A holistic regulatory approach to accelerated CMC development CMC Strategy Forum 2017 Seán Barry Ph.D Pharmaceutical Assessor Health Products Regulatory Authority Disclaimer: The opinions expressed are my own and do not necessarily represent those of the HPRA or EMA
Transcript
A holistic regulatory approach to accelerated CMC
development
CMC Strategy Forum 2017
Seán Barry Ph.DPharmaceutical AssessorHealth Products Regulatory Authority
Disclaimer: The opinions expressed are my own and do not necessarily represent those of the HPRA or EMA
- Adaptive pathway- Conditional approval- PRIME
- Accelerated approval- Fast track designation- Breakthrough therapy
Pathways with accelerated clinical development
Potential bottleneck due to constraints of CMC development
CMC considerations for accelerated development
• Despite the reduced timeframes, no compromise to patient safety is allowed. Therefore, product and process characterisation is required much earlier
• Full process validation studies incomplete?
• Only Phase 2 batches at time of filing?
• Launching from clinical site?
• Stability studies ongoing?
• Control strategy still evolving?
How should regulators and industry approach
accelerated quality development?
Accelerated clinical development programmes must strike a balance between regulatory flexibility and ensuring the critical aspects of CMC are not compromised
What are the solutions??
Use of models
Post-approval validation
Prior knowledge
PACMPs
Holistic approach
General CMC strategies for accelerated schemes *
• Some activities are started sooner than traditional development
• There is a greater focus on consistent supply post-launch rather than process optimization
• The QTPP and CQAs should be defined early• May need to fix cell line at Phase I in order to avoid the
need for comparability studies• Scale up from clinical to commercial material post-
approval, pre-launch with appropriate bridging data• Manufacture DP PPQ batches using clinical DS batches
prior to completion of DS PPQ batches
* Mostly relevant for recombinant proteins/mAbs etc.
ConcurrentValidation
Continuous process
verification
ProcessValidation
In-line/on-line /at-line controls & PAT Depends on level of process understanding Prior knowledge
PV conducted in parallel with routine production
Possible when there is a very strong benefit/risk
Ongoing process
verification
Relies on a protocol Indicates how process knowledge, control
strategy and characterisation methods will be used
Ongoing process verification -protocols
• Already routinely used for e.g. full scale validation of resin lifetime and reprocessing (e.g. re-filtration)
• Full realisation of leveraging data from post-validation studies will require wider use of protocols
• Protocol should indicate how process knowledge, control strategy and characterisation methods will be use to assess product quality throughout the lifecycle
• Protocol should include tests and acceptance criteria that will be used to further demonstrate that the process remains in a validated state
• Result of ongoing process verification activities should be available during inspection
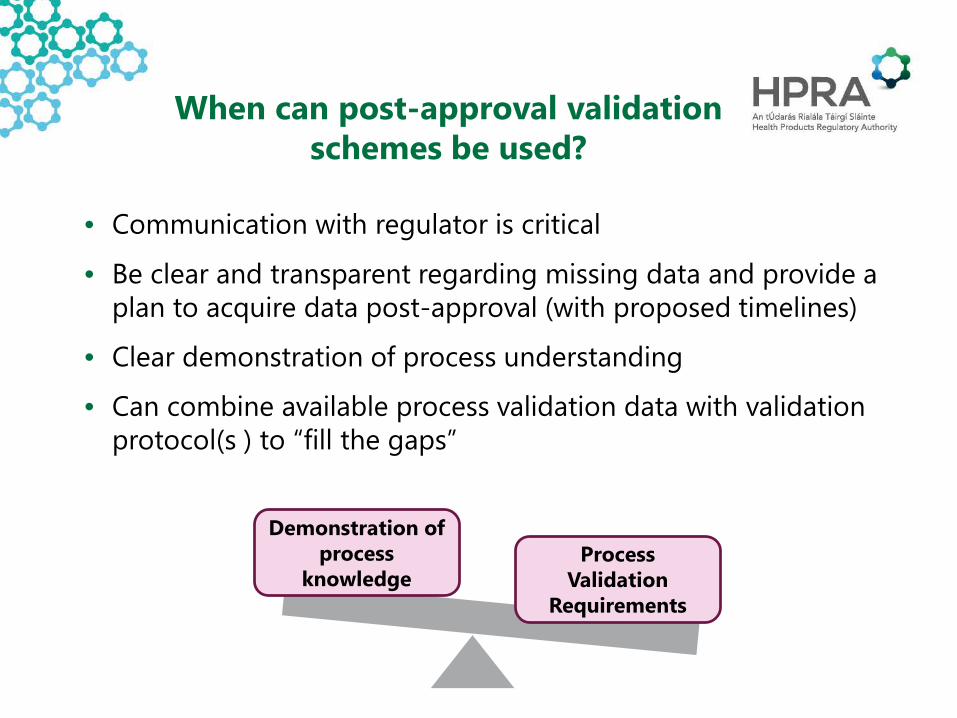
When can post-approval validation schemes be used?
Level of flexibility is case-by-case and depends on:
1. Seriousness of the indication and the level of unmet medical need
2. Product characteristics
3. Manufacturing processes complexity
4. Applicant’s risk assessment
5. Possibly the manufacturer’s PQS (ICH Q12)
When can post-approval validation schemes be used?
• Communication with regulator is critical
• Be clear and transparent regarding missing data and provide a plan to acquire data post-approval (with proposed timelines)
• Clear demonstration of process understanding
• Can combine available process validation data with validation protocol(s ) to “fill the gaps”
Demonstration of process
knowledgeProcess
ValidationRequirements
Small scale models• Greater use of models can reduce the subsequent process
validation burden
• Build into early development with a view to establishing the PARs early in the clinical programme
• Using small scale data to justify all PARs allows the PV batches to be run at set-point/NORs
• Even if process changes during development, many PARs will still be relevant, particularly true for scale-up when PARs are scale independent
• Additional data from prior knowledge could complement some gaps in small scale data
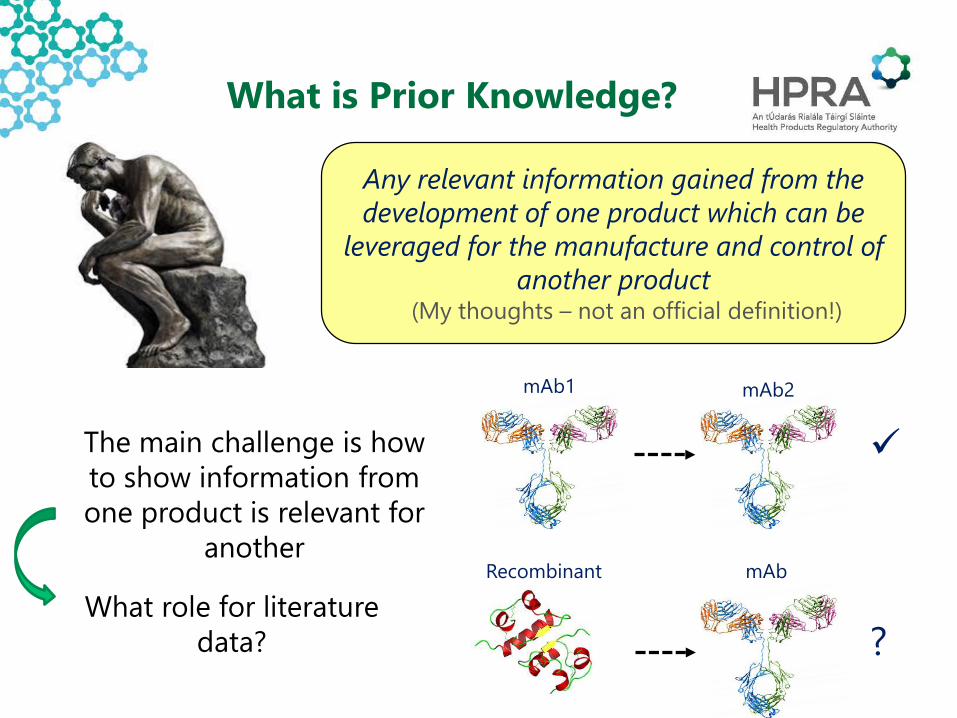
What is Prior Knowledge?
Any relevant information gained from the development of one product which can be
leveraged for the manufacture and control of another product
(My thoughts – not an official definition!)
The main challenge is how to show information from one product is relevant for
another
mAb1 mAb2
Recombinant mAb
?What role for literature
data?
Preliminary QTPP
Define CQAs
Define preliminary CPPs, IPCs, PARs
Small scale studies
Process validation
Prior Knowledge
Prior Knowledge
Prior Knowledge
Prior Knowledge
There are considerable possibilities for utilizing prior knowledge in the
development and control strategy
What to document in the file
Explain how prior knowledge was used in:
Establishing the input set points
Setting the outer boundaries of the process ranges
Defining the CPPs
Assigning CQAs ... (what about setting criticality of attributes where there is a different indication, dosage etc?).
Note: EMA has limited experience with the use of Prior Knowledge or platforms in the dossier, so any novel approach would benefit from early Scientific Advice
CPP = Critical process parameterKPP = Key process parameter
Prior knowledge
Product specific
Leveraging prior knowledge to define the criticality of process parameters
mAb1 mAb2 mAb3 mAb4
Buffer pH 6.5-7.5 6.2-7.2 6-8 6-8
Buffer conductivity 25-29 25-29 23-27 20-30
Load ratio 3.0-4.0 3.0-4.0 3.0-4.0 3.0-4.0
• For each combination of PARs (input) there is a known output (IPCs and other quality data from PV/small scale)
• Could a model be built for each unit operation to show that for a similar product and manufacturing process, the proposed operating ranges always result in a product of acceptable quality?
Can ranges from previous products be used to justify current ranges?
Range finding studies (PV/small scale)SEC, cIEX, yield, HCP, DNA ....
Standard IPC programme
Input
Output
Output
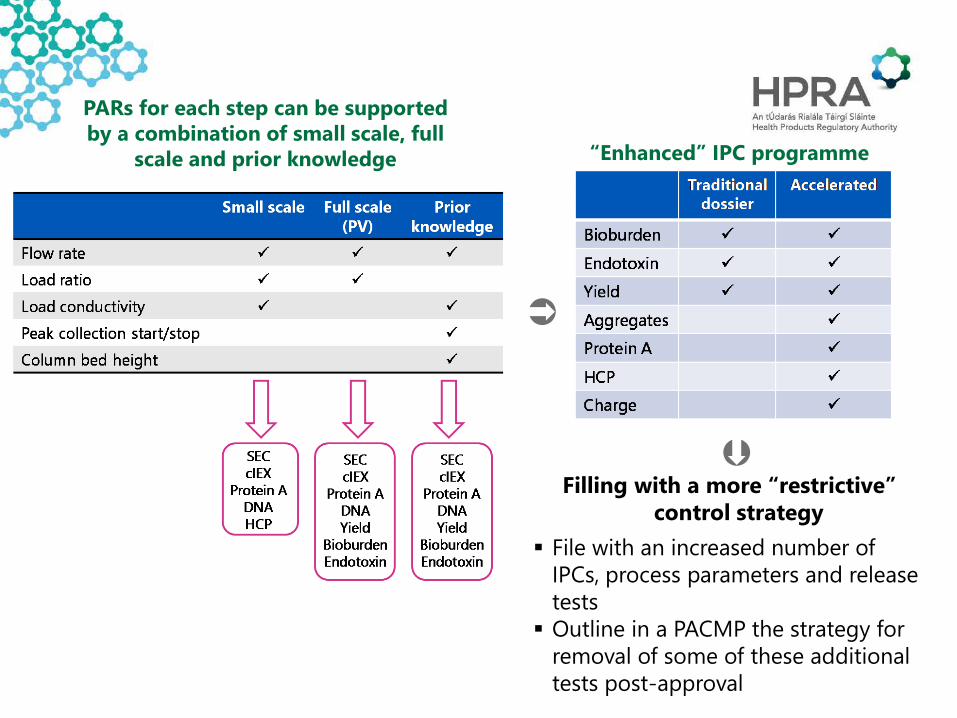
Filling with a more “restrictive” control strategy
File with an increased number of IPCs, process parameters and release tests Outline in a PACMP the strategy for
removal of some of these additional tests post-approval
PARs for each step can be supported by a combination of small scale, full
scale and prior knowledge “Enhanced” IPC programme
PV batches run at set point/NOR but product-
specific small scale data not available to challenge ranges
Small scale and full scale data from prior knowledge
Additional small scale/full scale data provided post approval to validate PARs
Example 2
Combination of ongoing process verification and prior knowledge
Example 1
Product-specific small scale data available but 3 full scale
PV batches not available
PARs justified based on small scale data and prior knowledge from full scale
Additional data from full scale PV batches provided post-approval
These approaches have yet to be seen in a dossier
Post approval change management protocols (PACMP)
In the context of the lifecycle approach, certain validation and/or upscaling/change activities may be agreed to be conducted post-marketing with the use of appropriate regulatory tools available (e.g., post approval change management protocols).
• Becoming more common, however full potential has not yet been realised
• Opportunity for the applicant to perform a gap analysis and present a plan to the regulator how the knowledge gap will be addressed post approval
• Untapped resource for expected bottlenecks in accelerated pathways
• Continues to be challenging – how to set clinically meaningful specs when only a few batches available?
• Wide specifications could be approved with commitment to revise after certain number of batches produced
• Balance between specs which ensure a safe & efficacious product and avoiding unnecessary batch rejection
• File with an increased testing panel, some of which can be removed post-approval as more batch data become available
• Data may not yet be available to support removal of testing of e.g. HCP, DNA, Protein A etc. from specs
Specifications and accelerated development
Theoretical example for setting specifications with few batches
Unlikely to be accepted
More likely to be accepted
May not be accepted
−PACMP submitted−Specifications revised via variation after 10
batches−Revised acceptance criteria will be based on
the following statistical approach....−Plan for how specs will be clinically qualified −Any OOT data will be investigated as part of
the PQS
Specifications will be revisited after more batch data is available
Some final thoughts on accelerated pathways from an assessor’s viewpoint
• An accelerated time line does not mean a premature dossier.
• Accelerated pathway products can be closer to late stage IMP however an accelerated dossier ≠ IMPD!
• Currently a “rolling review” model for quality/CMC is possible as part of the PRIME scheme but not other accelerated procedures engage early through scientific advice
• Full validation of methods required• All sections of Module 3 to be completed!
Conclusions Having less data at the time of approval should not compromise patient safety
Several avenues exist to address the bottleneck due to CMC development
A holistic approach utilizing the combined tools of post-approval validation, prior knowledge, small scale models and PACMPs could take CMC off the critical path
However there may be a trade between initial reduced manufacturing flexibility and earlier approval
Both industry and regulators should continue to explore new approaches to overcoming the CMC challenges of accelerated development