Applications of ^ O NMR Spectroscopy to Natural Products Chemistry David W. Boykin
INTRODUCTION The investigation of natural products chemistry has been and continues to be
an integral part of the development of Organic Chemistry. Of paramount importance to the study of natural products, whether isolated from plants, mammals, microorganisms, marine life or insects, is compound identification or, if a new substance, structure elucidation. Today, natural products chemists have an array of highly sophisticated instrumentation available to perform structure elucidation. Of the more powerful technologies mass spectrometry and nuclear magnetic resonance (NMR) spectroscopy are the most widely used. A combination of the many forms of 1 H and l^c NMR spectroscopy can provide extraordinary amounts of structural information and, arguably, in most cases is sufficient for characterization of new structures. The entire December 1992 issue of Magetic Resonance in Chemistry was devoted to articles using iH and 13c NMR spectroscopy for structure elucidation of natural products and serves to illustrate the power of this approach. Use of NMR spectroscopy of some of the more difficult to detect nuclei such as oxygen, nitrogen and sulfur is rapidly growing [1-3].
l^O NMR spectroscopy is emerging as a useful adjuvant to other spectroscopic methodologies for acquiring structural information. Oxygen, the most abundant element on earth, widely occurs in many types of natural products [4]. Detection of oxygen by NMR spectroscopy can be achieved for the l^O isotope. Because the l^o isotope is quadrupolar, its spin quantum number is 5/2, its natural abundance is 0.037% and its receptivity relative to ^^C is 0.06, it is sometimes considered to be difficult to study by NMR spectroscopy. However, by use of modem instrumentation and wise choices of experimental conditions (pulse repetition time, solvent, temperature and concentration), spectra with acceptable signal-to-noise ratio can be obtained generally in less than four hours (and often in less than one hour) for molecules with molecular weights of less than 300. A representative natural abundance l^o NMR spectrum for the natural product santonin obtained in approximately two hours on a Varian VXR 400 NMR spectrometer at 54.22 MHz is shown in Figure 1. Of course, enrichment with the l^O isotope, even at low levels(ca. 1%), dramatically reduces the instrument time required to obtain high quality data [5]. l^O NMR linewidths increase with molecular weight and currently the practicality of the experiment
550
becomes questionable for many compounds of molecular weight above 500. The purpose of this review is to provide an overview of the l^O NMR spectroscopy characteristics of organic molecules, to give illustrations of previous use of this method in natural products chemistry and , when possible , to offer some suggestions for future development of the methodology.
-CH3
o b
I I M T I r I t I t T T > I I I T r I I I I I I I > I 1 J I 1 I I I t I I r'"| i t i i | i i » i i i i i t f 7 t t r i T > t
500 400 300 200 100 0 PPM
Fig. 1. l^O NMR spectrum of santonin, 0.5M in acetonitrile at IS^Cj internal reference 2-butanone 558 ppm, external reference water 0 ppm.
551
BACKGROUND
During the last decade I ^ Q nuclear magnetic resonance (NMR) spectroscopy has emerged as a valuable method for organic chemists concerned with structure and conformation elucidation as well as for those interested in probing electronic distribution [1, 6-10]. Despite some of the problems with detection, the large chemical shift range for common functional groups (ca. 800 ppm) makes I ^ Q NMR spectroscopy useful for detecting subtle changes in structure. It has become well established that l^o NMR chemical shifts are more sensitive to structural changes than those of 13c and 15N [1,10]. Figure 2 illustrates the 17o NMR chemical shift range of some common functional groups.
FUNCTIONAL GROUPS
HgO (Internal)
HgO (External)
Sulphlnylimlnes Anhydrides (c-0)
Isocyanates
Anhydrides (C=0)
PiiBH3HEaH • • • • • • pUffl^HH
TW 900 800 700 600 500 400 300 200
17 0 NMR Chemical Shift (PPM) -100
Fig. 2. General ^ O NMR chemical shift ranges for important functional groups.
552
l^O NMR chemical shifts are perhaps best understood in terms of an additive dependency of the paramagnetic, GoP, and diamagnetic, Oo^ screening constants Equation (1) [8].
8o = aoP + aod (1)
A number of investigations have led to the conclusion that most ^^O NMR chemical shifts are largely dependent upon the paramagnetic term [1,8,9]. The description of Karplus and Pople is often used to describe the paramagnetic term, shown as Equation 2, for an oxygen nucleus (O) bound to another nucleus (X) [11].
GoP = -e2h2 / 2m2c2AE (r-3)2po[IQox] (2)
E is the "average excitation energy," frequently approximated as AE, the difference in energy between ground state and the first maximum in the electronic spectra, (r'3)2po is the inverse of the mean volume of 2p orbitals on oxygen, and Qox is the charge density bond order matrix. Even though these terms may be interrelated, a number of empirical correlations between 1^0 NMR chemical shifts and other data taken to represent various terms in the Karplus-Pople expression have been reported [8].
The major structural factors which influence l^O NMR chemical shifts are electronic, steric and hydrogen bonding. The influence of each of these factors will be briefly illustrated here; however, the reader is referred to several reviews for more in-depth coverage of these topics [1, 6-8]. Generally, double-bonded oxygen, with significant n bond character, is more sensitive to substituent effects than single bonded oxygen. Large changes in l^O NMR chemical shifts, both shielding and deshielding, can be observed by changes in the electronic and steric environment for a given functional group. Large shielding effects are observed for hydrogen bond formation to multiple bonded oxygen; smaller effects are observed for hydrogen bond formation to single-bonded oxygen.
Electronic effects A study of substituted acetophenones by Brownlee and co-workers [12] nicely
illustrates the effect of electronic factors on carbonyl 1^0 NMR chemical shifts. Substitution with the electron donating amino group results in shielding and introduction of the electron withdrawing nitro group produces deshielding of the acetophenone carbonyl l^O NMR signal (cf. 1-3). The l^O NMR chemical shift range for the carbonyl signal of the /^^fra-substituted acetophenones that was observed is approximately 50 ppm. The acetophenones l^O NMR chemical
553
shifts were correlated with a+ values, DSP values and by the more fundamental property the calculated n electron density[12]. Recently, the l^o NMR chemical shift of related carbonyl systems (methyleneindanones) covering an even wider chemical shift range (ca. 100 ppm) have been shown to be correlated with AMI calculated electron densities [13].
564 ppm
NHi NO2
1 2 3
Dahn and co-workers have systematically examined the effect of substituents on a wide range of carbonyl functional groups and have interpreted the magnitude of the rho values for carbonyl l^O NMR chemical shift-sigma plus correlations as a measure of the "electron-demand" or polarity for different classes of carbonyl groups [14-18]. Table 1 contains rho plus values for the more common carbonyl compounds which have been studied to date. The rho plus values range from approximately 29 for the highly electron deficient -COCF3 carbonyl group to ca. 7 for the more electron rich, non-hindered amide carbonyl system. The rho plus value noted for the N,N-dimethylbenzamides, ca. 2, reflects the large torsion angle of the functional group and is a classical example of a significant decrease in conjugation between aryl ring and functional group as a result of loss of planarity [19]. Dahn, et al. [15] have also shown that the l^o NMR chemical shift of the parent carbonyl compound is linearly related to the magnitude of the p+ value and the chemical shifts are thought to be dependent upon the electron density/bond order of the carbonyl group (Figure 3 is a plot analogous to that of Dahn). Deviations from the line were suggested to arise from other contributions to the chemical shift, probably the AE term of the Karplus Pople expression[15]. The values in Table 1 and Figure 3 reflect the electron demand of various functional groups that organic chemists have intuitively employed for decades; however, this work by Dahn, et al elegantly places them on a quantitative scale [14-18].
554
Table 1
Sensitivity of 4-X-Ar-COY I^Q N M R chemical shifts to substituents on the aryl
ring
Y
CF3
H
Br
CH3
CI
SEt
F
OCOAr
CHN2
OCH3
NH2
NHCH3
coo-N(CH3)2
5(X=H)^
544
562
513
549
491
489
353
386
440
337
326
318
265
348
p+b
29
26
24
22
20
16
14
13
10
8
7
7
5
2
Ref.
14
14
14
14
14
14
14
14
15
14
14
19
14
19
^ The carbonyl I ^ Q NMR chemical shift(ppm) for the parent compound in each series. b Rho plus value from correlation using Hammett Brown sigma plus constants.
555
600
g 500 H a a
13
u
z
400
I ^-^
200
SEt O
OCOAr
O
N(CH3)2
NHCH3
T "
Rho-Plus Values
Fig. 3. Plot of Carbonyl 170 NMR chemical shift yalues(ppin) for ArCOX versus rho-plus values for chemical shift sigma-plus correlations(see ref 15).
The l^o NMR chemical shifts of functional groups other than carbonyl ones, including aromatic nitro groups [20], pyridine N-oxides [21,22], phenols [23], and anisoles [24] have also been found to be predictably quite sensitive to electronic effects. Interestingly, the l^O NMR chemical shifts of aryl sulfones [25,26] and sulfoxides [25,27] are essentially insensitive to substituent effects.
556
Steric effects In addition to providing insights into electronic distribution, other important
structural information can be readily deduced from l^O NMR data. Conformational structure of both aromatic [28,29] and aliphatic series [30] of oxygen containing compounds has been studied extensively by 1^0 NMR methodology. However, more recently, work from our laboratories has shown that l^O NMR spectroscopy is a valuable method for detection of the effects of steric interactions on molecular structure for organic compounds. It is useful to divide the steric interactions into two categories: one involves systems in which steric interactions result in rotation of functional groups around single bonds to relieve van der Waals interactions, and the other deals with rigid systems in which steric interactions are partially accommodated by bond angle and bond length distortions.
van der Waals effects: conformationally flexible systems l^O NMR spectroscopy has been successfully used to gain information about
the conformational structure of both aromatic and aliphatic compounds. The conformation of functional groups attached to aromatic rings can be significantly influenced by introduction of bulky groups near such a functional group. Repulsive van der Waals interactions can be reduced by torsional rotation around the single bond connecting the functional group and the aromatic ring. Such interactions are often discussed in terms of steric inhibition of resonance. Large differences in l^O NMR chemical shifts noted for ortho and para nitrobenzenes in the pioneering work of Christ and Diehl were attributed to steric inhibition of resonance [28]. However, only in recent years have quantitative relationships between l^O NMR chemical shifts and functional group torsion angles been developed [6,7,31-33]. As was previously pointed out [1], the effect of torsion angle rotation on ^^O NMR chemical shift can potentially lead to three different results: (i) deshielding, (ii) shielding, or (iii) no change. A large number of examples of the deshielding effect have been observed [6,7,31-33], three cases in which shielding was noted have been reported [34-36] and one example in which no change on I ^ Q NMR chemical shift was observed on torsion angle rotation has been published [35]. Only the deshielding cases (illustrated by 2, 4-6) will be discussed in this review.
557
Good quantitative correlations between l^o NMR chemical shifts (deshielding) and torsion angles have been reported for aryl nitro compounds [31], aryl ketones [32,33], aryl acids [33], aryl esters [33], aryl acid chlorides [37], and aryl amides [33], and a more qualitative correlation has been noted for aryl sulfinyl compounds [38]. The 1^0 NMR carbonyl chemical shift for several different aryl systems plotted versus the torsion angle defined by the intersection of the plane of the carbonyl group and the plane of the aryl ring deduced from molecular mechanics (MM2) energy minimized structures are shown in Figure 4. These results are representative of the general relationships that have been developed between torsion angles and ^^O NMR chemical shifts. MM2 calculations predict that for many different carbonyl systems rotation around the single bond connecting a functional group to an aryl ring results in repulsive van der Waals energy of near zero [7]. Consequently, the change in electron density on the carbonyl oxygen as a result of torsion angle rotation is considered to be the major contributor to the observed ^7o NMR chemical shift variation. It is important to note that the origin of the deshielding shifts detected in conformationally mobile systems is fundamentally different from the origin of deshielding shifts observed for the rigid planar systems reviewed below.
558
700
Torsion Angle (Degrees)
Fig. 4. Plot of the carbonyl ^'O NMR chemical shift for aryl acids O (33), aryl amides^ (33), aryl esters # (33 ) , aryl acid chlorides ffl(37), and aryl ketones ^ (32) verus torsion angle between the aryl ring and the carbonyl functional group.
van der Waals effects: rigid planar systems Large downfield shifts of the N-oxide ^'O NMR signal for hindered
pyridine-N-oxides (7,8) and quinoline-N-oxides (9,11) systems in which torsion angle twist is not thought to be likely led to a broader study of rigid planer systems[39,40].
559
H j C r C H a CH3
341 ppm
o -
":)
.CH,
1 0
370 ppm
1 1 1 2
In an investigation of the 1^0 NMR properties for a series of 2- and 4-alkyl substituted pyridine-N-oxides[cf. 7,8], it was found that the 2-alkyl compounds were deshielded relative to the 4-alkyl isomers [39], Large deshielding effects were noted for highly hindered compounds such as 8. Quinoline N-oxides exhibited similar effects; the deshielding effect of the methyl group in 2-methylquinoline-N-oxide (10) is similar to that noted for 2-methylpyridine-N-oxide [40]. However, 8-methylquinoline-N-oxide (11) displayed a much larger effect, presumably as a consequence of the peri-like relationship between the methyl and N-oxide groups [40]. In contrast, a large upfield shift, a consequence of intramolecular hydrogen bonding, was noted for 8-hydroxyquinoline-N-oxide (12) (vide infra).
560
Several series of hindered carbonyl compounds were systematically investigated in an effort to understand the origin of the downfield shifts of the 17o NMR signals in rigid planar systems. Selected compounds and the corresponding 1^0 NMR data from these studies are shown below. A 27 ppm deshielding effect is noted (cf. 13 and 14) for the hindered isomer of the t-butylphthalide system [41,42]. MM2 calculations for these isomers predicted carbonyl aryl ring torsion angles of essentially zero. However, in the case of 14, the calculations predict moderate carbonyl bond angle deformations. The I ^ Q NMR data for a similar pair of compounds, the indanones 15 and 16, have been obtained [43]. The downfield position of the l^o NMR signal for 16 is viewed as a consequence of repulsive van der Waals interactions between the t-butyl group and the carbonyl oxygen. MM2 calculations for 16 reveal no torsion angle twist; however, bond angle deformations are predicted.
CHj
O 170 ppm
HaC^ CH3 346 ppm
.CH3
^
O 173 ppm
13
H3C.. CH3 541 ppm
CH3
1 6
Reasonable models (isomers) for unhindered phthalimides which would allow direct comparison of chemical shifts for the hindered phthalimides do not exist. Nevertheless, the progressive deshielding of the carbonyl l^o NMR shift of 17-20 as the N-alkyl groups become larger is consistent with increasing steric interaction and presumably increasing repulsive van der Waals interactions. MM2 calculations predict no torsion angle changes for these compounds but do suggest in-plane bonding deformations [44].
561
17
379 ppm
O
N - H
• <
O
Cc ^ \ ^
1 8
374 ppm
o
N - C H 3
-< 0
383 ppm
0
O: " " \ ^ = ^ CH3
0
1 9
394 ppm
0
f 1 ; N - 4 - C H 3 ^ ^ - ^ ^ CH3
0
2 0
The flavone pair (21 and 22) and the t-butylanthraquinone examples (23 and 24) show pronounced deshielding shifts for the hindered isomers (e.g., 50 ppm; compare 23 and 24). MM2 calculations for these sets of compounds also predict bond angle deformations without torsion angle twist.
451 ppm
21 22
523 ppm 573 ppm
23 24
The origin of the deshielding effects in these systems appears to be repulsive van der Waals interactions. Chesnut has shown for several heteroatom nuclei that
562
repulsive van der Waals interactions are responsible for deshielding shifts. For example, deshielding shifts of ^'^O, ^^N and 31p NMR data were correlated with repulsive van der Waals energies [45-47]. An approach for accessing this possibility for the effects observed for the carbonyl systems described above was developed which compared MM2 calculated van der Waals energies for hindered and unhindered isomers with l ^ o NMR chemical shift differences [7]. A plot of estimated "local" repulsive van der Waals energies versus 17o NMR chemical shift differences yielded a line with a slope of approximately 13 ppm/kcal [7]. This correlation was considered a reflection of the role of the r '3 term of the Karplus- Pople equation in determining the deshielding effects on ^^O NMR chemical shifts in these systems.
In summary, it is reiterated that large deshielding effects on 1^0 NMR chemical shifts can be observed in hindered carbonyl systems which are conformationally flexible as well as in rigid planar systems. The origin of the downfield effects in the two systems are different consequences of repulsive van der Waals interactions. Futhermore, semi-flexible systems have been studied and, in general, the observed deshielding effects could be quantitatively factored into torsion angle and rigid planar components [48].
Hydrogen bonding effects The significant influence of hydrogen bonding on carbonyl ^^O NMR
chemical shifts was noted by early workers in the field [28,49,50]. A 52 ppm shielding shift was observed for the acetone 1^0 NMR signal on intermolecular hydrogen bonding with water [50]. Large shielding shifts of carbonyl l^O NMR signals resulting from intramolecular hydrogen bonding have been reported for salicylaldehydes, ortho-hydroxy -acetophenones [51], and -quinones and related compounds [52,53]. Some of the earlier l^O NMR spectroscopy literature for intramolecular hydrogen bonding systems is confusing, since often the observed upfield shifts were described as if arising exclusively from hydrogen bonding. To determine the magnitude of the contribution from hydrogen bonding to an acceptor functional group l^Q NMR chemical shift, it is essential to factor out any contributions from electronic and/or torsion angle effects. A method of analysis was developed to deal with these factors during a study of 6^r//z^-amino-and amido-acetophenones [54]. The validity of the factoring method for determination of torsion angle contributions in conformationally flexible systems was confirmed by a study of rigid planar amino- and amido-fluorenones and anthraquinones [55]. This factoring approach has led to the deduction of A 5 H B
values (hydrogen bonding component to the functional group l^O NMR chemical shift) for a number of intramolecular hydrogen bonding systems [23,54-58].
563
Not surprisingly, the A5HB values generally depend on the basicity of the hydrogen bond acceptor and the acidity of the hydrogen bond donor. Figure 5 illustrates the average change in shielding shift (A8HB values) observed for aryl ketone acceptors as a function of acidity of the donor. Note that as the acidity varies from the moderately strong phenolic donor to the considerably weaker amino donor the shielding influence is reduced. Figure 6 contains average hydrogen bonding induced shielding data (A5HB values) for systems in which the hydrogen bond donor is held constant (phenolic) and the basic character of the acceptor is varied. In this case the magnitude of the shielding observed decreases with decreasing basicity of the acceptor functional group. Other factors such as the geometry of the intramolecular hydrogen bond have also been shown to be extremely important [59]. For 7-hydroxyindanones [59] where the hydrogen bond distance is significantly longer as a consequence of the geometric constraints of the 5-ring-6-ring fusion, the A8HB value is dramatically reduced to ca. 10 ppm in comparison to the average values of 52 ppm noted for shorter hydrogen bonding distances arising from 6-ring-6-ring fusion [56,60].
60
e a,
50 H
M 40
.s e o m
30 H
Ji 20
^ 10
ArOH
ArNH2
^
Donor Group
Fig. 5. Hydrogen-bonding shielding effects on ArCOR 170 NMR signals on intramolecular hydrogen bonding to various donors.
564
60-
s a a
"wD 5 0 -G
n
40H
30
iJ 20H
lOH
ArCOR
ArCOH
ArN02
ArCOOR
Acceptor Functional Group
Fig. 6. Hydrogen-bonding shielding effects on different functional group 170 NMR signals on intramolecular hydrogen bonding to a phenolic donor.
Interestingly, the effect on the 170 NMR chemical shift of multiple hydrogen bonding to a single carbonyl group acceptor appears to be roughly additive. For example, in systems capable of forming two intramolecular hydrogen bonds to a single carbonyl acceptor, the A6HB values observed are approximately twice those noted for systems which can form only a single hydrogen bond (60). These effects are apparent from 170 NMR data obtained from toluene solutions for 1-hydroxyanthraquinone (25), 1,8-dihydroxyanthraquinone (26) and 3-methyl-l,8-dihydroxyanthroquinone (27) [60].
88 ppm 456 ppm
OH O
91 ppm 395 ppm OH ^ HO
25
92 ppm 389 ppm OH ^ HO
27
565
A comparison of l^O NMR results for 2',4,-dihydroxyacetophenone (28) and 2',6'-dihydroxyacetophenone (29) from two different solvents, acetonitrile and toluene, lead to a better understanding of hydrogen bonding in these phenolic systems [56],
463 ppm
29
In both cases, 28 and 29, formation of two intramolecular hydrogen bonds to the carbonyl group is not possible for geometric reasons. Larger A5HB values were anticipated as a result of an expected intermolecular hydrogen bonding component to the chemical shift; however, in acetonitrile A5HB values of 50 and 51 ppm, consistent with values for monohydroxyacetophenones, were noted. In addition, a small (ca. 2 ppm) upfield shift of the signal for the internal reference, 2-butanone, was observed. These results suggest that the phenolic OH group not participating in intramolecular hydrogen bonding is primarily involved in intermolecular hydrogen bonding with the solvent (acetonitrile). The A 6 H B
values for 28 and 29 obtained from toluene solution are 55 and 57, in good agreement with the average value of 54 found for toluene solutions of other systems which can form only one intramolecular hydrogen bond. In the toluene solution, l^O NMR study of 28 and 29 revealed that the 2-butanone signals (internal reference) were shielded by 11-13 ppm. These results suggest that in toluene solution phenolic OH groups which are not participating in intramolecular hydrogen bonding are free to form intermolecular hydrogen bonds. Apparently, in the case of toluene as the solvent, phenolic hydrogen bonds with solvent are weak. Consequently, by performing l^o NMR spectroscopy measurements in toluene solution and by observing the chemical shift of an internal ketone reference, "free" phenolic groups can be detected.
It should be noted that proton to oxygen coupling is often directly observed for intramolecular hydrogen bonded phenolic systems in acetonitrile solutions [56,57]. Direct proton-to-oxygen coupling has also been observed for simple alkyl alcohols [61]. The magnitude of the coupling constant ^JOH appears to be fairly constant and these values are in agreement with those obtained by lineshape analysis [8,62]and from l^o INEPT experiments[63]. The values are
566
typically 80 ± 25 Hz (see Table 2). The U Q H values have been shown to be dependent upon concentration, temperature and solvent [61]. Currently, there exists only an incomplete understanding of the factors which influence UoH values; consequently, further investigations are needed.
Table 2
Representative values of directly observed proton to oxygen coupling (UoH) in
acetonitrile solution.
Compound
2'-hydroxyacetophenone
2'-hydroxy-4'-methoxyacetophenone
2'-hydroxypropiophenone
2-acetyl-1 -naphthol
methyl salicylate
ethyl salicylate
salicylaldehyde
5-acetoxy-2-hydroxybenzaldehyde
2-nitrophenol
2-methoxyphenol
3-pentanol
cyclopentanol
cyclohexanol
IJOH(HZ)
86
75
91
80
73
104
92
58
96
72
79
79
73
Ref.
56
56
56
56
57
57
23
23
58
23
61
61
61
^In each case the solvent was anhydrous acetonitrile and data were collected at
75°C
l^O NMR SPECTROSCOPY OF NATURAL PRODUCTS
A recent survey of application of NMR of chalcogen nuclei to natural products chemistry included some interesting examples of ^^o NMR results for
567
selected natural products [64]. A number of reports have now appeared describing the l^Q NMR properties of several natural product systems. Even more specialized usage of l^o NMR spectroscopy includes a study on coal distillates [65] and l^o NMR imaging of plant stems [66]. In this section of the chapter an overview of the l^o NMR chemical shift characteristics of some important classes of natural products will be presented. Occasionally, for illustrative purposes, 1^0 NMR spectra of selected natural products obtained in the author's laboratory will be included. Data for certain key compounds needed to facilitate discussion, also obtained in the authors laboratory, are included. The new data presented here were measured under our standard operating conditions [60] using acetonitrile as solvent and, except for artemisinin(vWe infra), at TS^C.
Ketones The ketone functional group appears in a wide variety of natural products
ranging from the simple terpenes to complex macrocycles and polyfunctional molecules. This section will focus on simple, naturally-occurring ketones, largely from the terpene family, which illustrate relationships between structure and ^^O NMR chemical shifts. Other more complex ketone containing structures will be examined later according to their natural product family classification, e.g. chromones, flavones, etc.
Double bonds in conjugation with the ketone function result in shielding of the l^O NMR carbonyl signal. The magnitude of the shielding shift is modest in acyclic systems(typically 5 ppm or less) whereas, in cyclic ones the shift is larger[67-70].Note the 12 ppm difference in l^o NMR chemical shift for 30 and 31, even larger differences are observed for 5-membered ring pairs[67]. Analysis of the apparently competing factors which cause these differences between cyclic and acyclic systems is incomplete. The effect of substituents on the l^O NMR chemical shifts of simple cyclohexenones (31-33) has been investigated [67]. Both a and p alkyl groups cause shielding of the cyclohexenone carbonyl signal (cf. 32 and 33 with 31).
30 31 32 33
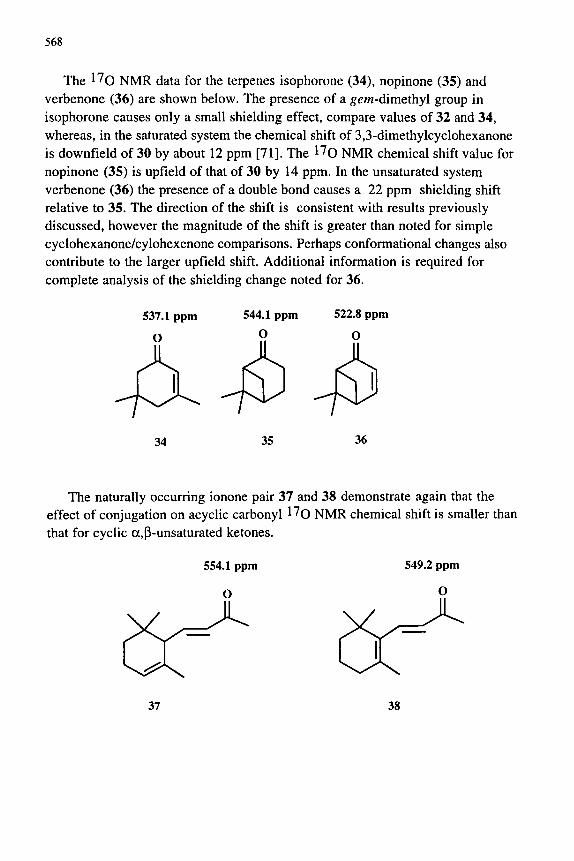
568
The 1^0 NMR data for the terpenes isophorone (34), nopinone (35) and verbenone (36) are shown below. The presence of a g^/n-dimethyl group in isophorone causes only a small shielding effect, compare values of 32 and 34, whereas, in the saturated system the chemical shift of 3,3-dimethylcyclohexanone is downfield of 30 by about 12 ppm [71]. The l^o NMR chemical shift value for nopinone (35) is upfield of that of 30 by 14 ppm. In the unsaturated system verbenone (36) the presence of a double bond causes a 22 ppm shielding shift relative to 35. The direction of the shift is consistent with results previously discussed, however the magnitude of the shift is greater than noted for simple cyclohexanone/cylohexenone comparisons. Perhaps conformational changes also contribute to the larger upfield shift. Additional information is required for complete analysis of the shielding change noted for 36.
34 35 36
The naturally occurring ionone pair 37 and 38 demonstrate again that the effect of conjugation on acyclic carbonyl l^o NMR chemical shift is smaller than that for cyclic a,p-unsaturated ketones.
37 38
569
A recent report describes the ^^o NMR chemical shifts of various oxidation products 39-42 of (+)-3-carene [72]. The chemical shifts of the two a,P-unsaturated ketones 39 and 40 are substantially shielded relative to the simple cyclohexenones 32 and 33. This shielding effect doubtlessly involves conjugation with the cyclopropane ring[68,73], however detailed analysis requires additional data to appropriately account for the influence of the ^em-dimethyl group. The quinone-like molecule 41 gives only one carbonyl signal, apparently the l^O NMR chemical shift of the two different oxygen atoms overlap. The carbonyl l^o NMR chemical shift for 41 is downfield of its analogs 39 and 40 consistent with the quinone-like structure(vWe infra)A^O NMR data for other bicyclic ketones related to naturally occurring ones have been reported[71].
518.8 ppm 571.7 ppm 546.8 ppm
39
Lactones A number of important lactones occur in nature and consequently an
understanding of l^o NMR chemical shift values for this family of cyclic esters may prove useful in characterization of such compounds. This section focuses on relatively simple lactones, other lactones such as courmarins are treated later as a separate class of natural products. l^O NMR results for a number of simple lactones have been published [74]. The signals for both the carbonyl group oxygen and the single-bonded oxygen are sensitive to substituents; however, no systematic variation with ring size is apparent(cf. 43-46). Substitution at the 5-position of the butyrolactone ring results in deshielding of the single-bonded oxygen l^o NMR signal, however the carbonyl resonance is not effected(cf. 44 and 47). Introduction of a double bond in conjugation with the carbonyl group leads to shielding of both lactone l^O NMR resonances(cf. 44 and 48), whereas introduction of a double bond in conjugation with the single-bonded oxygen results in deshielding of both signals(cf. 47 and 49).
570
348.5 ppm
O
241.0 ppm
340.5 ppm
178.5 ppm
367.0 ppm
o-o CI o o 167.0 ppm
377.0 ppm
43 44 45 46
340.3 ppm
O O
206.6 ppm
326.7 ppm
172.7 ppm
351.3 ppm
C o ^ > o 239.0 ppm
47 48 49
Data for the natural products sclareolide (50) (see Figure 7) and the ascaricide santonin (51) (see Figure 1) are consistent with that for simpler lactones. The carbonyl oxygen signals for 50 and 51 and the single-bonded
P 353 ppm
^O 243 ppm
CH,
509.3 ppm O
H,C CH, CH3 O-
a\CH3
188.8 ppm O 345.4 ppm
50 51
oxygen signal for 50 are substantially deshielded compared to 44. The changes in chemical shift are generally reasonable based upon substituent effects observed of simple lactones[74], however there are too many structural variations for which adequate models are unavailable to allow a detailed analysis of the l ^ o NMR chemical shifts of 50 and 51 .
571
I I t I M I I I I I I I 5 0 0
I I j I I I I I I I I I j r
4 0 0 3 0 0 I I I I I I I I I
200 T> ' ' » I I I 1 t I I I I I ) I I I
100 0 PPM
Fig. 7. 17o NMR spectrum of sclareolide (SO), 0.5 M in acetonitrile at 75^0, internal reference 2-butanone 558 ppm, external reference water 0 ppm.
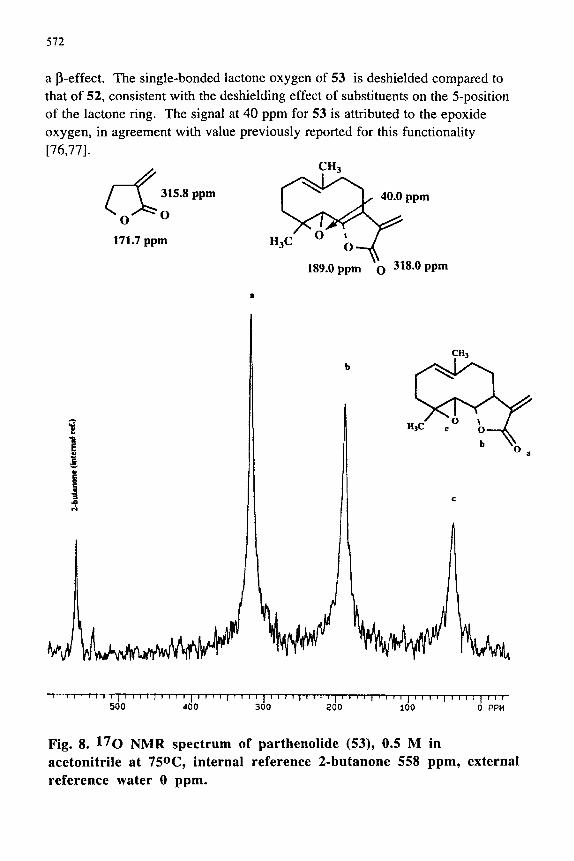
Butenolides constitute a major class of lactone natural products which have been shown to play important roles in cancer chemotherapy [75]. It was previously noted that introduction of a double bond in conjugation with the lactone carbonyl results in shielding of the carbonyl l^o NMR chemical shift; for example compare the carbonyl chemical shift of 44 and 48 [74]. However, l^Q NMR data for lactones with conjugated exocyclic double bonds do not appear to have been reported. The l^o NMR values for the simple butenolide 52 and parthenolide (53) (see Fig. 8) were obtained in acetonitrile solution. Note that the l^o NMR chemical shifts of the carbonyl groups of 52 and 53 are similar. Interestingly, the conjugation of an exocyclic double bond causes larger shielding than noted for an endocyclic double bond(cf. 48 and 52); perhaps , in part due to
572
a p-effect. The single-bonded lactone oxygen of 53 is deshielded compared to that of 52, consistent with the deshielding effect of substituents on the 5-position of the lactone ring. The signal at 40 ppm for 53 is attributed to the epoxide oxygen, in agreement with value previously reported for this functionality [76,77].
CH3
\ 315,8 ppm
o-^o 71.7 ppm H3C
^<^ -|
0 » 0 —
189.0 ppm
^ 40.0 ppm
Q 318.0 ppm
• 1 I I I I I [ I » [ I I I I I 1 I 1 I I i n I I I t I I I I » » I { I f I I j I I I I I ) F f I I I I I I I I I I I I 1 I 1
500 400 300 200 100 0 PPM
Fig. 8. 17o NMR spectrum of parthenolide (53), 0.5 M in acetonitrile at 750C, internal reference 2-butanone 558 ppm, external reference water 0 ppm.
573
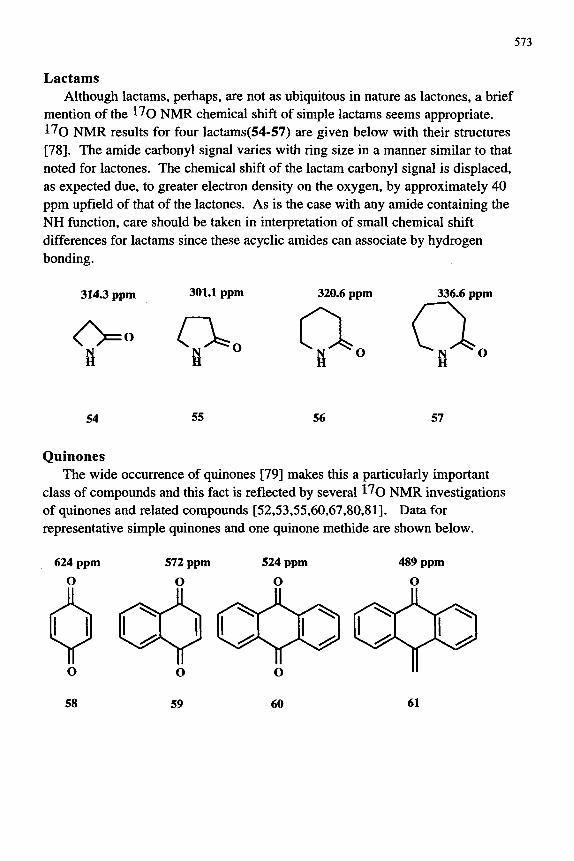
Lactams Although lactams, perhaps, are not as ubiquitous in nature as lactones, a brief
mention of the l^o NMR chemical shift of simple lactams seems appropriate. 17o NMR results for four lactams(54-57) are given below with their structures [78]. The amide carbonyl signal varies with ring size in a manner similar to that noted for lactones. The chemical shift of the lactam carbonyl signal is displaced, as expected due, to greater electron density on the oxygen, by approximately 40 ppm upfield of that of the lactones. As is the case with any amide containing the NH function, care should be taken in interpretation of small chemical shift differences for lactams since these acyclic amides can associate by hydrogen bonding.
314.3 ppm 301.1 ppm
O. B
o
320.6 ppm
0-O
336.6 ppm
54 55 56 57
Quinones The wide occurrence of quinones [79] makes this a particularly important
class of compounds and this fact is reflected by several ^^O NMR investigations of quinones and related compounds [52,53,55,60,67,80,81]. Data for representative simple quinones and one quinone methide are shown below.
624 ppm O
572 ppm
O
574
The chemical shift of the quinone ^^O NMR signal is shielded by fusion of aromatic rings, compare 59 and 60 with 58, consistent with greater delocalization of the carbonyl group's 7C-electron density. Interestingly, the 1^0 NMR chemical shift for the quinonemethide 61 is shielded by 35 ppm compared to anthraquinone which is consistent with differences in their calculated electron densities [81,82].
The observed chemical shifts for quinones studied to date suggest that the quinone-carbonyl group responds to substituent effects as do other carbonyl groups. The electronic effect of substituents on the carbonyl chemical shift appears normal (electron donators cause shielding; electron withdrawing groups produce deshielding). The ^^o NMR signals of hindered quinones are deshielded relative to non-hindered isomers. Intramolecular hydrogen bonding from phenolic OH groups cause large (ca. 50 ppm per H-bond) shielding shifts of quinone carbonyl l^o NMR signals(see 25-27).
A study of the biosynthesis of citrinin(62) in Aspergillus terreus used several isotopically labeled acetates including [l-13c, 17o]-acetate. The ^^O NMR spectrum of citrinin obtained from [l-l^c, l^oj-acetate gave three signals (148, 179 and 279 ppm) (83). These signals were assigned to 0-2 and to the oxygen atoms attached to C-8 and C-6, respectively. The structure of citrinin was described in terms of a tautomeric equilibrium involving the atoms at C-6 and C-8[83].
62
Coumarins and chromones A special family of lactones, the coumarins, which widely occur in plants[84],
have been investigated by ^^O NMR spectroscopy in some detail. Values for l^o NMR chemical shifts for representative members of the class are shown as 64 and 65; data for dihydrocoumarin 63 is given to illustrate the influence of conjugation on the l^O NMR signal for the lactone carbonyl group.
Tables 3 and 4 contain results from a study of a series of coumarins and furocoumarins in, 1,2-dibromoethane, which found that the lactone carbonyl signal consistently appeared near 350 ppm; more variability was seen for the single bonded oxygen, especially for the furocoumarins, typically appearing at 220 ppm [85]. Synthetic 3-aryl coumarins also exhibit 8(C=0) and 6(-0-) values near those mentioned above [86]. Recently, a study of a series of 7-substituted-4-methylcoumarins, with a wide range in electronic character of the substituents, demonstrated that the carbonyl signal is quite sensitive to substituent effects and that the l^O NMR chemical shift is reasonably well correlated with the carbonyl oxygen AMI estimated electron density [87].
Table 3 17o NMR chemical shifts (ppm) of coumarins in 1,2-dibromoethane at 70oC[85]
:ixx. No. 64 65a 66 67
Rl H H
CH3 H
R2 H OCH3 H CH3
5(C=0) 351 347 351 352
8(-0-) 219 225 220 221
^ Signal for OCH3 group 62 ppm.
576
Table 4 17o NMR chemical shifts (ppm) of furocoumarins (psoralens)
in 1,2-dibromoethane at 70T [ref. 85] R2
No.
68 69 70 7 1 72 7 3 74
Rl
H 0CH3 H OCH3 0CH3 H e
R2
H H 0CH3 0CH3 Br H H
R3
H H H H H d H
5(C=0)
352 350 351 351 352 351 351
5(-0-)a
219 200C 220 200C 200C
215 200C
5b(-0-
200 200C 202 200C 200C
207 200C
) 5(other)
14 45 25C
44
^ Lactone single bonded oxygen, b Furan single bonded oxygen. ^ Broad
overlapping signals. ^ ^ - " M
l^o NMR data for a few chromones were also reported earlier[88,89]. As can be seen from the data (from chloroform solutions ) given with the structures 66-68 the ketone chemical shift is sensitive to substitiuents like most aryl ketones. However, the vinyl ether signal is relatively insensitive to
66 67 68
577
substituents, except in the more complicated chromone visnagin(69). The ^^O spectra for visnagin and its analog khellin(70) are shown in Fig, 9. The chemical shift value differences between 69 and 70 are consistent with the change in structure (the addition of the 9-methoxy group in khellin). The carbonyl l^O
' \ \ \ i-fTnm-i 1 i M p ^ M p M t \-t M I p » M I I 1 < A I t M I \ M I v\\ x \ \ \ \ \ \ \ \y \ 300 200 — 500 400 100 0 PPM
Fig, 9. 17 o NMR spectrum of visnagin(69)(bottom) and khellin(70)(top) , 0,5 M in acetonitrile at 75^ C, internal reference 2-butanone 558 ppm, external reference water 0 ppm.
578
NMR chemical shifts, as expected, are essentially the same [only small effects due to ''meta "-substitution are anticipated]. The 5-methoxy signal is shielded by ca. 10 ppm in 70 as a result of the "para " -methoxy, consistent with results from anisoles[24]. Both ring oxygens are shielded due to the introduction of the 9-methoxy group in 70 as expected due to their "orr/zo"-relationship[90]. The 9-methoxy signal is significantly shielded in accord with results for the analogous methoxy group of l,2,3-trimethoxybenzene[90]( see also the aryl ether section below).
40.4 ppm 459.3 ppm
<fT \ 11
200.4 ppm
OCH3 0 vs ^ ^ 0 CH3
166.2 ppm
69
^
O'
192.7 ppm
30.9 ppm
OCH3
YST 0CH3
5.5 ppm
70
460.6 ppm
0
ll 11
0 CH3
154.2 ppm
A recent report describes the use of l^o NMR spectroscopy to differentiate between coumarins and their isomers, the chromones [91]. This study reports data for five isomeric pairs of coumarins and chromones in dimethylsulfoxide at lOOX. Results from two pair are shown as 71-74 to illustrate the difference between the data for the isomers. After investigating the use of several solvents these authors selected dimethylsulfoxide as the solvent of choice for their studies, despite the large solvent signal, due to the greater solubility of the coumarins and the chromones . These investigators suggest that the difference in chemical shift of the two different type carbonyl groups (enone and lactone) and the two different single-bonded oxygens (vinyl ether and lactone) can be reliably used to distinguish between chromones and coumarins. Previous investigators had noted the characteristic region of 1^0 NMR absorption for the coumarin and chromone systems; however, Nagasawa and co-workers[91] were first to recognize the importance of this approach for natural products structure elucidations. This method appears to be a valuable way to distinguish between courmarins and chromones, however as with all structural assignments based on chemical shift
579
differences, each case should be carefully examined and the effect of all substituents must be taken into account.
CH, CH,
H,C
339.7 ppm 213.7 ppm
CH, O
H,C
440.2 ppm 165.4 ppm
71 72
352.8 ppm 216.8 ppm
439.9 ppm 165.4 ppm
73 74
Substituents influence carbonyl I ^ Q N M R chemical shifts of the chromanones listed in Table 5, as they do other aryl ketones[89]. Introduction of a methoxy group "para" to the carbonyl in 76 causes shielding analogous to that for acetophenones[12].On the other hand, location of a methyl group in the 5-position causes a 25 ppm deshielding shift(cf.79).The deshielding shift was attributed to the peri interaction between the methyl and carbonyl groups[64,89]. It was suggested that the interaction results in loss of coplanarity of the aryl ring and the carbonyl group which would account for the deshielding. However, molecular mechanics calculations predict a carbonyl-aryl ring torsion angle of only 3 degrees, which is insufficient to account for a 25 ppm downfield shift[48] . Repulsive van der Waals interactions, as described earlier in this review, are likely to contribute to the observed downfield shift[48]. The large shielding shift of the carbonyl signal of 81, a consequence of intramolecular hydrogen bonding, is consistent with results from other hydrogen bonded systems [60]. Data are not available that would allow an estimation of the electronic effect of the OH group in the chromanone system, hence predicting the portion of the shielding effect solely attributable to hydrogen bonding is not currently possible. The cyclic oxygen signal for the chromanones is essentially unaffected by substituents.
580
except for 80 for which the 8-methyl group causes significant shielding, arising from a y-gauche interaction[89].
Table 5 ^^O NMR chemical shifts of substituted chromanones in chloroform at 55''C [89]
Flavones and flavanones Flavones and their partially saturated analogs, flavanones, appear widely in
plant sources and, consequently, continue to be extensively investigated[92]. A l^O NMR study of ten methoxy substitued flavones in chloroform solution has appeared[93]. However, the l^o NMR data shown below for some selected flavones and flavanones were obtained from acetonitrile solutions for these compounds. The difference in 1^0 NMR chemical shift for the flavone and flavanone carbonyl groups is large cf. 68 and 82. The shielding of 68 relative to 82 (ca. 80 ppm) is attributable to an increase in electron density on the carbonyl oxygen of 68 as a result of both oxy-enone and 2-phenyl conjugation. Introduction of a methoxy group in the 7-position of both systems(83 and 84) produces shielding of 13-18 ppm as expected of this electron donating group, based upon the early studies on acetophenones[12]. However, introduction of a methoxy group in position 5, which is electronically, but not sterically, equivalent to the 7-position produces deshielding shifts of 24 and 34 ppm(see 85 and 86).
581
The downfield shifts are consistent with repulsive van der Waals interactions between the 5-methoxy group and the carbonyi oxygen [48]. The I^Q N M R
O Ph
158 ppm
68 82
HaCO
66.1 ppm 158.6 ppm
H3CO' " ^ O Ph
65.1 ppm 95.5 ppm
83 84
chemical shift of the carbonyi group of the 5-hydroxyflavone 88 is 380 ppm , which represents substantial shielding and doubtlessly arises from a significant contribution from intramolecular hydrogen bonding. The isomeric 7-hydroxyflavone 87 is poorly soluble in acetonitrile. In order to obtain data for 87, enrichment by exchange with H2^^0 was necessary. The carbonyi signal for 87 appears at 432 ppm. Consequently, the hydrogen bond induced shift for the carbonyi signal of 87 is deduced to be 52 ppm. This hydrogen bonding shift is in agreement with those of other aryl carbonyi groups[60].
582
61.2 ppm 472.0 ppm
OCH3 O
62.5 ppm 543.6 ppm
OCH3 O
86
86.4 ppm 380.4 ppm
OH O
O Ph
87 88
Carbohydrates One of the major families of natural products, carbohydrates, which contain
multiple oxygen functions, present difficulties in acquiring interpretable natural abundance l^O NMR spectra. Application of 1^0 NMR to the study of carbohydrates is quite challenging not only due to the numerous alcohol and ether-like oxygens, which results in signal assignment difficulties, but also because of the general necessity of obtaining the data in water solution. Despite these difficulties significant progress is being made in the evaluation of the ^^O NMR spectroscopic properties of sugars by Lauterwein and co-workers[94,95]. Good quality spectra are obtained at high temperature (90'*C)[96], using 1^0-depleted water[96] and water suppression techniques[97]. Assignments have been made by use of shift reagents[95,98] and by a computational strategy which solves multiple linear equations using observed chemical shift differences between
583
signals for sugars and a model compound expressed as a sum of y and 6 shielding terms(94).
-10.6 ppm 1 HOfi;
H O ^ L6 ppm UQ ^
9.6 ppm
jC 63.7 ppm
I.O
9.6 ppm *
^OH 36.5 ppm 47.9 ppm
-9.9 ppm 1 6.3 ppm
HO|LC ^ 9 Q 6.3 ppm HO V l ' ' ^ - L N
HO j L g J ^ ^ ^ ^ 6.3 ppm 1
52.6 ppm
^ O H
1 44.0 ppm
89 90
-7.3 ppm H O
OH 34.4 ppm 50.5 ppm
91
The assignments for the l^o NMR signals of the common sugars D-glucose(89), D-mannose(90) and D-galactose(91) have been made based on chemical shift arguments, selective enrichment (or depletion), and shift reagent studies [94,98]. The l^O NMR chemical shift assignments for 89-91 are given with their structures[94]. Note primary OH signals (C-6) appear between -7 and -11 ppm, generally the secondary OH signals (C-2, C-3, C-4) overlap between 6 and 10 ppm (except for C-4 for galactose); however, the secondary OH's at the anomeric position (C-1) appear between 36 and 50 ppm, depending on the a or p configuration, [assignments to a and p resonances were not made]. The I ^ Q NMR chemical shift of the 6-membered ring oxygen for these sugars appears between 50 and 64 ppm.
584
-10.2 ppm 1
HOB 9.6ppmHO ^
HO-
9.6 ppm
iC 55.4 ppm
I.O
OHp 9.6 ppm *
0CH3 9.6 ppm
-11.4 ppm 1
HOB 8.6 ppm HO
HO-
8.6 ppm
^
8.6
50.4 ppm
uo
OH| ppm OCH3
8.6 ppm
92 93
-10.7 ppm 1
HOjL 10.8 ppm HO ' y
H 3 C O .
-10.7 ppm
iC
• ^
10.8
65.4 ppm
. 0
OHT' ppm 1
1
OH
37.7 ppm 48.9 ppm
94
The assignment of the l^O NMR chemical shifts of three O-methyl derivatives of D-glucose (92-94) have been made using similar approaches[94]. For both methyl a- and P- glucopyranosides 92 and 93 the methoxy signal overlaps the resonances of the secondary OHs (C-2,C-3,C-4); the coincidence of the four signals was inferred from signal intensity[94]. In the case of the 3-0-methyl-D-glucopyranose (94 ) the 3-methoxy signal overlaps that of the 6-hydroxy, again overlap was inferred from signal intensity [94].
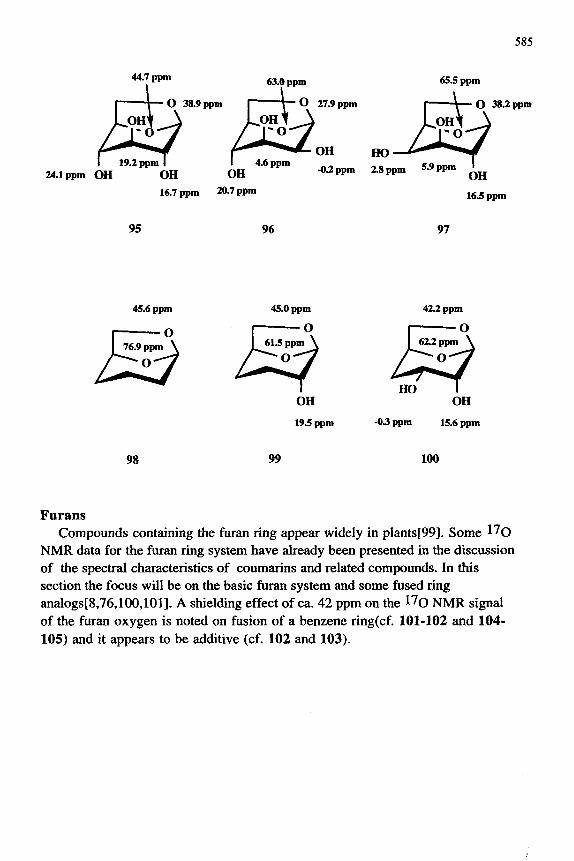
Recently, all eight diastereoisomeric 1,6-anhydro-P-D-hexopyranoses and three related model compounds have been studied in l^O-depleted water using l^O NMR spectroscopy methodology [94,97]. Chemical shift assignments for all resonances in the eleven compounds were made. The results for glucosan ( 95), mannosan( 96), and galactosan ( 97) as well as the three model compounds 98-100 are shown below.
585
44.7 ppm
I - t — O 38.9 ppm p
I 19.2 ppm I I 4. Sppm 24.1 ppm OH OH OH
16.7 ppm 20.7 ppm
95
63.0 ppm
\ - O 27.9 ppm
P 1.6 ppm
-0.2 ppm
H O — ^ 2.8 ppm
65.5 ppm
1 *V— O 38.2 ppm
M^ 5.9 ppm •
OH 16.5 ppm
96 97
42.2 ppm
I o I 62.2 ppm \
HO I OH
-03 ppm 15.6 ppm
98 99 100
Furans Compounds containing the furan ring appear widely in plants[99]. Some l^o
NMR data for the furan ring system have already been presented in the discussion of the spectral characteristics of coumarins and related compounds. In this section the focus will be on the basic furan system and some fused ring analogs[8,76,100,101]. A shielding effect of ca. 42 ppm on the l^O NMR signal of the furan oxygen is noted on fusion of a benzene ring(cf. 101-102 and 104-105) and it appears to be additive (cf. 102 and 103).
586
240 ppm 199 ppm
101 102 103
247.5 ppm 203 ppm
104 105
The l^O NMR chemical shift data for series of 2-substituted and 2,5-disubstituted furans failed to correlate with Hammett-type constants[101]. However, the data were well correlated with the parameter Q which depends, in part, upon the polarizability and ionization potential of the substituent.
Phenols and aryl ethers Numerous naturally occurring aromatic systems ranging from alkaloids to
lignans contain phenolic and aryloxy groups[102-105]. Several examples containing these functional groups have already been treated in connection with review of the ^^O NMR properties of coumarins, chromanones, etc.(v/d^ supra). The purpose of this section is to briefly review the l^Q NMR characteristics of such functional groups and, in particular, examine the chemical shifts of structures containing more than one of the single bonded oxygen functional groups, for example, systems which contain a 1,2,3-trimethoxyaryl ring. Multi-alkyloxyaryl rings appear in numerous important naturally occurring compounds such as podophyllotoxin, glyfoline and derivatives. As yet, none of the more complex structures have been studied by 1^0 NMR spectroscopy but much of the
587
needed basic results on simple phenols and multi-methoxy aryl have been reported. Additionally, also included in this section are l^o NMR results obtained in our laboratory on selected trimethoxyaryl systems which were selected to facilitate discussion.
1^0 NMR chemical shift vales of various phenolic compounds have been reported in earlier l^o NMR investigations[28,49,51,53,60,106], however a systematic study of phenols did not appear until recently [23 ]. Substituent electronic effects on the phenolic OH l^o NMR chemical shift is illustrated by the results for 108-110 and is similar to substituent effects on the methoxy signal of anisoles [24]. Electron donating groups causes shielding and electron withdrawing groups results in deshielding of the phenolic OH I ^ Q NMR chemical shift. Data for other phenols are given with the structures 111-114.
NH, NO2
108 109 110
OH OCH3
111 112 113 114
It is apparent by comparing the data for ortho and para isomers 111-112 and 113-114 that formation of an intramolecular hydrogen bond to the single bonded oxygen of both the OH and OCH3 groups causes shielding of the 1^0
588
NMR chemical shift. The hydrogen bond shift of 8-14 ppm is considerably smaller than noted for hydrogen bond induced chemical shift for carbonyl group 17o NMR chemical shifts(yide supra).
The influence of both electronic and steric factors on ^^O NMR chemical shifts of anisoles have been extensively studied[24,90]. The electronic effect of substituents is analogous to that of phenols which was previously discussed. The effect of orthO'SAkyl groups of varying bulk has been assessed and is illustrated in Table 6.
Table 6 1^0 NMR chemical shift data of methoxy groups of substituted anisoles in chloroform solution^ .
0CH3
Ri. JL .R2
No. Rl R2 5(ppm)
115 116 117 118 119
Me Et i-Pr t-Bu t-Bu
Me Et i-Pr t-Bu Me
16.5 16.7 13.5 27.3 27.0
^ Obtained at room temperature.
The deshielding shift for the highly hindered anisoles 118-119 relative to the less hindered ones 115-117 was suggested to arise from repulsive van der Waals interactions and/or bond angle deformation in these systems [90].
Results from studies on multi-methoxy aryl systems are indicated below with the structures 120-123. The greater shielding of the 1^0 NMR chemical shift for the ortho isomer 122 than that of the para isomer 121 was interpreted as arising from rotation of the methoxy groups from the plane of the aromatic ring as a result of repulsion between non-bonding electrons of the oxygen atoms[90]. Recent investigations employing GAUSSIAN-86 calculations and using the GIAO approach have resulted in the conclusion that the origin of the deshielding is repulsive van der Waals interactions between the two oxygen atoms[107,108]. An even greater shielding shift is noted for the signal of the 2-methoxy group of l,2,3,-trimethoxybenzene(123). Using the ortho-para equivalency assumption the
589
electronic effect of an ortho methoxy group is -8 ppm(cf. 120 and 121), consequently the chemical shift of 123 would be expected to be 32 ppm if only electronic effects were involved. The highly shielded value for for the 2-methoxy group of 123 is greater than two times the difference in chemical shifts of 121 and 122, presumably arising from even greater repulsive van der Waals interactions.
-6.1 ppm
37.1ppin OCH3 37.1 ppm
OCH3 H3CO^ 1 OCH3
OCH,
120 121 122 123
Data collected for this review in our laboratory from acetonitrile solutions for several substituted 1,2,3-trimethoxybenzenes are given below with their structures. The difference in chemical shift for 123 given below and that taken from the literature(see above) reflects the differences in solvent and temperature used for the two measurements. The ^^O chemical shift of the two equivalent methoxy groups(at the 3 and 5 positions) are essentially unaffected as the substituent at the 1-position is changed(124-128). In contrast, the signal for the methoxy group at the 4-position is influenced by the substituent at the 1-position . In the case of methyl and hydroxymethyl substitution(124 and 125) small shielding shifts consistent with those noted for anisoles[24] are observed. Substitution of vinyl and carbonyl groups(126-128) in the 1-position cause substantial downfield shifts of the 4-methoxy signal. The downfield shift of approximately 22 ppm for the l^O NMR signal for the 4-methoxy group of the natural product asarone(126) is significantly greater than the downfield shifts noted for the 4-methoxy signal for the carbonyl substituted compounds 127 and 128. In fact, the result is in contrast to that found on substitution of a vinyl group in the para position of anisole where a small shielding shift was noted[109]. The large downfield shift noted for the methoxy signal for 126 is consistent with increased double bond character of the 4-methoxy group(increased conjugation). If this is the case, a change in conformation of the 4-methoxy group might be expected which also would be likely to cause
590
conformational changes for the other two methoxy groups, however only small changes in the l^o NMR chemical shifts of the signal for the 3,5-dimethoxy groups are noted. Additional study is required for a more complete understanding of the factors which determine the ^^O NMR chemical shifts in these important 1,2,3-trimethoxyaryl systems.
1.3 ppm
OCH3 35.4 ppm .OCH3
-0.9 ppm
35.0 ppm OCH3 35.0 ppm
H3CO, JL .OCH3
-0.2 ppm
36.1ppm OCH3 26.1 ppm ' OCH3
5.0 ppm CH2OH
123 124 125
23.1 ppm
36.4 ppm
OCH3
126 127 128
Miscellaneous natural products A recent report describes the use of l^o NMR spectroscopy for confirmation
of stereochemistry of a dimethoxy derivative of abietic acid methyl ester [110]. The assignment of the axial stererochemistry to both of the methoxy groups in 129 is based upon ID and 2D ^H and ^^C NMR spectroscopic measurements.
591
l^O NMR chemical shifts and their assignment for 129 are shown with the structure below. The l^o NMR signals for the two methoxy groups in this diterpene apparently overlap at 1.6 ppm. The chemical shift of 1.6 ppm is viewed by the authors as consistent with values of methoxy groups in axial positions of simple cyclohexane ring systems and the assignment was made on that basis[l 10].
129 The antimalarial arteminisin(130) contains an endo cyclic peroxide group , a
functional group which rarely appears in natural products.
CH, 267.2 ppm „ V ^
H,C
88.0 ppm I H
n II CH3
371.1 ppm
130
Fig. 10 contains the l^o NMR spectrum of arteminisin obtained at 40oC in acetonitrile. The resonance for the peroxy group is readily recognized as the signal at 267 ppm due to its large line width, approximately 1900Hz[lll,112]. The lactone carbonyl signal appears at 370 ppm and the signal at 200 ppm is assigned to the lactone single bond oxygen. The remaining signal at 88 ppm is
592
attributable to the ketal-like oxygen and is in the region noted earlier for ketal type oxygens[76,113].
I I r [ f
4 0 0
I I I I I I t I
300 ' I ' l l
200 I r I I M f j F T | -
100 0 PPM
Fig. 10. 17o NMR spectrum of artemisinin (130), 0.5 M in acetonitrile at 40<>C, internal reference 2-butanone 558 ppm, external reference water 0 ppm.
Pyrylium salts provide the brilliant color in many flowers and consequently these cationic oxygen containing heterocyclic compounds have been extensively studied[l 14]. While numerous physical studies on these systems have been performed the first l^O NMR investigation was reported only recently[115]. ^^O NMR chemical shifts of a series of isobenzopyrylium salts were found to be sensitive to substitution at the 1- and 3-postion which is illustrated by the data given with the structures 131-134. It is interesting that a larger shielding shift is noted for replacement of the 3-methyl by a phenyl group than the substitution of
593
the 1-methyl group by a phenyl one (cf. 131-133). The origin of this difference is not clear, but it may be a result of different conformations of the phenyl group at the two different postions. The 1-phenyl group is likely to be more twisted out of the plane of the isobenzopyrylium ring than a phenyl group at the 3-position. The effect of substitution of phenyl groups appears to be approximately additive since the l^o NMR chemical shift value of 134 is shielded by 30 ppm compared to 131. If the effect of phenyl group substitution were exactly additive the shielding shift of 134 would have been predicted to be 27 ppm .
300 ppm 289 ppm 284 ppm 270 ppm
CH,
131 132 133 134
Several other natural products systems have been studied, some quite extensively, by l^o NMR methodology, however space constraints prohibt detailed discussion of these systems therefore only important leading references will be given. Extensive l^o NMR studies on amino acids and small peptides have been performed by Lauterwein and coworkers [116-119], by Fiat and coworkers [120-123] and others [124]. Several studies have used l^O-enriched dioxygen and carbon monoxide to study by I ^ Q NMR techniques the interactions of these biochemically important small molecules with various proteins [125-128]. A number of investigators have explored the properties and interactions of nucleic acid bases [129,130], nucleosides [131,132], nucleotides [133-138] and one report has appeared in which l^o NMR spectroscopy approaches were applied to the study of small molecule-DNA interactions [139]. A recent report describes the careful analysis of the effect of structure on l^o NMR chemical shifts of over forty hydroxyterpenoids [140]. A study of the 1^0 NMR spectroscopy of over thirty steroid ketones, acids, esters and alcohols enriched with l^O has recently appeared [141].
CONCLUSIONS The 17o NMR chemical shift patterns for the common and not so common
functional groups encountered in naturally occurring compounds provides a
594
Strong foundation for assisting with structure elucidation and analysis of stereochemical relationships. Even though most of the relationships developed between structure and 1^0 NMR chemical shift are empirical they are valuable predictive tools. Increasing interest in and the study of the origins of l^O NMR chemical shifts should place these relationships on a more solid theoretical foundation in the years ahead. The rapid growth in numbers of publications which apply ^^O NMR spectroscopy to natural product problems clearly indicates the growing importance of this methodology to the field.
ACKNOWLEDGMENTS Acknowledgment is made to the Donors of the Petroleum Research Fund,
administered by the American Chemical Society, for partial support of this research, to NSF (CHE-8506665), and most especially to my colleagues, whose names are cited in the appropriate publications in the reference section, without whom this chapter would not have been possible.
7. D.W. Boykin and A.L. Baumstark, Tetrahedron, 45 (1989) 3613-3651. 8. J.-P. Kintzinger, in: P. Diehl, E. Fluck, R. Kosfield (Eds), NMR-17,
Oxygen-17 and Silicon-29, Springer-Verlag, Berlin, 1981, pp 1-64. 9. J.-P. Kintzinger, in: P. Laszlo (Ed), NMR of Newly Accessible Nuclei,
Academic Press, New York, 1983, Vol. 2, pp 79-104. 10. W.G. Klemperer, in: J.B. Lambert and E.G. Riddell (Eds), Reidel,
Dordrecht, Holland, 1983, pp 245-260. n . M. Karplus and J.A. Pople, J. Chem. Phys., 38 (1963) 2803-2807. 12. R.T.C. Brownlee, M. Sadek and D.J. Craik, Org. Magn. Reson., 21 (1983)
616-620.
595
13. A. Kumar and D.W. Boykin, J. Mol. Struct., 296 (1993) 95-101. 14. H. Dahn, P. Pechy and V.V. Toan, Angew. Chem., Int. Ed. Engl., 29 (1990)
647-648. 15. H. Dahn and P. Pechy, J. Chem. Soc. Perkin Trans. 2, (1993) 67-70. 16. H. Dahn and P. Pechy, J. Chem. Soc. Perkin Trans. 2, (1991) 1721-1723. 17. H. Dahn and P. Pechy, J. Chim. Phys., 89 (1992) 1683-1687. 18. H. Dahn, P. Pechy and V.V. Toan, Magn. Reson. Chem., 28 (1990) 883-887. 19. J. Spychala and D.W. Boykin, J. Chem. Research, in press. 20. D.J. Craik, G.C. Levy and R.T.C. Brownlee, J. Org. Chem., 48 (1983)
1601-1606. 21. D.W. Boykin, A.L. Baumstark and P. Balakrishnan, Magn. Reson. Chem.,
23 (1985), 276-279. 22. M. Sawda, Y. Takai, S. Kimura and S. Misumi, Tetrahedron Letters, (1986),
3013-3016. 23. D.W. Boykin, S. Chandrasekaran and A.L. Baumstark, Magn. Reson. Chem.,
31 (1993)489-494. 24. M. Katoh, T. Suguwara, Y. Kawada and H. Iwamura, Bull. Chem. Soc. Jpn.,
52 (1977) 3475-3476. 25. S.A. Evans, Jr., in: D.W. Boykin (Ed), I ^ Q N M R Spectroscopy in Organic
Chemistry, CRC Press, Boca Raton, PL, 1991 Chapt. 10, pp 263-320. 26. J.W. Kelly and S.A. Evans, Jr., Magn. Reson. Chem., 25 (1987) 305-308. 27. G. Modena, U. Quintily and G. Scorrano, J. Am. Chem. Soc, 94 (1972)
202-208. 28. H.A. Christ and P. Diehl, Helv. Phys. Acta, 36 (1963) 170-182. 29. D.J. Sardella and J.B. Strothers, Can. J. Chem., 47 (1969) 3089-3092. 30. E.L. Eliel, K.-T. Liu and S. Chandrasekaran, Org. Magn. Reson., 21 (1983)
179-182. 31. P. Balakrishnan and D.W. Boykin, J. Org. Chem., 50 (1985) 3661-3663. 32. M.G. Oakley and D.W. Boykin, J. Chem. Soc. Chem. Commun., (1986) 439-
441. 33. A.L. Baumstark, P. Balakrishnan, M. Dotrong, C.J. McCloskey, M.G.
Oakley and D.W. Boykin, J. Am. Chem. Soc, 109 (1987) 1059-1062. 34. H. Cerfontain, C. Kruk, R. Rexwinkel and F. Stunnberg, Can. J. Chem., 65
26 (1988) 19-23. 36. D.W. Boykin, Spectroscopy Letters, 25 (1992) 1199-1205. 37. D.W. Boykin and A. Kumar, Spectroscopy Letters, 24 (1991) 723-731. 38. R. Benassi, U. Folli, D. larossi, A. Mucci, L. Schenetti and F. Taddei, Magn.
Reson. Chem., 28 (1990) 702-710. 39. D.W. Boykin, P. Balakrishnan and A.L. Baumstark, Magn. Reson. Chem.,
23 (1985) 695-697. 40. D.W. Boykin, P. Balakrishnan and A.L. Baumstark, J. Heterocycl. Chem.,
22 (1985) 981-984. 41. A.L. Baumstark, P. Balakrishnan and D.W. Boykin, Tetrahedron Letters,
(1986) 3079-3082.
596
42. D.W. Boykin, A.L. Baumstark, M.M. Kayser and CM. Soucy, Can. J. Chem., 65 (1987) 1214-1217.
43. D.W. Boykin, R.L. Hertzler, J.K. Delphon and E.J. Eisenbraun, J. Org. Chem., 54 (1989) 1418-1423.
44. A.L. Baumstark, M. Dotrong, M.G. Oakley, R. Stark and D.W. Boykin, J. Org. Chem., 52 (1987) 3640-3643.
45. S. Li and D.B. Chesnut, Magn. Reson. Chem., 24 (1986) 96-100. 46. S. Li and D.B. Chesnut, Magn. Reson. Chem., 23 (1985) 625-638. 47. D.B. Chesnut, D.W. Wright and B.A. Krizek, J. Mol. Structure, 190 (1988)
99-111. 48. D.W. Boykin, B. Dewprashad and E.J. Eisenbraun, J. Org. Chem., 55
(1990)425-429. 49. H.A. Christ, P. Diehl, H.R. Schneider and H. Dahn, Helv. Chim. Acta, 44
(1961) 865-880. 50. J. Reuben, J. Am. Chem. Soc, 91 (1968) 5725-5729. 51. T.E. St. Amour, M.I. Burgar, B. Valentine and D. Fiat, J. Am. Chem. Soc,
103(1981) 1128-1136. 52. S. Chandrasekaran, W.D. Wilson and D.W. Boykin, Org. Magn. Reson., 22
(1984) 757-760. 53. G. Jaccard and J. Lauterwein, Helv. Chim. Acta, 69 (1986) 1469-1485. 54. A.L. Baumstark, S.S. Graham and D.W. Boykin, J. Chem. Soc. Chem.
Commun., (1989) 767-768. 55. A.L. Baumstark, S.S. Graham and D.W. Boykin, Tetrahedron Letters (1990)
957-960. 56. D.W. Boykin, A.L. Baumstark and M. Beeson, J. Org. Chem., 56 (1991)
1969-1971. 57. D.W. Boykin and A. Kumar, J. Heterocycl. Chem., 29 (1992) 1-4. 58. D.W. Boykin, J. Mol. Struct., 295 (1993) 39-45. 59. D.W. Boykin and A. Kumar, J. Mol. Structure, in press. 60. A.L. Baumstark and D.W. Boykin, New J. Chem., 16 (1992) 357-360. 61. S. Chandrasekaran and D.W. Boykin, Heteroatom Chem., 3 (1992) 63-67. 62. C. Delseth, J.-P. Kintzinger, T.T.T. Nguyen and H. Neiderberger, Org.
Magn Reson., 11 (1978)97-110. 63. M. Schumacher and J. Lauterwein , J. Magn. Reson., 83 (1989) 97-110. 64. H. Duddeck, in: Atta-ur Rahman (Ed), Studies in Natural Products
Chemistry, Vol. 9, Elsevier, Amsterdam (1991), pp 109-126. 65. D.W. Grandy, L. Petrakis, D.C. Young and B.C. Gates, Nature, 308 (1984)
W.D. Lust, M. Mattingly and W. Kuhn, in: T.A. Baillie, J.R. Jones (Eds), "Synthesis and Applications of Isotopically Labelled Compounds, 1988." Proceedings of the Third International Symposium, Innsbruck, Austria, 17-21 July 1988, Elsevier, Amsterdam, 1989, pp 499-508.
67. D.W. Boykin, A.L. Baumstark, A. Mehdizadeh and M.K. Venkatramanan, Magn. Reson. Chem., 28 (1990) 305-310.
68. C. Delseth and J.-P. Kintzinger, Helv. Chim. Acta, 59 (1976) 466-475. 69. C. Delseth, T.T. T. Nguyen and J.-P. Kintzinger, Helv. Chim. Acta, 63
(1980) 498-503.
597
70. D.W. Boykin and A.L. Baumstark in: D.W. Boykin(Ed), 17o NMR Spectroscopy in Organic Chemistry, CRC Press, Boca Raton, FL, 1991, pp. 205-231.
71. J. K. Crandall, M.A. Centro, and S. Borresen, J.Org. Chem., 44 (1979)1184-1186.
72. E. Kolehmaninen, K. Laiha, M. Heinanen, K. Rissanen,R. Frohlich, J. Korvola, P. Manttari and R. Kauppinen, J. Chem. Perkin Trans. 2, (1993) 641-648.
73. D.W. Boykin, P. Balakrishnan and A. L. Baumstark, Magn. Reson Chem. 25 (1987) 248-250.
75. J.M. Cassady and M. Suffness, in: J.M. Cassady and J.D. Douros (Eds), "Anticancer Agents Based on Natural Product Models," Academic Press, New York, 1980, pp 201-269.
76. S. Chandrasekaran in: D.W. Boykin(Ed), l^o NMR Spectroscopy in Organic Chemistry, CRC Press, Boca Raton, FL, 1991, pp. 141-203.
77. J. P. Monti, R. Faure, A. Sauleau and J. Sauleau, Magn Reson. Chem., 24 (1986) 15-20 .
78. D.W. Boykin, D.W. SuUins, N. Phourhmady and E.J. Eisenbraun, Heterocycles, 29 (1989) 307-31.
80. A. L. Baumstark, M. Dotrong, R.R.Stark and D.W. Boykin ,Tetrahedron Letters, (1988) 2143-2146.
81. D.W. Boykin, A. Kumar and S.M. Shevchenko, Magn. Reson. Chem., 30 (1992) 16-19.
82. S. G. Semenov and S.M. Shevchenko, Zh. Org. Khim., 21 (1985) 368-372. 83. U. Sankawa, Y. Ebizuba, H. Noguchi, Y. Isikawa, S. Kitaghawa, T.
Yamamoto, T. Kobayashi, Y. litak and H. Seto, Tetrahedron, 39 (1983) 1061-1068.
84. R.D.H. Murray, J. Mendez and S. A. Brown, "The Natural Coumarins", John Wiley, New York, 1982.
85. H. Duddeck, D. Rosenbaum, M. Hani, A. Elgamal and N.M.M. Shalaby, Magn. Reson. Chem.,25 (1989) 489-491.
86. Y. Ming and D.W. Boykin, Heterocycles, 26 (1987) 3229-3231. 87. A. Kumar and D.W. Boykin, Indian J. Heterocyclic Chem., in press. 88. H. Duddeck and A. Levai, Magn. Reson. Chem., 30 (1992) 65-67. 89. H. Duddeck, J.C. Jaseberenyi, A. Levi, T. Timar and M.H.A. Elgamal,
Magn, Reson. Chem., 27 (1989) 170-172. 90. M.A. Wysocki, P.W. Jardon, G.J. Mains, E.J. Eisenbraun and D.W. Boykin,
Magn. Reson. Chem., 25 (1987) 331-334. 91. K. Nagasawa, Y. Higuchi, K. Ito, M. Imanari and N.Fujii, Chem. Pharm.
93. J.-C. Wallet, E. M. Gaydou and M.T. Cung, Bull. Liaision Groupe Polyphenols,(1990) 207-210.
94. J. Lauterwein, J.Schulte, M. Schumacher,and M. Cerny,, Magn. Reson. Chem.,30 (1992) 312-319.
95. I.P.Gerothanassis, J. Lauterwein and N. Sheppard, J. Magn. Reson., 48 (1982)431-446.
96. J. Lauterwein and LP. Gerothanasis, J. Magn. Reson. 51 (1983) 153-156. 97. J. Shulte and J. Lauterwein, Magn. Reson. Chem. 30 (1992) 334-337. 98. P.A.L. Gorin and M. Mazurek, Carbohydrate Research, 67 (1978) 479-483. 99. P.M. Dean, "Naturally Occurring Oxygen Ring Compounds",
Butterworths,London, 1963. 100. S. Chimichi, R. Nesi and F. DeSio, Org. Magn. Reson. 22 (1984) 55-
56. 101. E.L. Clennan and M.E. Mehrsheik-Mohammadi, Magn, Reson. Chem.,
23 (1985) 985-987. 102. D. C. Ayres and J.D. Loike, "Lignans", Cambridge University Press,
Cambridge, 1990. 103. E. Haslam, "Plant Polyphenols",Cambridge University Press,
Cambridge, 1989. 104. G.A. Cordell, "Introduction to Alkaloids", John Wiley and Sons, New
York,1981. 105. T.-L. Su and K. A. Wantanabe in: Attar-ur Rahman and F.Z.
Basha(Eds),Studies in Natural Products Chemistry,Vol. 13, Elsevier,Amsterdam, 1993, pp 347-432.
106. V.V. Lapachev, I.Y. Mainagashev, S.A. Stekhova, M. A. Fedotov, V.P. Krivopalov and V.P. Mamaev, J. Chem. Soc, Chem. Commun., (1985) 494-495.
107. J.C. Facelli, R.C. Contreras and M.F. Tufro, J. Mol. Structure(Theochem), 281 (1993) 61-66.
108. R.C. Contreras, R. R. Biekofsky, D. G. de Kowalewski, A. M. Orendt and J. C. Facelli, J. Phys. Chem., 97 (1993) 91-93.
109. G.A. Kalabin, D.F. Kushnarev, R.B. Valeyev, B.A. Torofimov and M.A. Fedotov, Org. Magn. Reson. 18 (1982) 1- 9.
110. H. Kahlig, P.P.Nimmerrichter and W. Robien, Monatshefte Chemie, 124(1993)71-75.
111. A. L. Baumstark, P.C. Vasquez and P. Balakrishnan, Tetrahedron Letters, (1985)4485-4488.
112. M.G. Zagorski, D. S. Allan, R.G. Salomon, E. L.Clennan, P.C. Heah, and R. P. L'Esperance, J. Org. Chem., 50(1985)4484-4490.
113. M. Manoharan and E. L. Eliel, Magn. Reson. Chem., 23 (1985) 225 -231.
114. A.T. Balaban, A. Dinculescu, G. N. Dorofenko, A.V. Koblik, U.V. Mezheritskii and W. Scroth,"Pyrylium Salts, Synthesis and Physical Properties", Supp. 2, Advances in Heterocyclic Chemistry, Academic Press 1982.
599
115. A. Saba, F. S. Sib and J-P. Aycard, Spectroscopy Letters, 26 (1993) 491-496.
116. R. Hunston, LP. Gerothanassis and J. Lauterwein, Org. Magn. Reson.,18 (1982) 120-121.
117. I. P. Gerothanassis, R. N. Hunston and J. Lauterwein, Magn. Reson. Chem., 23 (1985) 659-665.
118. J. Lauterwein, LP. Gerothanassis and R. N. Hunston, J. Chem. Soc, Chem. Commun., (1984) 367-369.
119. R. N. Hunston, LP. Gerothanassis and J. Lauterwein, J. Am. Chem. Soc.,107 (1985) 2654-2661.
120. J. Tritt-Goc and D. Fiat, Magn. Reson. Chem., 29 (1991) 156-163. 121. B. Valentine, A. Steinschneider, D. Dhawan, M. L Burgar, T. St.
Amour and D. Fiat, Int. J. Peptide Protein Res., 25 (1985) 56-68. 122. H. Gilboa, A. Steinschneider, B. Valentine, D. Dhawan and D. Fait,
Biochim Biophys. Acta, 800 (1984) 251- 257. 123. M.L Burgar and D. Fiat, J. Phys. Chem., 85 (1981) 502-510. 124. C. Sakarellos, LP. Gerothanassis, N. Birlirakis, T. Karayanis, M.
Sakarrllos-Daitsiotis and M. Marraund, Biopolymers, 28 (1989) 15-26. 125. H. C. Lee, K. Cummings, K. Hall, L. P. Hager and E. Oldfield, J. Biol.
Chem., 263 (1988) 16118-16124. 126. L P. Gerothanassis, M. Momenteau and B. Loock, J. Am. Chem. Soc,
111(1989)7006-7012. 127. K. D. Park, K. Guo, F. Adebodun,M. L. Chiu, S. G. Sligar and E.
Oldfield, Biochemistry, 30 (1991) 2333-2347. 128.1. P. Gerothanassis, B. Loock and M. Momenteau, J. Chem. Soc,
Chem.. Commun., (1992) 598-600. 129. M. I. Burgar, D. Dhawan and D. Fait, Org. Magn. Reson., 20 (1982)
184-190. 130. S. Chandresekaran, W. D. Wilson and D.W. Boykin, J. Org. Chem., 50
(1985)829-831. 131. J. A. Coderre, S. Mehdi, P.C. Demmou, R. Weber, D.D. Traficante and
J. A. Gerlt, J. Am. Chem. Soc, 103 (1981) 1870-1872. 132. H.M. Schwartz, M. MacCross and S. S. Danyluk, J. Am. Chem. Soc,
105(1983)5901-5911. 133. M.D. Tsai in: D. L. Purich(Ed), Methods of Enzymology, Academic
Press, New York, 87 (1982) 235- 279. 134. S. L. Huang and M. D. Tsai, Biochemistry, 21 (1982) 951-959. 135. M. Petershiem, V. W. Miner, J.A. Gerlt and J.H. Prestegard, J. Am.
Chem. Soc, 105 (1983) 6357-6358.
600
136. S. Mehdi and J.A. Gerlt, Biochemistry, 23 (1984) 4844-4852. 137. D.A. Wisner, C.A. Steginsky, Y.-J. Shyy and M.D. Tsai, J. Am. Chem.
Soc, 107(1985)2814-2815. 138. M. A. Reynolds, J.A. Gerlt, P.C. Demon, N. J. Oppenhiemer and G. L.
Kenyon, J. Am. Chem. Soc, 105 (1983) 6475-6481. 139. W.D. Wilson, Y.-H. Wang, S. Kusuma, S. Chandresekaran and D. W.
Boykin, Biophys. Chem., 24 (1986) 101-109. 140. E. Kolehmainen and K. Laihia, Magn. Reson. Chem., 31 (1993) 763-