Alfredo Santamaría E, M.D. Hepatólogo Pediatra Clínico y de Trasplantes. HUSVF - Medellín. Colegio Colombiano de Gastroenterología, Hepatología y Nutrición Pediátrica. COLGAHNP. [email protected]
Transcript
Alfredo Santamaría E, M.D. Hepatólogo Pediatra Clínico y de Trasplantes.
HUSVF - Medellín.
Colegio Colombiano de Gastroenterología, Hepatología y Nutrición Pediátrica. COLGAHNP.
Reducción de la formación o flujo de la bilis = retención de sustancias biliares dentro del hígado normalmente excretadas y/o eliminadas en el lumen intestinal.
(Bilirrubinas, Ac. biliares, colesterol).
COLESTASIS Colestasis e hiperbilirrubinemia NO son sinónimos.
NO {Bilirrubina directa > 2 mg/dl ó > 20% del total de Hiperbilirrubinemia} NO
Hiperbilirrubinemia conjugada (directa) (> 1,0 mg / dl, 17 mmol /L) se considera patológica y justifica evaluación (1A).
NINGUNA condición fisiológica cursa con hiperbilirrubinemia directa.
COLESTASIS
COLESTASIS Ictericia (signo principal).
Evidente cuando nivel de bilirrubina total
excede de 2,5 a 3,0 mg / dl (42-51 mmol /
L).
Bebé alimentado con fórmula con ictericia
después de 2 semanas de edad =
bilirrubina total y directa.
Amamantado puede hacerse seguimiento
clínico hasta las 3 semanas de edad (1A).
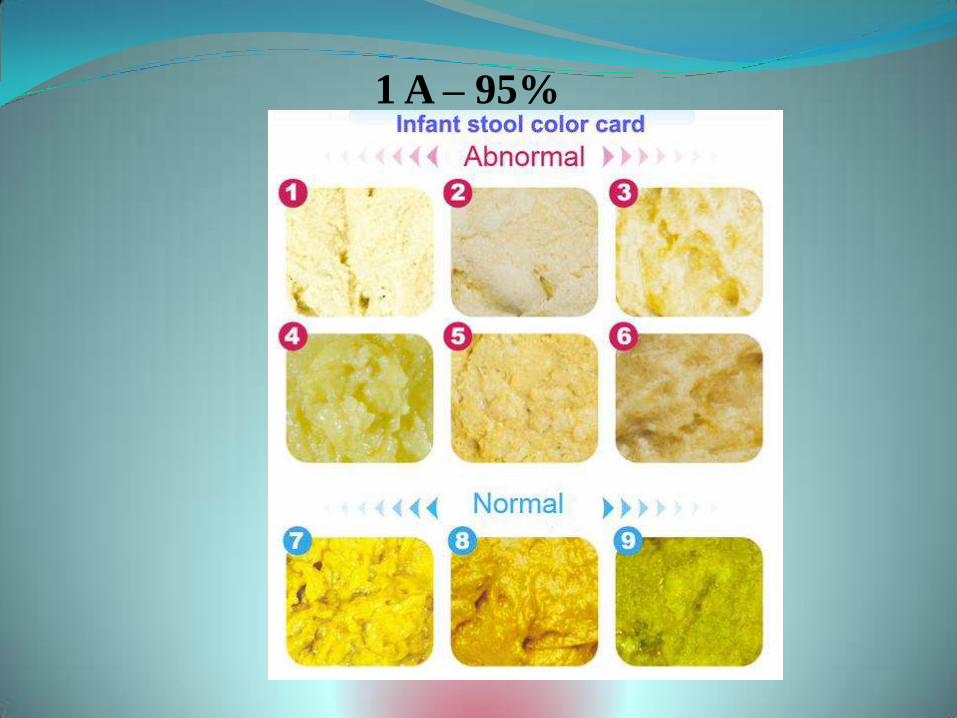
1 A – 95%
www.yellowalert.org
ETIOLOGIAENFERMEDAD HEPATICA COLESTATICA
1. S.Colestáticos intrafamiliares progresivos.
2. Atresia de vías biliares
3. Sindrome de Alagille(Pobreza de conductos biliares sindrómica)
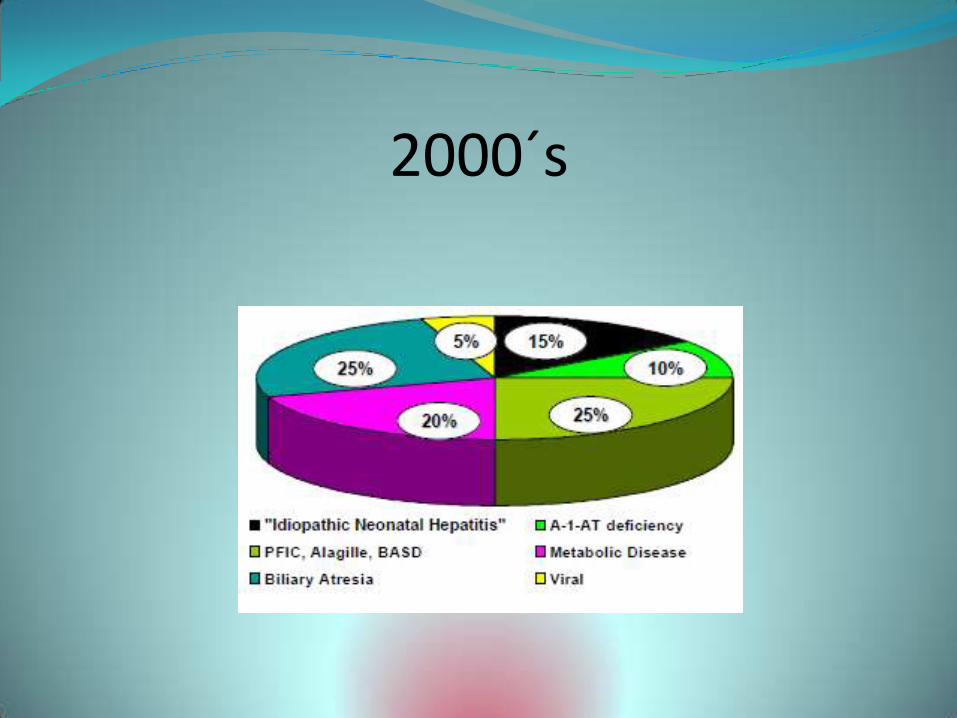
Mayoría de los niños que se presentaban con colestasis se clasificaban con Dx de “hepatitis neonatal idiopática”.
Claro reconocimiento de “diagnóstico” por defecto, debido a que la fisiopatología subyacente era desconocida.
“Hepatitis neonatal idiopática” era simplemente un término conveniente para un síndrome clínico que se manifiesta por ictericia prolongada en el recién nacido, en combinación con un variable pero definido cuadro histológico: hepatitis de células gigantes.
El término hepatitis no implicaba una causa específica o entidad diagnóstica.
80´s A finales de 1980 surgió una clara comprensión de la
fisiología hepatobiliar en la vida temprana y el fenómeno de la colestasis fisiológica causada por la inmadurez de la secreción de bilis.
Casos de “hepatitis neonatal familiar”: errores congénitos o defectos genéticos en algún proceso fundamental metabólico hepático o función excretora:
Transporte de membrana alterado.
Biosíntesis de ácidos biliares
Disfunción de organelo (1969: déficit de a-1 AT).
2000´s
Prevalencia desconocida.
En los últimos 15 años hay reportados en la literatura de 100 a 300 casos de deficiencia de 3 B HSD.
15-50 casos de déficit de delta-4-3 oxo- 5 B reductasa. (AKR1D1)
Dos (2) pacientes en Colombia (HPTU – HUSVF). AKR1D1.
DEFECTOS DE LA SINTESIS DE ÁCIDOS BILIARES
Jörg Jahnel, Evelyn Zöhrer, Lorenzo D'Antiga, Dominique Debray, Antal Dezsofi,
Dorothea Haas, Nedim Hadzic, Emmanuel Jacquemin, Thierry Lamireau, Giuseppe Maggiore, Pat McKiernan,
Michele Pinon, Henkjan Verkade, Ulrich Baumann,
Loreto Hierro, Valerie McLin, Björn Fischler, Emmanuel Gonzales
JPGN, in review
Resultados
Todos los 36 centros respondieron al cuestionario
16 centros de 11 países reportaron 63 pacientes
• 55 casos de déficit en HSD3B7
• 8 casos de déficit en AKR1D1
La mayoría de los pacientes fueron reportados en:
• Francia (n=27)
• Italia (n=12)
• Reino Unido (n=11)
En 8 países no se reporto ningún caso.
Lista de centros de participantes por país y distribución de pacientes
Pais Centro Pacientes Pais Centro Pacientes
AustriaVienna 1x 3β-HSD Lithuania Vilnius 0
Graz, Salzburg, Innsbruck 0
Netherlands
Amsterdam 1x Δ4-3-oxoR
Belgium Brussels 0 Groningen 1x 3β-HSD
Bulgaria Stara Sagora 0 Utrecht 0
Croatia Zagreb 0 Poland Warsaw 0
Denmark Copenhagen 0 Portugal Coimbra 1x 3β-HSD
FranceBicêtre , Paris
22x 3β-HSD3x Δ4-3-oxoR
Spain Madrid 2x 3β-HSD
Sweden Stockholm 1x 3β-HSD
Bordeaux 0Switzerland
Geneva 2x 3β-HSD
Necker/Bicêtre , Paris 2x 3β-HSD Chur, Basel 0
GermanyHeidelberg
1x 3β-HSD Turkey Ankara 0
1x Δ4-3-oxoR
UK
Birmingham 1x 3β-HSD
Wuppertal, Dresden 0 Bristol 0
Hannover 1x 3β-HSDLondon
8x 3β-HSD2x Δ4-3-oxoRHungary Budapest 0
Israel Tel Aviv 1x 3β-HSD
Italy
Bergamo 0
Padova8x 3β-HSD
1x Δ4-3-oxoR
Pisa 2x 3β-HSD
Torino 1x 3β-HSD
FISIOPATOLOGIA Hepatocelular : alteración en la formación de
bilis.
Obstructivo.
Retención de ácidos biliares endógenos y
colestasis = daño hepático progresivo.
Inducción de permeabilidad mitocondrial,
apoptosis y necrosis celular.
Colestasis neonatal, más frecuente.
ATRESIA DE VIAS BILIARES
ATRESIA DE VIAS
BILIARES Proceso inflamatorio idiopático.
PROGRESIVO. Conductos biliares intra
y extrahepáticos = fibrosis y
obliteración del tracto biliar y desarrollo
de cirrosis biliar.
1). Viral. 2).Disregulación inmune
incluso autoinmunidad. 3). Vascular. 4).
Morfogénesis defectuosa por
mutaciones genéticas. 5). Toxinas.
CUADRO CLINICO Manifestaciones primarias por acúmulo de