Ligase IVImmunodeficiency Due to Mutations in DNA A Severe Form of Human Combined
EhlFriedrich, Barbara Selle, Charlotte Niemeyer and Stephan Marzenna Orlowska-Volk, Detlev Schindler, WilhelmUlrich Pannicke, Elisabeth Nikolopoulos, Anke Peters, Anselm Enders, Paul Fisch, Klaus Schwarz, Ulrich Duffner,
A Severe Form of Human Combined Immunodeficiency Due toMutations in DNA Ligase IV1
Anselm Enders,* Paul Fisch,† Klaus Schwarz,‡ Ulrich Duffner,* Ulrich Pannicke,‡
Elisabeth Nikolopoulos,† Anke Peters,* Marzenna Orlowska-Volk,† Detlev Schindler,§
Wilhelm Friedrich,¶ Barbara Selle,� Charlotte Niemeyer,* and Stephan Ehl2*
DNA ligase IV (LigIV) deficiency was identified as the molecular basis for a severe form of combined immunodeficiency in twomicrocephalic siblings with cellular radiosensitivity. In one patient the diagnosis was made directly after birth, allowing analysisof the role of LigIV in the development of specific immune cells. Absolute numbers of B cells were reduced 100-fold and �� T cells10-fold, whereas �� T cells were normal. Spectratyping of all three cell populations showed a diverse repertoire, but sequencingof IgH V(D)J junctions revealed shorter CDR3 regions due to more extensive nucleotide deletions among D and J elements andfewer N nucleotide insertions. Clonal restriction of IgG-expressing, but not IgM-expressing, B cells and the lack of primary andsecondary lymph node follicles indicated impaired class switch recombination. Observations in the older sibling showed that thisrudimentary immune system was able to mount specific responses to infection. However, partial Ab responses and extensiveamplification of �� T cells could not prevent a life-threatening course of viral and bacterial infections, the development of anEBV-induced lymphoma, and immune dysregulation reflected by severe autoimmune cytopenia. Impaired generation of immunediversity under conditions of limited LigIV activity can cause a human SCID variant with a characteristic immunologicalphenotype. The Journal of Immunology, 2006, 176: 5060–5068.
T he V(D)J recombination process induces double-strandDNA breakage and rejoining. It forms the basis for thegeneration of diversity in the specific immune system (1).
Early lymphoid progenitors express the RAG-1 and RAG-2 pro-teins, which bind at the recombination signal sequences flankingthe genetic V, D, or J elements, induce double-strand DNA breaksto excise intervening DNA sequences, and generate molecularhairpin structures at the remaining (coding) ends. DNA rejoining isthen conducted by nonhomologous DNA end joining (NHEJ),3 adouble-strand break repair pathway active in all nucleated cells (2).As a first step in NHEJ, the Ku proteins bind to the DNA ends andassociate with the DNA-dependent protein kinase (PK) catalyticsubunit. The resulting DNA-PK complex associates with Artemisand activates its endonuclease activity. The Artemis/DNA-PKcomplex opens the hairpinned V, D, or J ends. End processing bythe endonuclease activity of Artemis in combination with tem-plate-dependent as well as template-independent polymerase ac-tivity generates junctional diversity. The DNA-dependent PK cat-alytic subunit then also acts as a scaffold for the x-ray repair cross-
complementing protein 4 (XRCC4)/ligase IV (LigIV) complex,which finally rejoins the DNA ends (reviewed in Ref. 3).
The key role for some of the enzymes involved in V(D)J re-combination for human lymphocyte development is well illus-trated in patients with primary immunodeficiencies (4). Patientswith null mutations in RAG-1, RAG-2, or Artemis completely lackB and T cells (5, 6) and present with SCID, a uniformly lethaldisorder unless treated by bone marrow transplantation or genetherapy (7). Sensitivity to ionizing irradiation differentiates Arte-mis-deficient patients from RAG-deficient patients (5), illustratinga more general role of the NHEJ pathway in the repair of double-strand DNA breaks. Patients with null mutations in the other genesinvolved in V(D)J recombination have not been reported. This mayin part be explained by their essential role in development. Micewith a targeted deletion of DNA LigIV die at an early embryonicstage, suggesting other essential roles of that enzyme in develop-ment (8).
Although the expected presentation of patients with null muta-tions in genes involved in V(D)J recombination is a T�B� SCID,patients with hypomorphic mutations may retain some recombi-nation activity and develop a rudimentary specific immune system(9, 10). Secondary modification by an infectious or self Ag canthen induce characteristic phenotypes such as Omenn syndrome(10–12), a disorder characterized by generalized erythroderma,hepatosplenomegaly, eosinophilia, elevated IgE, and oligoclonal Tcell expansion. Both hypomorphic Artemis and RAG mutationscan lead to Omenn syndrome (10, 13), indicating that the partiallydeveloped specific immunity and not the particular genetic defectmay be responsible for the phenotype. Thus, patients with hypo-morphic mutations can provide important insights on how the hu-man immune system develops and acts under limiting conditions.
Reports on six patients with hypomorphic mutations in LigIV(14, 15) have been published to date. The first patient developedleukemia at the age of 14 and was diagnosed on the basis of hisincreased sensitivity to radiation therapy (16). Another patient in a
*Department of Pediatrics and Adolescent Medicine, University of Freiburg,Freiburg, Germany; †Institute for Pathology, University of Freiburg, Freiburg, Ger-many; ‡Transfusion Medicine, University Hospital and Institute for Clinical Trans-fusion Medicine and Immunogenetics, Ulm, Germany; §Institute of Human Genetics,Wurzburg, Germany; ¶Children’s Hospital, University of Ulm, Ulm, Germany; and�St. Annastiftskrankenhaus, Ludwigshafen, Germany
Received for publication September 22, 2005. Accepted for publication January25, 2006.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by German Research Foundation Grant SFB620 forprojects A4 (to S.E.) and Z2 (to P.F.).2 Address correspondence and reprint requests to Dr. Stephan Ehl, Department ofPediatrics and Adolescent Medicine, University of Freiburg, Mathildenstrasse 1,79106 Freiburg. E-mail address: [email protected] Abbreviations used in this paper: NHEJ, nonhomologous end joining; LigIV, DNAligase IV; PK, protein kinase; XRCC4, x-ray repair cross-complementing protein 4.
recently published study also developed T cell leukemia at the ageof 4.5 years and showed a phenotype resembling Nijmegen break-age syndrome with microcephaly and growth retardation (17). Theother four patients were diagnosed between 9 and 48 years of agewith various abnormalities, including developmental delay, micro-cephaly, and pancytopenia (14). Although in two patients the pan-cytopenia was severe enough to warrant bone marrow transplan-tation, none of the patients has been described as being severelyimmunodeficient (18). Although the crucial role of LigIV for hu-man V(D)J recombination has been established in a gene-targetedhuman pre-B cell line (19) and in cell lines obtained from LigIV-deficient patients (20–23), the phenotypic consequences of LigIVdeficiency for human lymphocyte development and immunity toinfection remain poorly defined.
Here we describe the immunological profile of two siblings whowere found to be compound heterozygous for a null allele and ahypomorphic mutation in DNA LigIV and who presented withsevere immunodeficiency. One patient was identified after multiplesevere infections and the other was identified directly after birth,providing the opportunity to study the impact of LigIV deficiencyboth on early lymphocyte development and on immune defense.
Materials and MethodsInformed consent
Informed consent for the performed studies was obtained from the patient’sfamily in accordance with the guidelines of the local ethics committee.
Irradiation experiments
Primary diploid fibroblast-like cells were grown from skin biopsy speci-mens using standard cell culture techniques. Confluent, quiescent (G1
phase) cultures were trypsinized, and aliquots were irradiated at the indi-cated dosage levels using a 6-MV linear accelerator (Siemens) at a doserate of 2 Gy min�1. For colony survival studies, aliquots of trypsinizedfibroblasts were irradiated at 0, 0.5, 1, 1.5, 2, 3, 4, 6, and 8 Gy and seededin triplicate into petri dishes as described (24). Eagle’s MEM with 15%FBS was replaced every 3–4 days. After a growth period of 2 wk, colonies(�50 cells) were stained with 1% crystal violet in 20% ethanol. Means andSDs of the ratio of colony number relative to the number of seeded cellswere calculated individually and plotted as colony survival vs radiationdosage for the two patient and the two normal control strains. Dose-re-sponse curves were fitted to the linear quadratic model SF � exp(��X ��X2), where SF is the survival fraction, X the radiation dose, and � and �are fitted parameters. Curves and statistics were generated using the Originsoftware (MicroCal). For 72-h cell cycle analysis, fibroblasts irradiatedwith 0 and 1.5 Gy were seeded at a density of 4000 cells/cm2 into parallelcultures. Growth medium was Eagle’s MEM substituted with 15% FBS,1 � 10�4 M BrdU, and 1 � 10�4 M 2-deoxycytidine. After the growthperiod, the cells were trypsinized and stained with Hoechst 33258 andpropidium iodide. Bivariate histograms were recorded on an LSR flowcytometer (BD Biosciences) and analyzed quantitatively using MULTI2Dand MCYCLE software (25).
Genetic analysis
The LigIV gene was amplified with primer pairs yielding overlapping am-plimers. These amplimers were sequenced in both directions. Primer se-quences for amplification and sequencing are available on request([email protected]). Nucleotide and amino acid countsare according to RefSeq database sequences NM_002312 and NP_002303,respectively.
Immunophenotyping and IgH and T cell CDR3 spectratyping
Lymphocyte phenotyping was performed with four-color flow cytometryusing Abs from BD Pharmingen and Immunotech as described previously(26). TCR CDR3 spectratyping was performed as described by Pannetier etal. (27, 28) for analysis of the TCR �-chain. The primers and details forspectratyping of the TCR �-chains have been published (26). IgH spectra-typing was established according to previously published methods (29) andnew work (E. Nikodopoulos, and P. Fisch, manuscript in preparation). Inbrief, the CDR3 regions were amplified using 35 cycles of PCR with aforward (sense) primer specific for the V region family and a backward(antisense) primer for the constant region. In a second round, three cycles
of a primer extension reaction with a fluorescent (5� FAM-labeled) nestedantisense primer specific for the constant region was performed (runoffreaction). The products were then analyzed on an ABI 3100 capillary se-quencer (Applied Biosystems) to determine the length distribution of thefluorescent fragments. Clonal populations generally appear as sharp peaks,whereas polyclonal populations typically show Gaussian distributions butmay show one or several prominent peaks within the polyclonal peaks ifthe repertoire is dominated by a few clones. The PCR products were clonedinto the pCR2.1-TOPO vector (Invitrogen Life Technologies), amplified,and sequenced. Ig gene segments were identified using the internationalImMunoGeneTics (IMGT) database (imgt.cines.fr) (30). Gene junctionswere analyzed using the IMGT/JunctionAnalysis tool (31). All N nucleo-tide sequences of seven or more nucleotides were individually screened forthe presence of a second D element in both orientations. A homology of atleast 7 bp and appropriate chromosomal localization relative to the first Delement were required for identification of a second D element.
Histology
During placement of a central i.v. catheter, a cervical lymph node frompatient 2 was removed for histological analysis. The sample was fixed in4% buffered formalin, embedded in paraffin, cut at 4 �m, and stained withH&E. For immunohistochemistry, the paraffin sections were put on albu-minized glass slides following heat-mediated Ag retrieval. The primaryAbs were obtained from DakoCytomation and included CD20 (clone L26),CD3 (clone F7.2.38), CD4 (clone MT310), CD8 (clone C8/144B), CD21(clone 1F8), CD30 (clone Ber-H2), and CD68. Visualization of the Abs wasperformed using the ChemMate detection kit, AP/RED (DakoCytomation).
ResultsClinical course of two siblings with microcephaly,radiosensitivity, and severe immunodeficiency
A 2-year-old girl (P1) born to nonconsanguineous parents of Ger-man origin was admitted to our hospital with a history of chronicdiarrhea, failure to thrive, autoimmune cytopenia, and recurrentinfections. At birth the girl was microcephalic with no further dys-plastic stigmata. She developed mild intermittent diarrhea andgrowth delay. At 1 year of age she suffered from pneumococcalsepsis two weeks after measles, mumps, and rubella vaccination.Three months later she presented with severe neutropenia (80/�l)and thrombocytopenia (18,000/�l). PCR was positive for humanherpes virus 6 from bone marrow and peripheral blood. She hadvarying lymphopenia with reduced T cells, almost absent B cells,and normal NK cells. The thymus was small for her age butpresent. Motor development and cognitive development were nor-mal, and all developmental milestones had been achieved in time.At 20 mo of age she was diagnosed with persistent Norwalk virus-positive diarrhea with pronounced failure to thrive and the need forparenteral nutrition. Four months later, she was referred to ourhospital with high fever, cervical lymphadenopathy, hepatomeg-aly, and a pronounced necrotizing mucositis. PCR for EBV waspositive in peripheral blood (2,900 genome equivalents/ml) andbone marrow (640,000 genome equivalents/ml). Imaging studiesrevealed multiple ovaloid mass lesions in the brain and the lung. Abiopsy showed an EBV-encoded latent membrane protein 1-positive,monoclonal, diffuse, large cell non-Hodgkin B cell lymphoma. Inten-sive chemotherapy reduced the tumor mass but was followed by se-vere mucositis with oral and gastrointestinal bleeding. The patientdeveloped rapidly progressive aspergillosis and died during prepara-tive therapy for allogeneic stem cell transplantation.
While the first patient was being treated for the lymphoma, asister was born (P2) who was also microcephalic (Fig. 1). At 2 wkof age she had an absolute lymphocyte count of 936/�l with asimilar subset distribution as her older sister. At 4 mo of age shereceived a bone marrow transplant from a matched unrelated donorfollowing nonmyeloablative conditioning with fludarabine and
thiotepa. Three months after transplantation she developed a se-vere hemolytic-uremic syndrome, presumably triggered by cyclo-sporine that responded to plasmapheresis but that left her withcompensated renal insufficiency. She is presently clinically stable8 mo after transplantation with full chimerism and ongoing thymic
reconstitution. At 1 year of age she has a significant neurodevel-opmental delay.
Compound heterozygosity for LigIV mutations is the geneticbasis of the severe immunodeficiency
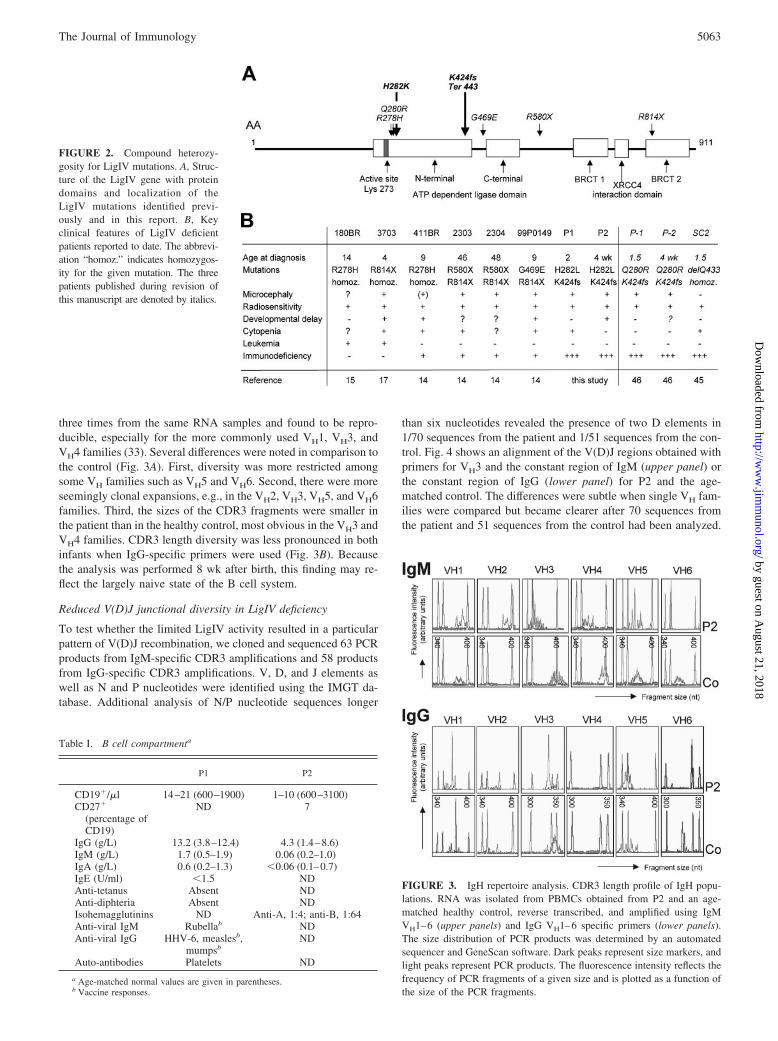
The lymphocyte subset distribution in the two patients (BlowT-lowNK�) suggested a genetic defect affecting both T and B celldifferentiation. After adenosine deaminase, RAG1/2, and Artemisdeficiency had been excluded by enzymatic testing or genomicsequencing, we considered LigIV syndrome. Two consistent fea-tures of LigIV syndrome are microcephaly and increased radio-sensitivity. Clinically, the B cell lymphoma and the toxicity ofchemotherapy observed in P1 were consistent with a defect inDNA repair. Fibroblasts strains from both siblings with LigIV de-ficiency proved significantly hypersensitive to ionizing irradiationregarding their markedly reduced long-term (14 d) clonogenic sur-vival (Fig. 1A). In short-term (72 h) growth studies, the patients’fibroblasts showed arrest in the G2 phase of the cell cycle follow-ing ionizing radiation (Fig. 1B). This finding explains the poorclonogenic survival. The degree of radiosensitivity was compara-ble to that of a paradigm genetic radiosensitivity disorder, ataxiatelangiectasia (32) (Fig. 1B). Both patients showed normal lengthdevelopment along the 25th to 50th percentile (Fig. 1C, upperpanels). By contrast, the head circumference was disproportionallysmall and was below the 3rd percentile in both patients (Fig. 1C,lower panel). Although no severe immunodeficiency had been re-ported in the six previously published patients (14, 15, 17, 18), wedecided to sequence the LIG4 gene. We identified a point mutation(1118A3T, resulting in H282L) on the maternal allele and a de-letion of five bases (1544AAAGA1548) on the paternal allele,leading to a frame shift and an altered amino acid sequence startingafter D423 and premature termination of translation after aminoacid 442 (Fig. 2A) in both patients. The parents were heterozygousfor these mutations.
Impaired B cell development, but remaining specific Abproduction in LigIV deficient patients
The absolute numbers of peripheral B cells were reduced �100-fold in both siblings (Table I). Nevertheless, serum Igs and specificAbs could be detected. When analyzed at 4 wk of age in the ab-sence of obvious infections, P2 had low amounts of IgM, whereasIgA was absent. IgG levels could not be interpreted due to remain-ing maternal Abs. Nevertheless, IgM hemagglutinins were detect-able. Moreover, 7% of the B cells expressed the memory markerCD27, similar to age-matched controls. P1 was analyzed after se-vere infections and showed surprisingly high levels of serum Igs.Total IgG and IgM were elevated, but IgA was in the normalrange. Despite three vaccinations, the patient did not have specificAbs to tetanus or diphtheria and showed only low titers to pneu-mococcus despite having undergone culture-proven sepsis. Bycontrast, significant IgG responses could be detected against themeasles and mumps vaccines and to the human herpes virus 6infection documented by PCR. Response to the rubella vaccinewas limited to IgM, and no Ab response could be detected againstEBV. In addition to these partial antimicrobial Ab responses, thepatient developed high titers of autoantibodies against platelets,resulting in severe thrombopenia.
Reduced BCR diversity in LigIV deficiency
To analyze the B cell repertoire generated under conditions oflimited LigIV activity, we performed CDR3 spectratyping of theVH genes using IgM- and IgG-specific primers in P2 and a healthyage-matched control. The spectratyping analysis was performed
FIGURE 1. Sensitivity to ionizing radiation in two patients with micro-cephaly and immunodeficiency. A, Colony survival. Dose vs radiation re-sponse curves of fibroblasts from the two patients, P1 (filled triangle) andP2 (inverted filled triangle), are compared with those of two random nor-mal controls, C1 (open circle) and C2 (closed circles), and studied in par-allel. Error bars denote one SD. B, G2 phase accumulation. 72 h afterirradiation with 1.5 Gy, fibroblasts were stained with Hoechst 33258 andpropidium iodide, and cell cycle status was determined by flow cytometry.The percentage of cells in G1/G2 phase is shown as a function of the ratioof cells in G2 phase among all cycling cells. The ratios from fibroblasts ofpatient P1 (filled triangle) and patient P2 (inverted filled triangle) arewithin the range of radiosensitive ataxia telangiectasia fibroblast strains(gray diamonds; n � 18), whereas normal control strains (gray circles; n �15) show a much lower ratio. The high noncycling cell fractions of thepatient cultures relative to positive (ataxia telangiectasia) and negative(normal) controls indicate their poor growth. C, Growth charts showingdevelopment of length and head circumference. The bold line representsthe 50th percentile for German children, and the lines beneath it show the25th, 10th, and 3rd percentiles.
5062 SEVERE IMMUNODEFICIENCY IN LIGASE IV SYNDROME
three times from the same RNA samples and found to be repro-ducible, especially for the more commonly used VH1, VH3, andVH4 families (33). Several differences were noted in comparison tothe control (Fig. 3A). First, diversity was more restricted amongsome VH families such as VH5 and VH6. Second, there were moreseemingly clonal expansions, e.g., in the VH2, VH3, VH5, and VH6families. Third, the sizes of the CDR3 fragments were smaller inthe patient than in the healthy control, most obvious in the VH3 andVH4 families. CDR3 length diversity was less pronounced in bothinfants when IgG-specific primers were used (Fig. 3B). Becausethe analysis was performed 8 wk after birth, this finding may re-flect the largely naive state of the B cell system.
Reduced V(D)J junctional diversity in LigIV deficiency
To test whether the limited LigIV activity resulted in a particularpattern of V(D)J recombination, we cloned and sequenced 63 PCRproducts from IgM-specific CDR3 amplifications and 58 productsfrom IgG-specific CDR3 amplifications. V, D, and J elements aswell as N and P nucleotides were identified using the IMGT da-tabase. Additional analysis of N/P nucleotide sequences longer
than six nucleotides revealed the presence of two D elements in1/70 sequences from the patient and 1/51 sequences from the con-trol. Fig. 4 shows an alignment of the V(D)J regions obtained withprimers for VH3 and the constant region of IgM (upper panel) orthe constant region of IgG (lower panel) for P2 and the age-matched control. The differences were subtle when single VH fam-ilies were compared but became clearer after 70 sequences fromthe patient and 51 sequences from the control had been analyzed.
FIGURE 3. IgH repertoire analysis. CDR3 length profile of IgH popu-lations. RNA was isolated from PBMCs obtained from P2 and an age-matched healthy control, reverse transcribed, and amplified using IgMVH1–6 (upper panels) and IgG VH1–6 specific primers (lower panels).The size distribution of PCR products was determined by an automatedsequencer and GeneScan software. Dark peaks represent size markers, andlight peaks represent PCR products. The fluorescence intensity reflects thefrequency of PCR fragments of a given size and is plotted as a function ofthe size of the PCR fragments.
a Age-matched normal values are given in parentheses.b Vaccine responses.
FIGURE 2. Compound heterozy-gosity for LigIV mutations. A, Struc-ture of the LigIV gene with proteindomains and localization of theLigIV mutations identified previ-ously and in this report. B, Keyclinical features of LigIV deficientpatients reported to date. The abbrevi-ation “homoz.” indicates homozygos-ity for the given mutation. The threepatients published during revision ofthis manuscript are denoted by italics.
First, there was significant clonal identity among IgG sequencesfrom the patient. Although among IgM sequences 23/24 controland 37/39 patient sequences were unique, only 15/31 IgG se-quences from the patient compared with 26/27 from the controlshowed a unique sequence. Thus, in contrast to IgM, the clonaldiversity of IgG-expressing B cells appeared to be significantlymore restricted in P2 than in the control. Second, N nucleotidesequences were used less often and, if used, were shorter in the
patient (Fig. 5A). This could be seen among both IgM and IgGsequences (Fig. 5 and data not shown). By contrast, the number ofsequences that harbored P nucleotides was similar in both infants(15/52 in P2 vs 16/49 in the control). Third, more deletions of theend nucleotides could be observed among the J and D elements ofthe patient. Fig. 5 shows the number of nucleotides deleted fromthe J elements (Fig. 5B) and the D elements (Fig. 5C) among theunique IgM and IgG sequences from P2 and the control.
FIGURE 4. VH3 junctional sequences among IgM� and IgG� B cells. PCR products from the IgH CDR3 spectratyping (see Fig. 3) were isolated, clonedinto the pCR2.1-TOPO vector, amplified, and sequenced. Sequences amplified using VH3- and IgM-specific (upper panel) or VH3- and IgG-specific primers(lower panel) are shown for P2 and a healthy age-matched control. The sequences are shown as displayed by the IMGT software. The dots in the alignedV and J regions as well as in the D regions reflect deletions of the germline end nucleotides. The far right column shows the number of amino acids inthe CDR3 region.
FIGURE 5. Fewer N nucleotides and more gene end deletions among J and D elements in LigIV deficiency. The unique VH3 junctional sequencesamplified using IgM- and IgG-specific primers from the patient (52 unique/70 total sequences) and the control (49 unique/51 total sequences) were alignedusing the IMGT software. A, The number of N nucleotides was determined in the individual sequences. B and C, J and D elements were compared withthe germline sequences, and the number of nucleotides deleted from 5� and 3� ends during V(D)J recombination was determined for the individualsequences. B, Number of nucleotides deleted from the J elements. C, Number of nucleotides deleted from the D elements. Data are displayed as box plotswith the median, 25/75th, and 10/90th percentiles indicated as horizontal bars.
5064 SEVERE IMMUNODEFICIENCY IN LIGASE IV SYNDROME
Absence of primary and secondary lymph node follicles
A cervical lymph node from P2 was removed. H&E staining re-vealed a poorly structured fibrotic node with pronounced lym-phopenia, lack of follicular areas, and interfollicular zones (Fig.6A). Immunohistochemical analysis using B cell Abs (CD20; Fig.6, B and C) showed a proper number of follicles (normal anlage)but of small size and with different severity of B cell depletion.The follicles lacked CD3� T cells but contained abundant CD68�
macrophages (Fig. 6D). These cells can frequently be observed inthe granuloma-like structures forming in lymph nodes of SCIDpatients. There were no secondary follicles, no centroblasts, and nosigns of germinal center activity as shown by negative staining forthe proliferation marker Ki-67 (data not shown).
Impaired T cell development but normal T cell repertoirediversity
The number of circulating CD4� and CD8� T cells was reducedin both patients with a reduced proportion of CD45RA� naive Tcells among total CD4� T cells. T cell proliferation assessed by[3H]thymidine incorporation was significantly reduced in responseto PHA and anti-CD3 stimulation, and there was no response totetanus in P1. Using a CFSE dilution, we could demonstrate sig-nificant proliferation of CD4� T cells obtained from P2 in re-sponse to PHA and anti-CD3/anti-CD28, whereas there was littleresponse from CD8� T cells (data not shown). All of the seven V�chains analyzed were used by T cells from P2, and there wereonly limited shifts compared with published controls (Fig. 7, Aand B). CDR3 spectratyping of the V� chain revealed a poly-clonal repertoire that resembled the diversity of an age-matchedcontrol (Fig. 7C).
Predominance of �� T cells
Both patients showed a predominance of �� T cells (Table II). Inthe absence of any obvious infection in P2, 29% of CD3� lym-phocytes expressed the �� TCR. Up to 89% of CD3� lymphocytesexpressed the �� TCR in P1 after a history of severe bacterial andviral infections. The diversity of �-chains was analyzed in T cellsfrom P2 by TCR CDR3 spectratyping analysis (Fig. 8). Significantgenetic diversity could be documented among the more commonV�1, V�2, and V�3 chains, whereas the more rare V�4, V�5, andV�6 chains showed an oligoclonal spectrum. The overall pattern ofCDR3 sizes was as diverse as that of an age-matched control (Fig.
8), arguing against Ag-driven oligoclonal expansion as the basisfor the �� T cell predominance in P2.
DiscussionIn this report LigIV deficiency has been identified as a new geneticcause for a variant of severe combined immunodeficiency in hu-mans. It adds to the wide phenotypic spectrum of LigIV syndrome(Online Mendelian Inheritance in Man no.601837), a previouslydescribed heterogenous disorder with radiosensitivity, growth fail-ure, microcephaly, developmental delay, predisposition to lym-phoma, and milder forms of immunodeficiency. In all patients de-scribed it is caused by hypomorphic LigIV mutations (14, 15, 17,34). This event is not unexpected, because mice with a targeteddeletion of LigIV die during embryonal development (8). Neitherof the LigIV mutations identified in this study have been reportedpreviously (Fig. 2A). The first mutation leads to a frameshift afterD423 and a premature stop after amino acid 442, presumably rep-resenting a null allele. The most similar mutation that has beencharacterized in detail is the R580X mutation, likely representinga null allele, because the protein is not stably expressed, does notinteract with XRCC4, and does not enter the nucleus (14). Thesecond most likely hypomorphic mutation (H282L) is close to theconserved ligase motif that includes the active site K273. It is inthe vicinity of the R278H mutation (15) that leads to 5–10% re-sidual adenylation and double-strand ligation activity (20, 22, 23).It has been reported that this activity is further reduced below 1%by the additional polymorphisms A3V and T9I (34). These poly-morphisms were not present in our patients. Because the functionalconsequences of the H282L mutation have not yet been studied byin vitro assays, we cannot exclude the possibility that additionaluncharacterized defects may also contribute to the impaired classswitch recombination.
FIGURE 6. Absence of primary and secondary lymph node follicles.Lymph node sections were either stained with H&E or immunostained withthe indicated mAbs.
FIGURE 7. Normal �� T cell repertoire. A and B, V� chain expressionamong CD4� (A) and CD8� T cells (B) as determined by flow cytometry.C, CDR3 length profile of the indicated V� populations. cDNA was pre-pared from PBMC RNA, amplified using V�-specific primers, and ana-lyzed by spectratyping (see legend to Fig. 3). The frequency of PCR frag-ments of a given size is plotted as a function of the size of the PCRfragment. Dark peaks represent size markers, and light peaks representPCR products.
Characteristic phenotypic features of most patients describedwith LigIV syndrome include microcephaly and radiosensitivity(Fig. 2B). Growth defects and radiosensitivity can also be observedin mice deficient in the NHEJ enzymes Ku (35–37), as well as inmice deficient in XRCC4 or LigIV, when bred on a p53-deficientbackground to overcome their embryonic lethality (38, 39). Al-though microcephaly has not been reported in these mice, exces-sive neuronal apoptosis has been observed (8, 38, 39), indicatingthe need for repair of double-strand breaks during nervous systemdevelopment. Neuronal cell loss in the brain is a known etiologicfactor for microcephaly in humans. The clinical outcome of thisneurological defect is, however, highly variable. Although LigIV-deficient mice die during embryonal development (8), the devel-opmental delay in patients with hypomorphic mutations rangesfrom no defects (patient 118BR) (15) to significant developmentaland mental delay (patient 99P0149) (14). This can only in part beexplained by the localization of the individual mutations, becauseour two siblings differed markedly in their cognitive developmentand motor development. Our observations also imply that the neu-rodevelopmental outcome cannot be predicted by the extent towhich a particular mutation impairs immunological development.
The crucial role of LigIV in DNA double-strand repair andNHEJ identified in mice (8) and human cell lines (19) predicted thepossibility of a SCID phenotype in human LigIV syndrome. Cy-topenia of several hemopoietic lineages and an increased suscep-tibility to infection have been reported in some LigIV-deficientpatients (14). However, despite various abnormalities in the fre-quency and fidelity of V(D)J recombination in vitro (14, 20–23),none of the mutations described to date caused a severe immuno-deficiency in vivo (14, 15). In plasmid-based assays, cell linesobtained from patients 180BR and 411BR only showed a 2–3-fold
reduction in the frequency of V(D)J recombination (14, 20, 23),whereas it was reduced 1000-fold in the gene-targeted LigIV-de-ficient human pre-B cell line N114P2 (19). In our patients, theLigIV mutations led to a significant impairment of lymphocytedevelopment. Obviously, the repertoire of peripheral B and T cellsdetected in the periphery represents the complex result of devel-opmental impairment, homeostatic proliferation, and peripheralmodification by infectious and other types of Ags. However, theyounger sibling was studied only a few weeks after birth withoutobvious infections, and the spectratyping analysis revealed alargely undisturbed repertoire. The severely reduced number ofcirculating B and T lymphocytes may, therefore, allow a roughestimation of the efficiency of V(D)J recombination. Interestingly,the impact was different on various cell populations. Although Bcell numbers were reduced �100-fold, there was only a 10-foldreduction in �� T cells and no reduction in �� T cells. This couldbe due to cell type-specific differences in LigIV requirements, aninterpretation that is supported by the finding that the repertoirediversity was more restricted among B cells than among T cells.An additional factor may be thymic selection. Under conditions oflimiting production of precursor cells, a relatively higher propor-tion of T cells may be able to pass selection in the thymus such thatthe lack of mature T cells may be relatively less pronounced thanthe lack of T cell precursors. The predominance of �� T cellsobserved in both patients may have been influenced by the lessstringent thymic selection of �� T cells that is probably not influ-enced by MHC restriction. Following the infections in P1, the ��
T cells may have been triggered and further expanded by microbialAgs recognized by the �� TCR (40–43) or other triggering mech-anisms of the innate immune system.
LigIV deficiency significantly affected the fidelity of the joiningreaction in V(D)J recombination. Previous studies (20) with trans-fected recombination substrates revealed signal joint, but not cod-ing joint, deletions during recombination in the 180BR cell line.The defects in cell lines from patients 2304 and 411BR were morepronounced and included deletions, lack of end protection, andreduced insertion of N nucleotides by TdT (14, 20, 21). In thesequences obtained from peripheral B cells of P2 we found sig-nificant deletions of D and J elements and reduced N nucleotideinsertion. This may suggest a disturbed interaction between LigIVand TdT, but a direct interaction between these two enzymes hasnot been proven biochemically. The increased number of nucleo-tide deletions could be due to lack of end protection by LigIV (21).Another possible explanation is that, due to limited LigIV activity,the DNA ends are accessible to nucleases such as Artemis for alonger period of time, leading to a relative overactivity of theseenzymes.
FIGURE 8. Normal �� T cell repertoire. CDR3 length profile of theindicated V� populations. cDNA was prepared from PBMC RNA, ampli-fied using V� specific primers, and analyzed by spectratyping (see legendto Fig. 3). The frequency of PCR fragments of a given size is plotted as afunction of the size of the PCR fragment. Dark peaks represent size mark-ers, and light peaks represent PCR products.
The severe immunodeficiency observed in our patients includedsusceptibility to life-threatening bacterial and viral infections, de-velopment of an EBV-positive B cell lymphoma, and severe au-toimmune cytopenia. These features developed in the presence ofnormal to elevated total levels of serum Igs, indicating that a sig-nificant number of Ab-producing plasma cells could be generatedfrom the few developing B cells. However, specific IgM and IgGresponses were only detected against some vaccines and infec-tions, presumably reflecting an additional defect in the periphery.In line with this interpretation, the frequent finding of identicalsequences showed that the repertoire diversity of IgG Ig receptorswas more limited than that of IgM. This could either reflect limitedhelper T cell activity or, more likely, indicate a role for LigIV inclass switch recombination. An altered pattern of recombination atthe S�-S� switch junctions has recently been observed in DNAobtained from patients 2303/4 and 411BR (44). The absence ofprimary and secondary lymph node follicles in P2 was also con-sistent with a role for LigIV in class switch recombination. Thecytopenia in our patients was not due to ineffective hematopoiesis(data not shown) but was caused by autoantibodies with peripheraldestruction. It remains unclear to what extent autoimmunity con-tributed to the cytopenia observed in the previously reported Li-gIV-deficient patients.
A predominance of �� T cells and Ab-mediated autoimmunitydespite minimal B cell and severely reduced �� T cell numbershas recently been reported as a characteristic phenotype of patientswith a SCID variant due to hypomorphic RAG mutations andCMV infection (26, 45). This report describes another molecularcause for this phenotype. Neither of our patients showed evidenceof CMV infection. It is conceivable that EBV infection could havea similar effect on the T cell repertoire. However, the predomi-nance of �� T cells was also observed in the “naive” youngersibling. As argued previously (26), it is therefore plausible to as-sume that limited V(D)J recombination activity may lead to a de-velopmental advantage for �� T cells. Under conditions of �� Tlymphopenia, herpes virus infections may then additionally am-plify these cells in the periphery.
During the course of revision of this work, three additional Li-gIV-deficient patients with severe immunodeficiency and radio-sensitivity have been published (Table II). A homozygous Q433mutation was detected in a developmentally normal, normoce-phalic 18-mo-old patient with a history of recurrent respiratoryinfections, chronic diarrhea, and failure to thrive (46). In the sec-ond study, compound heterozygosity for Q280R and K424F (thelatter was also present in our patients) was demonstrated in twomicrocephalic siblings presenting at 18 mo and at 4 weeks of agewith recurrent respiratory infections, failure to thrive, and pneu-mococcal sepsis (47). In both studies, the functional consequencesof the mutations were demonstrated by complementation assays.The cellular phenotype was similar with 0–20 B cells/�l, 200–400T cells/�l, and normal to diminished NK cells. �� T cells were notreported. All infants were severely hypogammaglobulinemic anddid not produce specific Abs. V(D)J junctional sequences wereassessed in bone marrow (46), in some T cell sequences, and insubstrates from in vitro recombination assays (47) with overallsimilar observations, i.e., increased deletions among D and J seg-ments, reduction of N nucleotides, and a shift toward microhomol-ogy-directed joining (46, 47). All three patients died, one duringconditioning and two within 2 mo after bone marrow transplanta-tion. Together, these three and our two patients firmly establishLigIV deficiency as yet another genetic defect that needs to beconsidered in the wide and still evolving differential diagnosis ofvariants of severe combined immunodeficiency in humans (7). Theclinical phenotype resembles “leaky” forms of RAG or Artemis
deficiency, and we speculate that this could also include Omennsyndrome (9, 13). Radiosensitivity and microcephaly are importantclues guiding rational genetic analysis in these patients.
AcknowledgmentsWe are grateful to Alain Fischer (Institut National de la Sante et de laRecherche Medicale, Paris, France) for pointing out microcephaly as animportant diagnostic clue for the identification of LigIV-deficient patients.We thank Ralf Kuppers (Institut fur Zellbiologie, Essen, Germany) forhelpful discussions on the sequence data. We thank all nurses and doctorsinvolved in the dedicated clinical care of the patients. The excellent tech-nical help by Annette Schult, Tatjana Kersten, Daniela Bukatz, andSylvia Koch is kindly appreciated.
DisclosuresThe authors have no financial conflict of interest.
References1. Dudley, D. D., J. Chaudhuri, C. H. Bassing, and F. W. Alt. 2005. Mechanism and
control of V(D)J recombination versus class switch recombination: similaritiesand differences. Adv. Immunol. 86: 43–112.
2. Lieber, M. R., Y. Ma, U. Pannicke, and K. Schwarz. 2004. The mechanism ofvertebrate nonhomologous DNA end joining and its role in V(D)J recombination.DNA Repair (Amst.) 3: 817–826.
3. Lieber, M. R., Y. Ma, U. Pannicke, and K. Schwarz. 2003. Mechanism andregulation of human non-homologous DNA end joining. Nat Rev. Mol. Cell Biol.4: 712–720.
4. de Villartay, J. P., A. Fischer, and A. Durandy. 2003. The mechanisms of immunediversification and their disorders. Nat. Rev. Immunol. 3: 962–972.
5. Moshous, D., I. Callebaut, R. de Chasseval, B. Corneo, M. Cavazzana-Calvo,F. Le Deist, I. Tezcan, O. Sanal, Y. Bertrand, N. Philippe, et al. 2001. Artemis,a novel DNA double-strand break repair/V(D)J recombination protein, is mutatedin human severe combined immune deficiency. Cell 105: 177–186.
6. Schwarz, K., G. H. Gauss, L. Ludwig, U. Pannicke, Z. Li, D. Lindner,W. Friedrich, R. A. Seger, T. E. Hansen-Hagge, S. Desiderio, et al. 1996. RAGmutations in human B cell-negative SCID. Science 274: 97–99.
7. Fischer, A., F. Le Deist, S. Hacein-Bey-Abina, I. Andre-Schmutz,G. de Saint Basile, J. P. de Villartay, and M. Cavazzana-Calvo. 2005. Severecombined immunodeficiency a model disease for molecular immunology andtherapy. Immunol. Rev. 203: 98–109.
8. Frank, K. M., J. M. Sekiguchi, K. J. Seidl, W. Swat, G. A. Rathbun, H. L. Cheng,L. Davidson, L. Kangaloo, and F. W. Alt. 1998. Late embryonic lethality andimpaired V(D)J recombination in mice lacking DNA ligase IV. Nature 396:173–177.
9. Moshous, D., C. Pannetier, R. de Chasseval, F. le Deist, M. Cavazzana-Calvo,S. Romana, E. Macintyre, D. Canioni, N. Brousse, A. Fischer, et al. 2003. PartialT and B lymphocyte immunodeficiency and predisposition to lymphoma in pa-tients with hypomorphic mutations in Artemis. J. Clin. Invest. 111: 381–387.
10. Villa, A., S. Santagata, F. Bozzi, S. Giliani, A. Frattini, L. Imberti, L. B. Gatta,H. D. Ochs, K. Schwarz, L. D. Notarangelo, et al. 1998. Partial V(D)J recom-bination activity leads to Omenn syndrome. Cell 93: 885–896.
11. Villa, A., C. Sobacchi, L. D. Notarangelo, F. Bozzi, M. Abinun,T. G. Abrahamsen, P. D. Arkwright, M. Baniyash, E. G. Brooks, M. E. Conley,et al. 2001. V(D)J recombination defects in lymphocytes due to RAG mutations:severe immunodeficiency with a spectrum of clinical presentations. Blood 97:81–88.
12. Omenn, G. S. 1965. Familial reticuloendotheliosis with eosinophilia. N. Engl.J. Med. 273: 427–432.
13. Ege, M., Y. Ma, B. Manfras, K. Kalwak, H. Lu, M. R. Lieber, K. Schwarz, andU. Pannicke. 2005. Omenn syndrome due to ARTEMIS mutations. Blood 105:4179–4186.
14. O’Driscoll, M., K. M. Cerosaletti, P. M. Girard, Y. Dai, M. Stumm, B. Kysela,B. Hirsch, A. Gennery, S. E. Palmer, J. Seidel, et al. 2001. DNA ligase IVmutations identified in patients exhibiting developmental delay and immunode-ficiency. Mol. Cell 8: 1175–1185.
15. Riballo, E., S. E. Critchlow, S. H. Teo, A. J. Doherty, A. Priestley, B. Broughton,B. Kysela, H. Beamish, N. Plowman, C. F. Arlett, et al.1999. Identification of adefect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol. 9:699–702.
16. Plowman, P. N., B. A. Bridges, C. F. Arlett, A. Hinney, and J. E. Kingston. 1990.An instance of clinical radiation morbidity and cellular radiosensitivity, not as-sociated with ataxia-telangiectasia. Br. J. Radiol. 63: 624–628.
17. Ben-Omran, T. I., K. Cerosaletti, P. Concannon, S. Weitzman, andM. M. Nezarati. 2005. A patient with mutations in DNA ligase IV: clinical fea-tures and overlap with Nijmegen breakage syndrome. Am. J. Med. Genet. A. 137:283–287.
18. O’Driscoll, M., A. R. Gennery, J. Seidel, P. Concannon, and P. A. Jeggo. 2004.An overview of three new disorders associated with genetic instability: LIG4syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair (Amst.) 3:1227–1235.
19. Grawunder, U., D. Zimmer, S. Fugmann, K. Schwarz, and M. R. Lieber. 1998.DNA ligase IV is essential for V(D)J recombination and DNA double-strandbreak repair in human precursor lymphocytes. Mol. Cell 2: 477–484.
20. Riballo, E., A. J. Doherty, Y. Dai, T. Stiff, M. A. Oettinger, P. A. Jeggo, andB. Kysela. 2001. Cellular and biochemical impact of a mutation in DNA ligaseIV conferring clinical radiosensitivity. J. Biol. Chem. 276: 31124–31132.
21. Smith, J., E. Riballo, B. Kysela, C. Baldeyron, K. Manolis, C. Masson,M. R. Lieber, D. Papadopoulo, and P. Jeggo. 2003. Impact of DNA ligase IV onthe fidelity of end joining in human cells. Nucleic Acids Res. 31: 2157–2167.
22. Wang, H., Z. C. Zeng, A. R. Perrault, X. Cheng, W. Qin, and G. Iliakis. 2001.Genetic evidence for the involvement of DNA ligase IV in the DNA-PK-depen-dent pathway of non-homologous end joining in mammalian cells. Nucleic AcidsRes. 29: 1653–1660.
23. Badie, C., M. Goodhardt, A. Waugh, N. Doyen, N. Foray, P. Calsou,B. Singleton, D. Gell, B. Salles, P. Jeggo, et al. 1997. A DNA double-strand breakdefective fibroblast cell line (180BR) derived from a radiosensitive patient rep-resents a new mutant phenotype. Cancer Res. 57: 4600–4607.
24. Kalb, R., M. Duerr, M. Wagner, S. Herterich, M. Gross, M. Digweed, H. Joenje,H. Hoehn, and D. Schindler. 2004. Lack of sensitivity of primary Fanconi’sanemia fibroblasts to UV and ionizing radiation. Radiat. Res. 161: 318–325.
25. Schindler, D., and H. Hoehn. 1999. Flow cytometric testing for syndromes withchromosomal instability. In Diagnostic Cytogenetics. R. D. Wegner, ed. Springer,Berlin, pp. 269–281.
26. Ehl, S., K. Schwarz, A. Enders, U. Duffner, U. Pannicke, J. Kuhr, F. Mascart,A. Schmitt-Graf, C. Niemeyer, and P. Fisch. 2005. A variant of severe combinedimmunodeficiency with specific immune responses and predominance of �� Tcells. J. Clin. Invest. 115: 3140–3148.
27. Pannetier, C., J. Even, and P. Kourilsky. 1995. T-cell repertoire diversity andclonal expansions in normal and clinical samples. Immunol. Today 16: 176–181.
28. Pannetier, C., J. P. Levraud, A. Lim, J. Even, and P. Kourilsky. 1998. The im-munoscope approach for the analysis of T cell repertoires. In The Antigen T CellReceptor: Selected Protocols and Applications. J. R. Oksenberg, ed. Chapman &Hall, London, pp. 287–325.
29. Kuppers, R. 2004. Molecular single-cell PCR analysis of rearranged immuno-globulin genes as a tool to determine the clonal composition of normal and ma-lignant human B cells. Methods Mol. Biol. 271: 225–238.
30. Lefranc, M. P., V. Giudicelli, Q. Kaas, E. Duprat, J. Jabado-Michaloud,D. Scaviner, C. Ginestoux, O. Clement, D. Chaume, and G. Lefranc. 2005.IMGT, the international ImMunoGeneTics information system. Nucleic AcidsRes. 33: D593–D597.
31. Monod, M. Y., V. Giudicelli, D. Chaume, and M. P. Lefranc. 2004. IMGT/JunctionAnalysis: the first tool for the analysis of the immunoglobulin and T cellreceptor complex V-J and V-D-J junctions. Bioinformatics 20(Suppl. 1):I379–I385.
32. Ford, M. D., L. Martin, and M. F. Lavin. 1984. The effects of ionizing radiationon cell cycle progression in ataxia telangiectasia. Mutat. Res. 125: 115–122.
33. Brezinschek, H. P., R. I. Brezinschek, and P. E. Lipsky. 1995. Analysis of theheavy chain repertoire of human peripheral B cells using single-cell polymerasechain reaction. J. Immunol. 155: 190–202.
34. Girard, P. M., B. Kysela, C. J. Harer, A. J. Doherty, and P. A. Jeggo. 2004.Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: theimpact of two linked polymorphisms. Hum. Mol. Genet. 13: 2369–2376.
35. Gu, Y., K. J. Seidl, G. A. Rathbun, C. Zhu, J. P. Manis, N. van der Stoep,L. Davidson, H. L. Cheng, J. M. Sekiguchi, K. Frank, et al. 1997. Growth retar-dation and leaky SCID phenotype of Ku70-deficient mice. Immunity 7: 653–665.
36. Nussenzweig, A., C. Chen, V. da Costa Soares, M. Sanchez, K. Sokol,M. C. Nussenzweig, and G. C. Li. 1996. Requirement for Ku80 in growth andimmunoglobulin V(D)J recombination. Nature 382: 551–555.
37. Ouyang, H., A. Nussenzweig, A. Kurimasa, V. C. Soares, X. Li,C. Cordon-Cardo, W. Li, N. Cheong, M. Nussenzweig, G. Iliakis, et al. 1997.Ku70 is required for DNA repair but not for T cell antigen receptor gene recom-bination in vivo. J. Exp. Med. 186: 921–929.
38. Gao, Y., Y. Sun, K. M. Frank, P. Dikkes, Y. Fujiwara, K. J. Seidl,J. M. Sekiguchi, G. A. Rathbun, W. Swat, J. Wang, et al. 1998. A critical role forDNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95:891–902.
39. Frank, K. M., N. E. Sharpless, Y. Gao, J. M. Sekiguchi, D. O. Ferguson, C. Zhu,J. P. Manis, J. Horner, R. A. DePinho, and F. W. Alt. 2000. DNA ligase IVdeficiency in mice leads to defective neurogenesis and embryonic lethality via thep53 pathway. Mol. Cell 5: 993–1002.
40. De Paoli, P., D. Gennari, P. Martelli, V. Cavarzerani, R. Comoretto, andG. Santini. 1990. Gamma delta T cell receptor-bearing lymphocytes during Ep-stein-Barr virus infection. J. Infect. Dis. 161: 1013–1016.
41. Dechanet, J., P. Merville, A. Lim, C. Retiere, V. Pitard, X. Lafarge, S. Michelson,C. Meric, M. M. Hallet, P. Kourilsky, et al. 1999. Implication of �� T cells in thehuman immune response to cytomegalovirus. J. Clin. Invest. 103: 1437–1449.
42. Merville, P., J. Dechanet, F. Berge, G. Bone-Mane, J. Taupin, P. Michel, P. Joly,M. Bonneville, L. Potaux, and J. Moreau. 2000. Cytomegalovirus infection inkidney allograft recipients is followed by a prolonged expansion of �� T lym-phocytes. Transplant Proc. 32: 357–359.
43. Rothenberg, M. E., W. E. Weber, J. A. Longtine, and D. A. Hafler. 1996. Cy-totoxic � � I lymphocytes associated with an Epstein-Barr virus-induced post-transplantation lymphoproliferative disorder. Clin. Immunol. Immunopathol. 80:266–272.
44. Pan-Hammarstrom, Q., A. M. Jones, A. Lahdesmaki, W. Zhou, R. A. Gatti,L. Hammarstrom, A. R. Gennery, and M. R. Ehrenstein. 2005. Impact of DNAligase IV on nonhomologous end joining pathways during class switch recom-bination in human cells. J. Exp. Med. 201: 189–194.
45. de Villartay, J. P., A. Lim, H. Al-Mousa, S. Dupont, J. Dechanet-Merville,E. Coumau-Gatbois, M. L. Gougeon, A. Lemainque, C. Eidenschenk,E. Jouanguy, et al. 2005. A novel immunodeficiency associated with hypomor-phic RAG1 mutations and CMV infection. J. Clin. Invest. 115: 3291–3299.
46. van der Burg, M., L. R. van Veelen, N. S. Verkaik, W. W. Wiegant,N. G. Hartwig, B. H. Barendregt, L. Brugmans, A. Raams, N. G. Jaspers,M. Z. Zdzienicka, et al. 2006. A new type of radiosensitive T-B-NK� severecombined immunodeficiency caused by a LIG4 mutation. J. Clin. Invest. 116:137–145.
47. Buck, D., D. Moshous, R. de Chasseval, Y. Ma, F. Le Deist,M. Cavazzana-Calvo, A. Fischer, J. L. Casanova, M. R. Lieber, andJ. P. de Villartay. 2005. Severe combined immunodeficiency and microcephaly insiblings with hypomorphic mutations in DNA ligase IV. Eur. J. Immunol. 36:224–235.
5068 SEVERE IMMUNODEFICIENCY IN LIGASE IV SYNDROME