doi.org/10.26434/chemrxiv.12783227.v1 A Mechanochemical Model for the Simulation of Molecules and Molecular Crystals Under Hydrostatic Pressure Tim Stauch Submitted date: 10/08/2020 • Posted date: 11/08/2020 Licence: CC BY-NC-ND 4.0 Citation information: Stauch, Tim (2020): A Mechanochemical Model for the Simulation of Molecules and Molecular Crystals Under Hydrostatic Pressure. ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.12783227.v1 A novel mechanochemical method for the simulation of molecules and molecular crystals under hydrostatic pressure, the eXtended Hydrostatic Compression Force Field (X-HCFF) approach, is introduced. In contrast to comparable methods, the desired pressure can be adjusted non-iteratively and molecules of general shape retain chemically reasonable geometries even at high pressures. The implementation of the X-HCFF approach is straightforward and the computational cost is practically the same as for a regular geometry optimization. Pressure can be applied by using any desired electronic structure method for which a nuclear gradient is available. The results of X-HCFF for pressure-dependent intramolecular structural changes in the investigated molecules and molecular crystals as well as a simple pressure-induced dimerization reaction are chemically intuitive and fall within the range of other established computational methods. Experimental spectroscopic data of a molecular crystal under pressure are reproduced accurately. File list (2) download file view on ChemRxiv Manuscript_X-HCFF.pdf (774.95 KiB) download file view on ChemRxiv SI_X-HCFF.pdf (221.81 KiB)
Transcript
doi.org/10.26434/chemrxiv.12783227.v1
A Mechanochemical Model for the Simulation of Molecules andMolecular Crystals Under Hydrostatic PressureTim Stauch
Submitted date: 10/08/2020 • Posted date: 11/08/2020Licence: CC BY-NC-ND 4.0Citation information: Stauch, Tim (2020): A Mechanochemical Model for the Simulation of Molecules andMolecular Crystals Under Hydrostatic Pressure. ChemRxiv. Preprint.https://doi.org/10.26434/chemrxiv.12783227.v1
A novel mechanochemical method for the simulation of molecules and molecular crystals under hydrostaticpressure, the eXtended Hydrostatic Compression Force Field (X-HCFF) approach, is introduced. In contrastto comparable methods, the desired pressure can be adjusted non-iteratively and molecules of general shaperetain chemically reasonable geometries even at high pressures. The implementation of the X-HCFFapproach is straightforward and the computational cost is practically the same as for a regular geometryoptimization. Pressure can be applied by using any desired electronic structure method for which a nucleargradient is available. The results of X-HCFF for pressure-dependent intramolecular structural changes in theinvestigated molecules and molecular crystals as well as a simple pressure-induced dimerization reaction arechemically intuitive and fall within the range of other established computational methods. Experimentalspectroscopic data of a molecular crystal under pressure are reproduced accurately.
File list (2)
download fileview on ChemRxivManuscript_X-HCFF.pdf (774.95 KiB)
download fileview on ChemRxivSI_X-HCFF.pdf (221.81 KiB)
A Mechanochemical Model for the Simulation ofMolecules and Molecular Crystals Under Hydrostatic
Pressure
Tim Stauch∗,1,2,3
1 University of Bremen, Institute for Physical and Theoretical Chemistry, Leobener Straße NW2, D-28359 Bremen, Germany2 University of Bremen, Bremen Center for Computational Materials Science, Am Fallturm 1, D-28359 Bremen, Germany3 University of Bremen, MAPEX Center for Materials and Processes, Bibliothekstraße 1, D-28359 Bremen, Germany
Abstract: A novel mechanochemical method for the simulation of molecules and molecular crystals
under hydrostatic pressure, the eXtended Hydrostatic Compression Force Field (X-HCFF) approach, is
introduced. In contrast to comparable methods, the desired pressure can be adjusted non-iteratively and
molecules of general shape retain chemically reasonable geometries even at high pressures. The imple-
mentation of the X-HCFF approach is straightforward and the computational cost is practically the same
as for a regular geometry optimization. Pressure can be applied by using any desired electronic structure
method for which a nuclear gradient is available. The results of X-HCFF for pressure-dependent in-
tramolecular structural changes in the investigated molecules and molecular crystals as well as a simple
pressure-induced dimerization reaction are chemically intuitive and fall within the range of other estab-
lished computational methods. Experimental spectroscopic data of a molecular crystal under pressure are
The field of high-pressure chemistry has seen tremendous progress within the past few decades.1 Pressure-
induced changes in the rate, mechanism and equilibrium of chemical reactions have opened up new av-
enues in chemical synthesis.2,3 Using diamond anvil cells,4 pressures of several hundred GPa have been
realized experimentally, meaning that pressures as high as in Earth’s core are now available in the labo-
ratory.5 Not surprisingly, the geometries of molecules,6–8 the lattice constants of crystals9–11 and crystal
phases12 have been found to change under pressure. However, many of the pressure-induced structural
changes of molecules and crystals can only be inferred indirectly by interpreting the concomitant changes
in the spectroscopic properties, e.g., the wavelengths and intensities of absorption.
As a result, the demand for computational tools that predict structural and spectroscopic properties of
molecules as well as chemical reactivity at high pressure is increasing. Methods that model the influence
of pressure on periodic chemical systems have been described in the realm of Molecular Dynamics13 and
ab initio Molecular Dynamics.14–16 At the single-molecule level, several electronic structure methods for
the simulation of molecules under pressure are available.17 A particularly notable example is the eXtreme
Pressure Polarizable Continuum Model (XP-PCM),18–20 in which pressure is applied to a molecule via a
surrounding medium. In XP-PCM, pressure is modeled by decreasing the size of the cavity inside which
the molecule is placed and simultaneously increasing the Pauli repulsion of the surrounding medium.
XP-PCM has been used, e.g., in the simulation of chemical reactions19–21 and for investigating changes
in spectroscopic,22,23 electronic24 and structural25 properties of molecules under pressure.
A straightforward alternative to model molecules under pressure has its roots in quantum mechano-
chemistry.26 The earliest quantum mechanochemical method for the application of pressure is the Gen-
eralized Force-Modified Potential Energy Surface (G-FMPES) model,27–29 which builds on the FMPES
scheme,30 a protocol to optimize molecular geometries under external forces. In G-FMPES, mechanical
2
forces compress a molecule towards its geometric centroid during a geometry optimization. A variation
of this method, the Hydrostatic Compression Force Field (HCFF) approach,31 differs from G-FMPES
mainly in the definition of the surface area and the calculation of the pressure that is applied to the
molecule. HCFF has been used to calculate the pressure required to trigger spin-crossover in octahedral
metal ligand complexes.31 A drawback of the quantum mechanochemical pressure models, however, is
that the pressure that is applied to the molecule cannot be defined a priori. Instead, the applied pressure is
calculated from empirical parameters that are specified by the user, meaning that the desired pressure has
to be adjusted iteratively. A second drawback is that in some cases, e.g. in extended molecules, unrea-
sonable molecular geometries are found by G-FMPES and HCFF due to the tendency of the molecule to
minimize its surface area under compression. Hence, both G-FMPES and HCFF work best for molecules
that can be approximated as spherical.
In this paper, a new member of the family of mechanochemical pressure models, the eXtended Hy-
drostatic Compression Force Field (X-HCFF), is presented. It builds on the HCFF approach, but avoids
the deficiencies of the previous mechanochemical pressure models in that 1) the user has full control over
the pressure that is applied to the molecule, and 2) the pressure is applied truly hydrostatically, i.e. the
compression of extended molecules can be simulated realistically. It is shown that structural parameters
calculated with the novel X-HCFF model are of the same quality of those calculated with previous com-
putational methods in both single molecules and molecular crystals and that experimental spectroscopic
parameters can be reproduced accurately in the investigated test systems. Moreover, a pressure-induced
chemical reaction is reproduced. The implementation of the X-HCFF approach is straightforward and
only requires a surface tessellation routine, which is typically implemented in connection with implicit
solvent models in many quantum chemical program packages, as well as a nuclear gradient code for the
desired electronic structure method. While the calculations presented in this paper were all carried out
with Density Functional Theory (DFT),32,33 wave-function based methods can of course be used instead.
3
The computational cost of an X-HCFF calculation is virtually identical to the cost of a regular, pressure-
free geometry optimization. The X-HCFF model will be available in a future release of the Q-Chem
program package.34
The rest of this paper is structured as follows: After establishing the theoretical foundation of the
X-HCFF approach in Section 2, the performance of the method is tested in Section 3. In particular,
comparisons are made against structural parameters of molecules under pressure calculated with HCFF
(Section 3.1) and XP-PCM (Section 3.2). The performance of X-HCFF for the description of molecular
crystals under pressure is tested by comparing against results calculated with periodic DFT (Section 3.3)
as well as against experimental Raman spectra under pressure (Section 3.4). Finally, the dimerization
of carbon dioxide under pressure is investigated in Section 3.5 to demonstrate the capability of X-HCFF
to describe a simple pressure-induced chemical reaction. Possible routes for future developments are
pointed out in Section 4.
2 Theory
2.1 Theoretical background of HCFF
As X-HCFF is based on the HCFF approach, the theoretical background of the latter shall be summarized
first. In HCFF mechanical forces pointing towards the non-mass-weighted molecular centroid are applied
on the nuclei to compress a molecule.31 As motivated in the literature for the closely related G-FMPES
approach,27–29 the force acting on each atom is potentially unique, with the largest force acting on the
outermost atom and the forces on the inner atoms being scaled by the distances of these atoms to the
geometric centroid of the system. Using this condition as well as the definition of the pressure, P =
F⊥/A, where F⊥ is the normal component of the force at the surface and A is the surface area, the
4
magnitude of the force fHCFFi pushing an atom i towards the molecular centroid is calculated in the
HCFF scheme as
|fHCFFi | = −Pguess · AvdW ·
rirmax
, (1)
where Pguess is a user-defined guess for the pressure that is applied to the molecule, AvdW is the molecular
van-der-Waals surface area, ri is the distance of atom i to the molecular centroid and rmax is the distance
of the outermost atom to the centroid. The van-der-Waals surface is used as the definition of the molecular
surface, since the surrounding medium, which applies pressure to the molecule, e.g. in a diamond-anvil
cell, is in contact with the molecule at its van-der-Waals surface. The forces fHCFFi are then added to
the nuclear gradient during a geometry optimization, leading to a compression of the molecule. As in
other quantum mechanochemical optimization techniques,35 the optimization converges if the externally
applied force (in this case all forces fHCFFi ) and the internal restoring forces cancel. The pressure acting
on the molecule has to be calculated a posteriori via
PHCFF =〈||f ||〉AvdW
, (2)
where 〈||f ||〉 is the average force acting on the atoms. Typically, Pguess overestimates PHCFF. Hence, the
pressure that is applied to the molecule cannot be specified directly and instead needs to be calculated
from a user-defined parameter, which is one of the major drawbacks of the mechanochemical models of
pressure that have been developed. Moreover, since the forces are acting towards the molecular centroid,
molecules tend to minimize their surface areas. As a result, unreasonable compressed geometries can
ensue31 and extended molecules can fold into sphere-like objects, which is not the expected behavior in
hydrostatic compression experiments.
5
2.2 The X-HCFF approach
Both problems are circumvented in the X-HCFF approach. The basic idea that pressure is mediated to
the molecule via its van-der-Waals surface, e.g. during hydrostatic compression in a diamond-anvil cell,
is carried over from HCFF to X-HCFF. In many quantum chemical program packages the van-der-Waals
surface is calculated by superimposing atom-centered spheres, removing the overlapping regions and
tessellating the surface using a Lebedev grid (Figure 1A).36 Instead of compressing the molecules towards
its centroid, in X-HCFF truly hydrostatic compression is achieved by applying the forces perpendicular
to the van-der-Waals surface in each tessellation point (Figure 1B).
Each surface tessera j has an area Aj and the van-der-Waals surface is calculated as AvdW =∑NTess
j Aj ,
where NTess is the number of tessellation points. In X-HCFF, the force fi,j acting on atom i that stems
from the pressure on the surface tessera j acts along the connecting line between atom i and tessera j and
points away from the latter (blue arrows in Figure 1B). Using again the definition of pressure, this force
can be calculated via
fi,j = −P · Aj ·(ri − rj)|ri − rj|
, (3)
where P is the pressure that is applied to the molecule, ri is the position of atom i and rj is the position
of tessera j. The net force acting on atom i is then calculated by summing up the contributions from each
surface tessera that lies on the van-der-Waals sphere around atom i (blue spheres in Figure 1B).
fi =NTess,i∑
j
fi,j (4)
When calculating the gradient contribution for an atom, the tesserae that lie on the van-der-Waals spheres
around the other atoms are ignored. Since the force contributions from tesserae on opposite sides of the
6
A
B
Figure 1: A) Van-der-Waals surface of the water molecule approximated by a Lebedev grid, leading to atessellated surface. B) Basic idea of the X-HCFF approach for the water molecule with a reduced numberof tessellation points, taking one of the hydrogen atoms as an example. The blue spheres represent thetessellation points created by the van-der-Waals sphere around the hydrogen atom under consideration,whereas the transparent spheres are the tessellation points associated with the other atoms in the molecule.Dotted lines connect the tessellation points to the hydrogen atom and the blue arrows depict the resultinggradient contributions. For clarity, the force contributions of only three tessellation points are shown.The thick black arrow represents the resulting force that arises when adding up each individual gradientcontribution for the hydrogen atom. Arrows are not drawn to scale.
7
atom cancel and there are no tessellation points along the connecting line between two atoms, the result-
ing force contribution pushes the atoms towards one another, leading to a compression of the molecule.
The forces calculated for each atom via eq. 4 are added to the nuclear gradient during a geometry op-
timization, which converges when the external force due to pressure and the internal restoring force of
the molecule cancel. Since the tessellation is carried out anew in each optimization step, the tessellation
field reacts to the updated molecular geometry. As a result, the forces applied to the atoms are adjusted
each time such that the input pressure P is applied to the molecule. However, the X-HCFF ansatz does
not strictly guarantee that the gradient contributions on all atoms add up to zero in all cases, which is re-
quired to prevent translation and rotation of the molecule. While in all investigated systems the resulting
net gradient contribution was extremely small, it was split up evenly and added to the nuclear gradient of
each atom.
Since the van-der-Waals radii are not scaled in the X-HCFF approach, the only empirical parameter of
the model is the number of tessellation points per atom. The dependence of the results on this parameter
is tested in Section 3.2.
3 Results and Discussion
3.1 Comparison between X-HCFF and HCFF
To showcase the difference between X-HCFF and HCFF, an aliphatic molecule is compressed using both
approaches. While HCFF was shown to deliver physically meaningful and chemically intuitive results
for molecules that can be circumscribed onto a sphere,31 the method performs poorly for butane under
a pressure of 50 GPa (Figure 2): Since strong forces point towards the molecular centroid and these
forces are strongest for the outermost atoms, the molecule tends to minimize its surface and folds into a
8
HCFF:
X-HCFF:
Figure 2: Molecular geometries for butane at a pressure of 50 GPa as calculated with the HCFF31 (top)and the X-HCFF (bottom) approaches at the PBE37/cc-pVDZ38 level of theory with 302 tessellationpoints for each atom.
sphere-like object. This situation is reminiscent of planets and stars, in which gravitational forces pull
each particle towards the center, resulting in (roughly) spherical shapes. The creation of a sphere-like
geometry is a general feature of HCFF and can also be observed in many other molecules, which limits
the applicability of the method.
The X-HCFF approach, on the other hand, is not afflicted with this disadvantageous feature. While
the bond lengths in the molecules are shortened by pressure and bond angles change (cf. the Supporting
Information, Tables S1 and S2), the extended shape of butane is retained at 50 GPa (Figure 2). This is due
to the fact that each atom is being pushed inwards orthogonally to the surface, leading to a compression
of the covalent bonds and not to the creation of a sphere-like geometry. Therefore, X-HCFF is more
generally applicable for the simulation of molecules under hydrostatic pressure than HCFF.
Figure 3: Dependence of the central C−C bond length (single bond) in trans-1,3-butadiene on the numberof tessellation points (NTess) per atom, calculated with X-HCFF at the B3LYP39–41/6-31G(d,p)42 level oftheory.
3.2 Structural parameters in trans-1,3-butadiene as a function of pressure
To judge the dependence of structural data calculated with X-HCFF on the number of tessellation points,
trans-1,3-butadiene is used as a test case. Here, values between 6 and 1202 tessellation points per atom
are adjusted. Focusing on the C−C single bond length in trans-1,3-butadiene, it can be observed that,
with the exception of the smallest number of tessellation points, the results converge quickly (Figure 3).
With increasing pressure the C−C bond length decreases with the same slope for all values between 26
and 1202 tessellation points per atom. Analogous results were obtained for all other investigated systems
during testing. This is a profitable feature of X-HCFF, because the number of tessellation points is the
only empirical parameter in the model and the results are virtually independent of the precise choice
of this parameter. As the cost of the calculation is influenced only insignificantly by the number of
tessellation points per atom, a value of 302 is used in the remainder of this paper.
Since trans-1,3-butadiene was treated previously with XP-PCM,18 this molecule provides a testing
10
ground for the performance of X-HCFF in comparison to another computational method for the simu-
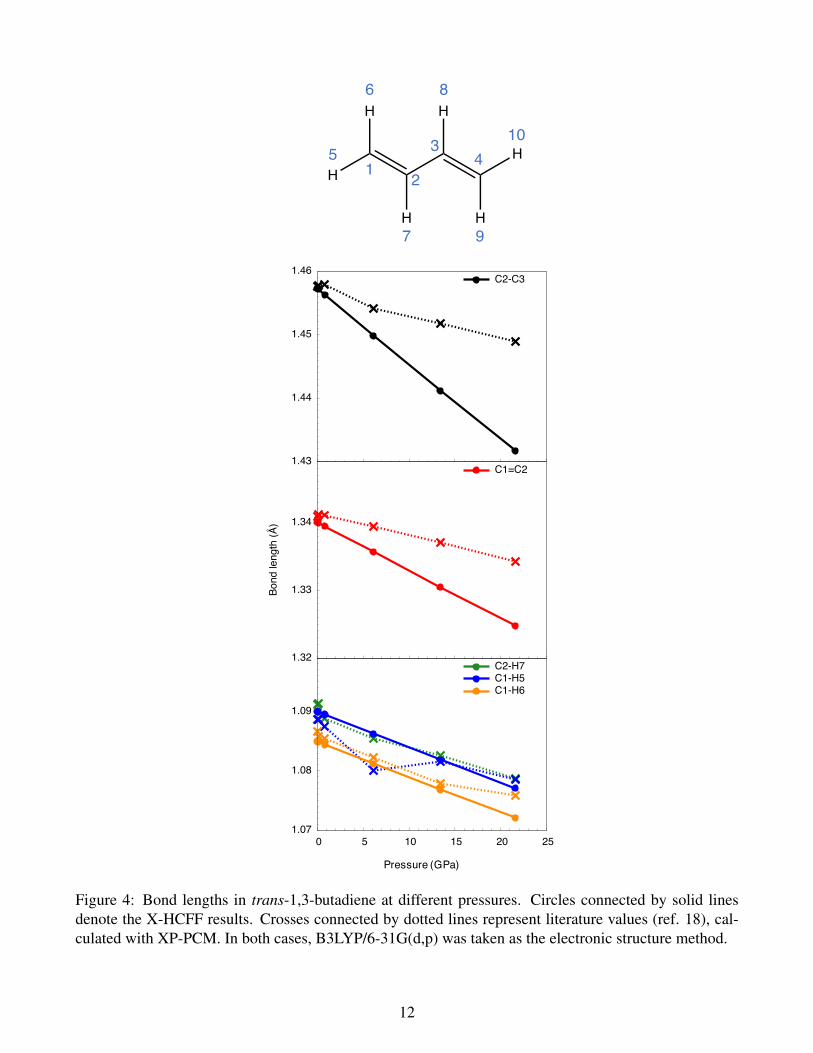
lation of molecules under pressure. Comparing the bond lengths in trans-1,3-butadiene calculated with
XP-PCM and X-HCFF (Figure 4), it becomes apparent that the pressure-induced decrease in carbon-
carbon bond lengths is more pronounced in X-HCFF than in XP-PCM. Also, the X-HCFF curve for the
central C−C single bond is smoother than in XP-PCM. Turning to the carbon-hydrogen bond lengths, the
results of X-HCFF and XP-PCM are more similar, with the difference that the bond lengths calculated
with X-HCFF again decrease more smoothly. In particular, X-HCFF finds a monotonous decrease of one
of the terminal C−H bond lengths (C1−H5 in Figure 4), while this is not the case for XP-PCM.
These results demonstrate that, using a single trans-1,3-butadiene molecule as an example, X-HCFF
affords continuous curves showing the progressive decrease of bond lengths with increasing pressure, in
agreement with chemical intuition, and that the decrease in bond lengths is proportional to the external
pressure. Extensive test calculations on a wide variety of molecules need to be carried out to judge how
general this trend is.
3.3 Structural parameters in a molecular crystal as a function of pressure
The performance of X-HCFF when treating molecular crystals is tested by calculating structural parame-
ters of 7-nitrotetrazolo [1,5]furazano[4,5-b]pyridine 1-oxide (NFP), for which reference values calculated
with a periodic implementation of DFT are available.43 The focus in the present study lies on intramolec-
ular geometric changes and not on changes in the lattice parameters, so that a single NFP molecule is
subjected to pressures between 0 and 100 GPa with the X-HCFF model. As can be seen in Figure 5,
the X-HCFF results can compete with those obtained via periodic DFT. Despite the differences in the
approaches and the applied electronic structure levels, which lead to deviations in the bond lengths at
0 GPa, many of the bond lengths in NFP decrease with roughly the same slope in both approaches. No-
11
H
H
H
H
H
H 1 2
345
6
7
8
9
10
1.43
1.44
1.45
1.46C2-C3
1.32
1.33
1.34
Bond
leng
th (Å
)
C1=C2
1.07
1.08
1.09
0 5 10 15 20 25
C2-H7 C1-H5 C1-H6
Pressure (GPa)
Figure 4: Bond lengths in trans-1,3-butadiene at different pressures. Circles connected by solid linesdenote the X-HCFF results. Crosses connected by dotted lines represent literature values (ref. 18), cal-culated with XP-PCM. In both cases, B3LYP/6-31G(d,p) was taken as the electronic structure method.
12
N N
O
N+
N
N N
H O-
N+
O
-O
1.15
1.2
1.25
1.3
1.35
1.4
1.45
1.5
0 20 40 60 80 100
Bond
leng
th (Å
)
Pressure (GPa)
C1-C5 C4-C5 N10-N12 C2-N11 N12-N13 N8-O14
NFP
179
16 6
4
5
1 72
3
11
15
8
14
12 13
10
Figure 5: Selected bond lengths in NFP at different pressures. Circles connected by solid lines denotethe X-HCFF values. Crosses connected by dotted lines represent literature values (estimated graphicallyfrom ref. 43), calculated with Density Functional Theory (DFT) for periodic systems.
13
table differences include the bond lengths N10−N12 and N8−O14, which remain almost constant and
even increase occasionally in the periodic calculations, whereas they decrease monotonously in X-HCFF.
It can be hypothesized that these differences can be traced back to crystal packing effects that are not cap-
tured by X-HCFF. However, one can state that the structural parameters calculated with X-HCFF for the
molecular NFP crystal are chemically intuitive and for many bond lengths the agreement with periodic
calculations is remarkable, which is an encouraging result considering that only a single molecule is
treated with X-HCFF. Nevertheless, in the future a careful calibration of the size of the system investi-
gated with X-HCFF, i.e. the number of molecules subjected to pressure in the geometry optimization,
needs to be carried out to determine the influence of the chemical environment in the crystal on the
structural parameters of an individual molecule.
3.4 Comparison with experimental Raman spectra under pressure
Although comparisons of structural data of single molecules and molecular crystals calculated with X-
HCFF and other approaches are helpful to judge the performance of the new method, the clearest crite-
rion for its reliability is the direct comparison with experimental data. Experimental structural data of
molecules and crystals under pressure are typically inferred from spectroscopic observables. A notable
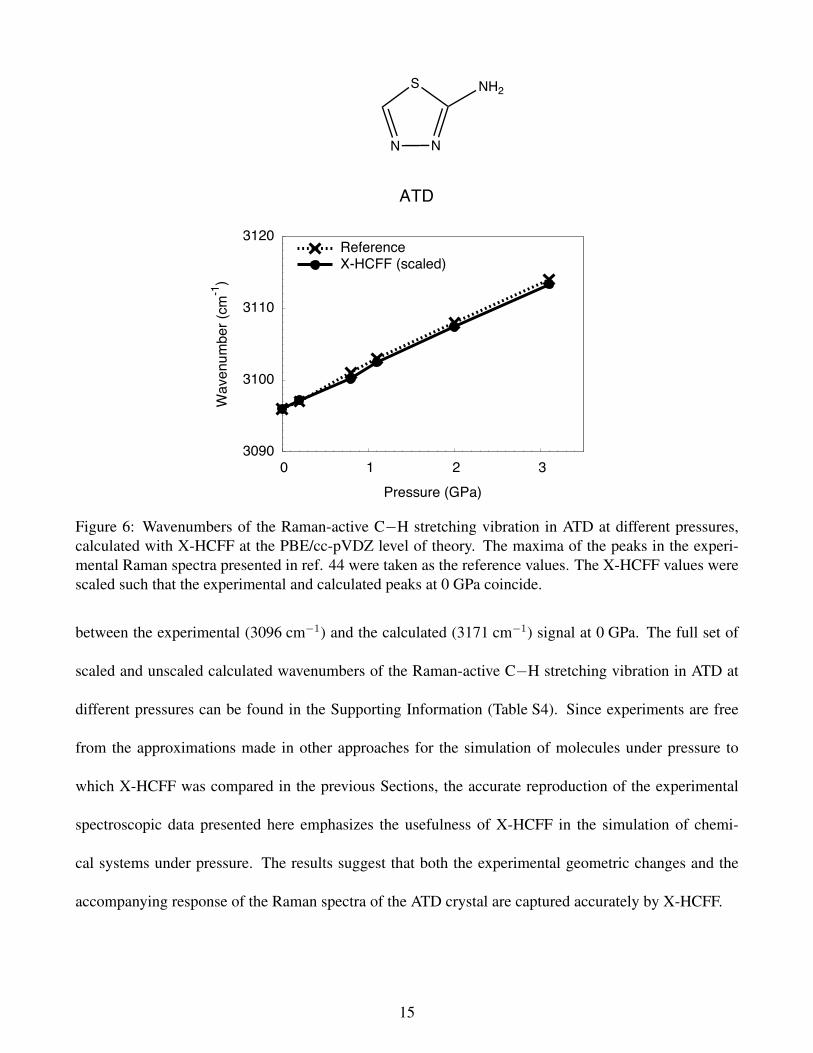
example is the pressure-induced increase in the wavenumber of the Raman-active C−H stretching vi-
bration in the molecular crystal 2-amine-1,3,4-thiadiazole (ATD), which has been reported recently.44 It
was found that the wavenumber of this vibration in the ATD crystal increases linearly with increasing
pressure.
Applying the same pressures as in the experiments to ATD with the X-HCFF approach at the PBE/cc-
pVDZ level of theory and subsequently calculating the Raman spectra yields virtually identical changes
in the Raman spectrum (Figure 6), provided that the calculated wavenumbers are scaled with the ratio
14
3090
3100
3110
3120
0 1 2 3
Wav
enum
ber (
cm-1
)
Pressure (GPa)
ReferenceX-HCFF (scaled)
ATD
N N
S NH2
Figure 6: Wavenumbers of the Raman-active C−H stretching vibration in ATD at different pressures,calculated with X-HCFF at the PBE/cc-pVDZ level of theory. The maxima of the peaks in the experi-mental Raman spectra presented in ref. 44 were taken as the reference values. The X-HCFF values werescaled such that the experimental and calculated peaks at 0 GPa coincide.
between the experimental (3096 cm−1) and the calculated (3171 cm−1) signal at 0 GPa. The full set of
scaled and unscaled calculated wavenumbers of the Raman-active C−H stretching vibration in ATD at
different pressures can be found in the Supporting Information (Table S4). Since experiments are free
from the approximations made in other approaches for the simulation of molecules under pressure to
which X-HCFF was compared in the previous Sections, the accurate reproduction of the experimental
spectroscopic data presented here emphasizes the usefulness of X-HCFF in the simulation of chemi-
cal systems under pressure. The results suggest that both the experimental geometric changes and the
accompanying response of the Raman spectra of the ATD crystal are captured accurately by X-HCFF.
15
3.5 Pressure-induced dimerization of carbon dioxide
To test the applicability of the X-HCFF approach when describing pressure-induced chemical reactions,
the dimerization of carbon dioxide under pressure is investigated as an example. This reaction has been
described previously using ab initio Molecular Dynamics (AIMD) simulations45 and has been proposed
as an intermediate step in the formation of polymeric CO2 under pressure.
Starting with a van-der-Waals complex of two CO2 molecules with a lateral distance of 3.15 A be-
tween each other, the application of pressure with X-HCFF at the PBE/cc-pVDZ level of theory leads to
a continuous decrease in the distance between the two CO2 molecules up to 90 GPa (Figure 7). Between
90 and 100 GPa, the distance between the CO2 molecules decreases sharply, marking the formation of
covalent bonds in the resulting CO2 dimer. A further increase in pressure leads to only a minor com-
pression of the C−O bonds within the four-membered ring of the CO2 dimer. Interestingly, carrying
out a pressure-free geometry optimization using the CO2 dimer created at 100 GPa as an input structure,
1
2
3
4
0 50 100 150
d (Å
)
Pressure (GPa)
O C O
O C O
O C
O
O
C O
d
d
Figure 7: Distance d between the carbon atom of a CO2 molecule and an oxygen atom of another CO2
molecule as a function of pressure, using X-HCFF at the PBE/cc-pVDZ level of theory. After dimeriza-tion occurs at approx. 100 GPa, d denotes the C−O bond length in the four-membered ring of the CO2
dimer.
16
the dimeric geometry is retained, emphasizing the metastable nature of the dimer that was described
previously.45 However, in the AIMD simulations the CO2 dimer was observed as a short-lived species
at pressures of 20 and 50 GPa, which is lower than in the X-HCFF calculations. A possible reason for
the higher pressure required to form the CO2 dimer using X-HCFF is that, in contrast to AIMD simu-
lations, thermal oscillations are not taken into account. Such fluctuations, however, lead to molecular
oscillations and could deliver crucial energy required to overcome the reaction barrier. Similar effects
have been discussed as the reason for the overestimation of rupture forces by static quantum chemical
methods when describing mechanochemical processes.26 Detailed calculations will need to be carried
out to reliably compare the pressures required to induce chemical reactions calculated by dynamic and
static simulations techniques.
The pressure-induced dimerization of CO2, which was successfully described with X-HCFF, demon-
strates that the predictive power of the new method is not limited to pressure-dependent structural and
spectroscopic properties, but also has high potential in describing chemical reactions under pressure.
In this case, a minimum on the potential energy surface under pressure, which was revealed by previ-
ous AIMD simulations, could be reproduced at the computational cost of a quantum chemical geometry
optimization.
4 Conclusions and Outlook
In this paper, the eXtended Hydrostatic Compression Force Field (X-HCFF), a novel mechanochemical
method for the simulation of molecules and molecular crystals under hydrostatic pressure, was intro-
duced. The X-HCFF method avoids common problems of related approaches and accurately reproduces
computational and experimental structural and spectroscopic reference data in a variety of test systems.
The straightforward implementation and the user-friendly and inexpensive computation make X-HCFF a
17
useful method for calculations of structural and spectroscopic properties as well as chemical transforma-
tions of molecules and molecular crystals under hydrostatic pressure.
In the future, careful benchmarks of various density functionals and wave-function based methods
will be carried out to determine the optimal electronic structure method that reproduces experimental
structural and spectroscopic data most accurately. Moreover, it is planned to leave the realm of small test
systems and evaluate the applicability of X-HCFF when treating crystallites. A particularly interesting
question is whether pressure-induced changes in lattice constants9–11 can be reproduced by using a quan-
tum mechanochemical method. The ability of X-HCFF to go beyond the treatment of single molecules
and deliver chemically meaningful results for the dimerization of carbon dioxide is very promising in
this regard. Another future route is provided by AIMD simulations, with which X-HCFF can be com-
bined easily, to simulate the time evolution of structural changes of molecules under pressure. This will
be particularly useful in modeling pressure-induced adsorption and desorption processes of, e.g., indus-
trially relevant gases on surfaces. Furthermore, more complex chemical reactions under pressure, e.g.
Diels-Alder reactions, will be treated with X-HCFF in the future.
Finally, it is important to note that in the X-HCFF approach molecules react towards hydrostatic
pressure only through force-induced compression of the nuclear scaffold. While this opens up the inter-
esting possibility of relating the strain induced by the compression with chemical reactivity,46 one must
acknowledge that pressure does not interact directly with the electrons, but only indirectly through an
altered arrangement of the nuclei. A computational method that simulates the influence of pressure on
molecules via interactions with the electron density is currently under development.
18
Acknowledgements
The author would like to thank M. Scheurer, University of Heidelberg, for helpful discussions and for
proofreading of the manuscript. The idea for this project was conceived during the work on a different
electronic structure method for the simulation of molecules under hydrostatic pressure, which is currently
being developed by M. Scheurer, A. Dreuw (University of Heidelberg), E. Epifanovsky (Q-Chem Inc.),
M. Head-Gordon (University of California, Berkeley) and the author of this paper.
References[1] McMillan, P. F. Chemistry at high pressure. Chem. Soc. Rev. 2006, 35, 855–857.
[2] Grochala, W.; Hoffmann, R.; Feng, J.; Ashcroft, N. W. The Chemical Imagination at Work in Very Tight Places. Angew.Chem. Int. Ed. 2007, 46, 3620–3642.
[3] Schettino, V.; Bini, R. Constraining molecules at the closest approach: chemistry at high pressure. Chem. Soc. Rev. 2007,36, 869–880.
[4] Li, B.; Ji, C.; Yang, W.; Wang, J.; Yang, K.; Xu, R.; Liu, W.; Cai, Z.; Chen, J.; kwang Mao, H. Diamond anvil cellbehavior up to 4 Mbar. Proc. Nat. Acad. Sci. U.S.A. 2018, 115, 1713–1717.
[5] Stixrude, L.; Cohen, R. E. High-Pressure Elasticity of Iron and Anisotropy of Earth’s Inner Core. Science 1995, 267,1972–1975.
[6] Casati, N.; Kleppe, A.; Jephcoat, A. P.; Macchi, P. Putting pressure on aromaticity along with in situ experimentalelectron density of a molecular crystal. Nat. Commun. 2016, 7, 10901.
[7] Chandrasekhar, M.; Guha, S.; Graupner, W. Squeezing Organic Conjugated Molecules - What Does One Learn? Adv.Mater. 2001, 13, 613–618.
[8] Kato, M.; Higashi, M.; Taniguchi, Y. Effect of pressure on the internal rotation angle of biphenyl in carbon disulfide. J.Chem. Phys. 1988, 89, 5417–5421.
[9] Puschnig, P.; Ambrosch-Draxl, C.; Heimel, G.; Zojer, E.; Resel, R.; Leising, G.; Kriechbaum, M.; Graupner, W. Pressurestudies on the intermolecular interactions in biphenyl. Synth. Met. 2001, 116, 327–331.
[10] Katrusiak, A. High-Pressure X-Ray Diffraction Study Of 2-Methyl-1,3-Cyclopentanedione Crystals. High Press. Res.1991, 6, 155–167.
[11] Cartz, L.; Srinivasa, S. R.; Riedner, R. J.; Jorgensen, J. D.; Worlton, T. G. Effect of pressure on bonding in blackphosphorus. J. Chem. Phys. 1979, 71, 1718–1721.
[12] Demontis, P.; Lesar, R.; Klein, M. L. New High-Pressure Phases of Ice. Phys. Rev. Lett. 1988, 60, 2284–2287.
[13] Nose, S.; Klein, M. L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076.
[14] Mugnai, M.; Cardini, G.; Schettino, V. Charge separation and polymerization of hydrocarbons at an ultrahigh pressure.Phys. Rev. B 2004, 70, 020101.
19
[15] Imoto, S.; Kibies, P.; Rosin, C.; Winter, R.; Kast, S. M.; Marx, D. Toward Extreme Biophysics: Deciphering the InfraredResponse of Biomolecular Solutions at High Pressures. Angew. Chem. Int. Ed. 2016, 55, 9534–9538.
[16] Marx, D.; Hutter, J. Ab Initio Molecular Dynamics. Basic Theory and Advanced Methods, 1st ed.; Cambridge UniversityPress: Cambridge, UK, 2009.
[17] Stauch, T. Quantum chemical modeling of molecules under pressure. Int. J. Quantum Chem. 2020, e26208.
[18] Cammi, R.; Verdolino, V.; Mennucci, B.; Tomasi, J. Towards the elaboration of a QM method to describe molecularsolutes under the effect of a very high pressure. Chem. Phys. 2008, 344, 135–141.
[19] Cammi, R. A New Extension of the Polarizable Continuum Model: Toward a Quantum Chemical Description of Chem-ical Reactions at Extreme High Pressure. J. Comput. Chem. 2015, 36, 2246–2259.
[20] Chen, B.; Hoffmann, R.; Cammi, R. The Effect of Pressure on Organic Reactions in Fluids - a New Theoretical Perspec-tive. Angew. Chem. Int. Ed. 2017, 56, 11126–11142.
[21] Fukuda, R.; Nakatani, K. Quantum Chemical Study on the High-Pressure Effect for [4 + 4] Retrocycloaddition ofAnthracene Cyclophane Photodimer. J. Phys. Chem. C 2019, 123, 4493–4501.
[22] Pagliai, M.; Cardini, G.; Cammi, R. Vibrational Frequencies of Fullerenes C60 and C70 under Pressure Studied with aQuantum Chemical Model Including Spatial Confinement Effect. J. Phys. Chem. A 2014, 118, 5098–5111.
[23] Fukuda, R.; Ehara, M.; Cammi, R. Modeling molecular systems at extreme pressure by an extension of the polarizablecontinuum model (PCM) based on the symmetry-adapted cluster-configuration interaction (SAC-CI) method: Confinedelectronic excited states of furan as a test case. J. Chem. Theory Comput. 2015, 11, 2063–2076.
[24] Rahm, M.; Cammi, R.; Ashcroft, N. W.; Hoffmann, R. Squeezing All Elements in the Periodic Table: Electron Config-uration and Electronegativity of the Atoms under Compression. J. Am. Chem. Soc. 2019, 141, 10254–10271.
[25] Caratelli, C.; Cammi, R.; Chelli, R.; Pagliai, M.; Cardini, G.; Schettino, V. Insights on the Realgar Crystal under Pressurefrom XP-PCM and Periodic Model Calculations. J. Phys. Chem. A 2017, 121, 8825–8834.
[26] Stauch, T.; Dreuw, A. Advances in Quantum Mechanochemistry: Electronic Structure Methods and Force Analysis.Chem. Rev. 2016, 116, 14137–14180.
[27] Subramanian, G.; Mathew, N.; Leiding, J. A generalized force-modified potential energy surface for mechanochemicalsimulations. J. Chem. Phys. 2015, 143, 134109.
[28] Jha, S. K.; Brown, K.; Todde, G.; Subramanian, G. A mechanochemical study of the effects of compression on a Diels-Alder reaction. J. Chem. Phys. 2016, 145, 074307.
[29] Todde, G.; Jha, S. K.; Subramanian, G. The effect of external forces on the initial dissociation of RDX (1,3,5-trinitro-1,3,5-triazine): A mechanochemical study. Int. J. Quantum Chem. 2017, 117, e25426.
[30] Ong, M. T.; Leiding, J.; Tao, H.; Virshup, A. M.; Martınez, T. J. First Principles Dynamics and Minimum EnergyPathways for Mechanochemical Ring Opening of Cyclobutene. J. Am. Chem. Soc. 2009, 131, 6377–6379.
[31] Stauch, T.; Chakraborty, R.; Head-Gordon, M. Quantum Chemical Modeling of Pressure-Induced Spin Crossover inOctahedral Metal-Ligand Complexes. ChemPhysChem 2019, 20, 2742–2747.
[32] Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, 864–871.
[33] Kohn, W.; Sham, L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140,1133–1138.
[34] Shao, Y. et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2014,113, 184–215.
20
[35] Kochhar, G. S.; Heverly-Coulson, G. S.; Mosey, N. J. Theoretical Approaches for Understanding the Interplay BetweenStress and Chemical Reactivity. Top. Curr. Chem. 2015, 369, 37–96.
[36] Lange, A. W.; Herbert, J. M. Polarizable Continuum Reaction-Field Solvation Models Affording Smooth PotentialEnergy Surfaces. J. Phys. Chem. Lett. 2010, 1, 556–561.
[37] Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77,3865–3868.
[38] Dunning, T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon andhydrogen. J. Chem. Phys. 1989, 90, 1007–1023.
[39] Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988,38, 3098–3100.
[40] Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of theelectron density. Phys. Rev. B 1988, 37, 785–789.
[41] Becke, A. D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377.
[42] Hehre, W. J.; Ditchfield, R.; Pople, J. A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaus-sian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261.
[43] Liu, H.; Wang, F.; Gong, X. DFT studies on 7-nitrotetrazolo [1,5]furazano[4,5-b]pyridine 1-oxide: Crystal structure,detonation properties, sensitivity and effect of hydrostatic compression. Struct. Chem. 2014, 25, 239–249.
[44] de Toledo, T. A.; da Costa, R. C.; Bento, R. R. F.; Pizani, P. S. Hydrostatic pressure and temperature effect on the Ramanspectra of the molecular crystal 2-amine-1,3,4-thiadiazole. J. Mol. Struct. 2018, 1156, 127–135.
[45] Tassone, F.; Chiarotti, G. L.; Rousseau, R.; Scandolo, S.; Tosatti, E. Dimerization of CO2 at High Pressure and Temper-ature. ChemPhysChem 2005, 6, 1752–1756.
[46] Stauch, T.; Dreuw, A. Quantum Chemical Strain Analysis For Mechanochemical Processes. Acc. Chem. Res. 2017, 50,1041–1048.
21
download fileview on ChemRxivManuscript_X-HCFF.pdf (774.95 KiB)
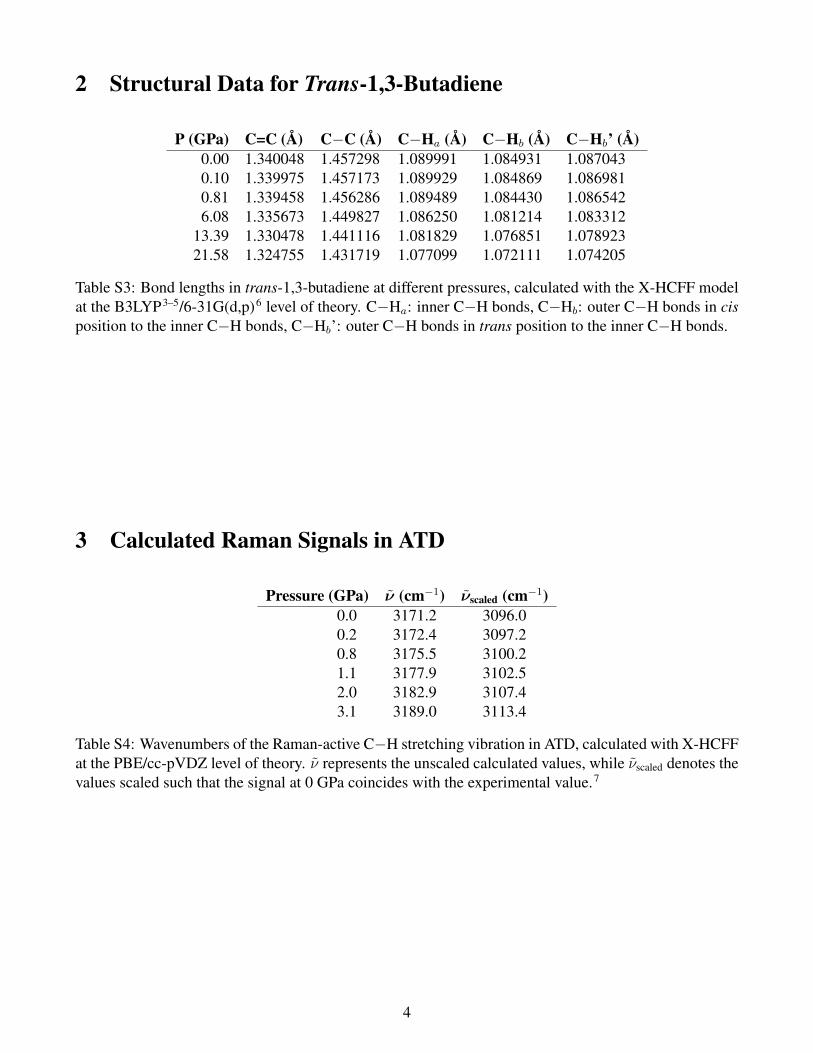
Table S3: Bond lengths in trans-1,3-butadiene at different pressures, calculated with the X-HCFF modelat the B3LYP3–5/6-31G(d,p)6 level of theory. C−Ha: inner C−H bonds, C−Hb: outer C−H bonds in cisposition to the inner C−H bonds, C−Hb’: outer C−H bonds in trans position to the inner C−H bonds.
Table S4: Wavenumbers of the Raman-active C−H stretching vibration in ATD, calculated with X-HCFFat the PBE/cc-pVDZ level of theory. ν represents the unscaled calculated values, while νscaled denotes thevalues scaled such that the signal at 0 GPa coincides with the experimental value.7
Table S5: Cartesian coordinates (A) of the carbon dioxide dimer used as the initial structure for thegeometry optimizations under pressure with X-HCFF.
5 References[1] J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868.
[2] T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023.
[3] A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100.
[4] C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789.
[5] A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377.
[6] W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261.
[7] T. A. de Toledo, R. C. da Costa, R. R. F. Bento and P. S. Pizani, J. Mol. Struct., 2018, 1156, 127–135.
5
download fileview on ChemRxivSI_X-HCFF.pdf (221.81 KiB)