ab initio Lattice Vibrations: Calculating the Thermal Expansion Coeffcient Felix Hanke & Martin Fuchs June 30, 2009 This afternoon’s plan • Phonons: harmonic vibrations for solids • Phonons: how • Thermodynamics with phonons • Obtain & assess phonons for Si • Calculate & understand thermal expansion practical exercise introductory talk
Transcript
ab initio Lattice Vibrations:Calculating the Thermal Expansion Coeffcient
Felix Hanke & Martin FuchsJune 30, 2009
This afternoon’s plan
• Phonons: harmonic vibrations for solids
• Phonons: how
• Thermodynamics with phonons
• Obtain & assess phonons for Si
• Calculate & understand thermal expansion
practical exercise
introductory talk
Recap: Molecular vibrations (non-periodic)
Newton’s equations for small displacements of atom !, coordinate i about PES minimum:
In symmetric matrix form
!!2["
m!"x!,i] =!
",j
1"
m!m"
#2E
#("x!,i)#("x",j)["mj"x",j ]
m!x!,i = F!,i({x",j})
m!x!,i ! 0 +!
",j
!2E
!("x!,i)!("x",j)"x",j
The harmonic lattice
D!",ij =!
R
eik·R!
mimj
!2E
!r(R=0),!i!r(R),"j
Translational symmetry (PBC’s) leads to k-dependenceOne eigenvalue problem for each k, giving 3Natoms modes
D is the dynamic matrix, contains information fromall supercells R, all pairs of atoms ! and ", and all pairs
of coordinates i and j:
!!2n(k)"n(k) = D(k)"n(k)
Phonon dispersion relation
Recall diatomic molecules:
2 atoms = 6 degrees of freedom
How about solids?
• 3 translations• 2 rotations• 1 vibration
! = 0! = 0! = !vib
Diamond fcc conventional cell
2 atoms per primitive cell = 6 phonon branches
a = 3.5Å
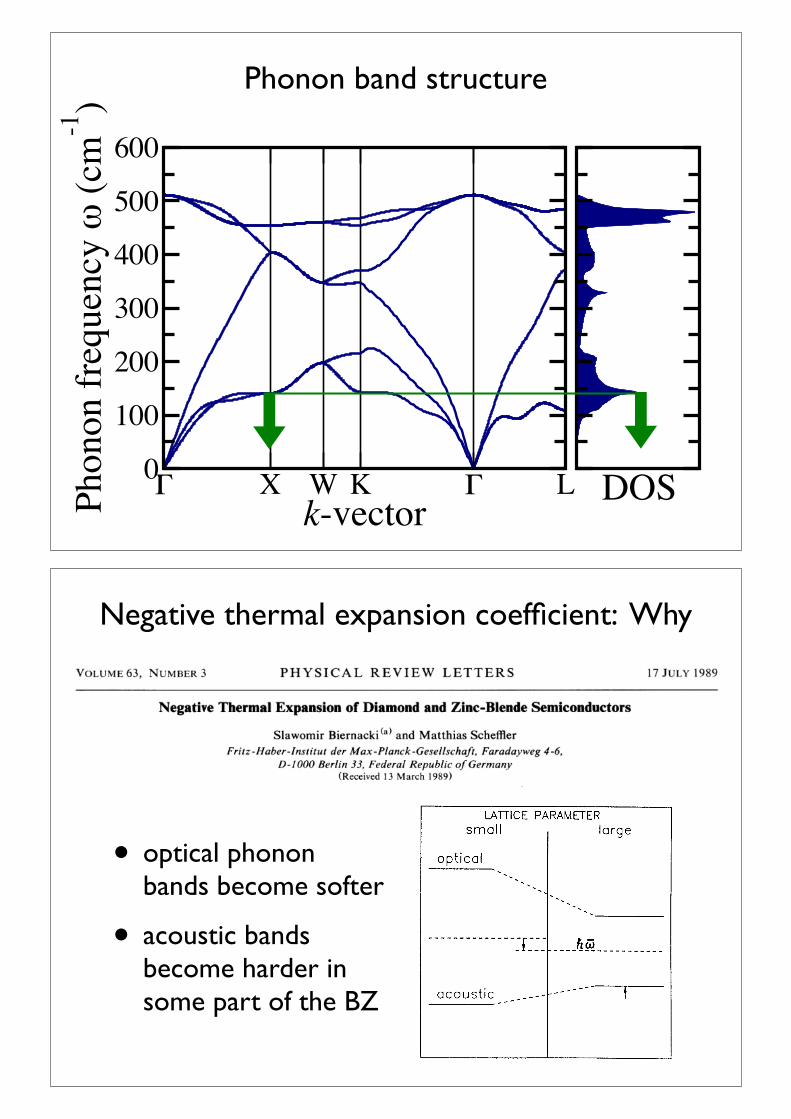
Phonon dispersion relation
! X W K ! Lk-vector
0
500
1000
1500
Fre
qu
ency
" (
cm-1
)
DOS0
500
1000
1500
acoustic branches
optical branches
Diamond fcc conventional cell
2 atoms per primitive cell = 6 phonon branches
a = 3.5Å
Phonon density of states
DOS = number of phonon modes per unit frequency per unit cell
smearing # makes it feasible to plot (same as electronic DOS!)
Converge smearing vs k-point sampling!
g(!) =!
n,k
"(! ! !n(k)) "!
n,k
exp"! (! ! !n(k))2
2#2
#
numerical computation: frequency interval [$+%$/2,$-%$/2] with discrete k-point sampling
g(!) =wk!2"
!
n,k
" !+!!/2
!!!!/2d! exp
#" (! " !n(k))2
2#2
$
The Direct Method
approximation: finite interaction distance, use finite supercells
D!",ij =!
R
eik·R!
mimj
!2E
!r(R=0),!i!r(R),"j
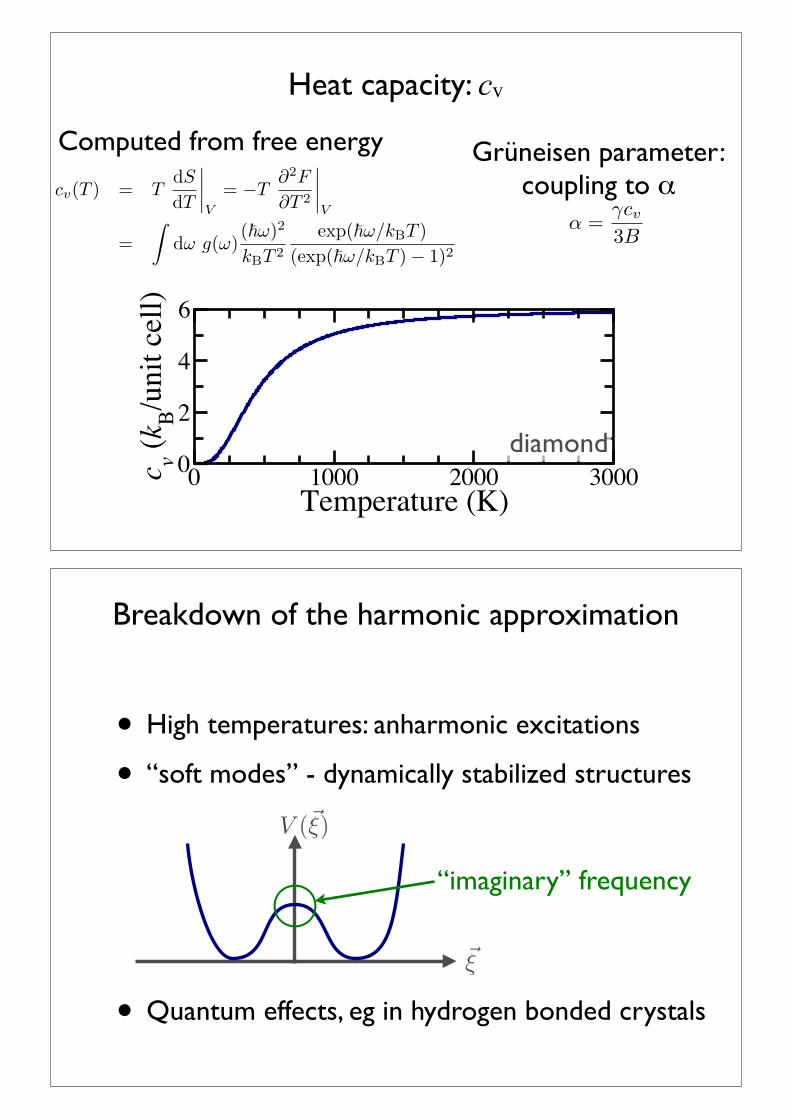
Harmonic approximation: Thermodynamics
Approximate thermal effects with independent harmonic oscillators: one for each mode
F =!
k,N
lnZ(k, N)
=!
k,N
"!!k,N
2+ kBT ln[1! exp(!!!k,N/kBT )]
#
Practically, one tends to use the density of states:
thermodynamic integration: slowly switching on anh terms from qh solution
Determine from self-consistently optimizing vacancy volume
Today’s tutorial: Thermal properties of Silicon
this talk afternoon exercise
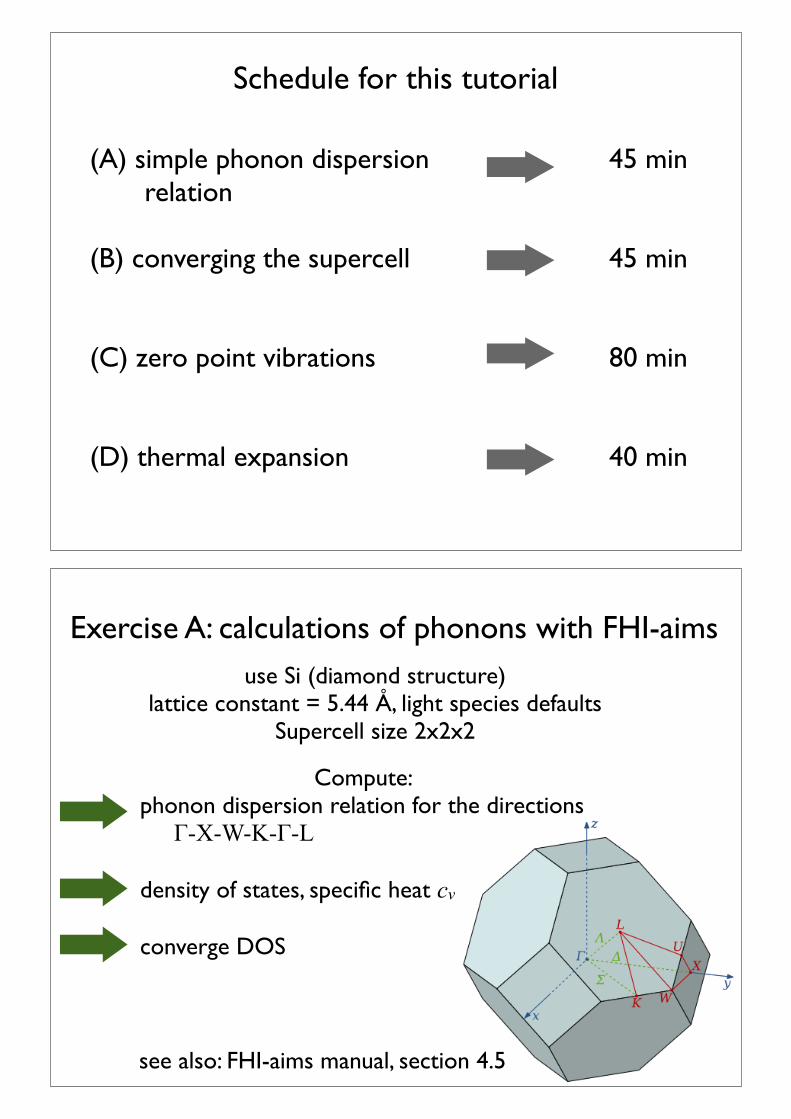
Schedule for this tutorial
(A) simple phonon dispersion relation
(B) converging the supercell
(C) zero point vibrations
(D) thermal expansion
45 min
45 min
80 min
40 min
Exercise A: calculations of phonons with FHI-aims
see also: FHI-aims manual, section 4.5
use Si (diamond structure)lattice constant = 5.44 Å, light species defaults
Supercell size 2x2x2
Compute: phonon dispersion relation for the directions "-X-W-K-"-L
density of states, specific heat cv
converge DOS
Exercise A: calculations of phonons with FHI-aims
Phonon calculation works similar to vibrations: run aims.phonons.workshop.mpi.pl in the directory containing your control.in and geometry.in files.
Please specify a SINGLE PRIMITIVE CELL ONLY!The script does all the necessary copying & displacing.
Attention
Outputphonon_band_structure.dat - same format as e-bandsphonon_DOS.dat - frequency, density of statesphonon_free_energy.dat - T, F(T), U(T), cv(T), Svib(T)output stream - status reportsphonon_workdir/ - working files & restart info
Exercise A: calculations of phonons with FHI-aims
Phonon dispersion calculation completely driven by phonon keyword in control.in (see FHI-aims manual, section 3.5)
k-grid: hand set to match supercell size, i.e. k_grid 6 6 6
Band structure:phonon band <start> <end> <Npoints> <sname> <ename>
working directory:/usr/local/aimsfiles/tutorial6/exercise_A
Exercise A: Solutions
! X W K ! Lk-vector
0
100
200
300
400
500F
requen
cy "
(cm
-1)
DOS0
200
400
0 200 400 600 800Temperature (K)
0
2
4
6
cv (
kB/u
nit
cel
l)
Exercise B: Supercell convergence
NOTE: We have provided partial control.in and geometry.in files as well as ALL of the required DFT
output for the remainder of this tutorial
Compute phonon dispersion and DOS for the supercell sizes 4x4x4, 6x6x6
Use the converged DOS settings from the last exercise.All necessary files are in directory/usr/local/aimsfiles/tutorial6/exercise_B
Exercise B: solutions2x2x2
4x4x46x6x6Ph
onon
freq
uenc
y !
(cm
-1)
k-vector
0
250
500
0
250
500
! X W K ! L0
250
500
Exercise B: solutions
0 200 400
Phonon frequency (cm-1
)
DO
S (
arb
un
its)
2x2x24x4x46x6x6
Exercise C: zero point vibrations
Using data for the lattice constants provided in the directory /usr/local/aimsfiles/tutorial6/exercise_C+D
(***) These exact settings are important for exercise D
For each lattice constant provided, calculate the total energy of a single unit cell using a 12x12x12 k-point grid. Store the result in the format 4x4x4_a*.***/output.single_point_energy
Again, for each lattice constant, calculate the phonon free energy & specific heat for a temperature range from 0K to 800K in 801 steps (***)
Extract the ZPE from the T=0 free energies from file phonon_free_energy.dat in each directory
produce a Murnaghan fit with and without ZPE and plot the two fits & the resulting lattice constants
Exercise C: solutions
5.30 5.35 5.40 5.45 5.50 5.55Lattice constant a (Å)
0
20
40
60
Ene
rgy
(meV
)
ZPEE(a), no ZPEE(a), incl ZPE
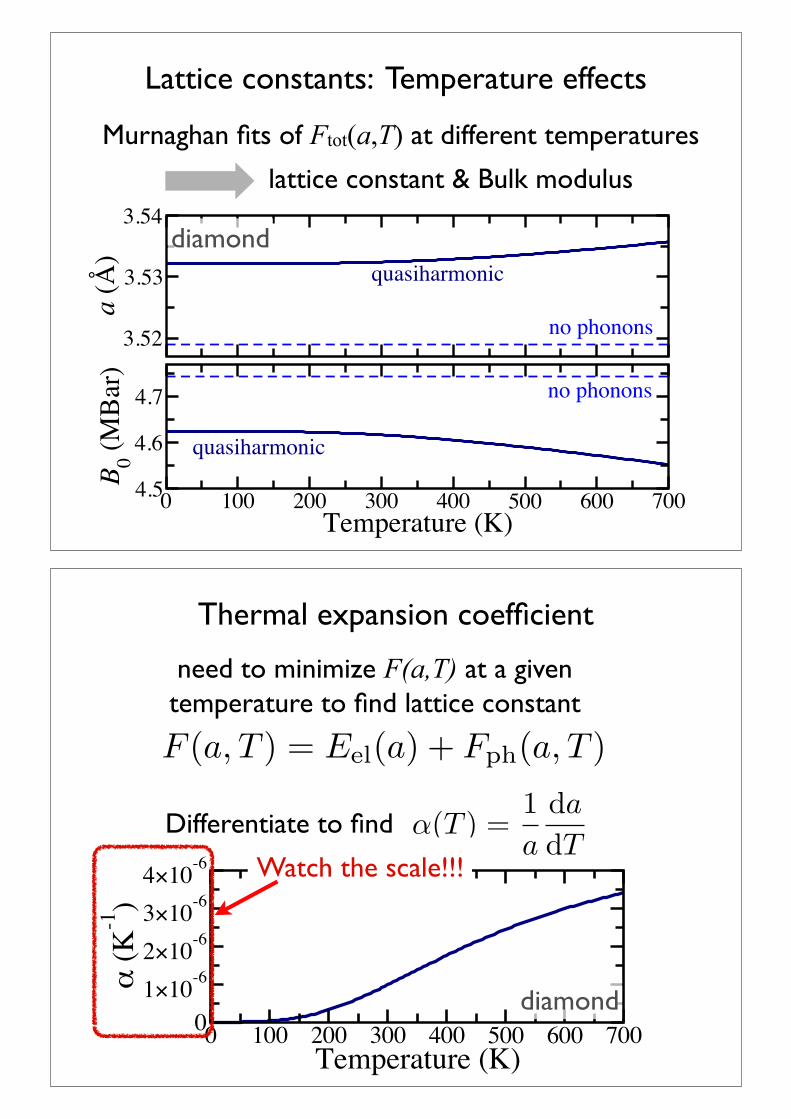
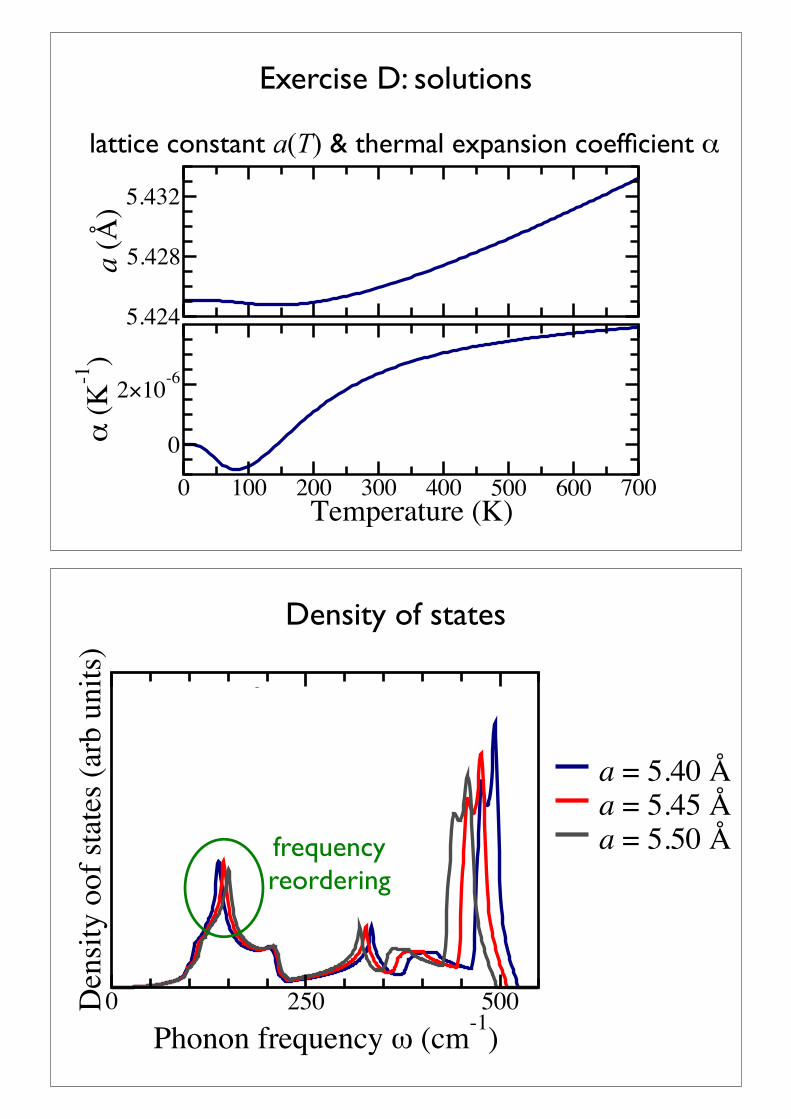
Exercise D: compute a(T), cv & !
Again, using the data provided directory/usr/local/aimsfiles/tutorial6/exercise_C+D
use the script eval_alpha.sh to calculate the lattice constant at different temperatures & the thermal expansion coefficient eval_alpha.sh has to be started in the directory exercise_C+D and requires the EXACT specifications of phonon free_energy from the last exercise.
find cv(T) at the optimal lattice constant a=5.44ÅCompare with previously calculated values