Page 1
AESGP Euro OTC News
Issue 286 | November 2016
Medicines
Pharmacovigilance
▪ PRAC Meeting - September / October 2016 6
▪ Explanatory Note to the GVP module VII 8
CMDh
▪ CMDh report - November 2016 8
Paediatrics
▪ Consultation on paediatric 2017 progress
report 10
Documents for comments
▪ WHO Draft Guidance on testing of SFFC
(QAS/15.634/Rev.2) 10
▪ Concept Paper on the revision of the “Guideline
on the Clinical investigation of medicinal
products for the treatment of migraine“ 10
Herbal news
▪ Draft EU monograph on species diureticae 11
More information: www.aesgp.eu/MAL17
Food
Food supplements
▪ Norway notifies Draft Regulation amending
the Regulation on Food Supplements 12
Food additives
▪ EFSA opinion on trimagnesium dicitrate anhydrous
(TMDC) to be used as a food additive in food
supplements in solid and chewable forms 13
Novel Food
▪ EFSA guidance documents published 13
▪ Final EU monograph on Harpagophytum
procumbens DC. and/or Harpagophytum
zeyheri Decne., radix 11
▪ Marisa Delbo elected new EMA HMPC chair 11
Page 2
Euro OTC News
Monthly news update by the Association of the European Self Medication Industry
Self-care: the first choice in health care 7 avenue de Tervuren, 1040 Brussels, Belgium Tel.: + 32 2 735 51 30 Fax: + 32 2 735 52 22 E-mail: [email protected] http://www.aesgp.eu
AESGP © 2016 | All rights reserved
2 AESGP Euro OTC News | Issue 286
▪ EFSA opinion on Safety of synthetic
L-ergothioneine (Ergoneine®) 14
Contaminants
▪ EFSA opinion on erucic acid in feed and food 14
DRVs
▪ EFSA Scientific Opinion on DRVs for vitamin D 15
EFSA
▪ Public consultation on EFSA terms of
Reference for a working group of the
Scientific Committee on Chemical mixtures 16
Food Chain
▪ AESGP attends the conference on “Food Chain
in the Digital Single Market” 17
▪ AESGP participates in the plenary meeting of
the Advisory Group on the Food Chain and
Animal and Plant Health 17
Medical devices
▪ Belgian classification of cranberry extract
based capsules as a medicine ruled invalid 19
WSMI General Assembly 2017 The Economics of Self Care
hosted by the Australian Self Medication Industry (ASMI) and the New Zealand Self Medication Industry (NZSMI)
AESGP Ingredients Directory
Page 3
Objectives of the conference
Self-care is nowadays recognised as a key element of healthcare. The European Commission’s
report on “Good governance of non-prescription medicines”, which was endorsed by all EU
Member States and relevant stakeholders, explains concrete ways forward. It paved the way to
the inclusion of specific recommendations in the first ever European Union Medicines Agencies
Network Strategy and multi-annual work programme calling for improved access to well-
established including non-prescription medicines. In addition, the Co-ordination Group for Mu-
tual Recognition and Decentralised Procedures (CMDh) advocates in its 2020 Strategy easier
access to non-prescription medicines and suggests a number of concrete actions in its Multi-
Annual Work Plan.
All this together is an excellent basis for discussing future priorities with the heads of the EU
authorities and their colleagues taking into account the proposals of the AESGP Self-Care Agen-
da 2020.
These proposals were recently complemented by a multi-annual work plan. It includes concrete
steps on how to make progress in three categories AESGP is following on the European level:
non-prescription medicines, food supplements and self-care medical devices.
Key proposals in the medicines’ sector include:
▪ Full functionality of the marketing authorisation / registration procedures for non-
prescription medicines
▪ Rigorous respect of timelines
▪ Ensure that the issues specific to non-prescription medicines in the Decentralised Mutual
Recognition Procedures are addressed in a special forum
▪ Reduce administrative burdens e.g. related to variations though the adequate use of
telematic systems and supported by the Regulatory Optimisation Group
The conference follows a long standing tradition to facilitate exchange between the Heads of
the EU Medicines Agencies and the European Self-Care industry. Full programme available at:
www.aesgp.eu/MAL17
Defining the priorities for the future development of self-care
AESGP Conference with the Heads of EU Medicines
Agencies (HMA) during the Maltese EU Presidency
3 AESGP Euro OTC News | Issue 286
MALTA | 20-21 February 2017
Page 4
4 AESGP Euro OTC News | Issue 286
Birgit Schuhbauer
Anthony Serracino Inglott
Ian Hudson
Catarina Andersson Forsman
Xavier de Cuyper
Christa Wirthumer-Hoche
Luca Pani
Hugo Hurts
Kristin Raudsepp
Belén Crespo
Christopher Fearne
Andrzej Ryś
Page 5
5 AESGP Euro OTC News | Issue 286
Karl Pall
Lilian Azzopardi
Lorraine Nolan
Thomas Senderovitz
Hubertus Cranz
Andreja Cufar
Practical information
Conference venue
Excelsior Grand Hotel Malta
Great Siege Road
Floriana FRN 1810
Malta
Registration
Registration can be made online or by using a
form that can be downloaded from the confer-
ence website: www.aesgp.eu/MAL17. We recom-
mend registration before 30 January 2017.
Helena Dalli
Guido Rasi
June Raine
Karl Broich
Page 6
Medicines
■ PRAC Meeting - September / October 2016
6 AESGP Euro OTC News | Issue 286
Pharmacovigilance
Signals
New signals detected from EU
spontaneous reporting systems:
esomeprazole
Scope: signal of gastric polyps
Discussion:
The Pharmacovigilance Risk As-
sessment Committee (PRAC) discus-
sed the cases of gastric polyps and
concurred that a plausible pathophy-
siologic mechanism could not be
excluded. In addition, a positive
dechallenge was observed in three
cases of fundic gland polyps (FGP).
PRAC Recommendation:
The PRAC agreed that the Rappor-
teur (Qun-Ying Yue, MPA, Sweden)
shall assess further data from Eu-
draVigilance and relevant literature
as well as available clinical guidance
regarding the potential risk of gastric
polyps associated with the pro-
longed use of esomeprazole and
other proton pump inhibitors (PPI). A
60-day timetable was recommended
for the assessment of this review
leading to a further PRAC recom-
mendation.
New signal detected from other
sources: proton pump inhibitors
(dexlansoprazole, esomeprazole,
lansoprazole, omeprazole, pantopra-
zole, rabeprazole)
Scope: signal of incident chronic
kidney disease (CKD) and progres-
sion to end stage renal disease
(ESRD)
Discussion: The PRAC discussed the
results from three large observatio-
nal studies (Lazarus et al., Xie et al.,
Arora et al.) on the risk of incident
CKD and progression to ESRD. The
MAHs should provide a proposal for
amending their product information
and/or RMP as applicable.
PRAC recommendation: (Rafe Suvar-
na, MHRA as Rapporteur)
The MAHs for pantoprazole-, lanso-
prazole-, dexlansoprazole-, rabepra-
zole-, omeprazole- and esomepra-
zole-containing products (Takeda,
Janssen-Cilag, Eisai, AstraZeneca)
should submit to the EMA, within 60
days, a cumulative review of all cases
of CKD and progression to ESRD,
including all MedDRA SOC investiga-
tions, related terms relevant to kid-
ney function and data on kidney
function measurements.
Others
PSUR procedure - Orlistat
Scope: Evaluation of a Periodic Safe-
ty Update Report Single Assessment
(PSUSA) procedure
PRAC recommendation:
Update of the Summary Product
Characteristics (SmPC) section 4.4
and 4.8. The package leaflet is up-
dated accordingly. The PRAC As-
sessment Report and the PRAC
recommendation are transmitted to
the EMA Committee for Human
Medicinal Products (CHMP) for
adoption of an opinion. In the next
Periodic Safety Update Report
(PSUR), the Marketing Autorisation
Holders (MAHs) should provide
detailed reviews of all cases of hepa-
totoxicity and nephrotoxicity with an
analysis of all serious cases belon-
ging to the MEdDRA SOC33
‘hepatobiliary disorders’ and Med-
DRA HLGT34 ‘nephropathies’, ‘renal
disorders’ and ‘urolithiasis’ respecti-
vely. In addition, the MAHs should
continue to closely monitor hepato-
toxicity and nephrotoxicity as major
safety concerns. The MAHs of Alli
and Xenical should submit to the
EMA, within 180 days, a detailed
cumulative review of all case reports
of nephrotoxicity from clinical trials,
post marketing surveillance and
literature. Moreover, they should
discuss the causal relationship bet-
ween orlistat and reports of nephro-
toxicity, taking into account potential
confounding factors (such as co-
morbidities) and the usage of these
drugs. Based on the analysis of the
cumulative review, the MAHs should
discuss any necessary amendments
to the product information, as ap-
propriate. The frequency of PSUR
submission should be revised from
yearly to three-yearly and the next
PSUR should be submitted to the
EMA within 90 days of the data lock
point. The list of Union reference
dates (EURD list) provided for under
Article 107c(7) of Directive 2001/83/
EC is updated accordingly. In addi-
tion, the PRAC agreed that no
further PSURs are required for pro-
ducts referred to in Articles 10(1),
10a, 14, 16a of Directive 2001/83/EC.
The EURD list is updated accordingly.
Page 7
7 AESGP Euro OTC News | Issue 286
PSUR procedure - Esmya (ulipristal
acetate)
Scope: Evaluation of a PSUSA proce-
dure
PRAC recommendation:
Based on the review of the data on
safety and efficacy, the risk-benefit
balance of Esmya (ulipristal acetate)
in the approved indication(s) re-
mains unchanged. The current terms
of the marketing authorisation(s)
should be maintained. In the next
PSUR, the MAH should provide a
detailed review on cases of hyper-
sensitivity. The MAH should discuss
causality for hypersensitivity reac-
tions and propose to update the
product information as applicable.
The MAH should submit to the EMA,
within 60 days, a detailed review on
arterial and venous thromboembolic
events (ATE/VTE). The MAH should
also discuss biological plausibility
based on the mechanism of action
of ulipristal acetate, focusing on the
role of oestrogen and progesterone.
The MAH should consider updating
the product information and RMP as
applicable. The next PSUR should be
submitted in accordance with the
requirements set out in the list of
Union reference dates (EURD list)
provided for under Article 107c(7) of
Directive 2001/83/EC.
PASS non-imposed in the marketing
authorisations - Ulipristal acetate
Scope: submission of the final results
of a prospective multicenter non-
interventional post-authorisation
safety study (PASS) of women treat-
ed with ulipristal acetate as preoper-
ative treatment of moderate to
severe symptoms of uterine fibroids.
The RMP is updated accordingly.
The PRAC assessment report will be
updated.
Applicant: Gedeon Richter Plc. PRAC
Rapporteur: Ulla Wändel Liminga
Public hearings – dry-run outcome
for discussion
As a follow-up to the public hearing
dry run exercise held in July 2016
within the framework of the PRAC
meeting to test the process and
procedures for public hearings by
using a fictional scenario of a safety
review (see PRAC minutes July 2016),
the EMA secretariat presented to the
PRAC some lessons learnt. Overall,
all aspects were implemented and
while some fine tuning is needed, no
major aspects were overlooked in
preparing for future public hearings
and the exercise confirmed that the
aims of the dry run were achieved.
As a next step, in line with the PRAC
work plan 2016 and based on the
work of the PRAC topic group, pro-
cedural and best practice guidance
for PRAC members on public hear-
ings’ will be discussed at PRAC in
November 2016.
Strategy on measuring the impact of
pharmacovigilance – draft reflection
paper on PRAC criteria to prioritise
collaborative impact research
PRAC lead: Marieke De Bruin, MBE,
NL
In line with the PRAC work plan 2016
as part of the ‘PRAC strategy on
measuring the impact of pharma-
covigilance activities’, based on the
reflection paper on ‘PRAC criteria for
prioritisation of collaborative impact
research’, the EMA Secretariat pre-
sented to the PRAC, based on the
EMA reviewers’ independently ap-
plied criteria, safety topics discussed
at PRAC since 2012 for which risk
minimisation measures were decid-
ed. Based on the criteria, the PRAC
considered some topics proposed by
the PRAC Interest Group (IG) for
potential impact research through
either a collaborative network study
or as an EMA funded study. Based
on the comments, further discussion
will be scheduled at PRAC in No-
vember 2016.
Effects tables in selected important
benefit/risk reviews – pilot phase for
discussion
PRAC lead: Rafe Suvarna, MHRA, UK
In line with the PRAC work plan 2016
and based on the CHMP experience,
the PRAC was presented with the
second effects table of the pilot that
was recently initiated to explore the
utility of such tables in post-
authorisation procedures. The PRAC
welcomed the initiative and encour-
aged further work on using effects
tables in selected important benefit-
risk reviews as part of the pilot exer-
cise at the level of the PRAC.
Annex 1. Renewals of the marketing
authorisation, conditional renewals
and annual reassessments
Ulipristal acetate (with risk manage-
ment Plan)
Applicant: Gedeon Richter Plc. PRAC
Rapporteur: Ulla Wändel Liminga,
MPA, Sweden
Scope: 5-year renewal of the market-
ing authorization
At its October meeting, the PRAC
discussed four ongoing safety re-
views which include the article 31
referral on paracetamol modified
released. The Committee did not
initiate or conclude a referral.
The PRAC also focused on the broad
spectrum of its responsibilities which
cover all aspects of the risk manage-
ment of the use of medicines. A
record of the discussions held will be
provided in the minutes of this
meeting, which will be published
following the next PRAC meeting at
the end of November.
The minutes of the September meet-
ing are available here and the high-
lights of the October meeting here.
Page 8
Co-ordination Group for Mutual Recognition and Decentralised Procedures-Human
■ CMDh report - November 2016
8 AESGP Euro OTC News | Issue 286
■ Explanatory Note to the GVP module VII
In the context of the Public Safety Update Reports
(PSUR) roadmap project, the Joint PRAC/CMDh recom-
mendation paper was finalised and adopted at the May
2016 PRAC and CMDh meeting, setting out the com-
mon understanding on the EU PSUR single assessment.
The recommendation paper identified that there were
areas where improvements could be made in terms of
the quality of PSUR submissions and consistency of the
PSUR assessments.
To this end, the Explanatory note to the GVP module VII
(which will ultimately lead to the update of GVP module
VII) has been elaborated by a drafting group composed
of Members from National Competent Authorities nom-
inated by PRAC and CMDh.
Members are asked to provide feedback on the draft
explanatory note to the GVP module VII by 15 Decem-
ber 2016 to allow sufficient time for the trade associa-
tions to coordinate an industry response to the EMA.
At its meeting on 7-9 November 2016, the CMDh has
elected Mrs Virginie Bacquet (FR) as chair of the revital-
ized non-prescription medicinal products Working
Group.
The meeting was also the last meeting of the Process
Improvement Working Party. The CMDh thanked the
WP and its long acting chair Christer Backman. Actions
from the work plan assigned to the working party will
be handled by dedicated working groups in the future.
Other items worth reporting include the following:
EU Work-Sharing Articles 45 & 46 of the Paediatric
regulation – Public Assessment Reports
The CMDh has agreed on a public assessment report for
paediatric studies submitted in accordance with Article
46 of the Paediatric Regulation for Pelargonium Syrup
(pelargonium sidoides root extract)
Certificate of suitability (CEP) suspensions at Zhe-
jiang Hisun Pharmaceutical Co., Ltd
The CMDh has been made aware that following the
issuing of a statement of GMP non-compliance, EDQM
has suspended CEPs for manufacturing sites of Zhejiang
Hisun Pharmaceutical Co., Ltd. (see EDQM CEP web-
page)
Marketing Authorisation Holders are advised to take the
necessary regulatory actions and follow the recommen-
dations stated in the Statement of Non-Compliance
issued by the Spanish Competent Authority in Septem-
ber 2016 (available on the EudraGMP website here;
search for inspection end date: 2016-06-04).
Revision of MRP/RUP flow chart
The CMDh agreed on an updated flow chart for MRP
and RUP procedures. The new flow chart foresees a
shorter period for comments by Member States at the
beginning of the procedure. Time periods for applicants
remain unchanged. The new flow chart also foresees
that RUPs can be closed by day 60 when no potential
serious risk to public health is raised/remains (MRPs
may be closed at day 60 when no potential serious risk
to public health or other concerns remain). The new
flow chart will be applicable to procedures submitted
after 31 January 2017.
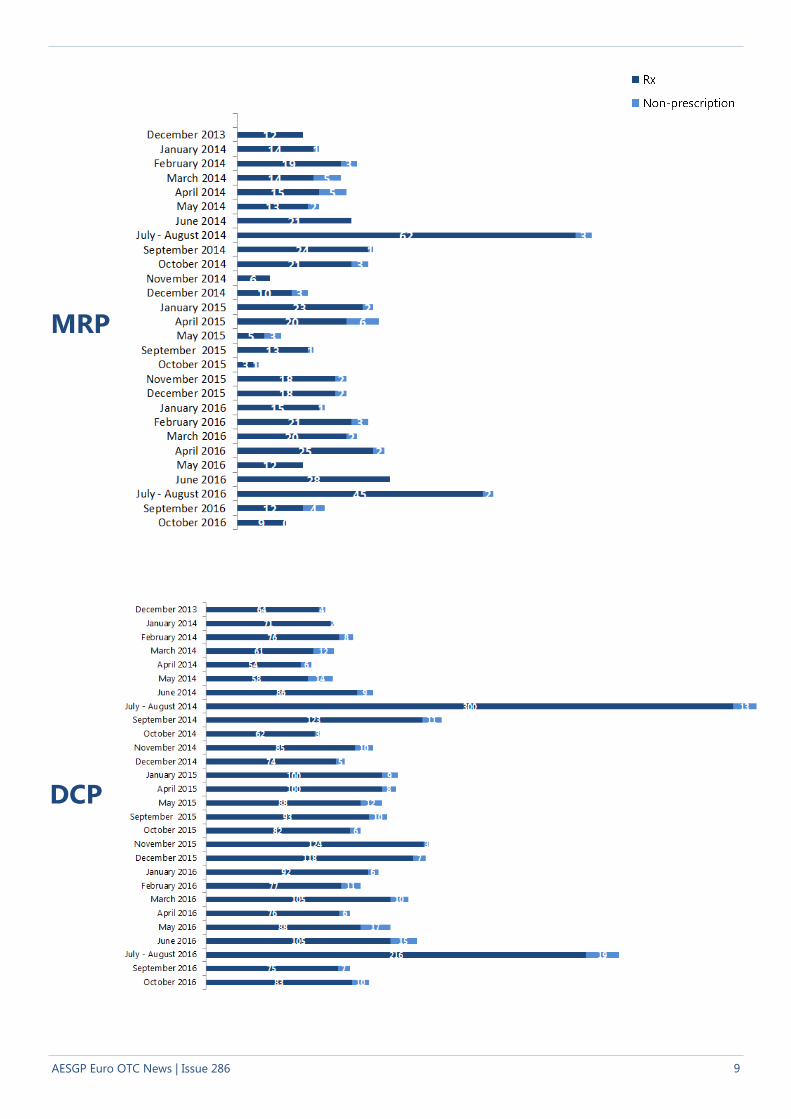
Statistics
In October 2016, 9 Mutual Recognition Procedures
(regarding 21 products) started. All these
procedures related to prescription-only medicinal
products in the reference Member State.
In October 2016, 93 Decentralised Procedures
(regarding 203 products) started. Out of the 93
procedures, 83 of these procedures related to pres-
cription-only medicinal products and 10 procedures
related to non-prescription medicinal products in
the reference Member State. Ninety one of those
procedures consisted of chemical substances, 1
biological and 1 herbal.
The above information as well as further details can be
found in the report from the CMDh meeting held on 7-9
November 2016.
Page 9
MRP
DCP
9 AESGP Euro OTC News | Issue 286
Page 10
10 AESGP Euro OTC News | Issue 286
Paediatrics
■ Consultation on paediatric 2017 progress report
The Regulation (EC) 141/2000 (so called Paediatric
Regulation) aimed to address a gap in knowledge on
how medicine should best be used by children, reduce
the level of off-label use and increase the number of
medicines specifically developed and tested for children. In 2013, the Commission published a first progress
report on the Paediatric Regulation and while it revealed
some promising signs of progress, it found that, due to
the length of medicinal products' development, it would
take at least 10 years to gain a full understanding of the
situation. The 10 year report to the European Commis-
sion can be downloaded here.
Under Article 50(3) of the Regulation, the Commission’s
second report is due in 2017. It should assess the Regu-
lation's impact on public health and businesses and is to
be presented to the European Parliament and the Coun-
cil. It is aimed at informing EU decision-makers about
the experience with the Regulation.
All stakeholders involved in the development, manufac-
ture and/or commercialisation of paediatric medicines,
as well as paediatric patient/parent groups, healthcare
professionals and academia are invited to provide their
views and feedback on the experience acquired with the
Paediatric Regulation to support the Commission in
drafting its second report.
The consultation document can be downloaded here. It
is also included as an attachment in this email for ease
of reference.
AESGP members are asked to send their input on
the consultation document by 29 January 2017 to the
AESGP offices.
Documents for comments
■ WHO Draft Guidance on testing of SFFC (QAS/15.634/Rev.2)
The World Health Organisation (WHO) working docu-
ment entitled “Draft Guidance on testing of ‘suspect’
spurious/falsely-labelled/falsified/counterfeit medi-
cines” (QAS/15.634-Rev.2)provides technical guidance
on laboratory testing of samples of suspected spurious/
falsely-labelled/falsified/counterfeit (SFFC) products
detected on the market of WHO Member States and
related aspects of sampling and reporting.
The Member State Mechanism on SSFFC medical pro-
ducts, created in 2012, makes recommendations to
support regulatory authorities to prevent, detect and act
against activities and behaviours that result in SSFFC
medical products. This document is intended to com-
plement the Member State Mechanism’s recommenda-
tions in accordance with resolution WHA67.20 on Regu-
latory system strengthening for medical products.
Any input on this draft WHO working document should
reach AESGP by 16 December 2016 using the table for
comments.
■ Concept Paper on the revision of the “Guideline on the Clinical investigation of medicinal products
for the treatment of migraine“
The Concept Paper on the revision of the “Guideline on
the Clinical investigation of medicinal products for the
treatment of migraine“ (EMA/CHMP/179671/2016) has
now been released for public consultation, following a
recommendation for a revision of the current guideline
by the Central Nervous System Working Party (CNSWP).
While the current guideline focusses on evidence
needed in support of treatment claims of acute mi-
graine attacks and migraine prophylaxis, it does not
Page 11
Herbal news
■ Draft EU monograph on species diureticae
The draft European Union herbal monograph on species
diureticae has been published for consultation. The
draft monograph addresses herbal substances which
may be used in combinations as THMP to increase the
amount of urine to achieve flushing of the urinary tract
as an adjuvant in minor urinary complaints.
The draft assessment report and draft list of references
are also available.
Members are asked to send their comments until 6
January 2017.
11 AESGP Euro OTC News | Issue 286
address the evidence needed to support a claim of
treatment of chronic migraine, which is a relatively new
concept. Recently a number of scientific advice
procedures have been considered for development
programs of medicinal products for the treatment of
chronic migraine. The other parts of the guideline still
apply and are up to date although a slight adaptation
may be discussed.
The proposed guideline will replace the current Guide-
line on clinical investigation of medicinal products in the
treatment of migraine (CPMP/EWP/788/01 Rev.1, 24
January 2007)
It is anticipated that a draft CHMP guidance document
will be released for consultation not later than Q4 2017.
Comments on this concept paper should reach AESGP
by 10 January 2017.
■ Marisa Delbo elected new EMA HMPC chair
Marisa Delbò, head of the Risk Management Office at
the Italian Medicines Agency (AIFA), was elected as the
new chair of the European Medicines Agency’s (EMA)
Committee on Herbal Medicinal Products (HMPC) at its
November meeting.
Maria Delbò was previously vice-chair of the HMPC
(2013-2016) and also vice-chair of the HMPC Working
Party on European Union Monographs and European
Union List (MLWP) (2011-2016).
The full press release is available here.
■ Final EU monograph on Harpagophytum procumbens DC. and/or Harpagophytum zeyheri Decne., radix
Further to its endorsement at the European Medicines
Agency’s (EMA) Committee on Herbal Medicinal Prod-
ucts (HMPC) July 2016 meeting, the final European
Union herbal monograph on Harpagophytum procum-
bens DC. and/or Harpagophytum zeyheri Decne., ra-
dix was released.
The monograph was adopted by consensus. The final
monograph is available on the “harpagophytum page”
under the ‘All documents’ tab, together with the HMPC
Opinion, the Final Assessment Report, the Final List of
references and the Overview of comments received.
Maria Delbò speaking at the AESGP Conference on “Herbal
(medicinal) products, food supplements and self-care medical
devices” in October 2014 in Brussels
Page 12
Food
Food supplements
■ Norway notifies Draft Regulation amending the Regulation on Food Supplements
Norway has notified a draft regula-
tion amending the Regulation on
Food Supplements to the European
Commission through the TRIS proce-
dure (*).
This draft text amends Regulation of
20 May 2004 No 755 on food supple-
ments setting the national maximum
limits for the amount of folic acid,
magnesium, vitamin C, calcium and
vitamin D in food supplements. It
separates maximum limits for young
children from 1 and up to 3 years old,
children from 3 and up to 11 years
old, adolescents from 11 and up to
18 years old, and adults from 18 years
old. It also introduces particular
requirements for labelling of food
supplements containing specific
amounts of folic acid, calcium, vita-
min C and vitamin D per daily portion
of consumption as recommended by
the manufacturer.
The measures requiring additional
mandatory food labelling have also
been notified according to Article 39
and 45 of Regulation (EU) No
1169/2011 on the provision of food
information to consumers.
The standstill period (**) will end on
13 February 2017. Further informa-
tion on the status of this notification
is available here.
Brief Statement of Grounds:
Norway implemented Directive
2002/46/EC on food supplements in
2004, and existing national maximum
limits for vitamins and minerals per
daily dose were then continued.
These national maximum limits are
set to ensure that food supplements
with vitamins and minerals are safe
for the entire healthy population.
However, since the existing maximum
limits are old, they are now being
revised. We have suggested establis-
hing separate maximum limits for
various age groups (young children,
children, adolescents, adults), in order
to make it possible to produce food
supplements adapted to different
consumer groups.
The existing maximum limits for
magnesium and calcium of 600 mg
and 1500 mg respectively, are propo-
sed reduced in line with the tolerable
upper intake levels (ULs) suggested
by the Norwegian Scientific Com-
mittee for Food Safety (NSCFS) and
updated intake data (see link below).
The existing maximum limits for folic
acid and vitamin D, are proposed to
be decreased for the youngest age
groups, while they are suggested
increased for the older age groups,
based on scientific opinions by the
NSCFS. The maximum limit for vita-
min C is proposed increased for all
age groups, based on a scientific
opinion by the NSCFS.
Differentiating the maximum limits
for separate age groups means that a
food supplement for some age
groups (usually adults) may contain
amounts of vitamins/minerals that
involve a health risk for younger age
groups, since Upper Limits (ULs) for
these age groups will be exceeded.
We have therefore deemed it essen-
tial requiring additional mandatory
food labelling, in order to protect
public health. Particular requirements
for labelling of food supplements
containing specific and high amounts
of folic acid, calcium, vitamin C and
vitamin D per daily portion, in one
measured small unit quantity, shall
ensure that the product reaches its
right age group and that consumers
are provided clear information that
the consumption of certain amounts
of vitamins or minerals might cause
adverse health effects for specific age
groups. The mandatory particulars
are deemed essential in order to
protect public health, in accordance
with Article 39 of Regulation (EU) No
1169/2011.
links to relevant scientific opinions by
the NSCFS for:
1) magnesium; http://www.vkm.no/
dav/f9ef23feb7.pdf ,
2) calcium and vitamin C; http://
www.vkm.no/dav/289a369b0c.pdf ,
3) folic acid; http://www.vkm.no/
dav/86f93d4c96.pdf and
4) vitamin D; http://vkm.no/
dav/422f24e6e0.pdf
12 AESGP Euro OTC News | Issue 286
*It allows the Commission and the Mem-
ber States of the EU to examine the techni-
cal regulations Member States intend to
introduce for products (industrial, agricul-
tural and fishery) and for Information So-
ciety services before their adoption. The
aim is to ensure that these texts are com-
patible with EU law and the Internal Market
principles. It applies in a simplified manner
to the European Free Trade Association
(EFTA) Member States which are signato-
ries to the Agreement on the European
Economic Area (EEA) and to Switzerland
and Turkey.
**Starting from the date of notification of
the draft, a 3-month standstill period –
during which the notifying Member State
cannot adopt the technical regulation in
question – enables the Commission and
the other Member States to examine the
notified text and to respond appropriately.
More information on this procedure and
what happen next can be found here.
i
Page 13
13 AESGP Euro OTC News | Issue 286
Food additives
■ EFSA opinion on trimagnesium dicitrate anhydrous (TMDC) to be used as a food additive in food
supplements in solid and chewable forms
EFSA has published its opinion on the Safety of trimagnesi-
um dicitrate anhydrous (TMDC) to be used as a food addi-
tive in food supplements in solid and chewable forms.
The Panel concluded that there was no safety concern from
the proposed typical use levels of TMDC as a stabiliser and
anticaking agent in food supplements, whereas at the
proposed maximum use levels, the resulting exposure to
magnesium in adults and the elderly at the high-level
would be above the upper level (UL).
TMDC is a magnesium salt of citric acid in the anhy-
drous form. Following oral ingestion, TMDC is expected
to fully and readily dissociate into magnesium and
citrate ions.
The toxicological data provided in support of the cur-
rent application were not in accordance with the Tier 1
requirement of the ‘ Guidance for submission for food
additive evaluations’; however, this was considered to
be justified by the Panel given that both magnesium
and citrate are natural constituents of the diet and also
the human body.
Exposure estimates to TMDC from its proposed use
were calculated for both typical and maximum use
levels alongside the resulting intake of magnesium and
citrate. At the proposed typical level, the intake of
magnesium resulting from the use of TMDC would be
below the tolerable upper level (UL) of 250 mg/day for
supplemental magnesium. In the worst case scenario of
high-level intakes of TMDC when used at the maximum
proposed use level of 120,000 mg/kg, the maximum
estimated intake of magnesium would be 311 mg/day
per day in the elderly and 389 mg/day in adults, in both
cases above the UL.
The Panel noted that the intake of both magnesium and
citrate resulting from the proposed use and use levels
of TMDC appears to be within the normal dietary intake
in the general population.
Recommendations
In the case of TMDC being used as a food additive in
food supplements containing a source of magnesium,
the total amount of magnesium that could be con-
sumed at a single eating occasion should be below the
UL of 250 mg/day. Therefore, risk managers may wish
to consider whether consumption advice and recom-
mended maximum daily dose of the TMDC being used
as a food additive in food supplements should be
provided to the consumers.
The Panel recommended lower specification limits for
lead, cadmium and arsenic, given that levels proposed
in the specifications could have a significant impact on
the exposure to these toxic elements.
The Panel recommended including in the specifications
a typical particle size distribution of TMDC under its
powder form.
An application has been introduced for the authorisation of the use
of TMDC as a stabiliser and/or anticaking agent in food supplements
in solid and chewable forms (food categories 17.1 and 17.3 of part E
of Annex II to Regulation (EC) No 1333/2008). The technological
function as a stabiliser/anti caking agent is reached by a dosage of 2
–12% TMDC of the final preparation.
Novel Food
■ EFSA guidance documents published
EFSA Panel on Dietetic Products, Nutrition and Allergies
(NDA) published its two guidance documents in the area of
novel foods:
Guidance on the preparation and presentation of an
application for authorisation of a Novel Food
Guidance on the preparation and presentation of a
notification for authorisation of Traditional Foods from
third countries
The final versions have been greatly modified compared
the draft versions subject to consultation earlier this year.
Most of the comments submitted by AESGP in April 2016
have been endorsed. Details of the comments received
may be found in the outcome of the public consultation
on:
Outcome of public consultation on the draft Guidance
on an application for authorisation of a Novel Food
Outcome of public consultation on the draft guidance
on traditional foods from third countries
i
Page 14
14 AESGP Euro OTC News | Issue 286
■ EFSA opinion on Safety of synthetic L-ergothioneine (Ergoneine®)
The EFSA Panel on Dietetic Products, Nutrition and
Allergies (NDA) published an opinion on the Safety of
synthetic L-ergothioneine (Ergoneine®) as a novel food
pursuant to Regulation (EC) No 258/97.
The Panel concludes that the novel food (NF), synthetic
L-ergothioneine (marketed as Ergoneine®), is safe
under the intended conditions of use as specified by the
applicant.
The NF that is the subject of the application is synthetic
L-ergothioneine (ET; marketed as Ergoneine®), which is
produced by a one-pot patented manufacturing pro-
cess. Chemically, ET is a derivative of thiolhistidine, i.e. 2
-thio-L-histidine-betaine. ET is naturally present in a
number of foodstuffs such as mushrooms, some varie-
ties of black and red beans, offal and cereals.
The applicant intends to use the NF in quantities of up
to 5 mg per serving in alcohol-free beverages, cereal
bars, milk, fresh dairy product s and chocolate. The
applicant also proposes to provide the NF as a food
supplement, with a recommended daily dose of up to
30 mg/day for adults and 20 mg/day for children.
The target population proposed by the applicant is
children above 3 years of age and the general adult
population, with the exception of pregnant and breast-
feeding women.
Considering the NOAEL of 800 mg/kg bw per day,
which was based on two subchronic toxicity studies in
rats, and the maximum estimated intake levels for L-
ergothioneine from all sources, the Panel concludes that
the margins of safety of 470 for adults (except pregnant
and breast feeding women) and of 216 for children
above 3 years of age are sufficient.
Contaminants
■ EFSA opinion on erucic acid in feed and food
EFSA Panel on Contaminants in the Food Chain (CONTAM Panel) has published its scientific opinion on the risks for
animal and human health related to the presence of erucic acid in feed and food.
Erucic acid is the trivial name of the fatty acid Z-13-docosenoic acid,
abbreviated as 22:1D13c, although it is more frequently found in the
literature as 22:1 n-9. Erucic acid is mainly present in the seeds of species
of the Brassicaceae, which includes important seed crops such as rape-
seed and mustards, and also important vegetable crops such as the
diverse group of kales, cabbages and turnips. Cultivars of Brassicaceae
with very low erucic acid content have been developed for seed oil pro-
duction for food and feed use in most countries, including the EU. Mus-
tard seed production is based on cultivars with high erucic acid content.
Erucic acid is also present at low concentrations in other food sources
such as fish. Erucic acid is present in Lorenzo’s oil, a drug used for thera-
py for ALD patients. Erucic acid doses range from 0.09 to 0.51 g/kg bw
per day.
According to the most common nomenclature for fatty acids, erucic acid
is abbreviated as 22:1 n-9 or 22:1 x-9. Erucic acid is a natural plant toxin
which is a contaminant according to the definition of the contaminant
provided in Council Regulation (EEC) No 315/93 as the presence of erucic
acid in food is the result of the agricultural production, more in particular
the choice of the variety.
The maximum levels for erucic acid (section 8.1) have been established in
Regulation(EC) 1881/2006, as amended by Commission Regulation (EU)
No 696/2014 for:
Vegetable oils and fats
Foods containing added vegetable oils and fats with the exception
of Infant formulae and follow-on formulae
Infant formulae and follow-on formulae
The appropriateness of setting a maximum level for erucic acid has been
highlighted by the Scientific Committee on Food (SCF) in its opinion
expressed on 17 September 1993 on essential requirements for infant
formulae and follow-on formulae. When the maximum levels of erucic
acid were established by Regulation (EC) 1881/2006, it was found neces-
sary to review the maximum levels in the future based upon an updated
risk assessment and also to consider the appropriateness of establishing
maximum levels for erucic acid in feed.
Reported occurrence in food supplements:
After the quality assessment of the analytical data on
erucic acid, a total of 12,444 food samples were available
to estimate dietary exposure. Before the occurrence data
were used to estimate dietary exposure, the data were
grouped at different FoodEx levels according to their
erucic acid levels and the number of samples reported
(Appendix C, Table C.5). This table shows occurrences in
the following food supplements categories but the num-
ber of samples is rather low (from 1 to 26):
Dietary supplements, unspecified
Mineral supplements
Protein and amino acids supplements
Supplements containing special fatty acids
(e.g.omega-3, essential fatty acids)
Plant extract formula
Algae formula (e.g. Spirulina, Chlorella)
Pollen-based supplement
i
Page 15
15 AESGP Euro OTC News | Issue 286
The Panel concluded (full conclusions from page 72 on-
wards):
For most humans, the main contributor to dietary expo-
sure to erucic acid was the food group ‘Fine bakery
wares’. In ‘Infants’, ‘Food for infants and small children’
was the main contributor to exposure.
The heart is the principal target organ for toxic effects
after exposure. Myocardial lipidosis was identified as the
critical effect for chronic exposure to erucic acid. This
effect is reversible and transient during prolonged expo-
sure.
A tolerable daily intake (TDI) of 7 mg/kg body weight
(bw) per day for erucic acid was established, based on a
no observed adverse effect level of 0.7 g/kg bw per day
for lipidosis in young rats and new born piglets. Mean
chronic exposure of the different groups of the popula-
tion did not exceed the TDI. The 95th percentile dietary
exposure level was highest in infants and other children,
ranging from1.3 to 7.4 mg/kg bw per day; the higher
level being at the level of the TDI. This may indicate a risk
for young individuals with high erucic acid exposure.
In pigs, levels of erucic acid are unlikely to represent a
health concern. However, for poultry, the small margin
between the lowest observed adverse effect level (LOAEL)
and the estimated exposure may indicate a health risk
where maximum inclusion rates are applied. Due to the
absence of adequate data, the risk for ruminants, horses,
fish and rabbits could not be assessed.
The Panel recommended:
To generate more analytical data on the occurrence
of erucic acid in relevant food and feed commodities
using sensitive and specific methods. Special atten-
tion should be paid to processed foods such as ‘Fine
bakery wares ’, ‘Food for infants and small children’
and ‘Composite foods ’
There should be more information on the levels in
animal-derived products (meat, milk and eggs) result-
ing from the transfer of erucic acid from animal feed.
There is a need for a repeated-dose toxicity study in
new born rats or pigs with pure erucic acid in order
to clarify the potential confounding effects of other
fatty acids present in the oil and to provide infor-
mation regarding the dose –response relationship.
Studies should be conducted on species differences
in the cardiac and hepatic metabolism of erucic acid.
Further studies are required to determine reference
points for target livestock animals and fish.
DRVs
■ EFSA Scientific Opinion on DRVs for vitamin D
EFSA Panel on Dietetic Products, Nutrition and Allergies
(NDA) has just published its scientific opinion on dietary
reference values (DRVs) for vitamin D.
EFSA’s assessment for European consumers follows that of
the UK’s Scientific Advisory Committee on Nutrition
(SACN) which has recommended DRVs for vitamin D for
the UK population. A joint explanatory note is attached to
EFSA’s scientific opinion that sets out the respective ap-
proaches taken by the two organisations in deriving DRV
values for Vitamin D – including methodology, the data
used and geographical area covered in the assessment.
In its opinion, the Panel concludes that Average Require-
ment (ARs) and Population Reference Intakes (PRIs) for
vitamin D cannot be derived for adults, infants and chil-
dren, and therefore defines Adequate Intake (AIs), for all
population groups. The DRVs for vitamin D are based on
the assumption of minimal exposure to the sun with re-
sulting limited levels of synthesised vitamin D.
The Panel considers that serum 25(OH)D concentration,
which reflects the amount of vitamin D attained from both
cutaneous synthesis and dietary sources, can be used as a
biomarker of vitamin D intake in adult and children popu-
lations with low exposure to UV-B irradiation and as a
biomarker of vitamin D status. The Panel notes that the
evidence on the relationship between serum 25(OH)D
concentration and the risk of musculoskeletal health out-
comes in (healthy) adults, infants and children, and some
adverse pregnancy-related health outcomes, is widely
variable. Several factors contribute to this, and also include
the large variation in the results from different laboratories
and assays used for measuring serum 25(OH)D concentra-
tions. Taking into account the overall evidence and uncer-
tainties, the Panel considers that a serum 25(OH)D con-
centration of 50 nmol/L is a suitable target value for all
population groups, in view of setting the AIs for vitamin D.
The Panel underlines that the meta-regression was done
on data collected under conditions of assumed minimal
cutaneous vitamin D synthesis. In the presence of cutane-
ous vitamin D synthesis, the requirement for dietary vita-
min D is lower or may even be zero.
Page 16
Summary of DRVs for vitamin D
The panel makes the following recommendations for
research:
There is a need for further research to study the re-
spective impact of vitamin D dietary intake and sun-
light exposure on serum 25(OH)D concentrations.
Future studies should investigate food-based strate-
gies to ensure adequate vitamin D intakes accounting
for latitude, sunlight exposure and diet.
Studies are needed that are specifically designed to
identify cut-off values for serum 25(OH)D concentra-
tion or other suitable biomarkers for vitamin D status
to derive DRVs for vitamin D for infants, children,
adults, pregnant and lactating women.
Standardised investigations are needed to assess
changes in musculoskeletal health outcomes (and
surrogate markers) in response to vitamin D2 and D3
intake, and in relation to serum 25(OH)D concentra-
tions.
The potential mechanisms of the cause and effect
relationships between vitamin D and nonmusculoskel-
etal health outcomes should be further explored.
There is a need for studies that assess the different
diets of infants, in particular those consuming infant or
follow-on formulas and processed cereal-based foods
fortified with vitamin D in addition to vitamin D sup-
plements.
More data on the effects of genotype and body fat
mass on vitamin D metabolism and the requirements
for vitamin D are warranted.
More precise data on total vitamin D concentration in
foods would also be useful. Studies investigating the
effect of 25(OH)D naturally occurring in foods on
serum 25(OH)D concentration are also suggested
The NDA Panel also published the Outcome of public
consultation on a Draft Scientific Opinion on Dietary
Reference Values for vitamin D
Age Adequate Intakes (µg/day)
7–11 months 10
1–3 years 15(a)
4–6 years 15(a)
7–10 years 15(a)
11–14 years 15(a)
15–17 years 15(a)
≥ 18 years(b) 15(a) (a): under conditions of assumed minimal cutaneous vitamin D synthesis. In the presence of endogenous cutaneous vitamin D synthesis (Section 2.3.1), the requirement for dietary vitamin D is lower or may be even zero. (b): including pregnancy and lactation.
(*) For ease of reference please see below the following defini-
tions:
Dietary Reference Values (DRVs) - quantitative reference values
for nutrient intakes for healthy individuals and populations which
may be used for assessment and planning of diets
Population Reference Intakes (PRI) - the level of (nutrient) in-
take that is adequate for virtually all people in a population group
Average Requirement (AR) - the level of (nutrient) intake that is
adequate for half of the people in a population group, given a
normal distribution of requirement
Adequate Intake (AI) - the value estimated when a Population
Reference Intake cannot be established because an average requi-
rement cannot be determined. An Adequate Intake is the average
observed daily level of intake by a population group (or groups)
of apparently healthy people that is assumed to be adequate.
More information can be found in the EFSA Scientific Opinion on
principles for deriving and applying Dietary Reference Values
(2010)
EFSA
■ Public consultation on EFSA terms of Reference for a working group of the Scientific Committee on Chemical mixtures
EFSA launched a Public consultation on the terms of
reference of the Scientific Committee Working Group on
“Harmonisation of risk assessment methodologies for
human health and ecological risk assessment of combi-
ned exposure to multiple chemicals”.
The working group will develop a draft guidance docu-
ment for human and ecological risk assessment of com-
bined exposure to multiple chemicals using existing
frameworks as starting points and tiered approaches for
each step (problem formulation, hazard identification,
hazard characterisation, exposure assessment, risk char-
acterisation).
Following input from this public consultation, the work-
ing group of the scientific committee will review the
contributions and consider them in developing the guid-
ance document. Once finalised, the draft guidance docu-
ment will be subject to another public consultation be-
fore its finalisation. Written comments might be sub-
mitted to EFSA via this link by 30 November 2016.
Human and ecological risk assessment of combined exposure to
multiple chemicals (“chemical mixtures”) poses a number of chal-
lenges to scientists, risk assessors and risk managers, particularly
because of the complexity of the problem formulation, the huge
number of chemicals involved, and the toxicological profiles and
exposure patterns of these chemicals in humans and species pre-
sent in the environment. The development of harmonised method-
ologies for combined exposure to multiple chemicals has been
identified by EFSA Scientific Committee as a key priority area for
EFSA and a number of EFSA panels and units have initiated activities
to support harmonisation of risk assessment methods for both the
human health and the ecological area.
i
i
16 AESGP Euro OTC News | Issue 286
Page 17
Food Chain
■ AESGP attends the conference on “Food Chain in the Digital Single Market”
AESGP attended on 9 November 2016 the conference
co-organised by the European Commission and Slovak
Presidency of the Council of the European Union on
“Food Chain in the Digital Single Market”. Among the
various presentations given, Ladislav MIKO (European
Commission, Deputy Director General Food Safety, DG
Health and Food Safety) gave a keynote speech entitled
‘Delivering on EU food safety and nutrition in 2050 –
Future challenges and policy’.
The IT services we are building today will have to adapt
to the longer term perspectives. Long term evolution of
IT tools will need further evolutions of the EU legal
bases. Reviewing regularly the implementing acts of our
base regulations is a realistic option. One of the con-
crete contributions to the Digital Single Market pre-
sented relates to the Paperless Food Chain Control.
Important and “Impacting” changes will be available
from tomorrow (electronic certification in a paperless
control environment starts in 2017). These changes can
already bring a revolution in the way control operations
are managed.
Regarding official eCommerce control of food, the
Commission revealed its action plan. The new Official
Control Regulation will allow online ‘mystery’ sampling
by national competent authorities (i.e. control authori-
ties do not need to identify themselves to purchase
online). Concluding that the legislation as it stands is
harmonized, complete and ready for Digital Single
Market but that the enforcement varies from a Member
State to another, the objective is now to get all Member
States on board in the enforcement of EU Agri-Food law
on internet sales of food so as to allow dedicated train-
ing for control staff and more coordinated actions. With
the examples of the German eCommerce control system
and the Slovakian digital integration official controls
allowing a quick dissemination of results, it is clear that
integration between national and EU environment is
critical for a more clever and predictive control system
foreseen for 2020 (IMSOC through the implementation
of the Official Control Regulation). Synergy in automat-
ed risk based approaches can raise efficiency in inter-
ception of non-compliant goods. Emerging challenges
for the official control are being tackled: The eCom-
merce is an example of how successful and beneficial is
the sharing of knowledge and experience between the
more advanced countries and the other EU Member
States.
International organisations are following the same path
as illustrated by the OIE Digital Action Plan and the IPPC
ePhyto pilot project; EU should join and coordinate
efforts with them.
New legal environment (eIDAS) and associated EU wide
architectures will help us to better interoperate and will
remove barriers to allow the streamlining of our import-
export processes.
International standardisation efforts, both at semantic
and technical level, have made very important progress
in these last years and are allowing our EU IT systems to
speak easily together as well as with non-EU commercial
partners. This is a key asset for future success.
The presentations given at the conference should be
shortly available on the conference webpage. A new
conference on this topic is expected to be held in Brus-
sels in October 2017.
■ AESGP participates in the plenary meeting of the Advisory Group on the Food Chain and Animal and
Plant Health
AESGP attended on Friday 25 November 2016 the ple-
nary meeting of the Advisory Group on the Food Chain
and Animal and Plant Health. Michael Scannell, Director
of Directorate D ‘Food chain: stakeholder and interna-
tional relations’ at DG SANTE was chairing the meeting.
Commission officials from the various DG SANTE’s
directorates and units presented successively the varied
topics on the agenda.
The Commission presented the state of play and next
steps of its strategy to combat antimicrobial resistance
(AMR). The Commission's 2011 Action Plan against the
rising threats from AMR contains 12 actions for imple-
mentation with EU Member States and identifies 7 areas
where measures are most needed:
17 AESGP Euro OTC News | Issue 286
Page 18
making sure antimicrobials are used appropriately
in both humans and animals
preventing microbial infections and their spread
developing new effective antimicrobials or alterna-
tives for treatment
cooperating with international partners to contain
the risks of AMR
improving monitoring and surveillance in human
and animal medicine
promoting research and innovation
improving communication, education and training
The evaluation of the Action Plan published in October
2016 by the Commission shows that this had a clear
added value acting as a symbol of political commit-
ment, stimulating several actions within Member States,
and has served to strengthen international cooperation.
The Action Plan has also provided a framework to guide
and coordinate activities on AMR at international level
in the area of monitoring and surveillance and on R&D.
The Commission will now continue and scale up its fight
against antimicrobial resistance (AMR), with the launch
in 2017 of a second Action Plan. Feedback on the re-
lated roadmap can still be provided through the online
form until December 2016. The new Action Plan will
take the form of a Commission communication to the
European Parliament and the Council will focus on
supporting Member States, particularly in establishing,
implementing and monitoring their National Action
Plans, bringing together EU funds and instruments in
order to promote innovation and research against AMR
and strengthening its leading role in global fora, nota-
bly within the international organisations and with
major trade partners.
The Commission elaborated further on its action plan
on Food& Feed eCommerce Control. An ad-hoc
Working Group of national authorities (e-commerce
network) has been created to get all Member States on
board in the enforcement of EU Agri-Food law on inter-
net sales of food so as to allow dedicated training for
control staff and more coordinated actions and will
meet for the second time in December 2016.
The proposal for SANTE actions in 2017 includes nota-
bly coordinated control actions on food supplements
and non-authorised novel foods.
As to the Official Control Regulation, the Commission
updated the group on the inter-institutional process,
providing an example of the text currently under revi-
sion with the Council and jurist linguists and presenting
the structure of the Regulation. This presentation was a
short follow-up to the ad-hoc Working Group meeting
on revision of the Official Control Regulation of Friday
30 September 2016 (to which AESGP participated). In
terms of timing, the text will be transmitted officially to
the Council early December for possible adoption by
Council and then Parliament (in Plenary) by March 2017.
More details will be provided at the next Working
Group meeting on revision of the Official Control Regu-
lation to be held once the regulation will be published.
On the implementing rules under Regulation (EU) No
1169/2011, related to origin information, the Commis-
sion presented the latest draft version. It covers all food
products and any statement relating to origin of food
products and clarifies any potential overlaps with other
legal frameworks. The text will soon be put to the feed-
back mechanism, i.e. open for comments from citizens
and stakeholders for 4 weeks.
The current legislative and risk assessment framework
and on the activities foreseen in 2017 in relation to food
contact materials (FCM) were presented. A draft mea-
sure on printed FCM including printing inks and FCM
that are printed may be prioritized in 2017-2018 follo-
wing a German notification raising health concerns.
Regarding the risk for human health of mineral oils
(Mineral Oil Hydrocarbons ‘MOH’ and Mineral Oil Satu-
rated Hydrocarbons ‘MOSH’) in food, the Commission is
to adopt and publish fairly soon a recommendation on
the monitoring of MOH in food and FCM. Since migra-
tion from FCMs such as recycled paper and board food
packaging could contribute significantly to the total
exposure, monitoring should include re-packaged food,
the packaging material and the presence of functional
barriers. The Commission highlighted continuing chal-
lenges in resources and in particular analytical metho-
dology. The results of the monitoring will feed into the
ongoing discussions on the need for a general maxi-
mum level for mineral oil in food.
18 AESGP Euro OTC News | Issue 286
Page 19
Medical devices
■ Belgian classification of cranberry extract based capsules as a medicine ruled invalid
Judgment of the Belgian Council of State invalida-
ting a decision from the Belgian Federal Agency for
Medicines and Health Products (FAMHP) classifying
a cranberry extract based capsules intended to pre-
vent and treat urinary tract infections as a medicinal
product
By a judgment delivered on 1 September 2016 in a case
opposing BV Medical Brands Innovations to the Belgian
State, the Belgian Council of State ruled that the FAMHP
decision taken on March 18, 2015 classifying the pro-
duct concerned (Cystiberry - capsules containing an
extract of whole cranberries) as a medicinal product for
human use was invalid.
Essentially, the Court ruled that the FAMHP decision
incorrectly implemented the ‘rule of doubt’ set in Article
2(2) of the Medicinal Products Directive 2001/83 and
did not comply with the criteria set out by the Court of
Justice of the EU (CJEU) with regard to the medicinal
product classification.
On the ‘rule of doubt’, the Belgian Council of State
recalled that – per CJEU case law (Case C‑140/07
Hecht‑Pharma) - Article 2(2) of Directive 2001/83 must
be interpreted as meaning that that directive does not
apply to a product in respect of which it has not been
scientifically established that it is a medicinal product by
function, without its being possible to exclude that possi-
bility. On that basis, FAMHP incorrectly concluded that
"where there is doubt about the status of the product, the
product must be regarded as a medicinal product."
The grounds of the contested FAMHP decision stated
that, even if questions would remain open as regards
the precise mechanism, the inhibitory effect of the
product concerned resulting from an interaction bet-
ween the product and the Fimbriae (the hair-like appen-
dages) on the bacterial cell wall of the E-coli bacteria
should be considered a pharmacological, immunologi-
cal or metabolic action.
The Belgian Council of State referred to the following
CJEU case-law:
Case C-308/11 (Chemische Fabrik Kreussler & Co.
GmbH v Sunstar Deutschland GmbH), in which the
Court pointed out at the outset that it is not apparent
either from Directive 2001/83 or from the guidance
document on the demarcation between the Cosmetic
Products Directive and the Medicinal Products Directive
that the molecules of the substance in question must
necessarily interact with a human cellular constituent in
order for it to be regarded as a substance which exerts a
‘pharmacological action’. (paragraph 29).
Case C‑140/07 (Hecht‑Pharma) recalling that it is appa-
rent from Article 1(2)(b) of Directive 2001/83 that the
substance in question must be capable of restoring, cor-
recting or modifying physiological functions by exerting a
pharmacological, immunological or metabolic action and
that that capability must have been scientifically establis-
hed.
According to the Belgian Council of State, FAMHP ap-
pears not to have carried out an assessment of the
product individually, taking into account the pharmaco-
logical properties as they can be established in the
present state of scientific knowledge and all the charac-
teristics of the product, including, inter alia, its composi-
tion, the manner in which it is used, the extent of its
distribution, its familiarity to consumers and the risks
which its use may entail. In other words, with regard to
the establishment of the pharmacological properties,
FAMHP has failed to scientifically establish in the con-
tested decision that the interaction between the pro-
duct and the Fimbriae (the hair-like appendages) on the
bacterial cell wall of the E-coli bacteria is capable of
exerting a pharmacological, immunological or metabolic
action in the meaning of Article 1(2)(b) of Directive
2001/83.
19 AESGP Euro OTC News | Issue 286
Page 20
20 AESGP Euro OTC News | Issue 286
Vienna 2017
AESGP Conference with the Heads of EU Medicines Agencies (HMA) during the Maltese EU Presidency
Agenda 2017
53rd AESGP Annual Meeting
30 May - 1 June
2017 AESGP Conference
on regulatory challenges for consumer health products
Brussels, Belgium
10-11 October