First-principles computational study of defect clustering in solid solutions of ThO 2 with trivalent oxides Vitaly Alexandrov Department of Chemical Engineering and Materials Science and NEAT ORU, University of California, Davis, California 95616, USA Department of Materials Science and Engineering, University of California, Berkeley, California 94720, USA Niels Grønbech-Jensen Department of Applied Science, University of California, Davis, California 95616, USA Alexandra Navrotsky Peter A. Rock Thermochemistry Laboratory and NEAT ORU, University of California, Davis, California 95616, USA Department of Chemical Engineering and Materials Science, University of California, Davis, California 95616, USA Mark Asta Department of Materials Science and Engineering, University of California, Berkeley, California 94720, USA Department of Chemical Engineering and Materials Science and NEAT ORU, University of California, Davis, California 95616, USA (Dated: July 16, 2018) The energetics of mixing and defect ordering in solid solutions of fluorite-structured ThO2 with oxides of trivalent cations (Sc, In, Y, Nd, La) are investigated by electronic density-functional- theory (DFT). Through DFT calculations of structures enumerated by lattice-algebra techniques, we identify the lowest-energy patterns for defect clustering for four separate dopant concentrations. The most stable structures are characterized by a repulsive interaction between nearest-neighbor vacancies on the oxygen sublattice. The enthalpies of formation with respect to constituent oxides are positive for all dopants considered, and show a tendency to decrease in magnitude as the size and electronegativity of the trivalent dopant decrease. Due to the small positive formation enthalpies and low oxygen-vacancy binding energy with La dopants, La2O3-ThO2 solid solutions are predicted to have relatively high ionic conductivities relative to those for the other aliovalent dopants considered. Our results are compared with those for the more widely studied ZrO2 and CeO2 fluorite-structured solid solutions with trivalent cations. I. INTRODUCTION Design of solid-state oxygen ion conductors, oxygen sensors and separation membranes 1–5 typically involves aliovalent cation doping of metal oxides to form charge- compensating oxygen vacancies. The observation of high oxygen conductivity in aliovalently doped fluorite- structured oxides has motivated extensive experimen- tal and theoretical research aimed at relating the struc- tural and chemical defect properties in these systems to their ionic conductivities. Recent studies of trivalent- doped zirconia, hafnia, ceria and thoria 6–12 have revealed a strong correlation between oxygen conductivity and the mixing thermodynamic properties. Specifically, en- thalpies of formation (ΔH f ) derived from calorimetric measurements tend to become more positive with dopant concentration (x) at low solute concentrations, in a man- ner consistent with a regular solution behavior; near some maximum composition (x max ), however, ΔH f is observed to level off or become less positive with further doping. 8,11,12 The value of x max is found to coincide with the maximum in measured ionic conductivity. 10 These experimental results suggest that the mobile oxygen va- cancies begin to strongly associate and form stable, rela- tivley immobile, clusters for dopant concentrations near and beyond x max . 7–9,12,13 However, the detailed nature of these defect clusters, and how they vary in different systems, remains incompletely understood. In the technologically important yttria-stabilized zirco- nia (YSZ) system, atomistic simulations 14–19 agree with neutron and X-ray diffraction experiments 7,20 that oxy- gen vacancies tend to align along the <111> direction as third nearest neighbors on the oxygen sublattice, and pre- fer to associate with the smaller host Zr ions rather than with the larger Y dopants. Further, DFT studies 14,15,18 have shown that these defect-clustering tendencies result in the stability of two ordered ground-state structures: the experimentally observed δ-Zr 3 Y 4 O 12 compound and an as-yet unobserved ZrO 2 -rich phase. Through studies of other trivalent and divalent dopants in ZrO 2 , it has been shown that the formation enthalpies of doped ZrO 2 compounds are most positive for small dopant ions. The results also demonstrate a tendency for oxygen vacancies to associate with the smaller cation species (Zr or dopant) arXiv:1009.1850v1 [cond-mat.mtrl-sci] 9 Sep 2010
Transcript
First-principles computational study of defect clustering in solid solutions of ThO2
with trivalent oxides
Vitaly AlexandrovDepartment of Chemical Engineering and Materials Science and NEAT ORU,
University of California, Davis, California 95616, USADepartment of Materials Science and Engineering,
University of California, Berkeley, California 94720, USA
Niels Grønbech-JensenDepartment of Applied Science, University of California, Davis, California 95616, USA
Alexandra NavrotskyPeter A. Rock Thermochemistry Laboratory and NEAT ORU,
University of California, Davis, California 95616, USADepartment of Chemical Engineering and Materials Science,
University of California, Davis, California 95616, USA
Mark AstaDepartment of Materials Science and Engineering,
University of California, Berkeley, California 94720, USADepartment of Chemical Engineering and Materials Science and NEAT ORU,
University of California, Davis, California 95616, USA(Dated: July 16, 2018)
The energetics of mixing and defect ordering in solid solutions of fluorite-structured ThO2 withoxides of trivalent cations (Sc, In, Y, Nd, La) are investigated by electronic density-functional-theory (DFT). Through DFT calculations of structures enumerated by lattice-algebra techniques,we identify the lowest-energy patterns for defect clustering for four separate dopant concentrations.The most stable structures are characterized by a repulsive interaction between nearest-neighborvacancies on the oxygen sublattice. The enthalpies of formation with respect to constituent oxidesare positive for all dopants considered, and show a tendency to decrease in magnitude as the size andelectronegativity of the trivalent dopant decrease. Due to the small positive formation enthalpies andlow oxygen-vacancy binding energy with La dopants, La2O3-ThO2 solid solutions are predicted tohave relatively high ionic conductivities relative to those for the other aliovalent dopants considered.Our results are compared with those for the more widely studied ZrO2 and CeO2 fluorite-structuredsolid solutions with trivalent cations.
I. INTRODUCTION
Design of solid-state oxygen ion conductors, oxygensensors and separation membranes1–5 typically involvesaliovalent cation doping of metal oxides to form charge-compensating oxygen vacancies. The observation ofhigh oxygen conductivity in aliovalently doped fluorite-structured oxides has motivated extensive experimen-tal and theoretical research aimed at relating the struc-tural and chemical defect properties in these systems totheir ionic conductivities. Recent studies of trivalent-doped zirconia, hafnia, ceria and thoria6–12 have revealeda strong correlation between oxygen conductivity andthe mixing thermodynamic properties. Specifically, en-thalpies of formation (∆Hf ) derived from calorimetricmeasurements tend to become more positive with dopantconcentration (x) at low solute concentrations, in a man-ner consistent with a regular solution behavior; nearsome maximum composition (xmax), however, ∆Hf isobserved to level off or become less positive with furtherdoping.8,11,12 The value of xmax is found to coincide withthe maximum in measured ionic conductivity.10 These
experimental results suggest that the mobile oxygen va-cancies begin to strongly associate and form stable, rela-tivley immobile, clusters for dopant concentrations nearand beyond xmax.7–9,12,13 However, the detailed natureof these defect clusters, and how they vary in differentsystems, remains incompletely understood.
In the technologically important yttria-stabilized zirco-nia (YSZ) system, atomistic simulations14–19 agree withneutron and X-ray diffraction experiments7,20 that oxy-gen vacancies tend to align along the <111> direction asthird nearest neighbors on the oxygen sublattice, and pre-fer to associate with the smaller host Zr ions rather thanwith the larger Y dopants. Further, DFT studies14,15,18
have shown that these defect-clustering tendencies resultin the stability of two ordered ground-state structures:the experimentally observed δ-Zr3Y4O12 compound andan as-yet unobserved ZrO2-rich phase. Through studiesof other trivalent and divalent dopants in ZrO2, it hasbeen shown that the formation enthalpies of doped ZrO2
compounds are most positive for small dopant ions. Theresults also demonstrate a tendency for oxygen vacanciesto associate with the smaller cation species (Zr or dopant)
arX
iv:1
009.
1850
v1 [
cond
-mat
.mtr
l-sc
i] 9
Sep
201
0
2
in a given system. In classical atomistic simulations ofthe energetics of defect cluster formation in trivalent-doped CeO2 (Ref.21,22) it has been demonstrated thatthe atomic geometry of defect clusters is controlled bya combination of electrostatic and elastic energy effects,the latter being dependent on the dopant ion size. Thebinding energies between dopant cation and oxygen va-cancies also have been shown to be a strong function ofthe dopant ion size, with the smallest dopants exhibitinggreatest binding energies with nearest-neighbor oxygenvacancies.
Solid solutions of ThO2 with aliovalent trivalentcations also can be utilized for solid electrolyte appli-cations, similar to doped zirconia and ceria. ThO2 alsohas been gaining increasing attention due to the renewedinterest in the Th–U fuel cycle which offers a numberof advantages over the U–Pu cycle including: higherthermal conductivity of ThO2 relative to UO2 based fu-els and lower concentrations of long-lived transuranicelements.23,24 The trivalent dopants Y, Nd and La areimportant products in the thorium-based nuclear cycle.Recently, Y2O3- and La2O3-doped ThO2 have been thesubject of calorimetric studies of mixing energetics.11 Asin doped CeO2 systems, the oxygen-ion conductivity inY2O3-doped ThO2 shows the aforementioned correlationbetween the maximum in formation enthalpy and oxygenconductivity.
The key to understanding the thermodynamics of de-fect clustering and its correlation with conductivity isto identify underlying interatomic interactions betweendopants and oxygen vacancies. In solid solutions of alio-valently doped oxides, electrostatic interactions betweenions with different charges and strain-mediated interac-tions due to the size mismatch between host and dopantatoms, and the presence of oxygen vacancies, are generalin nature. Thus, if these are the primary factors drivingdefect association, the nature of the defect clusters mightbe expected to be generic across fluorite-structured sys-tems, depending only on the charge and size of the aliova-lent dopant relative to the host. The analysis of drivingforces for defect clustering can be more complex if otherfactors play a significant role. For example, in the caseof YSZ, it has been suggested14,25,26 that the preferenceof Zr cations to be seven-fold coordinated by oxygen inthe lowest-energy monoclinic phase of ZrO2 leads to adriving force for association with oxygen vacancies in theeight-fold coordinated fluorite structure.
ThO2 is an interesting system from the standpoint ofstudying defect interactions in fluorite-structured com-pounds, as (in contrast to ZrO2) it is a system that dis-plays a stable fluorite structure in pure form, and (incontrast to CeO2) it displays a low equilibrium solubil-ity of oxygen vacancies and no tendency toward reduc-tion. In this study, we exploit first-principles DFT-basedcalculations combined with the lattice-algebra structureenumeration technique to study mixtures of ThO2 withtrivalent dopants. To investigate the energetics and thenature of defect clustering, we examine a set of dopants
TABLE I. The effective ionic radii of Shannon27 r (A) fortrivalent dopants in the octahedral coordinationa and Paulingelectronegativities χ.
Sc In Y Nd La
r 0.75 0.80 0.90 0.98 1.03
χ 1.36 1.78 1.22 1.14 1.10
a The ionic radius of the host Th4+ is 0.94 A and its Paulingelectronegativity is 1.3.
spanning a range of ionic sizes and electronegativities, asshown in Table I.
II. COMPUTATIONAL METHODOLOGY
First-principles calculations were carried out withinthe DFT plane wave formalism using the GGA-PBEexchange-correlation functional28 and the projector aug-mented wave (PAW) potentials29 as implemented in theVienna Ab initio Simulation Package (VASP).30,31 Ithas been demonstrated32 that the inclusion of Hubbard-type on-site electron interaction within the DFT +U approach33 for calculation of structural, elastic andelectronic properties of ThO2 changes the results onlymarginally as compared with pure DFT calculations. Weemploy the potentials labeled Th, O, Sc sv, In d, Y sv,Nd and La in the VASP PBE library; for Sc sv, Y svand In d these potentials include semicore states as va-lence electrons. In all calculations we employ a cutoffenergy of 500 eV which is 25% higher than the defaultvalue for the potentials chosen to ensure convergenceof the calculated stress tensors for volume relaxations.All atomic structure relaxations were performed start-ing from configurations of atoms in ideal fluorite posi-tions and optimizing cell volume, cell shape and all ionicpositions within a cell by applying a conjugate-gradientalgorithm until atomic forces were converged to a mag-nitude less than 0.1 meV/A. In these relaxation runs, weemployed the Monkhorst-Pack scheme34 to sample theBrillouin zone, using meshes with a density of at least4×4×4. Subsequent to the structural optimizations, anadditional static run for each final optimized geometrywas made using accurate tetrahedron integration withBlochl corrections,35 with a sufficiently dense sampling ofthe Brillouin zone to converge the total energy to betterthan 10−6 eV. For structures involving the trivalent Nddopant, which contains three valence f electrons, calcu-lations were performed spin-polarized, using a ferromag-netic arrangement of the local magnetic moments (for afew structures both ferromagnetic and antiferromagneticordering was considered, and the former was found to belower in energy).
In fluorite-structured ThO2 doped by oxides of triva-lent cations, one has to account for binary disorder onboth cation (Th4+, M3+, where M denotes the trivalent
3
FIG. 1. ThO2 cubic fluorite structure with two triva-lent dopants on the cation fcc sublattice and one charge-compensating oxygen vacancy on an anion simple cubic sub-lattice.
dopant species) and anion (O2−, Ovac) sublattices (seeFig. 1). In order to probe the configurational space andsample different possible atomic arrangements for givendopant concentrations, we employed a structure enumer-ation technique making use of the multicomponent multi-sublattice formalism previously developed to study alloysand recently applied to YSZ.14,15,18 We performed a com-plete structure enumeration in supercells consisting of upto nine primitive fcc unit cells (for the present study wefocus on compositions where we enumerated structuresonly up to six unit cells) using the Alloy Theoretic Auto-matic Toolkit (ATAT).36,37 These enumerated structureswere considered for dopant concentrations of x=0.33 and0.5 cation mole fraction. We also employed supercells,composed of 2×2×2 conventional fluorite unit cells (96atoms), to study defect energies at the more dilute com-positions of x=0.0625 and 0.125, corresponding respec-tively to a single vacancy with two dopant cations, andtwo vacancies with four dopants.
Formation enthalpies of solid solutions normalized percation can be expressed with respect to the constituentoxides as
∆Hf = E[Th1−xMxO2−0.5x]−(1−x)E[ThO2]−xE[MO1.5],
where E[ThO2] denotes the energy (per cation) offluorite-structured ThO2. Similarly, E[MO1.5] denotesthe energy (per cation) of Sc2O3, In2O3 and Y2O3 in theexperimentally-observed cubic bixbyite (C-type sesquiox-ide) structure, or Nd2O3 and La2O3 in the observed low-temperature hexagonal A-type structure. The energy percation for a solid solution with a cation dopant concen-tration x is denoted as E[Th1−xMxO2−x/2]. With the
above definition, positive values of ∆Hf correspond toan energetic tendency for phase separation of the solidsolutions into the respective constituent oxides.
TABLE II. Formation enthalpies (kJ/mol-cation) of the moststable defect associates for Th1−xMxO2−0.5x (M = Sc, Y,La) systems, as derived from calculations employing 2×2×2supercells of the host ThO2 conventional unit cell. The stabledefect clusters correspond to a nearest-neighbor M–Ovac–Mtriangular cluster.
x Sc Y La
Formation enthalpy
0.125 14.4 6.3 3.4
0.0625 8.8 3.4 2.0
Binding energy
0.0625 -1.9 (-1.9) -1.1 (-1.1) -0.2 (-0.6)
III. RESULTS
ThO2 compounds form in the cubic fluorite structurein which the cations form an fcc sublattice and anionsform a simple cubic sublattice (Fig. 1). Doping ThO2
by trivalent cations introduces one charge-compensatingoxygen vacancy per two trivalent dopants and leads toa coupled intra- and intersublattice interactions betweenall the species present (Th4+, M3+, O2−, Ovac). To in-vestigate the nature of these interactions, and the ge-ometries of the defect clusters that they favor energeti-cally, we investigate the formation energetics of a varietyof superstructures for relatively dilute and concentratedcompositions in sub-sections A and B, respectively.
A. Defect association in the dilute regime
In this section, we focus on systems with relatively di-lute dopant concentrations employing a 2×2×2 supercellof the host ThO2 crystal with one and two electricallyneutral M–Ovac–M units, corresponding to x=6.25% and12.5% dopant concentrations, respectively. We focuson a comparison of the energetics for under-sized Sc,intermediately-sized Y and over-sized La dopants (Ta-ble I). We examine a number of plausible defect arrange-ments and obtain only positive formation enthalpies inagreement with recent calorimetric data for Y and Ladopants.11
For the most dilute dopant concentration (6.25%) themost energetically stable clusters correspond to the com-pact triangle geometry involving M and Ovac as nearest-neighbor pairs. The calculated formation energies ofthese compact clusters are listed in Table II, and areseen to significantly decrease in magnitude from Sc toY to La. We also study the energetics of clustering be-tween two triplet defect clusters (12.5% defect level) byconsidering a number of different atomic arrangements.We find that the lowest-energy arrangements correspondto two nearest-neighbor triplet M–Ovac–M clusters, with
4
the two oxygen vacancies aligned as second neighbors onthe oxygen sublattice, i.e., along the <110> direction,with three dopant cations and one host thorium ion com-posing tetrahedron around the oxygen vacancy. The for-mation energies of these clusters are also listed in TableII and again show a trend of decreasing magnitudes for∆Hf going from Sc to Y to La.
We further estimate the binding energies between thetrivalent dopants and oxygen vacancies for the most sta-ble defect clusters. The binding energy here is defined asthe difference in energy between a system with a singledefect cluster and one with isolated defects, such thatnegative values indicate an energetically stable defectcluster. Due to the relatively small supercells used inthis study, we cannot derive highly accurate values forthese binding energies. Rather we explore trends by es-timating the energy of the isolated defects as the energyof the supercell in which the defects are separated as faras possible within the 2×2×2 supercell.
In the case of the M–Ovac–M triplet cluster, thebinding energy can be split into two parts correspond-ing to subsequent breaking of the first and the secondnearest-neighbor M–Ovas bond. This allows one to es-timate whether or not the dopant-vacancy interactionsare pairwise-additive in nature. The energy to break thefirst and second nearest-neighbor M–Ovac bonds is givenin the final row of Table II. For Sc and Y the interac-tions are found to be relatively short-range in nature,and the binding energies presented in Table II do in-dicate nearly pairwise-additive interactions between thedopant and oxygen vacancy (i.e., the energy to break thefirst and second nearest-neighbor M–Ovac bond is iden-tical within the precision of the DFT calculations). Inthe case of La, however, the interactions are found tobe much longer ranged; the interaction energy betweena La ion and a La–Ovac pair is found to vary apprecia-bly with distance out to the largest separation attainablein the 2×2×2 supercell. For the La dopant, it can beseen that the interactions between dopant and oxygenvacancy are not pairwise additive, as it requires nearlythree times the energy to break the second La–Ovac bondas it does the first. The longer-range an non-pairwise na-ture of the interactions between La and Ovac is likely amanifestation of elastically-mediated interactions, as su-percell calculations performed by constraining the ionsto ideal fluorite lattice sites yielded interaction rangesand pairwise-additive behavior very similar to that de-scribed above for the Y dopants. The fact that elasticinteractions can lead to long-range and non-pairwise in-teractions has been demonstrated by Bugaev et al.38 Itis nevertheless important to point out that the bindingenergies are particularly small for La system. More gen-erally, we note that the results in Table II show a trendtowards decreasing magnitude of the binding energies go-ing from Sc to Y to La.
−40
−20
0
20
40
60
80
100
120
140
Form
atio
n en
thal
py (k
J/m
ol−c
atio
n)
Sc Y La
phase−separating
compound−forming
fluorite frameworkfully−relaxed
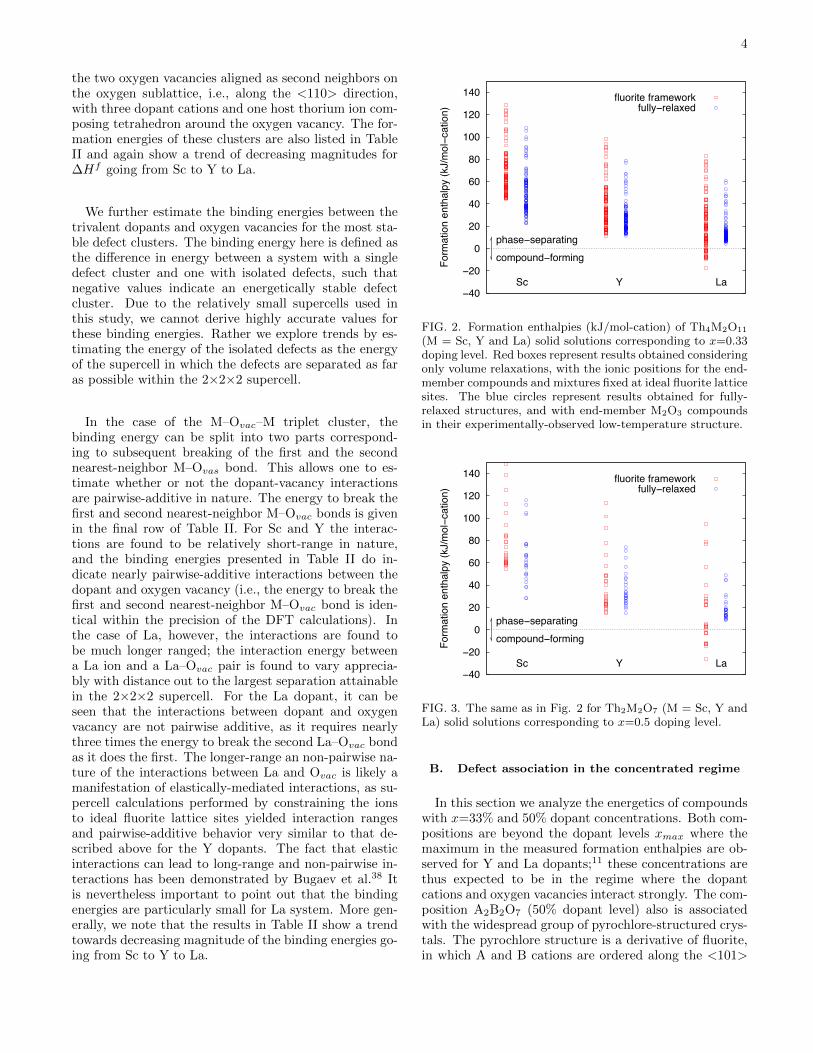
FIG. 2. Formation enthalpies (kJ/mol-cation) of Th4M2O11
(M = Sc, Y and La) solid solutions corresponding to x=0.33doping level. Red boxes represent results obtained consideringonly volume relaxations, with the ionic positions for the end-member compounds and mixtures fixed at ideal fluorite latticesites. The blue circles represent results obtained for fully-relaxed structures, and with end-member M2O3 compoundsin their experimentally-observed low-temperature structure.
−40
−20
0
20
40
60
80
100
120
140
Form
atio
n en
thal
py (k
J/m
ol−c
atio
n)
Sc Y La
phase−separating
compound−forming
fluorite frameworkfully−relaxed
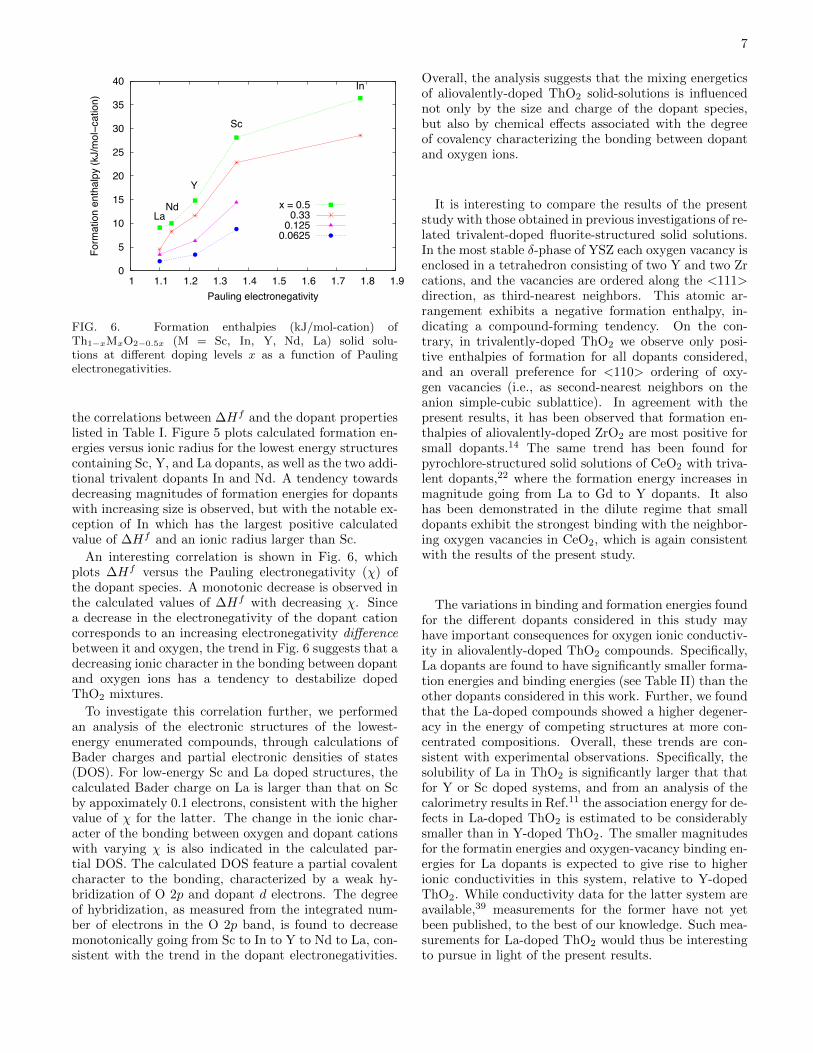
FIG. 3. The same as in Fig. 2 for Th2M2O7 (M = Sc, Y andLa) solid solutions corresponding to x=0.5 doping level.
B. Defect association in the concentrated regime
In this section we analyze the energetics of compoundswith x=33% and 50% dopant concentrations. Both com-positions are beyond the dopant levels xmax where themaximum in the measured formation enthalpies are ob-served for Y and La dopants;11 these concentrations arethus expected to be in the regime where the dopantcations and oxygen vacancies interact strongly. The com-position A2B2O7 (50% dopant level) also is associatedwith the widespread group of pyrochlore-structured crys-tals. The pyrochlore structure is a derivative of fluorite,in which A and B cations are ordered along the <101>
5
direction, and oxygen vacancies reside in the tetrahedralinterstices between adjacent B cations.
Compared to the dilute concentrations discussedabove, the number of distinct ionic configurations aremuch larger at the higher concentrations. In order todetermine the energetically preferred atomic arrange-ments, we performed an exhaustive structure enumer-ation for Th4M2O11 (33% dopant level) and Th2M2O7
(50% dopant level) compositions. The former enumera-tion yielded 117 symmetry-distinct configurations withunit cells containing 4 Th and 2 M cations, 11 oxy-gen ions and 1 oxygen vacancy, arranged over the sitesof the ideal fluorite structure. Similarly, the enumera-tion at 50% dopant concentration yielded 27 symmetry-distinct fluorite superstructures containing 2 Th and 2M cations, 7 O ions and 1 oxygen vacancy per unit cell.The calculated energies for each of these structures areplotted in Figs. 2 and 3 for x=0.33 and 0.5, respec-tively. In these plots, results labeled ”relaxed” corre-spond to formation energies computed as the energy dif-ference between a fully relaxed Th1−xMxO2−0.5x super-structure and the concentration-weighted average of theenergies of relaxed fluorite-structured ThO2 and M2O3
in its experimentally-observed low-temperature crystalstructure. The ”unrelaxed” results correspond to theanalogous energy difference, but using structures that areonly relaxed with respect to volume, constraining all ofthe ions to occupy the sites of an ideal fluorite lattice.
The results in Figs. 2 and 3 show a large spread inthe DFT-calculated formation enthalpies at each fixedcomposition. For the relaxed structures this spread in∆Hf between the lowest and highest energy structuresis largest for Sc (85.5 and 87.8 kJ/mol-cation at x=0.33and 0.5, respectively) and smallest for La (56.6 and 39.6kJ/mol-cation at x=0.33 and 0.5, respectively). All ofthe formation energies for the relaxed structures are pos-itive in Figs. 2 and 3, indicating a preference for phaseseparation at low temperatures, consistent with experi-mental measurements for La and Y.11 Further, the cal-culated values of ∆Hf decrease in magnitude going fromSc to Y to La. This trend is consistent with the resultsdiscussed above for the more dilute compositions, andwith measured solubility limits at T=1773 K, which arearound 50 mol% for La and 20 mol% for Y,11 and negli-gible in the case of Sc (the solubility of Sc has not beenmeasured to the best of our knowledge, but is expectedto be much smaller than for La and Y).
The differences in ∆Hf between the relaxed and unre-laxed structures plotted in Figs. 2 and 3 reflect a balancebetween two competing effects: (i) the stabilization ofthe Th1−xMxO2 compounds by atomic relaxation, and(ii) the stabilization of the end-member M2O3 compoundwhen it is allowed to change from an ideal fluorite latticeto its experimentally observed low-temperature crystalstructure. The first and second effects give rise to con-tributions that decrease and increase the value of ∆Hf ,respectively. For the Y dopant, with a size that is mostclosely matched to Th, the first and second effects are
FIG. 4. Illustration of the stable defect ordering pattern inTh1−xMxO2−0.5x solid solutions. The structure is character-ized by charge-neutral tetrahedra composed of one trivalentdopant M, two corner-shared M and one Th ion with the oxy-gen vacancies aligned as second nearest neighbors along the<110> direction.
comparable in magnitude for the low-energy structuresat each composition, such that the formation energies arenearly identical for relaxed and unrelaxed structures; thefirst effect is considerably larger than the second for thehigh-energy structures, leading to a decrease in the mag-nitude of ∆Hf for the relaxed versus unrelaxed struc-tures. For the under-sized Sc dopant, relaxation has theeffect of lowering the formation energy for all structuresconsidered, while for the over-sized La ion the low-energyand high-energy structures are respectively destabilizedand stabilized by atomic relaxations. It is interestingto note that in a recent analysis of measured formationenergies for La-doped ThO2 solid solutions,11 the mix-ing energy, defined as the formation energy of the solid-solution relative to fluorite-structured ThO2 and fluorite-structured La2O3, was computed to be negative, eventhough the measured values of ∆Hf relative to fluorite-structure ThO2 and A-type La2O3, are positive.11 Theseresults are qualitatively consistent with the present com-puted energies for La-doped ThO2, which show a changein the sign of the formation energy for relaxed versusunrelaxed low-energy structures, driven by the energydifference between fluorite and A-type La2O3 structures.
We consider next the qualitative features of the lowand high-energy ionic configurations enumerated in thisstudy. Table III summarizes the formation enthalpies ofthe most stable Th4M2O11 and Th2M2O7 compounds,along with their main structural features, including thedirection connecting oxygen-vacancy neighbors, and thenumber of cations of each type (Th and M) in the nearest-neighbor tetrahedron surrounding the vacancy. We in-clude such information for several of the lowest-energystructures for each dopant; some of the structures havenearly degenerate or close energies and therefore a num-ber of defect ordering patterns may be observed particu-larly in the presence of entropic effects at elevated tem-peratures.
We observe that the oxygen vacancies prefer to align as
6
TABLE III. Formation enthalpies of Th1−xMxO2−0.5x (M =Sc, Y, La) structures are listed for the lowest-energy fully-relaxed structures enumerated in this study. The structureslisted have formation energies within 2 kJ/mol-cation of thelowest energy structure for each dopant at each compositionconsidered. The third column lists the direction of orderingof oxygen-vacancy chains. The last column lists the num-ber of cations of host (Th) and dopant (M) species in the 4tetrahedral nearest-neighbor positions surrounding an oxygenvacancy.
Nearest Neighbors
x M ∆Hf Direction Th, M
0.5 Sc 28.1 <110> 1, 3
28.3 <110> 3, 1
Y 14.8 <110> 1, 3
La 9.1 <110> 1, 3
9.2 <110> 2, 2
10.1 <110> 1, 3
10.5 <110> 2, 2
0.33 Sc 22.9 <110> 1, 3
22.9 <110> 3, 1
23.4 <110> 1, 3
Y 11.7 <110> 1, 3
13.0 <110> 1, 3
13.2 <110> 1, 3
13.5 <110> 2, 2
13.6 <110> 1, 3
La 4.4 <112> 4, 0
6.1 <112> 2, 2
6.2 <112> 3, 1
second nearest neighbors along the <110> directions forall dopants with an exception of La for x=0.33 where thevacancies are ordered along the <112> direction. Theordering of oxygen vacancies along the<110> direction isalso observed for the La system at x=0.33, but at slightlyhigher energies (with ∆Hf ≥ 6.7 kJ/mol-cation). Ananalysis of nearest neighbors around the oxygen vacanciesshows that the only case in which the oxygen vacancy issurrounded completely by four host Th cations is for thelowest-energy La-doped ThO2 structure at x=0.33. In allother cases we see that the tetrahedra of cations aroundthe vacancy, as pictorially shown in Fig. 4, are composedof both host Th and dopant M ions, with a slight overallpreference for binding to trivalent dopant species.
For Y dopants at x=0.33 and 0.5, the lowest-energystructures are shown in a <110> projection in Fig. 7. Inthis projection, the smaller (red) circles denote <110>rows of oxygen ions containing no vacancies, while thedarker and lighter larger (blue) circles denote <110>columns of Y and Th cations, respectively; the black openboxes in the oxygen rows correspond to <110> rows ofoxygen vacancies. Both structures in Fig. 7 contain thesame structural motif, which, in the projected image in-
0
5
10
15
20
25
30
35
40
0.75 0.8 0.85 0.9 0.95 1 1.05
Form
atio
n en
thal
py (k
J/m
ol−c
atio
n)
Ionic radius (Å)
Sc
In
Y
Nd La
x = 0.50.33
0.1250.0625
FIG. 5. Formation enthalpies (kJ/mol-cation) ofTh1−xMxO2−0.5x (M = Sc, In, Y, Nd, La) solid solu-tions at different doping levels x as a function of ionic radius(A).
volves a column of oxygen vacancies located between atriangle of two Y and one Th ion. This motif is illustratedin a different representation in Fig. 4 where it is moreclearly apparent that the lowest-energy Y-doped ThO2
structures are characterized by a clustering of vacanciesas second neighbors on the anion sublattice (i.e., along<110>) directions, and a clear association of the oxy-gen vacancies with nearest-neighbor dopant cations. InFig. 4 the motif of the defect cluster involves tetrahedraconsisting of three Y and one Th cation, surrounding anoxygen vacancy; since these tetrahedra are arranged ina corner-sharing configuration the defect clusters are netcharge neutral. For the Th2Y2O7 structure (x=0.5) thelowest-energy structure shown in Fig. 7 (left) can also beidentified as being characteristic of the pyroclore struc-ture with the oxygen vacancies residing in the tetrahedralinterstice between adjacent Y dopants, aligned along the<110> direction.
The structures of the highest-energy configurationsfor Y-doped ThO2 considered in our calculations areillustrated in Fig. 8. These structures are character-ized by the location of rows of oxygen vacancies be-tween host Th ions, and thus contain large regions thatare locally charge imbalanced. The large energy differ-ence between these configurations and those characteris-tic of the lowest-energy structures demonstrates a strongpreference for association of oxygen-vacancy chains withdopant cation species in this system.
IV. DISCUSSION
The results in Figs. 2 and 3 and Tables II and III fea-ture a strong variation in the calculated values of ∆Hf asthe dopant species change from Sc to Y to La. To gainfurther insight to the origins of this trend, we explore
7
0
5
10
15
20
25
30
35
40
1 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9
Form
atio
n en
thal
py (k
J/m
ol−c
atio
n)
Pauling electronegativity
Sc
In
Y
NdLa
x = 0.50.33
0.1250.0625
FIG. 6. Formation enthalpies (kJ/mol-cation) ofTh1−xMxO2−0.5x (M = Sc, In, Y, Nd, La) solid solu-tions at different doping levels x as a function of Paulingelectronegativities.
the correlations between ∆Hf and the dopant propertieslisted in Table I. Figure 5 plots calculated formation en-ergies versus ionic radius for the lowest energy structurescontaining Sc, Y, and La dopants, as well as the two addi-tional trivalent dopants In and Nd. A tendency towardsdecreasing magnitudes of formation energies for dopantswith increasing size is observed, but with the notable ex-ception of In which has the largest positive calculatedvalue of ∆Hf and an ionic radius larger than Sc.
An interesting correlation is shown in Fig. 6, whichplots ∆Hf versus the Pauling electronegativity (χ) ofthe dopant species. A monotonic decrease is observed inthe calculated values of ∆Hf with decreasing χ. Sincea decrease in the electronegativity of the dopant cationcorresponds to an increasing electronegativity differencebetween it and oxygen, the trend in Fig. 6 suggests that adecreasing ionic character in the bonding between dopantand oxygen ions has a tendency to destabilize dopedThO2 mixtures.
To investigate this correlation further, we performedan analysis of the electronic structures of the lowest-energy enumerated compounds, through calculations ofBader charges and partial electronic densities of states(DOS). For low-energy Sc and La doped structures, thecalculated Bader charge on La is larger than that on Scby appoximately 0.1 electrons, consistent with the highervalue of χ for the latter. The change in the ionic char-acter of the bonding between oxygen and dopant cationswith varying χ is also indicated in the calculated par-tial DOS. The calculated DOS feature a partial covalentcharacter to the bonding, characterized by a weak hy-bridization of O 2p and dopant d electrons. The degreeof hybridization, as measured from the integrated num-ber of electrons in the O 2p band, is found to decreasemonotonically going from Sc to In to Y to Nd to La, con-sistent with the trend in the dopant electronegativities.
Overall, the analysis suggests that the mixing energeticsof aliovalently-doped ThO2 solid-solutions is influencednot only by the size and charge of the dopant species,but also by chemical effects associated with the degreeof covalency characterizing the bonding between dopantand oxygen ions.
It is interesting to compare the results of the presentstudy with those obtained in previous investigations of re-lated trivalent-doped fluorite-structured solid solutions.In the most stable δ-phase of YSZ each oxygen vacancy isenclosed in a tetrahedron consisting of two Y and two Zrcations, and the vacancies are ordered along the <111>direction, as third-nearest neighbors. This atomic ar-rangement exhibits a negative formation enthalpy, in-dicating a compound-forming tendency. On the con-trary, in trivalently-doped ThO2 we observe only posi-tive enthalpies of formation for all dopants considered,and an overall preference for <110> ordering of oxy-gen vacancies (i.e., as second-nearest neighbors on theanion simple-cubic sublattice). In agreement with thepresent results, it has been observed that formation en-thalpies of aliovalently-doped ZrO2 are most positive forsmall dopants.14 The same trend has been found forpyrochlore-structured solid solutions of CeO2 with triva-lent dopants,22 where the formation energy increases inmagnitude going from La to Gd to Y dopants. It alsohas been demonstrated in the dilute regime that smalldopants exhibit the strongest binding with the neighbor-ing oxygen vacancies in CeO2, which is again consistentwith the results of the present study.
The variations in binding and formation energies foundfor the different dopants considered in this study mayhave important consequences for oxygen ionic conductiv-ity in aliovalently-doped ThO2 compounds. Specifically,La dopants are found to have significantly smaller forma-tion energies and binding energies (see Table II) than theother dopants considered in this work. Further, we foundthat the La-doped compounds showed a higher degener-acy in the energy of competing structures at more con-centrated compositions. Overall, these trends are con-sistent with experimental observations. Specifically, thesolubility of La in ThO2 is significantly larger that thatfor Y or Sc doped systems, and from an analysis of thecalorimetry results in Ref.11 the association energy for de-fects in La-doped ThO2 is estimated to be considerablysmaller than in Y-doped ThO2. The smaller magnitudesfor the formatin energies and oxygen-vacancy binding en-ergies for La dopants is expected to give rise to higherionic conductivities in this system, relative to Y-dopedThO2. While conductivity data for the latter system areavailable,39 measurements for the former have not yetbeen published, to the best of our knowledge. Such mea-surements for La-doped ThO2 would thus be interestingto pursue in light of the present results.
8
FIG. 7. Lowest-energy atomic arrangements in Th1−xYxO2−0.5x solid solutions at x=0.5 (left) and 0.33 (right). The differentcircle symbols correspond to rows of O, Th and Y ions directed along the <110> direction (out of the plane of the page), whilethe open boxes correspond to rows of oxygen vacancies along the same direction. For clarity all atoms are displayed in theirideal fluorite positions.
FIG. 8. Highest-energy atomic arrangements inTh1−xMxO2−0.5x solid solutions at x=0.5 (left) and0.33 (right). The symbols have the same meanining as inFig. 7. For clarity all atoms are displayed in their perfectfluorite positions.
V. CONCLUSIONS
In this study we have employed DFT calculations tostudy formation energetics and defect-ordering tenden-cies in ThO2 compounds doped with the trivalent cationspecies: Sc, In, Y, Nd and La. The main results derivedfrom these calculations can be summarized as follows:
(1) Solid solutions of fluorite-structured ThO2 with ox-ides of trivalent cations (Sc, In, Y, Nd, La) exhibit pos-itive enthalpies of formation from constituent oxides, in-dicative of a phase-separating tendency across all dopantconcentrations considered (6.25, 12.5, 33.3 and 50%).This strongly suggests that there are no underlying stablelong-range ordered phases in these compounds. However,the large variation in energy for different dopant/vacancyarrangements suggests an appreciable driving force forshort-ranged ordering (clustering) in these solid solutionsat finite temperatures.
(2) For a given dopant concentration the formationenthalpies tend to decrease in magnitude as the size andelectronegativity of the trivalent dopant decrease. This
trend is consistent with available experimental data onsolubility limits which are highest for Nd- and La-dopedThO2.
(3) The lowest-energy atomic structures are character-ized by a repulsion of oxygen vacancies as nearest neigh-bors. Oxygen vacancies are found to prefer to align assecond nearest neighbors along the <110> direction forall of the dopants and concentrations considered, withthe exception of 33.3% La-doped ThO2 where they orderas more distant neighbors along the <112> direction.
(4) The solute-vacancy binding energies in ThO2 solidsolutions with under-sized Sc and intermediate-sized Ydopants are estimated to be on the order of a few kJ/mol-cation. For over-sized La dopants, this binding energy isestimated to be significantly smaller in magnitude.
(5) The small magnitude of the formation and bindingenthalpies for La-doped ThO2 solid solutions is expectedto result in an ionic conductivity that is higher in thissystem than for the other trivalent-doped systems con-sidered in this study.
VI. ACKOWLEDGEMENTS
This work was supported as part of the Materials Sci-ence of Actinides, an Energy Frontier Research Centerfunded by the U.S. Department of Energy, Office of Sci-ence, Office of Basic Energy Sciences under award num-ber de-sc0001089. The authors acknowledge fruitful dis-cussions with Dr. Tatiana Shvareva. We also acknowl-edge very helpful input from Dr. Axel van de Walle onthe application of the ATAT package in this work.