1 Amino Acids Adapted from NPTEL notes: http://nptel.ac.in/courses/104103071/32 and https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/proteins.htm#aacd1 Introduction In nature three kinds of polymers occur: (i) polysaccharides, (ii) proteins and (iii) nucleic acids. This section discusses proteins and peptides that are polymers of amino acids linked together by amide bonds. Hence proteins are polyamides. Amino acids are a type of organic acid that contain both a carboxyl group (COOH) and an amino group (NH 2 ) as shown in Scheme 1. Here, “R” denotes a group that forms the “side chain” of amino acids. Scheme 1 In the approximately 20 amino acids found in our bodies, what varies is the side chain. Some side chains are hydrophilic while others are hydrophobic. Since these side chains stick out from the backbone of the molecule, they help determine the properties of the protein made from them. The amino acids in our bodies are referred to as alpha-amino acids. The central carbon is in an alpha position in relation to the carbonyl carbon. In Scheme 2, you notice that the amino acid is drawn with the acidic hydrogen attached to the amine group. The carboxyl and amino groups of the amino acids can respectively donate a proton to and accept a proton from water. This exchange happens simultaneously in solution so that the amino acids form doubly ionized species, termed zwitter ions (from German zwei, meaning two) in solution. Amino acids in solution area always found this is the form. C C + NH 3 H O O - R zwitter ion Scheme 2
Transcript
1
Amino Acids Adapted from NPTEL notes: http://nptel.ac.in/courses/104103071/32 and
Introduction In nature three kinds of polymers occur: (i) polysaccharides, (ii) proteins and (iii) nucleic acids. This section discusses proteins and peptides that are polymers of amino acids linked together by amide bonds. Hence proteins are polyamides. Amino acids are a type of organic acid that contain both a carboxyl group (COOH) and an amino group (NH2) as shown in Scheme 1. Here, “R” denotes a group that forms the “side chain” of amino acids.
Scheme 1
In the approximately 20 amino acids found in our bodies, what varies is the side chain. Some side chains are hydrophilic while others are hydrophobic. Since these side chains stick out from the backbone of the molecule, they help determine the properties of the protein made from them.
The amino acids in our bodies are referred to as alpha-amino acids. The central carbon is in an alpha position in relation to the carbonyl carbon.
In Scheme 2, you notice that the amino acid is drawn with the acidic hydrogen attached to the amine group. The carboxyl and amino groups of the amino acids can respectively donate a proton to and accept a proton from water. This exchange happens simultaneously in solution so that the amino acids form doubly ionized species, termed zwitter ions (from German zwei, meaning two) in solution. Amino acids in solution area always found this is the form.
C C+NH3
H O
O-
R
zwitter ion
Scheme 2
2
α-Amino Acids
The structures of the 20 most common naturally occurring amine acids are shown in Scheme 3. They differ only in the side chain attached to the α-carbon.
Scheme 3
3
• Each amino acid contains an "amine" group, (NH2) and a "carboxylic acid" group (COOH) (shown in blue in the diagram).
• The blue colored group is invariant (does not change) in all the amino acids • The amino acids vary in their side chains (indicated in black in the diagram). • The first eight amino acids in the orange shaded area are nonpolar and
hydrophobic - Mnemonic: PRO ALA - VAL LEU – MET - PHE ILE - TRP • The other amino acids are polar and hydrophilic ("water loving"). • The two amino acids in the magenta box are acidic ("carboxylic" group in the side
chain) – GLU and ASP – Mnemonic: Use Acidic GLUe ASAP • The three amino acids in the light blue box are basic ("amine" group in the side
chain) – His, Lys and Arg. Mnemonic: His Arguments are Based on Lys • Glycine is a small amino acid with no asymmetric carbon. • Three aromatic amino acids are Phe, Tyr & Trp – Mnemonic: Try a roma(n)tic
Phe(ailed) Tr(i)p
Although there are many ways to classify amino acids, these molecules can be assorted into six main groups, on the basis of their structure and general chemical characteristics of their R groups.
Acidic and their Amide Aspartate, Glutamate, Asparagine, Glutamine
Ref: http://en.wikipedia.org/wiki/Amino_acid
Among them, ten are essential amino acids (Scheme 4). These amino acids are to be obtained from diets because we either cannot synthesize them at all or cannot synthesize them in adequate amounts. These are shown in their zwitterionic form below.
In 19 of the 20 naturally occurring α-amino acids, except glycine, the α-carbon is an asymmetric center. Thus, they can exit as enantiomers, and the most amino acids found in nature have L-configuration. Scheme 5 shows the Fischer projection of an amino acid with a carboxyl group on the top and the R group on the bottom of the vertical axis is an L-amino acid if the amino group is on the left and a D-amino acid if the amino group is one the right.
Scheme 5
Acid-Base Properties α-Amino Acids Amino acid has a carboxyl group and amino group, and each group can exist in an acidic or basic form, depending on the pH of the solution in that the amino acid is dissolved. In addition, some amino acids, such as glutamate, also contain ionizable side chain.
The pKa values of the carboxyl group and the protonated amino group of the amino acids approximately are 2 and 9, respectively (Scheme 6). Thus, both groups will be in their acidic forms in highly acidic medium (pH ~ 0). At pH 7, the pH of the solution is greater than the pKa of the carboxyl group, but less than the pKa of the protonated amino group. Hence, the carboxyl group will be in its basic form and the amino group in its acidic form (called Zwitter ion). In strongly basic medium (pH 11), both groups will
5
be in basic form. Thus, an amino acid can never exist as an uncharged compound, regardless of the pH of the medium.
Scheme 6
The Isoelectric Point (pI)
The isolectric point (pI) of an amino acid is the pH where it has no net charge. For example, the pI of an amino acid that does not possess an ionizable side chain is midway between its two pKa values (Scheme 7).
Scheme 7
In case of an amino acid that contains an ionizable side chain, the pI is the average of the pKa values of the similarly ionizing groups. For example, see pI of lysine (Scheme 8).
Scheme 8
6
Separation of Amino Acids
A. Electrophoresis
In this method, the amino acids can be separated on the basis of their pI values (Scheme 9). A few drops of a solution of amino acid mixture are applied to the middle of the piece of filter paper or to a gel. When this paper or gel is placed in a buffered solution between two electrodes and an electric field is applied, an amino acid having a pI greater than the pH of the medium will have an overall positive charge and will move toward the cathode. While an amino acid with a pI less than the pH of the buffer will have an overall negative charge and will move toward anode. In case of the molecules have the same charge, the larger one will migrate more slowly compared to that of the
smaller one during the electrophoresis. After the separation, the filterpaper is sprayed with ninhydrin and dried in a warm oven to give purple colored spot. The amino acids are identified by their location on the paper comparing with a standard.
The reaction of ninhydrin with an amino acid to form a purple colored product.
Scheme 9
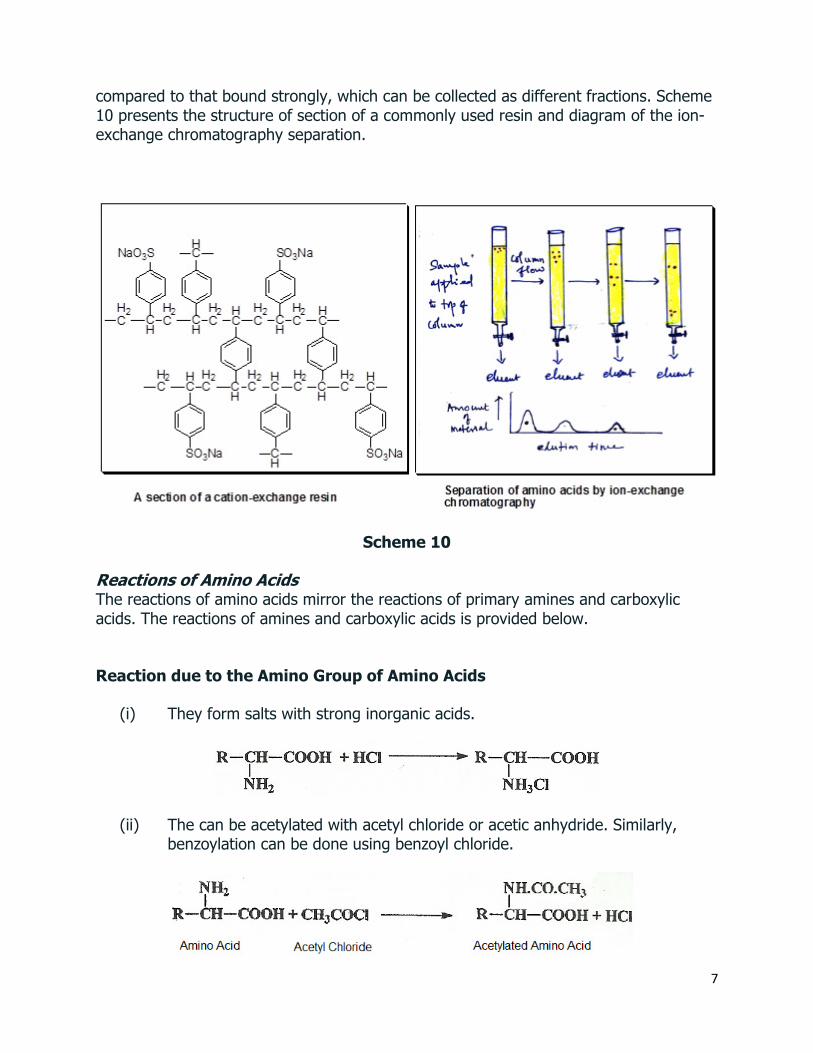
B. Ion-Exchange Chromatography: In this technique, a column is packed with an insoluble ion-exchange resin. Then, a solution of a mixture of amino acids is loaded on the top of the column, and eluted with aqueous solutions of increasing pH. Since the amino acids bind with the resin at different extent, during the elution, the weakly bound amino acid can flow faster
7
compared to that bound strongly, which can be collected as different fractions. Scheme 10 presents the structure of section of a commonly used resin and diagram of the ion-exchange chromatography separation.
Scheme 10
Reactions of Amino Acids The reactions of amino acids mirror the reactions of primary amines and carboxylic acids. The reactions of amines and carboxylic acids is provided below.
Reaction due to the Amino Group of Amino Acids
(i) They form salts with strong inorganic acids.
(ii) The can be acetylated with acetyl chloride or acetic anhydride. Similarly, benzoylation can be done using benzoyl chloride.
8
(iii) On treatment with nitrous acid, they form α-hydroxy acids. As one mole of nitrogen is eliminated (one half of N2comes from the amino acid) for each free amino group, this forms the basis of Van Skyke method for determination of free-NH2 groups in amino acids and proteins.
(iv) When treated with chloroform and alcoholic caustic potash they give carbylamine reaction.
(v) Amino acid with formaldehyde results in blockage of the amino group.
When excess formaldehyde is added, the dimethylol derivative of the amino acid is obtained, which is (CH2OH)2-CH(R)-COOH.
(vi) When treated with hydroiodic acid, the amino group is knocked off the molecule resulting in a carboxylic acid.
H2N-CH(R)-COOH + 3HI � R-CH2-COOH + NH4l + H2O
(vii) When treated with nitrosyl chloride, nitrogen is given out with the formation of chloro acid.
H2N-CH(R)-COOH + NOCl�Cl-CH(R)-COOH + N2 + H2O
For eg. Glycine nitrosylation provides chloroacetic acid.
(viii) Deamination of amino acids:
a- Oxidative deamination: produces α-keto acid and NH3.
9
b- Reductive deamination: produces fatty acid and NH3.
c- Hydrolytic deamination: produces hydroxy fatty acid and NH3
Reaction due to Carboxyl Group of Amino Acids
(i) Salt formation: with strong alkali, amino acids produce the corresponding salt.
(ii) Ester formation: with alcohols, amino acids produce the ester.
Reaction type: Nucleophilic Acyl Substitution
This reaction is also known as the Fischer esterification. Esters are obtained by refluxing the parent carboxylic acid with the appropriate alcohol with an acid catalyst. The equilibrium can be driven to completion by using an excess of either the alcohol or the carboxylic acid, or by removing the water as it forms.
10
(iii) Decarboxylation reaction: Amino acids undergo decarboxylation to produce primary amines when they are heated with barium hydroxide. This is the reaction used by the putrifaction processes by large intestine bacteria to form primary amines.
Apart from these reactions of amino acids, the general reactions of carboxylic acids as described below are also applicable.
(iv) Preparation of Acyl Chlorides
Reaction type: Nucleophilic Acyl Substiution
Acyl chlorides are prepared by treating the carboxylic acid with thionylchloride, SOCl2, in the presence of a base. Acyl chlorides are by far the most commonly encountered of the acyl halides.
(v) Preparation of Acid Anhydrides
Reaction type: Nucleophilic Acyl Substiution
Symmetrical anhydrides can be are prepared by heating the carboxylic acid. Symmetrical anhydrides are by far the most commonly encountered, e.g.acetic anhydride.
Esters can also be made from other carboxylic acid derivatives, especially acyl halides and anhydrides, by reacting them with the appropriate alcohol in the presence of a weak base.
(vi) Preparation of Amides
11
Reaction type: Nucleophilic Acyl Substiution
In general, it is not easy to prepare amides directly from the parent carboxylic acid as shown above. As shown in the reaction below, the acid will protonate the amine preventing further reaction since the carboxylate is a poor electrophile and the ammonium ion is not nucleophilic.
It is much easier to convert the carboxylic acid to the more reactive acyl chloride first.
(vii) Reduction of Carboxylic Acids
Reaction usually in Et2O or THF followed by H3O+work-ups
Reaction type: Nucleophilic Acyl Substiution then NucleophilicAddition Carboxylic acids are less reactive to reduction by hydride than aldehydes, ketones or esters. Carboxylic acids are reduced to primary alcohols.
Reaction type: Substitution Reagents most commonly are Br2 and either PCl3, PBr3 or red phosphorous in catalytic amounts. Carboxylic acids can be halogenated at the C adjacent to the carboxyl group. The product of the reaction, an α-bromocarboxylic acid can be converted via substitution reactions to α-hydroxy- or α-amino carboxylic acids.
(ix) Decarboxylation
12
Reaction type: Elimination Loss of carbon dioxide is called decarboxylation. Simple carboxylic acids rarely undergo decarboxylation. Carboxylic acids with a carbonyl group at the 3- (ie. β-) position readily undergo thermal decarboxylation, e.g. derivatives of malonic acid.
The Synthesis of α-Amino Acids Some of the common methods employed for the synthesis of α-amino acids follow:
Amination of α-Halo acids: The simplest method is the conversion of carboxylic acid into it’s α-bromo-derivative that can be reacted with ammonia to give α-amino acid (Schemes 11). This method is used for synthesis of neutral amino acids such as alanine and glycine.
Scheme 11
Gabriel’s Pthalimide Synthesis: Another method is reaction using pthalimide salts to produce an ester which upon hydrolysis produces the α-amino acids. This method of introducing an amino group with the Gabriel procedure provides a better yield when compared to that of the above described reaction with ammonia as an aminating agent.
Scheme 12
13
An example for Methionine synthesis is provided below in Scheme 12.
Modified forms of this method for synthesis of glycine and aspartic acid is provided below in Scheme 13 and 14.
Scheme 13
Scheme 14
Malonic Ester Synthesis Method is used to prepare leucine, isoleucine, norleucine, phenylalanine, methionine and proline. The method is generally used to prepare α-
14
bromo acid. The treatment of the acid with bromine in the presence of a small amount of phosphorus gives acid bromide which undergoes (electrophilic) bromination at the α-position via its enol tautomer. The resulting product exchanges with more of the acid to give α-bromo acid together with more acid bromide for the further bromination (Scheme 12)
The amino acid can also be synthesized with Hell-Volhard-Zelinski halogentation of unsubstituted acids. For eg. a modified form of malonic acid synthesis method for synthesis of phenylalanine is depicted in Scheme 15.
These are naturally occurring polymers in living systems. The polymers with molecular weights less than 10000 are termed weights are termed as proteins. The acidaffords the constituent α-amino acids. The Peptide Bond If the amine and carboxylic acid functional groups in amino acids amide bonds, a chain of amino acid units, called aand 17B) simple tetrapeptide structure is shown in the following diagram. By convention, the amino acid component retaining a free amine group is drawn end (the N-terminus) of the peptide chain, and the amino acid retaining a free carboxylic acid is drawn on the right (the Ccarboxylic acid functions on a peptide chain form a zwitterionic structure atisoelectric pH.
These are naturally occurring polymers in living systems. The polymers with molecular weights less than 10000 are termed as peptides and those with higher molecular weights are termed as proteins. The acid-catalyzed hydrolysis of peptides and proteins
amino acids.
If the amine and carboxylic acid functional groups in amino acids join together to form amide bonds, a chain of amino acid units, called a peptide, is formed (Scheme 17A and 17B) simple tetrapeptide structure is shown in the following diagram. By convention, the amino acid component retaining a free amine group is drawn
terminus) of the peptide chain, and the amino acid retaining a free carboxylic acid is drawn on the right (the C-terminus). As expected, the free amine and carboxylic acid functions on a peptide chain form a zwitterionic structure at
Scheme 17A
Scheme 17B
15
and https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/protein2.htm#aacd6
These are naturally occurring polymers in living systems. The polymers with molecular as peptides and those with higher molecular
catalyzed hydrolysis of peptides and proteins
join together to form , is formed (Scheme 17A
and 17B) simple tetrapeptide structure is shown in the following diagram. By convention, the amino acid component retaining a free amine group is drawn at the left
terminus) of the peptide chain, and the amino acid retaining a free terminus). As expected, the free amine and
carboxylic acid functions on a peptide chain form a zwitterionic structure at their
16
The conformational flexibility of peptide chains is limited chiefly to rotations about the bonds leading to the alpha-carbon atoms. This restriction is due to the rigid nature of the amide (peptide) bond. As shown in the following diagram (Scheme 18), nitrogen electron pair delocalization into the carbonyl group results in significant double bond character between the carbonyl carbon and the nitrogen. This keeps the peptide links relatively planar and resistant to conformational change. The color shaded rectangles in the lower structure define these regions, and identify the relatively facile rotations that may take place where the corners meet (i.e. at the alpha-carbon). This aspect of peptide structure is an important factor influencing the conformations adopted by proteins and large peptides.
Scheme 18 The Primary Structure of Peptides Because the N-terminus of a peptide chain is distinct from the C-terminus, a small peptide composed of different amino acids may have a several constitutional isomers. For example, a dipeptide made from two different amino acids may have two different structures. Thus, aspartic acid (Asp) and phenylalanine (Phe) may be combined to make Asp-Phe or Phe-Asp, remember that the amino acid on the left is the N-terminus. The methyl ester of the first dipeptide (Scheme 19) is the artificial sweetener aspartame, which is nearly 200 times sweeter than sucrose. Neither of the component amino acids is sweet (Phe is actually bitter), and derivatives of the other dipeptide (Phe-Asp) are not sweet.
Scheme 19
17
A tripeptide composed of three different amino acids can be made in 6 different constitutions, and the tetrapeptide shown above (composed of four different amino acids) would have 24 constitutional isomers. When all twenty of the natural amino acids are possible components of a peptide, the possible combinations are enormous. Simple statistical probability indicates that the decapeptides made up from all possible combinations of these amino acids would total 2010 ! Natural peptides of varying complexity are abundant. The simple and widely distributed tripeptide glutathione (first entry in the Table 2), is interesting because the side-chain carboxyl function of the N-terminal glutamic acid is used for the peptide bond. An N-terminal glutamic acid may also close to a lactam ring, as in the case of TRH (second entry). The abbreviation for this transformed unit is pGlu (or pE), where p stands for "pyro" (such ring closures often occur on heating). The larger peptides in the table also demonstrate the importance of amino acid abbreviations, since a full structural formula for a nonapeptide (or larger) would prove to be complex and unwieldy. The formulas using single letter abbreviations are colored red.
Table 2. Some Common Natural Peptides
18
The ten peptides listed in this table make use of all twenty common amino acids. Note that the C-terminal unit has the form of an amide in some cases (e.g. TRH, angiotensin & oxytocin). When two or more cysteines are present in a peptide chain, they are often joined by disulfide bonds (e.g. oxytocin & endothelin); and in the case of insulin, two separate peptide chains (A & B) are held together by such links. The different amino acids that make up a peptide or protein, and the order in which they are joined together by peptide bonds is referred to as the primary structure. The primary structure of a peptide or protein describes the sequence of amino acids in the chain. Insulin is the first protein whose amino acid sequence was determined. Scheme 20 presents the primary structure of a hexapeptide.
Scheme 20
From the examples shown in Table 1 above, it should be evident that it is not a trivial task to determine the primary structure of such compounds, even modestly sized ones. Complete hydrolysis of a protein or peptide, followed by amino acid analysis establishes its gross composition, but does not provide any bonding sequence information. Partial hydrolysis will produce a mixture of shorter peptides and some amino acids. If the primary structures of these fragments are known, it is sometimes possible to deduce part or all of the original structure by taking advantage of overlapping pieces. For example, if a heptapeptide was composed of three glycines, two alanines, a leucine and a valine, many possible primary structures could be written. On the other hand, if partial hydrolysis gave two known tripeptide and two known dipeptide fragments, as shown in Scheme 21, simple analysis of the overlapping units identifies the original primary structure. Of course, this kind of structure determination is very inefficient and unreliable. First, we need to know the structures of all the overlapping fragments. Second, larger peptides would give complex mixtures which would have to be separated and painstakingly examined to find suitable pieces for overlapping. It should be noted, however, that modern mass spectrometry uses this overlap technique effectively. The difference is that bond cleavage is not achieved by hydrolysis, and computers assume the time consuming task of comparing a multitude of fragments.
Scheme 21
19
Secondary Structure of Protein The various properties of peptides and proteins depend not only on their component amino acids and their bonding sequence in peptide chains, but also on the way in which the peptide chains are stretched, coiled and folded in space. Because of their size, the orientational options open to these macromolecules might seem nearly infinite. Fortunately, several factors act to narrow the structural options, and it is possible to identify some common structural themes or secondary structures that appear repeatedly in different molecules. These conformational segments are sometimes described by the dihedral angles Φ & Ψ, defined in the diagram in Scheme 22. Five factors that influence the conformational equilibria of peptide chains are: • The planarity of peptide bonds. Conformations are defined by dihedral angles Φ & Ψ. • Hydrogen bonding of amide carbonyl groups to N-H donors. • Steric crowding of neighboring groups. • Repulsion and attraction of charged groups. • The hydrophilic and hydrophobic character of substituent groups.
Scheme 22
The secondary structure describes how the segments of the backbone chain fold. These conformations are stabilized by H-bonding between the peptide groups-between NH of one amino acid residue and C=O group of another (Scheme 23). There are two predominant types of secondary structures as shown in Scheme 24: α-helix and β-pleated sheet.
Scheme 23
20
Scheme 24. A segment of a protein in: (a) an α-helix; (b) β-pleated sheet.
α-Helix:
The first type of secondary structure is α-helix, where the backbone coils around the long axis of the protein molecule. The substituents on the α-carbon of the amino acids protrude outward from the helix to minimize the steric hindrance. The H attached to amide nitrogen makes H-bonding with the carbonyl oxygen of an amino acid. The N-terminal residue is on the top, and the C-terminal residue is at the bottom of Scheme 24a. The alpha-helix is right-handed, which means that it rotates clockwise as it spirals away from a viewer at either end. Other structural features that define an alpha-helix are: the relative locations of the donor and acceptor atoms of the hydrogen bond, the number of amino acid units per helical turn and the distance the turn occupies along the helical axis. The first hydrogen bond (from the N-terminal end) is from the carbonyl group of the first amino acid residue and the N-H group of the fifth amino acid residue. Three amino acids, fall entirely within this turn. A careful analysis of the structure indicates there are 3.6 amino acid units per turn. The distance covered by the turn is 5.4 Å. Using the dihedral angle terminology noted above, a perfect α-helix has Φ = -58º and Ψ = -47º. In natural proteins the values associated with α-helical conformations range from -57 to -70º for Φ, and from -35 to -
21
48º for Ψ. The alpha helix is the most stable secondary structure and accounts for a third of the secondary structure found in most globular (non-fibrous) proteins.
β-Pleated Sheet:
The second type of secondary structure is the β-pleated sheet, in which the backbone is extended in a zigzag structure resembling pleats. The linear zig-zag conformation of a peptide chain may be stabilized by hydrogen bonding to adjacent parallel chains of the same kind. Bulky side-chain substituents destabilize this arrangement due to steric crowding, so this beta-sheet conformation is usually limited to peptides having a large amount of glycine and alanine. Steric interactions also cause a slight bending or contraction of the peptide chains, and this results in a puckered distortion (the pleated sheet). As shown in Scheme 24b, the adjacent chains may be oriented in opposite N to C directions, termed antiparallel. Using the dihedral angle terminology, an antiparallel β-sheet has Φ = -139º and a Ψ = 135º. Alternatively, the adjacent peptide chains may be oriented in the same direction, termed parallel. By convention, beta-sheets are designated by broad arrows or cartoons, pointing in the direction of the C-terminus as shown in Scheme 25.
Scheme 25
Tertiary Structure of Protein
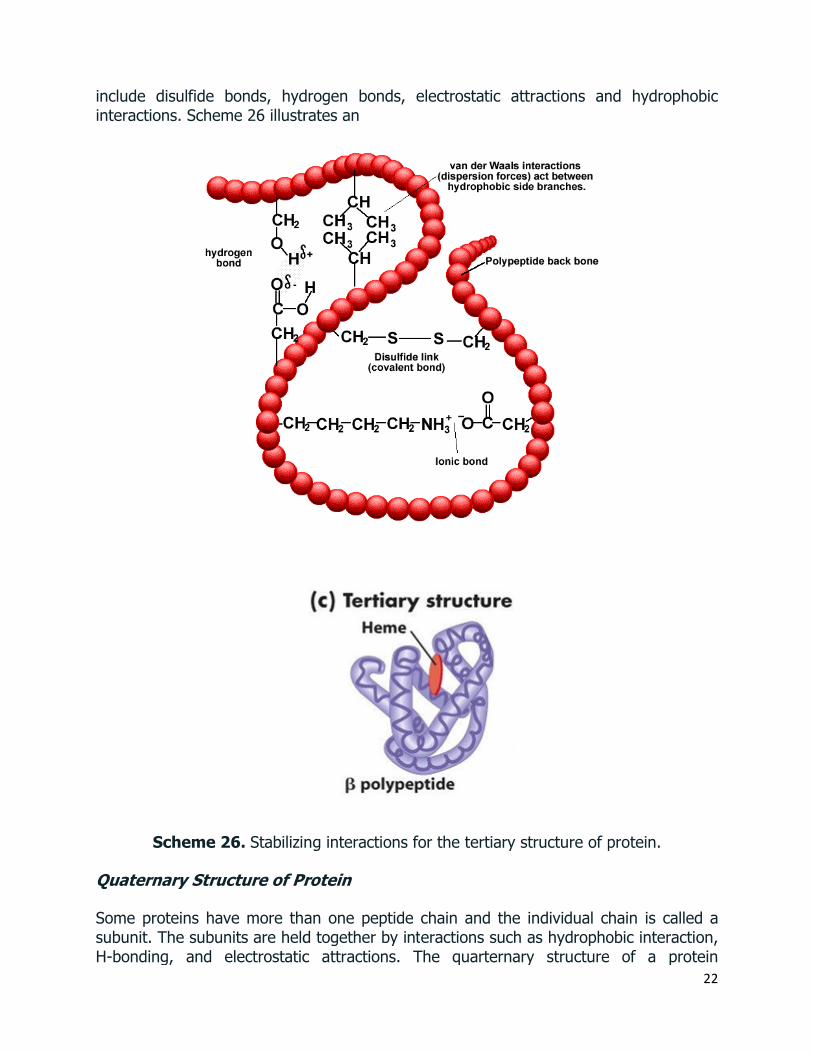
Most proteins and large peptides do not adopt completely uniform conformations, and full descriptions of their preferred three dimensional arrangements are defined as tertiary structures.
The tertiary structure of a protein describes the three-dimensional arrangement of all the atoms. In solution, proteins fold to maximize their stability through interactions
include disulfide bonds, hydrogen bonds, electrostatic attracinteractions. Scheme 26 illustrates an
Scheme 26. Stabilizing interactions for the tertiary structure of protein.
Quaternary Structure of Protein
Some proteins have more than one peptide chain and the individual chain is called asubunit. The subunits are held together by intH-bonding, and electrostatic attractions. The quarternary structure of a protein
include disulfide bonds, hydrogen bonds, electrostatic attractions and hydrophobic illustrates an
Stabilizing interactions for the tertiary structure of protein.
Quaternary Structure of Protein
Some proteins have more than one peptide chain and the individual chain is called asubunit. The subunits are held together by interactions such as hydrophobic int
bonding, and electrostatic attractions. The quarternary structure of a protein 22
tions and hydrophobic
Stabilizing interactions for the tertiary structure of protein.
Some proteins have more than one peptide chain and the individual chain is called a ractions such as hydrophobic interaction,
bonding, and electrostatic attractions. The quarternary structure of a protein
23
describes the way the subunits are arranged in space. Scheme 4 shows the structure of hemoglobin which is a tetrameric structural protein comprising two α and two β subunits.
Scheme 27. Quaternary Protein Structure: Three-Dimensional Arrangement of Subunits. (Ref: https://www.mun.ca/biology/scarr/Gr09-07d.html)
Determination of the structure of peptides
Acid hydrolysis of polypeptide to amino acids followed by analysis provides only the empirical formula of the polypeptide. It does not provide any amino acid sequence information. To obtain this, terminal residue analysis needs to be performed. Since the two termini of the peptide chains contain different groups, two different methods: N-terminus and C-terminus residue analysis can be performed.
N-Terminus Analysis – 2 methods, (i) Sanger and (ii) Edman methods are discussed below.
(i) Sanger Sequencing
24
A popular method is the Sanger method (Scheme 28) introduced by Frederick Sanger in 1945. This method uses 2,4-dinitrofluorobenzene (DNFB) which undergoes nucleophilic substitution by the free amino group to give N-dinitrophenyl (DNP) derivative. The N-terminal modified peptide is hydrolyzed to DNFB labeled N-terminus amino acid residue and remnant of the peptide chain. These components are isolated and identified with mass spectrometry to determine the N-terminal residue. The analysis is repeated with the remnant peptide to determine the 2nd N-terminus amino acid residue. A series of such analyses provide the sequence of amino acid residues starting from the N-terminus of the polypeptide chain.
Scheme 28 DNFB based Sanger Sequencing of N-terminus residue
(ii) Edman Sequencing
In 1950, Pehr Edman introduced another method utilizing phenylthioisocyanate, which reacts with the amino group of the N-terminal residue to produce a substituted thio urea as shown in Scheme 29 below. Mild hydrolysis with dilute HCl results in the
Scheme 29 Edman Sequencing of N-terminus residue in peptides
25
removal of the N-terminal residue as a phenylthiohydantoin. This can be identified with mass spectrometry and the remnant peptide can be subjected to further analysis to determine the 2nd N-terminal amino acid residue in the peptide.
C-Terminus Analysis – Carboxypeptidase method
The most successful method used for C-terminal analysis has been enzymatic rather than chemical method. Carboxypeptidase is an enzyme obtained from pancreas and this selectively hydrolyzes the peptide bond adjacent the free carboxylic acids, i.e. It cleaves the C-terminal amino acid residue, when leaving the rest of the peptide intact. The C-terminus residue can be analyzed.
Scheme 30 Carboxypeptidase Method
However this reaction cannot be terminated with limited proteolysis leading to cleavage of one residue. It leads to a mixture of polypeptide with one, two and/or more C-terminal residues lost from the C-terminus. So, a mixture of free carboxy terminus amino acids are produced. Careful analysis with mapping can lead to interpretation of C-terminus residue sequence in polypeptides.
Structure-Property Relationships
The compounds we call proteins exhibit a broad range of physical and biological properties. Two general categories of simple proteins are commonly recognized: Fibrous and Globular Proteins
Fibrous Proteins As the name implies, these substances have fiber-like structures, and serve as the chief structural material in various tissues. Corresponding to this structural function, they are relatively insoluble in water and unaffected by moderate changes in temperature and pH. Subgroups within this category include
(i) Collagens & Elastins, the proteins of connective tissues. tendons and ligaments. (ii) Keratins, proteins that are major components of skin, hair, feathers and horn. (iii) Fibrin, a protein formed when blood clots.
Globular Proteins
26
Members of this class serve regulatory, maintenance and catalytic roles in living organisms. They include hormones, antibodies and enzymes. They either dissolve or form colloidal suspensions in water. Such proteins are generally more sensitive to temperature and pH change than their fibrous counterparts.
Synthesis of Peptides
The aim of peptide synthesis is to design peptides that have identical amino acid sequence as that of naturally occurring peptides. This would require put-together a methodology to link amino acids in a predetermined sequence of predetermined length.
When linking two different types of amino acids, say carboxy group of one and amine group of another to form a peptide, one of the challenges is prevent the peptide bond formation between carboxy and amine groups of the same type. For eg., when formulating alanylglycyline, one should prevent formation of glycylglycine (digylcine) and alanylalanine (dialanine). This can be prevented by attaching an amine “blocking” group to alanine and a carboxy “blocking” group to glycine. This would force the formation of N-protected and C-protected alanylglyine.
The challenge lies in de-blocking the groups without hydrolyzing or modifying the peptide bond and side-chains of the amino acid residues. Also selective removal of the C-protecting group would make it amenable for adding a third amino acid residue to the growing chain.
Scheme 31
27
The strategy for peptide synthesis, as outlined below, should now be apparent. The following example shows a selective synthesis of the dipeptide Ala-Gly.
An important issue remains to be addressed. Since the N-protective group is an amide, removal of this function might require conditions that would also cleave the just formed peptide bond. Furthermore, the harsh conditions often required for amide hydrolysis might cause extensive racemization of the amino acids in the resulting peptide. This problem strikes at the heart of our strategy, so it is important to give careful thought to the design of specific N-protective groups. In particular, three qualities are desired:
1) The protective amide should be easy to attach to amino acids. 2) The protected amino group should not react under peptide forming conditions. 3) The protective amide group should be easy to remove under mild conditions.
Protection of Amino Group
A number of N-protective groups that satisfy these conditions have been devised; and two of the most widely used, carbobenzoxy (Cbz) and t-butoxycarbonyl (BOC or t-BOC), are described here.
Scheme 32
The reagents for introducing these N-protective groups are the acyl chlorides or anhydrides shown in the left portion of the above diagram. Reaction with a free amine function of an amino acid occurs rapidly to give the "protected" amino acid derivative shown in the center. This can then be used to form a peptide (amide) bond to a second amino acid. Once the desired peptide bond is created the protective group can be removed under relatively mild non-hydrolytic conditions. Cleavage of the reactive benzyl
28
or tert-butyl groups generates a common carbamic acid intermediate (HOCO-NHR) which spontaneously loses carbon dioxide, giving the corresponding amine.
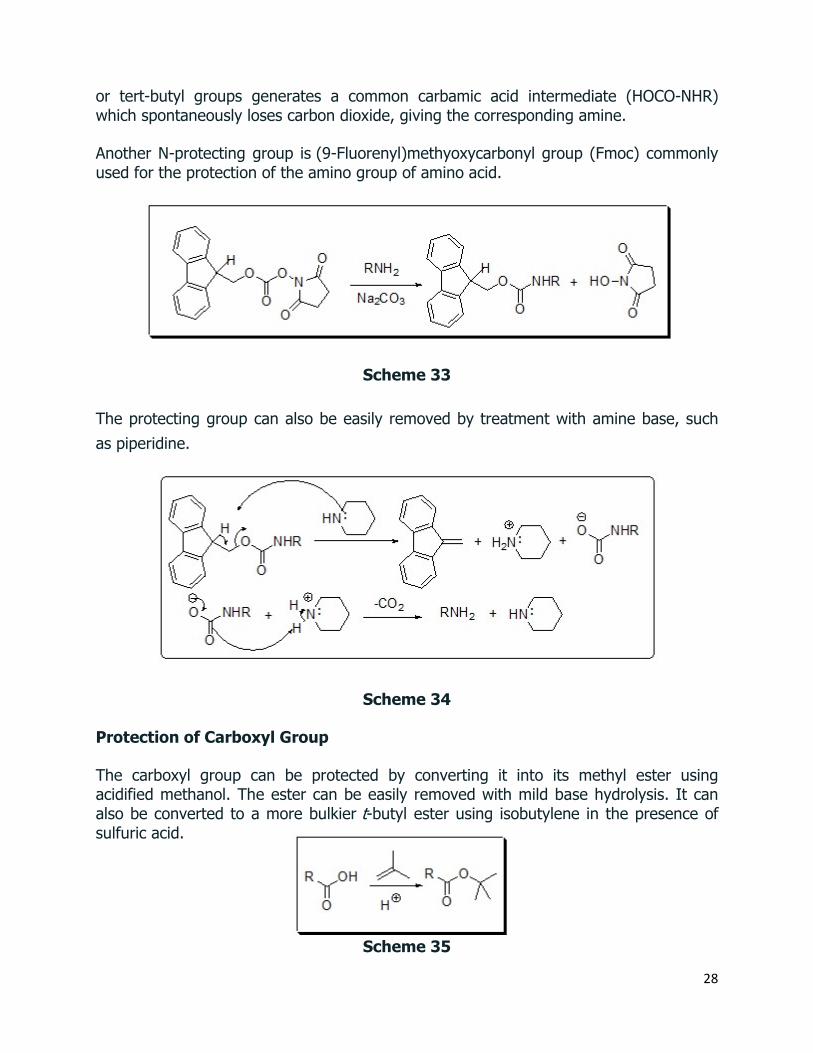
Another N-protecting group is (9-Fluorenyl)methyoxycarbonyl group (Fmoc) commonly used for the protection of the amino group of amino acid.
Scheme 33
The protecting group can also be easily removed by treatment with amine base, such
as piperidine.
Scheme 34
Protection of Carboxyl Group
The carboxyl group can be protected by converting it into its methyl ester using acidified methanol. The ester can be easily removed with mild base hydrolysis. It can also be converted to a more bulkier t-butyl ester using isobutylene in the presence of sulfuric acid.
Scheme 35
29
The protecting group can also be easily removed using mild acid hydrolysis.
Scheme 36
If the methyl ester at the C-terminus is left in place, this sequence of reactions may be repeated, using a different N-protected amino acid as the acylating reagent. Removal of the protective groups would then yield a specific tripeptide, determined by the nature of the reactants and order of the reactions.
The synthesis of a peptide of significant length (e.g. ten residues) by this approach requires many steps, and the product must be carefully purified after each step to prevent unwanted cross-reactions. To facilitate the tedious and time consuming purifications, and reduce the material losses that occur in handling, a clever modification of this strategy has been developed.
This procedure, known as the Merrifield Synthesis after its inventor R. Bruce Merrifield, involves attaching the C-terminus of the peptide chain to a polymeric solid, usually having the form of very small beads. Separation and purification is simply accomplished by filtering and washing the beads with appropriate solvents. The reagents for the next peptide bond addition are then added, and the purification steps repeated. The entire process can be automated, and peptide synthesis machines based on the Merrifield approach are commercially available. A series of equations illustrating the Merrifield synthesis may be viewed by clicking on the following diagram. The final step, in which the completed peptide is released from the polymer support, is a simple benzyl ester cleavage.
Merrifield Peptide Synthesis
30
Scheme 37
Two or more moderately sized peptides can be joined together by selective peptide bond formation, provided side-chain functions are protected and do not interfere. In this manner good sized peptides and small proteins may be synthesized in the laboratory. However, even if chemists assemble the primary structure of a natural protein in this or any other fashion, it may not immediately adopt its native secondary, tertiary and quaternary structure. Many factors, such as pH, temperature and inorganic ion concentration influence the conformational coiling of peptide chains. Indeed, scientists are still trying to understand how and why these higher structures are established in living organisms.
Denaturation
The natural or native structures of proteins may be altered, and their biological activity changed or destroyed by treatment that does not disrupt the primary structure. This denaturation is often done deliberately in the course of separating and purifying proteins. For example, many soluble globular proteins precipitate if the pH of the solution is set at the pI of the protein. Also, addition of trichloroacetic acid or the bis-amide urea (NH2CONH2) is commonly used to effect protein precipitation. Following denaturation, some proteins will return to their native structures under proper conditions; but extreme conditions, such as strong heating, usually cause irreversible change.
Some treatments known to denature proteins are listed in the following table.
Table 3: Denaturation of Proteins – Mechanism of Action
Denaturing Action Mechanism of Operation
Heat Hydrogen bonds are broken by increased translational and vibrational energy – eg. coagulation of egg white albumin on frying.
Ultraviolet Radiation Similar to heat – e.g. sunburn
Strong Acids or Bases Salt formation; disruption of hydrogen bonds – eg. skin blisters and burns, protein precipitation.
Urea Solution Competition for hydrogen bonds – eg. precipitation of soluble proteins.
Organic Solvents (e.g. ethanol & acetone)
Change in dielectric constant and hydration of ionic groups – eg. disinfectant action and precipitation of protein.
Agitation Shearing of hydrogen bonds - eg. beating egg white albumin into a meringue.
31
Not all proteins are easily denatured. As noted above, fibrous proteins such as keratins, collagens and elastins are robust, relatively insoluble, quaternary structured proteins that play important roles in the physical structure of organisms. Secondary structures such as the α-helix and β-sheet take on a dominant role in the architecture and aggregation of keratins. In addition to the intra- and intermolecular hydrogen bonds of these structures, keratins have large amounts of the sulfur-containing amino acid Cys, resulting in disulfide bridges that confer additional strength and rigidity. The more flexible and elastic keratins of hair have fewer interchain disulfide bridges than the keratins in mammalian fingernails, hooves and claws. Keratins have a high proportion of the smallest amino acid, Gly, as well as the next smallest, Ala. In the case of β-sheets, Gly allows sterically-unhindered hydrogen bonding between the amino and carboxyl groups of peptide bonds on adjacent protein chains, facilitating their close alignment and strong binding. Fibrous keratin chains then twist around each other to form helical filaments.
1
Carbohydrates I
11.1 Introduction
Carbohydrates are polyhydroxy aldehydes or ketones. They are primarily produced by plants and form a very large group of naturally occurring organic substances. Some common examples are cane sugar, glucose, starch, etc. They have general molecular formulas that make them appear to be hydrates of carbon, Cn(H2O)n , from where the name carbohydrate was derived. Carbohydrates are formed in the plants by photosynthesis from carbon dioxide and water in the presence of sunlight (Scheme 1).
Scheme 1
11.2 Classification
Carbohydrates are classified into two main classes, sugars and polysaccharides.
11.2.1 Sugars
Sugars are sweet crystalline substances that are soluble in water. These are further classified on the basis of their behavior on hydrolysis.
11.2.1.1 Monosaccharides
The simplest form of carbohydrates is the monosaccharide. 'Mono' means 'one' and 'saccharide' means 'sugar'. Monosaccharides are polyhydroxy aldehyde or ketone that cannot be hydrolyzed further to give simpler sugar. They may again be classified on the basis of the nature of carbonyl group.
• Polyhydroxy aldehydes are called aldoses. Example: Glucose
• Polyhydroxy ketones are called ketoses. Example: Fructose
2
The aldoses and ketoses are further divided based on the number of carbons present in their molecules, as trioses, tetroses, pentoses, hexoses etc. They are referred to as aldotrioses, aldotetroses, aldopentoses, aldohexoses, ketohexoses etc.
11.2.1.2 Oligosaccharides
Carbohydrates that produce two to ten monosaccharide units during the hydrolysis are called oligosaccharides. They can be further classified based on the number of monosaccharide units formed on hydrolysis.
Disaccharides: They give two monosaccharide units on hydrolysis, which may be the same or different. For example, sucrose on hydrolysis gives one molecule each of glucose and fructose, whereas maltose gives two molecules of glucose (Scheme 1).
Scheme 2
3
Trisaccharides: These carbohydrates yield three molecules of monosaccharides units on
hydrolysis (Scheme 2).
Scheme 3
11.2.1.3 Polysaccharides
These carbohydrates give a large number of monosaccharide units on hydrolysis. These monosaccharide units are joined together by oxide bridges. These linkages are called glycosidic linkages. The common and widely distributed polysaccharides correspond to the general formula (C6H10O5)n . Polysaccharides are not sweet in taste, so they are called non-sugars. Some common examples are starch, cellulose, glycogen, etc (Scheme 3).
Scheme 3
11.3 The D and L Notations
The notations D and L are used to describe the configurations of carbohydrates and amino acids. Glyceraldehyde has been chosen as arbitrary standard for the D and L notation in sugar chemistry. Because, this has an asymmetric carbon and can exist as a pair of enantiomers.
In a Fischer projection, the carbonyl group is always placed on the top position for monosaccharide. From its structure, if the –OH group attached to the bottom-most asymmetric center (the carbon that is second from the bottom) is on the right, then, the
4
compound is a D-sugar. If the –OH group is on the left, then, the compound is a L-sugar. Almost all sugars found in nature are D-sugar.
Like R and S , D and L indicate the configuration of an asymmetric carbon, but they do not indicate whether the compound rotates polarized light to the right or to the left. For example, D-glyceraldehyde is dextrorotatory, whereas D-lactic acid is levorotatory. In other words, optical rotation, like melting or boiling points, is a physical property of a compound, whereas “ R , S , D, and L” are conventions humans use to indicate the configuration of a molecule.
11.3.1 Configuration of the Aldoses
Aldotetrose : The structural formula of aldotetrose is CH2OH�CHOH�CHOH�CHO. There are four stereoisomers (two pairs of enantiomers). Those stereoisomers correspond to D- and L-erythrose and D- and L-threose.
Aldopentose : The structural formula of aldopentose is CH2OH�CHOH�CHOH�CHOH�CHO. Since it contains three asymmetric carbons and there are eight stereoisomers (four pairs of enantiomers) possible. Those are the D- and L-forms of ribose, arabinose, xylose and lyxose.
Aldohexose : They have four asymmetric center and therefore 16 stereoisomers (eight pairs of enantiomers). The four D-altopentose and eight D-aldohexose are shown below.
5
Configuration of the D-Aldoses
Configuration of the ketoses
Ketoses have the keto group in the 2-position with one less chiral carbon compared to aldoses. So, ketoses have a half number of stereoisomers compared to aldoses with same number of carbon atoms. For example, aldopentose has three chiral centers with eight stereoisomers, while ketopentose has two chiral centers with four stereoisomers. The configuration of D-2 ketones is illustrated by the following examples. Dihydroxyacetone may not be a sugar, but it is included as the ketose analog of glyceraldehyde.
6
Configuration of the Ketoses
Epimers are stereoisomers that differ in configuration of only one asymmetric carbon of enantiomers or diastereomers. Example, D-glucose and D-mannose are C-2 epimers and D-glucose and D-talose are C-3 epimers. D-fructose and D-tagatose are C-4 epimers of ketohexoses.
7
11.4 Cyclic Structure of Monosaccharides: Hemiacetal Formation
Aldoses contain an aldehyde group with several alcohol groups. The cyclic forms of D-glucose are six-membered hemiacetals formed by an intramolecular reaction of the –OH group at C-5 with the aldehyde group.
Cyclic structures of monosaccharides are named according to their five- or six-membered rings (Scheme 4-5). A six -membered cyclic hemiacetal is called a pyranose , derived from the name of the six-membered cyclic ether pyran . A five-membered cyclic hemiacetal is called a furanose , derived from the name of the five -membered cyclic ether furan . For example, the six-membered ring of glucose is called glucopyranose and the five-membered ring of fructose is called fructofuranose.
Scheme 5
8
Scheme 4
Haworth projection is "flattened" diagrams used to represent the stereochemistry carbohydrates. The six-membered ring of D-pyranose is represented as flat and is viewed edge on. The ring oxygen is always placed in the back right-hand corner of the ring, with the anomeric carbon (C-1) on the right-hand side and the primary alcohol group drawn up from the back left-hand corner (C-5).
• Groups on the right in a Fischer projection are down (below the ring) in a Haworth projection (Figure 1).
• Groups on the left in a Fischer projection are up (above the ring) in a Haworth projection.
Scheme 1
D-Furanose is viewed edge on, with the ring oxygen away from the viewer. The anomeric carbon is on the right-hand side of the molecule, and the primary alcohol group is drawn up from the back left-hand corner (Figure 2).
9
Figure 2
10
Carbohydrates II
11.5 Acylation and Alkylation of Monosaccharides
The –OH group of monosaccharide reacts with acetic anhydride or acetyl chloride to form esters. Similarly, ether can be prepared using methyl iodide/silver oxide. The nucleophilic character of –OH group is relatively poor, so silver oxide is used to increase the leaving tendency of the iodide ion in theSN2 reaction (Scheme 1).
Scheme 1
11.6 Glycosides
Glycosides are cyclic acetal form of sugars and the bond between the anomeric carbon and the alkoxy oxygen is called a glycosidic bond. They are prepared by the acid-catalyzed reaction of an alcohol with a pyranose or furanose.
Naming of glycosides is done by replacing the “ e ” of the sugar with “ ide ”. Example, a glycoside of glucose is glucoside and if pyranose or furanose name is used, the acetal is called pyranoside or furanoside. Both α - and β -glycoside obtained from the reaction of a
single anomer with an alcohol (Scheme 2).
11
Scheme 2
The reason for the formation of both glycosides is shown in Scheme 3. The protonation of the anomeric carbon –OH group followed by elimination of water gives a planar sp2 hybridized oxocarbenium ion. This can react with alcohol from both faces to give the β -glycoside and the α-glycoside.
Scheme 3
12
11.7 Anomeric Effect
When a pyranose or furanose ring closes, the hemiacetal carbon atom is converted from a flat carbonyl group to an asymmetric carbon. Depending on which face of the (protonated) carbonyl group is attacked, the hemiacetal –OH group can be directed either up or down. These two orientations of the hemiacetal –OH group give diastereomeric products called anomers, and the hemiacetal or acetal carbon atom is called the anomeric carbon atom. The preference of certain substituents bonded to the anomeric carbon for the axial position is called the anomeric effect. Ano is Greek for “upper”; thus, anomers differ in configuration at the upper-most asymmetric carbon. The anomeric carbon is the only carbon in the molecule that is bonded to two oxygen atoms. The anomer with the anomeric –OH group down (axial) is called the α -anomer, and the one with the anomeric –OH group up
(equatorial) is called the β -anomer (Scheme 4).
Scheme 4
In fructose, the α -anomer has the anomeric - OH group down, trans to the terminal –CH2OH group, while the β -anomer has it up, cis to the terminal –CH2OH group (Scheme 5).
Scheme 5
11.8 Mutarotation
Normally D-(+)-glucose has a melting point of 146°C. However, when D-(+)-glucose is crystallized by evaporating an aqueous solution kept above 98°C, a second form of D-(+)-glucose with a melting point of 150°C can be obtained. When the optical rotations of these two forms are measured, they are found to be significantly different, but when an aqueous solution of either form is allowed to stand, its rotation changes. The specific rotation of one form decreases and the other increases, until both solutions show the same value. For
13
example, a solution of α -D-(+)-glucose (mp 146°C) specific rotation gradually decreases
from an initial value of + 112.2° to + 52.7°, while The β -D-(+)-glucose (mp 150°C)
specific rotation gradually increases from an initial value of + 18.7° to + 52.7°. The three forms of glucose reach equilibrium concentrations with the specific rotation of +52.7. This change ("mutation") in the specific rotation toward equilibrium is called mutarotation (Scheme 6).
Scheme 6
11.9 Reducing and Non-reducing Sugars
The carbohydrates may also be classified as either reducing or non-reducing sugars. Cyclic acetals or ketals are not in equilibrium with their open chain carbonyl group containing forms in neutral or basic aqueous solutions. They cannot be oxidized by reagents such as Tollen's reagent (Ag+, NH3, OH-) or Br2. So, these are referred as non-reducing sugars. Whereas hemiacetals or hemiketals are in equilibrium with the open-chain sugars in aqueous solution. These compounds can reduce an oxidizing agent (eg. Br2), thus, they are classified as a reducing sugar.
11.10 Determination of Ring Size
The anomeric carbon can be found via methylation of the –OH groups, followed by hydrolysis. In the first step, all the –OH groups are transformed to –OCH3 groups with excess methyl iodide and silver oxide. The hydrolysis of the acetal then forms a hemiacetal in presence of acid. This pyranose structure is in equilibrium with its open-chain form. From
14
the open-chain form we can determine the size of the ring because the anomeric carbon attached –OH group is the one that forms the cyclic hemiacetal (Scheme 7).
Scheme 7
A monosaccharide's ring size can be determined by the oxidation of an acetal of the monosaccharide with excess periodic acid. The products obtained from periodate cleavage of a six-membered ring acetal are different from those obtained from cleavage of a five-membered ring acetal (Scheme 8-9).
Scheme 8
Scheme 9
15
11.11 Disaccharides
If the glycoside or acetol is formed by reaction of the anomeric carbon of a monosaccharide with OH group of another monosaccharide molecule, then the glycoside product is a disaccharide (Scheme 10).
Scheme 10
The anomeric carbon can react with any of the hydroxyl groups of another monosaccharide unit to form a disaccharide. Disaccharides can be categorized by the position of the hydroxyl group of another monosaccharide making up the glycoside.
Disaccharides have three naturally occurring glycosidic linkages
• 1-4' link: The anomeric carbon is bonded to oxygen on C-4 of second monosaccharide. • 1-6' link: The anomeric carbon is bonded to oxygen on C-6 of second monosaccharide. • 1-2' link: The anomeric carbons of the two monosaccharide unit are bonded through an
oxygen.
The “prime” superscript indicates that –OH group bonded carbon position of the second monosaccharide unit, α - and β -configuration given by based on the configuration at the
anomeric carbon of the first monosaccharide unit.
1-4' Glycosides : These represent the most common naturally occurring disaccharides. The linkage is between C-1 of one sugar subunit and C-4 of the other. For example, maltose is a disaccharide with two D-glucose units bearing 1,4'-glycosidic linkage. The stereochemistry of this linkage is α . So, the glycosidic linkage is called α -1,4'-glycosidic
linkage.
16
Cellobiose also contains two D-glucose subunits. The only difference from maltose is that the two glucose subunits are joined through a β -1,4'-glycosidic linkage.
Lactose , a disaccharide present in milk, contains D-galactose (non-reducing) and D-glucose (reducing) monosaccharide units. These units are hooked together by a β -1,4'-glycosidic
linkage.
1-6' Glycosides : The anomeric carbon of one unit hooked by the oxygen of the terminal carbon (C-6) of another monosaccharide unit. Example, gentiobiose is a sugar with two glucose units joined by a β -1,6'-glucosidic linkage.
17
1-2' Glycosides : The glycosidic bond is hooked between the two anomeric carbon of the monosaccharide units. For example, sucrose contains a D-glucose subunit and a D-fructose subunit, which have been joined by a glycosidic bond between C-1 of glucose (in the α -position) and C-2 of fructose (in the β- position).
11.12 Polysaccharides
Polysaccharides are carbohydrates that contain many monosaccharide units joined by glycosidic bonds. All the anomeric carbon atoms of polysaccharides are involved in acetal formation. So, polysaccharides do not react with Tollen's reagent, and they do not mutarotate.
Polysaccharides that are polymers of a single monosaccharide are called homopolysaccharides. If they made by more than one type of monosaccharide are called heteropolysaccharides. Example, a glucan is made by glucose units and galactan, which is made by galactose units. There are three important polysaccharides, which are starch, glycogen and cellulose.
Starch is a glucose polymer that is the principal food storage carbohydrate in plants. It is found in roots, rhizomes, seeds, stems, tubers and corms of plants, as microscopic granules having characteristic shapes and sizes. Most animals, including humans, depend on these plant starches for nourishment. The intact granules are insoluble in cold water, but grinding or swelling them in warm water causes them to burst.
18
The released starch consists of two fractions. About 20% is a water soluble material called amylose. The majority of the starch is a much higher molecular weight substance, consisting of nearly a million glucose units, and called amylopectin.
Amylose is a linear polymer of D-glucose units joined by α -1,4'-glycosidic linkages. They
are linear chains of several thousand glucose units joined by alpha C-1 to C-4 glycoside bonds. Amylose solutions are actually dispersions of hydrated helical micelles.
Amylopectin is a branched polymer of D-glucose units hooked by α -1,4'-glycosidic linkages and the branches are created by α-1,6'-glycosidic linkages. (branched networks
built from C-1 to C-4 and C-1 to C-6 glycoside links). It is essentially water insoluble. On an average, branches occur every twenty five glucose units.
Hydrolysis of starch, usually by enzymatic reactions, produces a syrupy liquid consisting largely of glucose. When cornstarch is the feedstock, this product is known as corn syrup. It is widely used to soften texture, add volume, prohibit crystallization and enhance the flavor of foods.It is a mixture of two components that can be separated on the basis of water solubility.
19
Glycogen functions as a carbohydrate storage form for animals. Like amylopectin, it is non-liner polymer of D-glucose units joined by α -1,4'-glycosidic linkages and α-1,6' -
glycosidic linkages at branches. The structure of glycogen is similar to that amylopectin, but it has more branches. On an average, branches occur every ten glucose units. The highly branched structure of glycogen provides many available glucose end groups for immediate hydrolysis to provide glucose needed for metabolism.
Cellulose serves as structural material in plants, providing structural strength and rigidity to plants. It is a linear polymer of D-glucose units joined by β -1,4'-glycoside bonds.
Humans and other mammals do not have the β -glucosidase enzyme needed to hydrolyze
cellulose, so they cannot obtain glucose directly from cellulose.
Synthetic Modification of Cellulose Cotton, probably the most useful natural fiber, is nearly pure cellulose. The manufacture of textiles from cotton involves physical manipulation of the raw material by carding, combing and spinning selected fibers. For fabrics the best cotton has long fibers, and short fibers or cotton dust are removed. Crude cellulose is also available from wood pulp by dissolving the lignan matrix surrounding it. These less desirable cellulose sources are widely used for making paper. In order to expand the ways in which cellulose can be put to practical use, chemists have devised techniques for preparing solutions of cellulose derivatives that can be spun into
20
fibers, spread into a film or cast in various solid forms. A key factor in these transformations are the three free hydroxyl groups on each glucose unit in the cellulose chain, --[C6H7O(OH)3]n--. Esterification of these functions leads to polymeric products having very
different properties compared with cellulose itself.
Cellulose Nitrate, first prepared over 150 years ago by treating cellulose with nitric acid, is the earliest synthetic polymer to see general use. The fully nitrated compound, --[C6H7O(ONO2)3]n--, called guncotton, is explosively flammable and is a component of smokeless powder. Partially nitrated cellulose is called pyroxylin. Pyroxylin is soluble in ether and at one time was used for photographic film and lacquers. The high flammability of pyroxylin caused many tragic cinema fires during its period of use. Furthermore, slow hydrolysis of pyroxylin yields nitric acid, a process that contributes to the deterioration of
early motion picture films in storage.
Cellulose Nitrate
Cellulose Acetate, --[C6H7O(OAc)3]n--, is less flammable than pyroxylin, and has replaced it in most applications. It is prepared by reaction of cellulose with acetic anhydride and an
acid catalyst. The properties of the product vary with the degree of acetylation. Some chain shortening occurs unavoidably in the preparations. An acetone solution of cellulose acetate may be forced through a spinneret to generate filaments, called acetate rayon, that can
be woven into fabrics.
Cellulose Acetate
Viscose Rayon, is prepared by formation of an alkali soluble xanthate derivative that can be spun into a fiber that reforms the cellulose polymer by acid quenching. The following general equation illustrates these transformations. The product fiber is called viscose
rayon.
ROH
NaOH
RO(-) Na(+) + S=C=S
RO-CS2
(-) Na(+) H3O
(+)
ROH
cellulose
viscose solution
rayon
21
Carbohydrates III
11.13 Reactions
Monosaccharides contain carbonyl functional group and alcohol functional groups, so it can be oxidized or reduced and can react with nucleophiles to form corresponding products.
11.13.1 Epimerization
In the presence of base, D-glucose may be converted into D-mannose via the removal of hydrogen at C-2 carbon followed by protonation of the enolate (Scheme 1).
Scheme 1
11.13.2 Enediol Rearrangement
The position of carbonyl group may shift via enediol intermediate under basic condition. For example, rearrangement of D-glucose gives D-fructose (Scheme 2).
Scheme 2
22
11.13.3 Reduction
The monosaccharide contains carbonyl group which can be reduced by the reducing agents such as NaBH 4 . Reduction of aldose forms one alditol and ketose forms two alditols (Scheme 3).
Scheme 3
11.13.4 Oxidation
• Bromine water oxidizes aldehyde functional group, but it cannot oxidize ketones or alcohols.
Therefore, aldose can be distinguished from ketose by observing reddish-brown colour of
bromine. The oxidized product is an aldonic acid (Scheme 4).
Scheme 4
• Tollen's reagent can oxidize both aldose and ketose to aldonic acids. For example, the enol
of both D-fructose and D-glucose, as well as the enol of D-mannose are same (Scheme 5).
23
Scheme 5
Both aldehyde and primary alcohol groups of an aldose are oxidized by strong oxidizing
agent such as HNO3. The oxidized product called an aldaric acid. Ketose also reacts with
HNO3 to give more complex product mixtures (Scheme 6).
Scheme 6
24
11.13.5 Osazone Formation
Aldose and ketose react with one equiv of phenylhydrazine to produce phenylhydrazones. In contrast, both C-1 and C-2 react with three equivalent of phenylhydrazine to form a bis-hydrazone known as an osazone (Scheme 7).
Scheme 7
The configuration at C-1 or C-2 is lost in the formation of osazone, C-2 epimers form identical osazones. For example, D-gluose and D-idose are C-2 epimers; both form the same osazone (Scheme 8).
Scheme 8
Ketose reacts with phenylhydrazine at C-1 and C-2 position to form osazone. D-Glucose and D-fructose form the same osazone (Scheme 9).
Scheme 9
25
11.13.6 The Ruff Degradation
Aldose chain is shortened by oxidizing the aldehyde to –COOH, then decarboxylation. In the Ruff degradation, the calcium salt of an aldonic acid is oxidized with hydrogen peroxide. Ferric ion catalyzes the oxidation reaction, which cleaves the bond between C-1 and C-2, forming an aldehyde. The calcium salt of the aldonic acid prepared from oxidation of an aldose with an aqueous solution of bromine and then adding calcium hydroxide to the reaction mixture (Scheme 10).
Scheme 10
11.13.7 The Kiliani–Fischer Synthesis
An aldose carbon chain can be increased by one carbon in a Kiliani–Fischer synthesis (Scheme 11). It is the opposite of Ruff Degradation reaction. This synthesis leads to formation of a pair of C-2 epimers.
Scheme 11
D-Erythrose gives the corresponding chain lengthened products D-ribose and D-arabinose (Scheme 12).
26
Scheme 12
Text Books
• P. Y. Bruce and K J R. Prasad, Essential Organic Chemistry , Pearson Education, New Delhi, 2008.
• R. R. Morrison, R. N. Boyd and S. K. Bhattacharjee, Organic Chemistry , Dorling Kindersley (India)
Pvt. Ltd, New Delhi, 2011.
• C. M. Loudon , Organic Chemistry , Oxford University Press, New Delhi, 2002.