58
PHATHOPHYSIOLOGY
| Date post: | 12-Aug-2015 |
| Category: |
Education |
| Upload: | free-education-for-all |
| View: | 141 times |
| Download: | 4 times |

PHATHOPHYSIOLOGY

BREATHING
• The main function is to oxygenate the hemoglobin and remove carbon dioxide from the blood.
• Into the pulmonary capillaries the venous blood receives O2 and releases CO2 becoming arteriosus.

O2O2
ATMOSPHERIC AIR
ATMOSPHERIC AIR
CO2CO2
TISSUESTISSUESLUNGSLUNGS

PLEURAL ANATOMY

AIRWAYS ANATOMY

In the conduction system the inspired air is heated, humidified and purified.This latter function is of fundamental importance; particles having a diameter greater than 5 mm are stopped before reaching the alveoli, retained by mucus and ejected toward the pharynx by mucociliary transport.
Particulate
Mucociliary transport is the mechanism by which the airways clears itself of secretions and trapped particulates. The two major components of this system are the mucous blanket and the ciliated epithelial cells.
Mucus
Cilia
Goblet cells
Columnar cells
Basement membrane
AIRWAYS FUNCTIONS



Dead space is the volume of air which is inhaled that does not take part in the gas exchange, either because it remains in the conducting airways, or reaches alveoli that are not perfused or poorly perfused. In other words, not all the air in each breath is available for the exchange of oxygen and carbon dioxide. Mammals breathe in and out of their lungs, wasting that part of the inspiration which remains in the conducting airways where no gas exchange can occur.
CONDUCTING ZONE
TRANSITIONALAL ZONE
RESPIRATORY ZONE
FUNCTIONAL ANATOMY
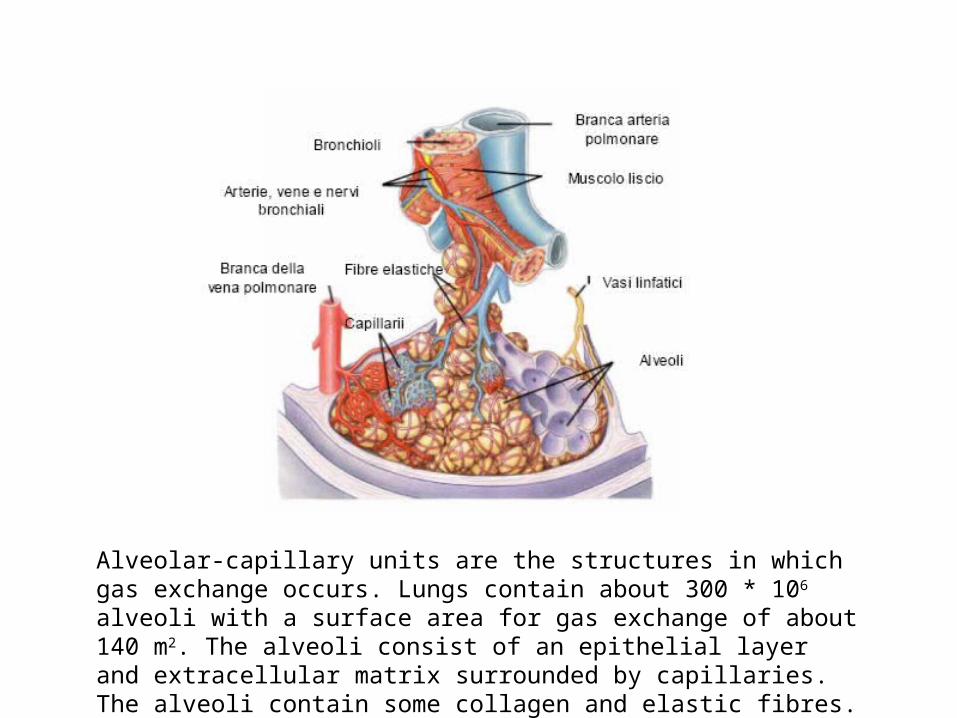
Alveolar-capillary units are the structures in which gas exchange occurs. Lungs contain about 300 * 106 alveoli with a surface area for gas exchange of about 140 m2. The alveoli consist of an epithelial layer and extracellular matrix surrounded by capillaries. The alveoli contain some collagen and elastic fibres.

Elastic fibres
Type II pneumocytes
Type I pneumocytes
Capillaries
Macrophages Endothelial cells
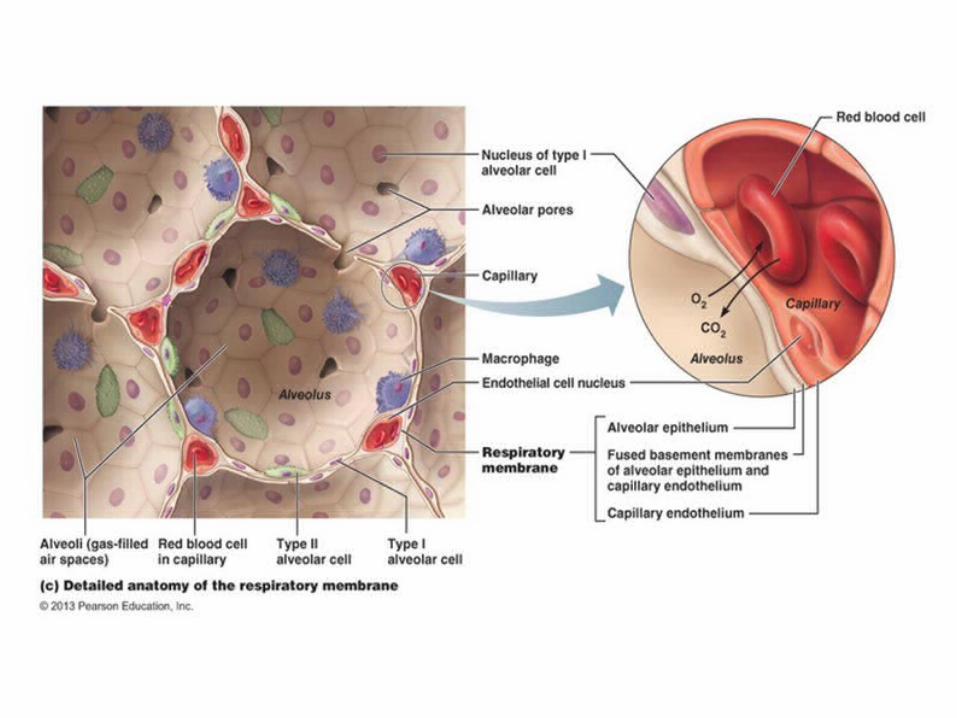
ALVEOLAR HYSTOLOGY
There are three major cell types in the alveolar wall (pneumocytes):
• Type I (Squamous Alveolar) cells that form the structure of an alveolar wall• Type II (Great Alveolar) cells that secrete pulmonary surfactant to lower the surface tension of
water and allows the membrane to separate, therefore increasing its capability to exchange gases. Surfactant is continuously released by exocytosis.
• Macrophages that destroy foreign material, such as bacteria.

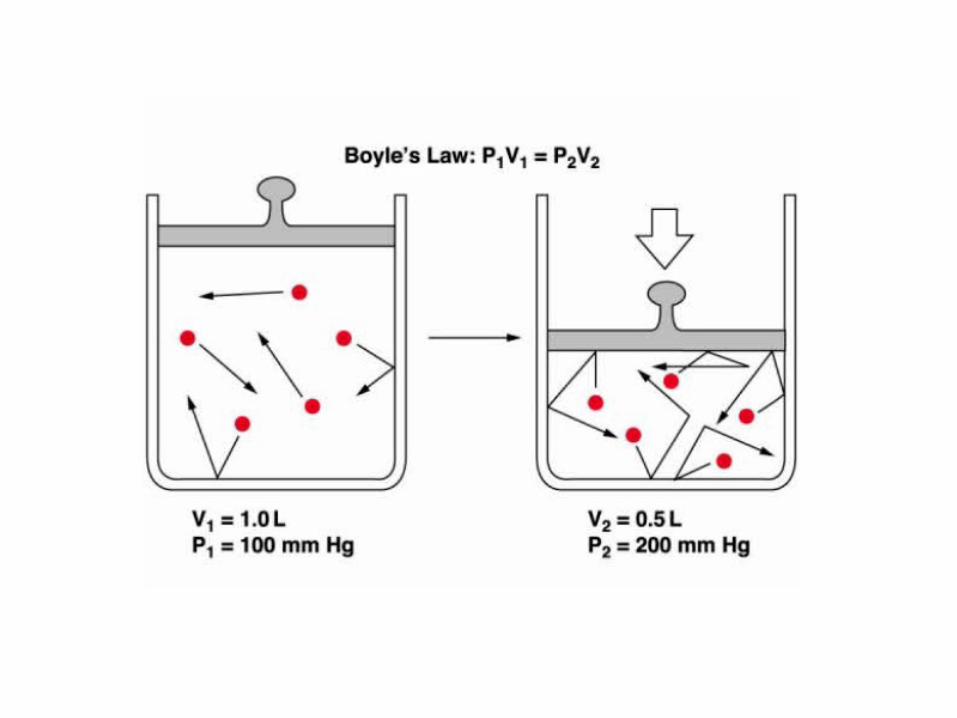
Inhalation (breathing in) is an active movement due to diaphragm and other
respiratory muscles contraction. These contractions cause the thoracic cavity to
increase in volume, thus decreasing the pressures within the lung according to
Boyle’s law. Intrapleural and Alveolar Pressures are related because pleural
space is an enclosed space and its volume can not be modified. This negative
pressure within the lungs acts as a Pressure Gradient, thus pulling air into the
lungs. As air fills the lungs, the negative alveolar pressure moves back towards
atmospheric pressure, and air flow into the lungs slows down. In contrast,
expiration (breathing out) is usually a passive process due to the elastic recoil
of the lung tissue (rich in elastic fibers). During exercise or other conditions
requiring hyperventilation expiration becomes an active process due to
muscular activity.
BREATHING MECHANISM


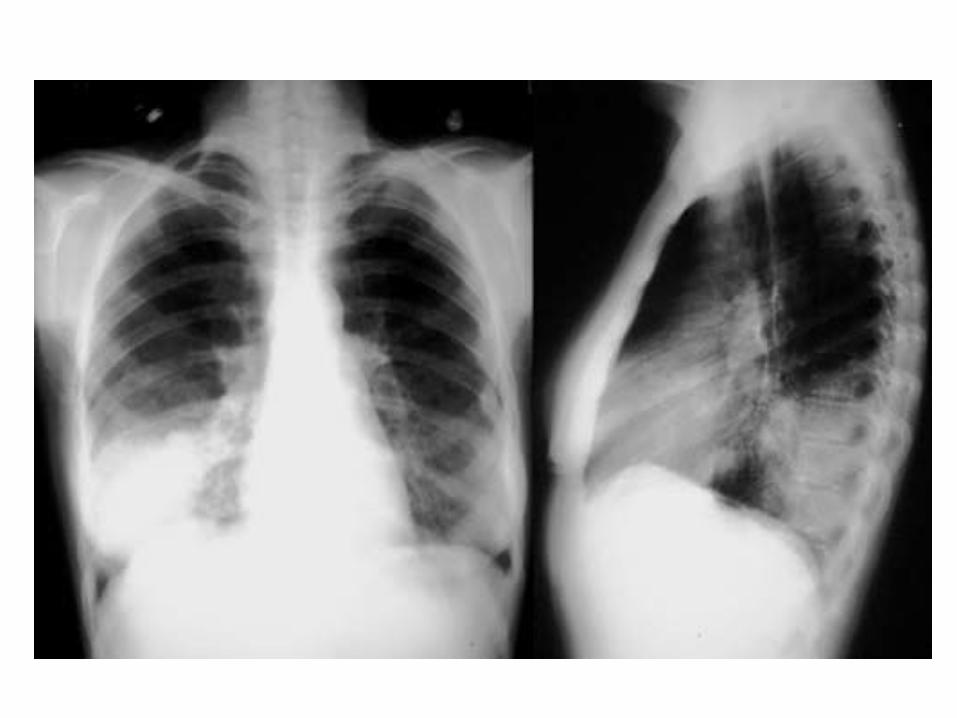
PNEUMONIA
DEFINITION: Infection interesting the lung (alveoli, distal airways and lung interstitium).
CLASSIFICATION:
Community acquired
Nosocomial


ETIOLOGY
• COMMUNITY ACQUIRED– Streptococcus Pneumoniae– Haemofilus Influenziae– Stafilococcus Aureus– Others (Mycoplasma, Clamidia, Legionella, etc.)
• HOSPITAL ACQUIRED– Pseudomonas Aeruginosa– Enterobacter– Klebsiella Pneumoniae

CLINICAL PICTURE
• Fever• Caugh without or with sputum• Shortness of breath• Chest pain (due to pleural involvment)• Physical examination (rales due to secretions)• Blood tests (inflammatory indexes, blood gases)• X-ray


4 MAIN HYSTOTYPES:
• NSLC (non-small lung cancer)– Squamous cell carcinoma– Adenocarcinoma– Large cell anaplastic carcinoma
• Small cell anaplastic carcinoma
LUNG CANCER
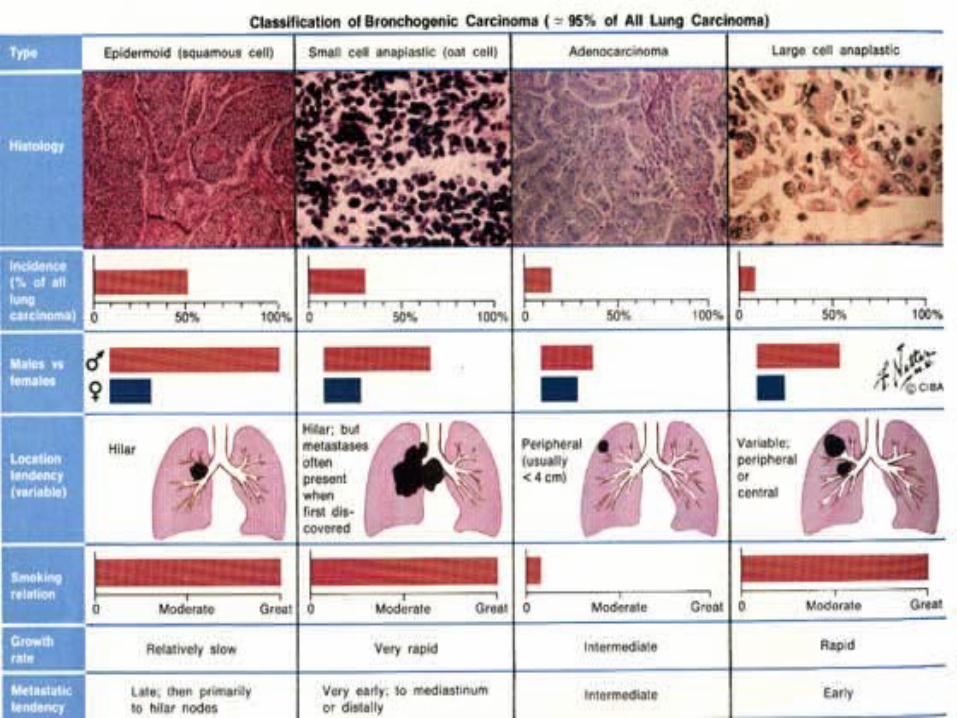
Adenocarcinomain the upper lobe of the
left lung

Large cell anaplastic carcinoma of the upper lobe of the right lung
with extensive involvement of hilar and carinal nodes.
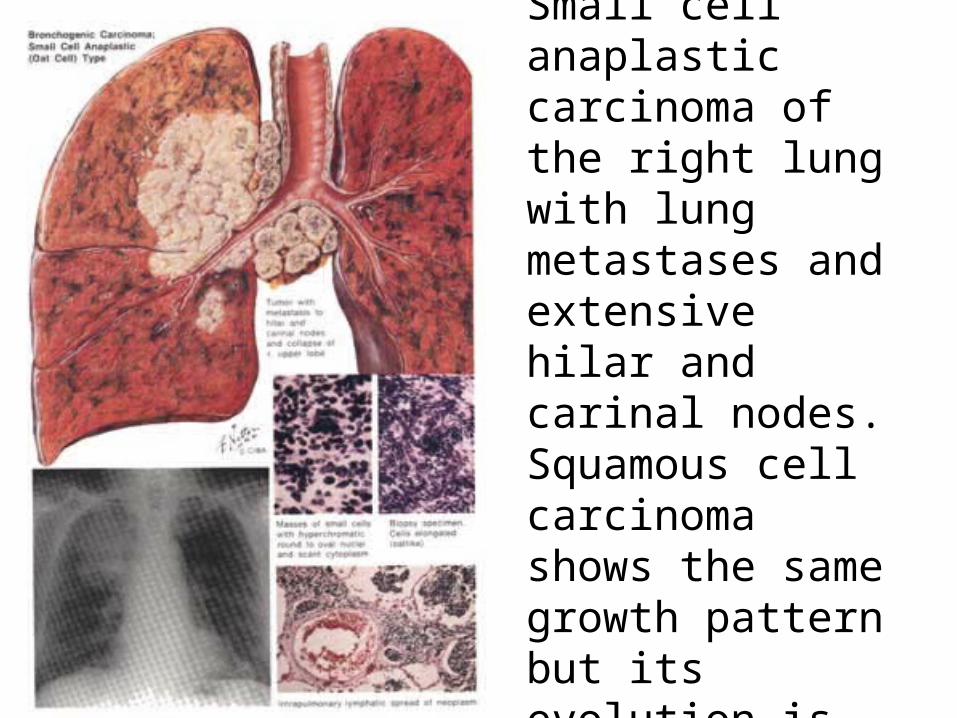
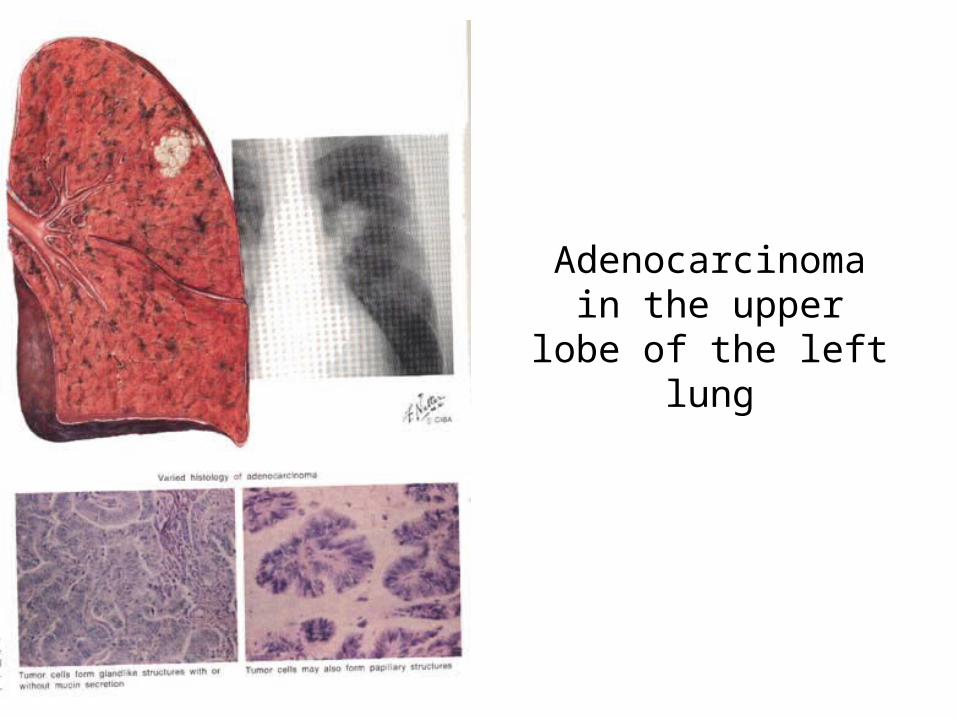
Small cell anaplastic carcinoma of the right lung with lung metastases and extensive hilar and carinal nodes.Squamous cell carcinoma shows the same growth pattern but its evolution is slower.
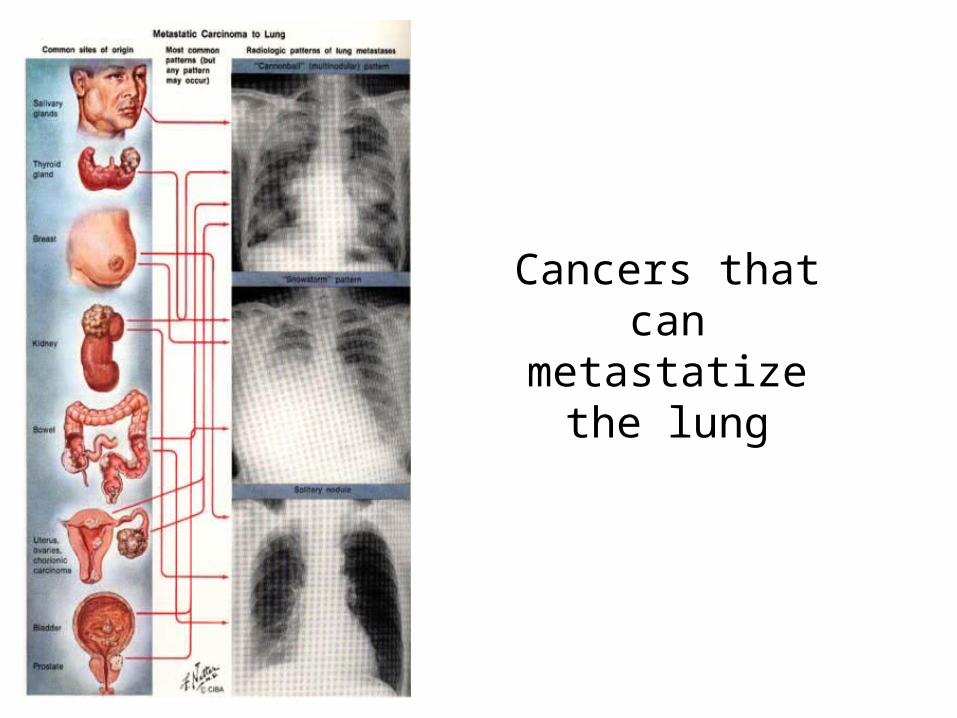
Cancers that can metastatize the lung

PLEURAL EFFUSION
• TRANSUDATE: heart failure, hypoalbuminemia.• EXUDATE: inflammatory fluid with high
concentration of protein, LDH and inflammatory cells (lymphocytes, granulocytes, macrophages, red blood cells or neoplastic cells). Causes: pleurisy, pneumonia, pulmonary embolism, lung cancer and pleural cancer.


THORACENTESIS
• Thoracentesis is a percutaneous procedure during which a needle is inserted into the pleural space and pleural fluid is removed through the needle
• Diagnostic thoracentesis refers to removal of a small volume of pleural fluid for analysis,
• Therapeutic thoracentesis refers to removal of a large volume (< 1L) of pleural fluid for relief of symptoms (.
Useful exams• Cell count• Biochemistry• Culture• Cytology

BRONCHOSCOPY
Bronchoscopy is the process of direct visualization of the tracheobronchial tree. Bronchoscopy is now performed almost exclusively with flexible fiberoptic instruments. The bronchoscopist is able to identify endobronchial pathology, including tumors, granulomas, bronchitis, foreign bodies, and sites of bleeding. Samples from airway lesions can be taken by several methods, including washing, brushing, and biopsy. Washing involves instillation of sterile saline through a channel of the bronchoscope. A portion of the liquid is collected by suctioning through the bronchoscope, and the recovered material can be analyzed for cells (cytology) or organisms (by standard stains and cultures). Brushing or biopsy of the surface of the lesion, using a small brush or biopsy forceps at the end of a long cable inserted through a channel of the bronchoscope, allows recovery of cellular material or tissue for analysis by standard cytologic and histopathologic methods. The bronchoscope can be used to sample material not only from the regions that can be directly visualized (i.e., the airways) but also from the more distal pulmonary parenchyma. With the bronchoscope wedged into a subsegmental airway, aliquots of sterile saline can be instilled through the scope, allowing sampling of cells and organisms even from alveolar spaces. This procedure is called bronchoalveolar lavage.

CYSTIC FIBROSIS

• Cystic fibrosis (CF) is a monogenic disorder that presents as a multisystem disease.
• The first signs and symptoms typically occur in childhood, but about 5% of patients in the United States are diagnosed as adults.
• Due to improvements in therapy, >46% of patients are now adults (≥18 years old) and 16.4% are past the age of 30.
• The median survival is >37.4 years for patients with CF; thus, CF is no longer only a pediatric disease.
• CF is characterized by chronic bacterial infection of the airways that leads to bronchiectasis and bronchiolectasis, exocrine pancreatic insufficiency and intestinal dysfunction, abnormal sweat gland function, and urogenital dysfunction.
CYSTIC FIBROSIS (CF)

The prevalence of CF varies with the ethnic origin of a population. CF is detected in approximately:
• 1 in 3000 live births in the Caucasian population of North America and northern Europe,
• 1 in 17,000 live births of African Americans• 1 in 90,000 live births of the Asian population of Hawaii.
The most common mutation in the CFTR gene (~70% of CF chromosomes) is a 3-bp deletion (a class II mutation) that results in an absence of phenylalanine at amino acid position 508 (ΔF508) of the CF gene protein product, known as CFTR. The large number (>1500) of relatively uncommon (<2% each) mutations identified in the CFTR gene makes genetic testing challenging.
EPIDEMIOLOGY

• Born/year in Italy: more than 200 people• Affected 5000-6000 people (40% adults)• Carriers: meore than 2.200.000 people• Median survaival: more than 35 years• CF is the most common autosomic recessive disease in caucasian population.
ITALIAN EPIDEMIOLOGY

CF is an autosomal recessive disease resulting from mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene located on chromosome 7. The mutations in the CFTR gene fall into five major classes. Classes I–III mutations are considered "severe," as indexed by pancreatic insufficiency and high sweat NaCl values. Class IV and V mutations can be "mild," i.e., associated with pancreatic sufficiency and intermediate/normal sweat NaCl values.
THE DISEASE
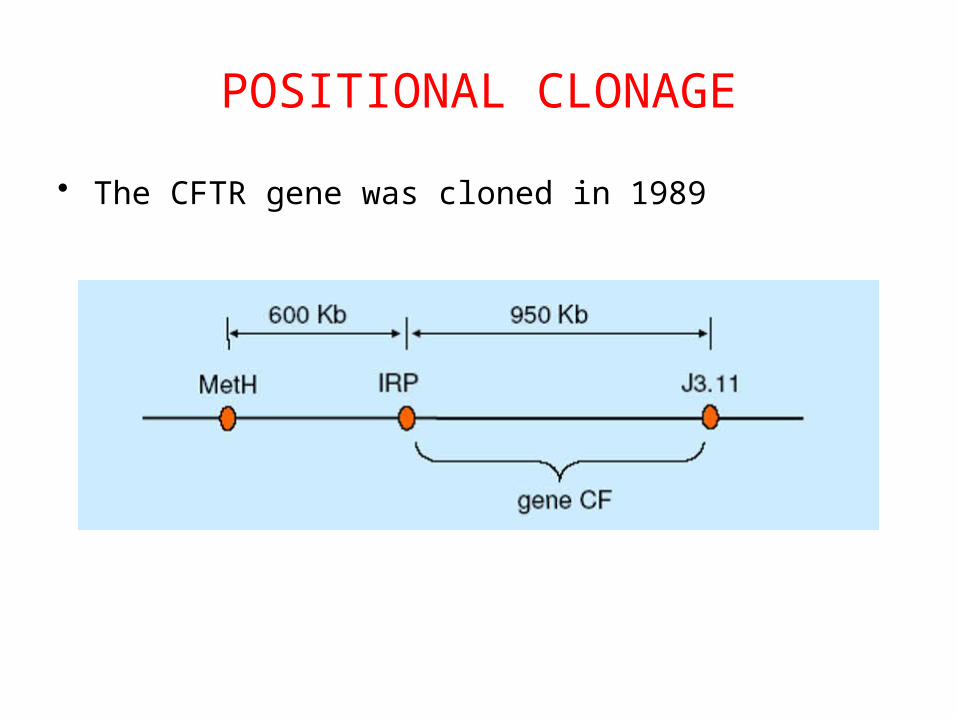
POSITIONAL CLONAGE
• The CFTR gene was cloned in 1989

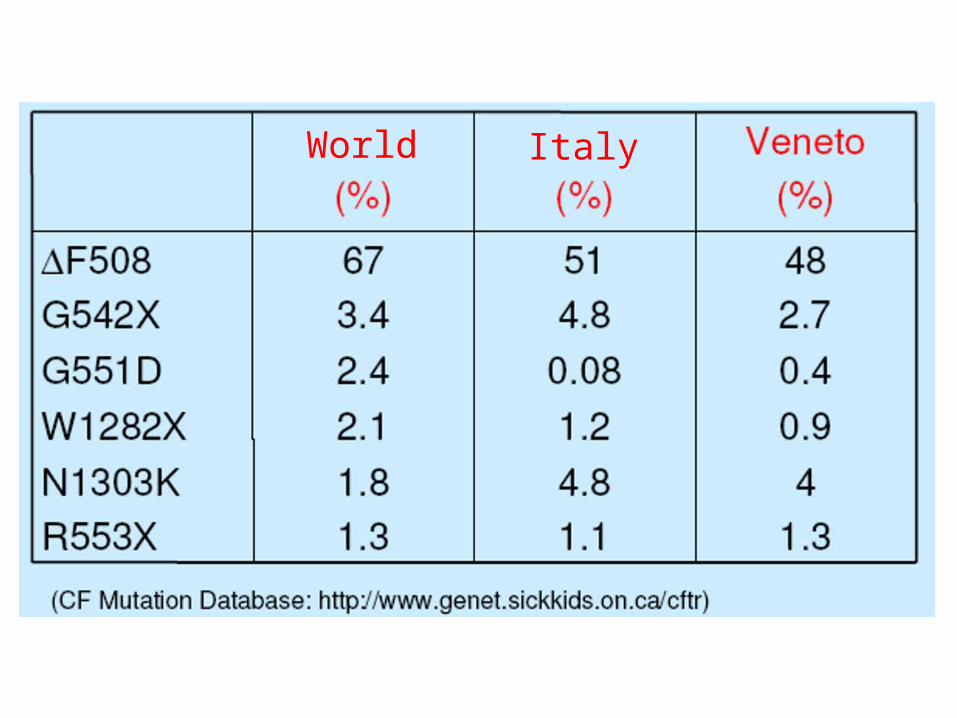
World Italy


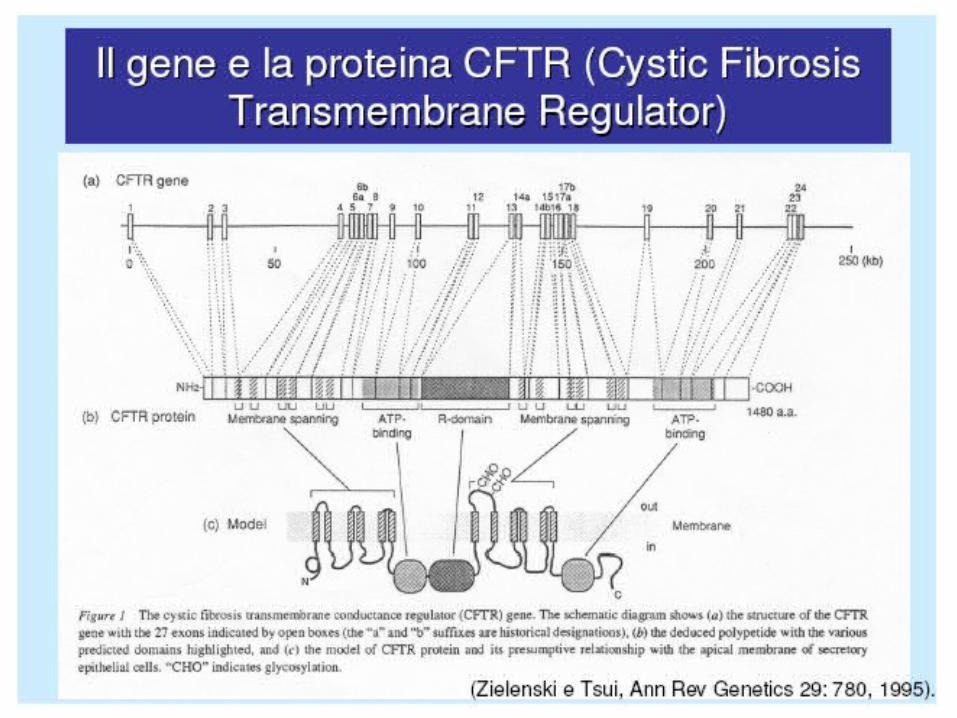
The CFTR protein is a single polypeptide chain, containing 1480 amino acids, that functions both as a cyclic AMP–regulated Cl− channel and as regulator of other ion channels. The fully processed form of CFTR localizes to the plasma membrane in normal epithelia. Biochemical studies indicate that the ΔF508 mutation leads to improper maturation and intracellular degradation of the mutant CFTR protein. Thus, absence of CFTR in the plasma membrane is central to the molecular pathophysiology of the ΔF508 mutation and other classes I–II mutations. Classes III–IV mutations produce CFTR proteins that are fully processed but are nonfunctional or only partially functional in the plasma membrane. Class V mutations include splicing mutations that produce small amounts of functional CFTR.
CFTR PROTEIN

The protein has six transmembrane domains, two intracellular “nucleotide binding folds” (NBFs), and a regulatory domain (R domain) containing a number of phosphorilation sites.
CFTR STRUCTURE



Schema describing classes of genetic mutations in CFTR gene and effects on CFTR protein/function. Note the ΔF508 mutation is a class II mutation and, like class I mutations, would be predicted to produce no mature CFTR protein in the apical membrane. CFTR, cystic fibrosis transmembrane conductance regulator.
Legend:
From: Chapter 259. Cystic FibrosisHarrison's Principles of Internal Medicine, 18e, 2012
From: Chapter 259. Cystic FibrosisHarrison's Principles of Internal Medicine, 18e, 2012

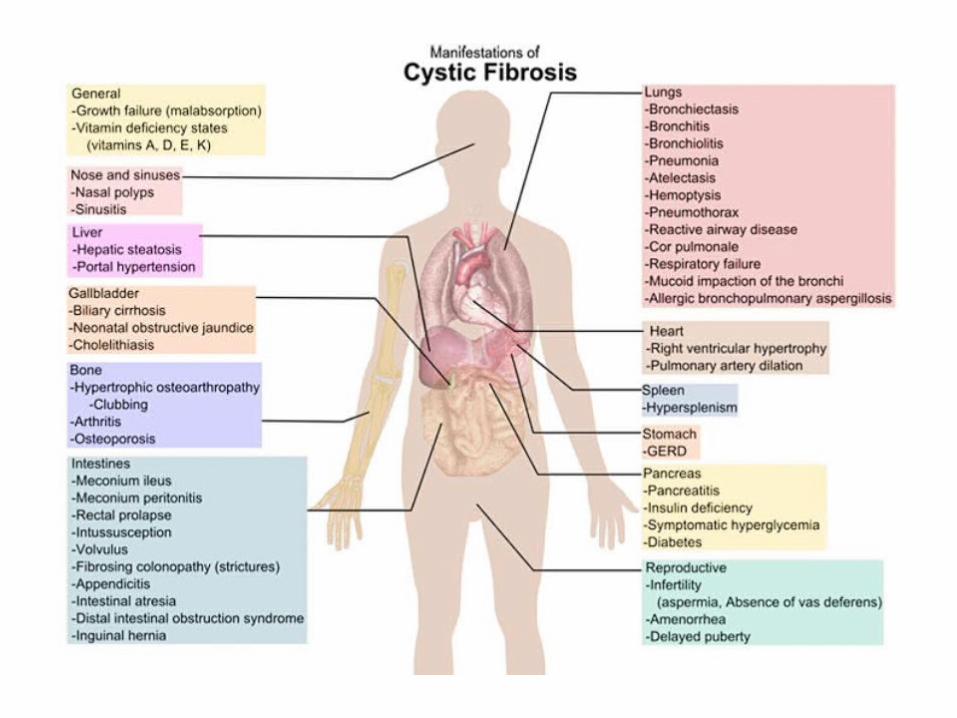
CLINICAL MANIFESTATIONS
Epithelial DysfunctionThe epithelia affected by CF exhibit different functions in their native state, i.e., some are volume-absorbing (airways and distal intestinal epithelia), and some are salt- but not volume-absorbing (sweat duct), whereas others are volume-secretory (proximal intestine and pancreas). Given this diversity of native activities, it is not surprising that CF produces organ-specific effects on electrolyte and water transport. However, the unifying concept is that all affected tissues express abnormal ion transport function.

Comparison of ion transport properties of normal (LEFT) and CF (RIGHT) airway epithelia. The vectors describe routes and magnitudes of Na+ and Cl− transport that is accompanied by osmotically driven water flow. The normal basal pattern for ion transport is absorption of Na+ from the lumen via an amiloride-sensitive epithelial Na+ channel (ENaC) composed of α, β, and γ ENaC subunits. This process is accelerated in CF. The capacity to initiate cyclic AMP–mediated Cl− secretion is diminished in CF airway epithelia due to abnormal maturation/dysfunction of the CFTR Cl− channel. The accelerated Na+ absorption in CF reflects the absence of CFTR inhibitory effects on Na+ channels. A Ca2+-activated Cl− channel, likely a product of the TMEM16a gene, is expressed in normal and CF apical membranes and can be activated by extracellular ATP. Horizontal arrows depict velocity of mucociliary clearance (μm/sec).

AIRWAYS

Recurrent infections of upper and lower airways• chronic sinusitis• nasal polyps• rhinorrhea• cough with mucopurulent very viscous sputum• recurrent bronchitis• gradual deterioration of respiratory function
Alternating periods of stability with exacerbationsThe bacteria involved are:
• Staphylococcus aureus ed Hemophilus influentiae during childhood
• Pseudomonas aeruginosa in adultsMore than 95% of deaths in CF patients are related to respiratory tract infections.
RESPIRATORY MANIFESTATIONS OF CF

• Pancreas: at this level the Cl-/HCO3- exchange is altered
resulting in reduced secretion of water and bicarbonate in pancreatic secretions; this is reflected in a non-complete excretion of pancreatic enzymes that determines the onset of chronic pancreatitis with gradual destruction of the parenchyma (cyst formation and fibrous tissue)• Bowel: the absorption of water and Na+ increases with meconium ileus in 10% of infants and frequent episodes of bowel obstruction in children and young adults.
OTHER MANIFESTATIONS OF CF

• Reproductive: occlusion of the deferens ducts in 95% of males with azoospermia. Female infertility due to mucous plug at the uterine cervix;
• Sweat Gland: excess sweat chloride, used as important diagnostic test.
OTHER MANIFESTATIONS OF CF

DIAGNOSIS
The large number of mutations described makes DNA analysis challenging in the diagnostic setting.
Clinical criteriaSweat test:The sweat glands secrete in their lumen high quantities of Na+ and Cl- which osmotically draw water .The sudorifics ducts are waterproof and have the task to absorb most of the electrolytes in order to avoid salts depletion.In the case of the CF the reabsorption of Cl- in the sudorific ducts is compromised and the concentration of Cl- in the sweat is very high compared to normal subjects (> 70 mEq /L)

GENE THERAPY
A possible application of gene therapy was hypotesized since1989, when it was sequenced and cloned the CFTR gene (Riordan et al., 1989);Transferring the CFTR gene into deficient CFTR cells lead, in vitro, to the production of Cl- channels;Theoretically transferring and making express airway cells of patients with CF the CFTR gene, should resolve the disease; gene therapy, in fact, addresses the basic effect rather than the downstream consequences of the disease. This approach has the potential to impact significantly on the natural history of the disease.

VECTORS
The transfection efficiency of naked DNA is so low that gene transfer agents (GTAs) have been designed to enhance entry to the cell/nucleus.
• viral (Adenovirus, Adeno-associated virus, Sendai virus, Lentivirus).
• non-viral, usually lipid-based, but also including nanoparticles (cationic liposomes, compacted DNA nanoparticles).
Limits: inflammatory response (mainly with pathogens viruses), host immune responses (that lead to absent gene expression after subsequent exposure)



















