Published: April 06, 2011 r2011 American Chemical Society 4774 dx.doi.org/10.1021/jp1097348 | J. Phys. Chem. B 2011, 115, 4774–4780 ARTICLE pubs.acs.org/JPCB A Theoretical Study of the Physicochemical Mechanisms Associated with DNA Recognition Modulation in Artificial Zinc-Finger Proteins Hirotoshi Mori* and Kaori Ueno-Noto † Division of Advanced Sciences, Ocha-dai Academic Production, Ochanomizu University, 2-1-1 Ohtsuka, Bunkyo-ku, Tokyo 112-8610, Japan 1. INTRODUCTION Zinc-finger (ZF) is a functional zinc protein that can bind DNAs with specific nucleic base sequences. The DNA-binding affinity of the ZF domain can be utilized for engineering DNA- binding functional proteins. 1-7 Many research groups have used the ZF framework to design artificial functional proteins with potential applications in medicine. 8-10 However, a more com- plete understanding of the basic physicochemical mechanism of DNA recognition by the ZF protein is now demanded. In terms of the engineering of functional proteins, the GAGA factor of Drosophila melanogaster is a particularly attractive ZF protein, since it uses a “single” Cys 2 /His 2 -type ZF domain for specific DNA sequence recognition. 11 A schematic representa- tion of the ZF domain in the GAGA factor is given in Figure 1. As shown in Figure 1, the ZF domain consists of two parts. The first is an R-helix structure, which is considered to bind DNA. The other is a β-hairpin region followed by a long amino acid tail containing regions named BR1 and BR2. Since the β-hairpin region is located behind the R-helix, the region has been considered to be less important than the R-helix for DNA recognition. However, in 2007, Dhanasekaran et al. reported an interesting experimental result that contradicts the chemical intuition. 4 They made three artificial ZFs (ZF β1F , ZF β2F , and ZF 2F ) from the wild-type GAGA factor of D. melanogaster. In the artificial ZFs, an amino acid residue in the β-hairpin region of the ZF domain was mutated singly (ZF β1F and ZF β2F ) or doubly (ZF 2F ), as shown in Figure 1b, c, and d, respectively. They then compared the artificial ZFs with the wild-type in terms of structural changes and DNA-binding affinities. 4 On the basis of circular dichroism (CD) spectra and gel mobility shift assay, they concluded the following two points: (1) the R-helix structure was well preserved in all mutants and (2) DNA-binding function was completely suppressed in the artificial ZFs, even though there was no amino acid mutation in the DNA-binding R-helix region. Their results suggest that regions other than the R-helix in the ZF domain play important roles in the biofunctional DNA binding of the ZF. However, the details of the physicochemical control mechanisms of DNA-binding ability have not yet been elucidated. The object of this study is to clarify the origin of the DNA- binding affinity modulation in the artificial GAGA factor ZF proteins from a theoretical point of view. Understanding the physical chemistry behind DNA-binding modulation is impor- tant for the artificial design of ZF biochemical function. On the basis of a comparison of experimental results with classical MD and ab initio electronic structure calculations, physicochemical differences between wild-type ZF and its mutants are discussed. 2. COMPUTATIONAL DETAILS To study the DNA-binding affinity of ZF and to understand the affinity modulation by amino acid point mutations, we performed two types of simulations: classical molecular dynamics (MD) and ab initio electronic structure calculations using the fragment molecular orbital (FMO) method. 12,13 The former was used to consider dynamic fluctuations of the biomolecular complexes in aqueous solution. The latter was used to treat intermolecular interactions between ZF and DNA in a more accurate quantum chemical level to support the classical MD results. On the basis of the classical MD simulation, binding free energies for the wild-type ZF-DNA complex and its mu- tant-DNA complexes were also compared. 2-1. Molecular Dynamics (MD). All the MD simulations were performed with the SANDER module of the AMBER 10 program package. 14 The force field used in our MD simulations was ff03 15 extended with the cationic dummy approach (CaDA) proposed by Pang. 16 The CaDA approach was applied to express Received: October 11, 2010 Revised: March 16, 2011 ABSTRACT: The DNA-binding ability of the zinc-finger (ZF) protein and the modulation of its affinity to DNA through amino acid mutations were theoretically investigated. Classical molecular dynamics and energy decomposi- tion analysis based on large-scale ab initio fragment molecular orbital calcula- tions were used to obtain the DNA binding affinities of wild-type and three mutant ZFs. Calculated binding free energies qualitatively well explained the DNA binding affinity modulation experimentally observed by Dhanasekaran et al. [Dhanasekaran, M.; et al., Biochemistry 2007, 46, 7506-7513]. It had been considered that only the R-helix domain in the ZF plays an important role in DNA recognition; however, our results clearly show that the N-terminal regions, BR1 and BR2, also play important roles in DNA recognition.
Transcript
Published: April 06, 2011
r 2011 American Chemical Society 4774 dx.doi.org/10.1021/jp1097348 | J. Phys. Chem. B 2011, 115, 4774–4780
ARTICLE
pubs.acs.org/JPCB
A Theoretical Study of the Physicochemical Mechanisms Associatedwith DNA Recognition Modulation in Artificial Zinc-Finger ProteinsHirotoshi Mori* and Kaori Ueno-Noto†
Division of Advanced Sciences, Ocha-dai Academic Production, Ochanomizu University, 2-1-1 Ohtsuka, Bunkyo-ku, Tokyo 112-8610, Japan
1. INTRODUCTION
Zinc-finger (ZF) is a functional zinc protein that can bindDNAs with specific nucleic base sequences. The DNA-bindingaffinity of the ZF domain can be utilized for engineering DNA-binding functional proteins.1-7 Many research groups have usedthe ZF framework to design artificial functional proteins withpotential applications in medicine.8-10 However, a more com-plete understanding of the basic physicochemical mechanism ofDNA recognition by the ZF protein is now demanded.
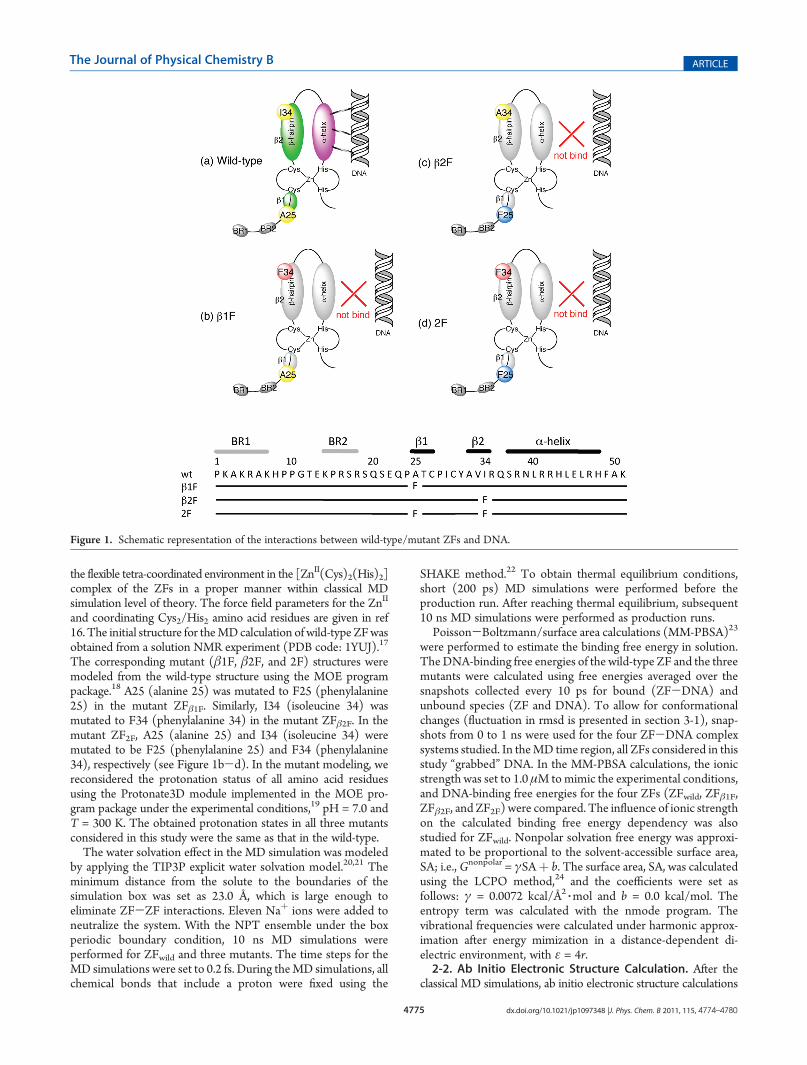
In terms of the engineering of functional proteins, the GAGAfactor of Drosophila melanogaster is a particularly attractive ZFprotein, since it uses a “single” Cys2/His2-type ZF domain forspecific DNA sequence recognition.11 A schematic representa-tion of the ZF domain in the GAGA factor is given in Figure 1. Asshown in Figure 1, the ZF domain consists of two parts. The firstis an R-helix structure, which is considered to bind DNA. Theother is a β-hairpin region followed by a long amino acid tailcontaining regions named BR1 and BR2. Since the β-hairpinregion is located behind the R-helix, the region has beenconsidered to be less important than the R-helix for DNArecognition. However, in 2007, Dhanasekaran et al. reportedan interesting experimental result that contradicts the chemicalintuition.4 They made three artificial ZFs (ZFβ1F, ZFβ2F, andZF2F) from the wild-type GAGA factor ofD. melanogaster. In theartificial ZFs, an amino acid residue in the β-hairpin region of theZF domain was mutated singly (ZFβ1F and ZFβ2F) or doubly(ZF2F), as shown in Figure 1b, c, and d, respectively. They thencompared the artificial ZFs with the wild-type in terms ofstructural changes and DNA-binding affinities.4 On the basis ofcircular dichroism (CD) spectra and gel mobility shift assay, theyconcluded the following two points: (1) theR-helix structure waswell preserved in all mutants and (2) DNA-binding function wascompletely suppressed in the artificial ZFs, even though therewas no amino acid mutation in the DNA-binding R-helix region.Their results suggest that regions other than theR-helix in the ZF
domain play important roles in the biofunctional DNA bindingof the ZF. However, the details of the physicochemical controlmechanisms of DNA-binding ability have not yet beenelucidated.
The object of this study is to clarify the origin of the DNA-binding affinity modulation in the artificial GAGA factor ZFproteins from a theoretical point of view. Understanding thephysical chemistry behind DNA-binding modulation is impor-tant for the artificial design of ZF biochemical function. On thebasis of a comparison of experimental results with classical MDand ab initio electronic structure calculations, physicochemicaldifferences between wild-type ZF and its mutants are discussed.
2. COMPUTATIONAL DETAILS
To study the DNA-binding affinity of ZF and to understandthe affinity modulation by amino acid point mutations, weperformed two types of simulations: classical molecular dynamics(MD) and ab initio electronic structure calculations using thefragment molecular orbital (FMO) method.12,13 The former wasused to consider dynamic fluctuations of the biomolecularcomplexes in aqueous solution. The latter was used to treatintermolecular interactions between ZF and DNA in a moreaccurate quantum chemical level to support the classical MDresults. On the basis of the classical MD simulation, binding freeenergies for the wild-type ZF-DNA complex and its mu-tant-DNA complexes were also compared.2-1.Molecular Dynamics (MD).All theMD simulations were
performed with the SANDER module of the AMBER 10program package.14 The force field used in our MD simulationswas ff0315 extended with the cationic dummy approach (CaDA)proposed by Pang.16 The CaDA approach was applied to express
Received: October 11, 2010Revised: March 16, 2011
ABSTRACT: The DNA-binding ability of the zinc-finger (ZF) protein and themodulation of its affinity to DNA through amino acid mutations weretheoretically investigated. Classical molecular dynamics and energy decomposi-tion analysis based on large-scale ab initio fragment molecular orbital calcula-tions were used to obtain the DNA binding affinities of wild-type and threemutant ZFs. Calculated binding free energies qualitatively well explained theDNA binding affinity modulation experimentally observed by Dhanasekaranet al. [Dhanasekaran, M.; et al., Biochemistry 2007, 46, 7506-7513]. It had beenconsidered that only the R-helix domain in the ZF plays an important role inDNA recognition; however, our results clearly show that theN-terminal regions,BR1 and BR2, also play important roles in DNA recognition.
4775 dx.doi.org/10.1021/jp1097348 |J. Phys. Chem. B 2011, 115, 4774–4780
The Journal of Physical Chemistry B ARTICLE
the flexible tetra-coordinated environment in the [ZnII(Cys)2(His)2]complex of the ZFs in a proper manner within classical MDsimulation level of theory. The force field parameters for the ZnII
and coordinating Cys2/His2 amino acid residues are given in ref16. The initial structure for theMDcalculation of wild-type ZFwasobtained from a solution NMR experiment (PDB code: 1YUJ).17
The corresponding mutant (β1F, β2F, and 2F) structures weremodeled from the wild-type structure using the MOE programpackage.18 A25 (alanine 25) was mutated to F25 (phenylalanine25) in the mutant ZFβ1F. Similarly, I34 (isoleucine 34) wasmutated to F34 (phenylalanine 34) in the mutant ZFβ2F. In themutant ZF2F, A25 (alanine 25) and I34 (isoleucine 34) weremutated to be F25 (phenylalanine 25) and F34 (phenylalanine34), respectively (see Figure 1b-d). In the mutant modeling, wereconsidered the protonation status of all amino acid residuesusing the Protonate3D module implemented in the MOE pro-gram package under the experimental conditions,19 pH = 7.0 andT = 300 K. The obtained protonation states in all three mutantsconsidered in this study were the same as that in the wild-type.The water solvation effect in the MD simulation was modeled
by applying the TIP3P explicit water solvation model.20,21 Theminimum distance from the solute to the boundaries of thesimulation box was set as 23.0 Å, which is large enough toeliminate ZF-ZF interactions. Eleven Naþ ions were added toneutralize the system. With the NPT ensemble under the boxperiodic boundary condition, 10 ns MD simulations wereperformed for ZFwild and three mutants. The time steps for theMD simulations were set to 0.2 fs. During theMD simulations, allchemical bonds that include a proton were fixed using the
SHAKE method.22 To obtain thermal equilibrium conditions,short (200 ps) MD simulations were performed before theproduction run. After reaching thermal equilibrium, subsequent10 ns MD simulations were performed as production runs.Poisson-Boltzmann/surface area calculations (MM-PBSA)23
were performed to estimate the binding free energy in solution.TheDNA-binding free energies of the wild-type ZF and the threemutants were calculated using free energies averaged over thesnapshots collected every 10 ps for bound (ZF-DNA) andunbound species (ZF and DNA). To allow for conformationalchanges (fluctuation in rmsd is presented in section 3-1), snap-shots from 0 to 1 ns were used for the four ZF-DNA complexsystems studied. In theMD time region, all ZFs considered in thisstudy “grabbed” DNA. In the MM-PBSA calculations, the ionicstrength was set to 1.0 μM tomimic the experimental conditions,and DNA-binding free energies for the four ZFs (ZFwild, ZFβ1F,ZFβ2F, and ZF2F) were compared. The influence of ionic strengthon the calculated binding free energy dependency was alsostudied for ZFwild. Nonpolar solvation free energy was approxi-mated to be proportional to the solvent-accessible surface area,SA; i.e.,Gnonpolar = γSAþ b. The surface area, SA, was calculatedusing the LCPO method,24 and the coefficients were set asfollows: γ = 0.0072 kcal/Å2
3mol and b = 0.0 kcal/mol. Theentropy term was calculated with the nmode program. Thevibrational frequencies were calculated under harmonic approx-imation after energy mimization in a distance-dependent di-electric environment, with ε = 4r.2-2. Ab Initio Electronic Structure Calculation. After the
classical MD simulations, ab initio electronic structure calculations
Figure 1. Schematic representation of the interactions between wild-type/mutant ZFs and DNA.
4776 dx.doi.org/10.1021/jp1097348 |J. Phys. Chem. B 2011, 115, 4774–4780
The Journal of Physical Chemistry B ARTICLE
were also performed to evaluate ZF-DNA interaction energieswith quantum chemical level. Since the complexes are thermallyfluctuating under physiological conditions, treatment of thefluctuations is also important in the ab initio calculations. Forour ab initio calculations, we picked up 50 snapshot structures ofeach ZF from the classical MD. Ideally speaking, an ab initio fullquantum molecular dynamics approach is best. However, such asimulation is computationally too expensive at the moment. Inthe snapshot structure selection procedure, the water layer within6 Å of the ZF-DNA complex was kept to include the explicitsolvent effect. Ab initio single-point energy calculations werethen performed for the water-solvated ZF-DNA complexmodels.
Even though the ZFs considered in this study are relativelysmall proteins, the numbers of atoms and electrons are still toolarge to apply a conventional ab initio molecular orbital ap-proach. In this study, a fragment molecular orbital (FMO)approach,12,13 which is a large-scale ab initio electronic structurecalculation method, was used to perform electronic structurecalculations. In our FMO calculations, we used a 2-body expan-sion scheme (FMO2) since our purpose is to reveal thequalitative differences between wild-type ZF-DNA interactionsand those of artificially designed ZFs.Hydrogen bonding and dispersion interactions are considered
to be significant to molecular recognition in the ZF-DNAcomplex, suggesting that consideration of the so-called “electro-nic correlation effect” is essential. In this study, an MP2 level ofperturbation approach was applied to consider the electroniccorrelations in the FMO scheme (FMO2-MP2).25,26 The basissets used in the calculations were 6-31G**27 for C, N, O, and Hand MCPdzp28-30 for S, P, Na, and Zn. The MCPdzp is arelativistic model core potential that can include scalar relativisticeffects and reduce CPU costs arising from the chemically inertcore electrons without loosing chemical accuracy.Using the above FMO scheme, we analyzed ZF-DNA inter-
actions decomposed into each amino acid-base pair level withthe PIEDA (pair interaction energy decomposition analysis)scheme.13 Physicochemical differences in the DNA-bindingabilities between the wild-type and corresponding artificial ZFswere investigated. All ab initio FMO calculations were performedusing the GAMESS quantum chemical program package.31
3. RESULTS AND DISCUSSION
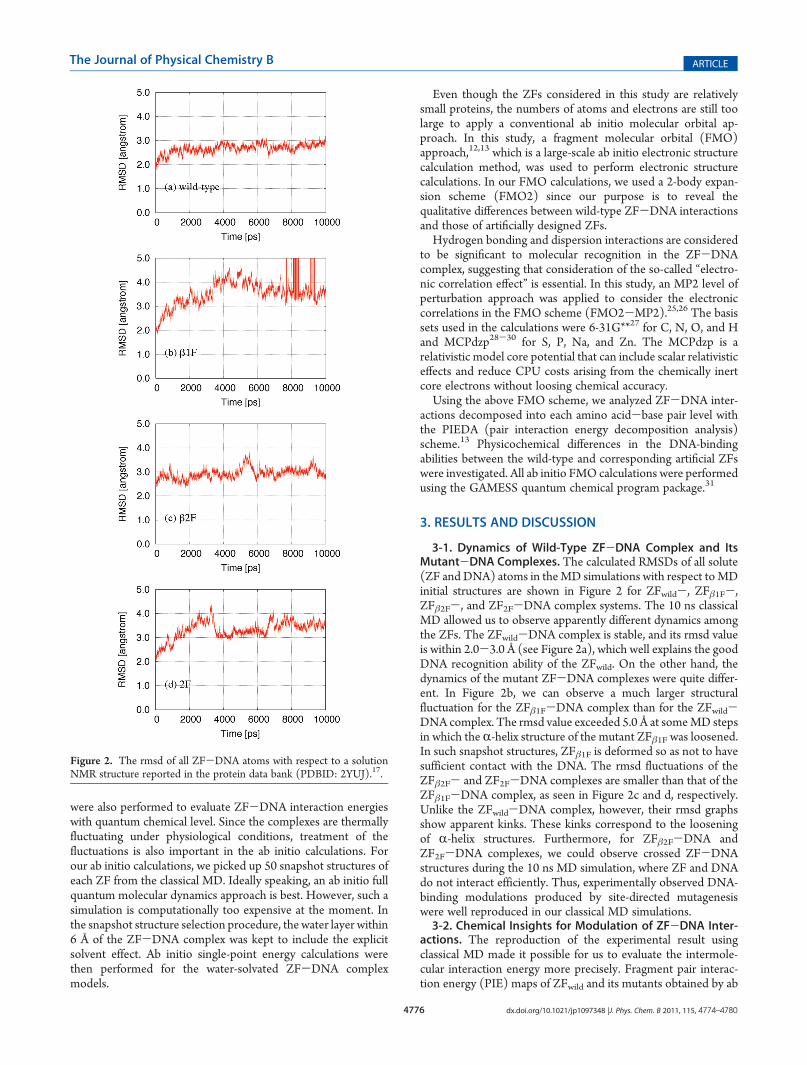
3-1. Dynamics of Wild-Type ZF-DNA Complex and ItsMutant-DNA Complexes. The calculated RMSDs of all solute(ZF and DNA) atoms in theMD simulations with respect toMDinitial structures are shown in Figure 2 for ZFwild-, ZFβ1F-,ZFβ2F-, and ZF2F-DNA complex systems. The 10 ns classicalMD allowed us to observe apparently different dynamics amongthe ZFs. The ZFwild-DNA complex is stable, and its rmsd valueis within 2.0-3.0 Å (see Figure 2a), which well explains the goodDNA recognition ability of the ZFwild. On the other hand, thedynamics of the mutant ZF-DNA complexes were quite differ-ent. In Figure 2b, we can observe a much larger structuralfluctuation for the ZFβ1F-DNA complex than for the ZFwild-DNA complex. The rmsd value exceeded 5.0 Å at someMD stepsin which the R-helix structure of the mutant ZFβ1F was loosened.In such snapshot structures, ZFβ1F is deformed so as not to havesufficient contact with the DNA. The rmsd fluctuations of theZFβ2F- and ZF2F-DNA complexes are smaller than that of theZFβ1F-DNA complex, as seen in Figure 2c and d, respectively.Unlike the ZFwild-DNA complex, however, their rmsd graphsshow apparent kinks. These kinks correspond to the looseningof R-helix structures. Furthermore, for ZFβ2F-DNA andZF2F-DNA complexes, we could observe crossed ZF-DNAstructures during the 10 ns MD simulation, where ZF and DNAdo not interact efficiently. Thus, experimentally observed DNA-binding modulations produced by site-directed mutagenesiswere well reproduced in our classical MD simulations.3-2. Chemical Insights for Modulation of ZF-DNA Inter-
actions. The reproduction of the experimental result usingclassical MD made it possible for us to evaluate the intermole-cular interaction energy more precisely. Fragment pair interac-tion energy (PIE) maps of ZFwild and its mutants obtained by ab
Figure 2. The rmsd of all ZF-DNA atoms with respect to a solutionNMR structure reported in the protein data bank (PDBID: 2YUJ).17.
4777 dx.doi.org/10.1021/jp1097348 |J. Phys. Chem. B 2011, 115, 4774–4780
The Journal of Physical Chemistry B ARTICLE
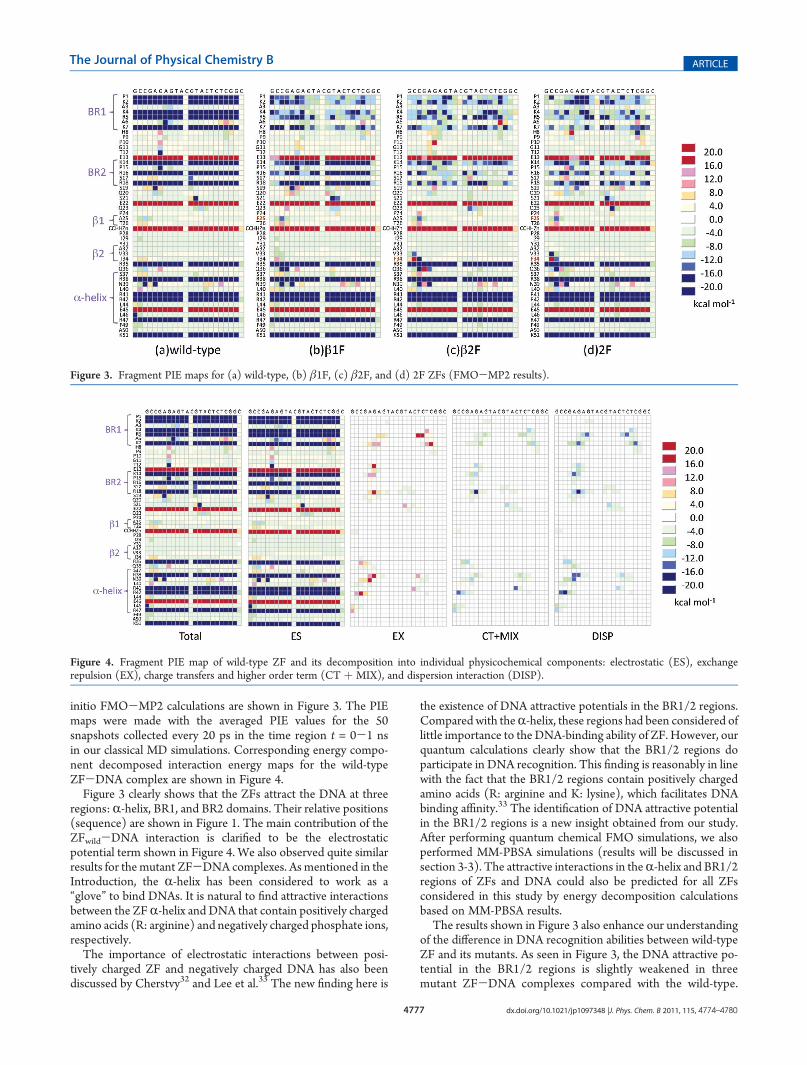
initio FMO-MP2 calculations are shown in Figure 3. The PIEmaps were made with the averaged PIE values for the 50snapshots collected every 20 ps in the time region t = 0-1 nsin our classical MD simulations. Corresponding energy compo-nent decomposed interaction energy maps for the wild-typeZF-DNA complex are shown in Figure 4.Figure 3 clearly shows that the ZFs attract the DNA at three
regions: R-helix, BR1, and BR2 domains. Their relative positions(sequence) are shown in Figure 1. The main contribution of theZFwild-DNA interaction is clarified to be the electrostaticpotential term shown in Figure 4. We also observed quite similarresults for the mutant ZF-DNA complexes. As mentioned in theIntroduction, the R-helix has been considered to work as a“glove” to bind DNAs. It is natural to find attractive interactionsbetween the ZFR-helix and DNA that contain positively chargedamino acids (R: arginine) and negatively charged phosphate ions,respectively.The importance of electrostatic interactions between posi-
tively charged ZF and negatively charged DNA has also beendiscussed by Cherstvy32 and Lee et al.33 The new finding here is
the existence of DNA attractive potentials in the BR1/2 regions.Compared with theR-helix, these regions had been considered oflittle importance to the DNA-binding ability of ZF. However, ourquantum calculations clearly show that the BR1/2 regions doparticipate in DNA recognition. This finding is reasonably in linewith the fact that the BR1/2 regions contain positively chargedamino acids (R: arginine and K: lysine), which facilitates DNAbinding affinity.33 The identification of DNA attractive potentialin the BR1/2 regions is a new insight obtained from our study.After performing quantum chemical FMO simulations, we alsoperformed MM-PBSA simulations (results will be discussed insection 3-3). The attractive interactions in theR-helix and BR1/2regions of ZFs and DNA could also be predicted for all ZFsconsidered in this study by energy decomposition calculationsbased on MM-PBSA results.The results shown in Figure 3 also enhance our understanding
of the difference in DNA recognition abilities between wild-typeZF and its mutants. As seen in Figure 3, the DNA attractive po-tential in the BR1/2 regions is slightly weakened in threemutant ZF-DNA complexes compared with the wild-type.
Figure 3. Fragment PIE maps for (a) wild-type, (b) β1F, (c) β2F, and (d) 2F ZFs (FMO-MP2 results).
Figure 4. Fragment PIE map of wild-type ZF and its decomposition into individual physicochemical components: electrostatic (ES), exchangerepulsion (EX), charge transfers and higher order term (CT þ MIX), and dispersion interaction (DISP).
4778 dx.doi.org/10.1021/jp1097348 |J. Phys. Chem. B 2011, 115, 4774–4780
The Journal of Physical Chemistry B ARTICLE
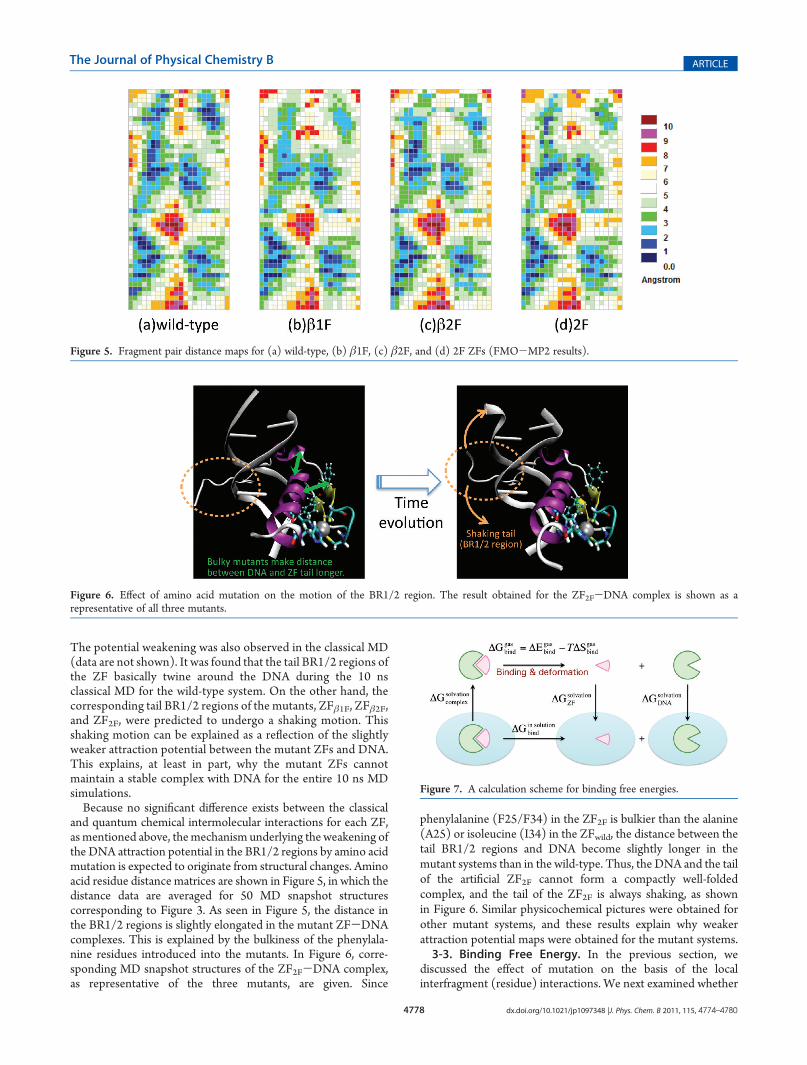
The potential weakening was also observed in the classical MD(data are not shown). It was found that the tail BR1/2 regions ofthe ZF basically twine around the DNA during the 10 nsclassical MD for the wild-type system. On the other hand, thecorresponding tail BR1/2 regions of the mutants, ZFβ1F, ZFβ2F,and ZF2F, were predicted to undergo a shaking motion. Thisshaking motion can be explained as a reflection of the slightlyweaker attraction potential between the mutant ZFs and DNA.This explains, at least in part, why the mutant ZFs cannotmaintain a stable complex with DNA for the entire 10 ns MDsimulations.Because no significant difference exists between the classical
and quantum chemical intermolecular interactions for each ZF,as mentioned above, themechanism underlying the weakening ofthe DNA attraction potential in the BR1/2 regions by amino acidmutation is expected to originate from structural changes. Aminoacid residue distance matrices are shown in Figure 5, in which thedistance data are averaged for 50 MD snapshot structurescorresponding to Figure 3. As seen in Figure 5, the distance inthe BR1/2 regions is slightly elongated in the mutant ZF-DNAcomplexes. This is explained by the bulkiness of the phenylala-nine residues introduced into the mutants. In Figure 6, corre-sponding MD snapshot structures of the ZF2F-DNA complex,as representative of the three mutants, are given. Since
phenylalanine (F25/F34) in the ZF2F is bulkier than the alanine(A25) or isoleucine (I34) in the ZFwild, the distance between thetail BR1/2 regions and DNA become slightly longer in themutant systems than in the wild-type. Thus, the DNA and the tailof the artificial ZF2F cannot form a compactly well-foldedcomplex, and the tail of the ZF2F is always shaking, as shownin Figure 6. Similar physicochemical pictures were obtained forother mutant systems, and these results explain why weakerattraction potential maps were obtained for the mutant systems.3-3. Binding Free Energy. In the previous section, we
discussed the effect of mutation on the basis of the localinterfragment (residue) interactions. We next examined whether
Figure 5. Fragment pair distance maps for (a) wild-type, (b) β1F, (c) β2F, and (d) 2F ZFs (FMO-MP2 results).
Figure 6. Effect of amino acid mutation on the motion of the BR1/2 region. The result obtained for the ZF2F-DNA complex is shown as arepresentative of all three mutants.
Figure 7. A calculation scheme for binding free energies.
4779 dx.doi.org/10.1021/jp1097348 |J. Phys. Chem. B 2011, 115, 4774–4780
The Journal of Physical Chemistry B ARTICLE
it was possible to explain the experimentally observed bindingaffinity suppression by the presence of mutants. To do this, wecalculated the binding free energies in the ZF-DNA complexesusing the MD results. The binding free energies were calculatedusing the following equation (its schematic calculation strategiesare given in Figure 7):
binding-free energy in an aqueous solution, interaction potentialenergy in a gas phase, solvent effect, and entropy term, respec-tively. With this scheme, we calculated the DNA binding freeenergies of ZFs in solution as a sum of three term contributions:binding energy in the gas phase, solvation free energy (solventeffect), and entropy term. The solvent effect term was calculatedusing the MM-PBSA method23 as implemented in AMBER.14
The entropy term was calculated by normal vibration analysiswith classical force field and harmonic approximations. Thissimulation level was adequate, since our object was not to obtainquantitative values but to qualitatively compare the binding freeenergies among wild-type and mutant systems. A similar ap-proach has been successfully used to compare DNA bindingaffinities among single, double, and triple zinc-finger ZFsystems.33 The calculated binding free energies obtained byclassical MD simulations are given in Table 1, in which twotypes of binding free energies using two different calculationschemes—single-trajectory and separate-trajectory—are shown.In the single-trajectory scheme, binding free energies of unboundspecies are calculated from the snapshots extracted from thesingle complex trajectory. On the other hand, in the separate-trajectory scheme, binding free energies of unbound species arecalculated from the separate trajectories of the unbound proteinand DNA.As seen in Table 1, for all affinity estimation schemes, we
obtained a larger DNA binding affinity for ZFwild than for ZFβ1F,ZFβ2F, or ZF2F. The DNA-binding affinities estimated withseparate-trajectories were smaller than those estimated withsingle-trajectories, and positive energies, which means no bind-ing, were obtained for mutant systems. The weaker binding freeenergies in the artificial ZFs correspond well to the slightweakening of the interaction between ZF and DNA discussedin the previous section. The theoretically predicted weakening ofDNA binding well explains the experimental observation re-ported by Dhanasekaran et al.Finally, it should be mentioned that the calculated DNA-
binding affinity reflects the ionic strength dependency of the bathsolutions, since there are differences between physiological con-ditions and the experimental ones used by Dhanasekaran et al.
(1.0 � 10-3 mM).4 The typical physiologically relevant con-centration of simple salt is about 100 mM of NaCl. Thecalculated DNA-binding affinities under several ionic strengthconditions are shown in Table 2. We found that ZFwild hasslightly weaker DNA-binding affinity (-6.7 kcal mol-1) at anionic strength of 100 mM than at the experimental ioniccondition (-10.4 kcal mol-1). The DNA-binding affinity ofZFwild decreases monotonically with increases in ionic strength.As already mentioned, the driving force of the DNA recognitionby ZF proteins is basically electrostatic attraction betweenpositively charged finger domains and negatively chargedDNA. The monotonic decrease in DNA-binding affinity is wellexplained by the electrostatic screening effect of salts.
4. CONCLUDING REMARKS
Using classical MD and ab initio FMO approaches, weinvestigated the chemical interactions in ZF-DNA complexes.In DNA recognition, attractive interactions in the R-helix as wellas in the BR1/2 regions of ZFs are very important. OurFMO-PIEDA analyses clearly show that the interactions inthe BR1/2 regions are weakened by the introduction of aminoacid mutations: A25f F25 or I34f F34. The weakening of theattractive potentials in the mutants is basically explained by thechange in the distance between ZF tail BR1/2 regions and thebound DNA. The distances are elongated due to the bulkiness ofthe phenylalanine residues introduced into the mutants. Reflect-ing the weakening of electrostatic interaction between positivelycharged BR1/BR2 regions and negatively charged DNA, bindingfree energies between ZFmutants andDNA are also estimated tobe weakened, which well explains the gel mobility shift assayresults obtained by Dhanasekaran et al. The weakness in theartificial ZF-DNA interactions results in larger fluctuations inthe artificial ZF-DNA complexes than in the wild-type complex.A monotonic decrease in DNA-binding affinity with increases inionic strength in the bath solvent was predicted for the ZF-DNAsystems.
Our study builds an important signpost for the future design ofDNA-binding functional proteins. Molecular simulations, classi-cal MD with a CaDA approach or large-scale ab initio electronicstructure calculation theory combined with a bioinformatic
Table 1. Binding Free Energy Calculated with Potential Energies by Classical MD Simulations Using an MM-PBSA SolvationModela
single trajectoryb -41.1( 2.8 -19.2( 3.0 -18.1( 2.7 -21.0( 3.1
separate trajectoryc -10.4( 6.2 10.6( 5.8 12.8( 6.3 8.6( 5.6aThe ionic strengths were set at 1.0 μM to mimic experimental conditions. bThe free energies of unbound species were calculated from the snapshotsextracted from the single complex trajectory. cThe free energies of unbound species were calculated from the separate trajectories of the unboundprotein and DNA.
Table 2. Influence of Ionic Strength on the CalculatedBinding Free Energy of Wild-Type ZF Using an MM-PBSASolvation Model
Present Addresses†Division of Chemistry, Center for Natural Sciences, College ofLiberal Arts and Sciences, Kitasato University, 1-15-1 Kitasato,Sagamihara, Kanagawa 228-8555, Japan.
’ACKNOWLEDGMENT
The authors are supported by the Ocha-dai Academic Produc-tion project run by the Japan Science and Technology Agency(JST). A part of the calculations reported here were performedusing computing resources in the Research Center for Computa-tional Science, Okazaki, Japan.
’REFERENCES
(1) Jantz, D.; Amann, B. T.; Gatto, G. J., Jr.; Berg, J. M. T. Chem. Rev.2004, 104, 789.(2) Durai, S.; Mani, M.; Kandavelou, K.; Wu, J.; Porteus, M. H.;
Chandrasegaran, S. Nucleic Acids Res. 2005, 33, 5978.(3) Dhanasekaran, M.; Negi, S.; Sugiura, Y. Acc. Chem. Res. 2006,
39, 45.(4) Dhanasekaran, M.; Negi, S.; Imanishi, M.; Sugiura, Y. Biochem-
istry 2007, 46, 7506.(5) Shiraishi, Y.; Imanishi, M.; Morisaki, T.; Sugiura, Y. Biochemistry
2005, 44, 2523.(6) Dhanasekaran, M.; Negi, S.; Imanishi, M.; Suzuki, M.; Sugiura, Y.
Biochemistry 2008, 47, 11717.(7) Negi, S.; Imanishi, M.; Matsumoto, M.; Sugiura, Y. Chem.—Eur.
J. 2008, 14, 3236.(8) Jamieson, A. C.;Miller, J. C.; Pabo, C. O.Nat. Rev. DrugDiscovery
2003, 2, 361.(9) Blancafort, P.; Segal, D. J.; Barbas, C. F., III. Mol. Pharmacol.
2004, 66, 1361.(10) Klug, A. FEBS Lett. 2005, 579, 892.(11) Pedone, P. V.; Ghirlando, R.; Clore, G. M.; Gronenborn, A. M.;
Felsenfeld, G.; Omichinski, J. Proc. Natl. Acad. Sci. U.S.A 1996, 93, 2822.(12) Fedorov, D. G.; Kitaura, K. J. Phys. Chem. 2007, 111, 6904.(13) Fedorov, D. G.; Kitaura, K. J. Comput. Chem. 2007, 28, 222.(14) Case, D. A.; Darden, T. A.; Cheatham, T. E., III; Simmerling,
C. L.; Wang, J.; Duke, R. E.; Luo, R.; Crowley, M.; Walker, R. C.; Zhang,W.; Merz, K. M.; Wang, B.; Hayik, S.; Roitberg, A.; Seabra, G.;Kolossv�ary, I.; Wong, K. F.; Paesani, F.; Vanicek, J.; Wu, X.; Brozell,S. R.; Steinbrecher, T.; Gohlke, H.; Yang, L.; Tan, C.; Mongan, J.;Hornak, V.; Cui, G.; Mathews, D. H.; Seetin, M. G.; Sagui, C.; Babin, V.;Kollman, P. A. AMBER 10, University of California: San Francisco,2008.(15) Duan, Y.; Wu, C; Chowdhury, S.; Lee, M. C.; Xiong, G.; Zhang,
W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; Caldwell, J.; Wang, J.;Kollman, P. J. Comput. Chem. 2003, 24, 1999.(16) Pang, Y.-P. J. Mol. Model. 1999, 5, 196.(17) Omichinski, J. G.; Pedone, P. V.; Felsenfeld, G.; Gronenborn,
A. M.; Clore, G. M. Nat. Struct. Biol. 1997, 4, 122.(18) MOE (The Molecular Operating Environment) Version 2009.10;
software available from Chemical Computing Group Inc.: 1010 Sher-brooke Street West, Suite 910, Montreal, Canada H3A 2R7; http://www.chemcomp.com.(19) Labute, P. Proteins: Struct. Funct. Bioinf. 2009, 75, 187.(20) Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey,
R. W.; Klein, M. L. J. Chem. Phys. 1983, 79, 926.
(21) Price, D. J.; Brooks, C. L. J. Chem. Phys. 2004, 121, 10096.(22) Miyamoto, S.; Kollman, P. A. J. Comput. Chem. 1992, 13, 952.(23) Wang, J.; Morin, P.; Wang, W.; Kollman, P. A. J. Am. Chem. Soc.
Chem. Phys. Lett. 2004, 396, 473.(26) Fedorov, D. G.; Kitaura, K. J. Chem. Phys. 2004, 121, 2483.(27) Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972,
M.; Miyoshi, E. J. Comput. Chem. 2007, 28, 2424.(29) Miyoshi, E.; Mori, H.; Hirayama, R.; Osanai, Y.; Noro, T.;
Honda, H.; Klobukowski, M.; Miyoshi, E. J. Chem. Phys. 2005,122, 074104.
(30) Osanai, Y.; Mon, M. S.; Noro, T.; Mori, H.; Nakashima, H.;Klobukowski, M.; Miyoshi, E. Chem. Phys. Lett. 2008, 452, 210.
(31) Schmidt, W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.;Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen,K. A.; Su, S. J.; Windus, T. L.; Dupuis, M. J. Comput. Chem. 1993,14, 1347.
(32) Cherstvy, A. G. J. Phys. Chem. B 2009, 113, 4242.(33) Lee, J.; Kim, J.-S.; Seok, C. J. Phys. Chem. B 2010, 114, 7662.