40 This arTicle has been peer-reviewed. Computing in SCienCe & engineering
H i g h - P e r f o r m a n c e C o m p u t i n g w i t h A c c e l e r a t o r s
Graphical processing units are now being used with dramatic effect to accelerate quantum chemistry calculations. However, early work exposed challenges involving memory bottlenecks and insufficient numerical precision. This research effort addresses those issues, proposing two new tools for accelerating matrix multiplications of arbitrary size where single-precision accuracy is not enough.
Accelerating Correlated Quantum Chemistry Calculations Using Graphical Processing Units
W ith the advent of modern quan-tum theory a century ago, sci-entists quickly realized that quantum mechanics could offer
a predictive theory of chemistry, revolutionizing the subject in the same way that Newton’s laws had transformed the study of classical mechanics. Over the intervening decades, we’ve witnessed an exponential increase in available computing power. Coupled with tremendous advances in theory and algorithmic methods, as well as the painstaking development of sophisticated pro-gram suites often consisting of millions of lines of code, the scope of phenomena now amenable to quantum chemistry’s predictive techniques is large and continually growing.
Modern computational chemistry has an im-portant role to play in many practical applications, such as the discovery of new drugs and industrial catalysts, and the development of new materials and technologies to meet global energy and en-vironmental challenges. Computational chemis-try’s widespread application has resulted in major efforts to reduce its computational cost: accurate quantum chemistry methods consume a signifi-cant fraction of computing resources at national laboratories.
As a result, we’re witnessing a new era in quan-tum chemistry code optimization that follows on the explosion of interest in using coprocessors such as graphical processing units. This interest in GPUs and other accelerators is largely driven by their combination of formidable performance and relatively low cost. But another key reason for their emergence in scientific fields was the release of Nvidia’s Compute Unified Device Architecture (CUDA) toolkit, which dramatically simplified code development for GPUs.
Already, computational chemists have be-gun using GPUs to accelerate molecular dy-namics and quantum Monte Carlo simulations, density-functional theory and self-consistent field calculations,1,2 as well as correlated quan-tum chemistry methods.3,4 Recent studies have
Mark A. Watson, Roberto Olivares-Amaya and Richard G. EdgarHarvard UniversityTomás AriasCornell UniversityAlán Aspuru-GuzikHarvard University
reported efficiency gains of between one and three orders of magnitude compared to conven-tional CPU implementations. Thus, state-of-the-art scientific applications are now possible that previously required extremely expensive and rare supercomputing facilities.
Many scientific applications require the accel-eration of linear algebra operations, which are quite well suited for GPU architectures. The CUDA software development toolkit includes an implementation of the basic linear algebra subprograms (BLAS) library ported to CUDA (Cublas). By simply replacing the BLAS *GEMM (general matrix multiply) routines with corre-sponding Cublas SGEMM (single-precision GEMM) calls to accelerate key matrix multiplications, our group was able to achieve a speedup of 4.3 times when calculating the RI-MP2 (resolution-of-the-identity second-order Møller-Plesset perturba-tion theory5,6) correlation energy of doeicosane (C22H46).3 This initial effort was one of the first quantum chemistry applications to leverage GPUs, but it revealed several issues for future work. For example, although modern GPU cards designed for research can have up to 4 Gbytes of RAM, consumer-level cards might have as little as 256 Mbytes. So, without some way to overcome the memory bottleneck, our first attempts to use GPUs to accelerate RI-MP2 calculations were limited to systems with 538 functions. Another is-sue is numerical precision. Most GPU cards cur-rently in use support only single-precision (SP) arithmetic. SP is generally insufficient to achieve chemical accuracy of 1 kilocalorie per mole (kcal/mol) in calculations on anything but the smallest and simplest systems because errors quickly accu-mulate for larger molecules. (Interestingly, in the computer science community, researchers have used algorithmic techniques to develop librar-ies that achieve precision beyond the hardware specification.7,8)
Here, we offer a detailed examination of this problem for the special case of SP GPU devices and applications to quantum chemistry. We de-scribe our efforts to develop tools for the GPU acceleration of correlated quantum chemistry cal-culations3,4 as well as address the limited GPU de-vice memory and how to achieve higher accuracy using only SP GPU operations.
Quantum chemistry theoryTraditional quantum chemistry strives to solve the time-independent Schrödinger equation,
ˆ ( ) ( ) ( ) ( ),H Er,R r,R R r,Ry y= (1)
where y(r, R) is the electronic wavefunction for a molecular system defined by the Hamilto-nian H (r, R) in terms of a set of coordinates for the electrons, r, and nuclei, R. E(R) is the total molecular energy, or potential energy surface. Equa-tion 1’s solution yields the electronic wavefunc-tion for a given nuclear configuration, and in turn the probability density for finding an electron at a given point in space. Exact solution is intrac-table, even numerically, for all but the simplest systems. This is because of the formidable nature of the many-body problem inherent in the form of H (r, R). Exact solutions are readily available only for one-electron systems, where there’s no electron-electron coupling.
Nevertheless, it’s a hallmark of quantum chem-istry that there’s a well-defined hierarchy of meth-ods for approximately solving Equation 1. Indeed, given sufficient computational power, a solu-tion might be improved systematically to yield a wavefunction of arbitrary numerical accuracy.
The simplest method in the hierarchy is a mean-field approach known as Hartree-Fock theory. In HF theory, we write the electronic wavefunction for an N-electron system as an anti symmetrized product of molecular orbitals, f(ri), which are wavefunctions for a single elec-tron under a one-electron Hamiltonian known as the Fock operator:
ˆ ( , ) ˆ( , ) [ ˆ ( ) ˆ ( )]./
f h Kj jj
N
r R r R r r= + −=
∑ 21
2
J
The Coulomb, ˆ ( )J j r , exchange, ˆ ( )K j r , and one-electron, ˆ ( )hj r , operators are given by
ˆ ( )
( ) ( ),
*
J jj j dr
r r
r rr=
′ ′
− ′′∫
f f
| |
ˆ ( ) ( )
( ) ( )( ),
*
K dj ij i
jr rr r
r rr rf
f ff=
′ ′
− ′′∫ | |
without some way to overcome the memory
bottleneck, our first attempts to use GpUs to
accelerate ri-Mp2 calculations were limited
to systems with 538 functions.
CISE-12-4-Watson.indd 41 04/06/10 12:01 PM
42 Computing in SCienCe & engineering
and
ˆ( , ) ( , ),h r R r R= − ∇ +12
2 v
respectively, where v(r, R) is the nuclear potential. The associated HF equations,
ˆ ( ) ( ),f i i iφ ε φr r=
permit a complete set of eigenfunctions. In the simplest case, where N is even, each of the N/2 orbitals corresponding to the lowest eigenvalues is associated with two electrons of opposite spin. These occupied orbitals are used to construct the many-electron wavefunctions; they’re labeled by the subscripts i, j, … . The remaining functions form the virtual orbitals and are indicated by the subscripts a, b, … .
Although in some implementations, the HF equations are solved numerically, the traditional approach is to expand the molecular orbitals (MOs) in a basis of atomic orbitals, {ha},
φ ηα α
αp p
N
cb
( ) ( )r r= ∑ .
The optimized coefficients, cap, and orbital ener-gies, ep, are determined from the secular equa-tions, which we can write in matrix notation as FC = SCe, where S is the atomic orbital (AO) overlap matrix, ⟨ha|hb⟩; Fis the AO Fock matrix, ⟨ha| f |hb⟩; C is the matrix of MO coefficients; and e is the diagonal matrix of MO energies. These are the celebrated Roothaan-Hall self-consistent field equations.
The HF solution (in a complete AO basis) gen-erally recovers more than 99 percent of the exact energy, which is remarkable. However, the accu-rate and efficient recovery of the neglected energy, or correlation energy, is quantum chemistry’s cen-tral challenge since an acceptable accuracy in the total energy is usually quoted as 1 kcal/mol for
predictive chemistry, while the correlation energy for two electrons in a doubly occupied orbital is about 25 kcal/mol.
Currently, density functional theory is the most popular approach for solving Equation 1 with electron correlation. DFT, which bypasses explic-it construction of the many-body wave function and focuses only on the much simpler 3D elec-tron density as the basic variable, is extremely useful because it favorably balances accuracy and efficiency.
Here, we examine another approach: MP2, a widely used and computationally efficient cor-related wavefunction-based method. MP2 can produce equilibrium geometries of comparable accuracy to DFT, and, in particular, it can cap-ture long-range correlation effects such as the dispersion interaction, where conventional DFT fails. For many weakly bound systems where DFT results are often questionable, MP2 is essentially the least expensive and most reliable alternative.
If HF theory can be viewed as a first-order solution to Equation 1, then MP2 theory is the second-order solution within the perturbation theory framework, where the many-electron Hamiltonian is partitioned as
ˆ ˆ ( ) ˆ .( )H f Hii
= +∑ r l 1
We’ve introduced an order parameter, l, to expand the energy and wavefunction,
E E E E= + + +( ) ( ) ( )0 1 2 2l l �
ψ ψ λψ λ ψ= + + +HF( ) ( )1 2 2 � ,
where yHF is the zero-order (HF) wavefunc-tion, and the zero and first-order energies are given by
E ii
( )0 = ∑e
and
E E H( ) ( )HF HF
ˆ .0 1+ = ⟨ ⟩λ ψ ψ| |
The second-order (MP2) correlation energy takes the form
E
ia jb ia jb ib ja
i j a
( )( ) [( ) ( )]
2
2 212=
+ −
+ − −
| | |
e e e eebijab∑ ,
Mp2 can produce equilibrium geometries of
comparable accuracy to density functional
theory, and, in particular, it can capture long-
range correlation effects such as the dispersion
interaction, where conventional dFT fails.
CISE-12-4-Watson.indd 42 04/06/10 12:01 PM
July/auguSt 2010 43
where the MO integrals,
( ) ( ),ij ab C C C Ci j a b| |= ∑ µ ν λ σµνλσ
µν λσ
are obtained by transforming the AO integrals,
( | )( ) ( ) ( ) ( )
µν λση η η ηµ ν=
−∫∫r r r r
r rr1 1 2 2
1 2
λ σ
| |d 11 2dr .
(2)
One way that we can considerably reduce the computational cost of MP2 calculations is to expand products of AOs as linear combinations of atom-centered auxiliary basis functions, P,
ρ η η ρµν µ ν µν µν( ) ( ) ( ) ( ) ( ),,r r r r r= ≈ = ∑� C PP
and therefore approximate Equation 2 in terms of only two- and three-index quantities as
( ) ( )( ) ( ).,
µν λσ µν λσ| | | |� = ∑ −
P Q
P P Q Q1
The idea is equivalent to an approximate insertion of the resolution of identity (RI),
I m m P P Q QP Qm
= ≈ ∑∑ −| | | | |)( )( ) ( ,,
1
from which the RI-MP2 name is derived.Our work is implemented in a development ver-
sion of Q-Chem 3.1,9 where the RI-MP2 corre-lation energy is evaluated in five steps.3 As we’ve shown in previous work, steps 3 and 4—where the approximate MO integrals are formed—were by far the most expensive operations for medium to large-sized systems, and require the matrix multiplications
( ) , ,ia jb B BiaQ jb QQ
|� ≈ ∑ (3)
and
B ia P P Qia QP
,/( )( ) .= −∑ | | 1 2
(4)
We’ll concentrate on these two operations here.
gpu acceleration of GEMMAs we now describe, our cleaving algorithm lets us accelerate matrix multiplications of arbitrary size on the GPU (assuming sufficient CPU memory). We also propose two different algorithms for the
MGEMM (mixed-precision GEMM) library (http://scigpu.org), using two different schemes to par-tition the matrices into simpler components.
cleaving GEMMsConsider the matrix multiplication C = AB,where Ais an (m × k) matrix, Bis a (k × n) matrix, and C is an (m × n) matrix. We can divide Ainto a column vector of r + 1 matrices
A
AA
A
=
0
1
�
r
,
where each entry Ai is a (pi × k) matrix, and
p mii
r=∑ .
In practice, all the pi will be the same, with the possible exception of pr, which will be an edge case.
In a similar manner, we can divide B into a row vector of s + 1 matrices B = (B0, B1 … Bs ), where each Bj is a (k × qj) matrix and
q njj
s=∑ .
Again, all the qj will be the same, with the pos-sible exception of qs. We then form the outer prod-uct of these two vectors:
C
AA
A
B B=
0
10�
r
( 11�Bs )
=
A B A B A BA B A B A B
A B A B
0 0 0 1 0
1 0 1 1 1
0
�
� �
s
s
r r s
.
Our cleaving algorithm lets us accelerate
matrix multiplications of arbitrary size on the
GpU (assuming sufficient cpU memory).
CISE-12-4-Watson.indd 43 04/06/10 12:01 PM
44 Computing in SCienCe & engineering
Each individual Cij = AiBj is a (pi × qj) matrix, and can be computed independently. Generaliz-ing this to a full *GEMM implementation, which includes the possibility of transposes being taken, is tedious but straightforward.
We’ve implemented this approach for the GPU as a complete replacement for *GEMM. We chose the pi and qj values such that each sub-multiplication fits within the currently available GPU memory. We stage each multiplication through the GPU and assemble the results on the CPU. This process is hidden from the user code, which simply sees a standard *GEMM call. We’re thus able to multiply matrices of arbitrary size using MGEMM, regardless of the available GPU memory.
Bitwise MGEMM algorithmConsider the partitioning of a double-precision (DP) floating-point number, A = m × 2k,
A A Au l≈ + ,
where Au and Al are SP numbers storing the uppermost nu, and the next lowest nl signifi-cant bits of m, respectively. We next consider the multiplication of two scalars, A and B. Applying the bitwise partitioning, we can ap-proximate the full DP multiplication as four SP multiplications,
AB A B A B A B A Bu u u l l u l l≈ + + + . (5)
Anticipating the performance of Equation 5, we can introduce an alternative approximation, involving only three SP multiplications,
AB A B A B A B Bu u u l l u l≈ + + +( ), (6)
where Bu + Bl is replaced by the SP cast of B. In general, we can expect the round-off error associ-ated with AlBu to be of the same order of magni-tude, on average, as the round-off error associated with Al(Bu + Bl).
Finally, we can generalize Equation 6 for the matrix multiplication,
AB A B A B A B B≈ + + +u u u l l u l( ), (7)
where, for each element of X ∈ {A, B},
X X Xij iju
ijl≈ + .
As above, we can consider the final term as ei-ther two separate multiplications, or as a single
multiplication where Bu + Bl is pre-summed. We can efficiently evaluate all the multiplications on the GPU using the Cublas SGEMM library rou-tines. We can then accumulate the results in DP on the CPU to yield the final approximation for AB.
For Equation 7 to be exact, in addition to the scalar multiplication issue, we have the additional round-off errors arising from the multiply-add operations. Specifically, if A is an M × K matrix and B is a K × N matrix, AB effectively con-sists of MN dot products of length K. As we ex-plore later in the benchmarks, as K increases or the range of magnitudes of Xij becomes wider, more round-off errors will occur due to the accumulation in SP.
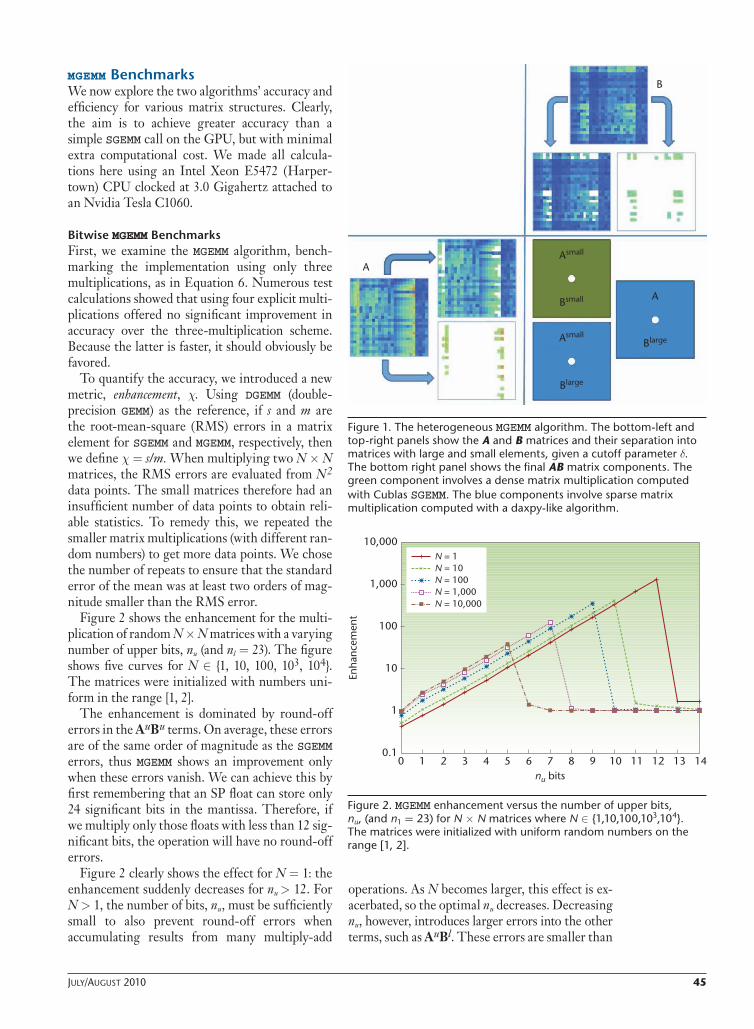
heterogeneous MGEMM algorithmA different way to improve precision is to consider the magnitude of the matrix elements from the outset. That is, we decompose the matrix multi-plication, C = AB, by splitting A and B into large and small components, giving us
where we’ve simply introduced a cutoff value, d, to define the split—that is, if |Xij| > d, we consider the element large; otherwise, we consider it small. AsmallBsmall consists entirely of small numbers, and we can run it with reasonable accuracy in SP on the GPU (using the cleaving approach, if needed). The other two terms contain large numbers and must run in DP to achieve greater accuracy. How-ever, because each of the large matrices will often be sparse, each term consists of a dense-sparse multiplication.
We store only nonzero terms of the Alarge and Blarge matrices, cutting the computational com-plexity significantly. Consider
′ =C A Bik ij jklarge .
Only a few Bjklarge will be nonzero, and we consider
each in turn. For a particular scalarB jklarge, only the
kth column of ′C will be nonzero and is equal to the product of B jk
large and the jth column of A. We can add the nonzero column vector ′Cik to the final result, C, and consider the next B jk
large.We can apply a similar process to AlargeBsmall (producing row vectors of C). Again, we can generalize this approach to a full *GEMM implementation includ-ing transposes. Figure 1 shows an illustration of the heterogeneous MGEMM algorithm.
CISE-12-4-Watson.indd 44 04/06/10 12:01 PM
July/auguSt 2010 45
MGEMM BenchmarksWe now explore the two algorithms’ accuracy and efficiency for various matrix structures. Clearly, the aim is to achieve greater accuracy than a simple SGEMM call on the GPU, but with minimal extra computational cost. We made all calcula-tions here using an Intel Xeon E5472 (Harper-town) CPU clocked at 3.0 Gigahertz attached to an Nvidia Tesla C1060.
Bitwise MGEMM BenchmarksFirst, we examine the MGEMM algorithm, bench-marking the implementation using only three multiplications, as in Equation 6. Numerous test calculations showed that using four explicit multi-plications offered no significant improvement in accuracy over the three-multiplication scheme. Because the latter is faster, it should obviously be favored.
To quantify the accuracy, we introduced a new metric, enhancement, c. Using DGEMM (double-precision GEMM) as the reference, if s and m are the root-mean-square (RMS) errors in a matrix element for SGEMM and MGEMM, respectively, then we define c = s/m. When multiplying two N × N matrices, the RMS errors are evaluated from N2 data points. The small matrices therefore had an insufficient number of data points to obtain reli-able statistics. To remedy this, we repeated the smaller matrix multiplications (with different ran-dom numbers) to get more data points. We chose the number of repeats to ensure that the standard error of the mean was at least two orders of mag-nitude smaller than the RMS error.
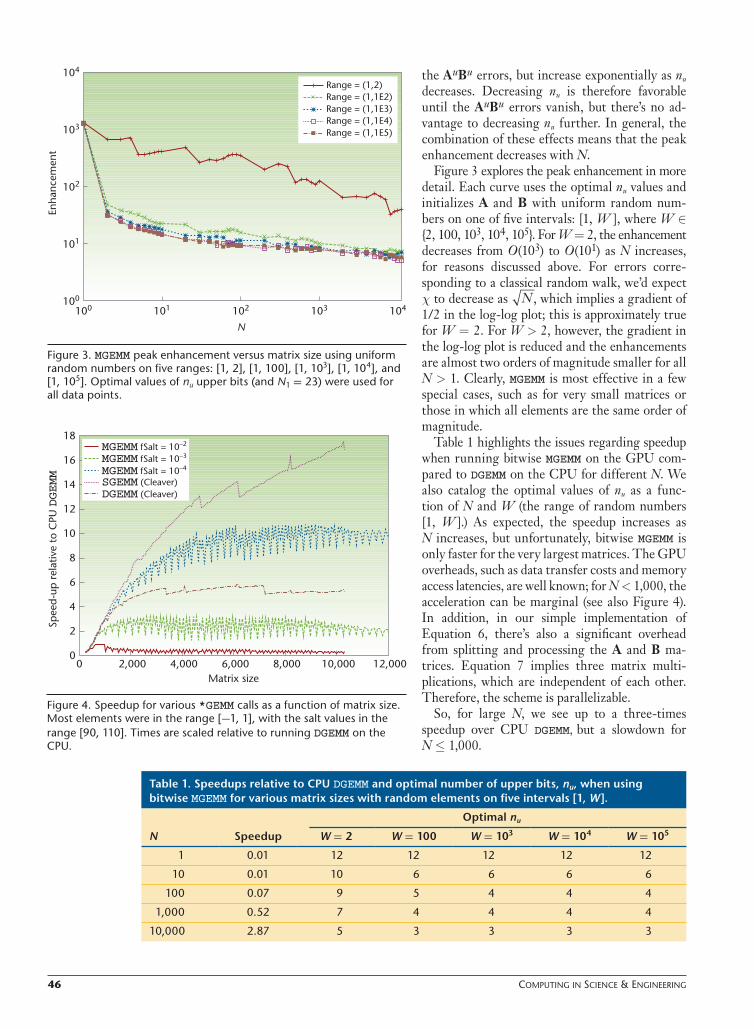
Figure 2 shows the enhancement for the multi-plication of random N × N matrices with a varying number of upper bits, nu (and nl = 23). The figure shows five curves for N ∈ {1, 10, 100, 103, 104}. The matrices were initialized with numbers uni-form in the range [1, 2].
The enhancement is dominated by round-off errors in the AuBu terms. On average, these errors are of the same order of magnitude as the SGEMM errors, thus MGEMM shows an improvement only when these errors vanish. We can achieve this by first remembering that an SP float can store only 24 significant bits in the mantissa. Therefore, if we multiply only those floats with less than 12 sig-nificant bits, the operation will have no round-off errors.
Figure 2 clearly shows the effect for N = 1: the enhancement suddenly decreases for nu > 12. For N > 1, the number of bits, nu, must be sufficiently small to also prevent round-off errors when accumulating results from many multiply-add
operations. As N becomes larger, this effect is ex-acerbated, so the optimal nu decreases. Decreasing nu, however, introduces larger errors into the other terms, such as AuBl. These errors are smaller than
Figure 2. MGEMM enhancement versus the number of upper bits, nu, (and n1 = 23) for N × N matrices where N ∈ {1,10,100,103,104}. The matrices were initialized with uniform random numbers on the range [1, 2].
0.1
1
10
100
1,000
10,000
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Enha
ncem
ent
nu bits
N = 1N = 10N = 100N = 1,000N = 10,000
Figure 1. The heterogeneous MGEMM algorithm. The bottom-left and top-right panels show the A and B matrices and their separation into matrices with large and small elements, given a cutoff parameter d. The bottom right panel shows the final AB matrix components. The green component involves a dense matrix multiplication computed with Cublas SGEMM. The blue components involve sparse matrix multiplication computed with a daxpy-like algorithm.
A
B
Asmall
Bsmall
Asmall
Blarge
Blarge
A
CISE-12-4-Watson.indd 45 04/06/10 12:01 PM
46 Computing in SCienCe & engineering
the AuBu errors, but increase exponentially as nu
decreases. Decreasing nu is therefore favorable until the AuBu errors vanish, but there’s no ad-vantage to decreasing nu further. In general, the combination of these effects means that the peak enhancement decreases with N.
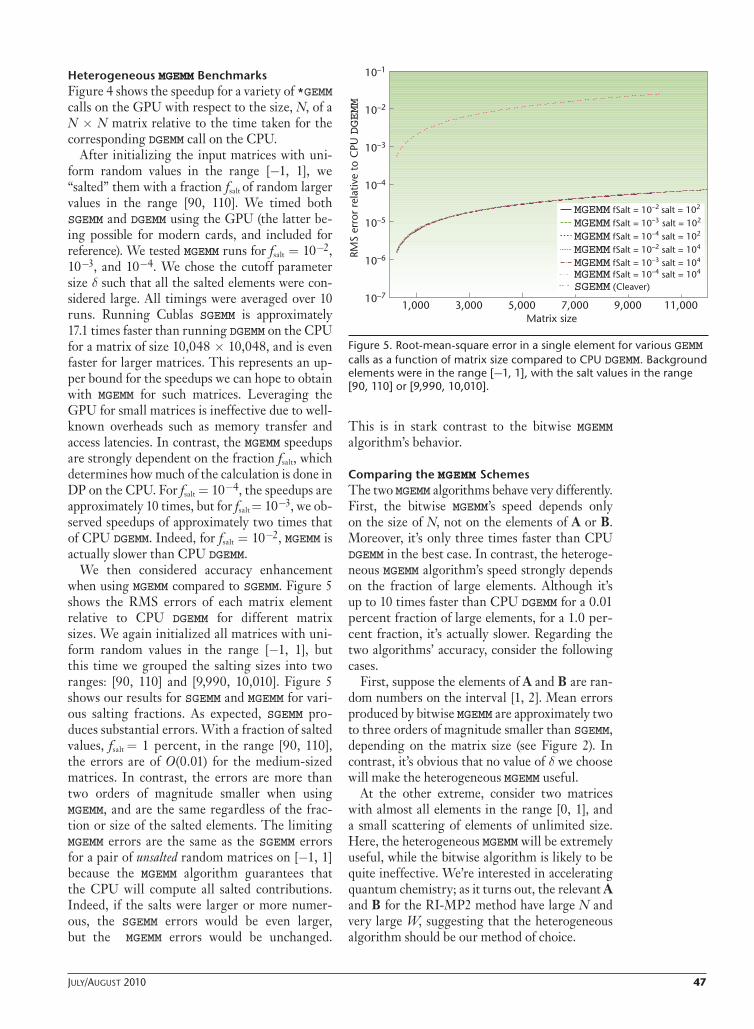
Figure 3 explores the peak enhancement in more detail. Each curve uses the optimal nu values and initializes A and B with uniform random num-bers on one of fi ve intervals: [1, W ], where W ∈{2, 100, 103, 104, 105}. For W = 2, the enhancement decreases from O(103) to O(101) as N increases, for reasons discussed above. For errors corre-sponding to a classical random walk, we’d expect c to decrease as N , which implies a gradient of 1/2 in the log-log plot; this is approximately true for W = 2. For W > 2, however, the gradient in the log-log plot is reduced and the enhancements are almost two orders of magnitude smaller for all N > 1. Clearly, MGEMM is most effective in a few special cases, such as for very small matrices or those in which all elements are the same order of magnitude.
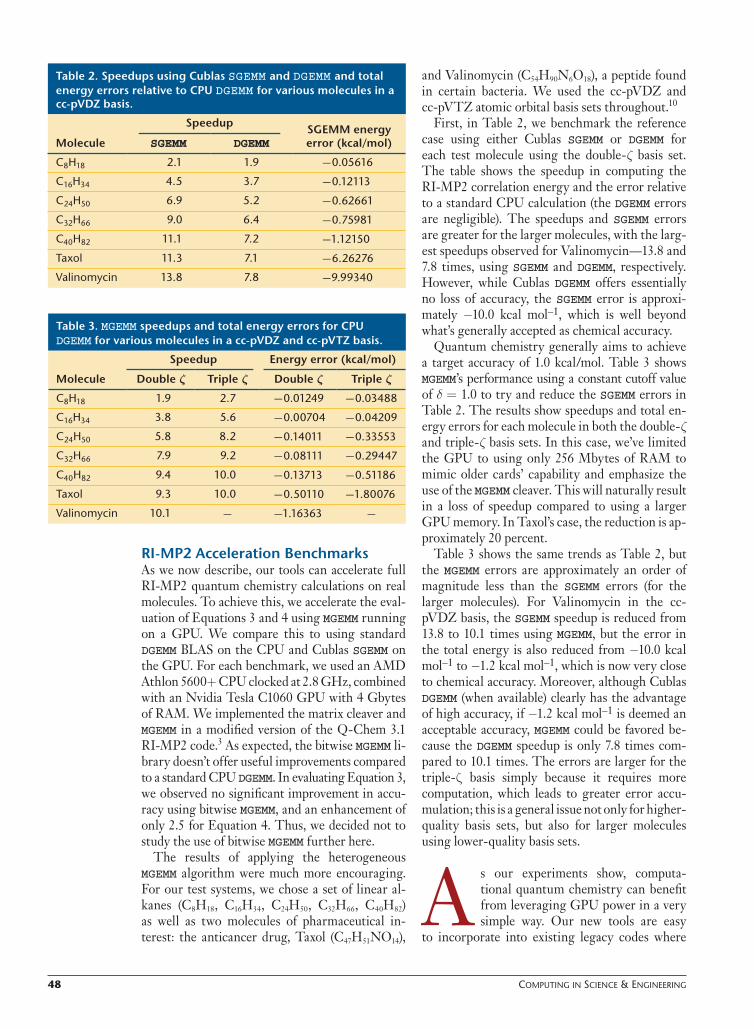
Table 1 highlights the issues regarding speedup when running bitwise MGEMM on the GPU com-pared to DGEMM on the CPU for different N. We also catalog the optimal values of nu as a func-tion of N and W (the range of random numbers [1, W ].) As expected, the speedup increases as N increases, but unfortunately, bitwise MGEMM is only faster for the very largest matrices. The GPU overheads, such as data transfer costs and memory access latencies, are well known; for N < 1,000, the acceleration can be marginal (see also Figure 4). In addition, in our simple implementation of Equation 6, there’s also a signifi cant overhead from splitting and processing the A and B ma-trices. Equation 7 implies three matrix multi-plications, which are independent of each other. Therefore, the scheme is parallelizable.
So, for large N, we see up to a three-times speedup over CPU DGEMM, but a slowdown for N ≤ 1,000.
Figure 4. Speedup for various *GEMM calls as a function of matrix size. Most elements were in the range [-1, 1], with the salt values in the range [90, 110]. Times are scaled relative to running DGEMM on the CPU.
table 1. speedups relative to cpu DGEMM and optimal number of upper bits, nu, when using bitwise MGEMM for various matrix sizes with random elements on fi ve intervals [1, w].
optimal nu
n speedup w = 2 w = 100 w = 103 w = 104 w = 105
1 0.01 12 12 12 12 12
10 0.01 10 6 6 6 6
100 0.07 9 5 4 4 4
1,000 0.52 7 4 4 4 4
10,000 2.87 5 3 3 3 3
Figure 3. MGEMM peak enhancement versus matrix size using uniform random numbers on fi ve ranges: [1, 2], [1, 100], [1, 103], [1, 104], and [1, 105]. Optimal values of nu upper bits (and N1 = 23) were used for all data points.
heterogeneous MGEMM BenchmarksFigure 4 shows the speedup for a variety of *GEMM calls on the GPU with respect to the size, N, of a N × N matrix relative to the time taken for the corresponding DGEMM call on the CPU.
After initializing the input matrices with uni-form random values in the range [-1, 1], we “salted” them with a fraction fsalt of random larger values in the range [90, 110]. We timed both SGEMM and DGEMM using the GPU (the latter be-ing possible for modern cards, and included for reference). We tested MGEMM runs for fsalt = 10-2, 10-3, and 10-4. We chose the cutoff parameter size d such that all the salted elements were con-sidered large. All timings were averaged over 10 runs. Running Cublas SGEMM is approximately 17.1 times faster than running DGEMM on the CPU for a matrix of size 10,048 × 10,048, and is even faster for larger matrices. This represents an up-per bound for the speedups we can hope to obtain with MGEMM for such matrices. Leveraging the GPU for small matrices is ineffective due to well-known overheads such as memory transfer and access latencies. In contrast, the MGEMM speedups are strongly dependent on the fraction fsalt, which determines how much of the calculation is done in DP on the CPU. For fsalt = 10-4, the speedups are approximately 10 times, but for fsalt = 10-3, we ob-served speedups of approximately two times that of CPU DGEMM. Indeed, for fsalt = 10-2, MGEMM is actually slower than CPU DGEMM.
We then considered accuracy enhancement when using MGEMM compared to SGEMM. Figure 5 shows the RMS errors of each matrix element relative to CPU DGEMM for different matrix sizes. We again initialized all matrices with uni-form random values in the range [-1, 1], but this time we grouped the salting sizes into two ranges: [90, 110] and [9,990, 10,010]. Figure 5 shows our results for SGEMM and MGEMM for vari-ous salting fractions. As expected, SGEMM pro-duces substantial errors. With a fraction of salted values, fsalt = 1 percent, in the range [90, 110], the errors are of O(0.01) for the medium-sized matrices. In contrast, the errors are more than two orders of magnitude smaller when using MGEMM, and are the same regardless of the frac-tion or size of the salted elements. The limiting MGEMM errors are the same as the SGEMM errors for a pair of unsalted random matrices on [-1, 1] because the MGEMM algorithm guarantees that the CPU will compute all salted contributions. Indeed, if the salts were larger or more numer-ous, the SGEMM errors would be even larger, but the MGEMM errors would be unchanged.
This is in stark contrast to the bitwise MGEMM algorithm’s behavior.
comparing the MGEMM schemesThe two MGEMM algorithms behave very differently. First, the bitwise MGEMM’s speed depends only on the size of N, not on the elements of A or B. Moreover, it’s only three times faster than CPU DGEMM in the best case. In contrast, the heteroge-neous MGEMM algorithm’s speed strongly depends on the fraction of large elements. Although it’s up to 10 times faster than CPU DGEMM for a 0.01 percent fraction of large elements, for a 1.0 per-cent fraction, it’s actually slower. Regarding the two algorithms’ accuracy, consider the following cases.
First, suppose the elements of A and B are ran-dom numbers on the interval [1, 2]. Mean errors produced by bitwise MGEMM are approximately two to three orders of magnitude smaller than SGEMM, depending on the matrix size (see Figure 2). In contrast, it’s obvious that no value of d we choose will make the heterogeneous MGEMM useful.
At the other extreme, consider two matrices with almost all elements in the range [0, 1], and a small scattering of elements of unlimited size. Here, the heterogeneous MGEMM will be extremely useful, while the bitwise algorithm is likely to be quite ineffective. We’re interested in accelerating quantum chemistry; as it turns out, the relevant A and Bfor the RI-MP2 method have large N and very large W, suggesting that the heterogeneous algorithm should be our method of choice.
Figure 5. Root-mean-square error in a single element for various GEMM calls as a function of matrix size compared to CPU DGEMM. Background elements were in the range [-1, 1], with the salt values in the range [90, 110] or [9,990, 10,010].
10–7
10–6
10–5
10–4
10–3
10–2
10–1
1,000 3,000 5,000 7,000 9,000 11,000
RMS
erro
r re
lativ
e to
CPU
DGEMM
Matrix size
MGEMM fSalt = 10–2 salt = 102
SGEMM (Cleaver)MGEMM fSalt = 10–4 salt = 104MGEMM fSalt = 10–3 salt = 104MGEMM fSalt = 10–2 salt = 104MGEMM fSalt = 10–4 salt = 102MGEMM fSalt = 10–3 salt = 102
CISE-12-4-Watson.indd 47 04/06/10 12:01 PM
48 Computing in SCienCe & engineering
ri-mp2 acceleration BenchmarksAs we now describe, our tools can accelerate full RI-MP2 quantum chemistry calculations on real molecules. To achieve this, we accelerate the eval-uation of Equations 3 and 4 using MGEMM running on a GPU. We compare this to using standard DGEMM BLAS on the CPU and Cublas SGEMM on the GPU. For each benchmark, we used an AMD Athlon 5600+ CPU clocked at 2.8 GHz, combined with an Nvidia Tesla C1060 GPU with 4 Gbytes of RAM. We implemented the matrix cleaver and MGEMM in a modifi ed version of the Q-Chem 3.1 RI-MP2 code.3 As expected, the bitwise MGEMM li-brary doesn’t offer useful improvements compared to a standard CPU DGEMM. In evaluating Equation 3, we observed no signifi cant improvement in accu-racy using bitwise MGEMM, and an enhancement of only 2.5 for Equation 4. Thus, we decided not to study the use of bitwise MGEMM further here.
The results of applying the heterogeneous MGEMM algorithm were much more encouraging. For our test systems, we chose a set of linear al-kanes (C8H18, C16H34, C24H50, C32H66, C40H82) as well as two molecules of pharmaceutical in-terest: the anticancer drug, Taxol (C47H51NO14),
and Valinomycin (C54H90N6O18), a peptide found in certain bacteria. We used the cc-pVDZ and cc-pVTZ atomic orbital basis sets throughout.10
First, in Table 2, we benchmark the reference case using either Cublas SGEMM or DGEMM for each test molecule using the double-ζ basis set. The table shows the speedup in computing the RI-MP2 correlation energy and the error relative to a standard CPU calculation (the DGEMM errors are negligible). The speedups and SGEMM errors are greater for the larger molecules, with the larg-est speedups observed for Valinomycin—13.8 and 7.8 times, using SGEMM and DGEMM, respectively. However, while Cublas DGEMM offers essentially no loss of accuracy, the SGEMM error is approxi-mately -10.0 kcal mol–1, which is well beyond what’s generally accepted as chemical accuracy.
Quantum chemistry generally aims to achieve a target accuracy of 1.0 kcal/mol. Table 3 shows MGEMM’s performance using a constant cutoff value of d = 1.0 to try and reduce the SGEMM errors in Table 2. The results show speedups and total en-ergy errors for each molecule in both the double-ζand triple-ζ basis sets. In this case, we’ve limited the GPU to using only 256 Mbytes of RAM to mimic older cards’ capability and emphasize the use of the MGEMM cleaver. This will naturally result in a loss of speedup compared to using a larger GPU memory. In Taxol’s case, the reduction is ap-proximately 20 percent.
Table 3 shows the same trends as Table 2, but the MGEMM errors are approximately an order of magnitude less than the SGEMM errors (for the larger molecules). For Valinomycin in the cc-pVDZ basis, the SGEMM speedup is reduced from 13.8 to 10.1 times using MGEMM, but the error in the total energy is also reduced from -10.0 kcal mol–1 to -1.2 kcal mol–1, which is now very close to chemical accuracy. Moreover, although Cublas DGEMM (when available) clearly has the advantage of high accuracy, if -1.2 kcal mol–1 is deemed an acceptable accuracy, MGEMM could be favored be-cause the DGEMM speedup is only 7.8 times com-pared to 10.1 times. The errors are larger for the triple-ζ basis simply because it requires more computation, which leads to greater error accu-mulation; this is a general issue not only for higher-quality basis sets, but also for larger molecules using lower-quality basis sets.
A s our experiments show, computa-tional quantum chemistry can benefi t from leveraging GPU power in a very simple way. Our new tools are easy
to incorporate into existing legacy codes where
table 2. speedups using cublas SGEMM and DGEMM and total energy errors relative to cpu DGEMM for various molecules in a cc-pVDZ basis.
speedup sgemm energy error (kcal/mol)molecule SGEMM DGEMM
C8H18 2.1 1.9 -0.05616
C16H34 4.5 3.7 -0.12113
C24H50 6.9 5.2 -0.62661
C32H66 9.0 6.4 -0.75981
C40H82 11.1 7.2 -1.12150
Taxol 11.3 7.1 -6.26276
Valinomycin 13.8 7.8 -9.99340
table 3. MGEMM speedups and total energy errors for cpu DGEMM for various molecules in a cc-pVDZ and cc-pVtZ basis.
speedup energy error (kcal/mol)
molecule Double ζ triple ζ Double ζ triple ζ
C8H18 1.9 2.7 -0.01249 -0.03488
C16H34 3.8 5.6 -0.00704 -0.04209
C24H50 5.8 8.2 -0.14011 -0.33553
C32H66 7.9 9.2 -0.08111 -0.29447
C40H82 9.4 10.0 -0.13713 -0.51186
Taxol 9.3 10.0 -0.50110 -1.80076
Valinomycin 10.1 - -1.16363 -
CISE-12-4-Watson.indd 48 04/06/10 12:01 PM
July/auguSt 2010 49
matrix multiplications involve a substantial frac-tion of the overall computational cost. Clearly, it’s possible to achieve more efficient GPU use in special cases by devoting time to rewriting and re-designing the algorithms for GPU architectures. However, this is often not a feasible option, es-pecially for a mature discipline such as quantum chemistry where we typically employ large pro-gram suites representing years or decades of cod-ing effort.
Traditionally, GPUs have had only SP sup-port, and most GPU cards worldwide currently lack DP capability. Indeed, we’re interested in us-ing commodity GPUs within a grid computing environment, such as that promoted by the Berkeley Open Infrastructure for Network Computing.11 GPUs in a typical BOINC client have no DP support, yet comprise a formidable resource worldwide. Nonetheless, DP devices and coprocessors are now available. Moreover, the trend seems to be toward the release of more advanced DP support, such as Fermi, the current code name for Nvidia’s next-generation GPU. Fermi will reportedly have a DP peak perfor-mance that’s only a factor of two less than the SP performance (for Nvidia’s C1060, the ratio is approximately 12).
It’s therefore valid to question the poten-tial of mixed-precision algorithms for next- generation GPU architectures. We believe this is an open issue. For example, practical calcula-tions on GPUs are typically bound by memory bandwidth, rather than raw operation count. Although DP cards are getting faster, this I/O bottleneck is likely to remain. In these cases, the transfer and processing of only SP data could ef-fectively double the performance compared to naive DP calculations. Overall, we believe that mixed-precision algorithms could remain im-portant for applications where the highest per-formance is a priority.
acknowledgmentsWe thank Leslie Vogt for helpful discussions and her contributions to the numerical benchmarks. The US National Science Foundation’s Cyber-Enabled Discovery and Innovation Award PHY-0835713 provided financial support, as did Nvidia; we also thank Conacyt and Fundación Harvard en México for additional financial support. Computations in this article were run on the Odyssey cluster sup-ported by the FAS Research Computing Group. We also thank the US National Nanotechnology Infra-structure Network Computation project for techni-cal support.
references1. K. Yasuda, “Two-Electron Integral Evaluation on the
Graphics Processor Unit,” J. Computational Chemistry,
vol. 29, no. 3, 2008, pp. 334–342.
2. I.S. Ufimtsev and T.J. Martinez, “Quantum Chemistry
on Graphical Processing Units, 1 Strategies for Two-
Electron Integral Evaluation,” J. Chemical Theory and
Computation, vol. 4, no. 2, 2008, pp. 222–231.
3. L. Vogt et al., “Accelerating Resolution-of-the-
Identity Second-Order Møller-Plesset Quantum
Chemistry Calculations with Graphical Processing
Units,” J. Physical Chemistry A, vol. 112, no. 10, 2008,
pp. 2049–2057.
4. R. Olivares-Amaya et al., “Accelerating Correlated
Quantum Chemistry Calculations Using Graphical
Processing Units and a Mixed Precision Matrix Multi-
plication Library,” J. Chemical Theory and Computa-
tion, vol. 6, no. 1, 2010, pp. 135–144.
5. T. Helgaker, P. Jørgensen, and J. Olsen, Molecular
Electronic-Structure Theory, John Wiley & Sons, 2000.
6. M. Feyereisen, G. Fitzgerald, and A. Komornicki,
“Use of Approximate Integrals in ab initio Theory,”
Chemical Physics Lett., vol. 208, nos. 5–6, 1993,
pp. 359–363.
7. X. Li et al., “Design, Implementation and Testing
of Extended and Mixed Precision BLAS,” ACM Trans.
Mathematical Software, vol. 28, no. 2, 2002,
pp. 152–205.
8. Y. Hida, X.S. Li, and D.H. Bailey, “Algorithms for
Quad-Double Precision Floating Point Arithmetic,”
Proc. 15th IEEE Symp. Computer Arithmetic, IEEE CS
Press, 2001, p. 155–162.
9. Y. Shao et al., “Advances in Methods and Algorithms
in a Modern Quantum Chemistry Program Package,”
Physical Chemistry Chemical Physics, vol. 8, 2006,
pp. 3172–3191.
10. T. Dunning Jr., “Gaussian Basis Sets for Use in Cor-
related Molecular Calculations,” J. Chemical Physics,
vol. 90, 1989, pp. 1007–1014.
11. J. Bohannon, “Distributed Computing: Grassroots
Mark a. watson is a postdoctoral fellow in the De-partment of Chemistry and Chemical Biology and the Initiative in Innovative Computing at Harvard University. His research interests include method development and high-efficiency implementations for quantum chemistry and density functional theory. Watson has a PhD in theoretical chemistry from the University of Cambridge. Contact him at mark. [email protected].
roberto Olivares-amaya is a PhD student in the Department of Chemistry and Chemical Biology at
CISE-12-4-Watson.indd 49 04/06/10 12:01 PM
50 Computing in SCienCe & engineering
Harvard University. His research interests include the study of structure, response, and reactivity of molecular systems studied by first-principles elec-tronic structure methods. Olivares-Amaya has an AM in chemistry from Harvard University and a BSc in chemistry from the National Autonomous Univer-sity of Mexico (UNAM). Contact him at olivares@ chemistry.harvard.edu.
richard G. edgar is a research associate at Harvard University’s Initiative in Innovative Computing, spe-cializing in adding GPU acceleration to a wide variety of codes. In addition to working on the SciGPU-GEMM library, his other projects include the Murchi-son Widefield Array data pipeline and the Freesurfer MRI image analysis software. Edgar has a PhD in theoretical astrophysics from the University of Cam-bridge. Contact him at [email protected].
Tomás arias is a professor in the Department of Physics at Cornell University. His research interests include quantitative prediction of macroscopic phe-nomena from first principles quantum mechan-ics calculations. His research involves fundamental developments in density functional theory, numer-ical methods, software design and parallel comput-ing principles, and rigorous methods linking first
principles electronic structure calculations to long-length scale phenomena. Current application inter-ests include intrinsic limits in nanoscale mechanical oscillators, fundamental processes in the growth of oxide materials for advanced electronic material, and ab initio electrochemistry with application to fuel cells and solar cells. Arias has a PhD in physics from the Massachusetts Institute of Technology. Con-tact him at [email protected] and http://people.ccmr.cornell.edu/~muchomas.
alán aspuru-Guzik is an associate professor in the Department of Chemistry and Chemical Biology at Harvard University. His research interests lie at the intersection of quantum information and com-putation and chemistry and electronic structure theory methods development, with an emphasis on renewable energy applications. Aspuru-Guzik has a PhD in theoretical physical chemistry from the University of California, Berkeley. Contact him at [email protected] and http://aspuru.chem.harvard.edu.
Selected articles and columns from IEEE Computer Society publications are also available for free at