptic loss in the hippocampus and neocortex much better correlates with the decline of
cognitive function in AD than the number of amyloid plaques.78
Phosphorylated tau in tangles
A42 in senile plaque
Astrocyte
Neuron
Capillary
Axon terminal
Figure 4. Schematic drawing of a neuron with an adjacent astrocyte and capillary.
Intracellular hyperphosphorylated tau in tangles and extracellular insoluble aggregates of
Aβ42 peptide within senile plaques are the characteristic hallmarks of AD.79
The core of SPs is consisted mainly of focal insoluble aggregates of amyloid beta peptide
42 (Aβ42) and is surrounded by dystrophic neurites, activated microglia, reactive astrocytes
and immune system proteins, to name a few. Aβ42 is a neurotoxic peptide and is the result of
alternate proteolytic cleavage of transmembrane amyloid precursor protein (APP) due to
increased activity of β- and γ-proteases towards α-proteases (Figure 5 on page 64).80
It is
believed that this altered processing of APP is the primary causative factor in the pathogenesis
of AD (amyloid cascade hypothesis). Aβ42 initially forms monofibrils, but then quickly
Page 75
61
aggregates into protofibrils and finally into insoluble extracellular Aβ pleated sheet
structures.81
β-Amyloid peptides (39-43 aminoacid residues) activate immune system
(complement, T-lymhocytes, microglia) and stimulate the release of chemokines and
cytokines. Inflammation surrounding Aβ plaques is therefore consistent feature of the AD in
brain and add to the pathologic cascade of the disease.82
Affected neurons are susceptible to
ischaemia, excitotoxicity and oxidative stress too, which quickens their apoptotic ruin. Aged
individual without AD have some density of SPs, though most often these are diffuse, not
compact as in AD, and are not disease-specific. The anatomic distribution of SP in AD is
widespread and higher order association cortex tends to have the highest density of SPs, and
primary motor and sensory cortex the least. It has been shown that soluble Aβ oligomer
intermediates are key contributors to Aβ mediated neurodegeneration. Thus, soluble Aβ
aggregates appeared to correlate much better with neuron loss and severity of AD than
insoluble high molecular weight Aβ fibrils within amyloid plaques.83–85
Neurofibrillary tangles (NFTs) consist of cross-linked paired helical filaments of
pathologically hyperphoshorylated form of a microtubule-associated protein, namely Tau
protein. This probably results from an imbalance in the activity and regulation of tau kinases
(like glycogensynthase kinase-3 and cyclin dependent kinase-5) and phosphatases (PP1,
PP2A, PP2B).86,87
Abnormally high levels of neurofibrillary tangles inside the neuron
destabilizes microtubules and disrupts axonal transport, which is believed to be directly
associated with neuronal death and disease progression.88
Braak H. and Braak E. have shown
that neurofibrillary pathology in the brain accumulates in a hierarchical topographic fashion,
with the transentorhinal cortex affected first (stages I and II), followed by the entorhinal
cortex, hippocampus and other limbic structures of the medial temporal lobe (stages III and
IV), and finally the neocortex (stages V and VI).89,90
The neocortex is involved in a
hierarchical fashion, the associative regions affected the most, whereas the primary sensory
and motor cortex are relatively spared. This corresponds to the clinical features of marked
impairment of memory and abstract reasoning, with preservation of vision and movement.
NFTs are also frequently present in certain subcortical nuclei such as the nucleus basalis,
limbic nuclei of the thalamus, amygdala, locus coeruleus, substantia nigra, and raphe nuclei of
the brainstem. The distribution, progression, and abundance of tangles are better proportional
to the severity of cognitive impairment and severity of AD than senile plaque pathology (tau
hypothesis). Most patients with AD are diagnosed at stage V or VI pathology, while those at
Page 76
62
lower stages are usually not demented. Interestingly, amyloid plaques and neurofibrillary
tangles are also observed in intellectually normal individuals too, but they are far more
abundant in patients with AD. Thus, although NFTs and SPs are considered to be the
characteristic pathologic changes of AD, they are not specific.
As stated above, AD is characterised by marked atrophy of the cerebral cortex,
hippocampus and subcortical brain regions. Subcortical brain regions include the basal
forebrain, the locus coeruleus, and the dorsal raphe nuclei, thus generating deficits of
acetylcholine, norepinephrine, and serotonin which contribute to the impairment of attention,
memory, mood, and behaviour. Numerous measurements on post-mortem AD brain tissues
have revealed a relatively selective degeneration of subcortical cholinergic neurons;
particularly nucleus basalis of Meynart, which is part of the magnocellular forebrain nuclei in
the basal forebrain that provide cholinergic innervation to the whole cortex. Choline
acetyltransferase (ChAT; acetylcholine synthesising enzyme) and acetylcholine esterase
(AChE; an enzyme within synaptic cleft that hydrolyses acetylcholine to choline and acetic
acid, thus preparing the synapse for the passage of a new impulse) activity in the neocortex
and hippocampus are reduced considerably (30 % - 70 %) in AD, which correlates positively
with the severity of dementia but not in other disorders such as a depression or schizophrenia.
Muscarinic receptor (mAChR; G-protein coupled) density is “notˮ affected, but pentameric
ionotropic nicotinic receptors (nAChR; mainly located presynaptically), particularly in the
cortex and hippocampus, are reduced. This discovery (1979) lead to the cholinergic
hypothesis which is the mainstay of the current symptomatic therapy approach. It states that
deficiency of acetylcholine (ACh) in affected areas is critical in the genesis of the symptoms.
Moreover, cholinergic neuronal loss correlates well with the severity of the disease.
Therefore, enhancement of cholinergic transmission in affected areas might compensate for
the cholinergic deficit.
1.4.3. Pharmaceutical management and research directions
For AD there is no known cure. Current drug treatments are just palliative in nature
(symptomatic treatment) and do not halt or even reverse the progression of the disease; hence,
they are far from ideal. The therapies under experimental and evaluation phases for the
treatment of AD have disease modifying and neuroprotective approaches. There are currently
Page 77
63
two licensed symptomatic treatments for AD: (A.) acetylcholinesterase inhibitors (AChEIs),
and (B.) memantine, the uncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist.
AChEIs augment acetylcholine (ACh) levels and its intrasynaptic residence time via
inhibition of acetylcholinesterase in the synaptic cleft, and therefore facilitates interaction
between ACh and the postsynaptic cholinergic receptors. Donepezil, rivastigmine, and
galantamine are approved in most countries worldwide for mild to moderately severe AD,
because clinical trials have demonstrated their benefits in cognitive functioning, activities of
daily living and behaviour.91
However, results in the long term have generally been
disappointing. Only patients with stable medical or psychiatric illnesses who adhere, tolerate
and respond to AChEI might experience modest cognitive improvements and small
symptomatic benefit.92
Memantine has been licensed for the treatment of moderate to severe AD. It is an
uncompetitive, moderate-affinity, and voltage dependent NMDA antagonist that selectively
blocks glutamate overactivation of these receptors. Interestingly, it exits the channel pore to
allow normal physiologic neurotransmission under conditions of learning and memory
formation. Thus, it protects neuron against glutamate-mediated excitotoxicity, because an
increase in extracellular glutamate can lead to overactivation of NMDA receptors, which
results in excessive Ca2+
influx through the receptor associated ion channel.91,92
Intracellular
calcium accumulation can induce a cascade of evens resulting in neuronal death by necrosis
or apoptosis.93
Enormous research attention is currently being directed at the strategies designed to modify
AD pathogenesis. These approaches are based on the recent advances in understanding AD
pathogenesis and are directed at slowing or halting the progression of the disease. Such
disease-modifying treatments, which target physiopathological biochemical pathways in AD,
include β-amyloid-lowering approaches, tau-based treatments (e.g. Tau phosphorylation
inhibitors), as well as neuroprotective and neurorestorative approaches. Anti-amyloid
therapies target reduction of amyloid production through inhibition or modulation of β- and γ-
secretases or enhance α-secretase activity. Other compounds aim to prevent the
oligomerization and fibrillization of Aβ (β-amyloid antiaggregants, and β-sheet breakers),
whereas other strategies include immunotherapies (β-amyloid vaccines and passive
immunization with anti-β-amyloid humanized monoclonal antibodies) endeavor to remove
Page 78
64
neurotoxic Aβ from the brain. According to the one of the most current versions of the
amyloid cascade hypothesis, amyloid-directed therapeutic strategies will only be effective
early in the disease, whereas latter in the disease tau-directed therapies may be a better
strategy of choice. Peroxisome proliferator-activated receptor γ agonists (PPAR-γ agonists),
metal chelators, and muscarinic M1 agonists also appear to diminish amyloid production.
Neuroprotective and neuroregenerative approaches include neurotrophic factors (e.g.
nonpeptidic neurotrophic factor enhancers), antioxidants (e.g. Coenzyme Q10, vitamin C),
anti-apoptotic agents (e.g. caspase inhibitors), astrocyte-modulating agents, NMDA-receptor
antagonists, and anti-inflammatory drugs, to name a few.94–96
Figure 5. Diagram of the cascade of events currently hypothesized to compromise the
physiopathology of AD. Sites for potential therapeutic interventions are designated with red
colour.
Page 79
65
The goal of disease-modifying therapy is to preserve neuronal integrity and synaptic
plasticity by reducing amyloid toxicity and neuronal vulnerability. The focus of disease-
modifying treatments is towards early and incipient AD with a view of preventing or
attenuating disease before symptoms occur. According to the current knowledge of the
disease physiopathology, interventions at the stage of mild to moderate AD may already be
too late. Furthermore, slowing progression in more severe stages of the disease might not be
desirable for patients, family members, or society as a whole. Though none are yet proven, it
might be foreseen that such treatments are on the edge of potential breakthrough. If
successful, they will be used in conjunction with existing symptomatic therapies.
1.4.4. The diagnosis of Alzheimer’s disease by PET
Though AD is the most common type of dementia, it still must be distinguished from other
causes of dementia, including vascular dementia (VaD), frontotemporal dementia (FTD),
corticobasal degeneration (CBD), dementia with Lewy body (DLB), and Parkinson's disease
dementia (PDD) among others. The detection of AD early in its clinical course can be quite
challenging, while identification later in its course is often more obvious.
Today, the diagnosis for possible and probable AD is based on clinical examination. A
number of common clinical features are specified within sets AD diagnostic criteria: memory
decline and impairment of at least one non-memory cognitive function (language, motor and
executive function, visuo-spatial skills) that is sufficiently severe to interfere with daily
function.63,79
The disease is confirmed by neuropsychological testing (memory testing and
assessment of intellectual functioning) and neurological examinations, which is crucial in
differential diagnosis of the disease. But for definitive diagnosis, histopathologic confirmation
is needed (microscopic examination of brain tissue pathologic changes), which is up to now
possible just by post-mortem examination (autopsy). Since there is no absolute qualitative
difference that distinguishes the brains of demented patients from those of non-demented
elderly individuals, the definitive diagnosis of AD depends on identifying quantitative
differences. Post-mortem neuropathologic examination of demented individuals frequently
demonstrates AD in combination with other pathologic changes most frequently with
cerebrovascular dementia (cerebral amyloid angiopathy), Lewy body disease or both (“Mixed
dementiaˮ).
Page 80
66
In preclinical AD individuals have measurable changes in the brain (injured or degenerated
nerve cells), cerebrospinal fluid (CSF) and/or blood biomarkers that indicate the earliest signs
of disease, but they not yet have developed symptoms such as memory lost.63
Thus, there is a
growing interest in measuring CSF biomarkers as their levels reflect the severity of symptoms
or disease progression in AD.97,98
The most promising biomarkers in CSF (β-amyloid and tau)
have high sensitivity for differentiating early AD, but lower specificity against other
dementias, which ranges between 60 and 90%. Combining a battery of CSF biomarkers may
improve diagnostic specificity.
Advances in a variety of structural and functional neuroimaging techniques allow the
greatest in vivo and non-invasive insight into brain structure and function, respectively. To
date, the best established methods for the detection and tracking AD include structural
magnetic resonance imaging (sMRI) measurements of regional and whole brain tissue
shrinkage, [18
F]FDG PET measurements of decline in the regional cerebral metabolic rate for
glucose, and PET measurements of fibrillar β-amyloid burden.99
For example, PET
radiotracers Pittsburgh compound-B (PIB)100,101
, [18
F]FDDNP102–104
and AmyvidTM
(vials
containing 500-1900 MBq/mL Florbetapir F18)105
which label SPs have been used in humans
and have demonstrated the characteristic retention in AD subjects that mirrors the pattern of
this hallmark known from post-mortem studies. Florbetapir F18 is the first and only FDA-
approved diagnostic PET tracer for estimation of β-amyloid neuritic plaque density in adult
patients with cognitive impairment who are being evaluated for AD and other causes of
cognitive decline. It is an adjunct to other diagnostic evaluations, because positive scan which
indicates moderate to frequent amyloid neuritic plaques does not establish a diagnosis of AD
or other cognitive disorder.
Page 81
67
CNNC
N
18F
[18F]FDDNP
OHO
HO
OH
18F
OH
[18F]FDG
Figure 6. PET images comparing temporal lobe uptake of [18
F]FDDNP, an β-amyloid binding
tracer, and [18
F]FDG, a marker of glucose metabolism, in a patient with AD (left) and a
control subject (right).106
[18
F]FDDNP and [18
F]FDG uptake are presented on a heat scale.
Note increased uptake and retention of [18
F]FDDNP (arrowheads) in temporal lobes of the
patient with AD, compared with those of the control subject. AD patient also demonstrates
typical decrease of [18
F]FDG or glucose metabolism in temporal (arrows) and parietal lobes
(not shown).
Thus, the potential of PET technique lies in its ability of in vivo non-invasive quantification
of AD pathology in preclinical phase. The combination of neuroimaging techniques with
blood or CSF biomarkers may in the future play a major part in establishing the diagnosis on
their own. However, although the new criteria and guidelines identify preclinical disease as a
stage of AD, they do not establish diagnostic criteria that doctors could use now. Rather, they
state that additional biomarker research and development in specific radiotracers are needed
before preclinical AD can be diagnosed.
Page 82
68
1.5. Vesicular acetylcholine transporter (VAChT) and the most
promising PET imaging tracers
Evidence from biopsy and autopsy samples suggests that degeneration of the cholinergic
neurons located in the basal forebrain nuclei (e.g. in the nucleus basalis of Meynert), the
major cholinergic output of the central nervous system (CNS), and their synapses in the
cerebral cortex and hyppocampus are among the earliest neurochemical changes identified in
AD, and precedes for several years clinical onset of the disease.107–110
Consequently, different
cholinergic synaptic elements are depleted in the cortex and subcortical brain areas, such as
α4β2 nicotinic ACh receptor, acetylcholine esterase (AChE), choline acetyltransferase
(ChAT), choline transporter (ChT) and vesicular acetylcholine transporter (VAChT).
VAChT is a presynaptic vesicular transmembrane glycoprotein and is responsible for
loading ACh into secretory vesicles making ACh available for secretion into synaptic cleft
(Figure 7).111,112
VAChT contains 12 transmembrane domains, and the gene coding for the
VAChT is embeded within the first intron of the ChAT's gene in all species examined,
suggesting that the expression of ChAT, regarded as reference standard for cholinergic
markers, is tightly coupled to that of VAChT.112–114
VAChT gained increasing interest in the
last years as a reliable cholinergic marker for in vivo imaging of cholinergic deficiencies using
PET or SPECT, because its reduced density and activity change in parallel fashion with
ChAT,115,116
and show a strong correlation with the onset, progression, and severity of the
AD.117
Thus, in vivo qualitative and quantitative non-invasive measurements of the mentioned
target with PET can provide valuable information on disease onset and progression with the
aim to obtain an early diagnosis and to better understand AD.
Page 83
69
Figure 7. Schematic illustration of a generalized cholinergic junction (ChT, choline
transporter; AcCoA, acetyl coenzyme A; ChAT, choline acetyltransferase; VAChT, vesicular
acetylcholine transporter; P, peptides; ATP, adenosine triphosphate; SNAPs, synaptosome-
associated proteins; VAMPs, vesicle-associated membrane proteins)118
Lipophilic aminoalcohol 2-(4-phenylpiperidino)cyclohexanol (vesamicol) is a well known
high affinity VAChT ligand with neuromuscular blocking properties as a result of
stereoselective, non-competitive (allosteric binding site) and reversible blockage of VAChT
(Figure 7 and 8).119–121
Vesamicol also binds with low affinity to α-122
and moderate to high
affinity to σ-receptors.123
Because of this low selectivity, vesamicol itself is not suitable for
PET or SPECT imaging in brain. However, it has been a useful lead for developing more
potent and selective ligands as potential SPECT and PET imaging probes aimed for in vitro,
ex vivo, and in vivo studies of AD by accurate mapping distribution and amount of VAChT in
the brain regions of interest (VAChT density: striatum > cortex > hippocampus >
hypothalamus > cerebellum).121,124–149
Results obtained from these numerous studies
confirmed that the VAChT binding site is stereoselective and that the (2R,3R)-enantiomers are
Page 84
70
generally more potent (lower Ki) than (2S,3S)-enantiomers.141,143
Benzovesamicol has been
found to be nearly equipotent to vesamicol, and is therefore one of the most explored class of
compounds.136
The different groups can be incorporated at the 5-position of the
benzovesamicol scaffold to modulate affinity, selectivity and pharmacokinetic properties of
potential VAChT tracers.136
PET radioligands for VAChT has been recently reviewed by
Giboureau et al.150
Poor selectivity over σ receptors (due to the structural similarity of σ
receptors and VAChT pharmacophores and the similar tissue distribution profiles), and
unfavourable pharmacokinetics (e.g. fast metabolism, low extraction from the blood, slow
brain kinetics) are the main reasons why only a few VAChT ligands are currently promising
radiotracers for the VAChT, though further validation is required to confirm their clinical
usefulness (Figure 8).150
N
HO
(1R,2R)-vesamicol
N
HO
HN
13CH3
(2R,3R)-[11C]MABV
N
HO
O
H2C
(2R,3R)-[18F]FEOBV
CH218F
N
HO
O
18F
(2R,3R)-[18F]-2-Hydroxy-3-(4-(4-fluorobenzoyl)piperidino)tetralin
N
HO
N
O
H
H
O
18F
(4aR,6R,7R,8aR)-[18F]FBMV
HO
123I
(2R,3R)-5-[123I]IBVM
N
HO
N
(2R,3R)-benzovesamicol
Page 85
71
Figure 8. Chemical structures of (1R,2R)-vesamicol, (2R,3R)-benzovesamicol and the most
promising PET and SPECT VAChT tracers, namely, (2R,3R)-[11
C]-5-(N-
methylamino)benzovesamicol ([11
C]MABV),151
(2R,3R)-5-[18
F]fluoroethoxybenzovesamicol
([18
F]FEOBV),134,135,143
(4-[18
F]fluorophenyl)((4aR,6R,7R,8aR)-7-hydroxy-6-(4-
phenylpiperidin-1-yl)hexahydro-2H-benzo[b][1,4]oxazin-4(3H)-yl)methanone
([18
F]FBMV),152
(2R,3R)-[18
F]-2-hydroxy-3-(4-(4-fluorobenzoyl)piperidino)tetralin,147
and
(2R,3R)-5-[123
I]iodobenzovesamicol ([123
I]IBVM).153
However, (2R,3R)-5-[123
I]IBVM is to date the only VAChT radioligand widely used in
human, and is therefore the lead VAChT tracer for SPECT.153–155
1.6. Synthesis of 5-aminobenzovesamicol (5-ABV) and its enantiomers
It should be noted that the synthesis of 5-aminobenzovesamicol (5-ABV) and its
enantiomers (Scheme 12), which have been previously described,134,136
were not performed in
this thesis, because there were enough of them left from the previous work by Giboureau et.
al.143
The first step of the synthesis of 5-ABV is the reduction of the commercial available
aminonaphthalene (1) by Birch reaction to 1,4-dihydronaphthalene-1-amine (2). The second
step is the protection of the amine function by trifluoroacetic anhydride to obtain
corresponding amide 3. The first two steps are almost quantitative. The third step is
epoxidation of 3 by reaction with m-chloroperoxybenzoic acid (m-CPBA) to produce the
corresponding epoxide 4 in ~75% yield. In the fourth step, the epoxide 4 is treated with the 4-
phenylpiperidine to obtain after in situ deprotection and subsequent chromatographic
purification two positional isomers 5-ABV and 8-ABV in ~25% and ~30% yield,
respectively. In the fifth step, enantiomers of racemic 5-ABV are separeted by using the
Mosher's acid chloride (MTPA-Cl), affording the corresponding diastereomers to allow their
chromatographic separation. After reductive cleavage of the MTPA groups using
diisobutylaluminium hydride (DIBAL-H), (2R,3R)-5-ABV and (2S,3S)-5-ABV are obtained
in 6-8 % overall yield in enantiomeric purity greater than 98% as determined by chiral HPLC.
Page 86
72
tBuOH, Et2O
Na, NH3
1 2
CF3
OO
CF3
O
benzene
3
4
OEt2O
Cl
O
OOH
NH
N
HO
5-ABV
O
Cl
CF3H3CO
EtOH, Et3N
(S)-MTPA-Cl
, DMAP/Et3N
HN
O
HN
O
N N
(2R,3R)-5-ABV (2S,3S)-5-ABV
1.
DIBAL-H
NH2 NH2 NHCOCF3
NHCOCF312
3
4
56
7
8
+ 8-ABV
2. Enantiomeric purification by chromatography
MTPA
MTPA MTPA
MTPA
H2N
HO
N
H2N
HO
N
DIBAL-H
(98 %) (100 %)
(75 %)
(30 %)
(70 %) (65 %)
(50 %) (65 %)
(25 %)H2N
Scheme 12. Synthesis of 5-ABV and its enantiomers.134,136
Page 87
73
2. AIMS AND SCOPE
Page 88
74
As discussed in Chapter 1.5, one of the best selective and specific high affinity radiotracer
to explore VAChT and one of the few experimental radiopharmaceuticals used in vivo in
human to obtain an early diagnosis of AD is 5-[123
I]-iodobenzovesamicol (5-[123
I]IBVM). 5-
[123
I]IBVM is SPECT radiotracer (Figure 8, page 70). We suppose that the fluorine derivative
5-fluorobenzovesamicol (5-FBVM) should be of similar affinity and selectivity for the
VAChT as 5-IBVM. This statement is based on the characteristic physico-chemical properties
of fluorine. Its small atomic size should not impose substantial steric hindrance at binding of
5-FBVM on VAChT, and its electronegativity is roughly similar to iodine's electronegativity.
In the case of successfully developed synthetic route towards 5-[18
F]FBVM, biological studies
could be done to prove the usefulness of 5-[18
F]FBVM as a radiotracer with the appropriate
pharmacokinetic and pharmacodynamic properties to selectively maps cholinergic brain areas
in vivo; providing a non-invasive means of safely and accurately evaluate the functional
integrity of cholinergic synapses in human using PET to obtain an early diagnosis of AD.
The main goal of the present PhD thesis is the synthesis of VAChT-selective ligand 5-
FBVM labelled with radioactive fluorine isotope F-18. To reach this goal, we have
established a plan based on following systematic and consecutive stages:
1. Choose appropriate synthetic method and find conditions via established theoretical
model to introduce non-radioactive fluorine isotope F-19 at the C-5 position of the 5-
aminobenzovesamicol (5-ABV).
HO
H2N
N
5-ABV
HO
F
N
5-FBVM
12
3
4
5 6
7
8 ?
2. QSAR study and in vitro VAChT binding affinity determination of 5-FBVM by
radioligand displacement study.
3. Choose and synthesize a suitable precursor of 5-FBVM for radiolabelling, and
establish theoretical model to introduce non-radioactive fluorine isotope F-19 at the C-
5 position of the benzovesamicol under conditions which can be transposed to
radiofluorination.
Page 89
75
HO
Precursor moiety
N
Benzovesamicol precursor
HO
F
N
5-FBVM
Under conditions which can be transposed to
radiofluorination
4. Develop improved method in order to acquire a variety of arylfluorides via one-pot
fluorination of the chosen type of the precursor under rapid and operationally simple
conditions.
Improved fluorination method
R
Precursor moiety
R
F
e.g. 5-FBVM
5. Examination of reaction parameters for radiofluorination of the developed
benzovesamicol precursor.
6. Synthesis of 5-[18
F]FBVM via radiofluorination of the developed benzovesamicol
precursor.
HO
Precursor moiety
N
Benzovesamicol precursor
HO
18F
N
5-[18F]FBVM
[18F]F-
Appropriate reaction conditions
The major challenge will be the radiofluorination of the corresponding benzovesamicol
precursor via aromatic nucleophilic fluorination, because benzovesamicol is a non-activated
system towards aromatic nucleophilic fluorination. Furthermore, the synthesis and isolation of
the target compound must be performed rapidly due to use of short-lived positron emitting
radionuclide F-18 (t1/2 ~ 110 min), which in the form of n.c.a. [18
F]fluoride is used in great
deficiency towards precursor and other reagents.
Page 90
76
3. RESULTS AND DISCUSSION
Page 91
77
3.1. Theoretical model for efficient one-pot fluoro-de-diazoniation
Balz-Schiemann reaction40
is a representative method and a broad scope method for the
regioselective nucleophilic introduction of fluorine into aromatic rings in preparative organic
syntheses. This is a deaminative fluorination type of reaction composed of three sequential
steps: (A.) diazoniation of primary aromatic amine in aqueous medium with sodium nitrite
(NaNO2) and fluoroboric acid (HBF4) at 0-5 oC to produce arenediazonium tetrafluoroborate
(ArN2+BF4
-), (B.) isolation and drying of ArN2
+BF4
- to avoid side formations of phenols and
biaryl ethers,156
and (C.) thermal fluorinated decomposition of ArN2+BF4
- (fluoro-de-
diazoniation).157,158
However, the method suffers from yield reproducibility problems,
because isolation and complete drying can be tedious and unsure (the method works only for
those diazonium tetrafluoroborates which can precipitate from aqueous medium), and
controlled thermal decomposition of ArN2BF4 is problematic.156,159
To overcome
reproducibility problems, simplify procedure, broaden substrate tolerance (e.g. anilines with
hydrophilic substituents, heterocyclic amines), improve safety (diazonium tetrafluoroborates
are toxic and potential explosive when perfectly dried), and to increase the yields, alternative
approaches based mostly on one-pot methodology (in situ fluoro-de-diazoniation without
isolation of arenediazonium salt) in organic and ionic solvents have been developed during
the last few decades.159–165
Page 92
78
Ar-NH2 + NaNO2 + 2 HX (aq.) ArN2+ X- + H2O + NaX
A.0-5 oC
Na+ -O-N=O + HX (aq.)
- NaX
Sodium nitrite
HO-N=OHX (aq.)
H2O+-N=O- H2O
N+=O N O+
B.
Nitrous acid(unstable)
Nitrosonium ion(nitrosyl cation)
NH2
+
R
H2N
R
- H+
+ H+
HN
R
N O
N-Nitrosamine(unstable)
N
R
Diazohydroxide
N
R
N OH2
- H2O
Aromatic diazonium ion
N O+
- H+
+ H+
C.
N O
N+ O
N OH N
R
NN
R
N
Scheme 13. (A.) Summary equation of formation of diazonium salt in acidified water solution
at 0-5 oC. (B.) Formation of nitrosonium ion/nitrosyl cation as diazonating agent. Nitrosonium
ion is a very weak electrophile due to resonance stabilisation. (C.) Mechanism of diazoniation
(N-nitrozination) of aniline.
Arenediazonium ions can undergo three types of reaction: (A.) Reactions of nucleophile at
N(2)-atom of diazonium moiety; (B.) Unimolecular nucleophilic substitution (SN1) via aryl
cation intermediate; (C.) One-electron reduction of diazonium moiety with further homolytic
dissociation of arenediazonium ion into aryl radical (Scheme 14). Understanding these
processes is pivotal to construct theoretical model for successful fluorination of 5-ABV via
modified Balz-Schiemann procedure.
A. Arenediazonium ions are soft electrophiles and coexist in cold solution with soft
nucleophiles. Stable diazo compound can be formed through attacking N(2)-atom by
soft nucleophile at enough elevated temperature. On the other hand, reaction with hard
nucleophile (e.g. hydroxide anion) gives corresponding unstable diazo compound that
undergoes decomposition to finally yield more stable product(s).
Page 93
79
B. Fluorination of anilines by Balz-Schiemann reaction is an example of unimolecular
nucleophilic aromatic substitution that proceeds via highly unstable and non-selective
aryl cation intermediate.
C. Arenediazonium ion can be subject to one-electron reduction if any present reagents or
solvent have lower redox potential than arenediazonium group. Consequently,
arendiazonium ion is decomposed via aryl radical, which is capable of abstracting a
hydrogen atom (e.g. from protic solvent) or another atom from a covalent bond (e.g.
Sandmeyer reaction).
Taken together, the diazonium ions can decompose both by ionic (heterolytic) as well as
radical (homolytic) paths.
Ar-N2
+
Ionic path(heterolytic decomposition) Radical path
- N2
Ar
Ar-X
Aryl cation
Arenediazonium ion
N(1) N(2)Ar+
Ion pair
One-electron transfer
N NAr+
XN NAr
Diazonium radical
Homolytic decomposition- N2
Ar.
Aryl radical
(g)
(g)
Protic solventAr-H Ar-Ar
X.
Ar-X
Ar.
X
+
. . .
Other radical reactions
X
X
Scheme 14. At elevated temperature aryldiazonium ion can decompose by ionic and/or
radical paths.166,167
The delicate decomposition path balance is crucially dependent on the reaction conditions
and the substitutents in the aromatic ring(s). More precisely, substituents (their mesomeric
and inductive effects) and their substitution pattern affect stability of the aryldiazonium ion
Page 94
80
and its decomposition intermediates, its redox potential and consequently its decomposition
temperature and paths.158,166,167
Substituents with electron withdrawing character (e.g. nitro
group) generally increase activation energy for de-diazoniation and are therefore rate
retarding, and vice versa for substituents with electron donating character. Moreover, the
redox potentials of the substituted aryl diazonium ions are highly useful in predicting the
nature of the de-diazoniation path. Aryl diazonium ions having electron-donating groups
increase electron density of the N(2)-diazonium atom and stabilize the diazonium cation,
thereby supressing their tendency to undergo one-electron reduction. On the other hand,
diazonium ions having electron-withdrawing substituents have a stabilising influence by
resonance on the diazenyl radical and increase the ease of their reduction.167
Redox potential of molecules is also strongly dependent on the selected solvent.
Table 2. Redox potentials vs normal hydrogen electrode (NHE) for halide ions in acetonitrile
(CH3CN) and water (H2O).167
reduced form of the redox couple
F -
Br -
I -
CH3CN H2O
2.4 3.6
1.2 2.0
1.1 2.2
Eo, V
As shown in Table 2, all halide ions have lower redox potential (easier to donate electron
and become oxidized) in acetonitrile than in water. This could be due to lower solvation
strength of CH3CN compared to H2O. The more is halide anion solvated less nucleophilic it is
and less efficiently it donates electron and become oxidized (higher redox potential).
According to the Table 2, the redox potential of fluoride is most likely not enough low for
diazonium ion to undergo a one-electron reduction by fluoride.
Thus, reaction conditions for successful (one-pot) fluoro-de-diazoniation should be
carefully chosen to promote aryl cation formation. The solvent, pH of the reaction medium,
nature of the counter ion and the presence of reducing agents and/or radical sources influence
arylfluorides yields. Choice of the solvent is one of the most important parameter to facilitate
fluoro-de-diazoniation.166,167
Chlorinated solvents such as chloroform (CHCl3,
tetrachloromethane (CCl4), and 1,2-dichlorobenzene have been reported to have beneficial
effect on arylfluride yields via probable enhancement of the ionic decomposition path.167
Page 95
81
Accoding to the studied properties of aryldiazonium ions, we can write parameters with
optimal properties for efficient one-pot fluoro-de-diazoniation reaction:
A. Solvent: Dissolving all the reagents with minimal solvation of fluoride anion in order
to preserve its nucleophilicity, non-nucleophilic, suitable high redox potential to avoid
reduction of the aryldiazonium ion and consequently suppress homolytic
decomposition path, to avoid formation of Ar-H (aprotic solvent) which can be
problematic to separate from Ar-F, high enough boiling point to decompose
aryldiazonium salt and to suppress radical decomposition path which is kinetically and
thermodynamically favoured to ionic path.
B. Temperature: Decomposition temperature of diazonium ion is unique to particular
diazonium ion and is strongly dependent on the substitution of aromatic ring(s).
C. Diazonating agent (source of nitrosonium ion): The best choice are alkyl nitrites (n-
butyl nitrite or t-butyl nitrite), although it is well known that they are mild reagents for
diazonation in organic solvents. They are soluble in organic solvents.
D. Source of fluoride anion: Should be soluble in organic solvent as boron trifluoride-
diethyl etherate (BF3xE2O), nitrosonium tetrafluoroborate (NO+BF4
-) or nitrosonium
hexafluorophosphate (NO+PF6
-). Alkali metal fluorides (CsF, KF, …) are not good
choice, because of their hygroscopicity and low solubility in organic solvents (long
reaction time and use of very high temperature can lead to thermal run-away and
multiplicity of products). Quaternary ammonium fluorides are very hygroscopic, but
have better solubility in organic dipolar aprotic solvents, such as N,N-
dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and acetonitrile (CH3CN),
than their alkali metal counterparts. They are generally commercially available in
hydrated states and for this reason are not the best choice. Only tetra-n-butyl
ammonium fluoride (TBAF) is commercially available as 1M solution in
tetrahydrofuran (THF). Silicium fluoride (SiF4) is toxic and thus not reagent of choice.
Page 96
82
3.2. Synthesis of 5-FBVM and its enantiomers via fluoro-de-diazoniation
We first performed one-pot fluoro-de-diazoniation of 5-ABV according to the literature
165
using 1,2-dichlorobenzene as the solvent, 1.2 eq. of t-butylnitrite as the diazonating agent and
1.5 eq. of boron trifluoride diethyl etherate (BF3xEt2O) as the fluoride source. As suggested, t-
butylnitrite was added portionwise to the hot (105 oC) reaction mixture of the solvent, 5-ABV,
and fluoride source, and left stirring for one hour. It is assumed that under this condition
amount of diazonium ion of starting aniline is very low, because the reaction temperature is
above the decomposition of diazonium salt, which probably have beneficial effect on the
arylfluoride (ArF) yields.165
After extraction and chromatographic purification, 5-FBVM was
obtained as a white powder in 16% yield. The product was analyzed by 1H-,
13C-nuclear
magnetic resonance (13
C-NMR) and mass spectrometry (MS).
N
HO
H2N
1,2-dichlorobenzene,
105 oC, 1h
5-ABV
1. 5-ABV2. 1.5 eq. BF3xEt2O3. 1.2 eq. t-butylnitrite N
HO
F16 % 5-FBVM
Scheme 15. Synthesis of 5-FBVM via one-pot fluoro-de-diazoniation of 5-ABV.
In Balz–Schiemann reaction, the mineral acid is used to generate nitrosonium ion ([NO]+),
and to protonate diazohydroxide, followed by elimination of water and formation of aryl-
diazonium ion (Scheme 13). In one-pot fluoro-de-diazoniation reaction using t-butylnitrite
(even weaker electrophile than nitrosonium ion) and BF3xEt2O is no protic acid to generate
nitrosonium ion from alkyl nitrite and to protonate diazohydroxide to form an aryldiazonium
ion. Accordingly, we proposed that the first rate-limiting step is nucleophilic attack of primary
aromatic amine on alkyl nitrite followed by the formation of diazohydroxide which is
complex with boron trifluoride molecule. Formation of the proposed complex probably
lowers activation energy (Ea) for breaking the CAr-N(1) bond at elevated temperature (second
rate-limiting step), and ArF is obtained via formation of diazonium fluoride and highly
unstable diazonium cation. Chlorinated solvents might stabilize this complex through weak
intermolecular electrostatic interaction, between the Cδ+
Ar-atom (diazohydroxide group exerts
negative inductive effect upon CAr-atom) and an electron pair on the chlorine atom of the
chlorinated solvent (Scheme 16). Besides their non-nucleophilic nature and high redox
Page 97
83
potential, this stabilization of the complex might be one of the reasons why chlorinated
solvents are the best choice for one-pot fluoro-de-diazoniation.
Ar NH2
Aniline
Alkyl nitrite
O N OR+
O
N OH
R
NHAr
Ar N N
H
O
Diazohydroxide
N-Nitrosamine
- ROH
Ar N N O
..
H
:
BFF
F
forming
O
Et Et
....
N N O
H
breaking
Complex
R Cl
_I
weak intermolecularelectrostatic interaction
..
.. ..
BF
F
F
ArN2 F- BF2OH - N2 (g)
First rate-limiting step
Second rate-limiting step
Ar
Ar-F
Aryl fluoride
F
Chlorinated solvent
Aryldiazonium fluoride
Aryl cation
Scheme 16. Proposed mechanism of formation arylfluoride (ArF) using aniline, alkylnitrite
and boron trifluoride etherate in chlorinated solvent under reflux.
Increasing the amount of t-butylnitrite and boron trifluoride (2 equivalents) did not increase
the 5-FBVM yield (12%).
We left reaction mixture of 5-ABV, 1.5 equivalent of BF3xEt2O, and 1.5 equivalent of t-
butylnitrite in dichloromethane (CH2Cl2) at room temperature overnight and afterwards
refluxed for 90 minutes. After the usual workup and chromatographic purification, (rac)-5-
FBVM was obtained in 25% yield. We also obtained (2R,3R)-5-FBVM and (2S,3S)-5-FBVM
by this procedure as white powder in 25% and 27% yield in two consecutive experiments,
respectively, starting from the corresponding (2R,3R)-5-ABV and (2S,3S)-5-ABV (Chapter
3.4, pages 88-98). The reaction mixture should be left at least four hours at cold to room
temperature, otherwise some unreacted 5-ABV was always detected after refluxing the
reaction mixture for 60-90 minutes.
Similarly, we detected considerable amount of unreacted 5-ABV with no formation of 5-
FBVM (thin layer chromatography, 1H-, and
13C-NMR) when 1.5 eq. of nitrosonium
tetrafluoroborate (NOBF4; diazonating agent and fluoride source), or 1.5 eq. of sodium
tetrafluoroborate (NaBF4) with 1.5 equivalent of t-butylnitrite, or 1.5 eq. tetra-n-
Page 98
84
butylammonium fluoride (TBAF 1M soln. in THF) with 1.5 eq. t-butylnitrite were used under
similar reaction conditions as described in previous paragraph. Failed attempts are probably
due to insolubility of NOBF4 and NaBF4 in chlorinated solvents, and much lower fluorinating
efficiency of TBAF compared to BF3xEt2O in this type of reaction.
Ionic liquid solvents are alternative reaction media of increasing interest in numerous types
of reactions; even in fluorination (Scheme 17).164,168–173
They are regarded as an
environmentally friendly reaction media (“green solvents”), in contrast to the volatile and
many times toxic organic solvents widely used in organic reactions. They can act as catalysts
by accelerating reaction rates (shorter reaction time and lower reaction temperature), enhance
the reactivity of reagents, improve reaction selectivity, reduce the formation of by-products
and facilitate catalyst recovery. According to the literature, fluorination method using ionic
solvents does not require strictly anhydrous conditions, which is in contrast what is generally
considered to be required for fluorination reactions. It is sometimes enough to use just
catalytic amounts of the solvent to complete reaction in relative short time. However, a major
disadvantage is their high cost that makes their regeneration an important issue.
Our attempt to perform one-pot fluoro-de-diazoniation of 5-ABV in ionic liquid, namely
[emim]x[OTf] (1-ethyl-3-methylimidazolium trifluoromethanesulfonate/triflate), using
nitrosonium tetrafluoroborate failed.164
It is important to note that 5-ABV was not soluble in
[emim]x[OTf] and that anion exchange between aryl diazonium cation and the cationic part of
the ionic solvent most probably prevented formation of 5-FBVM (Scheme 17).164,173
Page 99
85
CF3SO3N2 BF4
Aryl-diazonium tetrafluoroborate
N2
CF3SO3 BF4
Aryl-diazonium triflate
F
R R
O
S
O
O
CF3
Trifluoro-methanesulfonic acid aryl ester
RArylfluoride
R
- N2 (g)- N2 (g)heat heat
emim OTf emim BF4x x
N NEt Et
N NEt Et
CF3SO3 BF4
N NEt Et
N NEt Et
Scheme 17. Anion exchange (metathesis) between an aryl-diazonium ion and a cation of the
ionic solvent can (drastically) decrease yield of the desired arylfluoride, inspite of the low
nucleophilicity of the anion (e.g. triflate) of the ionic solvent. This phenomenom has special
importance in reactions involving short-lived reactive intermediates (aryl cation).
According to the anion exchange and the fact that ionic liquid is used in excess to fluoride
source, [emim]x[BF4] or [emim]x[PF6] should be the best choice. However, these solvents
can not be used in radiofluorination due to source of “cold’’ fluoride.
Page 100
86
3.3. Theoretical model for efficient fluoro-de-triazenation and synthesis
of 5-FBVM from the corresponding triazene precursor
1-Aryl-3,3-dialkyltriazenes (Ar-N=N-NR'R''), compounds having a diazoamino group, are
regarded as protected form of anilines and stable surrogates of aryldiazonium ions.44
After
treatment with the appropriate reagents, they are adaptable to numerous synthetic
transformations with wide applicability in chemical, medical and technological fields.174,175
Triazene group is stable towards electrophilic reagents, some oxidants and reductants, but not
towards acids at room or elevated temperature. Thus, their acid-triggered thermal
decomposition parallels that of diazonium ion reactions (Scheme 18).166,167,176
Consequently
the theoretical model for efficient fluoro-de-triazenation is almost the same as for fluoro-de-
diazoniation, except that the acid is present (Scheme 18).
Ar N(1) N(2) N(3)R2
HAAr N N NHR2 A
R2NHHA
R2NH2 A
Ar N N A
- N2 (g)
Ar
Ar F
F
AAr A
Solvent
Ar Solv
Ionic path
Ar
RedOx
Protic solvent, HA,triazene
Radical pathAr H
Ar Ar + ...
- N2 (g)
.
heat
Scheme 18. General competitive processes during fluoro-de-triazenation and fluoro-de-
diazoniation in the protic acid mediated decomposition of 1-arly-3,3-dialkyltriazenes.176
In
order to obtain arylfluoride in satisfactory yields, reaction conditions should be carefully
chosen that the radical decomposition path is suppressed and the ionic path maximized.
Aryltriazenes are safely and mostly readily prepared by coupling of a diazotized aniline
with amine or by the action of Grignard reagents on aryl azides. They can be stored for a long
period of time at cold (0-5 oC) protected from light.
177 Additionally, aryltriazenes can be
easily isolated, chromatographically purified, introduced in the early stages of the synthesis,
functionalized and thermally decomposed in the presence of protic acid in the latest stages if
the preceding reactions have been performed under non-acidic conditions.176,178
Thus, fluoro-
de-triazenation can be attractive means of forming 18
F-labelled fluoroaromatics by direct
nucleophilic substitution with [18
F]fluoride due to both the one-pot and rapid nature of
triazene transformation to fluoroarenes in order to obtain PET tracers with good specific
Page 101
87
activity. Although diaryliodonium salts have mainly been proven to be much more efficient
precursors for the introduction of [18
F]fluoride onto mostly simple non-activated aromatic
rings than fluoro-de-triazenation, preparation of complex diaryliodonium salt, such as from 5-
ABV, in a highly pure and stable form needed for radiofluorination can be very problematic.
Due to the reasons stated above, we decided to prepare corresponding 5-ABV triazene
precursor (5-TBV) and search for the fluoro-de-triazenation conditions that can be transposed
to radiolabelling.
The acid-triggered thermal decomposition of 1-aryl-3,3-dialkyltriazenes involves N(3)
protonation by a strong protic acid, followed by the rate-limiting heterolytic N(2)-N(3) bond-
cleavage at elevated temperature giving the corresponding diazonium ion and dialkylamine.
The N(3) protonation is a crucial step, because it competes with N(1) protonation but both are
more favourable than N(2) protonation. However, N(1) protonation does not induce triazene
decomposition, because it allows charge delocalisation over the three nitrogen atoms. In
contrast, N(3) protonation lengthens and destabilizes the N(2)-N(3) bond by inhibiting
delocalization across the triazene linkage. Electron-donating substituents on aryl ring facilitate
the N(3) protonation because electron-donation stabilizes the positive charge on N(3) and
therefore promote the N(2)-N(3) bond-breaking or diazonium ion formation.167,179,180
According to the Scheme 18 and chemical properties of the aryltriazenes and
aryldiazonium ions, the optimal properties of the used acid in fluoro-de-triazenation are the
following: (A.) strong acid to protonate and induce the cleavage of the triazene moiety; (B.)
solubility in chosen solvent (e.g. chlorinated solvents); (C.) non-nucleophilic conjugated base
(A-) with high redox potential to avoid the acid counterion substitueted byproduct (Ar-A)
formation and inducing radical decomposition pathway via reduction of the corresponding
diazonium ion intermediate.
We prepared the suitable triazene precursor 5-ABV-diethyltriazene (5-TBV) by the
standard synthetic procedure via diazoniation of the 5-ABV in the cold acidified water
mixture using sodium nitrite with subsequent addition of diethylamine to provide 5-TBV as a
white powder in almost quantitative yield (95%). (2R,3R)-5-TBV and (2S,3S)-5-TBV were
obtained by the same procedure as white powders in 96% and 92% yield, respectively,
starting from the corresponding (2R,3R)-5-ABV and (2S,3S)-5-ABV. We also prepared
triazene precursor (rac)-5-TBV in 88% yield by modified method using boron trifluoride
etherate and t-butylnitrite in CH2Cl2 with subsequent addition of diethylamine (Chapter 3.4).
Page 102
88
3.4. 3D QSAR study, synthesis, and in vitro evaluation of (+)-5-FBVM as
potential PET radioligand for the vesicular acetylcholine transporter
(VAChT)
Mitja Kovac a, Sylvie Mavel
a,*, Winnie Deuther-Conrad
b, Nathalie Méheux
a, Jana Glöckner
b, Barbara Wenzel
b, Marko Anderluh
c, Peter Brust
b, Denis Guilloteau
a, Patrick Emond
a
a Université François-Rabelais de Tours, INSERM U930, CHRU, Hôpital Bretonneau, Service
de Médecine Nucléaire, 37000 Tours, France
b Forschungszentrum Dresden-Rossendorf, Research Site Leipzig, Institute of
Radiopharmacy, Permoserstr. 15, 04318 Leipzig, Germany
c University of Ljubljana, Faculty of Pharmacy, Department of Pharmaceutical Chemistry,
Aškerčeva 7, 1000 Ljubljana, Slovenia
Published in: Bioorganic & Medicinal Chemistry 18 (2010) 7659-7667
Page 103
89
SUMMARY:
In this report we described the synthesis of 5-FBVM and its enantiomers via fluoro-de-
diazoniation from the corresponding 5-ABV. To demonstrate the suitability of triazene as
leaving group, the non-radioactive fluoro-de-triazenation of 5-TBV resulting in reasonably
25% yield of 5-FBVM was accomplished. We also performed three QSAR studies based on
32 vesamicol and benzovesamicol derivatives taking into account for the first time the
stereoselectivity of the VAChT binding site in order to predict the binding affinity of (2R,3R)-
and (2S,3S)-5-FBVM. Both enantiomers exhibited high in vitro VACHT binding affinites
determined by radioligand displacement studies, and were in the same range as 5-IBVM as
predicted by 3D QSAR studies. Only (2S,3S)-FBVM was selective enough over σ1 receptors
to warrant further investigation as a potential PET radioligand for in vivo mapping of
cholinergic nerve terminals.
STATEMENT: I declare, that nobody of co-authors has used the article 3D QSAR study,
synthesis, and in vitro evaluation of (+)-5-FBVM as potential PET radioligand for the
vesicular acetylcholine transporter (VAChT), published in the Bioorganic & Medicinal
Chemistry, for his/her own thesis.
Page 104
3D QSAR study, synthesis, and in vitro evaluation of (+)-5-FBVM as potentialPET radioligand for the vesicular acetylcholine transporter (VAChT)
Mitja Kovac a, Sylvie Mavel a,*, Winnie Deuther-Conrad b, Nathalie Méheux a, Jana Glöckner b,Barbara Wenzel b, Marko Anderluh c, Peter Brust b, Denis Guilloteau a, Patrick Emond a
a Université François-Rabelais de Tours, INSERM U930, CHRU, Hôpital Bretonneau, Service de Médecine Nucléaire, 37000 Tours, Franceb Forschungszentrum Dresden-Rossendorf, Research Site Leipzig, Institute of Radiopharmacy, Permoserstr. 15, 04318 Leipzig, Germanyc University of Ljubljana, Faculty of Pharmacy, Department of Pharmaceutical Chemistry, Aškerceva 7, 1000 Ljubljana, Slovenia
a r t i c l e i n f o
Article history:Received 18 May 2010Revised 26 May 2010Accepted 12 August 2010Available online 17 August 2010
Keywords:Benzovesamicol derivativeVAChTTriazeneFluoro-dediazoniation3D QSAR
a b s t r a c t
Located in presynaptic cholinergic nerve terminals, the vesicular acetylcholine transporter (VAChT) rep-resents a potential target for quantitative visualization of early degeneration of cholinergic neurons inAlzheimer’s disease using PET. Benzovesamicol derivatives are proposed as radioligands for this purpose.We report QSAR studies of vesamicol and benzovesamicol derivatives taking into account the stereose-lectivity of the VAChT binding site. Use of different data sets and different models in this study revealedthat both enantiomers of 5-fluoro-3-(4-phenyl-piperidin-1-yl)-1,2,3,4-tetrahydro-naphthalen-2-ol(5-FBVM) are promising candidates, with predicted VAChT affinities between 6.1 and 0.05 nM. The syn-thesis of enantiopure (R,R)- and (S,S)-5-FBVM and their corresponding triazene precursors for futureradiofluorination is reported. Both enantiomers exhibited high in vitro affinity for VAChT [(+)-5-FBVM:Ki = 6.95 nM and (�)-5-FBVM: Ki = 3.68 nM] and were selective for r2 receptors (�70-fold), only (+)-5-FBVM is selective for r1 receptors (�fivefold). These initial results suggest that (+)-(S,S)-5-FBVM warrantsfurther investigation as a potential radioligand for in vivo PET imaging of cholinergic nerve terminals.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
The degeneration of cholinergic neurons in the brain is one of themost significant neuropathological features in Alzheimer’s disease(AD) synapse disorder has been shown to precede the neurofibrillaryand neuritic aspects of AD during the course of the disease,1 and theloss of synaptic terminals correlates better with cognitive declinethan extracellular plaque load or loss of neurons.2 Located in presyn-aptic cholinergic nerve terminals, the vesicular acetylcholine trans-porter (VAChT) is postulated to be a valuable target for in vivoquantitative visualization of early neurodegenerative processes inAD by using molecular imaging techniques such as SPECT (SinglePhoton Emission Computed Tomography) and PET (Positron Emis-sion Tomography), PET being regarded as superior in terms of detec-tion efficiency, spatial resolution, and quantification. However,VAChT-specific tracer compounds labeled with the short-lived PETradionuclides 18F and 11C have not been developed for clinical useto date. Approaches to the development of tracers for VAChT imag-
ing by PET have focused on compounds such as benzovesamicols,3–6
trozamicols,7,8 or morpholino vesamicols,9,10 all based on the struc-ture of (�)-vesamicol, a drug that binds to a side allosteric to the act-eylcholine transport side with low-nanomolar affinity.11–14
However, (�)-vesamicol possesses moderate affinity to a-adreno-ceptors and nanomolar affinity to r-receptors.15 In combinationwith similar expression patterns of VAChT16 and r1-receptors17 inthe brain the latter cause marginal signal-to-background ratios inin vivo imaging by PET, one reason why so many VAChT radioligandshave failed in pre-clinical evaluation to date. On the other hand, thesuccessful application of the benzovesamicol-related SPECT ligand(�)-5-[123I]IBVM (5-iodo-3-(4-phenyl-piperidin-1-yl)-1,2,3,4-tet-rahydro-naphthalen-2-ol, Kd = 0.30 nM)18–21 has encouraged fur-ther research into the design of VAChT-specific PET tracers.Structure-affinity studies assessing the potential of iodobenzove-samicol derivatives for visualization of cholinergic terminals haverevealed that there is considerable bulk tolerance at differentstructural positions of the benzovesamicol molecule.18,22
Therefore we have recently synthesized and evaluated newbenzovesamicol derivatives21 and aza-analogs of trozamicol deriv-atives both in vitro and in vivo in animals models.23–25 Several 18F-labeled benzovesamicol derivatives have now been synthesizedincluding [18F]NEFA, [18F]FAA,6 [18F]FEOBV,5 and [18F]FPOBV3
(Table 1). However, except for [18F]FEOBV, which is currently
0968-0896/$ - see front matter � 2010 Elsevier Ltd. All rights reserved.doi:10.1016/j.bmc.2010.08.028
* Corresponding author. Address: Faculté de Pharmacie, Laboratoire de Biophy-sique Médicale et Pharmaceutique, 31 avenue Monge, 37200 Tours, France. Tel.: +332 47 36 72 40; fax: +33 2 47 36 72 24.
E-mail address: [email protected] (S. Mavel).
Bioorganic & Medicinal Chemistry 18 (2010) 7659–7667
Contents lists available at ScienceDirect
Bioorganic & Medicinal Chemistry
journal homepage: www.elsevier .com/locate /bmc
Page 105
under investigation in PET studies,4 these PET tracers have notbeen suitable for in vivo applications, possibly due to the metabolicsusceptibility of the F-carrying substituents, for example, amide inNEFA and alcoholate in FEOBV. In previous paper, FPOBV instabilityin vivo,3 possibly arising from the weak strength of the C(sp3)–Fbond (0.00079 kcal/mol, DiscoveryStudio2.5�, Accelrys Inc.) caus-ing de-fluorination is reported. To increase the metabolic stabilityand to reinforce the C–F bond strength by a C(sp2)–F bond(0.0628 kcal/mol, DiscoveryStudio2.5�, Accelrys Inc.), we havedeveloped the novel benzovesamicol analog 5-FBVM as a fluoroanalog of 5-IBVM (Table 1).
In 2008, Szymoszek et al.26 have published a first ComparativeMolecular Field Analysis (CoMFA) study using a partial leastsquares (PLS) algorithm for a set of 37 vesamicol derivatives, cov-ering three different structural types, to predict the binding affinityof vesamicol-type ligands from their respective molecular struc-ture. To expand these efforts, we have performed a further 3DQSAR study, which is based on 32 vesamicol and benzovesamicolderivatives (Table 1) in order to predict the binding affinity forthe new compound 5-FBVM. Furthermore, this study consideredfor the first time the stereoselectivity of the binding of vesamicolderivatives to the VAChT protein.
Moreover, we also wished to improve the accessibility of theradiolabeling procedure by radiofluorination of non-activated aro-matic cycles (i.e., without an electron withdrawing group in orthoor para position to the leaving group). Therefore, in this report wedescribe not only the synthesis of 5-FBVM via fluoro-dediazonia-tion based on the secondary amine 5-ABV, but also the synthesisof a suitable triazene precursor (5-TBV) for future 18F-labeling. Todemonstrate the suitability of triazene as leaving group, the non-radioactive fluorination of 5-TBV resulting in 5-FBVM wasaccomplished. Because of the stereoselective binding of vesamicolderivatives, 5-ABV was enantioseparated via Mosher ester
synthesis and provided the basis for the synthesis of enantiopure(+)-(S,S) and (�)-(R,R)-5-FBVM as well as (S,S) and (R,R)-5-TBV.
2. Results and discussion
2.1. QSAR study
The QSAR study is based on 32 derivatives which belong to theclasses of vesamicols (I) and benzovesamicols (II) (Table 1). Sixderivatives were synthesized and evaluated regarding VAChT affin-ity and specificity by in-house in vitro assays, the Ki values of theremaining compounds were taken from literature. Since bindingto the VAChT is known to be highly enantioselective (generally,the in vitro affinity for the VAChT of (�)-enantiomers is about 10times greater) we made three sets from the same overlay: Set 1for all 32 derivatives, Set 2 for the (�)-enantiomers (20 deriva-tives), and Set 3 for the (+)-enantiomers (12 derivatives). All thestructures were minimized under a CHARMm forcefield with a rootmean squared (RMS) difference of energy gradient reached0.1 kcal/mol Å (Discovery Studio� 2.5, Accelrys Inc., San Diego,CA). According to crystallographic findings on vesamicol deriva-tives10,27 and ABV (data not shown, structure see Table 1), thepiperidine ring is in a chair conformation, and is almost perpendic-ular to the cyclohexanol ring, with the hydroxyl in trans position asan equatorial conformer. The carbons C-9, C-11, and C-13 of piper-idine (see Table 1) and C-OH were chosen for the overlay. Ki valueswere taken from the literature, and pKi = �log Ki was used for theGenetic Function Approximation (GFA) algorithm.28 The QSAR pro-gram presents 179 physicochemical descriptors as different elec-tronic, spatial, shadow, shape, and thermodynamic indices. Afterseveral filters (analyzing the correlation matrix, eliminating thehighly correlated descriptors, and eliminating descriptors withtoo wide a range of training data (>700)), only 26 2D or 3D
Table 1Vesamicol analogs I and benzovesamicols II used for the QSAR studies
N
HO
N
HO
R R1
R2R3
5
8
I II
A CD
9
1113
R I Refa R1 R2 R3 II Refa
H (�)-vesamicol 21 H 5-I H (�)-5-IBVM 21(+)-vesamicol (+)-5-IBVM
o-CH3 (�)-oMV 30 H 5-N(Et)COCH2F H (�)-NEFA 50(+)-oMV
p-CH3 (�)-pMV 30 H 5-O(CH2)2F H (�)-FEOBV(+)-pMV (+)-FEOBV
o-I (�)-oIV 52 H 5-(CH2)3F H (�)-FPOBV 21(+)-oIV (+)-FPOBV
m-I (�)-mIV 52 H 5-OCH2CHCHI H (�)-AOIBV 21(+)-mIV (+)-AOIBV
p-I (�)-pIV 52 H 5-I and 8-OCH3 H (�)-MOIBV 21(+)-MOIBV
H 5-I CH2NH2 (�)-MAIBV 21(+)-MAIBV
H 6-I H (�)-6-IBVM 22H 7-I H (�)-7-IBVM 22H 8-I H (�)-8-IBVM 22p-I H H (�)-pI-BVM 22m-I H H (�)-mI-BVM 53
H 5-NH2 H (�)-ABV 50(+)-ABV
H H H (�)-BVM 50
a The data of binding affinities used for QSAR study were taken from these references.
7660 M. Kovac et al. / Bioorg. Med. Chem. 18 (2010) 7659–7667
Page 106
descriptors were retained as dependent on pKi. The definitions ofmean descriptors are given in Table 2. In the GFA model, linearor quadratic terms of descriptors were allowed in the selection.For the statistical parameters, the cross validated r2 is not the mostimportant, and the Friedman’s lack-of-fit (LOF) score, which evalu-ates the QSAR model by considering the number of descriptors aswell as the quality of fitness, is chosen: the lower the LOF, the lesslikely it is that the GFA model will fit the data. The significantregression is given by F, and the higher the value, the better themodel. The standard errors of regression coefficients are given inparentheses. GFA models were tested with 5-FBVM, with the excel-lent predictive sub-nanomolar Ki.
2.1.1. Set 1N = 32: (+) and (�)-enantiomers.Linear model form: pKi(+)/(�)pred = 79.61(±10.49) � 39.86
(±5.82) � JY + 0.99(±0.22) � Dipole_Z + 0.0185(±0.004) � Jurs_PN-SA_2 � 4.61(±1.45) � Shadow_nu + 3.28(±1.07) � Bond Energy�0.894(±0.348) � Van der Waals Energy.
Predicted affinity: pKi(+)-5-FBVMpred = 8.91 pKi(�)-5-FBVMpred =9.34.
The major contributing factors are JY > Shadow_nu > Bond En-ergy > Van der Waals Energy > Dipole_Z > Jurs_PNSA_2.
Quadratic model form: pKi(+)/(�)pred = 71.22(±9.53) � 32.92(±4.57)� JY + 1.03(±0.21)� Dipole_Z + 0.0160(±0.003)� Jurs_PNSA_2� 4.10(±1.28) � Shadow_nu � 0.73(±0.23) � Bond Energy � Vander Waals Energy.
Predicted affinity: pKi(+)-5-FBVMpred = 9.21 pKi(�)-5-FBVMpred =9.75.
2.1.2. Set 2N = 20: (�)-enantiomers.Linear model form: pK i(�)pred = 14.06(±3.67) + 0.0759
(±0.0120)�Molecular_SASA� 0.0766(±0.011)� E_ADJ_mag + 0.9205(±0.0834)� Jurs_PPSA_3� 38.88(±7.73)� Jurs_RPSA + 2.89(±1.13)�RadofGyration � 5.20(±0.76) � Shadow_nu � 66.45(±10.20)�Shadow_XZfrac + 27.27(±4.31)� Shadow_YZfrac� 0.335(±0.051)�Dihedral Energy.
Predicted affinity: pKi(�)-5-FBVMpred = 8.64.The major contributing factors are Shadow_XZfrac > Jurs_
RPSA > Shadow_YZfrac > Shadow_nu > RadofGyration � Jurs_PP-SA_3 > Dihedral Energy > Molecular_SASA � E_ADJ_mag.
Quadratic model form: pKi(�)pred = 26.21(±3.37)� 5.32(±0.85)JY � Shadow_nu � 0.0031(±0.0006)Dipole_Z � Jurs_PNSA_2� 0.0053(±0.001) Jurs_DPSA_3 � Dihedral Energy + 0.145(±0.02)Jurs_PPSA_3 � Shadow_nu.
Predicted affinity: pKi(�)-5-FBVMpred = 10.26.
2.1.3. Set 3N = 12: (+)-enantiomers.Linear model form: pKi(+)pred= 27.60(±0.02) + 3.42(±0.003)�
CHI_2� 1.73(±0.001)� CHI_V_3_P� 0.059(±0)� E_ADJ_mag + 19.66(±0.05)� JX� 30.19(±0.05)� JY� 0.654(±0.0003)� Dipole_Z � 9.64(±0.01) � Jurs_RPSA � 3.73(±0.007)� Shadow_XYfrac + 3.67(±0.002)� Bond Energy � 0.067(±0) � Dihedral Energy.
Predicted affinity: pKi(+)-5-FBVMpred = 8.22.The major contributing factors are JY > JX > Jurs_RPSA > Sha-
dow_XYfrac � Bond Energy � CHI_2 > CHI_V_3_P > Dipole_Z > Di-hedral Energy � E_ADJ_mag.
The best predictive values for 5-FBVM were obtained with thelinear model rather than the quadratic model. Furthermore, consid-ering the (�)-enantioselectivity of VAChT binding site, we focusedon (+)/(�) and (�) linear models. For set 1 ((+)/(�) model), the maindescriptor was JY (Balaban index which characterizes the shape of amolecule taking into account the relative covalent radius of theatoms of the model) with a mean value for the 32 compounds of1.50. As the coefficient in the equation was negative, the smallerthe value, the better the affinity for the VAChT (JY(�)-5-FBVM = 1.460compared to JY
(�)-IBVM= 1.447 and JY(�)-vesamicol = 1.579). For set 2 ((�)
model), the main descriptor was Shadow_XZfrac (a steric descrip-tor) with a mean value for the 20 compounds of 0.606. The negativecoefficient of shadow in the XZ plane indicated that a decrease inthe area of the molecular shadow in the XZ plane (Shadow_XZfrac(�)-5-FBVM = 0.577 compared to Shadow_XZfrac(�)-IBVM = 0.581 andShadow_XZfrac(�)-vesamicol = 0.611) was favorable for affinity forthe VAChT. The equations showed multiple occurrence of Jursdescriptors,29 suggesting the importance of charge distributionand surface areas, in particular for the Jurs_RPSA descriptor (totalpolar surface area divided by the total molecular solvent-accessiblesurface area) with a mean value for the 20 compounds of 0.0789.Since Jurs_RPSA depends on polar surface, fluorine derivativeshad the highest values and the coefficient in the equation wasnegative. For 5-FBVM the descriptor was the smallest of thefluorine derivatives (Jurs_RPSA(�)-5-FBVM = 0.128 compared toJurs_RPSA(�)-NEFA = 0.152 and Jurs_RPSA(�)-FEOBV = 0.130).
The 3D QSAR model (Discovery Studio� 2.5, Accelrys), definesthe critical regions (steric or electrostatic) affecting binding affin-ity. A contour plot of the electrostatic field region favorable(in blue) or unfavorable (red) for the VAChT affinity is shown inFigure 1 with the superposition of both stereoisomers. As reportedby the 3D QSAR modeling shown that substitutions near the 5-po-sition should increase the affinity for VAChT (as it was previouslysuggested by Szymoszek et al.26) as well as an electropositive sub-stituent (near the 8-position) as shown in Figure 2. A good ligandshould have strong Van der Waals attraction in the green areaand a polar group in the blue electrostatic potential area. Figure3 shows the differences obtained for the 3D QSAR model with po-sitive coefficients (in green) on a Van der Waals grid between the(�)-enantiomers model and the (+)-enantiomers model. Moreoverwe observed an overlap of a negative yellow area near the 5-posi-tion for (+)-enantiomer FBVM in the favorable positive green areaof (�)-enantiomers model, this could explain partially the stereo-specificity of VAChT binding site. With this good 3D model(N = 32) pKi(�)-5-FBVM = 9.72 and pKi(+)-5-FBVM = 8.24 with r = 0.922and r2 = 0.851 could be predicted, corresponding to the experimen-tal finding reported in Table 3 (pKi = 8.43 and 8.16, respectively).
A third QSAR study was performed based on Bayesian modeling(Discovery Studio� 2.5, Accelrys) which distinguishes ‘active’
r2 = 0.7425 r2 (adj) = 0.6807 r2 (pred) = 0.6101 RMS residualerror = 0.7419
FriedmanL.O.F. = 1.155
r2 = 0.7469 r2 (adj) = 0.6982 r2 (pred) = 0.6454 RMS residualerror = 0.7213
FriedmanL.O.F. = 1.028
r2 = 0.9591 r2 (adj) = 0.9223 r2 (pred) = 0.8523 RMS residualerror = 0.3716
FriedmanL.O.F. = 0.6274
r2 = 0.8346 r2 (adj) = 0.7904 r2 (pred) = 0.7359 RMS residualerror = 0.6105
FriedmanL.O.F. = 0.7662
r2 = 1.0000 r2 (adj) =1.0000
r2 (pred) =1.0000
RMS residualerror = 0.0004352
FriedmanL.O.F. = 1.844e�006
M. Kovac et al. / Bioorg. Med. Chem. 18 (2010) 7659–7667 7661
Page 107
ligands from baseline ligands. The Bayesian statistics assign theprobability for each individual descriptor of a molecule to be amember of an ‘active’ class. From the data set of 32 known ligands(Table 1), 81% were defined as ‘active’ or ‘inactive’, with a cutoff va-lue at a pKi = 7.69 (Ki <25 nM). From this effective model we tested(�) and (+)-5-FBVM to predict their activity. The results did not
distinguish these two enantiomers, they were both predicted as‘active’ with a probability of 82%. From the listing scores obtainedin this Bayesian model, the closest compound of FBVM was(�)oMV, which presented a Ki = 6.7 nM,30 that is, was close to theexperimental findings (Ki(�)-5-FBVM = 3.7 nM and Ki(+)-5-FBVM =6.9 nM, Table 3).
Table 2Description of the molecular properties used as descriptors in QSAR studies
Property Description
Molecular_SASA 2D surface area Total solvent accessible surface areaCHI-2 2D topological
descriptorsUnmodified molecular connectivity indices. This type of emphasizes different aspects of atom connectivity within amolecule—the amount of branching, ring, structures present and flexibility
CHI-V_3_P 2D topologicaldescriptors
E_ADJ_mag 2D topologicaldescriptors
JX 2D topologicaldescriptors
Highly discriminating descriptor. This Balaban indices characterize the shape of a molecule taking into accountelectronegativity of the atoms of the model
JY 2D topologicaldescriptors
Highly discriminating descriptor. This Balaban indices characterize the shape of a molecule taking into account relativecovalent radius of the atoms of the model
Shadow XZ frac Spatial Projection of molecular surface on a plan (XZ)Shadow XY frac Spatial Projection of molecular surface on a plan (XY)Shadow YZ frac Spatial Projection of molecular surface on a plan (YZ)Shadow_nu Spatial Ratio of largest to smallest dimension of projection of molecular surface on a planDipole Z 3D electronic
descriptorsDipole moment
Jurs_PNSA_2 Total charge weighted negative surface areaJurs_PPSA_3 Atomic charge weighted positive surface areaJurs_RPSA Relative polar surface area: total polar surface area divided by the total molecular solvent-accessible surface areaRadofGyration 3D molecular
propertiesAngle energy Molecular
propertiesConformational
Bond energy Molecularproperties
Conformational
Electrostaticenergy
Molecularproperties
Dihedral energy Molecularproperties
Van der Waalsenergy
Molecularproperties
Conformational
Figure 1. Isosurface of the 3D QSAR model coefficients on Electrostatic Potentialgrids with positive (in blue) and negative (in red) coefficients for the alignedmolecular structures of 32 (+)/(�)-enantiomers I and II in solid representation (A, C,and D visualize cycles as precise in Table 1).
Figure 2. Isosurface of the 3D QSAR model positive coefficients on Van der Waalsgrid (in green) and Electrostatic Potential grid (in blue) for (+)/(�)-enantiomermodel in solid representation with (�)-5-FEOBV.
7662 M. Kovac et al. / Bioorg. Med. Chem. 18 (2010) 7659–7667
Page 108
From these three QSAR studies, 5-FBVM is predicted to be a li-gand with nanomolar VAChT binding affinity, without significantdifferences between the two enantiomers.
2.2. Chemistry
Synthesis of the 5-ABV enantiomers which were the basis for allfluorinations has already been described.5,27,31
The Balz-Schiemann reaction (1927) is a well known process ofdeamine fluorination of aniline via the SN1 mechanism by the ac-tion of sodium nitrite (NaNO2), followed by thermal decompositionwith fluoroboric acid (HBF4). The mechanism of halogeno-dediazo-tization may take place via an ionic pathway (heterolytic decom-position)32,33 or via a radical pathway.34 An alkyl nitrite such asn-butylnitrite or t-butylnitrile, that is, soluble in organic solventscould be a good diazotizating agent according to the literature.35,36
Moreover they are very weak electrophiles (even weaker than thenitrosonium ion). The solvent, which is one of the key parametersfor a successful reaction, should be a chlorinated organic solventbecause it is aprotic and presents non-nucleophilic and non-oxi-dizing properties. Ionic liquid solvents are known to improve theyield of the Balz-Schiemann reaction.37,38 Because of the high re-dox potential of fluoride anion (e.g., F2/F� in CH3CN E = 2.4 V),the formation of a fluoroaryl bond proceeds generally via a hetero-lytic mechanism.39 Furthermore, the redox potential could be in-creased by solvation.39 The source of the fluorine atom isvariable: silicium fluoride (SiF4) is fairly toxic; alkali metal fluo-rides (CsF or KF) are hygroscopic with poor solubility; and quater-nary ammonium fluorides are very hygroscopic but are soluble inaprotic solvents. Other salts such as boron trifluoride-diethyletherate (BF3�Et2O) are excellent fluorinating agents35 and are sol-uble in chlorinated solvents in contrast to nitrosonium tetrafluoro-borate (NOBF4) or nitrosonium hexafluorophosphate (NOPF6). Thedecomposition temperature of the diazonium group depends onfurther substituents on the aromatic ring.40
We tested several fluorinating agents and reaction conditionssuch as (i) 1.5 equiv of NOBF4, CH2Cl2, 1 h reflux; (ii) 1.5 equiv ofNaBF4, 1.5 equiv t-BuONO, CH2Cl2, 1 h, rt; (iii) 1.5 equiv of(t-Bu)4NF, 1.5 equiv t-BuONO, CH2Cl2, 1 h, reflux; and (iv) 1.5 equivof BF3�Et2O, 1.2 equiv t-BuONO, C6H4Cl2, 1 h, 60 �C. Scheme 1presents the best results of one pot fluoro-dediazoniation of the pri-mary aromatic amine 5-ABV to obtained 5-FBVM in moderate yield(around 25% yield). The chlorinated organic solvents dichloroben-zene (method A)35 or CH2Cl2 (method B) were suitable for such
Table 3Affinities and selectivity of FEOBV, (rac)-5-IBVM, (rac)-5-ABV, and 5-FBVM for rVAChT, r1 and r2 (Ki values are given in means ± SD)
rVAChT Ki values, in nM Ratio of Ki values
r1 r2 r1/VAChT r2/VAChT
(�)-FEOBV 19.6 ± 1.1a 209 ± 94a n.d. 10.7 n.c.(+)-FEOBV 56.9 ± 4.8a 269 ± 37a n.d. 4.7 n.c.(rac)-5-IBVM 15.8 ± 5.42 266 ± 113a n.d 17 n.c.(rac)-5-ABV 40.6 ± 1.69 652 ± 60 n.d 16 n.c.(rac)-5-FBVM 10.9 ± 2.78 13.2 ± 5.84 229 ± 60a 1.21 21(+)-(S,S)-5-FBVM 6.95 ± 0.62 (pKi = 8.16) 38.1 ± 4.85 526b 5.48 76(�)-(R,R)-5-FBVM 3.68 ± 0.48 (pKi = 8.43) 3.57 ± 0.86 252 ± 13a 0.97 68
Ki values were determined by competition of derivatives against bound (�)-[3H]vesamicol under equilibrium at 22 �C, unless stated.n.d.: not determinated.n.c.: not calculated.All experiments were performed in triplicate (n P 3; an = 2; bn = 1).
Figure 3. Isosurface of the 3D QSAR model positive coefficients on Van der Waalsgrids in green and negative coefficients on Van der Waals grid in yellow: in solidrepresentation for (+)-enantiomers model, in quad mesh representation for (�)-enantiomers model with (+) and (�)-5-FBVM.
N
H2N
HO1.5 eq. BF3.Et2O, 1.2 eq. t -BuONO,C6H4Cl2
105°C, 1hN
F
HO
5-ABV 5-FBVM1) 1.5 eq. BF3.Et2O, 1.5 eq. t-BuONO,CH2Cl2 , RT, overnight2) reflux, 1.5h
Method A ; Yield 16%
Method B ; Yield 25%
Scheme 1. Synthesis of 5-FBVM from 5-ABV by one pot fluoro-dediazotization.
M. Kovac et al. / Bioorg. Med. Chem. 18 (2010) 7659–7667 7663
Page 109
one pot fluoro-dediazoniation. Increasing of the amount of nitrite orfluorine agent (2 equiv) did not increase the yield (12% yield).
Since this method is not useable for radiolabeling, we developeda further procedure to introduce a fluorine atom, in particular forfuture 18F-radiofluorination. Aryl-dialkyltriazenes (as a protectedform of aryldiazonium ions) have been extensively studied for dif-ferent purposes (for review, see Ref. 41). We synthesized triazene5-TBV precursors using two methods: the typical method via a dia-zonium salt quenched with diethylamine to provide 5-TBV inquantitative yield (method A, Scheme 2), and method B startedby the synthesis of a diazonium salt obtained by using t-butylni-trite in CH2Cl2 then quenched with diethylamine to provide 88%yield of 5-TBV. Triazenes can be decomposed by acidic-thermal-decomposition through diazonium ion to provide fluorinated42,43
or iodinated44 derivatives as described for certain radiolabelings.Two pathways are possible: photoinduced decomposition of 1-aryl-3,3-dialkyltriazenes or thermal decomposition via ionicpaths.39 Both processes start with cleavage of the N(2)–N(3)bond.34 Most studies agree on strong acid-catalyzed decomposi-tion of triazenes, involving fast and reversible protonation ofN(3), followed by the ‘slow’ heterolytic cleavage of the N(2)–N(3)bond to yield the corresponding diazonium ion and amine.39,45–47
Protonation of N(3) is a crucial step to decompose the triazenomoiety (competing with N(1) protonation): the partial atomcharges of N(1) and N(3) for 5-TBV were �0.138 and �0.0912,respectively (Discovery Studio� 2.5, CFF forcefield, Accelrys Inc),corresponding to the dipolar charge distribution of the triazenefunctional group.
Because the triazene moiety is decomposed through a diazo-nium ion, the theoretical model for acid-catalyzed thermal decom-position to yield the desired arylfluoride (fluoro-de-triazenation) isthe same as for fluoro-dediazoniation.48 This acid should not pres-ent a nucleophilic conjugated base (A�) to prevent competitionwith fluoride anion for aryl cation. Additionally, an acid with alow redox potential (e.g., trifluoroacetic acid) is desirable, to pre-vent reduction of the aryldiazonium ion via the radical pathwayand the formation of radicals.
For the synthesis of (±)-5-FBVM from the aryl-dialkyltriazene(±)-5-TBV we first tested TBFA in C6H4Cl2 or in CH2Cl2 at refluxand p-toluenesulfonic acid (PTSA) with a high redox potential
and non-nucleophilic properties. However, we observed the forma-tion of the corresponding aryl-p-toluenesulfonic ester analog. Incontrast, the reaction with triflic acid in CH2Cl2 was more success-ful, and we obtained 5-FBVM with 25% yield (Scheme 3). A chlori-nated organic solvent, especially CH2Cl2 (compared to C6H4Cl2),was a good solvent, possibly due to the potential stabilization ofthe diazonium-boron trifluoride complex intermediate. We alsotested several fluorinating agents, including tetra-n-butylammo-nium fluoride (TBAF), which is soluble in chlorinated solvents.Reaction of TBAF, CH2Cl2, and CF3SO3H under reflux was not suc-cessful (data not shown). The use of CsF in carbon tetrachloridewith triflic acid, as previously described for dimethyltriazene,49
failed in our case (data not shown), possibly due to the insolubilityof CsF and triflic acid in CCl4. Using KF in C2H4Cl2 at reflux was alsonot successful.
The enantiomeric purity and the optical rotation of (S,S)- and(R,R)-5-FBVM were checked by chiral HPLC by using an amylosebased column in RP mode (91% CH3CN/20 mM NH4OAc aq) and achiral detector. Under these conditions, (S,S)-5-FBVM was a (+)-enantiomer and (R,R)-5-FBVM a (�)-enantiomer.
2.3. In vitro evaluation
Vesamicol-derived ligands are generally insufficiently selectivetowards VAChT due to a non-negligible affinity to r receptors.Since these receptors are distributed in cholinergic brain areas,specific imaging of cholinergic deficiency is almost impossible withunselective compounds.
Binding affinities of 5-FBVM and reference compounds toVAChT and r receptor sites were determined in vitro with Ki val-ues presented in Table 3. 5-FBVM presents very good affinity forVAChT and, surprisingly, the (+)-(S,S) enantiomer showed almostthe same affinity as the (�)-(R,R) enantiomer (Ki(S,S)FBVM = 6.95 nMand Ki(R,R)FBVM = 3.68 nM), the affinity being better than for ABV(Ki = 6.95 nM), that is, known to be a good ligand.50 Furthermore,(+)-(S,S)-5-FBVM is selective for VAChT towards the r1 receptor(Ki(S,S) = 38.1 nM and Ki(R,R) = 3.75 nM), and both isomers are selec-tive for VAChT towards r2 receptors (Ki >200 nM). The affinity of 5-FBVM is in the same range as the affinity of 5-IBVM which is cur-rently regarded as the standard radiotracer.
N
H2N
HO1) 1.5 eq. NaNO2, HCl aq, 0°C, 0.5 h
N
Et2N-N=N
HO
5-ABV 5-TBV
2) NaHCO3 aq, 1.5 eq. Et2NH, 20°C, 1h
1) 1.5 eq. BF3.Et2O, CH2Cl2 ,1.5 eq. t-BuONO, 0°C, 2 h
2) NaHCO3 aq, 1.5 eq. Et2NH, 20°C, 1h
Method A; Yield 95%
Method B ; Yield 88%
Scheme 2. Synthesis of (S,S) and (R,R)-5-TBV.
N
F
HO
1) 3 eq. CF3SO3, CH2Cl2N
Et2N-N=N
HO
2) 1.5 eq. BF3.Et2O, 40°C, 1h
25%(rac)-5-FBVM(rac)-5-TBV
Scheme 3. Synthesis of (rac)-5-FBVM from triazene precursor (rac)-5-TBV.
7664 M. Kovac et al. / Bioorg. Med. Chem. 18 (2010) 7659–7667
Page 110
3. Conclusion
We obtained a good linear GFA model and a 3D QSAR modelwhich confirmed the spatial impact on affinity for VAChT via stericdescriptors and the Van der Waals coefficient. Each study predicteda good affinity of both 5-FBVM enantiomers.
All one pot acid-catalyzed thermal fluoro-dediazoniation reac-tions confirmed that parameters such as good solubility, non-nucleophilicity, and high redox potential of the reagents used, suchas triflic acid, are the most important conditions for successful flu-oro-detriazenation via the formation of diazonium ion. Using bor-on trifluoride etherate we succeeded in introducing fluorine on anaromatic nucleus. Dichloromethane proved to be the most oppor-tune solvent for one pot fluoro-dediazoniation. The in vitro evalu-ations confirmed the QSAR model where both enantiomersexhibited high affinity for VAChT [(+)-5-FBVM: Ki = 6.95 nM and(�)-5-FBVM: Ki = 3.68 nM]. The stereoisomers were selective to-wards r2 receptors (�70-fold), however, only (+)-5-FBVM is alsoselective for r1 receptors (�fivefold). Further experiments areneeded to improve the characterization of the pharmacologicaland pharmacokinetics profiles of this compound in order to deter-mine its potential use as an F-18-labeled imaging agent for studiesinvolving the cholinergic system.
4. Experimental section
4.1. Molecular modeling
Computational results were obtained using software programsfrom Accelrys Software Inc. The molecules were built and mini-mized in molecular package (Discovery Studio� 2.5.5, Accelrys,San Diego, CA) by CHARMm with CFF partial charge estimationmethod. The GFA model in QSAR protocol was used with a popula-tion size of 100 and 5000 maximum generations. All the parame-ters have been left to the system defaults. Two model forms havebeen used: linear or full quadratic. For the 3D QSAR model, the gridspacing was 1 Å.
4.2. Chemistry
NMR spectra were recorded on a Bruker DPX Avance 200 spec-trometer (200 MHz for 1H, 50.3 MHz for 13C). CDCl3 was used assolvent; chemical shifts are expressed in ppm relative to TMS asan internal standard. Mass spectra were obtained on a CG–MSHewlett Packard 5989A spectrometer (electronic impact at70 eV). The thin-layer chromatographic (TLC) analyzes were per-formed using Merck 60-F254 silica gel plates. Flash chromatographywas used for routine purification of reaction products using silicagel (230–400 mesh). Visualization was accomplished under UV orin an iodine chamber. All chemicals and solvents were of commer-cial quality and were purified following standard procedures. Ele-mental analyzes of new compounds were within ±0.5% oftheoretical values.
4-Phenylpiperidine, used in the synthesis of 5- and 8-amino-benzovesamicol (ABV),27 was obtained by alkaline fusion from 4-cyano-4-phenylpiperidine.31 Enantiomeric resolution of 5-ABVwas done by chromatography separation of diastereomeric N,O-bis-(�)-a-methoxy-a-trifluoromethylphenylacetyl (MTPA) deriva-tives followed by hydrolysis of Mosher esters.5 The optical purityof both (�)-ABV and (+)-ABV was checked on Chiracel OD column(4.6 � 250 nm, 10 lm particle, Daicel Chemical Industries Ltd, Ill-kirch France) with n-hexane/isopropanol (80/20) as eluent at a flowrate of 1.5 mL/min ((+)-ABV; tR = 11 min, (�)-ABV); tR = 13.5 min).
(�)-5-IBVM18,27 and (�)-FEOBV5 were synthesized as previ-ously described.
4.2.15-Fluoro-3-(4-phenyl-piperidin-1-yl)-1,2,3,4-tetrahydro-naphthalen-2-ol: 5-FBVM from 5-ABV4.2.1.1 Method A.
To a cold (0 �C) solution of (rac)-5-ABV (0.161 mg, 0.5 mmol) inanhydrous 1,2-dichlorobenzene (4 mL), boron trifluoride diethyletherate (BF3�O(CH2CH3)2) (0.064 mL, 0.75 mmol) was added. Thestirred reaction mixture was warmed to 105 �C and n-butylnitrite(0.07 mL, 0.6 mmol) was added. The mixture was warmed for1 h. After cooling to room temperature, the solution was quenchedwith water and extracted with EtOAc. The water phase was madebasic by Na2CO3 and was extracted once again with EtOAc. Thecombined organic extracts were dried over MgSO4 and concen-trated under reduced pressure. The crude product was purifiedby gradient flash chromatography (Al2O3, n-hexane/EtOAc 4/1 ton-hexane/EtOAc 1/1). (rac)-5-FBVM was obtained as a white pow-der in 16% yield.
4.2.1.2. Method B. To a cold (0 �C) solution of (rac)-5-ABV(0.161 mg, 0.5 mmol) in anhydrous dichloromethane (4 mL) borontrifluoride diethyl etherate (BF3�O(CH2CH3)2) (0.064 mL,0.75 mmol) and n-butylnitrite (0.095 mL, 0.75 mmol) were added.The mixture was stirred at room temperature overnight and after-ward heated to 60 �C for drying. After cooling, water was added tothis reaction residue. The water phase was made basic by NaHCO3
and was extracted with EtOAc for two times. The combined organicextracts were dried over MgSO4 and concentrated under reducedpressure. The crude product was purified by gradient flash chroma-tography (SiO2, n-hexane/EtOAc 4/1 to n-hexane/EtOAc 1/2). (rac)-5-FBVM was obtained as a white powder in 25% yield.
(+)-5-FBVM and (�)-5-FBVM were obtained by the same proce-dure as white powders in 27% and 25% yield, respectively, startingfrom the corresponding (+)-5-ABV and (�)-5-ABV.
1H NMR (CDCl3): d 1.75–1.96 (m, 4H, 4H-10), 2.53–3.08 (m, 9H,1H-11, 1H-1, 2H-4, 1H-3, 4H-9), 3.25 (dd, J = 5.4 Hz, J = 16 Hz, 1H-1), 3.85–3.99 (m, 1H-2), 4.34 (br s, OH), 6.86–6.97 (m, 1HAr), 7.10–7.17 (m, 2HAr), 7.26–7.41 (m, 5HAr).
13C NMR (CDCl3): d 19.0 (C-4), 33.7, 34.2 (2C-10), 37.6 (C-1),42.8 (C-11), 44.8, 53.4 (2C-9), 65.1 (C-2), 65.9 (C-3), 112.1 (2JC–F =22 Hz, C-6), 122.5 (2JC–F = 18 Hz, C-4a), 124.5 (4JC–F = 3 Hz, C-8),126.2 (CHAr), 126.7 (2CHAr), 127.1 (3JC–F = 8 Hz, C-7), 128.4 (2CHAr),136.5 (3JC–F = 8 Hz, C-8a), 145.9 (CAr), 160.9 (1JC–F = 243 Hz, C-5).
MS: m/z = 325 (14), 228 (74), 174 (35), 161 (18), 155 (16), 146(100), 133 (32), 56 (31).
Anal. Calcd for rac-C21H24FNO: C, 77.51; H, 7.43. Found: C,77.90; H, 3.91.
4.2.25-(3,3-Diethyltriaz-1-enyl)-3-(4-phenylpiperidin-1-yl)-1,2,3,4-tetrahydronaphthalen-2-ol: (rac)-5-ABV-diethyltriazene(5-TBV)4.2.2.1 Method A.
(rac)-5-ABV (256 mg, 0.8 mmol) was dissolved in 0.1 mL of 12 NHCl and the flask was immersed in an ice-bath (0–5 �C). A 1.5 equivof NaNO2 (82 mg) was added to the solution and the reaction mix-ture was stirred for 30 min. After neutralization with saturatedaqueous solution of NaHCO3, 1.5 equiv of diethyl amine (0.12 mL)was added to react for 1 h. The mixture was extracted three timeswith CH2Cl2. Combined organic extracts were dried over MgSO4
and concentrated under reduced pressure. The triazene (rac)-5-TBV was obtained as a white powder and was pure enough to beused without any purification (95% yield).
(+)-5-TBV and (�)-5-TBV were obtained by the same procedureas white powders in 92% or 96% yield, respectively, starting fromthe corresponding (+)-5-ABV and (�)-5-ABV.
4.2.2.2. Method B. (rac)-5-ABV (256 mg, 0.8 mmol) was dis-solved in CH2Cl2 (2 mL) and the flask was immersed in an ice-bath
M. Kovac et al. / Bioorg. Med. Chem. 18 (2010) 7659–7667 7665
Page 111
(0–5 �C). A 1.5 equiv of boron trifluoride diethyl etherate (0.15 mL)and 1.5 equiv of t-butylnitrite (0.14 mL) were added to the stirredsolution. After 2 h, the reaction mixture was neutralized with sat-urated aqueous solution of NaHCO3 and 1.5 equiv of diethyl amine(0.12 mL) was added and stirred for 1 h. The mixture wasquenched with water and extracted three times with CH2Cl2. Com-bined organic extracts were dried over MgSO4 and concentratedunder reduced pressure. The triazene (rac)-5-TBV was pure enoughto be used without any purification (88% yield).
1H NMR (CDCl3): d 1.34 (t, J = 7 Hz, 6H, 2CH3), 1.76–2.01 (m, 4H,2H-10, 2H-12), 2.48–3.08 (m, 8H, 1H-11, H-1, 2H-4, 2H-9, 2H-13),3.37 (dd, J = 5.6 Hz, J = 16 Hz, 1H-1), 3.45 (dd, J = 3.4 Hz, J = 15.5 Hz,1H-1), 3.83 (q, J = 7 Hz, 4H, 2CH2), 3.91–3.97 (m, 1H-2), 6.95 (d,J = 6.5 Hz, 1HAr), 7.15–7.37 (m, 7HAr).
13C NMR (CDCl3): d 12.6 (br s, 2CH3); 21.5 (C-4), 33.8, 34.3 (2C-10), 38.1 (C-1), 42.8 (C-11), 45.0, 53.5 (2C-9), 65.4 (C-2), 66.7 (C-3),114.0 (CHAr), 125.9, 126.1, 126.3 (3CHAr), 126.7 (2CHAr), 128.4(2CHAr), 129.4 (CAr), 134.6 (CAr), 146.1 (CAr), 148.9 (CAr).
MS: m/z = 307 (84), 174 (100), 129 (26), 117 (44), 115 (41), 91(49), 70 (33).
4.2.3. (rac)-5-Fluoro-3-(4-phenyl-piperidin-1-yl)-1,2,3,4-tetrahydro-naphthalen-2-ol: (rac)-5-FBVM from 5-triazene 5-TBV
To a solution of (rac)-5-TBV (0.162 g, 0.4 mmol) in anhydrousCH2Cl2 (5 mL) trifluoromethanesulfonic acid monohydrate(CF3SO3H�H2O) (0.18 mg, 1.2 mmol) dissolved in CH3CN (0.5 mL)was added. BF3�Et2O (boron trifluoride diethyl etherate, 0.075 mL,0.6 mmol) diluted in CH2Cl2 (0.2 mL) was then added to the stirredreaction mixture. The reaction mixture was heated to 60 �C for dry-ing over 1 h. After cooling, water was added to this reaction resi-due. The water phase was made basic by NaHCO3 and wasextracted with CH2Cl2. The combined organic extracts were driedover MgSO4 and concentrated under reduced pressure. The crudeproduct was purified by gradient flash chromatography (SiO2, n-hexane/EtOAc 4/1 to n-hexane/ EtOAc 1/2). (rac)-5-FBVM was ob-tained as a white powder in 25% yield.
4.3. Determination of optical rotation of (R,R)-5-FBVM and(S,S)-5-FBVM
The analytical separation of the 5-FBVM enantiomers by chiralHPLC was performed on a Reprosil Chiral-AM-RP column(250 � 4.6 mm), which is based on amylose-tris-(3,5-dimethyl-phenyl)-carbamate as chiral selector (Dr. Maisch-GmbH, Ger-many). The optical rotation was determined by using a chiraldetector (OR 2090 model from JASCO, Germany). In general, theOR detector operated under following conditions: range: 0.05, re-sponse: SLOW, gain: 10. The polarity of signal amplitudes obtainedby the chiral detector was checked with (�)-vesamicol as referencecompound. By using 91% CH3CN/20 mM NH4OAc aq and a flow rateof 1 mL/min the enantiomers were separated. With tR = 15.5 minthe (�)-(R,R)-5-FBVM eluted in front of the (+)-(S,S)-5-FBVM withtR = 23.5 min.
4.4. Receptor binding studies
For radioligand displacement studies, the test compounds weresolved in DMSO at 10 mM stock solutions. Serial dilutions in therange of 0.01 nM to 1 lM were obtained by further dilution inincubation buffer.
VAChT affinity was determined by radioligand displacementstudies on homogenates of PC12 cells stably transfected withrVAChT (Ali Roghani, Texas Tech University, Lubbock, TX, USA)by using (�)-[3H]vesamicol (Perkin Elmer; specific activity:1296 GBq/mmol). Assays were incubated in 50 mM TRIS–HCl, pH
7.4 at room temperature. Incubation was terminated after 60 minby filtration (GF-B filter, pre-incubated in 0.3% PEI at room temper-ature for 90 min; Brandel Cell harvester). Non-specific binding wasdetermined in the presence of 10 mM (±)-vesamicol.
r1 affinity was determined by radioligand displacement studieson homogenates of rat cortical membranes by using (�)-[3H]pen-tazocine (Perkin Elmer; specific activity: 1070 GBq/mmol). Assayswere incubated in 50 mM TRIS–HCl, pH 7.4 at room temperature.Incubation was terminated after 120 min by filtration (GF-B filter,pre-incubated in 0.3% PEI at room temperature for 90 min; BrandelCell harvester). Non-specific binding was determined in the pres-ence of 10 lM haloperidol.
r2 affinity was determined by radioligand displacement studieson homogenates of rat liver membranes by using (�)-[3H]DTG(Perkin Elmer; specific activity: 1147 GBq/mmol) in the presenceof 1 lM dextrallorphan (Roche) to block r1 binding of [3H]DTG. As-says were incubated in 50 mM TRIS–HCl, pH 7.4 at room tempera-ture. Incubation was terminated after 120 min by filtration (GF-Bfilter, pre-incubated in 0.3% PEI at room temperature for 90 min;Brandel Cell harvester). Non-specific binding was determined inthe presence of 10 lM haloperidol.
All assays were performed in triplicates at least three times. TheIC50-values were estimated by computational non-linear regres-sion analysis. Ki-Values were calculated according to Cheng andPrusoff.51
Acknowledgment
This work was supported by INSERM. This study was funded inpart by FEDER: ImAD project. We thank the ‘Département d’analy-ses Chimiques et S.R.M. biologique et médicale’ (Tours, France) forchemical analyzes.
References and notes
1. Bauer, J.; Hull, M.; Berger, M. Z. Gerontol. Geriatr. 1995, 28, 155.2. Terry, R. D.; Masliah, E.; Salmon, D. P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen,
L. A.; Katzman, R. Ann. Neurol. 1991, 30, 572.3. Giboureau, N.; Emond, P.; Fulton, R. R.; Henderson, D. J.; Chalon, S.; Garreau, L.;
Roselt, P.; Eberl, S.; Mavel, S.; Bodard, S.; Fulham, M. J.; Guilloteau, D.; Kassiou,M. Synapse 2007, 61, 962.
4. Kilbourn, M. R.; Hockley, B.; Lee, L.; Sherman, P.; Quesada, C.; Frey, K. A.;Koeppe, R. A. Nucl. Med. Biol. 2009, 36, 489.
5. Mulholland, G. K.; Jung, Y. W.; Wieland, D. M.; Kilbourn, M. R.; Kuhl, D. E. J. J.Labelled Compd. Radiopharm. 1993, 33, 583.
6. Rogers, G. A.; Stone-Elander, S.; Ingvar, M.; Eriksson, L.; Parsons, S. M.; Widén,L. Nucl. Med. Biol. 1994, 21, 219.
7. Efange, S. M.; Langason, R. B.; Khare, A. B.; Low, W. C. Life Sci. 1996, 58,1367.
8. Efange, S. M.; Khare, A.; Parsons, S. M.; Bau, R.; Metzenthin, T. J. Med. Chem.1993, 36, 985.
9. Sorger, D.; Scheunemann, M.; Vercouillie, J.; Grossmann, U.; Fischer, S.; Hiller,A.; Wenzel, B.; Roghani, A.; Schliebs, R.; Steinbach, J.; Brust, P.; Sabri, O. Nucl.Med. Biol. 2009, 36, 17.
10. Sorger, D.; Scheunemann, M.; Großmann, U.; Fischer, S.; Vercouille, J.; Hiller,A.; Wenzel, B.; Roghani, A.; Schliebs, R.; Brust, P.; Sabri, O.; Steinbach, J. Nucl.Med. Biol. 2008, 35, 185.
11. Parsons, S. M.; Bahr, B. A.; Rogers, G. A.; Clarkson, E. D.; Noremberg, K.; Hicks, B.W. Prog. Brain Res. 1993, 98, 175.
12. Bahr, B. A.; Clarkson, E. D.; Rogers, G. A.; Noremberg, K.; Parsons, S. M.Biochemistry 1992, 31, 5752.
13. Altar, C. A.; Marien, M. R. Synapse 1988, 2, 486.14. Bahr, B. A.; Parsons, S. M. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 2267.15. Efange, S. M.; Mach, R. H.; Smith, C. R.; Khare, A. B.; Foulon, C.; Akella, S. K.;
Childers, S. R.; Parsons, S. M. Biochem. Pharmacol. 1995, 49, 791.16. Ichikawa, T.; Ajiki, K.; Matsuura, J.; Misawa, H. J. Chem. Neuroanat. 1997, 13,
23.17. Phan, V. L.; Miyamoto, Y.; Nabeshima, T.; Maurice, T. J. Neurosci. Res. 2005, 79,
561.18. Jung, Y. W.; Van Dort, M. E.; Gildersleeve, D. L.; Wieland, D. M. J. Med. Chem.
1990, 33, 2065.19. Kuhl, D. E.; Minoshima, S.; Fessler, J. A.; Frey, K. A.; Foster, N. L.; Ficaro, E. P.;
Wieland, D. M.; Koeppe, R. A. Ann. Neurol. 1996, 40, 399.20. Sorger, D.; Schliebs, R.; Kampfer, I.; Rossner, S.; Heinicke, J.; Dannenberg, C.;
Georgi, P. Nucl. Med. Biol. 2000, 27, 23.
7666 M. Kovac et al. / Bioorg. Med. Chem. 18 (2010) 7659–7667
Page 112
21. Zea-Ponce, Y.; Mavel, S.; Assaad, T.; Kruse, S. E.; Parsons, S. M.; Emond, P.; Chalon,S.; Giboureau, N.; Kassiou, M.; Guilloteau, D. Bioorg. Med. Chem. 2005, 13, 745.
22. Jung, Y. W.; Frey, K. A.; Mulholland, G. K.; del Rosario, R.; Sherman, P. S.; Raffel,D. M.; Van Dort, M. E.; Kuhl, D. E.; Gildersleeve, D. L.; Wieland, D. M. J. Med.Chem. 1996, 39, 3331.
23. Assaad, T.; Mavel, S.; Emond, P.; Chalon, S.; Drossard, M. L.; Vergote, J.; Bodard,S.; Allouchi, H.; Besnard, J.-C.; Guilloteau, D. J. Labelled Compd. Radiopharm.2007, 50, 139.
24. Assaad, T.; Mavel, S.; Parsons, S. M.; Kruse, S.; Galineau, L.; Allouchi, H.;Kassiou, M.; Chalon, S.; Guilloteau, D.; Emond, P. Bioorg. Med. Chem. Lett. 2006,16, 2654.
25. Emond, P.; Mavel, S.; Zea-Ponce, Y.; Kassiou, M.; Garreau, L.; Bodard, S.;Drossard, M. L.; Chalon, S.; Guilloteau, D. Nucl. Med. Biol. 2007, 34, 967.
26. Szymoszek, A.; Wenzel, B.; Scheunemann, M.; Steinbach, J.; Schüürmann, G. J.Med. Chem. 2008, 51, 2128.
27. Rogers, G. A.; Parsons, S. M.; Anderson, D. C.; Nilsson, L. M.; Bahr, B. A.;Kornreich, W. D.; Kaufman, R.; Jacobs, R. S.; Kirtman, B. J. Med. Chem. 1989, 32,1217.
28. Rogers, D.; Hopfinger, A. J. J. Chem. Inf. Comput. Sci. 1994, 34, 854.29. Stanton, D. T.; Jurs, P. C. Anal. Chem. 1990, 62, 2323.30. Shiba, K.; Ogawa, K.; Ishiwata, K.; Yajima, K.; Mori, H. Bioorg. Med. Chem. 2006,
14, 2620.31. Berkoff, C. E.; Rivard, D. E.; Kirkpatrick, D.; Ives, J. L. Synth. Commun. 1980, 10,
939.32. Hashida, Y.; Landells, R. G. M.; Lewis, G. E.; Szele, I.; Zollinger, H. J. Am. Chem.
Soc. 1978, 100, 2816.33. Szele, I.; Zollinger, H. J. Am. Chem. Soc. 1978, 100, 2811.
34. Galli, C. Chem. Rev. 1988, 88, 765.35. Garel, L.; Saint-Jalmes, L. Tetrahedron Lett. 2006, 47, 5705.36. Doyle, M. P.; Bryker, W. J. J. Org. Chem. 1979, 44, 1572.37. Laali, K. K.; Gettwert, V. J. J. Fluorine Chem. 2001, 107, 31.38. Hubbard, A.; Okazaki, T.; Laali, K. K. J. Org. Chem. 2008, 73, 316.39. Satyamurthy, N.; Barrio, J. R.; Schmidt, D. G.; Kammerer, C.; Bida, G. T.; Phelps,
M. E. J. Org. Chem. 1990, 55, 4560.40. Yoneda, N.; Fukuhara, T. Tetrahedron 1996, 52, 23.41. Kimball, D. B.; Haley, M. M. Angew. Chem., Int. Ed. 2002, 41, 3338.42. Berridge, M. S.; Sayre, L. M.; Arora, P. K.; Terris, A. H.; Riachi, N. J.; Harik, S. I. J.
Med. Chem. 1993, 36, 1284.43. Pages, T.; Langlois, B. R. J. Fluorine Chem. 2001, 107, 321.44. Harmata, M.; Hong, X.; Zheng, P. Tetrahedron Lett. 2006, 47, 7343.45. Schmidt, B. F.; Snyder, E. J.; Carroll, R. M.; Farnsworth, D. W.; Michejda, C. J.;
Smith, R. H. J. Org. Chem. 1997, 62, 8660.46. Smith, R. H.; Wladkowski, B. D.; Mehl, A. F.; Cleveland, M. J.; Rudrow, E. A.;
Chmurny, G. N.; Michejda, C. J. J. Org. Chem. 1989, 54, 1036.47. Tewson, T. J.; Welch, M. J. J. Chem. Soc., Chem. Commun. 1979, 1149.48. Barrio, J. R.; Satyamurthy, N.; Ku, H.; Phelps, M. E. J. Chem. Soc., Chem. Commun.
1983, 443.49. Pages, T.; Langlois, B. R.; Bars, D. L.; Landais, P. J. Fluorine Chem. 2001, 107, 329.50. Rogers, G.; Kornreich, W.; Hand, K.; Parsons, S. Mol. Pharmacol. 1993, 44, 633.51. Cheng, Y.; Prusoff, W. H. Biochem. Pharmacol. 1973, 22, 3099.52. Shiba, K.; Yano, T.; Sato, W.; Mori, H.; Tonami, N. Life Sci. 2002, 71, 1591.53. Efange, S. M. N.; von Hohenberg, K.; Khare, A. B.; Tu, Z.; Mach, R. H.; Parsons, S.
M. Nucl. Med. Biol. 2000, 27, 749.
M. Kovac et al. / Bioorg. Med. Chem. 18 (2010) 7659–7667 7667
Page 113
99
3.5. Aromatic fluoro-de-triazenation with boron trifluoride diethyl
etherate under non protic acid conditions
Mitja Kovac a,b
, Marko Anderluh a, Johnny Vercouillie
a, Denis Guilloteau
a, Patrick Emond
a,
Sylvie Mavel a
a Université François-Rabelais de Tours, INSERM U930, CHRU, Hôpital Bretonneau,
Service de Médecine Nucléaire, 37000 Tours, France
b University of Ljubljana, Faculty of Pharmacy, Department of Pharmaceutical Chemistry,
Aškerčeva 7, 1000 Ljubljana, Slovenia
Published in: Journal of Fluorine Chemistry 147 (2013) 5-9
Page 114
100
SUMMARY:
A strong protic acid is required to transform triazeno moiety into diazonium group at
elevated temperature. Since the phenyl cation intermediate is highly reactive non-
discriminating species, and even the conjugate base of the used acid is considered non-
nucleophilic (e.g. triflate), fluoro-de-triazenation is often accompanied by the formation of the
acid counterion substituted byproduct (Ar-A);173,176
especially if the acid is used in excess.181
To circumvent this limitation, we examined the coordination chemistry of triazene
derivatives,182–184
and electron donor-acceptor (EDA) complexes between boron trifluoride
and methylated ammonia derivatives.185–189
Accordingly, we propose fluoro-de-triazenation
can be successfully accomplished by the only presence of boron trifluoride via complexation
at elevated temperature. Our hypothesis was first confirmed on the model precursor, 3,3-
diethyl-1-naphthyltriazene and after on simple para substitueted 3,3-diethyl-1-aryltriazenes,
by conventional and microwave heating. To prove that the method is applicable to obtain
more complex arylfluorides too, 5-FBVM was accomplished in high 72% yield under
appropriate microwave conditions in tetrachloromethane as the most opportune solvent.
STATEMENT: I declare, that nobody of co-authors has used the article Aromatic fluoro-de-
triazenation with boron trifluoride diethyl etherate under non protic acid conditions,
published in the Journal of Fluorine Chemistry, for his/her own thesis.
Page 115
Aromatic fluoro-de-triazenation with boron trifluoride diethyl etherate undernon-protic acid conditions
Mitja Kovac a,b, Marko Anderluh b, Johnny Vercouillie a, Denis Guilloteau a,Patrick Emond a, Sylvie Mavel a,*a Universite Francois-Rabelais de Tours, INSERM U930, CHRU, Hopital Bretonneau, Service de Medecine Nucleaire, 37000 Tours, Franceb University of Ljubljana, Faculty of Pharmacy, Department of Pharmaceutical Chemistry, Askerceva 7, 1000 Ljubljana, Slovenia
1. Introduction
Most of the pharmacologically active fluorinated drugs arearomatic, bearing a fluoro or a trifluoromethyl substituent [1].The efficient regioselective introduction of fluorine in electron-rich arenes under mild conditions continues to be a challenge[2,3]. Fluoro-de-triazenation (so called Wallach reaction) repre-sents one of the few regioselective nucleophilic routes yieldingarylfluorides (Ar-F) [4]. The triazenes have been known for morethan a century and have been studied for their versatility inorganic synthesis, especially after their biological activities werefirst reported in the beginning of the 1960s [5,6]. The 3,3-dialkyl-1-aryltriazenes (Ar–N55N–NR0R00) are regarded as pro-tected form of aryldiazonium ions and therefore their acid-triggered thermal decomposition parallels that of the corre-sponding diazonium ionic reactions [4,7–10]. The use of 3,3-dialkyl-1-aryltriazenes (Ar–N55N–NR0R00) has an essential ad-vantage over the aryldiazonium ions because of their solubilityin a number of anhydrous organic and ionic solvents [4,9,11].Moreover, they can be safely and readily prepared in moderateto high yields [5,12,13]. In parallel, solid-phase methodologieswere also applied for the synthesis and reactivity of resin-bound
triazenes [10,14,15]. The 3,3-dialkyl-1-aryltriazenes areregarded as protected form of aryldiazonium ions and thereforetheir acid-triggered thermal decomposition parallels that of thecorresponding diazonium ionic reactions. The reactivity ofaryltriazenes is well described, especially the competitionbetween ionic (heterolytic) and radical (homolytic) de-triazena-tion and de-diazoniation pathways during fluoro-de-diazonia-tion, mechanism strongly dependents upon the reactionconditions [4,7–10]. As protic acid is usually required todecompose aryltriazene, and since the formed phenyl cationintermediate is highly reactive, fluoro-de-triazenation is oftenaccompanied by the formation of a substantial amount of theacid counterion substituted byproduct (Ar-A) [9,11].
We have been investigating the fluoro-de-triazenation reactionas part of a program toward the synthesis of 5-fluoro-3-(4-phenylpiperidin-1-yl)-1,2,3,4-tetrahydro-naphthalen-2-ol (5-FBVM) [16]. Therein we have proposed that 3,3-diethyl-1-aryltriazene could be thermally decomposed and subsequentlyregioselectively fluorinated using boron trifluoride diethyl ethe-rate (BF3�Et2O) as both Lewis acid and fluorinating agent. Thus, themain advantage of the protocol is the avoidance of the competitiveformation of the unwanted compound Ar-A. Using 3,3-diethyl-1-naphthyltriazene as a model, the influence of different organicsolvents was studied. Furthermore, to demonstrate the versatilityof the method, a fluoro-de-triazenation was successfully per-formed using conventional and microwave heating on several 4-substituted 3,3-diethyl-1-aryltriazenes.
Journal of Fluorine Chemistry 147 (2013) 5–9
A R T I C L E I N F O
Article history:
Received 19 October 2012
Received in revised form 9 January 2013
Accepted 12 January 2013
Available online 21 January 2013
Keywords:
Fluorination
Triazene
Diazonium
Boron trifluoride
Arylfluoride
A B S T R A C T
Fluoro-de-triazenation of 3,3-diethyl-1-aryltriazenes can be achieved by conventional or under
microwave heating in carbon tetrachloride, in the presence of boron trifluoride diethyl etherate without
any protic acid to avoid corresponding unwanted byproduct formation.
� 2013 Elsevier B.V. All rights reserved.
* Corresponding author at: Faculte de Pharmacie, INSERM U930, 31 Avenue
Monge, 37200 Tours, France. Tel.: +33 2 47 36 72 40; fax: +33 2 47 36 72 24.
E-mail address: [email protected] (S. Mavel).
Contents lists available at SciVerse ScienceDirect
Journal of Fluorine Chemistry
jo ur n al h o mep ag e: www .e lsev ier . c om / loc ate / f luo r
0022-1139/$ – see front matter � 2013 Elsevier B.V. All rights reserved.
http://dx.doi.org/10.1016/j.jfluchem.2013.01.012
Page 116
2. Results and discussion
2.1. Preparation of 3,3-diethyl-1-aryltriazenes
All 3,3-diethyl-1-aryltriazenes 2a-g were prepared according toknown procedure [13], involving diazotization of aromatic amines1a-g with sodium nitrite in acidic aqueous medium at 0–5 8Cfollowed by addition of diethylamine to yield the correspondingtriazenes 2a-g (Table 1).
2.2. Fluoro-de-triazenation with polyphosphoric acid and boron
trifluoride diethyl etherate
Triflic acid (TfOH) is not an optimal acid for aromatic fluoro-de-triazenation. Besides its incompatibility with the acid-sensitivefunctional groups, substantial amounts of aryl triflate (Ar-OTf) isusually produced [9,11]. Polyphosphoric acid (PPA) presents aweaker nucleophilic conjugated base (A�) and with a suitableredox potential to avoid the radical decomposition pathways [4].Refluxing by using chloroform (CHCl3) instead of dichloromethane(CH2Cl2), and by using PPA in the presence of BF3�Et2O instead ofTfOH, the yield of 3a (5-FBVM) was increased from 25% aspreviously described [16] to 34% yield (Table 2). We used 3,3-diethyl-1-naphthyltriazene 2b as a model precursor for 5-FBVM, asin CHCl3, 3b was obtained in 28% yield compared to only 5% inCH2Cl2. PPA looked promising, although it has never been used influoro-de-triazenation before. Because of its polymeric nature, weassumed its counter anion (A�) is a non-nucleophilic species thatwould prevent the formation of the Ar-A byproduct.
In the presence of PPA, we observed that the triazenes 2a,b werestable before the addition of BF3�Et2O. With this observation inhand, along with the theoretical study of the coordinationchemistry of triazene derivatives [17–19], and electron donor–acceptor (EDA) complexes between BF3 and methylated ammoniaderivatives [20–22], we proposed that the fluorination of the 3,3-dialkyl-1-aryltriazenes was possible using BF3�Et2O only, asdepicted by the proposed reaction mechanism in Scheme 1.
The first rate limiting step is probably the reversible coordina-tion of the BF3, due to its electrophilicity and affinity towardnitrogen, at N(3) triazene which destabilizes the N(2)–-N(3) bondby inhibiting delocalization across the triazene linkage [23] and,therefore, induces its breakage, expelling BF2NEt2 followed in thesecond step by the formation of the aryldiazonium ion (Scheme 1).As suggested in the literature, the ‘‘reorganization energy’’ presentduring the pyramidalization (sp2–sp3) of the boron atom probablycontributes to a reduction in the exothermicity of the complexa-tion reaction [20–22]. Thus, boron trifluoride plays two rolesduring the reaction: (i) it serves as an electron acceptor towardaryltriazenes to enhance the reactivity of the N(2)–N(3) bond; (ii) itserves as a fluoride source for the regioselective fluorination of thearene.
In spite of a previous work of Saeki et al. [24] where after severaltentative experiments, ‘‘1-aryltriazenes did not react with borontrifluoride in the absence of either the palladium catalyst orboronic acid’’, this work gives proofs that fluoro-de-triazenation
can be successfully accomplished without any protic acid orcatalyst and only in the presence of BF3�Et2O.
2.3. Fluoro-de-triazenation of 3,3-diethyl-1-aryltriazenes by boron
trifluoride diethyl etherate in various solvents
As shown in Table 3 where 2b was selected as the modelprecursor, fluorination of 2b-g was carried out using 1.5equivalents of BF3�Et2O at reflux temperature under argon for1 h in various organic solvents.
By increasing the reaction temperature, reduced cleavage of 2b,leading to naphthalene 4 as ‘‘traceless’’ byproduct, was suppressed,while the ionic reaction was favored (Table 3). Interesting, incarbon tetrachloride (CCl4), no radical reaction was observed since4 was not detected. On the other hand, the radical decompositionpathway was favored over the ionic one in tetrahydrofuran (THF),as THF could act as a reducer as suggested in the literature [4,9,25].On the contrary, redox potential of CH3CN is not in favor of anyreduction of 2b or its aryldiazonium ion. But, as proposed andrationalized by Pages et al. [9], 4 could also be the result of redoxprocess(es) between non-complexated triazene and diazoniumion. Furthermore, solvent competition was observed with t-butanol (t-BuOH), besides 35% of 3b, formation of 1-t-butox-ynaphthalene was observed (data not shown).
Table 1Synthesis of 3,3-diethyl-1-aryltriazenes 2a-g. .
1. HCl 37 %, 2. NaNO2 aq. , 0 - 5 oC, 45 min
3. NHEt2, Na2CO 3 aq. , 0 - 5 oC to rt, 1 h1a-g 2a-g
Ar-N=N-NEt2Ar NH2
Ar-NH2 Triazene (isolated yield %)
1a [16]
N
HO
H2N
2a (69)
1b 1-Naphthylamine 2b (34)
1c 4-Me-C6H4NH2 2c (76)
1d 4-NO2-C6H4NH2 2d (47)
1e 4-O-C4H9-C6H4NH2 2e (53)
1f 4-I-C6H4NH2 2f (31)
1g 4-CN-C6H4NH2 2g (56)
Table 2Fluoro-de-triazenation using polyphosphoric acid and boron trifluoride diethyl
etherate. .
Ar N N NEt2
PPA, BF3.Et2O
CH2Cl2 or CHCl3, reflux, Ar, 1hAr F
2a-b 3a-b
Triazene Arylfluoride Yield in CH2Cl2 (%) Yield in CHCl3 (%)
2a 3a 34 39
2b 3b 5 28
Ar N(1)
BF
F
F
N(2) N(3)Et2 Ar N(1) N(2) N(3)Et2
BF
F
F
Ar N N F
- N2 (g)
Ar F
EtO Et
- BF2NEt2,
Scheme 1. Proposed reaction mechanism of fluoro-de-triazenation with 3,3-diethyl-1-aryl triazene and BF3�Et2O.
M. Kovac et al. / Journal of Fluorine Chemistry 147 (2013) 5–96
Page 117
From the solvent effect studies, CCl4 and t-BuOH were chosen asprime solvents to extend the method to (4-substituted phenyl)-triazenes 2c-g (Table 3). Carrying reactions in CCl4 and t-BuOH didnot afford the corresponding Ar-F derivative under chosen reactionconditions for 2c,d and 2e. Especially for 2d and 2e, no triazenedecomposition in both examined solvents was detected. In general,the low reactivity of triazenes is reflected in their high temperatureof decomposition (>130 8C) [8] and correlates with diazoniumsalts decomposition (>90 8C) [26], and so is responsible for poorreproducible yields of fluoro-de-diazoniation. On the other hand,2f underwent fluoro-de-triazenation successfully in CCl4 and t-BuOH with yields 30% and 17% of 1-fluoro-4-iodobenzene 3f,respectively. Conducting reactions in t-BuOH, 4-fluorobenzonitrile3g was produced in 23% yield (Table 3). Unreacted triazeneprecursor 2g was found after reflux in CCl4 in the quenchedmixture. Noteworthy, 1-t-butoxy-4-iodobenzene and 4-t-butox-ybenzonitrile were formed in t-BuOH (data not shown), comingfrom solvent competition.
2.4. Microwave assisted fluoro-de-triazenation
Based on the observation that the main problem of the protocolis the high temperature of triazene decomposition, we performed afluoro-de-triazenation by microwave irradiation to compareresults with conventional heating conditions in CCl4 and t-BuOH.
As predicted, microwave irradiation (MW) facilitated thefluorination of 2a-g using 1.5 equivalent of BF3�Et2O in CCl4 andt-BuOH under anhydrous conditions at 110 8C for 10 min (Table 4).For unreacted compounds in conventional heating, the reactionsucceeded, and afforded 27% in CCl4 and 17% in t-BuOH of 3d(Table 4). No reducing products derived from radical pathways
were detected in both solvents. As 4-nitrophenyl-cation iselectronically more destabilized than the corresponding arene-diazonium ion, we assumed that rapid access to 110 8C anduniform MW dielectric heating allowed immediate formation ofaryl cation after triazene complexation which is crucial for asuccessful fluorination. Efficient MW-assisted fluorination wasalso observed with 2e in CCl4 to give 3e in 64% yield, but only a 15%yield was observed in t-BuOH (Table 4). The reaction in t-BuOH of3c did not yield the corresponding fluoroarene, but rather 1-t-butoxy-4-methylbenzene (data not shown). Yields of the 3b underMW in both solvents were comparable to the yields obtained underconventional heating. MW fluoro-de-triazenation of 2g in CCl4 alsoafforded 41% yield of 3g and comparable 16% yield to conventionalheating in t-BuOH. Under these conditions, reactions affordedsignificantly higher yields of fluorovesamicol derivative 3a fromprevious 25% to 72% in CCl4 and 54% in t-BuOH.
3. Conclusion
We have performed studies toward rapid and synthetically usefulfluorination to obtain 1-fluoronaphthalene, 4-substituted-1-fluoro-benzenes and validated the method by obtention of 5-fluoro-3-(4-phenylpiperidin-1-yl)-1,2,3,4-tetrahydro-naphthalen-2-ol from thecorresponding 3,3-diethyl-1-aryltriazene precursors in CCl4 withonly boron trifluoride diethyl etherate complex. This advantagedistinguishes the present method from numerous precedents whereprotic acid was necessary. The use of the controlled MW heatingcould be implemented successfully leading to significant improve-ment in the reaction efficiency and reaction time in cases whereconventional heating in CCl4 and t-BuOH has failed.
4. Experimental
4.1. General information
Column chromatography was used for routine purification ofreaction products using silica gel (Merck 60; 0.015–0.040 mm).Preparative TLC was carried out using 20 cm � 20 cm glass backedsilica gel F254 plates, 1 mm thickness. High-pressure liquidchromatography (HPLC) was carried out on a Beckman systemGold with XBridgeTM column (5 mm, 4.6 mm � 150 mm), at a flowrate of 1 mL/min, and a 254 nm ultraviolet detector (0.1 MNH4
+CH3COO�/CH3CN, 55/45). 1H and 13C NMR spectra wererecorded in a CDCl3 solution on a Bruker DPX Avance spectrometer(300 MHz for 1H, 75 MHz for 13C, and 282 MHz for 19F) at 298 K. 1Hand 13C chemical shifts (d) are expressed as part per million (ppm)relative to TMS as an internal standard and 1H spin-spin couplingconstants (J) are given in Hertz (Hz). HRMS data were recorded on a
Table 3Solvent influence on the yields of 3b-g in fluoro-de-triazenation of 2b-g. Ratio of ionic pathway (3b) were compared to radical pathway (4). .
BF3.Et2O
reflux, Ar, 1 h2b-g 3b-g
solventAr-N=N-NEt2 Ar-F + Ar-H
4
Triazene Aryl-fluoride Yield in CH2Cl2 (%) Yield in CHCl3 (%) Yield in
CCl4 (%)
Yield in THF or CH3CN (%) Yield in
t-BuOH (%)
2b 3b 8
3b/4a: �1/2
31
3b/4a: �1/1
59
4: not obs.bTrace
4: main pdt c
35
4: not obs.b
2c-e 3c-e – – 0 – 0
2f 3f – – 30 – 17
2g 3g – – 0 – 23
a Crude molar ratio determined by HPLC analysis, 4 was naphthalene.b Naphthalene was not observed.c Naphthalene was the main product.
Table 4Microwave assisted fluoro-de-triazenation of 2a-g. .
BF3.Et2O
solvent, Ar, 10 min2a-g 3a-g
MW / 110 oC
Ar-N=N-NEt2 Ar-F
Triazene Arylfluoride Isolated yield
in CCl4 (%)
Isolated yield in t-BuOH (%)
2a 3a 72 54
2b 3b 52 21
2c 3c 9 0
2d 3d 27 17
2e 3e 64 15
2f 3f 42 32
2g 3g 41 16
M. Kovac et al. / Journal of Fluorine Chemistry 147 (2013) 5–9 7
Page 118
Thermo Scientific Q-Exactive; analyses were done by infusion at1400 resolution. Microwave syntheses were carried out on aDiscover1 monomode reactor from CEM Corporation. All reagents,chemicals and solvents were used as received from commercialsuppliers.
Compounds 2a [16], 2c [13], 2d [27], and 2g [13] werecharacterized as previously described. The spectral data offluoroaryl derivatives 3b-d, f, g were identical to those obtainedfrom the commercial product, and could be observed by websoftware SciFinder1 (Copyright� 2012 American Chemical Socie-ty) in «Experimental Properties» from the CAS REGISTRY.
4.2. General procedure for preparation of 3,3-diethyl-1-aryltriazenes
2a-g
In a round bottom flask, arylamine 1a-g (35 mmol), aqueoushydrochloric acid (37%, 8.7 mL, 105 mmol) and water (90 mL) wereadded. The resulting mixture was cooled to 0–5 8C and a chilledwater solution of NaNO2 (25 mL, 2.96 g of NaNO2, 42 mmol) wasadded dropwise while maintaining the temperature below 5 8C.The mixture was left to stir for 45 min at 0–5 8C, after which it wasneutralized by the dropwise addition of a cold saturated solution ofNa2CO3 (10 mL) and diethylamine (5.4 mL, 52.5 mmol) in chilledwater (30 mL). After stirring for 1 h, the reaction mixture wasextracted with CH2Cl2 (3� 90 mL). Combined organic phases weredried over MgSO4 and concentrated under reduced pressure to givea crude residue which was chromatographically purified to furnish2a-g. Triazenes 2a-g were stored at �20 8C, in the dark to avoiddecomposition.
4.2.1. 3,3-Diethyl-1-naphthyltriazene 2bCompound 2b was purified by column chromatography (EtOAc/
n-heptane, 1/3) to give 2b (34%) as a red oil; 1H NMR (CDCl3): d 1.38(t, J = 7.1 Hz, 6H), 3.90 (q, J = 7.1 Hz, 4H), 7.45–7.51 (m, 4HAr), 7.66(dd, J = 6.6 Hz, J = 2.6 Hz, 1HAr), 7.82–7.86 (m, 1HAr), 8.60–8.63 (m,1HAr);
13C NMR (CDCl3): d 11.1 (brs, 2CH3), 42.0 (brs, 2CH2), 111.4,123.7, 124.8, 125.1, 125.7, 126.0, 127.6, 129.4, 134.3, 146.5; HRMS:calculated [M+H]+ for C14H17N3: 228.14952, found: 228.15182.
4.2.2. 3,3-Diethyl-1-(4-butoxyphenyl)triazene 2eCompound 2e was purified by column chromatography (EtOAc/
n-heptane, 1/5) to give 2e (53% yield) as a red oil; 1H NMR (CDCl3):d 1.00 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 6H), 1.47–1.57 (m, 2H),1.75–1.82 (m, 2H), 3.75 (q, J = 7.2 Hz, 4H), 3.98 (t, J = 7.2 Hz, 2H),6.89 (d, J = 6.9 Hz, 2HAr), 7.38 (d, J = 6.9 Hz, 2HAr);
13C NMR (CDCl3):d 11.2 (brs, 2CH3), 13.8, 19.2, 31.3, 42.2 (brs, 2CH2), 67.9, 114.6(2CHAr), 121.2 (2CHAr), 144.9, 156.9; HRMS: calculated [M+H]+ forC14H23N3O: 250.19139, found: 250.19389.
4.2.3. 3,3-Diethyl-1-(4-iodophenyl)triazene 2fCompound 2f was purified by column chromatography (EtOAc/
n-heptane, 1/6) to give 2f (31% yield) as a red-orange oil; 1H NMR(CDCl3): d 1.28 (t, J = 7.2 Hz, 6H), 3.77 (q, J = 7.2 Hz, 4H), 7.19 (d,J = 8.7 Hz, 2HAr), 7.64 (d, J = 8.7 Hz, 2HAr);
13C NMR (CDCl3): d 11.4(brs, 2CH3), 48.4 (brs, 2CH2), 88.9, 122.4 (2CHAr), 137.6 (2CHAr),150.8; HRMS: calculated [M+H]+ for C10H14IN3: 304.03052, found:304.03350.
4.3. Typical procedure for the reaction of 3,3-diethyl-1-aryltriazene
2a, 2b with polyphosphoric acid and boron trifluoride diethyl etherate
In CH2Cl2 or CHCl3 (5 mL), 5-fluoro-3-(4-phenylpiperidin-1-yl)-1,2,3,4-tetrahydro-naphthalen-2-ol 2a (100 mg, 0.25 mmol) wasdissolved. This solution was added to polyphosphoric acid 115%(70 mg), under argon atmosphere in a dry two-necked roundbottom flask. The mixture was left stirring for 5 min at room
temperature followed by BF3�Et2O (0.047 mL, 0.375 mmol) addi-tion. After refluxing for 1 h, the reaction mixture was quenchedwith a saturated Na2CO3 aqueous solution (5 mL), extracted withCH2Cl2 (3� 5 mL) and dried over MgSO4. After removal of thesolvent under reduced pressure, the residue was purified bycolumn chromatography (EtOAc/n-heptane, 1/5) to give 3a.
Compound 3b was obtained from the same procedure from 2b(114 mg, 0.5 mmol), in CH2Cl2 or CHCl3, leading to 3b (4 or 20 mg)in 5% or 28% yield, respectively. The spectral data of 3b wereidentical to those obtained from the commercial product.
4.3.1. 5-Fluoro-3-(4-phenyl-piperidin-1-yl)-1,2,3,4-
tetrahydronaphthalen-2-ol: 3aWhite powder (34% yield in CH2Cl2, 39% yield in CHCl3).
Identical spectral data to those described in literature [16]. 19FNMR (CDCl3) d �117.30 ppm (1F).
4.4. General procedure for the reaction of 3,3-diethyl-1-aryltriazene
2b-g with boron trifluoride diethyl etherate
Under argon atmosphere, 3,3-diethyl-1-aryltriazene 2b-g(0.50 mmol) was dissolved in 5 mL of solvent (CH2Cl2, CHCl3,CCl4, THF, CH3CN, or t-BuOH) in a dry two-necked round bottomed.While stirring, BF3�Et2O (0.10 mL, 0.75 mmol) was added and thereaction was left to stir for 5 min, at room temperature. Thereaction mixture was then refluxed under argon for 1 h. Aftercooling, saturated Na2CO3 aqueous solution (5 mL) was added tothe crude mixture and extracted with CH2Cl2 (3� 5 mL). Thecombined organic extracts were dried over MgSO4 and concen-trated under reduced pressure. The yields of 3b were determinedby HPLC from standards (8% yield in CH2Cl2, 31% yield in CHCl3, 59%yield in CCl4, negligible yields in THF and CH3CN, and 35% yield in t-BuOH). Yields of 3f and 3g were determined by 1H NMR from thecrude products (3f: 30% yield in CCl4 and 17% yield in t-BuOH; 3g:23% yield in t-BuOH). The spectral data of 3b, 3g, 3f and 4 wereidentical to commercial products.
4.5. General procedure for the microwave-assisted fluorination of 3,3-
diethyl-1-(4-substituted aryl)triazenes 2
Vial (10 mL) containing magnetic stirring bar was charged with2b-e (1.3 mmol), 4 mL of the solvent (CCl4 or t-BuOH) and BF3�Et2O(0.25 mL, 1.95 mmol) under an argon atmosphere. The microwavetube was immediately sealed with a silicon septum and placed inthe microwave (MW) cavity (Discover1, CEM) and subjected toMW irradiation for 10 min at 110 8C (ramp time 2 min, 20 W). Aftercooling to room temperature, the reaction mixture was quenchedwith saturated Na2CO3 aqueous solution (5 mL) and extracted withCH2Cl2 (3� 5 mL). After drying over MgSO4 and removal of thesolvent under reduced pressure, the crude residues 3c-e werepurified by column chromatography (n-heptane 100% to EtOAc/n-heptane, 1/5) to obtain pure products (yields in CCl4: 3b: 52%; 3c:9%; 3d: 27%; 3e: 64%; 3f: 42%; 3g: 41% and yields in t-BuOH: 3b:21%; 3d: 17%; 3e: 15%; 3e: 32%; 3g: 16%). The spectral data of 3cand 3d were identical to commercial products.
Compound 3a was obtained from the same procedure from 2a(30 mg, 0.074 mmol), in CCl4 and t-BuOH, leading, after purifica-tion on preparative TLC (EtOAc/n-heptane, 1/4), to 3a (17 and13 mg) in 72% and 54% yield, respectively.
4.5.1. 1-Butoxy-4-fluorobenzene 3ePale yellow oil (64% yield in CCl4, 15% yield in t-BuOH); 1H NMR
(CDCl3) d 0.96 (t, J = 7.4 Hz, 3H), 1.43–1.52 (m, 2H), 1.71–1.78 (m,2H), 3.90 (t, J = 6.5 Hz, 2H), 6.79–6.97 (m, 4HAr);
13C NMR (CDCl3): d13.9, 19.2, 31.3, 68.3, 115.4 (d, J = 8 Hz, 2CHAr), 115.7 (d, J = 23 Hz,2CHAr), 155.9, 156.5 (d, J = 250 Hz); 19F NMR (CDCl3) d �152.08;
M. Kovac et al. / Journal of Fluorine Chemistry 147 (2013) 5–98
Page 119
HRMS: calculated [M+H]+ for C10H13FO: 169.10232, found:169.10398.
Acknowledgements
This work was supported by Egide for graduate grant (PHCPROTEUS, 2012 n8 26502QF). The authors would also like toacknowledge Slovenian Research Agency for financial support ofSlovenian-French bilateral collaboration (project no. BI-FR/12–13-PROTEUS-007). This work was supported by INSERM. We thank Dr.Nabyl Merbouh (SFU) for the critical reading of the manuscript. Wethank the ‘‘Departement d’Analyses Chimiques et S.R.M. Biologi-que et Medicale’’ (PPF, Tours, France) for the chemical analyses andMrs. Nathalie Meheux for her technical assistance.
References
[1] (a) K.L. Kirk, J. Fluorine Chem. 127 (2006) 1013–1029;(b) D. O‘Hagan, Chem. Soc. Rev. 37 (2008) 308–319, and references cited therein;(c) T. Yamazaki, T.Taguchi, I.Ojima,Uniqueproperties of fluorineand theirrelevanceto medicinal chemistry and chemical biology, in: Fluorine in Medicinal Chemistryand Chemical Biology, John Wiley & Sons, Ltd., Chichester, UK, 2009, pp. 1–46.
[2] T. Furuya, J.E.M.N. Klein, T. Ritter, Synthesis 11 (2010) 1804–1821.[3] S.J. Tavener, J.H. Clark, J. Fluorine Chem. 123 (2003) 31–36.[4] N. Satyamurthy, J.R. Barrio, D.G. Schmidt, C. Kammerer, G.T. Bida, M.E. Phelps, J.
Org. Chem. 55 (1990) 4560–4564.[5] D.B. Kimball, M.M. Haley, Angew. Chem. Int. Ed. 41 (2002) 3338–3351.
[6] V.I. Nifontov, N.P. Bel’skaya, E.A. Shtokareva, Pharm. Chem. J. 27 (1993)652–665.
[7] P.S.J. Canning, K. McCrudden, H. Maskill, B. Sexton, J. Chem. Soc., Perkin Trans. 2(1999) 2735–2740.
[8] M. Dobele, S. Vanderheiden, N. Jung, S. Brase, Angew. Chem. Int. Ed. 49 (2010)5986–5988.
[9] T. Pages, B.R. Langlois, J. Fluorine Chem. 107 (2001) 321–327.[10] P.J. Riss, S. Kuschel, F.I. Aigbirhio, Tetrahedron Lett. 53 (2012) 1717–1719.[11] C.-K. Chu, J.-H. Kim, D.W. Kim, K.-H. Chung, J.A. Katzenellenbogen, D.Y. Chi, Bull.
Korean Chem. Soc. 26 (2005) 599–602.[12] S. Brase, T. Muller, Sci. Synth. 31b (2007) 1845–1872.[13] H. Ku, J.R. Barrio, J. Org. Chem. 46 (1981) 5239–5241.[14] S. Brase, M. Schroen, Angew. Chem. Int. Ed. 38 (1999) 1071–1073.[15] F. Stieber, U. Grether, H. Waldmann, Angew. Chem. Int. Ed. 38 (1999) 1073–1077.[16] M. Kovac, S. Mavel, W. Deuther-Conrad, N. Meheux, J. Glockner, B. Wenzel, M.
Anderluh, P. Brust, D. Guilloteau, P. Emond, Bioorg. Med. Chem. 18 (2010)7659–7667.
[17] C. GuhaRoy, R.J. Butcher, S. Bhattacharya, J. Organomet. Chem. 693 (2008)3923–3931.
[18] J. Iley, R. Moreira, E. Rosa, J. Chem. Soc., Perkin Trans. 2 (1991) 81–86.[19] X.-H. Xie, J.-Y. Chen, W.-Q. Xu, E.-X. He, S.-Z. Zhan, Inorg. Chim. Acta 373 (2011)
276–281.[20] T.A. Ford, J. Mol. Struct. (THEOCHEM) 897 (2009) 145–148.[21] F. Gaffoor, T.A. Ford, Spectrochim. Acta Part A 71 (2008) 550–558.[22] J.M. Miller, M. Onyszchuk, Can. J. Chem. 41 (1963) 2898–2902.[23] F. Rakotondradany, C.I. Williams, M.A. Whitehead, B.J. Jean-Claude, J. Mol. Struct.
(THEOCHEM) 535 (2001) 217–234.[24] T. Saeki, E.-C. Son, K. Tamao, Org. Lett. 6 (2004) 617–619.[25] C. Ruchardt, E. Merz, B. Freudenberg, H.-J. Opgenorth, C.C. Tan, R. Werner, Chem.
Soc. Spec. Publ. 24 (1970) 8–10.[26] L. Garel, L. Saint-Jalmes, Tetrahedron Lett. 47 (2006) 5705–5708.[27] N. Satyamurthy, J.R. Barrio, J. Org. Chem. 48 (1983) 4394–4396.
M. Kovac et al. / Journal of Fluorine Chemistry 147 (2013) 5–9 9
Page 120
106
3.6. Examination of reaction parameters for radio-fluoro-de-triazenation
of 5-TBV
As discussed in Chapter 3.3., protic acid (HA) is one of the most important parameter
required to decompose aryltriazene. In radiofluorination, [18
F]fluoride is in great deficiency
(nanomolar scale) versus triazene precursor and auxiliary acid ([18
F]F-/triazene or protic acid
ratio ~10-4
).190
This encouraged us to search for the strong acid, devoided of nucleophilicity of
its conjugated base (A-) with high enough redox potential not to reduce diazonium ion after
thermal heterolytic decomposition of protonated triazene moiety. Polyphosphoric acid (PPA)
looked promising, although has never been used in fluoro-de-triazenation reactions before. It
is a clear very hygroscopic viscous liquid. Its composition is designated by the percentage
content of phosphoric anhydride (PA, P2O5). This is very flawed, because molar content of
PPA not just depends on the PA content but on the number of fractions and fractional
composition of homologs. Storage, time and frequent usage have great influence on PPA
composition too.191,192
As molarity was not provided by supplier, we designated its use as a
mass of PPA per volume of the used solvent or per mass of the precursor.
According to the 1-NF yield shown in Table 3, PPA 115 (~83% PA content) (Entry 3) is
more appropriate protic acid for fluoro-de-triazenation than triflic acid (Entries 1 and 2),
probably due to its polymeric structure which precludes formation of the acid counterion
substituted byproduct (Ar-A). It should be noted that naphthalene, determined by 1H-NMR
analysis, was the main product in Entries 1 and 2 pointed to radical decomposition path
predominated over ionic one.
Table 3. Fluoro-de-triazenation of 3,3-diethy-1-naphthyltriazene (1-NT) using different
sources of fluoride and acid at reflux in chlorinated solvents.
N N NEt25 mL solvent,
fluoride source, acid
reflux, 1h
F
3,3-diethyl-1-naphthyl triazene(1-NT)
1-fluoronaphthalene(1-NF)
naphthalene(1-NH)
under Ar,
Page 121
107
Entry Precursor Solvent Fluoride source Acid 1-NF Yield (%)a
1 250 mg 1-NT CH2Cl2 1 eq. KF/K222 1.5 eq. TfOH Negligible 1-NF,
1-NH >> 1-NF
2 250 mg 1-NT CHCl3 1 eq. TBAF 1M
soln. in THF
1.5 eq. TfOH 1-NF ~3%,
1-NH >> 1-NF
3 250 mg 1-NT CHCl3 1 eq. TBAF 1M
soln. in THF
216 mg PPA/100
mg 1-NT
1-NF ~7%,
1-NH/1-NF ~1/1
a Crude yield of 1-NF and 1-NF/1-NH ratio were estimated from
1H-NMR data
In the next step, we examined PPA stoichiometry on the 1-NF yield. Table 4 clearly
indicates that for complete 1-NT decomposition 144 mg of PPA per 100 mg of 1-NT was
necessary or 29 mg of PPA per 1 mL CHCl3. Higher amount of the acid did not lower the 1-
NF yield considerably, although more naphthalene was formed (Table 4, Entries 2-3 and 5-7).
A little better yields of the desired arylfluoride were obtained in more concentrated reaction
mixtures (Table 4, Entries 2 and 3) pointed to interfacial processes were important due to low
solubility of the used acid in organic solvents.
Table 4. Examination of PPA stoichiometry and concentration on 1-fluoronaphthalene yield.
N N NEt2CHCl3,
1 eq. TBAF 1M soln. in THF, PPA 115
reflux, 60 min
F
1-NT 1-NF 1-NH
Entry Precursor CHCl3 PPA 115 1-NF Yield (%)a 1-NH/1-NF
b
1 250 mg 1-NT 20 mL 72 mg/100mg 1-NT
9 mg/1mL
~90% 1-NT as
unreacted reactant /
2 250 mg 1-NT 5 mL 144 mg/100mg 1-NT
72 mg/1mL
12% ~1.5
3 250 mg 1-NT 5 mL 216 mg/100mg 1-NT
108 mg/1mL
7% ~2.5
Page 122
108
4 100 mg 1-NT 5 mL 77 mg/100mg 1-NT
15 mg/1mL
1%
(~25% 1-NT)
~5.5
5 100 mg 1-NT 5 mL 144 mg/100mg 1-NT
29 mg/1mL
3% ~2.5
6 100 mg 1-NT 5 mL 216 mg/100mg 1-NT
43 mg/1mL
3% ~4
7 100 mg 1-NT 5 mL 288 mg/100mg 1-NT
58 mg/1mL
2% ~5
a Crude yields of 1-NF were estimated from the
1H-NMR data.
b 1-NF/1-NH
ratios were estimated from the
1H-NMR data.
In order to find the most appropriate fluoride source for fluoro-de-triazenation, we
examinated various fluoride sources routinely used in radiochemistry. As shown in Table 5,
fluoro-de-triazenation was successful only by using TBAF as the fluoride source. Attempts by
using CsF/18-Crown-6 and KF/18-Crown-6 complexes may failed due to the ability of 18-
Crown-6 to complex the aryldiazonium ion.193,194
With this regard solubilization of the
fluoride is reduced since parts of the cryptand can react with aryldiazonium ion instead of
mobilizing the potassium or cesium fluoride, thus enhancing thermal stability of complexed
aryldiazonium ion and giving much lower fluorination yields or complete failure. In this
regard, NaF/15-Crown-5 is supposed to be a better choice.195
Table 5. Examination of fluoride sources routinely used in radiofluorination.
N N NEt2 1 eq. fluoride source,
5mL CHCl3, reflux, 60 min
F
100 mg 1-NT 1-NF
216 mg PPA 115
Entry Fluoride source 1-NF Yield (%)a
1 TBAF 1M soln.
in THF
3%
2 CsF/18-Crown-6 /
Page 123
109
3 KF/18-Crown-6 /
a Crude yields of 1-NF were estimated from the
1H-NMR data.
As PPA 115 is highly viscous polymer and practically insoluble in chlorinated solvents,
we decided to use less dense PPA 105 (~76% PA) for more convenient handling during the
study of thermal decomposition of 5-TBV in the presence of different amounts of PPA 105
without source of fluoride in CCl4 (Scheme 19). Reaction medium or decomposition of 5-
TBV was analysed by high-pressure liquid chromatography (HPLC). Precursor decomposed
completely within 20 minutes at 90 oC in the presence of 10 μL PPA mixture. We repeated
this reaction 3 times to meet a reproducibility issue. 5-TBV was also decomposed completely
within 10 minutes using 10 μL of PPA mixture. Such short decomposition time of 5-TBV is
well suited with the half-life of fluorine-18.
N
HO
1 mg 5-TBV
200 L CCl4, 90oC, 20 min
10-40 L PPA mixture(1g PPA 105 in 1 mL CH3CN)
Decomposition products
NNEt2N
Scheme 19. Study of 5-TBV time-decomposition under different amounts of PPA 105 but
without the presence of fluoride source.
Page 124
110
3.7. Radiochemistry
3.7.1. Preparation of [18
F]TBAF using TRACERlab™
FX F-N synthesizer
TRACERlab™
FX F-N is an automated versatile synthesizer for production of 18
F-tracers
(18
O-water trapping, nucleophilic substitution, hydrolysis, purification and formulation) via
nucleophilic substitution with [18
F]fluoride trapped from [18
O]-enriched water.
Figure 9. TRACERlab™
FX F-N module.
As usually, [18
F]fluoride anion was produced by the proton irradiation of an 18
O-enriched
water target (
18O(p,n)
18F). After irradiation, [
18F]fluoride was transferred to the TRACERlab
™
FX F-N synthesiser. To obtain [18
F]fluoride in a “nakedˮ or dehydrated form suitable for
aromatic nucleophilic substitution reactions, [18
F]fluoride was separated from the 18
O-
enriched water via anion exchange resin (Waters Sep-Pak® Accell Plus QMA) as described in
literature.196
Elution of the [18
F]fluoride was achieved with 0.6 mL 0.075 M (45 μmol) tetra-n-
butylammonium hydrogen carbonate (TBAHCO3) water solution. Poorly nucleophilic
hydrogen carbonate counter-ion in TBAHCO3 was utilised to prepare [18
F]TBAF under
weakly basic conditions (pH ~8), because [18
F]fluoride is easily rendered non-nucleophilic by
protonation. Azeotropic drying was performed with anhydrous acetonitrile in a glassy carbon
reactor with magnetic stirrer and retractable needle. By this way 5.71 GBq [18
F]TBAF/mL
CH3CN was prepared.
Page 125
111
[18F]F- in 18O-encriched water
F-18 separation
cartridge
CH3CN CH3CN H2O
TBA-HCO3
Glassy carbon reactor
Figure 10. Schematic presentation of [18
F]TBAF/mL CH3CN preparation in TRACERlab™
FX F-N synthesiser. The eluant vial (V1) contained 0.6 mL 0.075 M tetra-n-butylammonium
hydrogen carbonate water solution, the second vial (V2) 2 mL anhydrous CH3CN for
azeotropic evaporation (drying step), the third vial (V3) 1 mL anhydrous CH3CN to obtain
[18
F]TBAF/mL CH3CN, and the fourth vial (V4) H2O to rinse the reactor.
Page 126
112
3.7.2. N.c.a. 18
F-radiofluoro-de-triazenation of 5-TBV
We carried out n.c.a. 18
F-radiofluoro-de-triazenation of 5-TBV under conventional and
microwave heating in chlorinated solvents using n.c.a. [18
F]TBAF as the [18
F]fluoride source,
and PPA 105 as the protic acid to induce triazene moiety decomposition at elevated
temperature (Scheme 20). Upon reaction completion, the reaction mixture was quenched with
triethylamine and sample of the crude product analysed by analytical HPLC system using an
analytical HPLC column (CH3CN/0.1 M NH4OAc 30/70, 1 mL/min). It should be noted that
temperature in the microwave reactor did not reach more than 50 oC.
N
HO
2 mg 5-TBV
200 L CHCl3 or CCl4
10 L PPA mixture
NNEt2N
N
HO
5-[18F]FBVM 18F
Thermal heating: 90 oC, 15 min
or
MW: Tset point = 90 oC, 90 W, 10 min
[18F]TBAF,
(1 g PPA 105 in 1 mL CH3CN),
Scheme 20. N.c.a. fluoro-de-triazenation of 5-TBV under conventional heating or/and
microwave (MW) irradiation in chlorinated solvents.
Figure 11. Analytical chromatogram for 5-TBV (tR 10.8 min) (UV detector set at 254 nm).
0,0 2,0 4,0 6,0 8,0 10,0 12,0 15,0
-20
0
20
40
60
80
100
120
140
16020110609 #17 [modified by Administrateur] 5-TBV UV_VIS_2mAU
min
1 - 10,855
WVL:254 nm
Page 127
113
Figure 12. Analytical chromatogram of the crude 5-FBVM (tR 7.6 min) (UV detector set at
254 nm).
Figure 13. Analytical chromatogram of coinjected crude 5-FBVM (tR 7.7 min) and 5-TBV (tR
11.2 min) (UV detector set at 254 nm).
0,0 2,5 5,0 7,5 10,0 12,5 15,0 17,5 20,0
-10,0
0,0
10,0
20,0
30,0
40,0
50,0
60,020110609 #14 [modified by Administrateur] 5-FBV UV_VIS_2mAU
min
1 - 7,591
2 - 11,828
3 - 13,729
WVL:254 nm
0,0 2,0 4,0 6,0 8,0 10,0 12,0 14,0 16,0 18,2
-10
0
10
20
30
40
50
60
70
80
9020110609 #15 5-FBV + 5-TBV UV_VIS_2mAU
min
1 - 7,239
2 - 7,725
3 - FDDNP - 8,8704 - 9,262
5 - 11,155
6 - 13,822
7 - 16,330
WVL:254 nm
Page 128
114
No. Ret.Time Height Area
min mV mV min
8 2,95 1048,217 1415,607
9 2,99 12,797 0,304
10 4,88 6,511 0,225
11 7,67 18,715 13,670
12 7,85 2,097 0,165
13 7,95 0,906 0,029
14 8,93 18,218 21,615
15 9,10 1,795 0,274
16 9,32 1,074 0,038
17 9,78 1,402 0,052
18 9,95 11,180 15,894
Total:
1155,232 1472,320
Scheme 14. Radiation and analytical chromatograms (UV detector set at 254 nm),
respectively, after microwave irradiation in CHCl3 using 32 MBq [18
F]TBAF per attempt.
0,0 2,0 4,0 6,0 8,0 10,0 12,0 14,0 16,4
-200
0
200
400
600
800
1 000
1 2002012-07-26 #5 essai RadioactivitymV
min
1 - 0,1052 - 0,8563 - 0,9404 - 1,0905 - 1,1416 - 1,6607 - 2,033
8 - 2,9509 - 2,991
10 - 4,878
11 - PhFprop eth - 7,67212 - 7,84713 - 7,95314 - 8,93015 - 9,10016 - 9,31517 - 9,77918 - 9,94919 - 9,99820 - 10,21421 - 10,72822 - 10,96223 - 11,20424 - 11,69725 - 11,83426 - 12,91427 - 13,15028 - 13,40729 - 13,56130 - 14,05931 - 14,45432 - 14,55033 - 14,82534 - 14,93635 - 15,11336 - 15,24737 - 15,53938 - 15,996
0,0 2,0 4,0 6,0 8,0 10,0 12,0 14,0 16,4
-50
100
200
300
4502012-07-26 #5 essai UV_VIS_3mAU
min
1 - 2,193
2 - 4,280
3 - 5,4404 - 6,007
5 - 9,373 6 - 10,9937 - 11,053
8 - 13,533
WVL:254 nm
Page 129
115
5-TBV was decomposed completely under conventional heating or microwave irradiation
using 10 μL of PPA mixture (1g PPA 105 in 1 mL of CH3CN) in CHCl3 and CCl4. However,
only microwave irradiation (Tset point = 90 oC, 90 W, 10 min) of 2 mg 5-TBV solubilized in 200
μL of CHCl3 using 32 MBq of [18
F]TBAF and 10 μL of PPA mixture may provided trace of
the 5-[18
F]FBVM (Figure 14).
To improve the 5-[18
F]FBVM yield via n.c.a. fluoro-de-triazenation of 5-TBV further
attempts are needed. Possibilities for optimization are to further optimize reaction time and
microwave irradiation conditions by increasing the power and the temperature set point
especially when the reaction is conducted in CCl4.
Page 130
116
4. EXPERIMENTAL
Page 131
117
In this section just missing experimental procedures are provided. The others have been
described in the two articles enclosed (Chapter 3.4, pages 96-97 and Chapter 3.5, pages 99-
105).
4.1. General information
The thin-layer chromatographic (TLC) analyzes were performed on Merck 60-F254 silica gel
plates. High-pressure liquid chromatography (HPLC) and radio-HPLC were carried out on a
Dionex system with Luna® Phenyl-Hexyl column from Phenomenex
® (5 μm, 4.6 x 250 mm),
at a flow rate of 1 mL/min, and a 254 nm ultraviolet detector (CH3CN/0.1 M NH4OAc
30/70). 1H and
13C NMR spectra were recorded in a CDCl3 solution on a Bruker DPX Avance
spectrometer (300 MHz for 1H, 75 MHz for
13C). No carrier added aqueous [
18F]fluoride ion
was produced in a biomedical cyclotron (GE Healthcare, petTrace), and [18
F]TBAF produced
in a TRACERlab™
FX F-N synthesiser. Microwave syntheses were carried out on a
Discover® monomode reactor from CEM Corporation using 10 mL reactor vial and IR
temperature sensor. All reagents, chemicals and solvents were used as received from
commercial suppliers.
4.2. Chemistry
4.2.1. One-pot fluoro-de-diazoniation of 5-aminobenzovesamicol (5-ABV) in 1,2-
dichlorobenzene (page 82)
A dry two-necked flask was charged with the solution of 5-ABV (224 mg, 0.7 mmol) in
1,2-dichlorobenzene (2 mL). Boron trifluoride etherate (0.13 mL, 1.05 mmol) was added at
room temperature, then the mixture was heated at 105 oC, and tert-butyl nitrite (0.11 mL, 0.84
mmol) was added. The medium was maintained at reaction temperature for 1 h. After cooling,
a saturated aqueous solution of Na2CO3 (3 mL) was added then the crude mixture was
extracted with CH2Cl2 (3 x 3 mL). Collected organic phases were dried over MgSO4, filtered
and evaporated under reduced pressure. The crude product was purified by gradient flash
chromatography (SiO2, n-hexane/EtOAc 4/1 to n-hexane/EtOAc 1/2). 5-FBVM was obtained
as a white powder in 16% yield.
For corresponding analysis see Chapter 3.4, page 96 (page of the article 7665).
Page 132
118
4.2.2. One-pot fluoro-de-diazoniation of 5-aminobenzovesamicol (5-ABV) in
ionic liquid (pages 84-85)
Nitrosonium tetrafluoroborate (114 mg, 0.96 mmol) was added to [emim]x[TfO] ionic
liquid (1.8 g, 6.8 mmol) and the mixture was cooled to 0-5 oC in an ice bath. 5-ABV (256 mg,
0.8 mmol) was added slowly, and the mixture stirred for 30 min at 0-5 oC, and during the
night at room temperature. Then, the reaction mixture was heated at 70-80 oC for 1 h, and
after cooling, extracted with ether (3 x 2 mL). Based on 1H and
13C NMR analysis, the ether
phase contained no 5-FBVM.
4.2.3. Fluoro-de-triazenation of 3,3-diethyl-1-naphthyltriazene (1-NT) using
KF/Kryptofix®
complex and triflic acid (TfOH) in dichloromethane (pages
106-107)
A dry two-necked flask was charged with KF (64 mg, 1.1 mmol, Sigma-Aldrich®
≥ 99%)
and Kryptofix®
2.2.2. (415 mg, 1.1 mmol, Merck Millipore) under an argon atmosphere. A
solution of triflic acid (0.15 mL, 1.65 mmol, Sigma-Aldrich® ≥ 99%) in CH3CN (0.5 mL) and
a solution of 3,3-diethyl-1-napthyltriazene (250 mg, 1.1 mmol) in CH2Cl2 (5 mL) were added,
kept under argon. After 5 min stirring, the mixture was refluxed 1 h. After reaction, a
saturated aqueous solution of Na2CO3 (5 mL) was added then the crude mixture was extracted
with CH2Cl2 (3 x 5 mL). Collected organic phases were dried over MgSO4, filtered and
evaporated under reduced pressure. The residue was analysed by 1H and
13C NMR to give 1-
fluoronaphthalene in negligible yield after comparison the spectral data with those obtained
from the commercial product.
1H NMR (CDCl3): δ ppm 7.21-7.28 (m, 1HAr), 7.44-7.51 (m, 1HAr), 7.60-7.72 (m, 3HAr),
7.93-7.95 (m, 1HAr), 8.21-8.25 (m, 1HAr); 13
C NMR (CDCl3): δ ppm 109.4 (d, J = 19.8 Hz),
120.6 (d, J = 5.2 Hz), 123.6 (d, J = 4.1 Hz), 123.7 (d, J = 16.5 Hz), 125.6 (d, J = 8.4 Hz),
126.2, 126.8, 127.5, 134.9, 158.8 (d, J = 252 Hz).
Page 133
119
4.2.4. Fluoro-de-triazenation of 3,3-diethyl-1-naphthyltriazene (1-NT) using
tetra-n-butylammonium fluoride (TBAF) and triflic acid (TfOH) in
chloroform (pages 106-107)
3,3-diethyl-1-napthyltriazene (250 mg, 1.1 mmol) was dissolved in 5 mL of CHCl3 in a dry
two-necked flask. A solution of triflic acid (0.15 mL, 1.65 mmol, Sigma-Aldrich® ≥ 99%) in
CH3CN (0.5 mL) was added. After 5 min stirring at RT, 1 M solution of TBAF in THF (1.1
mL, 1 equiv, TBAF 1 M soln. in THF, Sigma-Aldrich®) was added, and the resulting mixture
was then refluxed for 1 h. After cooling, saturated Na2CO3 aqueous solution (5 mL) was
added to the crude mixture and extracted with CH2Cl2 (3 x 5 mL). The combined organic
extracts were dried over MgSO4 and concentrated under reduced pressure. Yield of 1-
fluoronaphthalene was determined by 1H NMR from the crude product (~3%), and the
spectral data of the product were identical to the commercial product.
4.2.5. Fluoro-de-triazenation of 3,3-diethyl-1-naphthyltriazene (1-NT) using
tetra-n-butylammonium fluoride (TBAF) and polyphosphoric acid (PPA) in
chloroform (pages 106-107)
A dry two-necked flask was charged with PPA (540 mg, polyphosphoric acid reagent
grade, 115% H3PO4 basis, Sigma-Aldrich®
) and a solution of 3,3-diethyl-1-napthyltriazene
(250 mg, 1.1 mmol) in CHCl3 (5 mL) was added. After 5 min stirring at RT, 1 M solution of
TBAF in THF (1.1 mL, 1 equiv, TBAF 1 M soln. in THF, Sigma-Aldrich®) was added, and
the resulting mixture was then refluxed for 1 h. After cooling, saturated Na2CO3 aqueous
solution (5 mL) was added to the crude mixture and extracted with CH2Cl2 (3 x 5 mL). The
combined organic extracts were dried over MgSO4 and concentrated under reduced pressure.
Yield of 1-fluoronaphthalene was determined by 1H NMR from the crude product (7%), and
the spectral data of the product were identical to the commerical product.
Page 134
120
4.2.6. General procedure for the fluoro-de-triazenation of 3,3-diethyl-1-
naphthyltriazene (1-NT) with different amounts of polyphosphoric acid
(PPA) (pages 107-108)
A dry two-necked flask was charged with PPA (77 mg, 144 mg, 180 mg, 216 mg, 288 mg,
360 mg, 540 mg polyphosphoric acid reagent grade, 115% H3PO4 basis, Sigma-Aldrich®) and
a solution of 3,3-diethyl-1-napthyltriazene (100 mg, 0.44 mmol or 250 mg, 1.1 mmol) in
CHCl3 (5 mL or 20 mL) was added. After 5 min stirring at RT, 1 M solution of TBAF in THF
(0,44 ml or 1.1 mL, 1 equiv, TBAF 1 M sol. in THF, Sigma-Aldrich®) was added, and the
resulting mixture was then refluxed for 1 h. After cooling, saturated Na2CO3 aqueous solution
(5 mL or 20 mL) was added to the crude mixture and extracted with CH2Cl2 (3 x 5 mL or 3 x
20 mL). The combined organic extracts were dried over MgSO4 and concentrated under
reduced pressure. Yield of 1-fluoronaphthalene was determined by 1H NMR from the crude
product, and the spectral data of the product were identical to the commercial product.
4.2.7. General procedure for the fluoro-de-triazenation of 3,3-diethyl-1-
naphthyltriazene (1-NT) with different fluoride sources (pages 108-109)
A dry two-necked flask was charged with PPA (216 mg, polyphosphoric acid reagent
grade, 115% H3PO4 basis, Sigma-Aldrich®
) and a solution of 3,3-diethyl-1-napthyltriazene
(100 mg, 0.44 mmol) in CHCl3 (5 mL) was added. After 5 min stirring at RT, an alkaline
fluoride salt (CsF, KF)/18-Crown-6 complex (1 equiv) dissolved in CH3CN (3 mL) or 1 M
solution of TBAF in THF (0,44 ml, 1 equiv, TBAF 1 M sol. in THF, Sigma-Aldrich®) was
added, and the resulting mixture was then refluxed for 1 h. After cooling, saturated Na2CO3
aqueous solution (5 mL) was added to the crude mixture and extracted with CH2Cl2 (3 x 5
mL). The combined organic extracts were dried over MgSO4 and concentrated under reduced
pressure. Yield of 1-fluoronaphthalene was determined by 1H NMR from the crude product,
and the spectral data of the product were identical to the commercial product.
Page 135
121
4.2.8. General procedure for the de-triazenation of 5-TBV with polyphosphoric
acid (PPA) (page 109)
Vial (1 mL) was charged with 5-TBV (1 mg, 2.5 μmol), CCl4 (0.2 mL), and a mixture of
PPA in CH3CN (5, 10, 20, 40 μL mixture of 1 g PPA, 105% H3PO4 basis, in 1 mL CH3CN).
The vial was immediately sealed with a septum and the resulting mixture heated at 90 oC for
20 min. After reaction, the medium was analysed by HPLC (CH3CN/0.1 M NH4OAc 30/70, 1
mL/min, Luna 5u Phenyl-Hexyl column from Phenomenex®, 250 x 4.6 mm).
4.3. Radiochemistry
4.3.1. Preparation of [18
F]TBAF using TRACERlab™
FX F-N synthesizer (pages
110-111)
No carrier added aqueous [18
F]fluoride ion was produced in a biomedical cyclotron (GE
Healthcare, petTrace), by irradiation of 2 mL water target using a 14 MeV proton beam on >
95% [18
O]-enriched water by the 18
O(p,n)18
F nuclear reaction.
After irradiation, [18
F]fluoride in [18
O]-enriched water was transferred to the TRACERlab™
FX F-N synthesizer, placed in a closed lead-shielded hot cell, and passed through an anion
exchange resin (Waters Sep-Pak® Accell Plus QMA). Trapped [
18F]fluoride ions were then
eluted from the Sep-Pak®
cartridge and transferred to the reactor vessel using 0.6 mL 0.075 M
solution TBAHCO3 as an eluent solution. Water was removed by azeotropic evaporation with
acetonitrile (2 x 0.5 mL) at 90 °C under stream of nitrogen and under vacuum. By adding 1
mL CH3CN to the dry residue, a solution of 5.71 GBq [18
F]TBAF in 1 mL CH3CN was
prepared.
4.3.2. General procedure for the thermal n.c.a. 18
F-radiofluoro-de-triazenation
of 5-TBV (page 112)
Vial (1 mL) was charged with 8-TBV (2 mg, 5 μmol), CCl3 or CCl4 (0.2 mL), a mixture of
PPA in CH3CN (10 μL mixture of 1 g PPA, 105% H3PO4 basis, in 1 mL CH3CN), and
[18
F]TBAF (10 and 25 μL solution of [18
F]TBAF in 1 mL CH3CN, 47 and 24 MBq per run,
respectively). The vial was immediately sealed with a septum and the resulting mixture heated
at 90 oC for 15 min. After reaction, the reaction mixture was quenched with NHEt2 (30 μL),
Page 136
122
and analysed by radio-HPLC (CH3CN/0.1 M NH4OAc 30/70, 1 mL/min, Luna 5u Phenyl-
Hexyl column from Phenomenex®, 250 x 4.6 mm).
4.3.3. General procedure for the microwave-assisted n.c.a. 18
F-radiofluoro-de-
triazenation of 5-TBV (pages 112-115)
The microwave tube (1 mL) was charged with 5-TBV (2 mg, 5 μmol), CHCl3 or CCl4 (0.2
mL), a mixture of PPA in CH3CN (10 μL mixture of 1 g PPA, 105% H3PO4 basis, in 1 mL
CH3CN), and [18
F]TBAF (10 and 25 μL solution of [18
F]TBAF in 1 mL CH3CN, 32 and 24
MBq per run, respectively). The microwave tube was immediately sealed with a septum and
placed in the microwave (MW) cavity (Discover®, CEM) and subjected to MW irradiation for
10 min at temperature set point 90 o
C (90 W). After reaction, the reaction mixture was
quenched with NHEt2 (30 μL), and analysed by radio-HPLC (CH3CN/0.1 M NH4OAc 30/70,
1 mL/min, Luna 5u Phenyl-Hexyl column from Phenomenex®, 250 x 4.6 mm).
Page 137
123
5. CONCLUSION
Page 138
124
In the present thesis, we have synthesized, by fluoro-de-diazoniation using tert-butyl nitrite
as diazonating agent, and boron trifluoride etherate as fluorinating agent, (rac)-5-FBVM and
its corresponding enantiomers (2R,3R)- and (2S,3S)-5-FBVM in around 25% yields. QSAR
studies based on 32 vesamicol and benzovesamicol derivatives taking into account the
stereoselectivity of the VAChT binding site were performed. As predicted by 3D QSAR
studies, both enantiomers exhibited high in vitro VAChT binding affinities determined by
radioligand displacement studies, and were in the same range as 5-IBVM which is the
reference compound (the only SPECT radioligand for VAChT used in human). However,
only (2S,3S)-FBVM was selective over σ1 receptors to warrant further investigation as a
potential PET VAChT radioligand for in vivo mapping of cholinergic nerve terminals to
obtain an early diagnosis of Alzheimer's disease. We also demonstrated, that fluoro-de-
triazenation of the corresponding triazene precursor 5-TBV can be a potential method to
introduce radioactive isotope fluorine-18 on the non-activated benzovesamicol scaffold. With
this work we have fulfilled the first three goals of the thesis (Chapter 2 - Aims and Scope,
pages 73-75).
Furthermore, during theoretical examination of the coordination properties of aryltriazenes
and boron trifluoride, we have proved that fluoro-de-triazenation is possible by the use of
boron trifluoride etherate as the sole two roles reagent. In this manner, competitive formation
of the acid counterion substituted byproduct was eliminated and 5-FBVM yield was greatly
increased (72%) in tetrachloromethane under optimized microwave conditions. To
demonstrate versatility of the method, successful fluoro-de-triazenation was accomplished on
different para-substituted aryltriazenes under conventional and microwave heating. With this
work, the fourth goal of the thesis has been fulfilled.
As radiofluorinated boron trifluoride or boron tetrafluoride are not optimal for
radiofluorination due to dilution of specific radioactivity, we have examinated different
reaction parameters with the main focus on the acid to trigger triazene decomposition but with
no interference in the radiofluorination step (the fifth goal of the thesis). According to the
results in the non-radioactive attempts, polyphosphoric acid (PPA) proved to be the most
suitable one, although has never been used in this type of reaction before. Radiofluorination
of (rac)-5-TBV using PPA and tetra-n-butylammonium [18
F]fluoride ([18
F]TBAF) as a source
of no-carrier-added [18
F]fluoride in chloroform under microwave conditions may afforded
trace of the 5-[18
F]FBVM (the sixth goal of the thesis). This encouraging result warrants to
Page 139
125
optimise 5-[18
F]FBVM yield via fluoro-de-triazenation or other precursors, such as
diaryliodonium salt, allowing fluorionation on non-activated aryl systems.
Page 140
126
6. REFERENCES
Page 141
127
1. Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthesis of 11C, 18F, 15O, and 13N
Radiolabels for Positron Emission Tomography. Angew. Chem. Int. Ed., 47, 8998-9033
(2008).
2. Wood, K.A.; Hoskin, P.J.; Saunders, M.I. Positron Emission Tomography in
Oncology: A Review. Clin. Oncol., 19, 237-255 (2007).
3. Bengel, F.M.; Higuchi, T.; Javadi, M.S.; Lautamäki, R. Cardiac Positron Emission
Tomography. J. Am. Coll. Cardiol., 54, 1-15 (2009).
4. Di Carli, M.F.; Dorbala, S.; Meserve, J.; El Fakhri, G.; Sitek, A.; Moore, S.C. Clinical
Myocardial Perfusion PET/CT. J. Nucl. Med., 48, 783-793 (2007).
5. Zimmer, L.; Luxen, A. PET radiotracers for molecular imaging in the brain: Past,
present and future. NeuroImage, 61, 363-370 (2012).
6. Burns, H.D.; Hamill, T.G.; Eng, W.S.; Francis, B.; Fioravanti, C.; Gibson, R.E.
Positron emission tomography neuroreceptor imaging as a tool in drug discovery, research
and development. Curr. Opin. Chem. Biol., 3, 388-394 (1999).
7. Lever, J.R. PET and SPECT Imaging of the Opioid System: Receptors, Radioligands
and Avenues for Drug Discovery and Development. Curr. Pharm. Des., 13, 33-49 (2007).
8. Lin, M.; Shon, I.H.; Lin, P. Positron emission tomography: current status and future
challenges. Inter. Med. J., 40, 19-29 (2010).
9. McQuade, P.; Rowland, D.J.; Lewis, J.S.; Welch, M.J. Positron-Emitting Isotopes
Produced on Biomedical Cyclotrons. Curr. Med. Chem., 12, 807-818 (2005).
10. Blodgett, T.M.; Meltzer, C.C.; Townsend, D.W. PET/CT: Form and Function1.
Radiology, 242, 360-385 (2007).
11. Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular Imaging with PET. Chem.
Rev., 108, 1501-1516 (2008).
12. O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F
bond. Chem. Soc. Rev., 37, 308-319 (2008).
13. Zhou, P.; Zou, J.; Tian, F.; Shang, Z. Fluorine Bonding - How Does It Work In
Protein-Ligand Interactions? J. Chem. Inf. Model., 49, 2344-2355 (2009).
14. Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib.
Med. Chem., 22, 527-540 (2007).
15. Hunter, L. The C-F bond as a conformational tool in organic and biological chemistry.
Beilstein J. Org. Chem., 6, 1-14 (2010).
Page 142
128
16. Ehrenkaufer, R.E.; Potocki, J.F.; Jewett, D.M. Simple Synthesis of F-18-Labeled 2-
Fluoro-2-Deoxy-D-Glucose: Concise Communication. J. Nucl. Med., 25, 333-337 (1984).
17. Eskola, O.; Grönroos, T.; Bergman, J.; Haaparanta, M.; Marjamäki, P.; Lehikoinen, P.;
Forsback, S.; Langer, O.; Hinnen, F.; Dollé, F.; Halldin, C.; Solin, O. A novel electrophilic
synthesis and evaluation of medium specific radioactivity (1R,2S)-4-
[18F]fluorometaraminol, a tracer for the assessment of cardiac sympathetic nerve integrity
with PET. Nucl. Med. Biol., 31, 103-110 (2004).
18. Mukherjee, J.; Yang, Z.Y.; Das, M.K.; Brown, T. Fluorinated benzamide neuroleptics-
III. Development of (S)-N-[(1-allyl-2-pyrrolidinyl)methyl]-5-(3-[18F]fluoropropyl)-2,3-
dimethoxybenzamide as an improved dopamine D-2 receptor tracer. Nucl. Med. Biol., 22,
283-296 (1995).
19. Hashizume, K.; Tamakawa, H.; Hashimoto, N.; Miyake, Y. Single-step synthesis of
[18F]haloperidol from the chloro-precursor and its applications in PET imaging of a cat’s
brain. Appl. Radiat. Isot., 48, 1179-1185 (1997).
20. Hamacher, K.; Hamkens, W. Remote controlled one-step production of 18F labeled
butyrophenone neuroleptics exemplified by the synthesis of n.c.a. [18F] N-
methylspiperone. Appl. Radiat. Isotop. 46, 911-916 (1995).
21. Piert, M.; Machulla, H.J.; Picchio, M.; Reischl, G.; Ziegler, S.; Kumar, P.; Wester,
H.J.; Beck, R.; McEwan, A.J.; Wiebe, L.I.; Schwaiger, M. Hypoxia-Specific Tumor
Imaging with 18F-Fluoroazomycin Arabinoside. J. Nucl. Med., 46, 106-113 (2005).
22. Chang, C.W.; Chou, T.K.; Liu, R.S.; Wang, S.J.; Lin, W.J.; Chen, C.H.; Wang, H.E. A
robotic synthesis of [18F]fluoromisonidazole ([18F]FMISO). Appl. Radiat. Isotop., 65,
682-686 (2007).
23. Kemp, D.D.; Gordon, M.S. Theoretical Study of the Solvation of Fluorine and
Chlorine Anions by Water. J. Phys. Chem. A, 109, 7688-7699 (2005).
24. Schlyer, D.J.; Bastos, M.A.V.; Alexoff, D.; Wolf, A.P. Separation of [18F]fluoride
from [18O]water using anion exchange resin. Int. J. Rad. Appl. Instrum. A, 41, 531-533
(1990).
25. Cai, L.; Lu, S.; Pike, V.W. Chemistry with [18F]Fluoride Ion. Eur. J. Org. Chem., 17,
2853-2873 (2008).
26. Jewett, D.M.; Toorongian, S.A.; Mulholland, G.K.; Watkins, G.L.; Kilbourn, M.R.
Multiphase extraction: Rapid phase-transfer of [18F]fluoride ion for nucleophilic
radiolabeling reactions. Int. J. Rad. Appl. Instrum. A, 39, 1109-1111 (1988).
Page 143
129
27. Briard, E.; Pike, V.W. Substitution-reduction: an alternative process for the [18F]N-
(2-fluoroethylation) of anilines. J. Labelled Compd. Radiopharm., 47, 217-232 (2004).
28. Kim, D.W.; Ahn, D.S.; Oh, Y.H.; Lee, S.; Kil, H.S.; Oh, S.J.; Lee, S.J.; Kim, J.S.;
Ryu, J.S.; Moon, D.H.; Chi, D.Y. A New Class of SN2 Reactions Catalyzed by Protic
Solvents: Facile Fluorination for Isotopic Labeling of Diagnostic Molecules. J. Am. Chem.
Soc. 128, 16394-16397 (2006).
29. Kim, D.W.; Jeong, H.J.; Lim, S.T.; Sohn, M.H. Recent Trends in the Nucleophilic
[18F]-radiolabeling Method with No-carrier-added [18F]fluoride. Nucl. Med. Mol.
Imaging, 44, 25-32 (2010).
30. Pike, V.W. PET radiotracers: crossing the blood-brain barrier and surviving
metabolism. Trends Pharmacol. Sci., 30, 431-440 (2009).
31. Baumann, C.A., Mu, L., Wertli, N.; Krämer, S.D.; Honer, M.; Schubiger, P.A.;
Ametamey, S.M. Syntheses and pharmacological characterization of novel thiazole
derivatives as potential mGluR5 PET ligands. Bioorg. Med. Chem., 18, 6044-6054 (2010).
32. Hussainy, R.A.H.; Verbeek, J.; Born, D.; Molthoff, C.; Booij, J.; Herscheid, J.D.M.
Synthesis, biodistribution and PET studies in rats of 18F-Labeled bridgehead fluoromethyl
analogues of WAY-100635. Nucl. Med. Biol., 39, 1068-1076 (2012).
33. Beuthien-Baumann, B.; Hamacher, K.; Oberdorfer, F.; Steinbach, J. Preparation of
fluorine-18 labelled sugars and derivatives and their application as tracer for positron-
emission-tomography. Carbohydr. Res., 327, 107-118 (2000).
34. Dollé, F.: Helfenbein, J.; Hinnen, F.; Mavel, S.; Mincheva, Z.; Saba, W.; Schöllhorn-
Peyronneau, M.-A., Valette, H.; Garreau, L.; Chalon, S.; Halldin, C.; Madelmont, J.C.;
Deloye, J.B.; Bottlaender, M.; Le Gailliard, J.; Guilloteau, D.; Emond, P. One-step
radiosynthesis of [18F]LBT-999: a selective radioligand for the visualization of the
dopamine transporter with PET. J. Labelled Compd. Radiopharm., 50, 716-723 (2007).
35. Ahmed, N.; Langlois, R.; Rodrigue, S.; Bénard, F. Van Lier, J.E. Automated synthesis
of 11β-methoxy-4,16α-[16α-18F]difluoroestradiol (4F-M[18F]FES) for estrogen receptor
imaging by positron emission tomography. Nucl. Med. Biol., 34, 459-464 (2007).
36. Hashizume, K.; Hashimoto, N.; Miyake, Y. Rapid and Efficient Synthesis of High-
Purity Fluorine-18 Labeled Haloperidol and Spiperone via the Nitro Precursor in
Combination with a New HPLC Separation Method. Bull. Chem. Soc. Jpn., 70, 681-687
(1997).
Page 144
130
37. Dollé, F. Fluorine-18-Labelled Fluoropyridines: Advances in Radiopharmaceutical
Design. Curr. Pharm. Des., 11, 3221-3235 (2005).
38. Ding, Y.-S.; Liang, F.; Fowler, J.S.; Kuhar, M.J.; Carroll, F.I. Synthesis of
[18F]norchlorofluoroepibatidine and its N-methyl derivative: new PET ligands for
mapping nicotinic acetylcholine receptors. J. Labelled Compd. Radiopharm., 39, 827-832
(1997).
39. Dolci, L; Dollé, F.; Valette, H.; Vaufrey, F.; Fuseau, C.; Bottlaender, M.; Crouzel, C.
Synthesis of a fluorine-18 labeled derivative of epibatidine for in vivo nicotinic
acetylcholine receptor PET imaging. Bioorg. Med. Chem. 7, 467-479 (1999).
40. Balz, G.; Schiemann, G. Über aromatische Fluorverbindungen, I.: Ein neues Verfahren
zu ihrer Darstellung. Ber. Dtsch. Chem. Ges., 60, 1186-1190 (1927).
41. Wallach, O. Ueber das Verhalten einiger Diazo- und Diazoamidoverbindungen. Justus
Liebigs Ann. Chem., 235, 233-255 (1886).
42. Merritt, E.A.; Olofsson, B. Diaryliodonium Salts: A Journey from Obscurity to Fame.
Angew. Chem. Int. Ed., 48, 9052-9070 (2009).
43. Nozaki, T.; Tanaka, Y. The preparation of F18-labelled aryl fluorides. Int. J. Appl.
Radiat. Isot., 18, 111-119 (1967).
44. Gross, M.L.; Blank, D.H.; Welch, W.M. The triazene moiety as a protecting group for
aromatic amines. J. Org. Chem. 58, 2104-2109 (1993).
45. Kook, C.S.; Reed, M.F.; Digenis, G.A. Preparation of fluorine-18-labeled haloperidol.
J. Med. Chem. 18, 533-535 (1975).
46. Berridge, M.S.; Sayre, L.M.; Arora, P.K.; Terris, A.H.; Riachi, N.J.; Harik, S.I. Design
and synthesis of 18F-labeled neurotoxic analogs of MPTP. J. Med. Chem. 36, 1284-1290
(1993).
47. Kilbourn, M.R.; Welch, M.J.; Dence, C.S.; Tewson, T.J.; Saji, H.; Maeda, M. Carrier-
added and no-carrier-added syntheses of [18F]spiroperidol and [18F]haloperidol. Int. J.
Appl. Radiat. Isot., 35, 591-598 (1984).
48. Coenen, H.H.; Ermert, J. Direct Nucleophilic 18F-Fluorination of Electron Rich
Arenes: Present Limits of No-Carrier-Added Reactions. Curr. Radiopharm., 3, 163-173
(2010).
49. Lancer, K.M.; Wiegand, G.H. The ortho effect in the pyrolysis of iodonium halides. A
case for a sterically controlled nucleophilic aromatic (SN) substitution reaction. J. Org.
Chem., 41, 3360-3364 (1976).
Page 145
131
50. Grushin, V.V. Carboranylhalonium ions: from striking reactivity to a unified
mechanistic analysis of polar reactions of diarylhalonium compounds. Acc. Chem. Res., 25,
529-536 (1992).
51. Chun, J.-H.; Lu, S.; Lee, Y.-S.; Pike, V.W. Fast and High-Yield Microreactor
Syntheses of ortho-Substituted [18F]Fluoroarenes from Reactions of [18F]Fluoride Ion
with Diaryliodonium Salts. J. Org. Chem., 75, 3332-3338 (2010).
52. Martín-Santamaría, S.; Carroll, M.A.; Carroll, C.M.; Carter, C.D.; Pike, V.W.; Rzepa,
H.S.; Widdowson, D.A. Fluoridation of heteroaromatic iodonium salts-experimental
evidence supporting theoretical prediction of the selectivity of the process. Chem.
Commun., 649-650 (2000).
53. Ermert, J.; Hocke, C.; Ludwig, T.; Gail, R.; Coenen, H.H. Comparison of pathways to
the versatile synthon of no-carrier-added 1-bromo-4-[18F]fluorobenzene. J. Labelled
Compd. Radiopharm., 47, 429-441 (2004).
54. Moon, B.S.; Kil, H.S.; Park, J.H.; Kim, J.S.; Park, J.; Chi, D.Y.; Lee, B.C.; Kim, S.E.
Facile aromatic radiofluorination of [18F]flumazenil from diaryliodonium salts with
evaluation of their stability and selectivity. Org. Biomol. Chem., 9, 8346-8355 (2011).
55. Dollé, F.; Hinnen, F.; Emond, P.; Mavel, S.; Mincheva, Z.; Saba, W.; Schöllhorn-
Peyronneau, M.-A.; Valette, H.; Garreau, L.; Chalon, S.; Halldin, C.; Helfenbein, J.;
Legaillard, J.; Madelmont, J.-C.; Deloye, J.-B.; Bottlaender, M.; Guilloteau, D.
Radiosynthesis of [18F]LBT-999, a selective radioligand for the visualization of the
dopamine transporter with PET. J. Labelled Compd. Radiopharm., 49, 687-698 (2006).
56. Steiniger, B.; Wuest, F.R. Synthesis of 18F-labelled biphenyls via SUZUKI cross-
coupling with 4-[18F]fluoroiodobenzene. J. Labelled Compd. Radiopharm., 49, 817-827
(2006).
57. Wüst, F.R.; Höhne, A.; Metz, P. Synthesis of 18F-labelled cyclooxygenase-2 (COX-2)
inhibitors via Stille reaction with 4-[18F]fluoroiodobenzene as radiotracers for positron
emission tomography (PET). Org. Biomol. Chem., 3, 503-507 (2005).
58. Wüst, F.R., Kniess, T. Synthesis of 4-[18F]fluoroiodobenzene and its application in
sonogashira cross-coupling reactions. J. Labelled Compd. Radiopharm., 46, 699-713
(2003).
59. Ross, T.L.; Ermert, J.; Hocke, C., Coenen, H.H. Nucleophilic 18F-Fluorination of
Heteroaromatic Iodonium Salts with No-Carrier-Added [18F]Fluoride. J. Am. Chem. Soc.,
129, 8018-8025 (2007).
Page 146
132
60. Scott, P.J.H.; Shao, X. Fully automated, high yielding production of N-succinimidyl 4-
[18F]fluorobenzoate ([18F]SFB), and its use in microwave-enhanced radiochemical
coupling reactions. J. Labelled Compd. Radiopharm., 53, 586-591 (2010).
61. Berchtold, N.C.; Cotman, C.W. Evolution in the Conceptualization of Dementia and
Alzheimer’s Disease: Greco-Roman Period to the 1960s. Neurobiol. Aging, 19, 173-189
(1998).
62. Boller, F.; Forbes, M.M.; History of dementia and dementia in history: An overview.
J. Neurol. Sci., 158, 125-133 (1998).
63. Alzheimer's Association. 2012 Alzheimer’s disease facts and figures. Alzheimer’s &
Dementia, 8, 131-168 (2012).
64. Hsiungin, G.-Y.R. In: Atlas of Alzheimer’s Disease; Feldman, H.H., Ed.; Informa
Healthcare: United Kingdom, 27-40 (2007).
65. Scarmeas, N.; Stern, Y. Cognitive Reserve and Lifestyle. J. Clin. Exp. Neuropsyc., 25,
625-633 (2003).
66. Bird, T.D. Genetic Factors in Alzheimer’s Disease. New Engl. J. Med., 352, 862-864
(2005).
67. Leduc, V.; Jasmin-Bélanger, S.; Poirier, J. APOE and cholesterol homeostasis in
Alzheimer’s disease. Trends Mol. Med., 16, 469-477 (2010).
68. Mackenziein, I.R. In: Atlas of Alzheimer’s Disease; Feldman, H.H., Ed.; Informa
Healthcare: United Kingdom, 71-82 (2007).
69. Kaye, J.A.; Swihart, T.; Howieson, D.; Dame, A.; Moore, M.M.; Karnos, T.;
Camicioli, R.; Ball, M.; Oken, B.; Sexton, G. Volume loss of the hippocampus and
temporal lobe in healthy elderly persons destined to develop dementia. Neurology, 48,
1297-1304 (1997).
70. Fox, N.C.; Crum, W.R.; Scahill, R.I.; Stevens, J.M.; Janssen, J.C.; Rossor, M.N.
Imaging of onset and progression of Alzheimer’s disease with voxel-compression mapping
of serial magnetic resonance images. Lancet., 358, 201-205 (2001).
71. Förstl, H.; Zerfass, R.; Geiger-Kabisch, C.; Sattel, H.; Besthorn, C.; Hentschel, F.
Brain atrophy in normal ageing and Alzheimer’s disease. Volumetric discrimination and
clinical correlations. Br. J. Psychiatry, 167, 739-746 (1995).
72. Qadi N.; Alipour, S.; Beattie, B.L. In: Atlas of Alzheimer’s Disease; Feldman, H.H.,
Ed.; Informa Healthcare: United Kingdom, 59-70 (2007).
Page 147
133
73. Guillozet, A.L.; Weintraub, S.; Mash, D.C.; Mesulam, M.M. Neurofibrillary tangles,
amyloid, and memory in aging and mild cognitive impairment. Arch. Neurol.. 60, 729-736
(2003).
74. Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: the amyloid cascade hypothesis.
Science, 256, 184-185 (1992).
75. Cummings, J.L. Alzheimer’s Disease. New Engl. J. Med., 351, 56-67 (2004).
76. Reitz, C. Alzheimer’s Disease and the Amyloid Cascade Hypothesis: A Critical
Review. Int. J. Alzheimers Dis., 2012, 1-11 (2012).
77. Knopman, D.S.; Parisi, J.E.; Salviati, A.; Floriach-Robert, M.; Boeve, B.F.; Ivnik,
R.J.; Smith, G.E.; Dickson, D.W.; Johnson, K.A.; Petersen, L.E.; McDonald, W.C.; Braak,
H.; Petersen, R.C. Neuropathology of Cognitively Normal Elderly. J. Neuropath. Exp.
Neurology. Neur., 62, 1087-1095 (2003).
78. Hardy, J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J.
Neurochem., 110, 1129-1134 (2009).
79. Qadi, N.; Feldman, H.H. In: Atlas of Alzheimer’s Disease; Feldman, H.H., Ed.;
Informa Healthcare: United Kingdom, 41-58 (2007).
80. Sahasrabudhe, S.R.; Brown, A.M.; Hulmes, J.D.; Jacobsen, J.S.; Vitek, M.P.; Blume,
A.J.; Sonnenberg, J.L. Enzymatic generation of the amino terminus of the beta-amyloid
peptide. J. Biol. Chem., 268, 16699-16705 (1993).
81. Hilbich, C.; Kisters-Woike, B.; Reed, J.; Masters, C.L.; Beyreuther, K. Aggregation
and secondary structure of synthetic amyloid βA4 peptides of Alzheimer’s disease. J. Mol.
Biol., 218, 149-163 (1991).
82. Akiyama, H.: Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.;
Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; Finch, C.E.; Frautschy, S.; Griffin, W.S.;
Hampel, H.; Hull, M.; Landreth, G.; Lue, L.; Mrak, R.; Mackenzie, I.R.; McGeer, P.L.;
O'Banion, M.K.; Pachter, J.; Pasinetti, G.; Plata-Salaman, C.; Rogers, J.; Rydel, R.; Shen,
Y.; Streit, W.; Strohmeyer, R.; Tooyoma, I.; Van Muiswinkel, F.L.; Veerhuis, R.; Walker,
D.; Webster, S.; Wegrzyniak, B.: Wenk, G.; Wyss-Coray, T. Inflammation and
Alzheimer’s disease. Neurobiol. Aging, 21, 383-421 (2000).
83. Crouch, P.J.; Harding, S.-M.E.; White, A.R.; Camakaris, J.; Bush, A.I.; Masters, C.L.
Mechanisms of Aβ mediated neurodegeneration in Alzheimer’s disease. Int. J. Biochem.
Cell Biol., 40, 181-198 (2008).
Page 148
134
84. Maltsev, A.V.; Bystryak, S.; Galzitskaya, O.V. The role of β-amyloid peptide in
neurodegenerative diseases. Ageing Res. Rev., 10, 440-452 (2011).
85. Ferreira, S.T.; Klein, W.L. The Aβ oligomer hypothesis for synapse failure and
memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem., 96, 529-543 (2011).
86. Stoothoff, W.H.; Johnson, G.V.W. Tau phosphorylation: physiological and
pathological consequences. Biochim. Biophys. Acta, 1739, 280-297 (2005).
87. Froelich-Fabre, S.; Bhat, R.V. Mechanisms of tauopathies. Drug Discov. Today, 1,
391-398 (2004).
88. Trojanowski, J.Q.; Schmidt, M.L.; Otvos, L.; Arai, H.: Hill, W.D.; Lee, V.M.
Vulnerability of the neuronal cytoskeleton in aging and Alzheimer disease: widespread
involvement of all three major filament systems. Annu. Rev. Gerontol. Geriatr., 10, 167-
182 (1990).
89. Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta
Neuropathol., 82, 239-259 (1991).
90. Braak, H.; Braak, E. Evolution of the neuropathology of Alzheimer’s disease. Acta
Neurol. Scand., 165, 3-12 (1996).
91. Upadhyaya, P.; Seth, V.; Ahmad, M. Therapy of Alzheimer’s disease: An update. Afr.
J. Pharm. Pharmacol. , 4, 408-421 (2010).
92. Qadi, N.; Assaly, M.; Lee, P.E. In: Atlas of Alzheimer’s Disease; Feldman, H.H., Ed.;
Informa Healthcare: United States of America, 105-124 (2007).
93. Meldrum, B.; Garthwaite, J. Excitatory amino acid neurotoxicity and
neurodegenerative disease. Trends Pharmacol. Sci., 11, 379-387 (1990).
94. Tayeb, H.O.; Yang, H.D.; Price, B.H.; Tarazi, F.I. Pharmacotherapies for Alzheimer’s
disease: Beyond cholinesterase inhibitors. Pharmacol. Ther., 134, 8-25 (2012).
95. Salloway, S.; Mintzer, J.; Weiner, M.F.; Cummings, J.L. Disease-modifying therapies
in Alzheimer’s disease. Alzheimer’s Dement., 4, 65-79 (2008).
96. Kurz, A.; Perneczky, R. Novel insights for the treatment of Alzheimer’s disease. Prog.
Neuropsychopharmacol. Biol. Psychiatry, 35, 373-379 (2011).
97. Parnetti, L.; Lanari, A.; Amici, S.; Gallai, V.; Vanmechelen, E.; Hulstaert, F. CSF
phosphorylated tau is a possible marker for discriminating Alzheimer’s disease from
dementia with Lewy bodies. Neurol. Sci., 22, 77-78 (2001).
Page 149
135
98. Wallin, Å.K.; Blennow, K.; Andreasen, N.; Minthon, L. CSF Biomarkers for
Alzheimer's Disease: Levels of β-Amyloid, Tau, Phosphorylated Tau Relate to Clinical
Symptoms and Survival. Dement. Geriatr. Cogn. Disord., 21, 131-138 (2006).
99. Reiman, E.M.; Jagust, W.J. Brain imaging in the study of Alzheimer’s disease.
NeuroImage, 61, 505-516 (2012).
100. Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.;
Bergström, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; Ausén, B.; Debnath, M.L.;
Barletta, J.; Price, J.C.; Sandell, J.; Lopresti, B.J.; Wall, A.; Koivisto, P.; Antoni, G.;
Mathis, C.A.; Långström, B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh
Compound-B. Ann. Neurol., 55, 306-319 (2004).
101. Price, J.C.; Klunk, W.E.; Lopresti, B.J.; Lu, X.; Hoge, J.A.; Ziolko, S.K.; Holt, D.P.;
Meltzer, C.C.; DeKosky, S.T.; Mathis, C.A. Kinetic modeling of amyloid binding in
humans using PET imaging and Pittsburgh Compound-B. J. Cereb. Blood Flow Metab.,
25, 1528-1547 (2005).
102. Liu, J.; Kepe, V.; Zabjek, A.; Petric, A.; Padgett, H.C.; Satyamurthy, N.; Barrio, J.R.
High-yield, automated radiosynthesis of 2-(1-{6-[(2-[18F]fluoroethyl)(methyl)amino]-2-
naphthyl}ethylidene)malononitrile ([18F]FDDNP) ready for animal or human
administration. Mol. Imaging Biol., 9, 6-16 (2007).
103. Teng, E.; Kepe, V.; Frautschy, S.A.; Liu, J.; Satyamurthy, N.; Yang, F.; Chen, P.P.;
Cole, G.B.; Jones, M.R.; Huang, S.C.; Flood, D.G.; Trusko, S.P.; Small, G.W.; Cole, G.M.;
Barrio, J.R. [F-18]FDDNP microPET imaging correlates with brain Aβ burden in a
transgenic rat model of Alzheimer disease: effects of aging, in vivo blockade, and anti-Aβ
antibody treatment. Neurobiol. Dis., 43, 565-575 (2011).
104. Shin, J.; Kepe, V.; Barrio, J.R.; Small, G.W. The merits of FDDNP-PET imaging in
Alzheimer’s disease. J. Alzheimers Dis., 26, 135-145 (2011).
105. Yang, L.; Rieves, D.; Ganley, C. Brain Amyloid Imaging - FDA Approval of
Florbetapir F18 Injection. New Engl. J. Med., 367, 885-887 (2012).
106. Petrella, J.R.; Coleman, R.E.; Doraiswamy, P.M. Neuroimaging and Early Diagnosis
of Alzheimer Disease: A Look to the Future. Radiology, 226, 315-336 (2003).
107. Samuel, W.; Terry, R.D.; DeTeresa, R.; Butters, N.; Masliah, E. Clinical correlates of
cortical and nucleus basalis pathology in alzheimer dementia. Arch. Neurol., 51, 772-778
(1994).
Page 150
136
108. Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen,
L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse
loss is the major correlate of cognitive impairment. Ann. Neurol., 30, 572-580 (1991).
109. Scheff, S.W.; DeKosky, S.T.; Price, D.A. Quantitative assessment of cortical synaptic
density in Alzheimer’s disease. Neurobiol. Aging, 11, 29-37 (1990).
110. DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s
disease: Correlation with cognitive severity. Ann. Neurol., 27, 457-464 (1990).
111. Eiden, L.E.; Schäfer, M.K.-H.; Weihe, E.; Schütz, B. The vesicular amine transporter
family (SLC18): amine/proton antiporters required for vesicular accumulation and
regulated exocytotic secretion of monoamines and acetylcholine. Pflugers Arch., 447, 636-
640 (2004).
112. Erickson, J.D.; Varoqui, H.; Schäfer, M.K.; Modi, W.; Diebler, M.F.; Weihe, E.;
Rand, J.; Eiden L.E.; Bonner, T.I.; Usdin, T.B. Functional identification of a vesicular
acetylcholine transporter and its expression from a “cholinergic” gene locus. J. Biol. Chem.
269, 21929-21932 (1994).
113. Roghani, A.; Feldman, J.; Kohan, S.A.; Shirzadi, A.; Gundersen, C.B.; Brecha, N.;
Edwards, R.H. Molecular cloning of a putative vesicular transporter for acetylcholine.
Proc. Natl. Acad. Sci. U.S.A., 91, 10620-10624 (1994).
114. Bejanin, S.; Cervini, R.; Mallet, J.; Berrard, S. A unique gene organization for two
cholinergic markers, choline acetyltransferase and a putative vesicular transporter of
acetylcholine. J. Biol. Chem., 269, 21944-21947 (1994).
115. Usdin, T.B.; Eiden, L.E.; Bonner, T.I.; Erickson, J.D. Molecular biology of the
vesicular ACh transporter. Trends Neurosci., 18, 218-224 (1995).
116. Gilmor, M.L.; Counts, S.E.; Wiley, R.G.; Levey, A.I. Coordinate expression of the
vesicular acetylcholine transporter and choline acetyltransferase following
septohippocampal pathway lesions. J. Neurochem., 71, 2411-2420 (1998).
117. Gilmor, M.L.; Erickson, J.D.; Varoqui, H.; Hersh, L.B.; Bennett, D.A.; Cochran, E.J.;
Mufson, E.J.; Levey, A.I. Preservation of nucleus basalis neurons containing choline
acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild
cognitive impairment and early Alzheimer’s disease. J. Comp. Neurol., 411, 693-704
(1999).
118. Katzung, B.G. In: Basic and Clinical Pharmacology; Katzung, B.G.; Masters, S.;
Trevor, A.J., Ed.; McGraw-Hill Medical: United States of America, 11st Ed., 81 (2009).
Page 151
137
119. Brittain, R.T.; Levy, G.P.; Tyers, M.B. The neuromuscular blocking action of 2-(4-
phenylpiperidino) cyclohexanol (AH 5183). Eur. J. Pharmacol., 8, 93-99 (1969).
120. Prior, C.; Marshall, I.G.; Parsons, S.M. The pharmacology of vesamicol: An inhibitor
of the vesicular acetylcholine transporter. Gen. Pharmacol., 23, 1017-1022 (1992).
121. Rogers, G.A.; Kornreich, W.D.; Hand, K; Parsons, S.M. Kinetic and equilibrium
characterization of vesamicol receptor-ligand complexes with picomolar dissociation
constants. Mol. Pharmacol., 44, 633-641 (1993).
122. Wannan, G.; Prior, C.; Marshall, I.G. α-Adrenoceptor blocking properties of
vesamicol. Eur. J. Pharmacol., 201, 29-34 (1991).
123. Efange, S.M.; Mach, R.H.; Smith, C.R.; Khare, A.B.; Foulon, C.; Akella, S.K.;
Childers, S.R.; Parsons, S.M. Vesamicol analogues as sigma ligands: Molecular
determinants of selectivity at the vesamicol receptor. Biochem. Pharmacol., 49, 791-797
(1995).
124. Bando, K.; Naganuma, T.; Taguchi, K.; Ginoza, Y.; Tanaka, Y.; Koike, K.; Takatoku,
K. Piperazine analog of vesamicol: In vitro and in vivo characterization for vesicular
acetylcholine transporter. Synapse, 38, 27-37 (2000).
125. Bando, K.; Taguchi, K.; Ginoza, Y.; Naganuma, T.; Tanaka, Y.; Koike, K.; Takatoku,
K. Synthesis and evaluation of radiolabeled piperazine derivatives of vesamicol as SPECT
agents for cholinergic neurons. Nucl. Med. Biol., 28, 251-260 (2001).
126. Efange, S.M.N.; Khare, A.; Parsons, S.M.; Bau, R.; Metzenthin, T. Nonsymmetrical
bipiperidyls as inhibitors of vesicular acetylcholine storage. J. Med. Chem., 36, 985-989
(1993).
127. Efange, S.M.; Michelson, R.H.; Knusel, B.; Hefti, F.; Boudreau, R.J.; Thomas, J.R.;
Tennison, J.R. Synthesis and biological evaluation of radioiodinated N-2-(4-piperidyl)ethyl
benzamides. Nucl. Med. Biol., 20, 527-538 (1993).
128. Efange, S.M.N.; Khare, A.B.; Foulon, C.; Akella, S.K.; Parsons, S.M.
Spirovesamicols: Conformationally Restricted Analogs of 2-(4-
Phenylpiperidino)cyclohexanol (Vesamicol, AH5183) as Potential Modulators of
Presynaptic Cholinergic Function. J. Med. Chem., 37, 2574-2582 (1994).
129. Efange, S.M.; Mach, R.H.; Khare, A.; Michelson, R.H.; Nowak, P.A.; Evora, P.H. p-
[18F]fluorobenzyltrozamicol ([18F]FBT): Molecular decomposition-reconstitution
approach to vesamicol receptor radioligands for positron emission tomography. Appl.
Radiat. Isot., 45, 465-472 (1994).
Page 152
138
130. Efange, S.M.N.; Kamath, A.P.; Khare, A.B.; Kung, M.-P.; Mach, R.H.; Parsons, S.M.
N-Hydroxyalkyl Derivatives of 3β-Phenyltropane and 1-Methylspiro[1H-indoline-3,4‘-
piperidine]: Vesamicol Analogues with Affinity for Monoamine Transporters. J. Med.
Chem., 40, 3905-3914 (1997).
131. Efange, S.M.N.; Khare, A.B.; Mach, R.H.; Parsons, S.M. Hydroxylated
Decahydroquinolines as Ligands for the Vesicular Acetylcholine Transporter: Synthesis
and Biological Evaluation. J. Med. Chem., 42, 2862-2869 (1999).
132. Efange, S.M.; Nader, M.A.; Ehrenkaufer, R.L.; Khare, A.B.; Smith, C.R.; Morton,
T.E.; Mach, R.H. (+)-p-([18F]fluorobenzyl)spirotrozamicol {(+)-[18F]Spiro-FBT}:
synthesis and biological evaluation of a high-affinity ligand for the vesicular acetylcholine
transporter (VAChT). Nucl. Med. Biol., 26, 189-192 (1999).
133. Efange, S.M.; von Hohenberg, K.; Khare, A.B.; Tu, Z.; Mach, R.H.; Parsons, S.M.
Synthesis and biological characterization of stable and radioiodinated (±)-trans-2-hydroxy-
3-P{4-(3-iodophenyl)piperidyl}-1,2,3,4-tetrahydronaphthalene (3′-IBVM). Nucl. Med.
Biol., 27, 749-755 (2000).
134. Mulholland, G.K.; Jung, Y.-W.; Wieland, D.M.; Kilbourn, M.R. Kuhl, D.E. Synthesis
of [18F]fluoroethoxy-benzovesamicol, a radiotracer for cholinergic neurons. J. Labelled
Compd. Radiopharm., 33, 583-591 (1993).
135. Mulholland, G.K.; Wieland, D.M.; Kilbourn, M.R.; Frey, K.A.; Sherman, P.S.; Carey,
J.E.; Kuhl, D.E. [18F]fluoroethoxy-benzovesamicol, a PET radiotracer for the vesicular
acetylcholine transporter and cholinergic synapses. Synapse, 30, 263-274 (1998).
136. Rogers, G.A.; Parsons, S.M.; Anderson, D.C.; Nilsson, L.M.; Bahr, B.A.; Kornreich,
W.D.; Kaufman, R.; Jacobs, R.S.; Kirtman, B. Synthesis, in vitro acetylcholine-storage-
blocking activities, and biological properties of derivatives and analogs of trans-2-(4-
phenylpiperidino)cyclohexanol (vesamicol). J. Med. Chem., 32, 1217-1230 (1989).
137. Rogers, G.A.; Stone-Elander, S.; Ingvar, M.; Eriksson, L.; Parsons, S.M.; Widén, L.
18F-labelled vesamicol derivatives: Syntheses and preliminary in vivo small animal
positron emission tomography evaluation. Nucl. Med. Biol., 21, 219-230 (1994).
138. Shiba, K.; Mori, H.; Matsuda, H.; Tsuji, S.; Tonami, N.; Hisada, K. In vivo
characterization of radioiodinated 2-(4-phenylpiperidino)cyclohexanol (vesamicol)
analogs: Potential radioligand for mapping presynaptic cholinergic neurons. Nucl. Med.
Biol., 22, 823-828 (1995).
Page 153
139
139. Shiba, K.; Mori, H.; Matsuda, H.; Tsuji, S.; Kuji, I.; Sumiya, H.; Kinuya, K.; Tonami,
N.; Hisada, K.; Sumiyosi, T. Synthesis of radioiodinated analogs of 2-(4-
Phenylpiperidino)cyclohexanol (vesamicol) as vesamicol-like agent. Nucl. Med. Biol., 22,
205-210 (1995).
140. Staley, J.K.; Mash, D.C.; Parsons, S.M.; Khare, A.B.; Efange, S.M. Pharmacological
characterization of the vesamicol analogue (+)-[125I]MIBT in primate brain. Eur. J.
Pharmacol., 338, 159-169 (1997).
141. Zea-Ponce, Y.; Mavel, S.; Assaad, T.; Kruse, S.E.; Parsons, S.M.; Emond, P.; Chalon,
S.; Giboureau, N.; Kassiou, M.; Guilloteau, D. Synthesis and in vitro evaluation of new
benzovesamicol analogues as potential imaging probes for the vesicular acetylcholine
transporter. Bioorg. Med. Chem., 13, 745-753 (2005).
142. Jung, Y.-W.; Frey, K.A.; Mulholland, G.K.; del Rosario, R.; Sherman, P.S.; Raffel,
D.M.; Van Dort, M.E.; Kuhl, D.E.; Gildersleeve, D.L.; Wieland, D.M. Vesamicol Receptor
Mapping of Brain Cholinergic Neurons with Radioiodine- Labeled Positional Isomers of
Benzovesamicol1. J. Med. Chem., 39, 3331-3342 (1996).
143. Giboureau, N.; Emond, P.; Fulton, R.R.; Henderson, D.J.; Chalon, S.; Garreau, L.;
Roselt, P.; Eberl, S.; Mavel, S.; Bodard, S.; Fulham, M.J.; Guilloteau, D.; Kassiou, M. Ex
vivo and in vivo evaluation of (2R,3R)-5-[18F]-fluoroethoxy- and fluoropropoxy-
benzovesamicol, as PET radioligands for the vesicular acetylcholine transporter. Synapse,
61, 962-970 (2007).
144. Kilbourn, M.R.; Hockley, B.; Lee, L.; Koeppe, R.A. Successful [18F]FEOBV
imaging of the VAChT in monkey brain. NeuroImage, 41, Supplement 2, T93 (2008).
145. Sorger, D.; Schliebs, R.; Kämpfer, I.; Rossner, S.; Heinicke, J.; Dannenberg, C.;
Georgi, P. In vivo [125I]-iodobenzovesamicol binding reflects cortical cholinergic
deficiency induced by specific immunolesion of rat basal forebrain cholinergic system.
Nucl. Med. Biol., 27, 23-31 (2000).
146. Emond, P.; Mavel, S.; Zea-Ponce, Y.; Kassiou, M.; Garreau, L.; Bodard, S.; Drossard,
M.L.; Chalon, S.; Guilloteau, D. (E)-[125I]-5-AOIBV: a SPECT radioligand for the
vesicular acetylcholine transporter. Nucl. Med. Biol., 34, 967-971 (2007).
147. Tu, Z.; Efange, S.M.; Xu, J.; Li, S.; Jones, L.A.; Parsons, S.M.; Mach, R.H. Synthesis
and in Vitro and in Vivo Evaluation of 18F-Labeled Positron Emission Tomography (PET)
Ligands for Imaging the Vesicular Acetylcholine Transporter. J. Med. Chem., 52, 1358-
1369 (2009).
Page 154
140
148. Efange, S.M.; Khare, A.B.; von Hohenberg, K.; Mach, R.H.; Parsons, S.M.; Tu, Z.
Synthesis and in Vitro Biological Evaluation of Carbonyl Group-Containing Inhibitors of
Vesicular Acetylcholine Transporter. J. Med. Chem., 53, 2825-2835 (2010).
149. Assaad, T. Synthesis and characterization of novel benzovesamicol analogs. Turk. J.
Chem., 35, 189-200 (2011).
150. Giboureau, N.; Som, I.M.; Boucher-Arnold, A.; Guilloteau, D.; Kassiou, M. PET
Radioligands for the Vesicular Acetylcholine Transporter (VAChT). Curr. Top. Med.
Chem., 10, 1569-1583 (2010).
151. Kilbourn, M.R.; Jung, Y.W.; Haka, M.S.; Gildersleeve, D.L.; Kuhl, D.E.; Wieland,
D.M. Mouse brain distribution of a carbon-11 labeled vesamicol derivative: Presynaptic
marker of cholinergic neurons. Life Sci., 47, 1955-1963 (1990).
152. Sorger, D.; Scheunemann, M.; Vercouillie, J.; Grossmann, U.; Fischer, S.; Hiller, A.;
Wenzel, B.; Roghani, A.; Schliebs, R.; Steinbach, J.; Brust, P.; Sabri, O. Neuroimaging of
the vesicular acetylcholine transporter by a novel 4-[18F]fluoro-benzoyl derivative of 7-
hydroxy-6-(4-phenyl-piperidin-1-yl)-octahydro-benzo[1,4]oxazines. Nucl. Med. Biol., 36,
17-27 (2009).
153. Mazère, J.; Prunier, C.; Barret, O.; Guyot, M.; Hommet, C.; Guilloteau, D.; Dartigues,
J.F.; Auriacombe, S.; Fabrigoule, C.; Allard, M. In vivo SPECT imaging of vesicular
acetylcholine transporter using [123I]-IBVM in early Alzheimer’s disease. NeuroImage,
40, 280-288 (2008).
154. Kuhl, D.E.; Koeppe, R.A.; Fessler, J.A.; Minoshima, S.; Ackermann, R.J.; Carey,
J.E.; Gildersleeve, D.L.; Frey, K.A.; Wieland, D.M. In vivo mapping of cholinergic
neurons in the human brain using SPECT and IBVM. J. Nucl. Med., 35, 405-410 (1994).
155. Barret, O.; Mazère, J.; Seibyl, J.; Allard, M. Comparison of noninvasive
quantification methods of in vivo vesicular acetylcholine transporter using [123I]-IBVM
SPECT imaging. J. Cereb. Blood Flow Metab., 28, 1624-1634 (2008).
156. Roe, A. In: Organic Reactions; Adams, R., Ed.; John Wiley & Sons Inc.: New York,
Vol. 5, 193-228 (1949).
157. Swain, C.G.; Rogers, R.J. Mechanism of formation of aryl fluorides from
arenediazonium fluoborates. J. Am. Chem. Soc., 97, 799-800 (1975).
158. Koval’chuk, E.P.; Reshetnyak, O.V.; Kozlovs’ka, Z.Y.; Błażejowski, J.;
Gladyshevs’kyj, R.Y.; Obushak, M.D. Mechanism of the benzenediazonium
tetrafluoroborate thermolysis in the solid state. Thermochim. Acta, 444, 1-5 (2006).
Page 155
141
159. Yoneda, N.; Fukuhara, T. Facile preparation of aromatic fluorides by deaminative
fluorination of aminoarenes using hydrogen fluoride combined with bases. Tetrahedron,
52, 23-36 (1996).
160. Doyle, M.P.; Bryker, W.J. Alkyl nitrite-metal halide deamination reactions. 6. Direct
synthesis of arenediazonium tetrafluoroborate salts from aromatic amines, tert-butyl nitrite,
and boron trifluoride etherate in anhydrous media. J. Org. Chem., 44, 1572-1574 (1979).
161. Yong, H.K.; Chun, H.L.; Ki, Y.C. Facile direct conversion of amino-heterocycles to
fluoro-heterocycles using t-butylthionitrite or t-butylthionitrate with sodium
tetrafluoroborate. Tetrahedron Lett., 31, 3019-3022 (1990).
162. Milner, D.J. Fluoroaromatics from Arylamines, a Convenient One-Pot Conversion
Using Nitrosonium Tetrafluoroborate. Synth. Commun., 22, 73-82 (1992).
163. Shinhama, K.; Aki, S.; Furuta, T.; Minamikawa, J. Facile Conversion of
Arenediazonium Salts to the Corresponding Fluoroarenes Using Boron Trifluoride Diethyl
Ether Complex. Synth. Commun., 23, 1577-1582 (1993).
164. Laali, K.K.; Gettwert, V.J. Fluorodediazoniation in ionic liquid solvents: new life for
the Balz–Schiemann reaction. J. Fluorine Chem., 107, 31-34 (2001).
165. Garel, L.; Saint-Jalmes, L. One-pot fluoro-de-diazoniation of anilines in organic
medium. Tetrahedron Lett., 47, 5705-5708 (2006).
166. Canning, P.S.J.; McCrudden, K.; Maskill, H.; Sexton, B. Rates and mechanisms of
the thermal solvolytic decomposition of arenediazonium ions. J. Chem. Soc., Perkin Trans.
2, 2735-2740 (1999).
167. Satyamurthy, N.; Barrio, J.B.; Schmidt, D.G.; Kammerer, C.; Bida, G.T.; Phelps,
M.E. Acid-catalyzed thermal decomposition of 1-aryl-3,3-dialkyltriazenes in the presence
of nucleophiles. J. Org. Chem., 55, 4560-4564 (1990).
168. Xue, H.; Verma, R.; Shreeve, J.M. Review of ionic liquids with fluorine-containing
anions. J. Fluorine Chem., 127, 159-176 (2006).
169. Kim, D.W.; Jeong, H.-J.; Lim, S.T.; Sohn, M.-H.; Chi, D.Y. Facile nucleophilic
fluorination by synergistic effect between polymer-supported ionic liquid catalyst and tert-
alcohol reaction media system. Tetrahedron, 64, 4209-4214 (2008).
170. Fuchigami, T.; Tajima, T. Highly selective electrochemical fluorination of organic
compounds in ionic liquids. J. Fluorine Chem., 126, 181-187 (2005).
Page 156
142
171. Das, S.; Chandrasekhar, S.; Yadav, J.S.; Grée, R. Ionic liquids as recyclable solvents
for diethylaminosulfur trifluoride (DAST) mediated fluorination of alcohols and carbonyl
compounds. Tetrahedron Lett., 48, 5305-5307 (2007).
172. Kim, H.W.; Jeong, J.M.; Lee, Y.S.; Chi, D.Y.; Chung, K.H.; Lee, D.S.; Chung, J.K.;
Lee M.C. Rapid synthesis of [18F]FDG without an evaporation step using an ionic liquid.
Appl. Radiat. Isot., 61, 1241-1246 (2004).
173. Chu, C.-K.; Kim, J.-H.; Kim, D.W.; Chung, K.-H.; Katzenellenbogen, J.A.; Chi, D.Y.
Aromatic Fluorination by Decomposition of Triazenes in Ionic Liquids. Bull. Korean
Chem. Soc., 26, 599-602 (2005).
174. Nifontov, V.; Bel’skaya, N.; Shtokareva, E. The reactivity and mechanism of action
of triazenes (review). Pharm. Chem. J., 28, 687-706 (1994).
175. Kimball, D.B.; Haley, M.M. Triazenes: A Versatile Tool in Organic Synthesis.
Angew. Chem. Int. Ed., 41, 3338-3351 (2002).
176. Pages, T.; Langlois, B.R. Fluorination of aromatic compounds from 1-aryl-3,3-
dimethyltriazenes and fluoride anions in acidic medium: 1. A model for 18F labelling. J.
Fluorine Chem., 107, 321-327 (2001).
177. Nifontov, V.I.; Bel’skaya, N.P.; Shtokareva, E.A. Manufacture methods of
synthesizing triazenes (review). Pharm. Chem. J. 27, 652–665 (1993).
178. Threadgill, M.D.; Stevens, M.F.G. Selective Reactions in the Triazene Series; I.
Reduction of 1-(4-Acetylphenyl)-triazenes. Synthesis, 1983, 289-291 (1983).
179. Hameed, A.J. Study of the structural and electronic properties of the 1-phenyl-3,3-
pentamethylenetriazenes. J. Mol. Struct., 725, 97-101 (2005).
180. Rakotondradany, F.; Williams, C.I.; Whitehead, M.A.; Jean-Claude, B.J. Semi-
empirical PM3 treatment of diaryl and dialkyl triazene decomposition in acid media. J.
Mol. Struct., 535, 217-234 (2001).
181. Yoneda, N.; Fukuhara, T.; Mizokami, T.; Suzuki, A. A Facile Preparation of Aryl
Triflates. Decomposition of Arenediazonium Tetrafluoroborate Salts in
Trifluoromethanesulfonic Acid. Chem. Lett., 20, 459-460 (1991).
182. GuhaRoy, C.; Butcher, R.J.; Bhattacharya, S. Rhodium complexes of 1,3-
diaryltriazenes: Usual coordination, N-H bond activation and, N-N and C-N bond
cleavage. J. Organomet. Chem., 693, 3923-3931 (2008).
Page 157
143
183. Xie, X.; Chen, J.; Xu, W.; He, E.; Zhan, S. Design, synthesis and reactivity with
dichloro-bis(triphenylphosphine) platinum(II) of two triazenide compounds. Inorg. Chim.
Acta, 373, 276-281 (2011).
184. Iley, J.; Moreira, R.; Rosa, E. Triazene drug metabolites. Part 10. Metal-ion catalysed
decomposition of monoalkyltriazenes in ethanol solutions. J. Chem. Soc., Perkin Trans. 2,
81-86 (1991).
185. Gaffoor, F.; Ford, T.A. The vibrational spectra of the boron halides and their
molecular complexes: Part 10. The complexes of boron trifluoride with ammonia and its
methyl derivatives. An ab initio study. Spectrochim. Acta A Mol. Biomol. Spectrosc., 71,
550-558 (2008).
186. Ford, T.A. The vibrational spectra of the boron halides and their molecular
complexes. Part 12: The complexes of boron trifluoride with methyl fluoride and methyl
chloride. An ab initio study. J. Mol. Struct., 897, 145-148 (2009).
187. Nxumalo, L.M.; Andrzejak, M.; Ford, T.A. The structure of the boron trifluoride-
ammonia complex: a Fourier transform matrix isolation infrared spectroscopic and ab
initio molecular orbital study. Vib. Spectrosc., 12, 221-235 (1996).
188. Franca, C.A.; Diez, R.P.; The Lewis acidity of boron trihalides revisited. J. Argent.
Chem. Soc., 97, 119-126 (2009).
189. Miller, J.M.; Onyszchuk, M. The Relative Acceptor Power of Boron Trihalides and
Borane Toward Trimethylamine. Can. J. Chem. 41, 2898-2902 (1963).
190. Pages, T.; Langlois, B.R.; Bars, D.L.; Landais, P. Fluorination of aromatic
compounds from 1-aryl-3,3-dimethyltriazenes and fluoride anions in acidic medium: 2.
Synthesis of (S)-[18F]-3-fluoro-α-methylphenylalanine. J. Fluorine Chem. 107, 329-335
(2001).
191. Platonov, V. Properties of polyphosphoric acid. Fibre Chem., 32, 325-329 (2000).
192. Downing, R.G.; Pearson, D.E. The Apparent Acidity Function (H0) of
Polyphosphoric Acid. J. Am. Chem. Soc., 83, 1718-1721 (1961).
193. Bartsch, R.A.; Chen, H.; Haddock, N.F.; Juri, P.N. Stabilization of aryldiazonium
ions by crown ether complexation. J. Am. Chem. Soc., 98, 6753-6754 (1976).
194. Krane, J.; Skjetne, T. Studies of crown ether complexes aryldiazonium ion
complexes. Tetrahedron Lett., 21, 1775-1778 (1980).
Page 158
144
195. Knöchel, A.; Zwernemann, O. Development of a no-carrier-added method for 18F-
labelling of aromatic compounds by fluorodediazonation. J. Labelled Compd.
Radiopharm., 38, 325-336 (1996).
196. Hamacher, K.; Coenen, H.H. Efficient routine production of the 18F-labelled amino
acid O-(2-[18F]fluoroethyl)-l-tyrosine. Appl. Radiat. Isot., 57, 853-856 (2002).
Page 159
Abstract
Deficiencies in vesicular acetylcholine transporter (VAChT) are among the earliest neuronal
changes preceding clinical symptoms of Alzheimer's disease, and show a strong correlation
with the severity of dementia. As (2R,3R)-5-IBVM is the lead and the only SPECT
radioligand for VAChT human imaging, we synthesized by fluoro-de-diazoniation its fluoro
analog 5-FBVM with corresponding enantiomers, and confirmed by 3D QSAR and in vitro
studies that both enantiomers of 5-FBVM are of the same order affinity as 5-IBVM.
Furthermore, we greatly improved 5-FBVM yield via fluoro-de-triazenation of the
corresponding triazene precursor 5-TBV using boron trifluoride etherate under non-protic
acid conditions in tetrachloromethane under optimized microwave irradiation. By testing
different reaction parameters in numerous experimental attempts to find fluoro-de-triazenation
conditions which can be transposed to radiofluorination, we may accomplished 5-[18
F]FBVM.
This encouraging result warrants to optimize 5-[18
F]FBVM yield via promising methods
obtained in cold chemistry.
Keywords: Alzheimer's disease, VAChT, PET, 5-FBVM, 5-TBV, triazene, fluoro-de-
triazenation.
Résumé
Les déficiences en transporteur vésiculaire de l'acétylcholine (VAChT) sont l'un des
symptômes précoces de perte neuronale lors de la maladie d'Alzheimer, perte fortement
corrélée avec la gravité de la démence associée. Comme le (2R,3R)-5-IBVM est le radioligand
de référence du VAChT utilisé en imagerie TEMP, la synthèse par fluoro-de-diazenation a
conduit à son analogue fluoré, le 5-FBVM, ainsi qu’à ses énantiomères. Par étude 3D-QSAR,
confirmée par évaluation in vitro, chaque énantiomère du 5-FBVM montre une affinité pour
le VAChT similaire au 5-IBVM. D'autres travaux ont permis d'améliorer le rendement en 5-
FBVM par fluoro-de-triazénation du précurseur triazène, le 5-TVB, en utilisant seulement de
l’éthérate de trifluorure de bore qui joue le double rôle d’acide de Lewis et d’agent fluorant,
dans le tétrachlorure de carbone, sous irradiation micro-onde. L’optimisation de la fluoro-de-
triazénation en étudiant différents paramètres expérimentaux compatibles avec un
radiomarquage a permis d’obtenir le 5-[18
F]FBVM. Ce résultat encourageant devrait conduire
à l’obtention du 5-[18
F]FBVM.