Clay Minerals (1990) 25, 289-301 A MOSSBAUER STUDY OF GREEN RUST PRECIPITATES: I. PREPARATIONS FROM SULPHATE SOLUTIONS A. H. CUTTLER, V. MAN, T. E. CRANSHAW* AND G. LONGWORTH* Polytechnic South West, Plymouth, Devon PL4 8AA, and *AEA Technology, Harwell, Didcot, Oxfordshire, OXI10RA, UK (Received 7 August 1989; revised 28 December 1989) A B S T R A C T : The preparation of green rusts from sulphate solutions and representative M6ssbauer spectra are described. As the samples oxidized readily, attention focused on the M6ssbauer parameters at liquid nitrogen and helium temperatures. The spectra recorded at 77 K could be fitted satisfactorily with one ferrous iron quadrupole doublet with a separation of 2.93 mms 1 and one ferric iron quadrupole doublet with a separation of 0.45 rams -1. In some spectra a ferric iron magnetic hyperfine of strength 49.2 T was also apparent. At 4.2 K, the ferrous iron exhibited a hyperfine splitting with a field of 12.4 T whilst the ferric iron exhibited a hyperfine splitting with a field of strength 50-4 T. The ratio of ferrous to ferric ions was 2.25 + 0.25 at 77 K and at 4.2 K, and -1.6 with a large variation at room temperature. The liquid helium spectra did not always give a good chi-squared fit, the main reason being attributed to relaxation. The line-width of the ferrous iron site at 77 K is slightly larger than that for iron metal and could be explained by a variation in the number of near Fe3+ neighbours at different Fee+ sites, consistent with the assumption that the ferrous iron site is in the hydroxide sheet. The effect of different numbers of Fe2+ and Fe3+ neighbours probably contributed to the increase in line-widths at 4.2 K compared with those at 77 K. The ferrous iron doublet is marginally different to those of chloride and hydroxy- carbonate green rusts and the aluminium analogues. Green rusts are unstable compounds containing a mixture of ferrous and ferric iron and were first described by Keller (1948) who produced a chloride and a sulphate green rust. The ratio of ferrous to ferric iron was found to lie between 0.8 and 4-0. A study by Bernal et al. (1959) of iron oxyhydroxides, which included green rusts, identified two forms of the sulphate species as well as the chloride form. These green rusts were prepared "by the partial oxidation of ferrous iron solutions" but the method was not described in sufficient detail to enable repeat studies to be made. Misawa et al. (1973, 1974) made a study by ultra-violet spectroscopy and chemical analysis of the aerial oxidation of neutral and slightly alkaline ferrous sulphate solutions. An intermediate green complex was observed which was shown to be a precursor of green rust II and on that basis the ferrous to ferric ratio was estimated to be about unity. Gancedo et al. (1976) studied the corrosion products of iron in aqueous solutions of ammonium nitrate and found the room temperature M6ssbauer quadrupole doublets to have parameters similar to those of synthetic green rusts I and II, namely, 2.0 and 2-37 mms -1, respectively. It was noted that the ferrous to ferric iron ratios were 1.88 and 0.63, respectively. McGill et al. (1976) in a study of the corrosion of cast iron in carbonate solutions reported the formation of a green rust compound which gave similar lines to green rust I but which was structurally different. Brindley & Bish (1976) pointed out that this was 1990 The Mineralogical Society
Transcript
Clay Minerals (1990) 25, 289-301
A M O S S B A U E R S T U D Y OF G R E E N R U S T P R E C I P I T A T E S : I. P R E P A R A T I O N S F R O M S U L P H A T E
S O L U T I O N S
A . H . C U T T L E R , V . M A N , T . E . C R A N S H A W * AND G . L O N G W O R T H *
Polytechnic South West, Plymouth, Devon PL4 8AA, and * AEA Technology, Harwell, Didcot, Oxfordshire, OXI10RA, UK
(Received 7 August 1989; revised 28 December 1989)
A B S T R A C T : The preparation of green rusts from sulphate solutions and representative M6ssbauer spectra are described. As the samples oxidized readily, attention focused on the M6ssbauer parameters at liquid nitrogen and helium temperatures. The spectra recorded at 77 K could be fitted satisfactorily with one ferrous iron quadrupole doublet with a separation of 2.93 mms 1 and one ferric iron quadrupole doublet with a separation of 0.45 rams -1. In some spectra a ferric iron magnetic hyperfine of strength 49.2 T was also apparent. At 4.2 K, the ferrous iron exhibited a hyperfine splitting with a field of 12.4 T whilst the ferric iron exhibited a hyperfine splitting with a field of strength 50-4 T. The ratio of ferrous to ferric ions was 2.25 + 0.25 at 77 K and at 4.2 K, and -1.6 with a large variation at room temperature. The liquid helium spectra did not always give a good chi-squared fit, the main reason being attributed to relaxation. The line-width of the ferrous iron site at 77 K is slightly larger than that for iron metal and could be explained by a variation in the number of near Fe 3+ neighbours at different Fe e+ sites, consistent with the assumption that the ferrous iron site is in the hydroxide sheet. The effect of different numbers of Fe 2+ and Fe 3+ neighbours probably contributed to the increase in line-widths at 4.2 K compared with those at 77 K. The ferrous iron doublet is marginally different to those of chloride and hydroxy- carbonate green rusts and the aluminium analogues.
G r e e n rusts are uns table c o m p o u n d s conta in ing a mix tu re o f fer rous and ferr ic i ron and
were first descr ibed by Kel le r (1948) w h o p r o d u c e d a ch lor ide and a su lpha te g reen rust.
The ra t io of fer rous to ferr ic i ron was found to lie b e t w e e n 0.8 and 4-0. A s tudy by Be rna l et
al. (1959) of i ron oxyhydrox ides , which inc luded g reen rusts, ident i f ied two forms of the
su lphate species as well as the chlor ide fo rm. These g reen rusts were p r e p a r e d "by the
par t ia l ox ida t ion of fer rous i ron so lu t ions" bu t the m e t h o d was no t desc r ibed in sufficient
deta i l to enab le r epea t studies to be made .
Misawa et al. (1973, 1974) m a d e a s tudy by u l t ra-v io le t spec t roscopy and chemica l
analysis of the aerial ox ida t ion of neu t ra l and slightly a lkal ine fer rous su lpha te solut ions.
A n i n t e rmed ia t e g reen complex was o b s e r v e d which was shown to be a p recu r so r of g reen
rust I I and on that basis the fer rous to ferr ic ra t io was e s t ima ted to be abou t unity.
G a n c e d o et al. (1976) s tudied the cor ros ion p roduc t s of i ron in a q u e o u s solut ions o f
a m m o n i u m ni t ra te and found the r o o m t e m p e r a t u r e M6ssbaue r q u a d r u p o l e double t s to
have pa r ame te r s similar to those o f synthet ic g reen rusts I and II , namely , 2.0 and
2-37 m m s -1, respect ively . It was no t ed that the fer rous to ferr ic i ron ra t ios w e r e 1.88 and
0.63, respect ively . McGi l l et al. (1976) in a s tudy of the cor ros ion of cast i ron in ca rbona te
solut ions r e p o r t e d the fo rma t ion of a g reen rust c o m p o u n d which gave similar l ines to g reen
rust I bu t which was s t ructural ly di f ferent . Br ind ley & Bish (1976) po in t ed ou t that this was
�9 1990 The Mineralogical Society
290 A . H . Cuttler et al.
likely to be a member of the pyroaurite group (which is based on a mixture of divalent and trivalent ions), giving rise to a carbonate-hydroxide. Taylor (1973) also proposed that the green rusts belonged to this class which Allmann (1968) had analysed in detail.
Taylor & MacKenzie (1980) made a study of the analogous ferrous iron-aluminium compounds. In this study they used oxygen-free conditions and controlled the ratio of ferrous iron to aluminium enabling quantitative evaluations to be made of the reaction and its products. In these studies by Taylor & Mackenzie (1980), the sulphate, chloride and carbonate compounds were prepared and stated to be "relatively stable". Titration data and X-ray diffraction (XRD) data were given but no M6ssbauer studies were made. This method appeared suitable, and was adapted, for a pure iron solution (Taylor, 1980). A later study by Murad & Taylor (1984) of the FeZ+Fe 3+ and FeZ+A13+ forms included M6ssbauer spectroscopy at room temperature and at 120 K, and two ferrous quadrupole doublets were observed in both compounds with two ferric doublets in the ferric species but only one ferric doublet in the aluminium species. The ferrous iron quadrupole doublets were 2.89 and 2-23 mms -1 for the ferrous-ferric species, and 2-89 and 2.48 mms -1 for the ferrous iron- aluminium species. A structure for the green rusts was proposed with tentative assignments of the ferrous doublets to specific sites.
A recent paper by Olowe et al. (1989) described the preparation by aerial oxidation of a ferrous sulphate-sodium hydroxide mixture with an initial pH of 7.8, the reaction being stopped when the pH fell to 7.2 (after about 2 h). By measuring the M6ssbauer parameters at Various temperatures between 77 K and 460 K, they concluded that the spectra could be fitted using three ferrous doublets and two ferric doublets. The outer two ferrous doublets with quadrupole splittings of 2.96 mms 1 and 2.90 at 77 K were not resolved. The third ferrous doublet with line separation 2.63 mms -1 was partially resolved only for the higher velocity line. The ferric doublets were not resolved either from each other or from the low- velocity components of the ferrous lines. The outer ferrous doublets were interpreted as arising from the hydroxide layer and the inner ferrous doublet to "a sulphate environment". The ferric component has been interpreted as arising from amorphous ferric hydroxide.
The present work is part of a comprehensive study of green rusts synthesized in aqueous chloride, sulphate, nitrate and carbonate solutions using both ferric and aluminium hydroxides to precipitate ferrous iron from solution. Most of this work, described by Man (1987), covered infra-red spectroscopy, XRD, M~ssbauer studies, and surface area and titration measurements. The similar methods of preparation of the sulphate and chloride precipitates are described in detail, and the M6ssbauer spectra of the sulphate green rusts are presented and their significance discussed.
M E T H O D
The method of preparation for the sulphate green rusts was essentially the same as that of Taylor & Mackenzie (1980) but with the substitution of ferric iron for aluminium, i.e. the method required the preparation of separate solutions of ferrous and ferric iron in oxygen- free conditions, taking each above pH 7 before mixing. The pH was maintained by the addition of an alkaline solution of either sodium or potassium hydroxide. The ratio of ferrous to ferric iron was at least 5 : 1 to ensure that not all ferrous iron would be adsorbed from the solution according to the range of values given by Keller (1948). Ferrous sulphate was used as the source of divalent iron and either ferric nitrate or ferric chloride as the
M6ssbauer study o f green rust 291
trivalent iron. Identical parameters were obtained for the products from the different ferric salts.
The ferric iron was prepared in a conical flask as a 0.08 M solution of 50 ml volume under oxygen-free conditions. The solution was stirred magnetically and nitrogen or argon bubbled at a rate of 200 ml per min. The top of the flask was sealed with parafilm and the temperature monitored. In most experiments the temperature was in the range 22 to 27~ The alkalinity of the solution was increased gradually to pH 7 when the ferric iron had "gelled" and was dark red in colour. The alkali solutions were 0.1 or 0.01 i sodium or potassium hydroxide and were introduced through the parafilm using a burette. These concentrations were lower than used by Taylor & Mackenzie (1980) but were chosen to reduce the precipitation of ferrous hydroxide in the second and third stages of the experiment. After the solution had reached pH 7, the burette was removed and the flask resealed, flushing with nitrogen (argon) to prevent the solution from settling.
The second stage consisted of the preparation, under oxygen-free conditions, of a 200 ml 0.1 M solution of ferrous sulphate in a flask sealed with parafilm and flushed with nitrogen or argon. In some cases mixing was entirely by nitrogen (argon) flushing, and in a few, magnetic stirring was also included. No attempt was made to remove any trace contamination of the nitrogen gas by oxygen. With a flow rate of 200 ml per min the total consumption of gas was between 24 1 and 48 1 which at the p.p.m, level gave an oxygen content of the order of 100/~g. It was calculated that this quantity would not give rise to appreciable oxidation of the ferrous iron and this was supported by evidence from similar experiments with the analogous ferrous iron-aluminium solutions. The ferrous iron solution was taken to pH 7 by the addition of alkali as with the ferric iron. In some experiments the pH was raised rapidly, in others slowly, to permit the solution to come to equilibrium. When pH 7 was reached, the ferric solution was added causing the pH to fall slightly. The pH was adjusted to 7-7-2 by the addition of alkali and maintained at the chosen level until no more alkali was consumed (usually after two or more hours). The concentrations of ferrous iron or ferric iron were occasionally reduced but never such that the ratio of ferrous to ferric iron fell below five. Cuttler et al. (1984) have reported such experiments in which the surface area and the ferrous : ferric ratio of the end product were the parameters most affected.
When the consumption of alkali had ceased, the solution was transferred under nitrogen gas to a separating flask which was then stoppered. The precipitate was allowed to settle, samples were withdrawn into centrifuge tubes, stoppered, and spun to concentrate the precipitate. A sample was then prepared for M6ssbauer spectroscopy by placing a small quantity of the "paste" between two polyethylene discs mounted in a perspex holder which was wrapped with sellotape in both directions to provide a seal against oxidation. This sample was then placed immediately in liquid nitrogen to reduce the possibility of further oxidation. At the same time, samples were taken for X R D analysis to confirm the formation of green rusts. Some samples were stored in liquid nitrogen for analysis at a later time and for transport to A E A Technology, Harwetl for the liquid helium measurements, as an earlier sample not treated in this way underwent some degradation on transport.
Most MOssbauer spectra were recorded on an Inotech 5200 multichannel analyser using a velocity generator similar to that described by Clark et al. (1967). The measurements at liquid helium temperature and a few at 77 K and 293 K were recorded at A E A Technology, Harwell. The spectra were folded in all instances using 256 channels at Polytechnic South West, and 512 channels at A E A Technology, Harwell. The source was 57Co in a rhodium
292 A . H . Cuttler et al.
matrix and isomer shifts have been quoted relative to iron metal at room temperature. The linearity and stability of both systems was better than one part in three hundred.
D A T A A N A L Y S I S
The M6ssbauer spectra were analysed on a Prime 850 computer using an interactive graphics programme. The error graph and z-squared values were used as the indicators for the fits. Spectra recorded at 77 K and 293 K presented no problems in analysis as the quadrupole doublets did not significantly overlap each other or the magnetic hyperfine component for which the parameters could be obtained easily. However, the 4.2 K spectrum contained overlapping ferrous and ferric spectra, both showing magnetic hyperfine splitting, and in addition there appeared to be some relaxation of the ferrous hyperfine field. In view of these uncertainties and difficulties encountered in obtaining satisfactory x-squared values, the analyses were performed to extract approximate parameters paying attention to the fitting of the main lines. The problem of line identification was alleviated by the resemblance of the ferrous iron spectrum to that of ferrous hydroxide (Miyamoto et al., 1967, or Greenwood & Gibb, 1971). Initial estimates of the ferrous iron parameters were made following the procedure outlined by van Dongen Torman et al. (1975). More exact solutions were obtained b3~ stripping the ferric iron components using distribution functions followed by interactive fitting of the ferrous iron hyperfine spectrum using calculated line positions from solutions of the energy Hamiltonian close to the estimated parameters. The intensities were determined from the paper by Kundig (1967). The only restriction was on the electric quadrupole component which was kept at the same value as at 77 K since this value emerged from the initial estimates. To give a better fit it was necessary to add a ferrous iron quadrupole doublet. Whilst the z-squared values were acceptable in two of the low-temperature spectra, in the other two they remained high. Examination of the error graphs indicated no particularly outstanding features, only a general decrease in transmission. This decrease and the ferrous iron doublet were attributed to relaxation processes, and more exact refinements were not considered appropriate in view of the problems associated with the overlap of the spectra.
R E S U L T S A N D D I S C U S S I O N
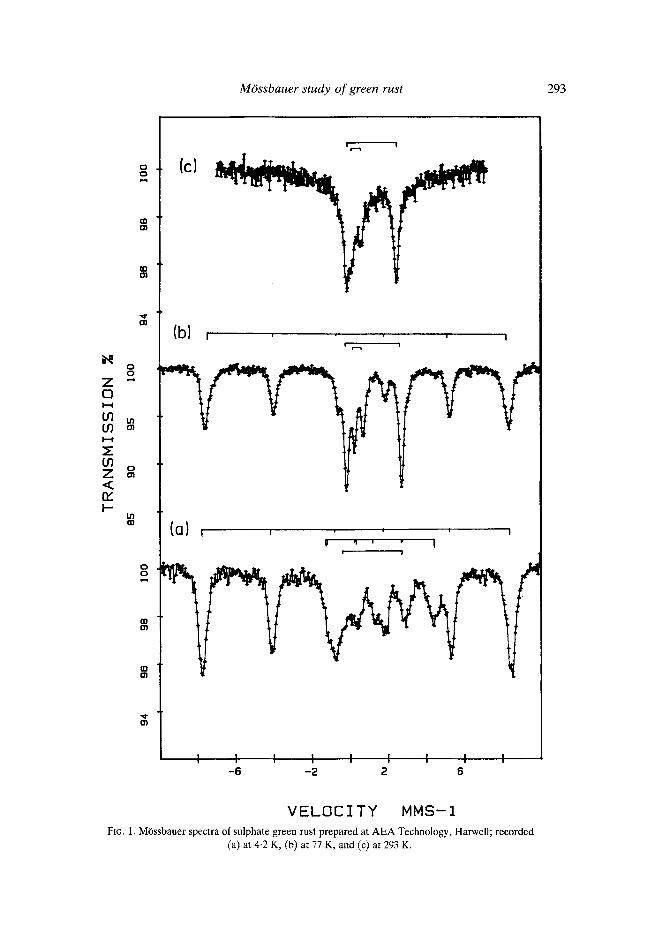
The M6ssbauer spectra at 293 K, 77 K and 4-2 K of a sample prepared at AEA Technology, Harwell are shown in Fig. 1, and the averaged parameters from a number of samples are given in Table 1. Figs. l(a) and l(b) show the presence of a magnetic hyperfine component in the spectrum at 4-2 K and at 77 K. This magnetic hyperfine component showed considerable relaxation at 293 K (Fig. lc). The interpretation of this latter spectrum is a problem due to the oxidation of the sample during measurement, as inferred from a change both in the colour and in the ferrous:ferric iron ratio. Thequadrupole doublet appeared to be slightly smaller in magn.itude than that at 77 K but greater than the value of 2.3 mms -1 reported by Gancedo et al. (1976). The spreads in the ferric iron magnetic hyperfine components at 77 K and at 4.2 K are similar, the lower temperature giving a slightly greater average value which appears to be similar to the value for goethite (Forsythe et al., 1968) but larger than that of the initial ferric gel.
The ferrous : ferric iron ratios of the paramagnetic components of a large number of green rusts lay between 2-0 and 2.5 and in most samples close to the average of 2-25. The
MOssbauer study of green rust 293
N
Z Q H
t~
H
LO
Z < n~ k-
(c)
II1
I0
0 0
0 0
P~
113
0 0
( b ) , , ,
( G ) ~ t p . , r I
l
I I I I I I I I I - 6 - 2 2 6
VELOCITY MMS-I FxG. 1. MOssbauer spectra of sulphate green rust prepared at AEA Technology, Harwell; recorded
(a) at 4-2 K, (b) at 77 K, and (c) at 293 K,
294 A . H . Cuttler et al.
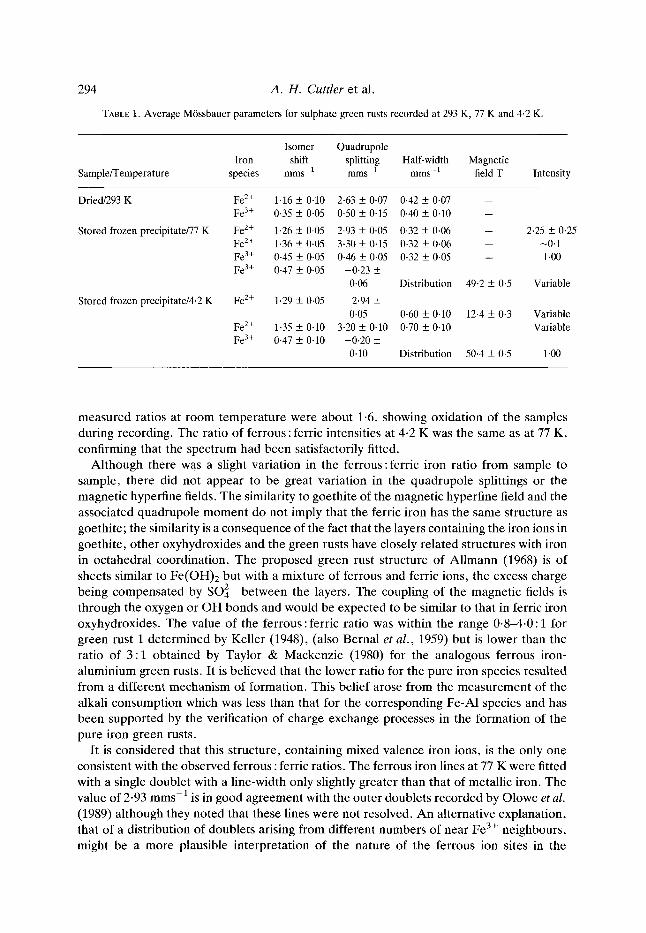
TABLE 1. Average Mrssbauer parameters for sulphate green rusts recorded at 293 K, 77 K and 4.2 K.
Isomer Quadrupole Iron shift splitting Half-width Magnetic
Sample/Temperature species mms 1 rams 1 mms-1 field T Intensity
Fe 2+ 1-35 _+ 0.10 3.20 • 0.10 0.70 • 0.10 Variable Fe 3+ 0-47 _+ 0.10 -0.20 •
0 .10 Distribution 50.4 + 0-5 1.00
measured ratios at room temperature were about 1.6, showing oxidation of the samples during recording. The ratio of ferrous : ferric intensities at 4-2 K was the same as at 77 K, confirming that the spectrum had been satisfactorily fitted.
Although there was a slight variation in the ferrous:ferric iron ratio from sample to sample, there did not appear to be great variation in the quadrupole splittings or the magnetic hyperfine fields. The similarity to goethite of the magnetic hyperfine field and the associated quadrupole moment do not imply that the ferric iron has the same structure as goethite; the similarity is a consequence of the fact that the layers containing the iron ions in goethite, other oxyhydroxides and the green rusts have closely related structures with iron in octahedral coordination. The proposed green rust structure of Al lmann (1968) is of sheets similar to Fe(OH)2 but with a mixture of ferrous and ferric ions, the excess charge being compensated by SO]- between the layers. The coupling of the magnetic fields is through the oxygen or OH bonds and would be expected to be similar to that in ferric iron oxyhydroxides. The value of the ferrous : ferric ratio was within the range 0-8-4.0 : 1 for green rust 1 determined by Keller (1948), (also Bernal et al., 1959) but is lower than the ratio of 3 :1 obtained by Taylor & Mackenzie (1980) for the analogous ferrous iron- aluminium green rusts. It is believed that the lower ratio for the pure iron species resulted from a different mechanism of formation. This belief arose from the measurement of the alkali consumption which was less than that for the corresponding Fe-A1 species and has been supported by the verification of charge exchange processes in the formation of the pure iron green rusts.
It is considered that this structure, containing mixed valence iron ions, is the only one consistent with the observed ferrous : ferric ratios. The ferrous iron lines at 77 K were fitted with a single doublet with a line-width only slightly greater than that of metallic iron. The value of 2.93 mms -1 is in good agreement with the outer doublets recorded by Olowe et al.
(1989) although they noted that these lines were not resolved. A n alternative explanation, that of a distribution of doublets arising from different numbers of near Fe 3+ neighbours, might be a more plausible interpretation of the nature of the ferrous ion sites in the
MOssbauer study o f green rust 295
hydroxide sheet. Such a distribution would appear to have only a small influence on the crystal field at the nucleus under consideration. There is no evidence for the third ferrous doublet observed by Olowe et al. (1989). The variation in the nature of the near neighbours might also affect the magnetic properties of the iron ions. This could account for the increase in the widths of the ferrous and the ferric absorption lines at 4-2 K compared with those at 77 K although, for the ferrous component, there appears to be some relaxation of the magnetic field, which might mask the effect of non-equivalent sites.
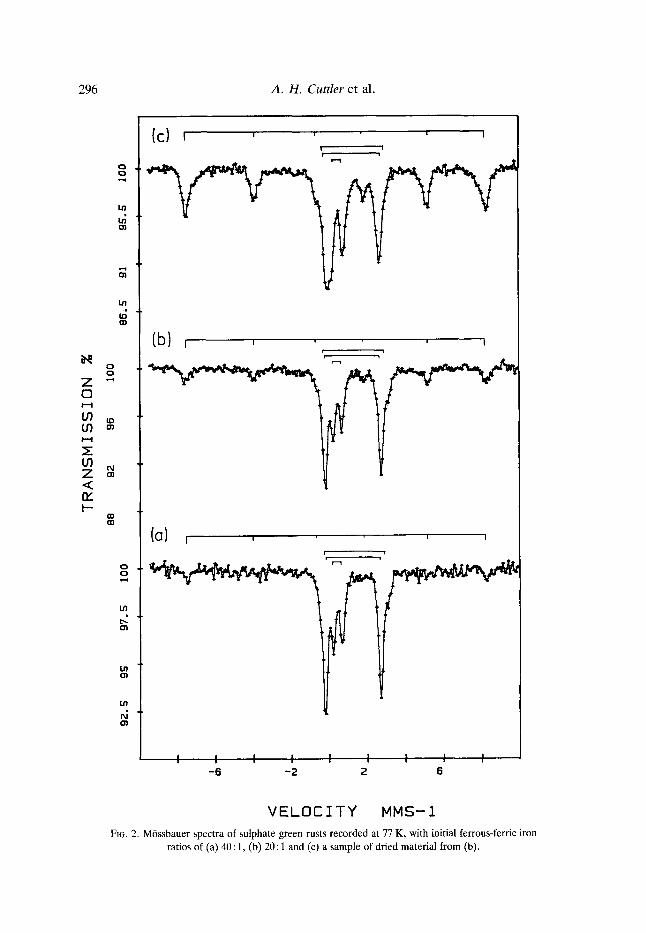
The intensity of the magnetic hyperfine component relative to the ferrous iron component at 77 K varied from sample to sample. Fig. 2(a) and 2(b) shows two samples, prepared at Polytechnic South West, in which the magnetic hyperfine component is almost non-existent or very small compared with the spectrum in Fig. l(b). The spectrum in Fig. 2(c) is from a sample washed in de-oxygenated water and vacuum-dried, and shows a decrease in the transmission of the paramagnetic ferric iron component relative to that of the ferrous iron component. It would appear that the oxidation of the ferrous iron has contributed also to an increase in the magnetically ordered ferric component. The variability in the intensity of this magnetic component indicates that it is not essential for the green rust structure; indeed green rusts prepared at concentrations <0.05 M do not show such a component. It is believed that the ferric magnetic hyperfine spectrum arises from partial ordering of the ferric gel before charge exchange processes have redistributed all the adsorbed ferrous iron throughout the material. The narrow line-widths imply either that there is a single site for the Fe 2+ ions, or that there is a distribution of sites with differing numbers of Fe 2+ and Fe 3+ neighbours. If this is the case, then the effects of the near neighbours must be small. This interpretation would have implications for the interpre- tation of other iron compounds as well as the interpretation of the spectra of hydroxy- carbonate green rusts by Murad & Taylor (1984). Unpublished studies on similar compounds by the authors indicated a mixed hydroxide/carbonate system. The quadrupole splittings of sulphate green rusts at 77 K are consistently larger than those of green rusts prepared using solutions of ferrous chloride or ferrous carbonate. The difference is small (0.10 mms 1 or less) and might be used, with care, to distinguish between such materials.
There is some evidence for a frozen ferrous sulphate solution. Occasionally, there is a slight asymmetry on the high-energy side of the ferrous iron line at 2.4 mms- 1 as can be seen in Fig. 2(a) and 2(b) although these are exceptions and account for less than 5% of the ferrous iron. The quadrupole splitting for frozen solutions is 3.2 mms-1 and the isomer shift is 1.36 mms -1 (Deszi et al., 1965). The presence of this doublet is probably due to a short spin and the use of the unwashed precipitate to reduce oxidation.
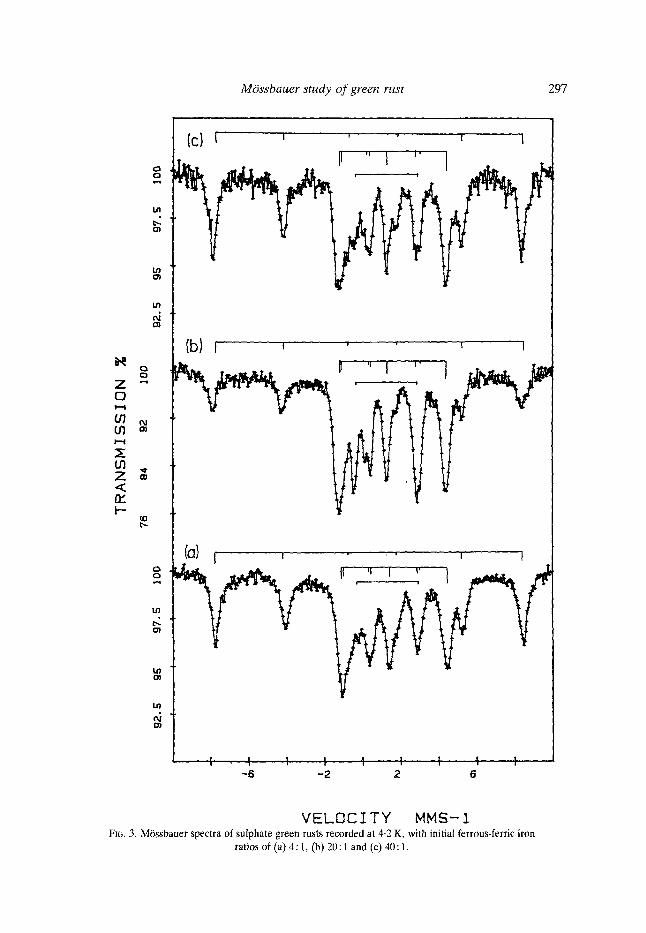
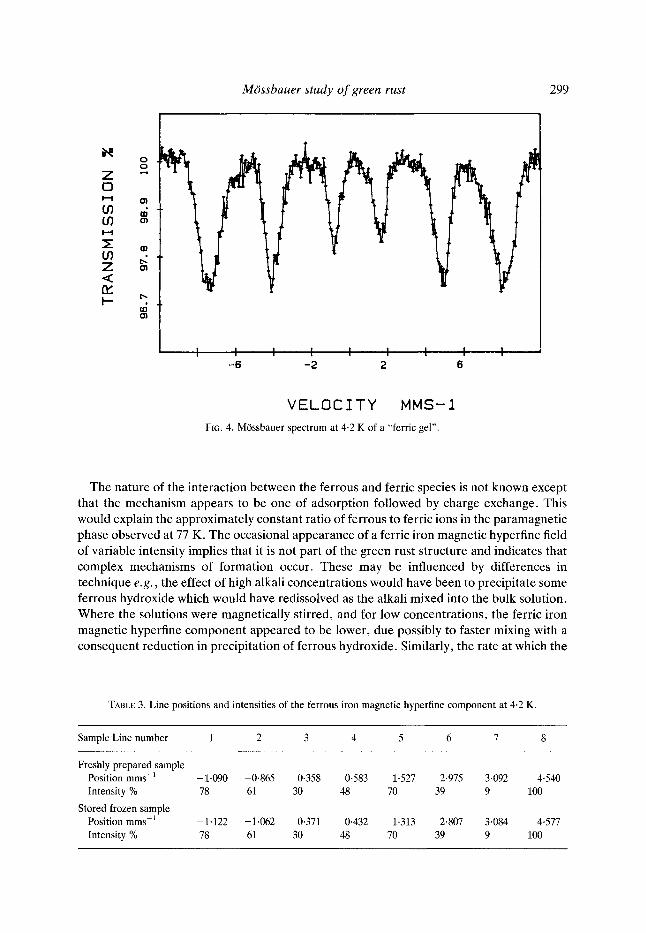
Fig. 3 shows three M6ssbauer spectra at 4.2 K. The spectrum in Fig. 3(a) was prepared at AEA Technology, Harwell and gave a magnetic hyperfine field for the ferrous iron of 12.2 T. The spectra in Fig. 3(b) and 3(c) correspond to those in Fig. 2(a) and 2(b) and gave a magnetic hyperfine field for the ferrous iron of 12-5 T. The spectrum of the sample prepared at A E A Technology was recorded immediately after preparation, whereas all other samples were prepared and stored at 77 K for a week or more before the M6ssbauer spectra were measured. It is thought that this difference is not significant as it is within experimental error although it is just possible that it resulted from ageing processes in the sample. Table 2 lists the relevant data concerning the magnetic field, the quadrupole field, the angles and the asymmetry parameter. These confirm that the ferrous iron is in an environment similar to that of ferrous hydroxide, Fe(OH)2 (Miyamoto et al., 1967; Greenwood & Gibb, 1971) and sheet-silicates (Coey, 1988). The addition of a ferrous doublet with a splitting of 3.2 mms 1
296 A . H . Cuttler e t al.
Z o H
H 5"
Z
n~
Ln
u;
I.n
0 0
0 0
( C I ) I ' ' J
Ln
~z
r
C ) I , , , I
I
( b ) , ,
I I I I I I I I I - 6 - 2 2 6
V E L O C I T Y MMS-I
FIG. 2. M6ssbauer spectra of sulphate green rusts recorded at 77 K, with initial ferrous-ferric iron ratios of (a) 40 : 1, (b) 20 : 1 and (c) a sample of dried material from (b).
MOssbauer study of green rust 297
(b) l ,
Z o
t~ Z
n, b-
0
Ul
L~
U~
d
Q 0
C) r
I
t n
O)
U~
O)
(cl i i
" [ I I
s I
a I
I I I I 1 I ~ 1 "I -- -8 -2 2 6
V E L O C I T Y MMS-I FIG. 3. MOssbauer spectra of sulphate green rusts recorded at 4-2 K, with initial ferrous-ferric iron
ratios of (a) 4:1, (b) 20 : I and (c) 40 : 1,
298 A . H . Cuttler et al.
TABLE 2. M6ssbauer parameters of the ferrous component at 4.2 K for a fresh sample, and for samples stored at 77 K.
Isomer Quadrupole Half- Magnetic Asymmetry shif t splitting width field parameter Angles Intensity
and an isomer shift of 1.25 mms -1 was necessary to fit most spectra. Whilst these values were similar to those of frozen sulphate solutions (Deszi et al., 1965), this did not appear to be the explanat ion as in most samples no such double t was observed at 77 K. This doublet has been in terpre ted as arising from a fast re laxat ion process associated with the ferrous iron magnetic hyperfine field. This relaxation would give values similar to the averages of the first four and the last four lines in Table 3 and has been shown in Fig. 3 with the other components . In some spectra where the value of x-squared remained high, it appeared that there was a broad, featureless, residual spectrum which was at t r ibuted to magnetic relaxat ion for which no model was available.
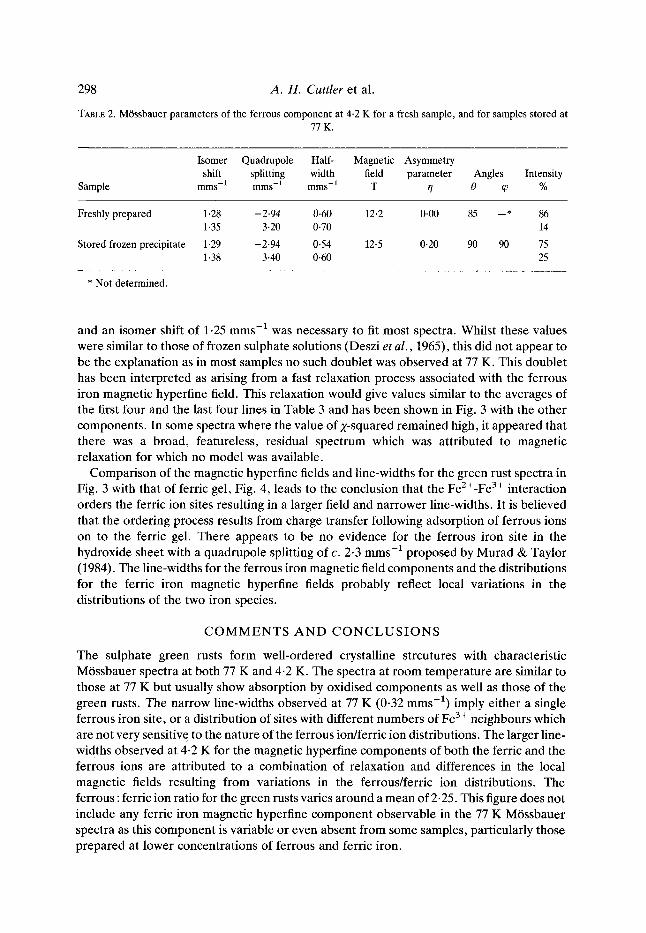
Compar ison of the magnetic hyperfine fields and line-widths for the green rust spectra in Fig. 3 with that of ferric gel, Fig. 4, leads to the conclusion that the FeZ+-Fe 3+ interact ion orders the ferric ion sites resulting in a larger field and nar rower line-widths. It is bel ieved that the order ing process results from charge transfer following adsorpt ion of ferrous ions on to the ferric gel. There appears to be no evidence for the ferrous iron site in the hydroxide sheet with a quadrupole splitting of c. 2.3 rams -1 proposed by Murad & Taylor (1984). The line-widths for the ferrous iron magnet ic field components and the distr ibutions for the ferric iron magnetic hyperfine fields probably reflect local variations in the distr ibutions of the two iron species.
C O M M E N T S A N D C O N C L U S I O N S
The sulphate green rusts form wel l -ordered crystalline strcutures with characteristic M/)ssbauer spectra at both 77 K and 4.2 K. The spectra at room tempera ture are similar to those at 77 K but usually show absorpt ion by oxidised components as well as those of the green rusts. The narrow line-widths observed at 77 K (0-32 mms -1) imply ei ther a single ferrous iron site, or a distr ibution of sites with different numbers of Fe 3+ neighbours which are not very sensitive to the nature of the ferrous ion/ferric ion distributions. The larger line- widths observed at 4.2 K for the magnetic hyperfine components of both the ferric and the ferrous ions are a t t r ibuted to a combinat ion of re laxat ion and differences in the local magnetic fields resulting from variations in the ferrous/ferric ion distributions. The ferrous : ferric ion ratio for the green rusts varies around a mean of 2.25. This figure does not include any ferric iron magnet ic hyperfine component observable in the 77 K M6ssbauer spectra as this component is variable or even absent from some samples, part icularly those p repared at lower concentrat ions of ferrous and ferric iron.
MOssbauer study of green rust 299
Z 0 H
tO lO H
Z <
0 ,-o
.J
I I I I I I I I I -6 -2 2 6
VELOCITY MMS-I
FIG. 4. MOssbauer spectrum at 4.2 K of a "ferric gel".
The nature of the interaction between the ferrous and ferric species is not known except that the mechanism appears to be one of adsorption followed by charge exchange. This would explain the approximately constant ratio of ferrous to ferric ions in the paramagnetic phase observed at 77 K. The occasional appearance of a ferric iron magnetic hyperfine field of variable intensity implies that it is not part of the green rust structure and indicates that complex mechanisms of formation occur. These may be influenced by differences in technique e.g., the effect of high alkali concentrations would have been to precipitate some ferrous hydroxide which would have redissolved as the alkali mixed into the bulk solution. Where the solutions were magnetically stirred, and for low concentrations, the ferric iron magnetic hyperfine component appeared to be lower, due possibly to faster mixing with a consequent reduction in precipitation of ferrous hydroxide. Similarly, the rate at which the
TABLE 3. Line positions and intensities of the ferrous iron magnetic hyperfine component at 4.2 K.
f e r rous i r on s o l u t i o n was t a k e n to p H 7 w o u l d h a v e a f fec ted t he q u a n t i t y of bas ic sal ts
f o r m e d . T h e s e sub t l e t i e s m a k e it i m p o s s i b l e to def ine t he p rec i se m e c h a n i s m or
m e c h a n i s m s o f f o r m a t i o n .
T h e M 6 s s b a u e r q u a d r u p o l e sp l i t t ing for t h e s u l p h a t e g r e e n rus ts a p p e a r s to b e sl ightly
l a rge r t h a n t h o s e for s imi la r g r e e n rus t s p r e p a r e d f r o m ch lo r ide a n d c a r b o n a t e so lu t ions .
W h i l s t t he p a r a m e t e r s a t 77 K m i g h t b e u s e d as a gu ide to the p r e s e n c e of g r e e n rus ts , t he
s imi lar i ty of t h e s e to o t h e r m i n e r a l s c o n t a i n i n g i r on m i g h t l imi t t h e i r appl icab i l i ty . T h e use
of d i f f e r ence t e c h n i q u e s for spec t r a of r e d u c e d a n d ox id i sed s amp le s m i g h t p r o v i d e t he
n e c e s s a r y i n f o r m a t i o n .
A C K N O W L E D G M E N T S
The authors wish to thank Mr L. Becker of AEA Technology (formerly AERE), Harwell for his assistance in running the M6ssbauer equipment and recording the spectra at 4.2 K. Dr Cuttler wishes also to acknowledge the receipt of a Nuffield Foundation Small Science Grant to cover travel and liquid helium costs.
R E F E R E N C E S
ALLMANN R. (1968) The crystal structure of pyroaurite. Acta Cryst. B24, 972-977. BERNAL J.D., DASGUPTA D.R. & McKAY A.L. (1959) The oxides and hydroxides of iron and their structural
interrelationships. Clay Miner. Bull. 4, 15-30. BR1NDLEY G.W. & B1SH D.L. (1976) Green rust; a pyroaurite type structure. Nature 273, 353. CLARK P.E., NICHOL A.W. & CARLOW J.S. (1967) A precision velocity generator for M6ssbauer experiments. J. Sci.
Inst. 44, 1001-1004. COEY J.M.D. (1988) Magnetic properties of iron in soil iron oxides and clay minerals. Pp. 397-462 in: Iron in Soils
and Clay Minerals (J.W. Stucki, B.A. Goodman & U. Schwertmann, editors). NATO ASI Series, D. Reidel Publishing Co., Dordrecht.
CUTrLER A.H., GLASSON D.R. & MAN V. (1984) Vacuum balance and related studies of green and red rusts. Thermochim. Acta 82, 231-240.
DESZI I., KESZTYHELYI L. & POCS L. (1965) M6ssbauer effect on some iron salts in ice. Phys. Letters 14, 14-16. FORSYTH J.B., HEDLEY I.G. & JOHNSON C.E. (1968) The magnetic structure and hyperfine field of goethite (0:-
FeOOH). J. Phys. C1, 179-188. GANCEDO J.R., MARTINEZ M.L. & OTON J.M. (1976) A M6ssbauer study of corrosion products of iron with
ammonium nitrate in aqueous solutions. J. Phys. 37 C6, 297-299. GREENWOOD N.N. & GISR T.C. (1971) M6ssbauer Spectroscopy, pp. 256-257. Chapman & Hall, London. KELLER G. (1948) Thesis, Bern. KUNDIG W. (1967) Evaluation of M6ssbauer Spectra for 57Fe. Nucl. Inst. Methods 48, 219-228. MAN V. (1987) The characteristics of synthetic and natural hydrous iron oxides in aqueous environments. PhD thesis,
Polytechnic South West, Plymouth, UK. McGILL I.R., MCENANEY B. & BOYLE A.J.F. (1976) Crystal structure of green rust formed by corrosion of cast iron.
Nature 259, 200-201. MISAWA T., HASHIMOTO K. & SH1MODA1RA S. (1973) Formation of the Fe(II)-Fe(III) intermediate green complex on
oxidation of ferrous iron in neutral and slightly alkaline sulphate solutions. J. Inorg. Nucl. Chem. 35, 4167-4174.
MISAWA T., HASHIMOTO K. & SHIMODAIRA S. (1974) The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature. Corrosion Sci. 14, 1311-149.
MIYAMOTO H., SHINJO T., BANDO Y. & TAKADA T. (1967) The M6ssbauer effect of iron-57 in ferrous hydroxide. J. Phys. Soc. Japan 23, 1421.
MURAD E. & TAYLOR R.M. (1984) The M6ssbauer spectra of hydroxycarbonate green rusts. Clay Miner. 19, 77-83. OLOWE A.A., G~NIN J.M.R. & BAUER P.H. (1989) MOssbauer effect evidence of a ferrous sulphate layer in the
structure of green rust 2 and its atmospheric oxidation. Hyperfine Interactions 46, 437-443. TAYLOR H.F.W. (1973) The crystal structure of some double hydroxide minerals. Mineral. Mag. 39, 377-389. TAYLOR R.M. (1980) Formation and properties of Fe(II)-Fe(III) hydroxy-carbonate and its possible significance in
soil formation. Clay Miner. 15, 369-382.
MOssbauer study o f green rust 301
TAYLOR R.M. & MCKENZIE R.M. (1980) The influence of aluminium on iron oxides. VI. The formation of Fe(II)-AI(III) hydroxy-chlorides, -sulfates and -carbonates as new members of the pyroaurite group and their significance in soils. Clays Clay Miner. 28, 179-187.
VAN DONGEN TORMAN J., JAGANNATHAN R. • TROOSTER J.M. (1975) Analysis of 57Fe MOssbauer hyperfine spectra. Hyperfine Interactions 1, 135-144.