A Mutation in the �3 Subunit of the Cardiac SodiumChannel Associated With Brugada ECG Phenotype
Dan Hu, MD, PhD; Hector Barajas-Martinez, PhD; Elena Burashnikov, BS; Michael Springer, MD;Yuesheng Wu, MD; Andras Varro, MD, PhD; Ryan Pfeiffer, BS; Tamara T. Koopmann, PhD;
Jonathan M. Cordeiro, PhD; Alejandra Guerchicoff, PhD;Guido D. Pollevick, PhD; Charles Antzelevitch, PhD
Background—Brugada syndrome, characterized by ST-segment elevation in the right precordial ECG leads and thedevelopment of life-threatening ventricular arrhythmias, has been associated with mutations in 6 different genes. Weidentify and characterize a mutation in a new gene.
Methods and Results—A 64-year-old white male displayed a type 1 ST-segment elevation in V1 and V2 duringprocainamide challenge. Polymerase chain reaction–based direct sequencing was performed using a candidate geneapproach. A missense mutation (L10P) was detected in exon 1 of SCN3B, the �3 subunit of the cardiac sodium channel,but not in any other gene known to be associated with Brugada syndrome or in 296 controls. Wild-type (WT) and mutantgenes were expressed in TSA201 cells and studied using whole-cell patch-clamp techniques. Coexpression ofSCN5A/WT�SCN1B/WT�SCN3B/L10P resulted in an 82.6% decrease in peak sodium current density, acceleratedinactivation, slowed reactivation, and a �9.6-mV shift of half-inactivation voltage compared with SCN5A/WT�SCN1B/WT�SCN3B/WT. Confocal microscopy revealed that SCN5A/WT channels tagged with green fluorescent protein arelocalized to the cell surface when coexpressed with WT SCN1B and SCN3B but remain trapped in intracellularorganelles when coexpressed with SCN1B/WT and SCN3B/L10P. Western blot analysis confirmed the presence ofNaV�3 in human ventricular myocardium.
Conclusions—Our results provide support for the hypothesis that mutations in SCN3B can lead to loss of transport andfunctional expression of the hNav1.5 protein, leading to reduction in sodium channel current and clinical manifestationof a Brugada phenotype. (Circ Cardiovasc Genet. 2009;2:270-278.)
Key Words: Brugada syndrome � arrhythmia � ion channels � SCN3B � protein trafficking
Brugada syndrome (BrS) is a cardiac channelopathycharacterized by ST-segment elevation or the appear-
ance of accentuated J waves in the right precordial leads (V1to V3) of the ECG and the development of life-threateningpolymorphic ventricular tachycardia. The ECG characteris-tics of the BrS are dynamic and often concealed but can beunmasked by potent sodium channel blockers.1,2
Clinical Perspective on p 278BrS has been associated with mutations in 6 different
genes. Mutations in SCN5A (Nav1.5, BrS1) have been re-ported in 11% to 28% of BrS probands, CACNA1C (Cav1.2,BrS3) in 6.7%, CACNB2b (Cavß2b, BrS4) in 4.8% andmutations in glycerol-3-phophate dehydrogenase 1-like en-zyme gene (GPD1L, BrS2), �1-subunit of sodium channel(SCN1B, BrS5), and MiRP2 (KCNE3; BrS6) are much morerare.3–8 These genetic defects lead to development of BrS
secondary to either a loss of function of sodium (INa) orL-type calcium (ICa) channel current, or a gain of function oftransient outward current (Ito). Thus, �72% of BrS probandsremain genotype negative. Here, we report the identificationof another gene associated with the BrS phenotype caused byloss of function of INa secondary to a mutation in the ß3-subunitof the cardiac sodium channel, encoded by SCN3B.
Methods
Mutation AnalysisGenomic DNA was prepared from peripheral blood lymphocytes ofthe patient. Genomic DNA was extracted from peripheral bloodleukocytes using a commercial kit (Gentra System, Puregene, Va-lencia, Calif). All known exons of the Brugada-susceptibility genes(SCN5A, IRX5, SCN1B, SCN3B, CaCNB2B, CACNA1C, KCNE2,KCNE3, and GPD1L) were amplified with intronic primers andsequenced in both directions to probe for mutations, with the use of
Received October 15, 2008; accepted April 20, 2009.From the Masonic Medical Research Laboratory (D.H., H.-B.M., E.B., Y.W., R.P., J.M.C., A.G., G.D.P., C.A.), Utica, NY; Cardiology Department
(M.S.), University of Louisville, Louisville, Ky; Heart Failure Research Center (T.T.K.), Academic Medical Center Amsterdam, The Netherlands;Department of Pharmacology and Pharmacotherapy (A.V.), University of Szeged, Szeged, Hungary; and Division for Cardiovascular Pharmacology(A.V.), Hungarian Academy of Sciences, Szeged, Hungary.
an ABI PRISM 3100-Avant Automatic DNA sequencer (AppliedBiosystem, Foster City, Calif). The sequence primers of SCN3B areshown in Table 1 (reference sequence, NM_018400). One hundredtwenty individuals, matched by race and ethnic background, with nohistory of cardiac arrhythmias were used as controls.
Site-Directed Mutagenesis and Transfection of theTSA201 Cell LineFor patch-clamp study, site-directed mutagenesis was performedwith QuikChange (Stratagene, La Jolla, Calif) on full-length humanwild-type (WT) and mutant SCN3B cDNA cloned in pIRES2-DsRed-Express (RFP) vector, the WT SCN1B cloned in pIRES2-AcGFP1 vector, and the WT SCN5A cloned in pcDNA3.1. SCN3Bwas a gift from Dr Takashi Tokino (Japan). In the case of traffickingstudies, human WT SCN5A cDNA cloned in pcDNA3.1 with fusiongreen fluorescent protein (GFP) at the C-terminus was coexpressedwith, the WT SCN1B cloned in pRC-CMV, and WT and mutatedSCN3B cloned in pIRES2 with RFP. The mutated plasmid wassequenced to ensure the presence of the mutation without spurioussubstitutions.
Sodium channels were expressed in a modified human embryonickidney cell line, TSA201. Briefly, transient transfection usingfugene6 (Roche Diagnostics, Indianapolis, Ind) was carried out withSCN5A, SCN1B, and SCN3B (WT or mutant) with a molar ratio of1:1:1. The cells were grown in GIBCO Dulbecco’s minimumessential medium (No. 10566, Gibco, Invitrogen, Carlsbad, Calif)with FBS (No. 16000) and antibiotics (No. 15140) on polylysinecoated 35-mm culture dishes (Cell�, Sarstedt, Newton, NC). Cellswere placed in a 5% CO2 incubator at 37°C for 24 to 48 hours beforepatch-clamp study. It is noteworthy that previous studies havedemonstrated that no endogenous SCN5A, and its �-subunits areexpressed in the TSA201 cell line.9
Patch-Clamp MethodMembrane currents were measured using whole-cell patch-clamptechniques. All recordings were obtained at room temperature (20°Cto 22°C) using an Axopatch 200B amplifier equipped with aCV-201A head stage (Axon Instruments Inc, Foster City, Calif).Measurements were started 10 minutes after obtaining the whole-cellconfiguration to allow the current to stabilize. The holding potentialwas maintained at �120 mV. Macroscopic whole cell Na� currentwas recorded by using bath solution perfusion containing (inmillimoles per liter) 130 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2, 2.8sodium acetate, 10 HEPES, 10 glucose (pH 7.3 with NaOH).Tetraethylammonium chloride (5 mmol/L) was added to the buffer toblock TEA-sensitive native currents. Patch pipettes were fabricatedfrom 1.5-mm OD borosilicate glass capillaries (Fisher Scientific,Pittsburgh, Pa). They were pulled using a gravity puller (ModelPP-830, Narishige Corp, Japan) to obtain resistances between 0.8and 2.8 mol/L � when filled with a solution containing (in mmol/L)5 NaCl, 5 KCl, 130 CsF, 1.0 MgCl2, 5 EGTA, 10 HEPES, and 5TEA (pH 7.2 with CsOH). Currents were filtered with a 4 poleBessel filter at 5 kHz and digitized at 50 kHz. Series resistance waselectronically compensated at �80%.
INa was elicited by depolarizing pulses ranging from �90 to �30mV in 5-mV increments with a holding potential of �120 mV. Peakcurrents were measured and INa densities (pA/pF) were obtained bydividing the peak INa by the cell capacitance obtained. Activation
properties were determined from I/V relationships by normalizingpeak INa to driving force and maximal INa, and plotting normalizedconductance versus Vm. Voltage-dependence of steady-state inacti-vation was obtained by plotting the normalized peak current (40-mstest pulse to �20 mV after a 1000-ms conditioning pulse from �140to �60 mV with the holding potential of �120 mV) versus Vm. Theactivation and steady-state inactivation curves were fitted to the Boltz-mann equation, I/Imax�1/(1�exp((V�V1/2)/k), to determine themembrane potential for half-maximal inactivation V1/2 and the slopefactor k. Pulses for recovery from inactivation were of 20-msduration. Peak current elicited during the second pulse was normal-ized to the value obtained during the initial test pulse. It wasanalyzed by fitting data to a double exponential function: I(t)/Imax�Af�(1�exp(�t/�f))�As�(1�exp(�t/�s)), where Af and As arethe fractions of fast and slow inactivating components, respectively,and �f and �s are their time constants.
All data acquisition and analysis were performed using pCLAMPversion 9.2 (Axon Instruments), Excel (Microsoft, Redmond, Wash),and Origin 7.5 (Microcal Software, Northampton, Mass).
Localization of Na� ChannelsWe assessed channel trafficking using Na� channels �-subunit(SCN5A) tagged with GFP. Confocal microscopy was used tolocalize the channels and identify trafficking defects. Briefly, cellswere grown on polylysine-coated glass bottom 35-mm culture dishesand studied 48 hours posttransfection. Experiments were performedon an Olympus FluoView laser-scanning confocal microscope(Olympus, Orangeburg, NY) and images were acquired with Fluo-View acquisition software program on a personal computer. GFP-labeled cells were analyzed in the xyz configuration. An argon laserprovided the excitation light at 488 nm and the emission light wascollected at 520 nm in photomultiplier tube 1. Transmission imagewas acquired in photomultiplier tube 2. Fluorescence signals werecollected with either a 40� or a 60� oil-immersion objective lens.xy frame was set to 512�512 pixels and laser intensity was set to 6%power. The z axis was changed in �0.50-�m increments bycomputer control through the entire volume of the cell. To quantifythe membrane expression of Nav1.5 fluorescence intensity at theplasma membrane region (2 �m) and the entire cell area in themiddle xy image of the z series stack was measured and the ratio ofperipheral to total cell area fluorescence was calculated. Analysis ofGFP-labeled cells was performed using both FluoView and Image Jsoftware.
Western Blot AnalysisMembrane proteins (50 �g/lane, except for TSA201-SCN3B 0.5�g/lane) were run on 5% to 15% gradient linear SDS-PAGE.BIO-RAD 161 to 0374 Precision Plus Protein Dual Color Standards10 �/lane was used as a molecular marker reference. Rabbitanti-human SCN3B polyclonal antibody at 1:500 (ab48552, Abcam,Cambridge, Mass) was used to detect bands in membrane proteinsfrom untransfected TSA201 cells (negative control), SCN3B trans-fected TSA201 cells (positive control), and from 2 human leftventricular samples.
Statistical AnalysisData are presented as mean�SEM. A 2-tailed Student t test was usedfor statistical comparison of 2 groups and ANOVA coupled withStudent-Newman-Keuls test for comparison of 3 or more groups(SigmaStat, Jandel Scientific Software, San Rafael, Calif). Differ-ences were considered statistically significant at a value of P�0.05.
ResultsIndex CaseThe study was approved by the regional institutional reviewboard. The proband, a 64-year old white man (German,Swedish, and Native American descent), presented with aresting ECG displaying a slight ST-segment elevation and
Table 1. Oligonucleotide Primers for Genetic Analysis ofNav�3 Subunits
Exon Forward Primer (5 to 3) Reverse Primer (5 to 3)
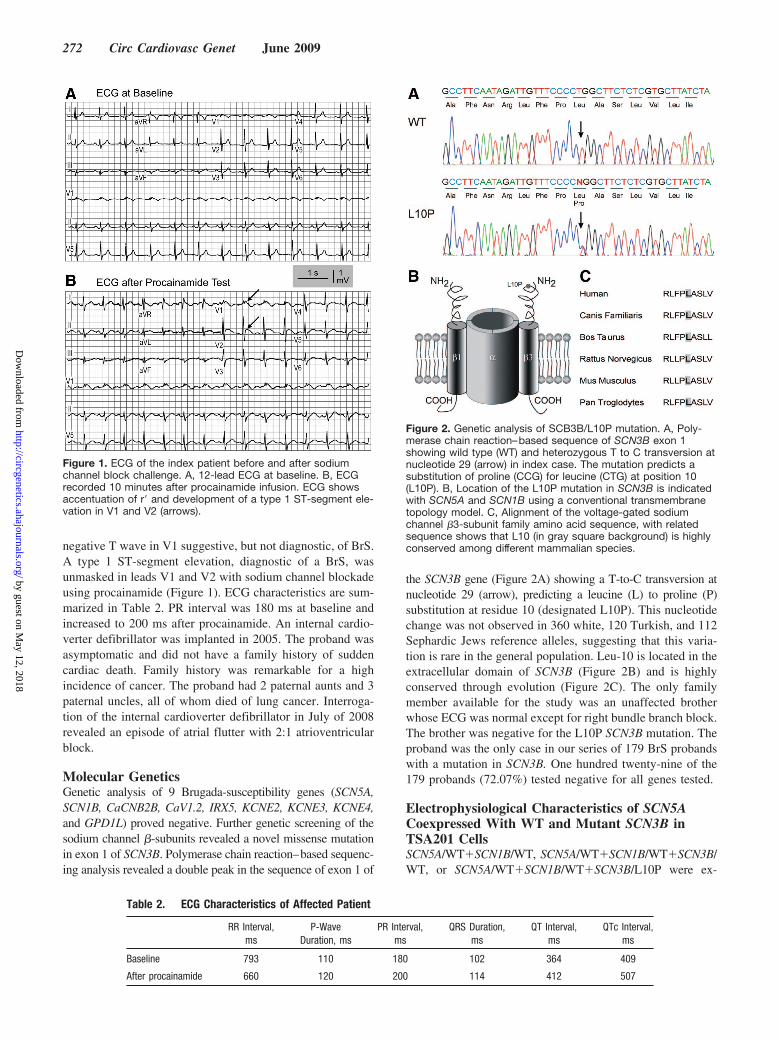
negative T wave in V1 suggestive, but not diagnostic, of BrS.A type 1 ST-segment elevation, diagnostic of a BrS, wasunmasked in leads V1 and V2 with sodium channel blockadeusing procainamide (Figure 1). ECG characteristics are sum-marized in Table 2. PR interval was 180 ms at baseline andincreased to 200 ms after procainamide. An internal cardio-verter defibrillator was implanted in 2005. The proband wasasymptomatic and did not have a family history of suddencardiac death. Family history was remarkable for a highincidence of cancer. The proband had 2 paternal aunts and 3paternal uncles, all of whom died of lung cancer. Interroga-tion of the internal cardioverter defibrillator in July of 2008revealed an episode of atrial flutter with 2:1 atrioventricularblock.
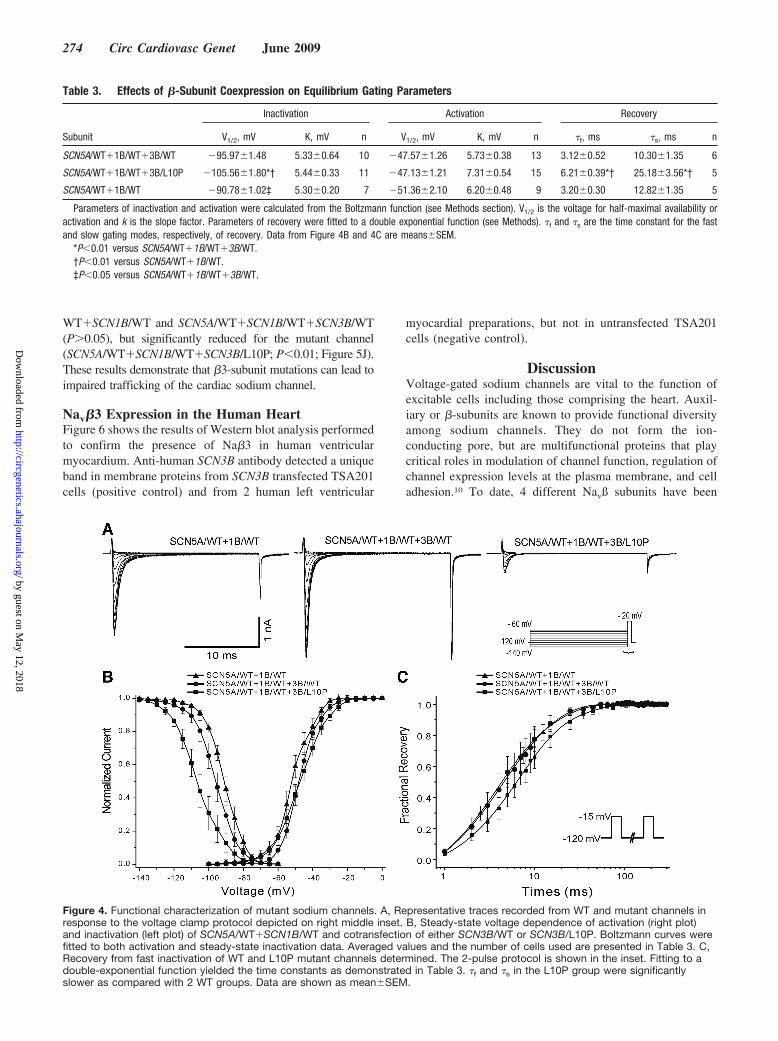
Molecular GeneticsGenetic analysis of 9 Brugada-susceptibility genes (SCN5A,SCN1B, CaCNB2B, CaV1.2, IRX5, KCNE2, KCNE3, KCNE4,and GPD1L) proved negative. Further genetic screening of thesodium channel �-subunits revealed a novel missense mutationin exon 1 of SCN3B. Polymerase chain reaction–based sequenc-ing analysis revealed a double peak in the sequence of exon 1 of
the SCN3B gene (Figure 2A) showing a T-to-C transversion atnucleotide 29 (arrow), predicting a leucine (L) to proline (P)substitution at residue 10 (designated L10P). This nucleotidechange was not observed in 360 white, 120 Turkish, and 112Sephardic Jews reference alleles, suggesting that this varia-tion is rare in the general population. Leu-10 is located in theextracellular domain of SCN3B (Figure 2B) and is highlyconserved through evolution (Figure 2C). The only familymember available for the study was an unaffected brotherwhose ECG was normal except for right bundle branch block.The brother was negative for the L10P SCN3B mutation. Theproband was the only case in our series of 179 BrS probandswith a mutation in SCN3B. One hundred twenty-nine of the179 probands (72.07%) tested negative for all genes tested.
Electrophysiological Characteristics of SCN5ACoexpressed With WT and Mutant SCN3B inTSA201 CellsSCN5A/WT�SCN1B/WT, SCN5A/WT�SCN1B/WT�SCN3B/WT, or SCN5A/WT�SCN1B/WT�SCN3B/L10P were ex-
Figure 1. ECG of the index patient before and after sodiumchannel block challenge. A, 12-lead ECG at baseline. B, ECGrecorded 10 minutes after procainamide infusion. ECG showsaccentuation of r and development of a type 1 ST-segment ele-vation in V1 and V2 (arrows).
Table 2. ECG Characteristics of Affected Patient
RR Interval,ms
P-WaveDuration, ms
PR Interval,ms
QRS Duration,ms
QT Interval,ms
QTc Interval,ms
Baseline 793 110 180 102 364 409
After procainamide 660 120 200 114 412 507
Figure 2. Genetic analysis of SCB3B/L10P mutation. A, Poly-merase chain reaction–based sequence of SCN3B exon 1showing wild type (WT) and heterozygous T to C transversion atnucleotide 29 (arrow) in index case. The mutation predicts asubstitution of proline (CCG) for leucine (CTG) at position 10(L10P). B, Location of the L10P mutation in SCN3B is indicatedwith SCN5A and SCN1B using a conventional transmembranetopology model. C, Alignment of the voltage-gated sodiumchannel �3-subunit family amino acid sequence, with relatedsequence shows that L10 (in gray square background) is highlyconserved among different mammalian species.
pressed in TSA201 cells to assess the effects of the mutation onsodium channel function. Figure 3A shows macroscopic cur-rents recorded from these channels together with the current-voltage relationships. Maximum peak inward current oc-curred at a potential of �35 mV for all channel types.Coexpression of SCN3B/WT with SCN5A/WT�SCN1B/WTincreased peak current density from �281.3�62.3 pA/pF to�402.8�93.2 pA/pF (n�9 and 13, respectively; P�0.05between 2 groups). Coexpression of SCN3B/L10P resulted ina marked decrease in peak sodium current density to�70.2�14.5 pA/pF (n�25; 17.4% of SCN5A/WT�SCN1B/WT�SCN3B/WT and 25.0% of SCN5A/WT�SCN1B/WTcurrent density; P�0.05 for each; Figure 3B). Coexpressionof SCN3B/L10P produced total loss of function in 40% ofcells studied (10 of 25 cells, Figure 3A).
The half-inactivation voltage (V1/2) of mutant INa channels(SCN5A/WT�SCN1B/WT�SCN3B/L10P) was 14.8 and 9.6mV more negative than those of SCN5A/WT�SCN1B/WTand SCN5A/WT�SCN1B/WT�SCN3B/WT channels, respec-tively (P�0.01, respectively; Table 3 and Figure 4B). Steady-state activation, obtained after applying the step protocol in insetof Figure 3A, was similar among the 3 groups (Figure 4B).Recovery from inactivation, measured using a standard double-paired pulse protocol, was similar in the 2 control groups but
slower in the mutant channels (P�0.01, respectively; Table 3and Figure 4C). The L10P mutation caused a shift in thevoltage dependence of steady-state inactivation and slowedrecovery from inactivation, thus serving to further reducesodium channel availability.
Trafficking of SCN5A Coexpressed WithSCN3B/L10P Mutant in TSA201 CellsTo evaluate whether the loss of function caused by theSCN3B/L10P mutation is due in part to a trafficking defect, westudied GFP-fusion-SCN5A/WT coexpressed with SCN1B/WTalone, or combined with either SCN3B/WT or SCN3B/L10P. xyzscans of SCN5A/WT�SCN1B/WT on the confocal microscoperevealed both a central and peripheral pattern of staining sug-gesting localization of the channel in the cell membrane andintracellular organelles (Figure 5A through 5C). Protein expres-sion of SCN5A/WT channels was enhanced when bothSCN1B/WT and SCN3B/WT were added (Figure 5D through5F). In contrast, SCN3B/L10P resulted in internal stainingconsistent with intracellular compartmentalization with noevidence of plasma-member staining (Figure 5G through 5I),suggesting that channels were trapped in the endoplasmicreticulum and/or Golgi complex. The ratio of peripheral tototal cell area fluorescence intensity was similar for SCN5A/
Figure 3. Effect of SCN3B mutation on sodiumchannel current recorded in TSA201 cells. A,Representative sodium current traces inTSA201 cells expressing SCN5A/WT�SCN1B/WTalone or cotransfected with SCN3B/WT or SCN3B/L10P. All were recorded 48 hours after transfection.Upper right and left panel shows WT ofSCN5A�SCN1B and SCN5A�SCN1B�SCN3B.Two SCN3B/L10P traces are shown in thelower grouping, 1 in which there was partialloss of function (1) and another in which therewas total loss of function (2). The inset showsthe voltage-clamp protocol employed. B,Current-voltage relationship for SCN5A/WT�SCN1B/WT (n�9), SCN5A/WT�SCN1B/WT�SCN3B/WT (n�13), and SCN5A/WT�SCN1B/WT�SCN3B/L10P (n�25) current den-sity. Peak current density was significantlyreduced for SCN5A/WT�SCN1B/WT�SCN3B/L10P when compared with SCN5A/WT�SCN1B/WT and SCN5A/WT�SCN1B/WT�SCN3B/WT (values are mean�SEM;P�0.01 for each).
WT�SCN1B/WT and SCN5A/WT�SCN1B/WT�SCN3B/WT(P0.05), but significantly reduced for the mutant channel(SCN5A/WT�SCN1B/WT�SCN3B/L10P; P�0.01; Figure 5J).These results demonstrate that �3-subunit mutations can lead toimpaired trafficking of the cardiac sodium channel.
Nav�3 Expression in the Human HeartFigure 6 shows the results of Western blot analysis performedto confirm the presence of Na�3 in human ventricularmyocardium. Anti-human SCN3B antibody detected a uniqueband in membrane proteins from SCN3B transfected TSA201cells (positive control) and from 2 human left ventricular
myocardial preparations, but not in untransfected TSA201cells (negative control).
DiscussionVoltage-gated sodium channels are vital to the function ofexcitable cells including those comprising the heart. Auxil-iary or �-subunits are known to provide functional diversityamong sodium channels. They do not form the ion-conducting pore, but are multifunctional proteins that playcritical roles in modulation of channel function, regulation ofchannel expression levels at the plasma membrane, and celladhesion.10 To date, 4 different Navß subunits have been
Table 3. Effects of �-Subunit Coexpression on Equilibrium Gating Parameters
Parameters of inactivation and activation were calculated from the Boltzmann function (see Methods section). V1/2 is the voltage for half-maximal availability oractivation and k is the slope factor. Parameters of recovery were fitted to a double exponential function (see Methods). �f and �s are the time constant for the fastand slow gating modes, respectively, of recovery. Data from Figure 4B and 4C are means�SEM.
*P�0.01 versus SCN5A/WT�1B/WT�3B/WT.†P�0.01 versus SCN5A/WT�1B/WT.‡P�0.05 versus SCN5A/WT�1B/WT�3B/WT.
Figure 4. Functional characterization of mutant sodium channels. A, Representative traces recorded from WT and mutant channels inresponse to the voltage clamp protocol depicted on right middle inset. B, Steady-state voltage dependence of activation (right plot)and inactivation (left plot) of SCN5A/WT�SCN1B/WT and cotransfection of either SCN3B/WT or SCN3B/L10P. Boltzmann curves werefitted to both activation and steady-state inactivation data. Averaged values and the number of cells used are presented in Table 3. C,Recovery from fast inactivation of WT and L10P mutant channels determined. The 2-pulse protocol is shown in the inset. Fitting to adouble-exponential function yielded the time constants as demonstrated in Table 3. �f and �s in the L10P group were significantlyslower as compared with 2 WT groups. Data are shown as mean�SEM.
described (SCN1B, SCN2B, SCN3B, and SCN4B),11–15 andshown to play a critical role in cell adhesion, signal trans-duction, channel expression at the plasma membrane, andvoltage dependence of channel gating.16–19 All are detectablein cardiac tissue.9 �1A, a splice variant of SCN1B, isexpressed in embryonic brain and adult heart in rat.20 Thedistribution and expression level of sodium channel �- and�-subunits in human and canine hearts has not been not wellcharacterized.
Sodium Channel �-Subunits Modulate SodiumChannel in HeartBecause of their significant role in modulating channelexpression and function, genes that encode cardiac channel�-subunit proteins are attractive candidates for ion chan-nelopathies like BrS.21 The role of �1-subunits have been
studied most extensively. �1-coexpression has been reportedto have no observable effect on SCN5A function,22,23 to resultin increased sodium current density with no detectable effectson channel kinetics or voltage dependence,24,25 to modulatechannel sensitivity to lidocaine block with subtle changes inchannel kinetics and gating properties26 as well as to shift thevoltage dependence of steady-state inactivation27–29 or alterthe rate of recovery from inactivation.29,30
Previous studies involving coexpression of SCN5A with �3have reported (1) increased current density, a depolarizingshift in the voltage dependence of inactivation, and anincreased rate of recovery from inactivation in Xenopusoocytes,30 or (2) a hyperpolarizing shift of inactivation,slowed recovery from inactivation, and reduced late sodiumchannel current, without any change in peak current densityin Chinese hamster ovary-K1 cells.29 This study involving
Figure 5. Trafficking of GFP-tagged SCN5Achannel proteins. TSA201 cells were trans-fected with GFP-tagged-SCN5A/WT cotrans-fected with SCN1B/WT and either SCN3B/WTor SCN3B/L10P. All GFP signals were recorded48 hours after transfection. Photomicrographsshow the phase contrast light transmissionimage (left), stacked serial confocal fluorescentimage (center), and a single fluorescent confo-cal image from the center of the same TSA201cell. A through C, SCN1B/WT�SCN5A/WT-GFP; staining in evident in both the peripheryand the center of the cell, suggesting thatSCN5A channels are trafficked to the cell mem-brane. D through F, Cotransfected withSCN1B/WT and SCN3B/WT; expression ofSCN5A-GFP channels is enhanced throughout.G through I, Cotransfected with SCN3B/L10P;fluorescence is localized in the perinuclearregion of the cell suggesting failure of mutantchannels to traffic to the plasma membrane. J,Quantitative analysis of relative level of mem-brane expression of enhanced GFP-taggedSCN5A in the plasma membrane. Fluorescenceintensity of SCN5A staining of the plasmamembrane (outermost 2 �m) is plotted as aratio of total cell area fluorescence. The ratiowas similar for SCN5A/WT�SCN1B/WT (n�22)and SCN5A/WT�SCN1B/WT�SCN3B/WT(n�26), but much reduced for SCN5A/WT�SCN1B/WT�SCN3B/L10P (n�44).**P�0.01, mean�SEM.
TSA201 cells, shows that coexpression of �3 with SCN5Aand �1 increases peak sodium current density, shifted thevoltage dependence of channel availability in a hyperpolar-izing direction without significantly changing the voltagedependence of activation or recovery kinetics (Figures 3 and4, Table 3).
Sodium Channel �-Subunits and ArrhythmiasAlthough mutations in Nav1 �-subunits have been associatedwith inherited diseases, including LQT3, BrS, progressiveconduction disease, and atrial standstill,31 to date only 2 genesencoding sodium channel �-subunits have been associatedwith human cardiac disease.7,32
It has long been appreciated that SCN5A mutations asso-ciated with LQTS and BrS are modulated by coexpression ofSCN1B.33 BrS is known to be caused by a reduction in INa.The role of �1-subunits to exacerbate the loss of functionproduced by R1232W/T1620 mol/L mutations in SCN5A inpatients with BrS was demonstrated by Wan et al.34
Mutations in �1-subunits (Nav�1 and Nav�1b) have re-cently been shown to be associated with a combined BrS andcardiac conduction disease phenotype in humans.7 Mutationin �4-subunit have been reported to be associated with anincrease in late INa giving rise to the LQT3 variant of thelong-QT syndrome.32
In this study, we provide evidence that SCN3B is aBrS-susceptibility gene. An L10P missense mutation in ahighly conserved residue, absent in 240 reference alleles, isshown to produce a major reduction in INa secondary to bothfunctional and trafficking defects in cardiac sodium channelexpression. It is well established that there are 4 fullyconserved cysteine residues, labeled C2, C21, C24, and C96in ß1- and ß3-subunits.15 The disulfide bonds between C2 andC24, C21 and C96 are believed to correspond to the interac-tion site for �-subunit association. Moreover, the former bondis also responsible for forming the Ig fold for ß1 and ß3, adisruption of which can cause an inherited epilepsy syn-drome. Our mutation (L10P) is located near C2, which may
affect the interaction of the ß3-subunit with the sodiumchannel complex, and lead to the phenomenon that weobserved. Little is known about cardiac sodium channel �-and �-subunits trafficking in vivo. A study using overexpres-sion of fluorescent-tagged SCN5A, �1, and �2 in HEK293cells suggested that SCN5A and �2 are transported separatelyto the plasma membrane whereas SCN5A and �1 form acomplex in the endoplasmic reticulum that may facilitateplasma membrane trafficking.35 In PC12 and Chinese hamsterovary-K1/Nav1.5 cells, enhanced GFP-tagged �extracellulardomain (extracellular domain deletion) �3 mutant showedinternal staining with little plasma membrane staining, andenhanced GFP-tagged �internal cardioverter defibrillator (in-tracellular domain deletion) �3-mutant showed no evidenceof surface staining but labeled an internal highly reticulatedcompartment that suggests endoplasmic reticulum. Theseresults indicate that a mutation in the extracellular domaincan impair trafficking of SCN3B to the membrane and thatdeletion of the intracellular domain totally disrupts traffickingof the �-subunit. Our results suggest that WT �3 plays a rolein facilitating SCN5A transport to the plasma membrane,because a mutation in the extracellular domain of �3 iscapable of disrupting trafficking of SCN5A to the plasmamembrane.
SCN3B and CancerAn interesting aspect of the family studied was that manyfamily members died of lung cancer. SCN3B levels areupregulated in human cancer cell lines by DNA damagingagents, as well as by overexpression of tumor suppressor p53,a transcription factor that induces growth arrest and/or apo-ptosis in response to cellular stress. Introduction of theSCN3B gene into T98G and Saos2 cells potently suppressedcolony formation, and adenovirus-mediated transfer ofSCN3B induced apoptosis when combined with anticanceragents. These results suggest that SCN3B mediates a p53-dependent apoptotic pathway and may be a candidate forgene therapy combined with anticancer drugs.36 A morerecent study discovered that SCN3B is a candidate cancergene, which could affect ion-channel transport.37 From theabove point of view, it is tempting to speculate that the L10Pmutation in SCN3B may also promote cancer and thuscontribute to death of mutant carriers in this family. Thishypothesis remains to be tested.
Limitations of the StudyA genotype-phenotype correlation between the �3-mutationand BrS phenotype is hampered by the high incidence ofcancer deaths in this family, which may be caused by thesame mutation in the ß3-subunit. Only 2 family memberswere available for study. A native American cohort was notavailable for the study as a control group.
Another limitation of the present study is the fact thatcharacterization of the SCN3B mutation were carried out in aheterologous mammalian expression system, creating condi-tions that may be different from those encountered in vivo asfar as a contribution of ß2- or ß4-subunits, or other compo-nents of the Na channel macromolecular complex. Despitethese limitations, the electrophysiological characteristics of
Figure 6. Nav�3 expression in human ventricular myocardium.Rabbit anti-human SCN3B antibody detects bands in mem-brane proteins from SCN3B transfected TSA201 cells (positivecontrol) and from 2 human left ventricular myocardial prepara-tions, but not in untransfected TSA201 cells (negative control).
the mutant channel are concordant with the BrS phenotype,and in combination with the clinical data supports a causalrelationship between the L10P mutation in SCN3B and thedisease.
Summary and ConclusionOur results provide evidence in support of the hypothesis thatmutations in the SCN3B-encoded Nav�3 subunit constituteanother pathogenic mechanism responsible for developmentof the BrS phenotype secondary to a loss of function ofcardiac sodium channel current.
AcknowledgmentsWe thank Judy Hefferon and Robert J. Goodrow, Jr, for technicalassistance and Susan Bartkowiak for maintaining our genetic database.
Sources of FundingThis work was supported by grant HL 47678 (to C.A.) from theNational Institutes of Health and the Grand Lodges of New YorkState and Florida.
DisclosuresNone.
References1. Brugada P, Brugada J. Right bundle branch block, persistent ST segment
elevation and sudden cardiac death: a distinct clinical and electrocardio-graphic syndrome: a multicenter report. J Am Coll Cardiol. 1992;20:1391–1396.
2. Antzelevitch C. Brugada syndrome. PACE. 2006;29:1130–1159.3. Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza
D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Ant-zelevitch C, O’Brien RE, Schultze-Bahr E, Keating MT, Towbin JA,Wang Q. Genetic basis and molecular mechanisms for idiopathic ven-tricular fibrillation. Nature. 1998;392:293–296.
4. Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C,Borggrefe M, Haverkamp W. Sodium channel gene (SCN5A) mutationsin 44 index patients with Brugada syndrome: different incidences infamilial and sporadic disease. Hum Mutat. 2003;21:651–652.
5. London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S,Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ,Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, WeissR, Dudley SC Jr. Mutation in glycerol-3-phosphate dehydrogenase 1 likegene (GPD1-L) decreases cardiac Na� current and causes inherited ar-rhythmias. Circulation. 2007;116:2260–2268.
6. Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC,Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P,Bonaros EP Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Ober-heiden R, Bhatia A, Hsu LF, Haissaguerre M, Schimpf R, Borggrefe M,Wolpert C. Loss-of-function mutations in the cardiac calcium channelunderlie a new clinical entity characterized by ST-segment elevation,short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449.
8. Delpon E, Cordeiro JM, Nunez L, Thomsen PEB, Guerchicoff A, Pol-levick GD, Wu Y, Kanters JK, Larsen CT, Burashnikov A, ChristiansenM, Antzelevitch C. Functional effects of KCNE3 mutation and its role inthe development of Brugada syndrome. Circ Arrhythmia Electrophysiol.2008;1:209–218.
9. Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T,Catterall WA. Distinct subcellular localization of different sodiumchannel a and b subunits in single ventricular myocytes from mouse heart.Circulation. 2004;109:1421–1427.
10. Isom LL, De Jongh KS, Catterall WA. Auxiliary subunits ofvoltage-gated ion channels. Neuron. 1994;12:1183–1194.
11. McClatchey AI, Cannon SC, Slaugenhaupt SA, Gusella JF. The cloningand expression of a sodium channel beta 1-subunit cDNA from humanbrain. Hum Mol Genet. 1993;2:745–749.
12. Isom LL, De Jongh KS, Patton DE, Reber BFX, Offord J, CharbonneauH, Walsh K, Goldin AL, Catterall WA. Primary structure and functionalexpression of the b1 subunit of the rat brain sodium channel. Science.1992;256:839–842.
13. Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF,Scheuer T, Catterall WA. Structure and function of the b 2 subunit ofbrain sodium channels, a transmembrane glycoprotein with a CAM motif.Cell. 1995;83:433–442.
14. Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D,Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, CurtisR. Sodium channel beta4, a new disulfide-linked auxiliary subunit withsimilarity to beta2. J Neurosci. 2003;23:7577–7585.
15. Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD,Hughes J, Richardson PJ, Mizuguchi K, Jackson AP. beta 3: an additionalauxiliary subunit of the voltage-sensitive sodium channel that modulateschannel gating with distinct kinetics. Proc Natl Acad Sci USA. 2000;97:2308–2313.
16. Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL. Sodium channelbeta subunits mediate homophilic cell adhesion and recruit ankyrin topoints of cell-cell contact. J Biol Chem. 2000;275:11383–11388.
17. Meadows L, Malhotra JD, Stetzer A, Isom LL, Ragsdale DS. The intra-cellular segment of the sodium channel beta 1 subunit is required for itsefficient association with the channel alpha subunit. J Neurochem. 2001;76:1871–1878.
18. Patton DE, Isom LL, Catterall WA, Goldin AL. The adult rat brain beta1 subunit modifies activation and inactivation gating of multiple sodiumchannel alpha subunits. J Biol Chem. 1994;269:17649–17655.
19. Isom LL. Sodium channel b subunits: anything but auxiliary. Neuroscientist.2001;7:42–54.
20. Kazen-Gillespie KA, Ragsdale DS, D’Andrea MR, Mattei LN, RogersKE, Isom LL. Cloning, localization, and functional expression of sodiumchannel beta1A subunits. J Biol Chem. 2000;275:1079–1088.
21. Meadows LS, Isom LL. Sodium channels as macromolecular complexes:Implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005;67:448–458.
22. Makita N, Bennett PB Jr, George AL Jr. Voltage-gated Na� channel b1subunit mRNA expressed in adult human skeletal muscle, heart, and brainis encoded by a single gene. J Biol Chem. 1994;269:7571–7578.
23. Yang JS, Bennett PB, Makita N, George AL, Barchi RL. Expression ofthe sodium channel beta 1 subunit in rat skeletal muscle is selectivelyassociated with the tetrodotoxin-sensitive alpha subunit isoform. Neuron.1993;11:915–922.
24. Qu Y, Isom LL, Westenbroek RE, Rogers JC, Tanada TN, McCormickKA, Scheuer T, Catterall WA. Modulation of cardiac Na� channelexpression in Xenopus oocytes by beta 1 subunits. J Biol Chem. 1995;270:25696–25701.
25. Nuss HB, Chiamvimonvat N, Perez-Garcia MT, Tomaselli GF, MarbanE. Functional association of the beta 1 subunit with human cardiac (hH1)and rat skeletal muscle (mu 1) sodium channel alpha subunits expressedin Xenopus oocytes. J Gen Physiol. 1995;106:1171–1191.
26. Makielski JC, Limberis JT, Chang SY, Fan Z, Kyle JW. Coexpression ofb 1 with cardiac sodium channel alpha subunits in oocytes decreaseslidocaine block. Mol Pharmacol. 1996;49:30–39.
27. Malhotra JD, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN,Brosius FC, Kass RS, Isom LL. Characterization of sodium channel a-and b-subunits in rat and mouse cardiac myocytes. Circulation. 2001;103:1303–1310.
28. An RH, Wang XL, Kerem B, Benhorin J, Medina A, Goldmit M, KassRS. Novel LQT-3 mutation affects Na� channel activity through inter-actions between alpha- and beta1-subunits. Circ Res. 1998;83:141–146.
29. Ko SH, Lenkowski PW, Lee HC, Mounsey JP, Patel MK. Modulation ofNav1.5 by b1- and b3-subunit co-expression in mammalian cells. PflugersArch. 2005;449:403–412.
30. Fahmi AI, Patel M, Stevens EB, Fowden AL, John JE III, Lee K, PinnockR, Morgan K, Jackson AP, Vandenberg JI. The sodium channel beta-subunit SCN3b modulates the kinetics of SCN5a and is expressed het-erogeneously in sheep heart. J Physiol. 2001;537:693–700.
31. Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlapsyndromes: different faces of SCN5A mutations. Trends CardiovascMed. 2008;18:78–87.
32. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B,Valdivia C, Ueda K, Canizales-Quinteros S, Tusie-Luna MT, Makielski JC,
Ackerman MJ. SCN4B-encoded sodium channel {beta}4 subunit in con-genital long-QT syndrome. Circulation. 2007;116:134–142.
33. Makita N, Shirai N, Wang DW, Sasaki K, George AL Jr, Kanno M,Kitabatake A. Cardiac Na� channel dysfunction in Brugada syndrome isaggravated by b1-subunit. Circulation. 2000;101:54–60.
34. Wan X, Wang Q, Kirsch GE. Functional suppression of sodium channelsby b1-subunits as a molecular mechanism of idiopathic ventricular fibril-lation. J Mol Cell Cardiol. 2000;32:1873–1884.
35. Zimmer T, Biskup C, Bollensdorff C, Benndorf K. The beta1 subunit but notthe beta2 subunit colocalizes with the human heart Na� channel (hH1)already within the endoplasmic reticulum. J Membr Biol. 2002;186:13–21.
36. Adachi K, Toyota M, Sasaki Y, Yamashita T, Ishida S, Ohe-Toyota M,Maruyama R, Hinoda Y, Saito T, Imai K, Kudo R, Tokino T. Identifi-cation of SCN3B as a novel p53-inducible proapoptotic gene. Oncogene.2004;23:7791–7798.
37. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Man-delker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C,Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF,Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Pap-adopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensuscoding sequences of human breast and colorectal cancers. Science.2006;314:268–274.
CLINICAL PERSPECTIVEBrugada syndrome is an inherited cardiac arrhythmia syndrome associated with a high incidence of sudden cardiac arrest.This disorder has previously been linked to mutations in 6 different genes: SCN5A, GPD1L, CACNA1c, CACNB2b, SCN1B,and KCNE3. This study provides evidence that a mutation in SCN3B, encoding the �3-subunit of the sodium channel, cancause a loss of function in INa leading to a Brugada phenotype. The genes thus far associated with Brugada syndrome leadto either a loss of function in sodium or calcium channel current (INa and ICa) or to a gain of function in transient outwardcurrent potassium current (Ito). The decrease in inward current or increase in outward current cause a shift in the balanceof current flowing during the early phases of the cardiac action potential leading to accentuation of the action potentialnotch in the epicardium but not in the endocardium. The resultant transmural gradient leads to an accentuation of theelectrocardiographic J wave, manifest also as an ST-segment elevation, and the development of phase 2 reentry andpolymorphic ventricular tachycardia. This Brugada syndrome phenotype is most commonly limited to the right precordialleads because Ito is usually most prominent in the right ventricular outflow tract. Understanding the genetic basis for theBrugada syndrome may assist with the diagnosis and ultimately with the approach to therapy.
is online at: Circulation: Cardiovascular Genetics Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer information about this process is available in the
requested is located, click Request Permissions in the middle column of the Web page under Services. FurtherCenter, not the Editorial Office. Once the online version of the published article for which permission is being
can be obtained via RightsLink, a service of the Copyright ClearanceCirculation: Cardiovascular Geneticsin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions: