29
Application Manual KF - Titration

Application Manual
KF - Titration

1
KF Titration Application Manual
Contents Page
1 Introduction............................................................................................................................................. 12 Main part................................................................................................................................................. 1
2.1 Basics of KF titration ....................................................................................................................... 14.1.1 The chemical reaction.............................................................................................................. 14.1.2 Side reactions .......................................................................................................................... 24.1.3 The influence of temperature and reaction medium ................................................................ 34.1.4 Titration curves ........................................................................................................................ 54.1.5 Volumetry and coulometry ....................................................................................................... 64.1.6 „Setting“ the titration................................................................................................................. 7
2.2 Working methods of KF titration...................................................................................................... 84.2.1 Sample properties.................................................................................................................... 8
2.2.2.1 Working with liquid samples ............................................................................................. 82.2.2.2 Working with solid samples .............................................................................................. 92.2.2.3 Dry-heating of samples using the oven ............................................................................ 9
4.2.2 Adaptation to the sample matrix ............................................................................................ 102.2.2.1 Working with other temperatures.................................................................................... 102.2.2.2 Setting the pH value ....................................................................................................... 11
2.3 Working specifications .................................................................................................................. 124.3.1 Titer determination ................................................................................................................. 124.3.2 Use of single-component reagents ........................................................................................ 134.3.3 Use of two-comonent systems............................................................................................... 14
2.4 Error and their consequences....................................................................................................... 154.4.1 The titration takes too long .................................................................................................... 154.4.2 The titration solution turns brown........................................................................................... 154.4.3 Detected water contents is too low ........................................................................................ 154.4.4 Detected water contents is too high....................................................................................... 164.4.5 Brown titration solution is leaking .......................................................................................... 164.4.6 Titration proceed fast and without stopping ........................................................................... 164.4.7 Wrong output of the result ..................................................................................................... 16
2.5 Validation of the KF titration .......................................................................................................... 164.5.1 Validation scheme and evaluation of general features.......................................................... 174.5.2 Inspection of testing means ................................................................................................... 184.5.3 Tests to be made ................................................................................................................... 19
2.6 KF titration and normative documents .......................................................................................... 212.7 KF titration and quality assurance................................................................................................. 23
Installation Qualification............................................................................................................................... 27

1
1 Introduction
The water contents or the humidity is an important factor for many products, since it influences the product properties or the quality of the products. Butter, for instance, contains up to 16 % of water. If the water content is too high, butter no longer meet the legal regulations. In this way, the microbiological composi-tion is exposed to considerably higher risks. If the water content is too low, the manufacturer simply gives away money. The determination of the water content, in addition to its mere necessity, is in many cases an important commercial factor to reduce costs.
There are countless applications in which the water determination is inevitable: The pourability of prod-ucts, the processing of plastics in injection moulding, or else, tablets will disintegrate if they are too dry, or they will deliquesce if they contain too much humidity. This list could be continued without ever reaching an end.
In this context, the water contents may be located in the ppm range, such as is the case with insulating oil, or in a high percentage range, for instance in the case of alcoholic extracts of natural matters (plants). Karl Fischer titration with its methods can cover this wide sector without any problems.
Karl Fisher reaction was published in 1935 by Karl Fischer as a method of water determination. He used pyridine, iodine, methanol, and SO2 as single-component compounds for his determinations. The underly-ing reaction equation was based on Bunsen’s equation. Stoichiometry of the reaction equation, however, was corrected in subsequent publications.
A titration can only be performed if the end point of the reaction is clearly marked or if the reaction is re-corded in the form of a titration curve and the transformation can be calculated on this basis. In the case of Karl Fischer titration the end of the titration can be seen in the colour change to brown, caused by the io-dine surplus. The own colour of the reaction, however, does not change in the course of the titration. Whereas the first titration clearly shifts from almost colourless to dark brown, the colour shift of many titra-tions may be from dark yellow to brown. This is based on the assumption that a multitude of samples is being titrated in one and the same solution. This is common practice today.
Modern titrators recognise the end of titration in an electrochemical way. In the case of Schott titrators, the indication is done in a bi-amperometric process. A voltage is applied between two platinum pins. As long as water is present in the solution, only iodide is present in the solution, and no current is flowing. As soon as the water is titrated away, the solution contains a small iodine surplus. The oxidation of the iodide to io-dine occurring at the anode, and the reduction of the iodine to iodide occurring at the cathode is reversible and indicated by a flow of current.
2 Main part
2.1 Basics of KF titration
4.1.1 The chemical reaction
The precise nature of the mechanism of Karl Fischer reaction was the subject of tedious discussions. Even the stoichiometry of the reaction was not clear for a long time.
In principle, titration would not be possible with it. Recently, the mechanism has been the object of several investigations which resulted in the following reaction equation:
ROH + SO2 + R´N [R´NH]SO3R
H2O + J2 + [R´NH]SO3R + 2 R´N [R´NH]SO4R + 2 [R´NH]JEquation 1: KF equation according to [1]
Legend:
ROH An alcohol, for instance, methanol, ethanol, ethylene-glycol-mono-ethyl-ether
R´N A basic solution, for instance imodazol (formerly often pyridine)
The investigations of the mechanism show a change in stoichiometry if work is done in other solvents. The addition of other solvents should therefore be limited to 50 volume %.

2
The basic components of the reaction are as follows: Alcohol Base SO2
Iodine
A distinction in 2 fundamental working techniques is made: Either all components are combined within one titration reagent (single-component reagent), or else, alcohol, base, and SO2 are patterned in a solvent component and titrated with iodine in alcohol (two-component reagent). Since, in contrast to the single-component reagent, base and SO2 are present in a surplus in the case of the two-component reagent, and since the system is moreover buffered, the reaction is clearly quicker. To achieve this effect for the single-component reagent, too, an additional auxiliary solvent is added in some cases.
The single-component reagent is mainly used to enable special samples to be dissolved by a variation of the solvent. With many polar samples, formamide is added, whereas chloroform is added with many non-polar samples. It is also possible to add long-chained alkanes or alcohols. Any addition of other solvents to alcohol should not exceed 50 volume %.
Intense investigations show the pH dependency of the reaction, and this is to be expected before the background of the reaction equation. The optimum pH is between pH 6 and 7. If the pH is lower, the reac-tion speed will slow down clearly. And if the pH value is higher than pH = 8.0, side reactions will give the il-lusion of an excessive consumption.
Therefore please note: When determining acids, check the pH value and add imidazol if required.
When determining bases, neutralise using benzoic acid or salicylic acid.
4.1.2 Side reactions
In the given circumstances, KF titration is water selective to a far-reaching extent. Nevertheless, there are of course a number of restrictions caused by a series of side reactions. These side reactions can be clas-sified according to the following types:
a) Reactions producing water
b) Reactions requiring water
c) Redox side reactions of the iodine
d) External water
Some examples will be quoted here. In many cases, the side reactions can be avoided by corresponding working conditions or an appropriate selection of the reagents.
a) Reactions producing water
The best known water-producing reaction is the formation of acetals and ketals. The alcohol required for this reaction and used as the solvent reacts with carbonyl groups (C=0) under formation of water. This wa-ter is also captured in the titration. The titration seems to be endless. The result is a high permanent "drift".
Alcohol Base SO2
Iodine
Single-component reagent 2-component reagent
Alcohol Base SO2
Iodine Alcohol
Solvent Titration agent
Methanol
Solvent Titration agent

3
The formation of acetals or ketals can be avoided, however, if special reagents for aldehydes and ketones are used. An alternative possibility would consist in working at a reduced temperature. In this case, work is done at temperatures below 0°C. The ionic KF reaction proceeds almost unaffected, while the formation of acetals or ketals is clearly slowed down. The lower temperature limit is determined by the viscosity of the pattern and the settlement by crystallisation of side components.
Under certain circumstances, aldehydes may also enter into a bi-sulfite addition with SO2. If the pH value is correctly set, this reaction should be largely suppressed. This reaction would not produce or bind any water, but reduce the available SO2 quantity.
Other reactions involving the formation of water include the reaction of carbonyles with amines to form Schiff’s bases, the formation of enamines, and the esterification of acids with the alcohol of the solvent. If methanol-containing solvents are used, the risk is reduced.
In the case of certain neutralisation reactions with the bases, water giving the illusion of an increased wa-ter contents may be formed.
b) Reactions requiring water
Ester decomposition would be a typical reaction requiring water. A temperature decrease should solve the problem. This problem is hardly of any practical importance, since most esters do not react under the con-ditions of KF titration.
c) Redox side reactions of the iodine
This involves a series of possible reactions. Peroxides react with iodine [1, 2]. Other oxidants such as chlorine, nitrogen oxides, and dichromate have to be reduced before use. Reductives such as ascorbic acid, tin (II)-salts, and mercaptan have to be oxidised prior to titration. [1, 2] contains a description of nu-merous side reactions and possible solutions.
d) External water
External water is the most frequent source of error with KF titration. There are various possible causes for its entrance into the titration cell. As a matter of course, the solvent or the pattern component must be dry-titrated first. This process is referred to as conditioning. If water still enters the titration cell, it is assigned as external water together with the sample.
External water enters the titration cell in one of the following ways: A titration cell is not tight. External particles, for instance, may be stuck in the ground surfaces. Defective O-rings at the screwed connections of the titration cell may be another possibility. The septum for the addition of liquid samples is not tight and worn out. The molecular sieve in the drying tube for pressure equalisation is used up and has to be dried. The pumping system contains damp air. The air for the addition of the solvent should also be dried us-
ing a molecular sieve.
4.1.3 The influence of temperature and reaction medium
The Karl Fischer reaction is a fast reaction. It depends much less on the temperature than a lot of the side reactions. The acetal and ketal formation, for instance, is reduced by a decrease of the temperature to such a degree that a Karl Fisher water determination is still possible in many cases. By suitable cooling facilities, the temperatures can be reduced down to – 30 to – 40 °C. The possible cooling agents include: dry ice with a matching organic solvent, ice/salt mixtures, or the connection of a cryostat to a temperature-controllable titration vessel. The increased duration of the titration with temperatures as low as these re-mains practical if work is done with two-component reagents.
Titration at increased temperatures is mainly used to improve and accelerate the process of dissolving a sample or to extract the water from a sample more speedily. The temperatures can be regulated to40 – 50 °C without any problems. If the temperature is further increased, a cooler is to be used in the posi-tion of the drying tube. The drying tube is then placed on top of the cooler. Temperature equalisation is simply done by using a small, heatable magnetic stirrer which is placed under the titration cell.

4
The titer of the titration agent will change if the working temperatures clearly differ from 20 °C. Literature quotes a titer change of 1 % per 10 °C [2]. In principle, it is recommended to determine the titer under the same conditions under which the sample is titrated.
In addition to the variation of the temperature, the use of different solvents enables an adaptation to the matrix. Considering that the alcohol participates in the chemical reaction, it cannot be fully substituted. The alcohol components used include: methanol, ethanol, 2-chloroethanol, ethylene-glycol-monomethyl ether. Owing to its solubility properties, methanol can be used on a broad basis, but is classified as poisonous (poison class 3). Likewise, some of the alternatives are poisonous, and if used, the required safety meas-ures have to be taken. They are indicated on the reagent bottles (R and S sentences).
The alcohol component should be contained by a minimum of 50 % in the titration cell. Other solvents are added to improve or enable the solubility of samples:
Solvents Application Observations
Formamide Polar substances,sugar, salts, proteines
Please observe hazards information and safety advice! Formamide can cause damage to the unborn child!
Chloroform,dichloromethane
Fats/greases, oils,hydrocarbons
Please observe hazards information and safety advice!
Long-chained alcohols, 1-propanol, 1-butanol, decanol
Fats/greases, oils,hydrocarbons
Please observe hazards information and safety advice!
Alkanes (ligroines,petrol ether)
Fats/greases, oils,hydrocarbons
Please observe hazards information and safety advice!
Ethylene glycol Absorption of gases Please observe hazards information and safety advice!
Acetic acid Amines, inorganiccompounds
Please observe hazards information and safety advice!
Toluene, Xylene Plastics, lubricating fats/greases, oils
Please observe hazards information and safety advice!
The use of formamide and chloroform accelerates titration, but may change the titer in the process. In this case, too, it is generally valid that the titer has to be determined under the same conditions under which the sample is later on titrated.

5
4.1.4 Titration curves
KF titration is controlled by the magnitude of
the current flow between the platinum pins of the electrode [µA] the reagent consumption [ml] the duration of the titration [s]
The current curve is required for the indication of the titration end. In addition, the titration control tries to ti-trate as quickly as possible after a check at the beginning of titration, in order to hit the endpoint of thetitration as accurately as possible when the end of the titration is approaching.
KF titration current curve
0
5
10
15
20
25
0 1 2 3 4 5
Titration agent [ml]
Str
om
[µ
A]
Fig. 2: Current curve of a KF titration
Figure 2 shows an indication curve showing the consumption on the x axis and the current in µA on they axis. The almost completely flat course of the current curve with a sudden rise towards the end of titra-tion is typical. Considering that this curve gives only little information on the reaction, it makes sense in practice to record that time versus consumption. On the x axis, the time is recorded, whereas the y axis shows the consumption as a function of time (Figure 3).
KF titration curve
0
1
2
3
4
5
20 40 60 80 100 120 140
Titration duration [s]
con
sum
pti
on
[m
l]
Fig. 3: Current curve of a KF titration
As you can see from the curve, approx. 95 % of the titration agent has been dosed within approx. 50 seconds after approx. 20 seconds of solving or extraction time. In the course of the remaining 70 seconds, titration was performed carefully and in minute quantities in order to achieve a result as accu-rate as possible. Titration will end as soon as no more reagent is being titrated within a defined period of time (mostly 10 or 20 seconds).
The following criteria are indicative of a "good" titration curve:Steep rise means fast titration (too flat = too slow) A round, but not too steady bow without bends (too round = too slow; bend = overtitration) A flat course, parallel with the x axis (further rise = drift).
Flat, parallel to x-axis
Not too sharp bend
Strong slope
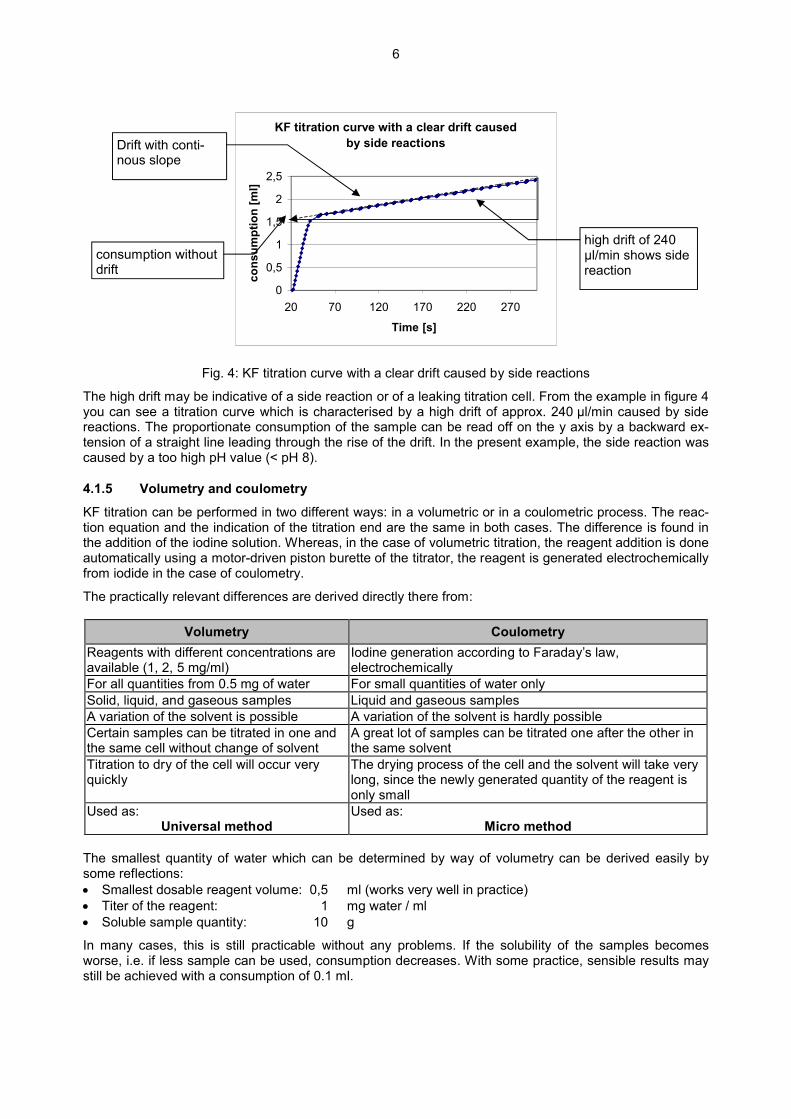
6
KF titration curve with a clear drift caused by side reactions
0
0,5
1
1,5
2
2,5
20 70 120 170 220 270
Time [s]
co
ns
um
pti
on
[m
l]
Fig. 4: KF titration curve with a clear drift caused by side reactions
The high drift may be indicative of a side reaction or of a leaking titration cell. From the example in figure 4 you can see a titration curve which is characterised by a high drift of approx. 240 µl/min caused by side reactions. The proportionate consumption of the sample can be read off on the y axis by a backward ex-tension of a straight line leading through the rise of the drift. In the present example, the side reaction was caused by a too high pH value (< pH 8).
4.1.5 Volumetry and coulometry
KF titration can be performed in two different ways: in a volumetric or in a coulometric process. The reac-tion equation and the indication of the titration end are the same in both cases. The difference is found in the addition of the iodine solution. Whereas, in the case of volumetric titration, the reagent addition is done automatically using a motor-driven piston burette of the titrator, the reagent is generated electrochemically from iodide in the case of coulometry.
The practically relevant differences are derived directly there from:
Volumetry Coulometry
Reagents with different concentrations are available (1, 2, 5 mg/ml)
Iodine generation according to Faraday’s law,electrochemically
For all quantities from 0.5 mg of water For small quantities of water onlySolid, liquid, and gaseous samples Liquid and gaseous samplesA variation of the solvent is possible A variation of the solvent is hardly possibleCertain samples can be titrated in one and the same cell without change of solvent
A great lot of samples can be titrated one after the other in the same solvent
Titration to dry of the cell will occur very quickly
The drying process of the cell and the solvent will take very long, since the newly generated quantity of the reagent is only small
Used as:Universal method
Used as:Micro method
The smallest quantity of water which can be determined by way of volumetry can be derived easily by some reflections: Smallest dosable reagent volume: 0,5 ml (works very well in practice) Titer of the reagent: 1 mg water / ml Soluble sample quantity: 10 g
In many cases, this is still practicable without any problems. If the solubility of the samples becomes worse, i.e. if less sample can be used, consumption decreases. With some practice, sensible results may still be achieved with a consumption of 0.1 ml.
high drift of 240 µl/min shows side reaction
consumption without drift
Drift with conti-nous slope

7
The following table shows the relationships between water contents, sample quantity under the above conditions:
Sample quatity[g]
Consumption[ml]
Titer[mg/ml]
Water content[ppm]
Water content[%]
0.05 10 5 1 000 000 1000.1 5 5 250 000 251 5 5 25 000 2.51 5 2 10 000 1.01 1 2 2 000 0.21 1 1 1 000 0.11 0.5 1 500 0.05
10 0.5 1 50 0.005
So the smallest water contents which can be titrated volumetrically are approx. 50 ppm (with TL alpha plus 5 10 ppm), the highest is 100 %.
4.1.6 “Setting” the titration
In principle, KF titration requires three different "methods":
Titer determination to establish the exact concentration of the titration agent. By way of a titration us-ing an accurate standard, the exact concentration of the titration agent is determined. The concentra-tion of the titration agent is subject to changes over time. The titer determination should be repeated approx. every week.
Blank value: Determination for working with the oven or for longer opening times of the titration cell. A blank value is particularly required for samples which are heated to dry using an oven. As a result of the gas flow through the oven, atmospheric water is entrained, thus leading to a blank value. If the sample is heated to dry for approx. 10 minutes, the blank value of approx. 10 minutes should be de-ducted from the titration result.
Sample titration for the determination of the water contents of the samples.This is the actual method for determining the water in a sample.
These "methods" are distinguished by the formula, but not by the setting of the parameters. The settings of the parameters involve:
Breaking current in [µA]; if this current is flowing between the indicator electrodes, the end criterion has been reached.
The default setting is 20 µA. Values from 10 – 30 µA make sense. Switch-off time in [s]; if the breaking current has exceeded this switch-off time, the titration will be
ended. The default setting is 10 s. Time spans between 0 – 10 s make sense. With a few special appli-
cations, e.g. applications involving the introduction of gases, other settings may make sense. Drift in [µl/min]; titration will be ended as soon as the addition of the titration agent falls short of the set
drift value. The default setting is 10 µl/min. Values from 3 – 30 µl/min make sense. Applied voltage between the platinum electrodes in [mV]: As a rule, this value does not have to be
changed. The default setting is 100 mV. Values between 20 – 150 mV make sense. An adaptation to the sample matrix is made possible by the extraction time in [s], to ensure a complete
water yield of the sample. Values between 10 and 600 s make sense. An adaptation to the sample matrix is made possible by the maximum duration of the titration in [s]. In
this way, a KF titration can also be made possible with a high drift. A titration should not last longer than 10 minutes.
The min. titration duration in [s] will accelerate the extraction of the water, without there being the risk that the titration is cancelled prematurely. The extraction behaviour is improved by a continuous addi-tion of the iodine.
For all of these parameters, meaningful settings are defaulted on the TitroLine KF Titrator, so that hardly any change of these values is required. All settings should possibly be the same for a certain type of ap-plication, since otherwise the results cannot be compared exactly. The titer may change by differing set-tings of the parameters.

8
2.2 Working methods of KF titration
4.2.1 Sample properties
The working methods of KF titration are highly dependent on the properties of the sample. Working with gaseous, liquid, low-viscous, high-viscous, and solid samples is distinguished by: Sample input Solubility Sample quantity Sample volume Water yield
2.2.2.1 Working with liquid samples
In general, liquid samples are injected through a septum into the titration cell using a one-way syringe. The size of the syringe depends on the sample volume. Syringes from 1 ml to 20 ml can be used. The needles of these syringes have a diameter from 0 .6 to 1.2 mm. The thinner needles are only available for low-viscous samples, whereas the thicker needles should be used for samples with a higher viscosity. The thicker the needle, the higher will be the degree of the effect on the septum. In this case, it has to be re-placed sooner. The length of the needles is between 50 and 90 mm. In this way it can be ensured that no drops remain adhered to the walls. The needles should not be immersed into the solvent.
Particularly high-viscous samples, such as oils, are transferred into the titration cell without a needle, di-rectly, and without the use of a septum.
The septum consists of a polymer which has to have the three following major properties: The proper size. It has to close again directly after being pierced. This point can only be realised with certain restrictions. The silicon discs used have to be replaced on a
regular basis.
We recommend the use of original spare parts.
As a rule, the samples have to be weighed in using an analytical weighing-balance.The working process is as follows: Put a 150 ml beaker glass on the balance. Draw up the liquid sample into the syringe. Put the needle protector on again. Place the syringe with the needle up into the beaker glass. Gauge the weighing-balance. Carefully take the syringe from the balance, then remove the needle protector. Inject the sample through the septum into the titration vessel. Remove the syringe from the septum, put on the needle protector again. Place the syringe with the needle up in the beaker glass on the weighing-balance. Read off the weight as a negative number on the weighing-balance, then enter it as an absolute value
on the titrator.
Working with sample volumes is unusual. When drawing up the sample into the syringe, please proceed carefully and slowly to avoid air (including air-borne humidity) being unnecessarily drawn through the samples.
Figure 5 shows the sample opening with the septum and a syringe:
SeptumNeedle
Cover
Plug
Fig. 5: Sample input through septum

9
2.2.2.2 Working with solid samples
The following working methods are common for solid samples: Direct sample input by opening the plug in the cover of the titration vessel Use of a solid lock
Direct sample input by opening the plug is certainly the simplest way of introducing solid samples in the KF titration vessel. During the opening time of the cell, humidity of the ambient air may lead to a blank value. This blank value, however, can be determined by way of a separate titration and then be automati-cally taken into account in arithmetic form. The smaller the water quantity to be determined, the less fa-vourable is this method of sample addition. As a rule, the blank value should not be greater than the water quantity to be determined.
The samples may be weighed with a weighing boat made of glass or aluminium to be transported to the ti-tration vessel. After weighing, the samples are no longer touched by hand.
Using the solid lock, it is possible to re-condition after the titration vessel was opened, prior to introducing the sample in the solvent. The sample quantity is limited to small sample quantities in the range of one gram. The working sequence is based on the following scheme: The sample is weighed inside the solid lock. Pull the outer part of the solid lock over the inner part containing the sample, until the part containing
the sample is fully closed. Open the titration vessel, then place the outer part of the solid lock with the built-in NS 19 ground sur-
face in the opening. Start conditioning. Push down the inner part, so that the sample can be dissolved away. Start titration with a longer extraction time for solving the sample. As an alternative, a minimum titration
duration can be specified.
Working with the solid lock may involve a higher drift.This can be taken into account accordingly in the titration methods.
2.2.2.3 Dry-heating of samples using the oven
In many cases a direct determination is impossible because: The samples are insoluble (plastics). The samples release the water only in heat. The samples enter into side reactions.
The samples are placed in the oven, heated up, and the water being released is transferred to the titration vessel by dried gas. In this process, the major quantity of water is released at the beginning. The following graph shows the way in which the water contained in the sample is released. At the end of titration, the added quantity is limited by the drift value. On the graph, a side reaction is indicated by a high residual drift. The x axis shows the duration of the titration, the first y axis shows the drift, the second y axis shows the consumption.
After approx. five minutes, the major part of the water is released. We recommend a dry-heating time of 10 minutes. This time is set as a waiting time on the titrator. Subsequently, the water quantity absorbed in the solvent is titrated within a short period of time. If the preceding drift is not reached again, or almost not reached again, this is indicative of a side reaction.

10
With the titration in the graph it was proven that thermal decomposition leads to the generation of formal-dehyde, which reacts with the methanol in the titration cell to acetal, with water being separated.
Water yield
0
50
100
150
200
250
300
1 6 11Time [min]
Wa
ss
era
bg
ab
e [
µl/
min
]
0
0,2
0,4
0,6
0,8
1
1,2
1,4
Ve
rbra
uc
h [
ml]
The set-up and the connection of the oven will be illustrated by the figure below:
The gases to be preferred include inert gases, such as nitrogen, since side reactions, e.g. oxidations, cannot occur.
The set-up and the connection of the oven will be illustrated by the figures below.
4.2.2 Adaptation to the sample matrix
The sample matrix has a major influence on the water determination. Direct water titration is not possible in all cases. Some samples release the water only very slowly or, under certain circumstances, do not re-lease it at all. The possibilities for getting the water out of the samples in a definable form include, for in-stance: The variation of the temperature Comminution Internal or external extraction Variation of the solvent Setting the pH value Oven
2.2.2.1 Working with other temperatures
A variation of the temperature of KF titration is simple method to: Slow down temperature-dependent side reactions in such a manner that the KF titration can run off
without disturbance Improve the input of volatile substances Accelerate the extraction of the water contained in samples at a higher temperature Accelerate titration
At low temperatures, many organic reactions go off clearly slower. KF titration is not that temperature-dependent. This means, for instance, that many ketones and aldehydes can still be titrated which would tend to disturbing side reactions at normal temperature. Cooling may be done by the external application of ice or by organic solvents with dry ice.
One problem of cooling is the condensation of atmospheric air during the process of opening the cell. It is therefore recommendable to work with a syringe through the septum or, in the case of solid samples, with a solid lock.
N2 Drying Oven Titration cell

11
Working at raised temperatures is opposed to this. The high air temperature enables a better extraction of water from the samples. Moreover, the reaction is speeded up. The simplist way of heating is the use of a heatable magnetic stirrer. Working without an additional cooler is possible in a temperature range from 40 – 50 °C. Methanol boils at approx. 65 °C. A reflux cooler can be set into the NS 14.5 ground opening of the dry air capillary. Subsequently, the desiccant tube is set on top of the cooler.
Pre-drying of the cooler in the drying oven is not required. If overtitration occurs during conditioning, for in-stance caused by a short-term removal of the electrode from the solution, the entire titration vessel plus the cooler can be "rinsed" with this solution. Particular care should be taken in this process. All apertures are to be closed (with plugs). Please observe all safety regulations applicable to the handling of the chemicals being used and the work in the laboratory. After the "rinsing" process, the plugs have to be re-placed by desiccant tubes!
Particular care should be taken to ensure the thorough drying of the desiccant. Best results were achieved using a molecular sieve of the 0.3 nm size, which was dried in the drying oven at approx. 250 °C for at least three hours. A possibly present indicator reacts clearly too late. Replacing and drying on a regular basis will ensure a low drift inside the titration vessel.
2.2.2.2 Setting the pH value
In Chapter 2.1.1 point, reference is already made to the pH-dependency of KF titration. The KF reaction requires a base to be functioning at all. The original composition used by Karl Fischer included pyridine, allegedly because it was just standing on the shelf. Pyridine has a pKs value of 5.25 and thus leads to a solvent which has a too low pH value for a fast reaction. Imidazol, which is often used today, has a pKs value of 6.95, and this means that a favourable pH value for a speedy titration is set. Other bases with comparable pKs values are also possible. The use of imidazol as a base for KF titration is under patent protection.
If the pH value is too high, side reactions will lead to incorrect results. With a too low pH value, titration will take too long. It is therefore essential to estimate of, or better verify, the pH value for a specific type of sample. Most pH electrodes contain an aqueous electrolyte which may give the illusion of an incorrect wa-ter content of the sample.

12
2.3 Working specifications
4.3.1 Titer determination
Method No. 1
Application Titer determination of KF titration agents
Equipment TitroLine KF Titrator or TitroLine alpha TM KF Titration Stand (TL KF scope of delivery) TZ 1770 Titration vessel (TL KF scope of delivery) TZ 1106 Double platinum electrode (TL KF scope of delivery) TZ 1721 Solid lock 5 ml one-way syringe with needle length approx. 60 mm plus needle protector 3 or 4-digit weighing-balance Beaker glass
Reagents Titration agents: Single- or two-component titration agents Pattern: Methanol, Composolver, or solvent depending on the titration agent Standard: Depending on the standard, please refer to table
Sample preparation
Sample input Through the septum using a syringe and needle Using a solid lock Directly, by opening the plug Connection of an oven Introduction of gas
Description The “titer determination” method is selected, and the calculation factor is checked.Liquid standard:The standard is weighed in the one-way syringe (water in a microliter syringe). The consumption to be expected should be approx. 5 ml.With reagent 5 mg water/ml this would be:Standard 10 mg water / ml: 2.500 gStandard 5 mg water / g: 5.000 gTare the weighing balance, transfer the sample through the septum in the titration vessel, then weigh the empty syringe again. Enter the weight on the titrator. Start titration.
Solid standard:Tare the weighing-balance. The standard (approx. 160 mg of sodium tartrate-hydrate) is weighed in the inner part of the solid lock using the weighing-balance. Carefully push the inner part of the solid lock into the outer part until the standard disappears completely in the lock and cannot fall off. The closed solid lock is then placed in the opening of the titration vessel, start conditioning. Upon completion of the conditioning process, push the inner part fully down so it can dissolve. Place the weighed-in quantity in the titrator, then start titration.
Calculation Liquid standard:Concentration titration agent [mg/ml] = weighed-in quantity [g] * standard [mg wa-ter/g] * factor 1 / ((consumption [ml] – blank value [ml]) * factor 2)
Water:Concentration titration agent [mg/ml] = weighed-in quantity [mg]((consumption [ml] – blank value [ml]) * factor 2)
Sodium tartrate-hydrate:Concentration titration agent [mg/ml] = weighed-in quantity [g] * 1000 * 0.1566((consumption [ml] – blank value [ml]) * factor 2)
Parameter End value: 20 µASwitch-off time: 10 secPotential: 100 mV

13
4.3.2 Use of single-component reagents
Method No. 2
Application Titration with single-component reagents
Equipment TitroLine KF Titrator or TitroLine alpha TM KF Titration Stand TZ 1770 Titration vessel TZ 1106 Double platinum electrode TZ 1721 Solid lock 5 ml one-way syringe with needle length approx. 60 mm plus needle protector 3 or better 4-digit weighing-balance Glass beaker
Reagents Titration reagent: Single-component reagent Pattern: Methanol, composolver or Combi solvent and mixtures
Sample preparation Solid sample have to be put into small pieces, liquid sample are put into a syringe, samples with higher viscosities require a syringe without a needle.
Sample input Through the septum using a syringe and needle Using a solid lock Directly, by opening the plug Connection of an oven Introduction of gas
Description The „sample titration“method is selected, and the calculation factor is checked.Liquid and high-viscous standards:Weigh sample with syringe. Tare balance, put sample through septum into the titra-tion vessel and weigh back empty sysringe, take the negative weight of the sample and start titration.
Solid standard:Tare the weighing-balance. The sample in the inner part of the solid trap is weighed and the weight is noted. Carefully push the inner part of the solid lock into the outer part until the standard disappears completely in the lock and cannot fall off. The closed solid lock is then placed in the opening of the titration vessel, start condition-ing. Upon completion of the conditioning process, push the inner part fully down so it can dissolve. Place the weighed-in quantity in the titrator, then start titration.
Calculation % Water = (consumption [ml] – blank value [ml]) * 100 /(weighed-in quantity [g] * 1000)
Parameters End value: 20 µAEnd point delay: 10 secPotential: 100 mV
Notice Make titre under the same conditions as sample titration (same solvent).
For fats etc. add Chloroform (up to 50%) or other fat solving media.
For polar sample add Formamide (up to 40%).

14
4.3.3 Use of two-component systems
Method No. 3
Application Titration with two-component systems
Equipment TitroLine KF Titrator or TitroLine alpha TM KF Titration Stand TZ 1770 Titration vessel TZ 1106 Double platinum electrode TZ 1721 Solid lock 5 ml one-way syringe with needle length approx. 60 mm plus needle protector 3 or better 4-digit weighing-balance Glass beaker
Reagents Titration agent: two-component agent Pattern: Two-component agent ATTENTION: Solvent contains components, which are necessay for the
chemical reaction. Only use recommended combinations!
Sample preparation Solid sample have to be put into small pieces, liquid sample are put into a syringe, sample with higher viscosity required a syringe without needle
Sample input Through the septum using a syringe and needle Using a solid lock Directly, by opening the plug Connection of an oven Introduction of gas
Description Liquid and high-viscous standards:Weigh sample with syringe. Tare balance, put sample through septum into the ti-tration vessel and weigh back empty sysringe, tale the negative weight of the sample and start titration.
Solid standard:Tare the weighing-balance. The sample in the inner part of the solid trap is weighed and the weight is noted. Carefully push the inner part of the solid lock into the outer part until the standard disappears completely in the lock and cannot fall off. The closed solid lock is then placed in the opening of the titration vessel, start conditioning. Upon completion of the conditioning process, push the inner part fully down so it can dissolve. Place the weighed-in quantity in the titrator, then start titration.
Calculation % Water = (sonsumption [ml] – blind value [ml])* 100 /(weighed-in quantity [g] * 1000)
Parameters End value: 20 µASwitch-off time:10 secPotential: 100 mV
Notice Make titre under the same conditions as sample titration (same solvent).
Additions of other solvents are possible only in limited amounts.
The titration is very fast. Sample has to be solved before the start of the titra-tion. In case of samples with slow water delivery stop time has to be set to longer times.

15
2.4 Error and their consequences
4.4.1 The titration takes too long
If the titration takes too long (> 5 minutes), something is not in order in most cases. The causes may be of a quite trivial nature. The following list shows causes for an excessive duration of titration and provides in-formation on how to detect and eliminate the individual causes.
Wrong solventIn particular with the two-component reagents, it happens from time to time that, for instance, methanol alone is used instead of a correct solvent including the required reaction partners. KF titration cannot take place.
Cell open or leaking
The drift is extraordinarily high. If its general value is around 4 µl/min, and suddenly it is in the range of 10 µl/min, this is a frequent cause of error.
Titration agent used up
Trivial, yet not uncommon. The dark brown bottles and the light-protected units make it difficult to see whether there is enough reagent contained in the bottle or in the unit.
Side reactions
In addition to the actual KF reaction, a side reaction is taking place which is also consuming titration agent. Towards the end, the drift remains constant on a certain value, for instance at 20 µl/min.
Chemicals too old
A “best before” date which should not be exceeded is printed on most reagents. If their use cannot be avoided, the titration should be observed closely.
No more drying effect of desiccantThe molecular sieve should be re-dried on a regular basis (> = 250 °C over more than three hours). Used molecular sieve makes itself noticeable by an above-normal drift (e.g. 6 instead of 4).
4.4.2 The titration solution turns brown
If the titration solution turns brown, one of the following problems is present as a rule: Overtitration
The titration reaches the end point too early, and therefore overtitration occurs. In order to determine the overtitration quantity, use the syringe to introduce drops of standard through the septum until a dis-coloration takes place. Count the drops, then determine the water contents for the drop number sepa-rately.
Microtip has come offThe titration reaches the end point too early, and therefore overtitration occurs. In order to determine the overtitration quantity, use the syringe to introduce drops of standard through the septum until a dis-coloration takes place. Count the drops, then determine the water contents for the drop number sepa-rately.
No water in sampleIf the titration begins with no water being contained in the sample, (insignificant) overtitration will occur as a matter of course.
4.4.3 Detected water contents is too low
There are many causes for incorrect titration results. Below are just a few examples of causes of errors.Wrong titer
The titer was not determined correctly or has changed.
Wrong reagentThe new reagent has a different factor, or it is a different titration agent.
Sample not dissolved (or only partially)If the sample is not fully dissolved, then only a part of the water can be determined.
Sample not comminutedIf samples are not disintegrated the water in the sample may often not be released to a sufficient degree.
Titration conditions are not idealUnder non-optimum titration conditions (polarity, solvent, pH value, temperature), it may e.g. happen that too little water is found in the sample.

16
4.4.4 Detected water contents is too high
Side reactionsA side reaction will always lead to an excessive value (please refer to the side reactions chapter).
Wrong titerThe titer was not determined correctly, or it has changed.
Wrong reagentThe titer was not determined correctly, or it has changed.
Sample not homogenousA part of the sample with a higher water contents may have been taken.
Incorrect formulaThe results may be incorrect owing to wrong blank values, titer. Is “sample titration” actually the current method? An incorrect result unit was selected.
4.4.5 Brown titration solution is leaking
LeakageAfter cleaning a unit it may happen from time to time that the hoses are not tightened firmly enough. A thread may have been damaged as a result of an excessive tightening of the hoses.
Microtip offThe micro titration tip with the valve is only plugged on the hose. It may have been pushed out, e.g. by clogging. The risk of clogging is particularly high after longer periods without operation.
Overpressure or vacuum within the systemIf the openings in the titration vessel are clogged, a vacuum may build up in the titration vessel during the evacuation process. Likewise, an overpressure may occur during the dosage of the solvent or the ti-tration agent.
4.4.6 Titration proceeds fast and without stopping
With two-component system: no solventWith two-component reagents the solvent has to contain the SO2, base, and alcohol components, oth-erwise the KF reaction cannot take place. Please make sure that the matching components are being used.
Excessive sample quantityThe sample contains so much water that the consumption is too high. The titration should be repeated with a smaller sample quantity after sucking the sample off.
Excessive water contentsThe sample contains so much water that the consumption is too high. The titration should be repeated with a smaller sample quantity after sucking the sample off.
Contaminated solventsThe solvents contain so much water that the conditioning process consumes too much reagent and lasts too long. The solvents should be pre-dried. This also improves their water extraction properties.
4.4.7 Wrong output of the result
Incorrect formulaAn incorrect output unit was selected.
Wrong methodInstead of the sample titration mode, titration was performed in the blank value or titer determination mode.
2.5 Validation of the KF titration
The sounding of the term might imply truth. This would mean that “validation” would be supposed to en-sure the veracity of an analysis result. However, Latin “validere” means “to correspond”, and this leads to the logic, official definition, DIN EN ISO 8402:
“Confirmation on the basis of an investigation and by the provision of an objective proof of the fact that the special requirements imposed on a particular, intended use are met".
Proof: provable information based on facts gathered by way of observation, measurement, a test, or in a similar form.

17
4.5.1 Validation scheme and evaluation of general features
The validation of a method requires a series of validation features which are identical for all methods. The individual items, however, are not of the same importance in each case. So it is, for instance, that the de-tection limit may certainly be particularly significant with regard to a method of trace analytics, whereas it is most likely to be irrelevant with regard to the contents determination in the high % range. In the follow-ing table, the validation features are evaluated including comments.
Validation feature Importance Significance for titration
Accuracy ++ the strong point of titration
Correctness ++ to be evidenced for each method
Traceability + as a rule, no sample preparation is necessary
Linearity + titration is a method with a high degree of linearity. The con-centration range being used should have been verified.
Selectivity o usually of no importance
Robustness ++ importance for the applicability to other methods
Detection limit o usually omitted
Determination limit o The range in which samples occur is defined and checked. As a rule, however, the sample is adapted to achieve a uniform consumption.
Titration analysis methods
Chemical reaction Reagent and dosage Indication
Applicability Titer stability of the titration agent Sensor functions according to specifications
Stoichiometry Accurate dosability with accurate volumes of the burette
Sensor is suitable for the indication of the chemical transformation (selectivity, sensi-tivity)
Reaction speed Accurately known composition and contents of the reagent
End of the chemical reaction
Side reactions Equivalent transformation can be read off or calculated from the curve
Chemical reaction Reagent and dosage Indication
Reagent Research andpublication
Ensures composition
Manufact. Patents Ensures durability
Device Experience Ensures accurate dosing Ensures indication function
Applications Declaration of conformity Declaration of conformity
Manufac-turer
Manufacturer’s test certificate Manufacturer’s test certificate
User Validation Titer determination Curve analysis
Inspection of testing means Inspection of testing means
18
4.5.2 Inspection of testing means
The manufacturer of a titration device warrants the proper functioning of his device. He verifies this, for in-stance, on the bases of the criteria contained in the appropriate tables (table 4 to 8). The user can adopt these features for his own testing-means inspection or make a selection which he considers useful for his own purposes.
Table 4: Visual inspection
Display Check segments: completeness, brightness, uniformity; check special characters
Plug connec-tions
Corrosion, solidity, bending, completeness, damage to the plug casings
Switches + pots Corrosion, smooth running, latching and end positions, mechanical stability,inscriptions
Casing Damage, impaired function
Cylinder, piston Damage, wear and tear, dull glass, glass surfaces (interior and exterior)
Piston rod Corrosion, damage
Seals Liquid leakage, cracks, deformation
Hoses Bends, damage, connection / threading, liquid leakage, air intake
Internal structure General condition
Printed boards Damage, in particular corrosion of the printed circuits, the components, the soldered points, the plugs
Functioning -mechanical sys-tem
Corrosion of and damage to the driving system, especially the spindle, play of the me-chanical system
Table 5: Interfaces
RS-232-C Functional check: Transmission of data to a test PC using terminal program
others Practical functional check, e.g. SGH interface with connected SGH device
Recorder output Measurement of 3 voltages, distributed across the range
Controller input Practical functional check with the corresponding device
Stirrer connection Practical functional check with connected stirrer
Centronics Connect printer, transmit 5 values
Table 6: Electronics
Measurement amplifier Inspection and readjustment according to alignmentinstructions
Controlling behaviour Visual inspection of the safety-relevant connections
Electrical safety Measurement based on standards
Table 7: Dialogue systemKeyboard function Menu scrolling, check of the function keys
Parameter input Menu scrolling with a stop at 5 positions previously de-termined at random. At these positions: Input of meaning-ful parameters. Check whether the values/characters dis-played correspond to those entered.
19
Table 8: Additionally with QA inspections
Certificate A quality certificate according to DIN 55350-18-4.2.2 is issued. This document can be used in quality management systems according to DIN EN ISO 9000 ff. The certificate must only be issued by persons being nominated by the management and the quality management department.
Spindle travel To be measured in three points using an altimeter: 10 %, 50 %, 100 %,variance comparison
Spindle force Measurement of the maximum in a force-sensing device
pH/mV ranges Variance comparison of the measurement deviation in 3 points each
Temperature range
as pH/mV range
Dead-Stop range as pH/mV range
Volume accordingDIN
Weighing with water according to DIN 12 650, Part 5, at 10 %, 50 %, 100 %,variance comparison
With regard to titration, the accurate dosage of the reagent is unquestionably the most important point. This is checked according to DIN 12650, Part 6. In the course of three dosing processes with approx. 10 %, 50 %, and 100 %, each value must not deviate by more than 0.3 % (or as per manufacturer’s speci-fication) from the desired value, referred to the total volume of the cylinder of the burette.
The new ISO 8655 (under preparation) requires 10 identical volumes each for nominal volume, 50 % nominal volume and the smallest selectable volume or 10 % of the nominal volume. The correctness of the volume with a 10 ml burette has to be ± 0.20 %, the precision has to be ± 0.10 per cent of the com-mand volume
The difference is found in the detail: whereas none of the single values must drop out according to the DIN, the news ISO makes weighing across a Gauss curve in which an individual value actually may be a "maverick".
Considering that most of the motor piston burettes do not have to be calibrated, a volumetric inspection according to these norms is only seldom required. As a matter of experience, visual inspections at shorter intervals make sense. The more comprehensive volumetric inspection should be performed once per year or at shorter intervals if the device is subject to great load or if a deviation has been detected.
A practice-oriented alternative (i.e. not according to any normative regulation) consists in a titer determina-tion using a standard or a titrimetic standard in which different titration volumes are used. In this process the volumes are selected in such a way that the same quantity range as with the samples is recorded and, at the same time, different positions of the piston burette are present.
In this way the electronics and many other items including the sensor are inspected automatically as well. The individual elements have a different influence on the different types of titration. For this reason, valida-tion is discussed at this point on the basis of two examples. If specific particularities of the testing-means supervision are of any significant importance, a special reference will be made.
4.5.3 Tests to be made
Test list including example:
Procee-ding
Elements What has to be done
Precon-ditions
Reactions A sample is titrated, the titration curve is analysed. Chapter XX shows the major fea-tures of a Karl Fischer titration curve. If the expected contents are smaller or greater than the first result, one has to reckon with a side reaction.
Reagent anddosing
A multiple determination (e.g. 10-fold) is performed using a standard, a titrimetric standard (di-sodium-tartrate dihydrate), or water (with microliter syringe, weigh if nec-essary). If different quantities are being used, which cover different burette volumes as well, this means that the proper functioning of the titrator is largely ensured. An additional testing-means supervision is only required if problems occur or if a basic inspection of the titrator is to be performed.
20
Procee-ding
Elements What has to be done
Precon-ditions
Tightness of the ti-tration vessel
The titration vessel has to be tight since the air contains a considerable amount of humidity. This is indicated by the drift. The drift should be less than 5 µg of water per minute. The drift will be greater if the oven is being used. The drift can be read off di-rectly on the device, or it is easily obtainable by waiting for one hour after conditioning (drying of the solvent), followed by a start of a titration.The drift is calculated as follows:Drift [ µg/min] = consumption [ml] * titer [mg water/ml] * 1000 / 60 [min]
Indication The proper functioning of the indication can by be established easily:
Remove the double platinum electrode from the titration vessel
Start titration: no current is flowing, or no voltage is being indicated
Use a metal office clip to short-circuit the two platinum pins: a current is flowing, and titration will disrupt after the switch-off time.
If this test is functioning, the indication system is working properly.
Validation Accuracy Multiple determination of a sample (e.g. 10-fold),determination of mean value and standard deviation
Correct-ness
In the course of each determination, a defined quantity of water (or standard) is added several times to the sample. This quantity must be retraced completely. Mean value and standard deviations of the individual retraced quantities are noted. Less water is indicative of an incomplete transformation. A higher water contents indicates a drift or side reactions..
The tests for linearity inspection are analysed. The straight line must show a slope (if the command consumption is recorded versus the actual consumption) of approx. 1.00. The straight line must lead through the zero point. The correlation co-efficient should be close to 1.00. Deviations indicate a drift, side reactions, or in-complete transformations.
Comparison to the standard the water contents and properties of which are known and which behaves identically to the sample. The standard has to be certified.
Retracing In the course of each determination, a defined quantity of water (or standard) is added several times to the sample. This quantity must be retraced completely. Mean value and standard deviations of the individual retraced quantities are noted.
Other samples with a known contents are added to the sample with a known con-tents. This sample quantity must be the retraced completely.
Linearity A number of samples with an increasing contents is titrated. In this process, for in-stance, 10 titrations are performed for each contents. The smallest and the greatest water contents should correspond to the practical requirements. The straight line must show a slope (if the command consumption is recorded versus the actual con-sumption) of approx. 1.00. The straight line must lead through the zero point. The cor-relation coefficient should be close to 1.00. Mean value and standard deviation of the individual titrations are calculated.
Selectivity Karl Fischer titration is selective for water. The method is tested for almost all types of samples. Side reactions have to be excluded (correctness, linearity, retracing).
Robust-ness
Robustness may be determined in the form of an inter-laboratory test.
All the relevant paramters of Karl Fischer titration are varied:Electrode, solvent quantities, temperature, steering speed, titration paramters,titration device
Detection limit
The detection limit is no criterion in contents determination. It can be defined similar to the determination limit.
Determi-nation limit
The determination limits result from linearity inspection. The smallest and greatest sample volumes which are located inside the linear range and meet the required standard deviation are defined as determination limits.

21
2.6 KF titration and normative documents
KF titration is contained in numerous normative documents. The following table will give you an overview of these standards.
Document number Title
BS EN ISO 10101-1 NATURAL GAS – DETERMINATION OF WATER BY THE KARL FISCHER METHOD – PART 1. INTRODUCTION
BS EN ISO 10101-2 NATURAL GAS – DETERMINATION OF WATER BY KARL FISCHER METHOD – PART 2. TITRATION PROCEDURE
BS EN ISO 10101-3 NATURAL GASES – DETERMINATION OF WATER BY THE KARL FISCHER METHOD PART 3. COULOMETRIC PROCEDURE
DIN 10252 ANALYSIS OF TOBACCO AND TOBACCO PRODUCTS; DETERMINATION OF WATER CONTENT; KARL FISCHER METHOD
DIN 53715 DETERMINATION OF WATER CONTENT OF PLASTICS BY THE KARL FISCHER METHOD
DIN 53979 TEST OF AIDS FOR DRY-CLEANING; DETERMINATION OF THE WATER CONTENT ACCORDING TO THE METHOD OF KARL FISCHER
DIN EN ISO 10101-1 NATURAL GAS – DETERMINATION OF WATER BY THE KARL FISCHER METHOD – PART 1. INTRODUCTION
DIN EN ISO 10101-2 NATURAL GAS – DETERMINATION OF WATER BYTHE KARL FISCHER METHOD – PART 2. TITRATION PROCEDURE
DIN EN ISO 10101-3 NATURAL GAS – DETERMINATION OF WATER BY KARL FISCHER METHOD – PART 3. COULOMETRIC PROCEDURE
DS PD 3201 DETERMINATION OF WATER BY THE KARL FISCHER METHOD. DETERMINATION OF WATER IN KETONES
EN 60814 (BS) INSULATING LIQUIDS – OIL-IMPREGNATED PAPER AND PRESSBOARD –DETERMINATION OF WATER BY AUTOMATIC COULOMETRIC KARL FISCHER TITRATION
IEC 60814 DETERMINATION OF WATER IN INSULATING LIQUIDS BY AUTOMATIC COULOMETRIC KARL FISCHER TITRATION
IP 356 WATER IN CRUDE OILS BY VOLUMETRIC KARL FISCHER TITRATION ISO 760 DETERMINTN.WATER-KARL FISHER METHOD (GENERAL METHOD)ISO 10101-1 NATURAL GAS – DETERMINATION OF WATER BY THE KARL FISCHER
METHOD – PART 1: INTRODUCTION ISO 10101-1 FRENCH NATURAL GAS – DETERMINATION OF WATER BY THE KARL FISCHER
METHOD – PART 1: INTRODUCTION ISO 10101-2 NATURAL GAS – DETERMINATION OF WATER BY THE KARL FISCHER
METHOD – PART 2: TITRATION PROCEDURE ISO 10101-2 FRENCH NATURAL GAS – DETERMINATION OF WATER BY THE KARL FISCHER
METHOD – PART 2: TITRATION PROCEDURE ISO 10101-3 NATURAL GAS – DETERMINATION OF WATER BY THE KARL FISCHER
METHOD – PART 3: COULOMETRIC PROCEDURE ISO 10101-3 FRENCH NATURAL GAS – DETERMINATION OF WATER BY THE KARL FISCHER
METHOD – PART 3: COULOMETRIC PROCEDURE ISO 10336 CRUDE PETROLEUM – DETERMINATION OF WATER – POTENTIOMETRIC
KARL FISCHER TITRATION METHOD ISO 10336 FRENCH CRUDE PETROLEUM – DETERMINATION OF WATER – POTENTIOMETRIC
KARL FISCHER TITRATION METHOD ISO 10337 CRUDE PETROLEUM – DETERMINATION OF WATER – COULOMETRIC
KARL FISCHER TITRATION METHOD ISO 10337 FRENCH CRUDE PETROLEUM – DETERMINATION OF WATER – COULOMETRIC
KARL FISCHER TITRATION METHOD ISO 10362-2 FRENCH CIGARETTES – DETERMINATION OF WATER IN SMOKE CONDENSATES –
PART 2: KARL FISCHER METHOD ISO 11021 ESSENTIAL OIL – DETERMINATION OF WATER CONTENT – KARL
FISCHER METHOD ANSI C59.53 WATER IN INSULATING LIQUIDS (KARL FISCHER METHOD)

22
Document number TitleAPI MPMS C10 S7 *E MEASUREMENT STANDARDS CHAPTER 10: SEDIMENT AND WATER
SECTION 7: STANDARD TEST METHOD FOR WATER IN CRUDE OILS BY KARL FISCHER TITRATION (VOLUMETRIC)
ASTM D 1123 TEST METHOD FOR WATER IN ENGINE COOLANT CONCENTRATE BY THE KARL FISCHER REAGENT METHOD
ASTM D 1364 STANDARD TEST METHOD FOR WATER IN VOLATILE SOLVENTS (KARL FISCHER REAGENT TITRATION METHOD)
ASTM D 1533 STANDARD TEST METHODS FOR WATER IN INSULATING LIQUIDS (KARL FISCHER REACTION METHOD)
ASTM D 1744 TEST METHOD FOR DETERMINATION OF WATER IN LIQUID PETROLEUM PRODUCTS BY KARL FISCHER REAGENT
ASTM D 4017 STANDARD TEST METHOD FOR WATER IN PAINTS AND PAINT MATERIALS BY KARL FISCHER METHOD
ASTM D 4377 WATER IN CRUDE OILS BY POTENTIOMETRIC KARL FISCHER TITRATIONASTM D 4928 WATER IN CRUDE OILS BY COULOMETRIC KARL FISCHER TITRATION ASTM D 5530 TEST METHOD FOR TOTAL MOISTURE OF HAZARDOUS WASTE FUEL BY
KARL FISCHER TITRIMETRYASTM E 203 TEST METHOD FOR WATER USING VOLUMETRIC KARL FISCHER
TITRATIONASTM E 700 WATER IN GASES USING KARL FISCHER REAGENT ASTM F 1214 WATER SOLUBILITY IN LIQUID PETROLEUM BY KARL FISCHER BS 2511 METHODS FOR THE DETERMINATION OF WATER (KARL FISCHER
METHOD)BS 3156 S11.3 SS11.3.1
ANALYSIS OF FUEL GASES - PART 11. METHODS FOR NON-MANUFACTURED GASES - SECTION 11.3 DETERMINATION OF WATER IN NATURAL GAS BY THE KARL FISCHER METHOD - SUBSECTION 11.3.1 INTRODUCTION
BS 3156 S11.3 SS11.3.2
ANALYSIS OF FUEL GASES - PART 11. METHODS FOR NON-MANUFACTURED GASES - SECTION 11.3 DETERMINATION OF WATER IN NATURAL GAS BY THE KARL FISCHER METHOD - SUBSECTION 11.3.2 TITRATION METHOD
BS 3156 S11.3 SS11.3.3
ANALYSIS OF FUEL GASES - PART 11. METHODS FOR NON-MANUFACTURED GASES - SECTION 11.3 DETERMINATION OF WATER IN NATURAL GAS BY THE KARL FISCHER METHOD - SUBSECTION 11.3.3 COULOMETRIC METHOD
BS 4993 P5 METHODS OF TEST FOR ALUMINIUM FLUORIDE FOR INDUSTRIAL USE -DETERMINATION OF MOISTURE CONTENT (KARL FISCHER METHOD)
BS 5202 P15 METHODS FOR CHEMICAL ANALYSIS OF TOBACCO AND TOBACCO PRODUCTS - PART 15. DETERMINATION OF WATER IN SMOKE CONDENSATE OF CIGARETTES (KARL FISCHER METHOD)
BS 5202 P19 METHODS FOR CHEMICAL ANALYSIS OF TOBACCO AND TOBACCO PRODUCTS - PART 19. DETERMINATION OF WATER CONTENT (KARL FISCHER METHOD)
BS 5711 P8 DETERMINATION OF WATER CONTENT: KARL FISCHER METHODBS 5752 P13 METHODS OF TEST FOR COFFEE AND COFFEE PRODUCTS - PART 13.
ROASTED GROUND COFFEE: DETERMINATION OF MOISTURE CONTENT [KARL FISCHER METHOD (REFERENCE METHOD)]
BS 6725 METHOD FOR DETERMINATION OF WATER IN LIQUID DIELECTRICS BY AUTOMATIC COULOMETRIC KARL FISCHER TITRATION
BS 684 P2 S2.1 METHODS OF ANALYSIS OF FATS AND FATTY OILS - PART 2: OTHER METHODS - SECTION 2.1: DETERMINATION OF WATER BY THE KARL FISCHER METHOD
ISO 11021 FRENCH ESSENTIAL OIL - DETERMINATION OF WATER CONTENT - KARL FISCHER METHOD
ISO 11817 ROASTED GROUND COFFEE - DETERMINATION OF MOISTURE CONTENT - KARL FISCHER METHODS (REFERENCE METHOD)
ISO 11817 FRENCH ROASTED GROUND COFFEE - DETERMINATION OF MOISTURE CONTENT - KARL FISCHER METHODS (REFERENCE METHOD)

23
Document number TitleISO 4317 SURFACE-ACTIVE AGENTS AND DETERGENTS - DETERMINATION OF
WATER CONTENT - KARL FISCHER METHODISO 6488-1 TOBACCO - DETERMINATION OF WATER CONTENT - PART 1:
KARL FISCHER METHODISO 6488-1 FRENCH TOBACCO - DETERMINATION OF WATER CONTENT - PART 1:
KARL FISCHER METHODISO 8534 ANIMAL AND VEGETABLE FATS AND OILS - DETERMINATION OF WATER
CONTENT - KARL FISCHER METHODISO 8534 FRENCH ANIMAL AND VEGETABLE FATS AND OILS - DETERMINATION OF WATER
CONTENT - KARL FISCHER METHODJIS K0113 GENERAL RULES FOR METHODS OF POTENTIOMETRIC,
AMPEROMETRIC COULOMETRIC, AND KARL-FISCHER TITRATIONS
On the Internet, these normative documents or standards can be investigated and also ordered as titles or abstracts. The following addresses are intended to give you a starting point for your own excursion to the Internet:
ASTM http://www.astm.orgInformation Services GmbH http://www.global.ihs.comDIN-Normen http://www.din-normen.deVDE http://www.vde-verlag.deTechnischer Fachbuch-Vertrieb http://www.tfv.ch
Further working specifications are contained in the Phamakopoeen (Ph. Eur. II; DAB 96, USP XXI).
2.7 KF titration and quality assurance
As a rule, KF titration is used within the framework of quality assurance. Many aspects have already been mentioned in the validation section, such as the testing-means supervision.In order to enable the account for the current trends, it is not intended to list the features of all different quality systems. We rather include some important organisations including their Internet addresses.
Some features, however, should be noted here:ISO 9000: A testing-means supervision made ensure the eligibility for use of a KF titrator. In the preceding chapter as well as in the annex you will find some practical information. The attributality to a national or other standard is important. KF titration offers some certified standards. The scope of delivery of the TitroLine KF Titrator contains standards of this type. The manufacturer of the reagents will offer you de-tailed information in this context.
GLP/GMP This is focused on the contents and form of the documentation regarding the use of theTitroLine KF Titrator. The comprehensibility of the result is the most important requirement in this context.
Validation: Please refer to chapter XX
For the GLP and GMP range you will find a number of good tips on the Internet compiled by the DGGF association.
DGGF (Deutsche Gesellschaft für Gute Forschungspraxis)http://www.dggf.de
BARQA (British Association for Research Quality Assurance) http://www.barqa.com
BIRA (British Institute of Regulatory Affairs) http://www.bira.org.uk
DIA (Drug Information Association) http://www.diahome.org
ESRA (European Society of Regulatory Affairs), viele Links zu internationalen Behörden http://www.esra.org
ISQA (International Society of Quality Assurance) http://www.quality.org/isqa.info.txt

24
Almost all industrial association meanwhile have set up their own homepages. As a rule, you will find there information on the industry being served as well as on the economic importance plus links to the member companies.
International
ICH (International Conference on Harmonisation)
http://www.ifpma.org/ich1.html
Die Homepage der international abgestimmten Arzneimittelprüfrichtlinien
Deutschland
BAH (Bundesfachverband der Arzneimittel-Hersteller, Bonn)
http://www.bah-bonn.de
BPI (Bundesverband der Pharmazeutische Industrie, Frankfurt)
http://www.bpi.de
VCI (Verband der chemischen Industrie, Frankfurt)
http://www.chemische-industrie.de
EU (European Union)http://europa.eu.int/eur-lex The wording of all major laws of the European Union can be obtained here. You will get free access to publications of the gazette of the EU from the past 20 days as well as to the European Treaties, consoli-dated versions of the currently applicable EU law, and judgements of the European Court, and this in all official languages of the EU. They are updated on a daily basis.
OECD (Organisation for Economic Co-Operation and Development, Paris, Frankreich) http://www.oecd.org/ehsA brief English text on the OECD activities in the range of GLP as well as the complete English wording of the new edition of the "OECD Principles of GLP" and the consent documents can be found at http://www.oecd.org/ehs/glp.htm
US-FDA (U.S. Food & Drug Administration)http://www.fda.gov/fdahomepage.html
Food and Drug AdministrationCenter for Drugs and BiologicsOffice of Drug Research and Review (HFN-100)5600 Fishers LaneRockville, Maryland 20857(301-443-4330)
D-BgVV (Bundesinstitut für gesundheitlichen Verbraucherschutz und Veterinärmedizin, Berlin)http://www.bgvv.de Die GLP-Bundesstelle (z.Zt. ohne eigene Homepage) ist Teil des Fachbereichs 8 'Chemikalienbewertung'.
D-UBA (Umweltbundesamt, Berlin)http://www.umweltbundesamt.de
US-EPA (U.S. Environmental Protection Agency)http://www.epa.gov

25
Installation-Qualification (IQ)
Place of Titrator:Company name:Laboraty:StreetZip Code:City:Fone:Fax:E-Mail:
IQ done by:Company name:Laboraty:StreetZip Code:City:Fone:Fax:E-Mail:
Users of titrator:Name:
Fone:E-Mail:
Name:Fone:E-Mail:
Name:Fone:E-Mail:
Type of Qualification:Qualification XRe-QualificationRe-Qualification after Changes
Building up and Starting
Implementation of Methods:Name: Titer ReagentApplication: Titre Determination with Standard 10 mg water/g
Name:Application:
Name:Application:
Training and Introduction:Done by:Trained Persons:

26
General Conditions:Place:Power supply:Light:Athmosphere:Temperature:Humidity:
Components:TitratorTL KF: XSerial Number:Version Software:TM KF: XSerial Number:
Comments:
Electrical connection of the InstrumentPower Supply
Basic Settings
DocumentationTL KF:TM KF:
Qualification Method
Method Titre
General Description
Titre Determination of Reagent with standard 10.00mg Water/g
Equipment Titrator TL KF TM KF Titration vessel TZ 1720 Electrode TZ 1106 Balance 0.0001 g resolution
Reagents One Component Reagent 5 mg water/ml
Methanol dried or:
Two Component Reagent 5 mg water/ml
Solvent
Standard 10 mg Water/ml

27
Sample amount About 2 ml weighted exactly with 4 digits
Sample input Fill a syringe with about 2 ml of standard, Put it onto the balance and tare the bal-ance. Put the standard into the titration cell and weigh the empty syringe back.
Description The titration starts with conditioning. Wait until the drift value is less than 10 µl/min!! Stop conditioning. After sample input, start the titration and the drift at the end of the titration. It should be similar than before the titration.
Calculation Titer = Weight * 10.00 / ml (10.00 is the mg water value for every gram)
Numbers of titra-tion
5
Statistic RSD > 0,5 %
Documentation GLP Report
Parameter Settings
End criteria 20 µA, 10 sec, no drift, 10 sec start time, selection of the correct reagents
Results of Qualification
Results
Nr.Titration
Weight of Standard in[g]
[ml] Titer Remarks
12345 (Mean)
RSD
The IQ was successful. All criteria are fulfilled.
Date:
____________________________________ __________________________________
Annexis:
Printouts of parameter and results

















