26
Anterior Horn Cell Anterior Horn Cell Disorders Disorders Shekhar J.Lamdhade Shekhar J.Lamdhade Consultant Neurologist Consultant Neurologist Al Amiri Hospital Al Amiri Hospital
| Date post: | 31-Dec-2015 |
| Category: |
Documents |
| Upload: | reginald-charles |
| View: | 232 times |
| Download: | 4 times |

Anterior Horn Cell DisordersAnterior Horn Cell Disorders
Shekhar J.LamdhadeShekhar J.Lamdhade
Consultant NeurologistConsultant Neurologist
Al Amiri HospitalAl Amiri Hospital

Anterior Horn Cell Anterior Horn Cell DisordersDisorders
* Classification* Classification
* Amyotrophic Lateral * Amyotrophic Lateral Sclerosis (ALS) - clinical Sclerosis (ALS) - clinical manifestations, manifestations, - - differential diagnosis, differential diagnosis, - - treatmenttreatment

Anterior horn ≈ Motor NeuronAnterior horn ≈ Motor NeuronDisorders Disorders ≈ Degeneration (MND)≈ Degeneration (MND)
MNDMND – Progressive degeneration of – Progressive degeneration of neurons of motor cortex, brain neurons of motor cortex, brain stem, and spinal cordstem, and spinal cord
Manifested by Weakness, atrophy Manifested by Weakness, atrophy (LMN) and corticospinal tract (LMN) and corticospinal tract signs (UMN) signs (UMN)
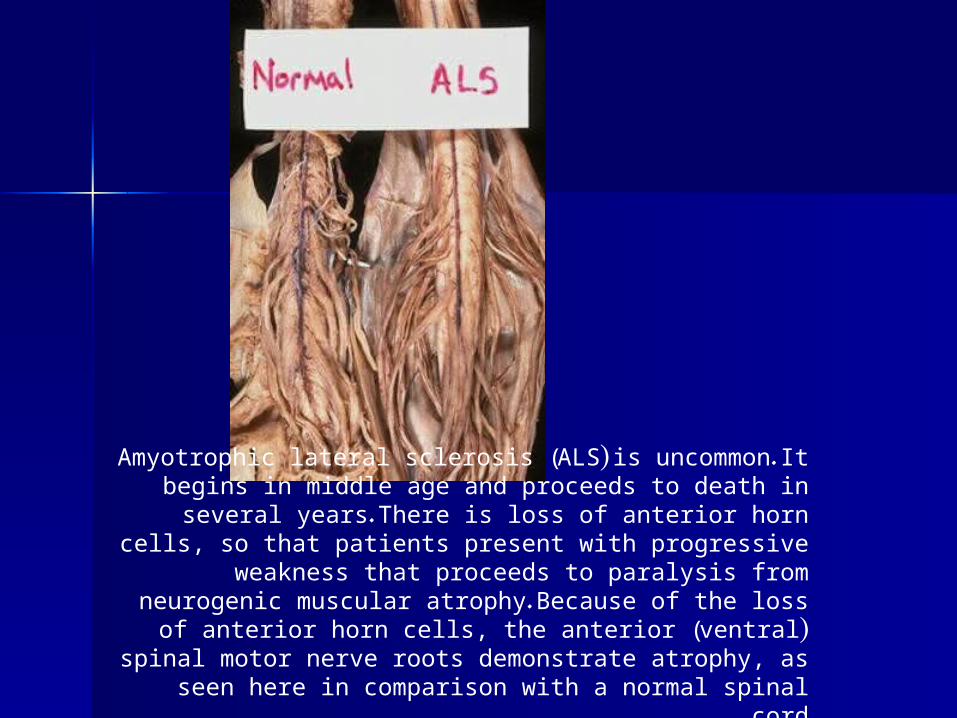
Amyotrophic lateral sclerosis (ALS) is uncommon. It begins in middle age and proceeds to death in several years. There is loss
of anterior horn cells, so that patients present with progressive weakness that proceeds to paralysis from neurogenic muscular atrophy. Because of the loss of anterior horn cells, the anterior
(ventral) spinal motor nerve roots demonstrate atrophy, as seen here in comparison with a normal spinal cord .

Types: SMA (Spinal muscular Types: SMA (Spinal muscular atrophy)atrophy) (Congenital form)(Congenital form)
SMA - I - Infantile SMA - I - Infantile SMA- II - ChildhoodSMA- II - Childhood SMA III - AdolescenceSMA III - AdolescenceC/F: floppy baby, generalized poor C/F: floppy baby, generalized poor
muscular development, sluggish muscular development, sluggish reflexes.reflexes.
Delayed motor mile stones, feeding & Delayed motor mile stones, feeding & respiratory difficulties, recurrent respiratory difficulties, recurrent aspirations aspirations death.death.
Defect: Mutation in SMN geneDefect: Mutation in SMN gene

Motor Neuron secretes Trophic factorMotor Neuron secretes Trophic factor

Motor Neuron DiseaseMotor Neuron Disease
TypesTypes
1.1. Amyotrophic Lateral Sclerosis Amyotrophic Lateral Sclerosis ( ALS)( ALS)
2.2. Progressive spinal muscular Progressive spinal muscular atrophyatrophy
3.3. Progressive bulbar palsyProgressive bulbar palsy
4.4. Primary lateral sclerosisPrimary lateral sclerosis

1.1. Amyotrophic Lateral SclerosisAmyotrophic Lateral Sclerosis (ALS) (ALS)
Most frequent form: Muscular atrophy Most frequent form: Muscular atrophy and hyper and hyper
reflexia.reflexia.
2.2. Progressive spinal muscular atrophy:Progressive spinal muscular atrophy:
Diffuse muscular weakness and Diffuse muscular weakness and atrophy - specially of spinal muscles.atrophy - specially of spinal muscles.

3.3. Progressive bulbar palsy:Progressive bulbar palsy: Weakness & wasting in muscles Weakness & wasting in muscles innervated by motor nuclei of innervated by motor nuclei of lower brain stem – jaw, face, lower brain stem – jaw, face, tongue, pharynx and larynx.tongue, pharynx and larynx.
4.4. Primary lateral sclerosis:Primary lateral sclerosis: Predominant spastic weakness, Predominant spastic weakness, hyperreflexia, Babinski sign. LMN hyperreflexia, Babinski sign. LMN signs none or very late.signs none or very late.

ALS ( Amyotrophic Lateral ALS ( Amyotrophic Lateral Sclerosis)Sclerosis)
Charcot – 1869Charcot – 1869
Incidence : 0.4-1.76/100,000Incidence : 0.4-1.76/100,000
M=FM=F
World wideWorld wide
Some clustering of cases in Kii Some clustering of cases in Kii peninsula of Japan & Guam.peninsula of Japan & Guam.
10% familial cases, AD type 10% familial cases, AD type

ALS – Clinical featuresALS – Clinical features
Weakness begins in one limb, distally.Weakness begins in one limb, distally.
Unexplained tripping – mild foot dropUnexplained tripping – mild foot drop
Difficulty in using fingers – using key.Difficulty in using fingers – using key.
Unexplained cramps & fasciculations: Unexplained cramps & fasciculations: shoulder – arm- fore arm,shoulder – arm- fore arm,
thighs-leg-footthighs-leg-foot

ALS – Clinical featuresALS – Clinical features
Wasting of small muscles of hands : Wasting of small muscles of hands : Abductors, adductors, extensors more Abductors, adductors, extensors more
weak than flexors.weak than flexors. - - CLAW HANDCLAW HAND..DTRDTR: hyper/ hypo , Babinski sign + ve,: hyper/ hypo , Babinski sign + ve,Preserved abdominal reflexesPreserved abdominal reflexesLateLate – involvment of neck, pharynx – involvment of neck, pharynx
and larynx.and larynx.CourseCourse : Progressive, 90% die in 6 : Progressive, 90% die in 6
yearsyears

The hallmark of motor neuron disease is denervation and atrophy of muscle due
to loss of spinal motor neurons (Courtesy of Samar Hasnain)

PBP (Progressive bulbar palsy)PBP (Progressive bulbar palsy)
25% patients starts as PBP.25% patients starts as PBP.
Dysarthria, absent gag reflex. Dysphagia Dysarthria, absent gag reflex. Dysphagia – unable to swallow food/liquids.– unable to swallow food/liquids.
Atrophy of tongue, Jaw jerk brisk or Atrophy of tongue, Jaw jerk brisk or absent.absent.
Pseudo bulbar signs – pathological Pseudo bulbar signs – pathological laughter or cry.laughter or cry.
Weak Respiratory Muscles: breathing Weak Respiratory Muscles: breathing difficulty, aspiration pneumonias, difficulty, aspiration pneumonias,
Respiratory failure in 2-3 yearsRespiratory failure in 2-3 years

Tongue in ALS:Weak with little wastingAt rest & Attempted protrusion

PMA (Progressive muscular PMA (Progressive muscular atrophy)atrophy)
M:F – 4:1M:F – 4:1Slower course, better prognosis in young Slower course, better prognosis in young
onset patients.onset patients.Symmetrical weakness, absent or Symmetrical weakness, absent or
sluggish reflexessluggish reflexesD/D: Motor neuropathyD/D: Motor neuropathy – immune – immune
mediated, paraproteinemia, IgM mediated, paraproteinemia, IgM antibodies against GM ganglioside.antibodies against GM ganglioside.
Focal conduction blocks, sensory Focal conduction blocks, sensory abnormalities on EMG/ NCSabnormalities on EMG/ NCS

PLS (Progressive lateral PLS (Progressive lateral sclerosis)sclerosis)
Rare. Rare.
Pure spastic involvement. 5-6 Pure spastic involvement. 5-6 decadedecade
LL UL bulbar involvement.LL UL bulbar involvement.
- -- -
* No ocular muscle involvement.* No ocular muscle involvement.
* No bladder impairment.* No bladder impairment.

D/DD/D
** Multiple sclerosis (MS)Multiple sclerosis (MS) – optic – optic nerve, cerebellum, bladder, nerve, cerebellum, bladder, sensory features.sensory features.
Diagnostic - CSF and MRI.Diagnostic - CSF and MRI.
**Slow compression of spinal cordSlow compression of spinal cord – – meningioma, syryngomyelia – MRI.meningioma, syryngomyelia – MRI.

D/DD/D
**Lyme diseaseLyme disease – rare, serum antibodies. – rare, serum antibodies.
**HIV myelopathyHIV myelopathy – history/other features – history/other features / serum Antibodies / MRI./ serum Antibodies / MRI.
**B12 deficiencyB12 deficiency – sensory involvement/ – sensory involvement/ Nerve conduction abnormalities, low Nerve conduction abnormalities, low serum b12.serum b12.
** Inclusion body myositisInclusion body myositis – muscle – muscle biopsy biopsy

D/DD/D
Bulbar weaknessBulbar weakness ::
**Myasthenia gravisMyasthenia gravis -- fatigability, serum Ach fatigability, serum Ach receptor antibodies, positive Tensilon receptor antibodies, positive Tensilon test, decremental pattern on EMG/NCStest, decremental pattern on EMG/NCS
**Polio myelitis -Polio myelitis - Acute viral illness, juvenile Acute viral illness, juvenile onset, epidemic, abnormal CSF, positive onset, epidemic, abnormal CSF, positive serum antibodies.serum antibodies.
**Muscular dystrophyMuscular dystrophy - - High CK, muscle High CK, muscle biopsybiopsy

Pathogenesis:Pathogenesis:
Largely unknownLargely unknown
SOD 1 mutation (superoxide SOD 1 mutation (superoxide dysmutase)dysmutase)
Excitotoxic glutaminergic activity Excitotoxic glutaminergic activity and mitochondrial dysfunctionand mitochondrial dysfunction

Treatment : Mainly SupportiveTreatment : Mainly Supportive
RILUZOLERILUZOLE : Anti glutaminergic : Anti glutaminergic
: used in bulbar onset : used in bulbar onset patients patients
: prolongs disease by 3- 4 : prolongs disease by 3- 4 months months
The only specific drugThe only specific drug..

Treatment :Treatment :
SpasticitySpasticity : Baclofen, Tizanidine, : Baclofen, Tizanidine, ValiumValium
Physiotherapy, breathing exercises.Physiotherapy, breathing exercises.Respiratory support :Respiratory support : BIPAP, BIPAP,
tracheostomy, ventilatory supporttracheostomy, ventilatory support
Feeding :Feeding : Nasogastric (NG) tube, Nasogastric (NG) tube, PEG ( Percutaneous PEG ( Percutaneous
endoscopic gastrostomy) endoscopic gastrostomy)


Renowned scientist Stephen Hawking suffers from amyotrophic lateral sclerosis. (Reproduced by permission of
AP/Wide World Photos( (

Thank you.Thank you.

















