Approximate Labelled Subtree Homeomorphism Based on : “Approximate Labelled Subtree Homeomorphism” R. Y. Pinter, O.Rokhlenko, D. Tsur, M. Ziv-Ukelson “Alignment of Metabolic Pathways” R. Y. Pinter, O. Rokhlenko, E. Yeger- Lotem, M. Ziv-Ukelson
Transcript
Approximate Labelled Subtree Homeomorphism
Based on: “Approximate Labelled Subtree Homeomorphism”
R. Y. Pinter, O.Rokhlenko, D. Tsur, M. Ziv-Ukelson
“Alignment of Metabolic Pathways” R. Y. Pinter, O. Rokhlenko, E. Yeger-Lotem,
M. Ziv-Ukelson
The general Idea
Biological Problem
Converting into terms of computer science problem
Finding the solution
Reverting back to Biological terms
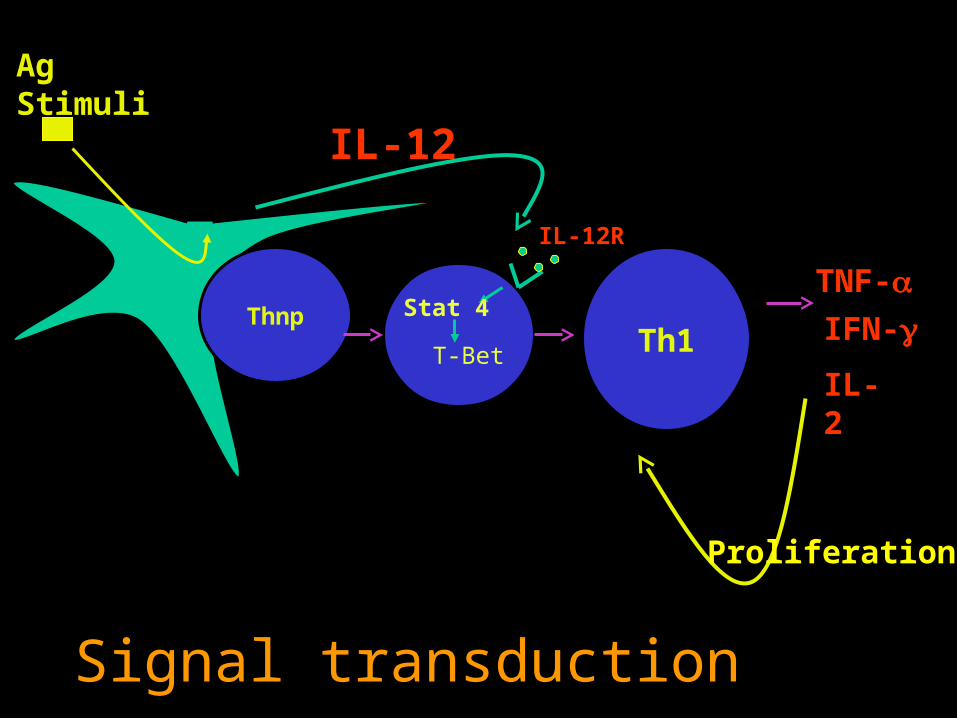
Metabolism
IL-2Th1
TNF-IFN-
Proliferation
IL-12
Ag Stimuli
Thnp
IL-12R
T-Bet
Stat 4
Signal transduction
Why pathways?
Metabolic and regulatory pathways
have biological importance. These pathways are evolutionary
conserved.
What do we want to do?
Compare one metabolic pathway of a
certain organism against the same
metabolic pathways in other
organisms. Compare a metabolic pathway against
other metabolic pathways in the same
organism.
How do we do (it)?
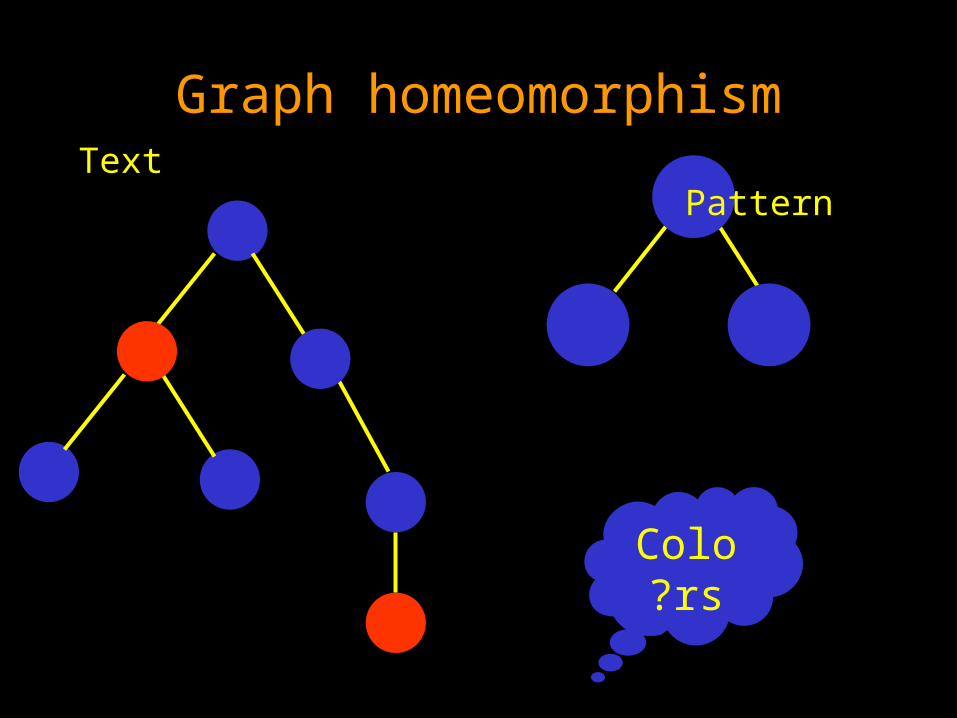
The subtree homeomorphism problem:
Given a pattern tree P and a text tree T, find a subtree of T which is isomorphic to P or decide that there is no such tree.
Degree 2 node can be deleted from the text tree.
?
Graph homeomorphism Text
Pattern
Colors
?
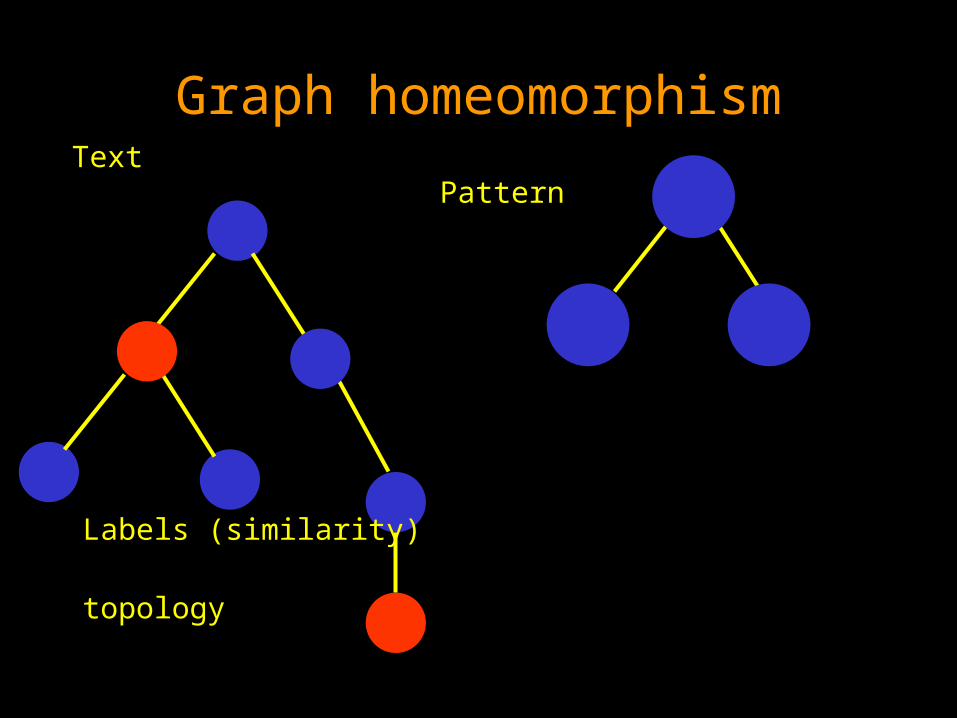
Graph homeomorphism Text
Pattern
Graph homeomorphism Text Pattern
Labels (similarity)
topology
Back to 2nd semester… An unrooted tree is an undirected, acyclic,
connected graph (T=(VT,ET((
A rooted tree is a triple Tr=(VT,ET,r( where
(VT,ET( is an unrooted tree, and r is some vertex
in V which is called the root. The root node of the
tree implies the direction for all the edges in the
graph. A multi-source tree is an acyclic, directed graph,
whose underlying undirected graph is a tree.
Back to 2nd semester…
A tree is said to be ordered if the relative order of its subtree in each node is fix. Otherwise a tree is unordered.
for “ordered”
Problem:
What are we allowed to do?
Taking into account both label similarity
and topology. We are permited to delete vertexes
from the text tree. We are NOT permited to delete vertexes
from the pattern tree.
What we “gonna” see today:
Rooted unorderedO(m2n + mn log n)
Unrooted unorderedO(m2n + mn log n)
Directed multi source unordered
O(m2n + mn log n)
Rooted orderedO(mn)
Some definitions:
Let Δ denote a predefined node-to-node similarity score table.
Let D denote a predefined score for deleting a
node from a tree (usually a penalty). A mapping M from T1 to T2 is a partial one to one
map from the nodes of T1 to the nodes of T2 that preserves the ancestor relations of the nodes.
Our problem:
Let M be a mapping from T1 to T2 . The Labelled Subtree Homeomorphic Similarity Score of
M[T1,T2] is:
LSH (M[T1,T2]) = D (|T1|-|T2|) + ∑ (u,v) ∈ M Δ]u,v]
Given two undirected labeled trees P and T, We want to find a mapping M and a subtree t of T, such that:
LSH (M [t,P]) is maximal.
Scoring
Text Pattern
Score
-1-1
+2-2
-2+2Score:2
Score:2
Score:5
Dynamic programming
vu
x1 x2
y3y2y1
TP
x1x2…u…
y1w11w12w1m
y2w21w22w1m
y3w31w32w1m
…
vwn1wn1wnm
RScore[u,v] is the maximum between two terms:
The node-to-node similarity value Δ[v,u] plus the sum of the weights of the matched edges in the maximal assignment over G. This term is only compute if c(u) ≤ c(v) (otherwise: -∞).
The weight RScore[yi,u] for the comparison of u and the best scoring child yi of v, updated with the penalty for deleting v.
C(u) is the number of the children of u
RScore[u,v] - example
Pattern Text
score matrix
deletion = -1
ab
u10-∞v9
deletion
8
w8
deletion
12
ab
u1010
v5-2
w33
a
b
u
v
w
The assignment problem
Let G be a bipartite graph G = (V = X U Y,E) with weights w (x,y) for all edges. The assignment problem is to compute a matching M (list of monogamic pairs) such that: The size of M is maximal among all the
matchings. From all the matchings above, The sum of
the weights is maximal.
Solving the assignment problem
Reduction from the assignment problem to the min cost max flow problem. We’ll construct G’ which contains G(V,E) with the following changes:
Two more vertexes: s,t Edges from s to X and from Y to t, while w (s,x) = 0, w (y,t) =0 The cost of the other edges in E is –w (x,y) The capacity of all edges is 1
What is it? Among all the maximal flows we’ll choose the
cheapest
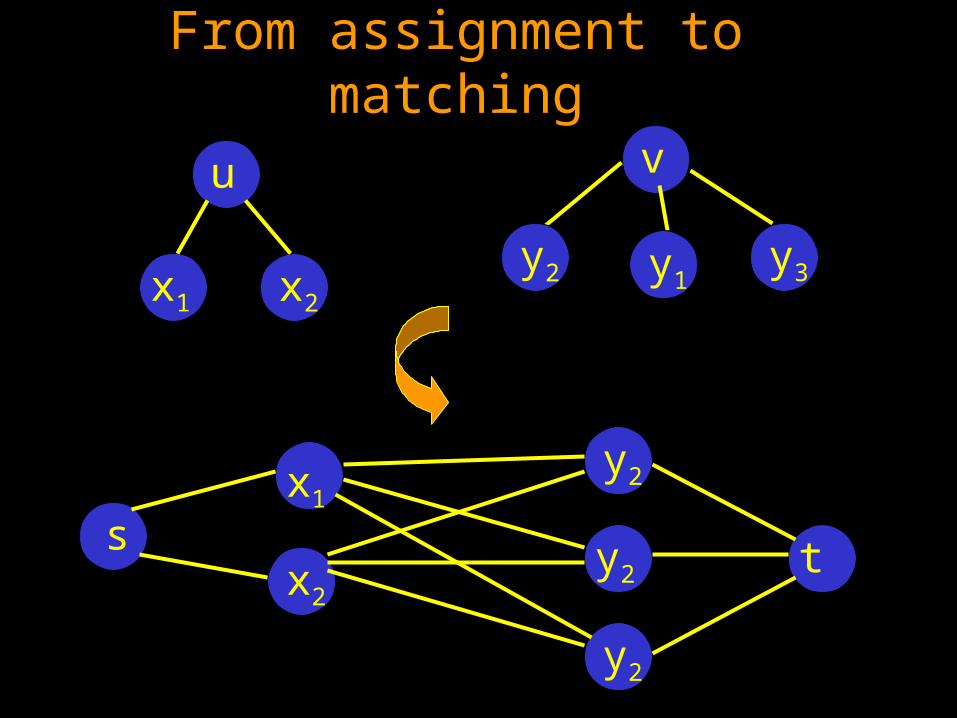
From assignment to matching
u
x1 x2
v
y3y1y2
x1
x2
y2
y2
y2
s t
Time complexity analysis
Edmonds and Karp’s algorithm: O(EV*logV)
Fredman and Tarjan: O(VE + V2logV) (independent of the edges cost)
Gabow and Tarjan: O(V1/2Elog(VC) where the input costs are integers and in the range [-C,….,C] (the similarity assumption)
Reminder…
What did we have so far?
Motivation“Advanced” homeomorphism: labels
and topologyScoring and deletionDynamic programmingMatchingQuestions?
The algorithm for rooted unordered trees:
Input: Rooted trees T = (VT,ET,r) and
P = (VP,EP,r’ )).
Output: The root of the subtree t of T which has the highest similarity score to P, (and homeomorphic to P).
for each node u of P in postorder dofor each node v of T in postorder do
if u is leaf thenif v is leaf then
RScore(v, u) = Δ [v,u]else
RScores(v,u) = ComputeScores (v,u)end if
elseif Level(u) > Level(v)
then RScore(v, u) = -∞else RScores(v,u) = ComputeScores (v,u)
end if ; end if; end for; end for
Dynamic programming
Node to node score
Delete from the pattern
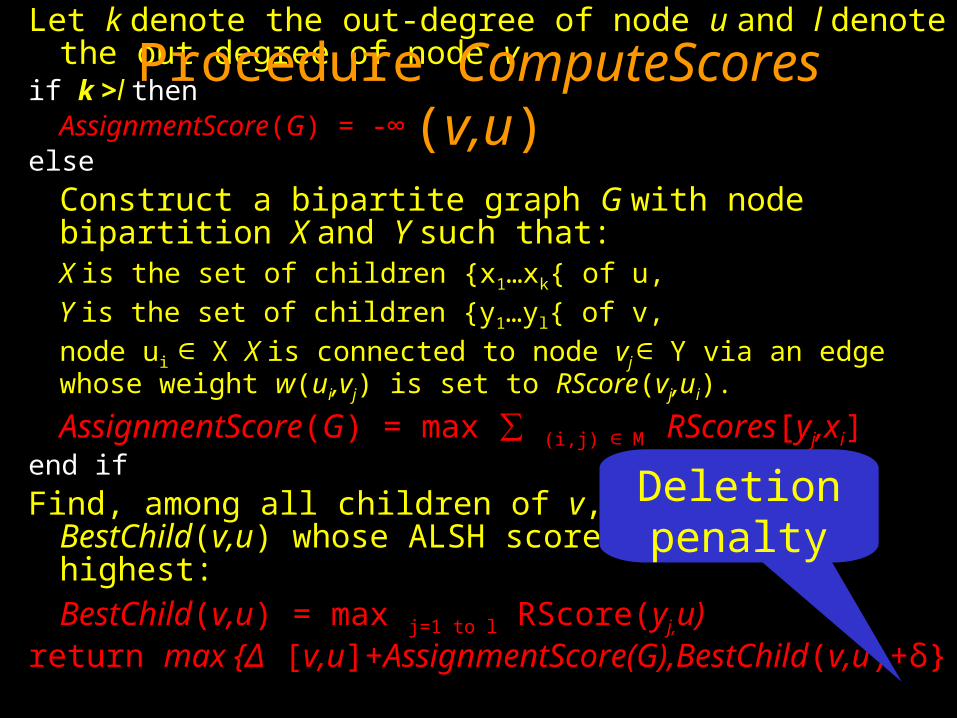
Let k denote the out-degree of node u and l denote the out degree of node v
if k >l thenAssignmentScore(G) = -∞
else
Construct a bipartite graph G with node bipartition X and Y such that: X is the set of children {x1…xk{ of u,
Y is the set of children {y1…yl{ of v,
node ui ∈ X X is connected to node vj ∈ Y via an edge whose weight w(ui,vj) is set to RScore(vj,ui).
AssignmentScore(G) = max ∑ (i,j) ∈ M RScores[yj,xi]end if
Find, among all children of v, the node BestChild(v,u) whose ALSH score with u is highest: BestChild(v,u) = max j=1 to l RScore(yj,u)
return max {Δ [v,u]+AssignmentScore(G),BestChild(v,u)+δ}
Procedure ComputeScores (v,u)
Deletion penalty
Time complexity analysis
Observation 1:
∑u =1 to m c(u) = m-1
∑v =1 to n c(v) = n-1
The number of the vertexes in the pattern
Time complexity analysisThe weighted assignment is computed once
for each pair u,v u T, v PIn a bipartite graph there are c(v)+c(u) nodes
and c(v)c(u) edges. Based on Fredman and Tarjan the time complexity is:
O(∑u=1 to m ∑ v=1 to n)c(u)2)c(v)+c(u)c(v) log (c(v))
= (observation 1)
O(∑u=1 to m c(u)2)n+c(u)n log n) = (observation 1)
O(m2n + mn log n)
Unrooted unordered trees:
The problem: each vertex in both the text tree and the pattern tree can be the root.
The naïve solution: choose an arbitrary node r of T to get a rooted tree. Next, for each u P compute rooted ALSH between Pu and Tr.
Time complexity: O(m3n+m2n log n)
2nd try: Select an arbitrary node r in T as the root For each internal node in T (in post order)
and for each node in P compute an
“improved” matching problem
2nd try: Select an arbitrary node r in T as the root For each internal node in T (in post order)
and for each node in P compute an
“improved” matching problem
2nd try: Select an arbitrary node r in T as the root For each internal node in T (in post order)
and for each node in P compute an
“improved” matching problem
General idea for keeping the time complexity
Find the best match between the children {x1,..,xn) of v∈T and {y1,…,ym} of u∈P.
After computing the best match and removing a node xi (which act as the parent of u) there is a way to find the optimal matching between {x1,…,xn}\xi and {y1,…,ym} in O(d(u)c(v)+c(v) log c(v))
The total time complexity for computing all assignments between v and u: O(d(u)2c(v)+d(u)c(v) log c(v))
Time complexity
Observation 2: The sum of vertex degrees in an unrooted tree P is
∑u =1 to m d(u) = 2m-2
We’ve study that at
Combinatorics
Time complexity – continue…
O((∑u =1 to m ∑v =1 tond(u)2c(v))+d(u)c(v) log c(v))
=
O((∑u =1 to m d(u)2n +d(u)n log n)
=
O(m2n + mn log n)Observation 1
Observatin 2
Up the tree…
For each vertex v∈T, u∈P and xi∈ neighbors (u), UScore[v,u, xi[ is the maximal LSH between a subtree pu,xi of P and a corresponding homeomorphic subtree of tv,r if one exists.
otherwise, UScore[v,u,xi] is set to -∞
A subtree in P which his root is u and the
root’s parent is xi
UScore[u,v,xi] is the maximum between two
terms: The node-to-node similarity value Δ[v,u] plus
the sum of the weights of the matched edges in the maximal assignment over Gi. This term is only compute if d(u) - 1 c(v)
(otherwise: -∞). The weight UScore[yi,u,xi] for the comparison
of u and the best scoring child yi of v, updated with the penalty for deleting v.
d(u) is the degree of u
And if ‘u’ is the root…
We have to compute an additional entry
UScore[v,u,Φ]. This entry represent the fact that u
might be the root of P. The root of P will be node u such that:
UScore[v,u,Φ] is maximal.
Multi-source graphs
DAG = Directed Acyclic Graph. A multi-source tree is a DAG whose its
underlying structure is an unrooted,
unordered trees.
Multi-source graph - example pattern text
UScore[u,v,r’] = -∞
r’ r
u v
Multi-source graphs & alignment
We’ll use the algorithm for the unrooted unordered tress.
We’ll filter out subtree alignments that map together edges of conflicting direction.
We’ll split the bipartite graph G = {X U Y,E} into two different graphs: one correspond to macthing of incoming-edge neighbors of u and v and the other for matching outgoing edge neighbors.
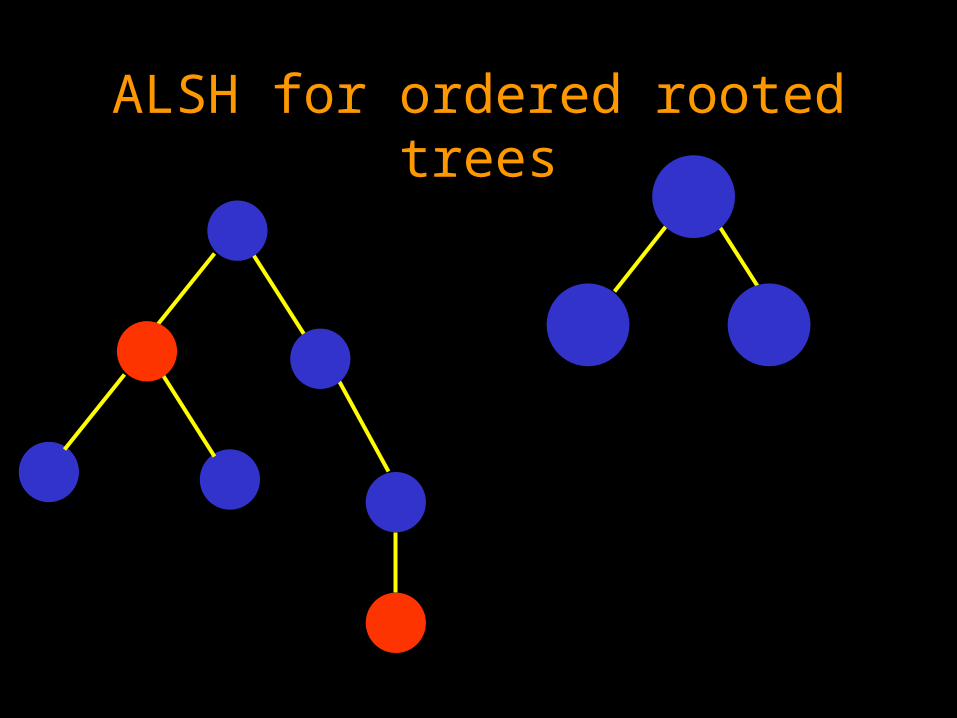
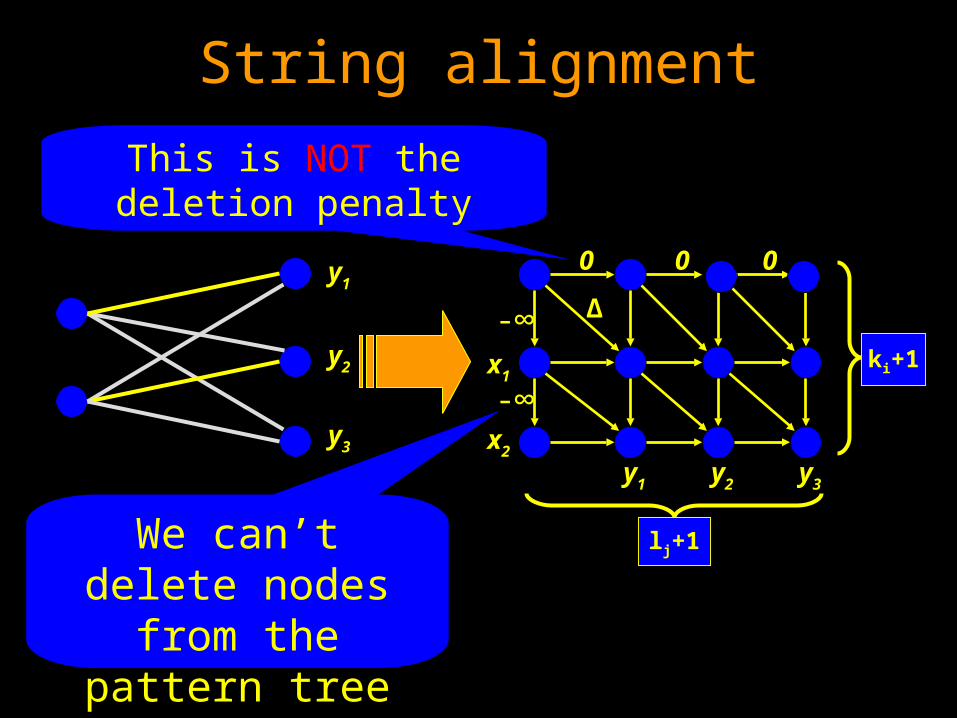
ALSH for ordered rooted trees
Solving ALSH for ordered rooted trees
Maximum weighted matching problem on ordered bipartite graphs, where no edges are allowed to cross.
Given a pattern string X, a source Y, and a character to character similarity table Δ[∑X, ∑Y], find among all |X|-sized subsequences of Y the subsequence Q which is most similar to X, that is, the sum ∑i=1 to |X| Δ[Qi,Xi] is maximized.
String alignment
y3
y2
y1
ki+1
y1 y2 y3
x1
x2
lj+1
-∞
-∞
0 0 0
∆
We can’t delete nodes from the
pattern tree
This is NOT the deletion penalty
Time complexity for rooted ordered
For each node pair (v∈T,u∈P), the time complexity of the assignmentb is
O(c(u)c(v)) (dynamic programming)
∑u =1 to m ∑v =1 to n O(c(v) c(u)) =
∑v =1 to n O(m c(v)) =
O(m n)Observation 1
The tool: MetaPathwayHunter
What can it do?
A pathway against a pathway - 5 best alignments.
A pathway against a directory of pathways – 5 best alignment for pathway in the directory (sorted by score).
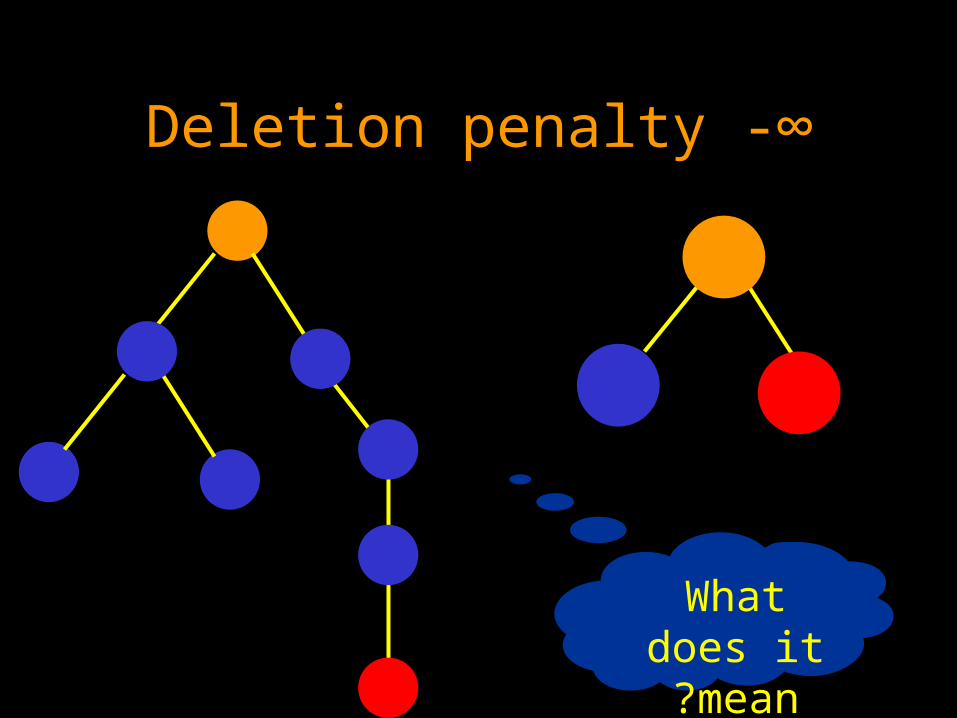
Two extreme cases of deletion penalty
Assuming the similarity score is negative (≤ 0)
• Deletion penalty 0: always worth deleting
• Deletion penalty -∞ : never worth deleting
Deletion penalty 0
What does it mean?
Deletion penalty -∞
What does it mean?
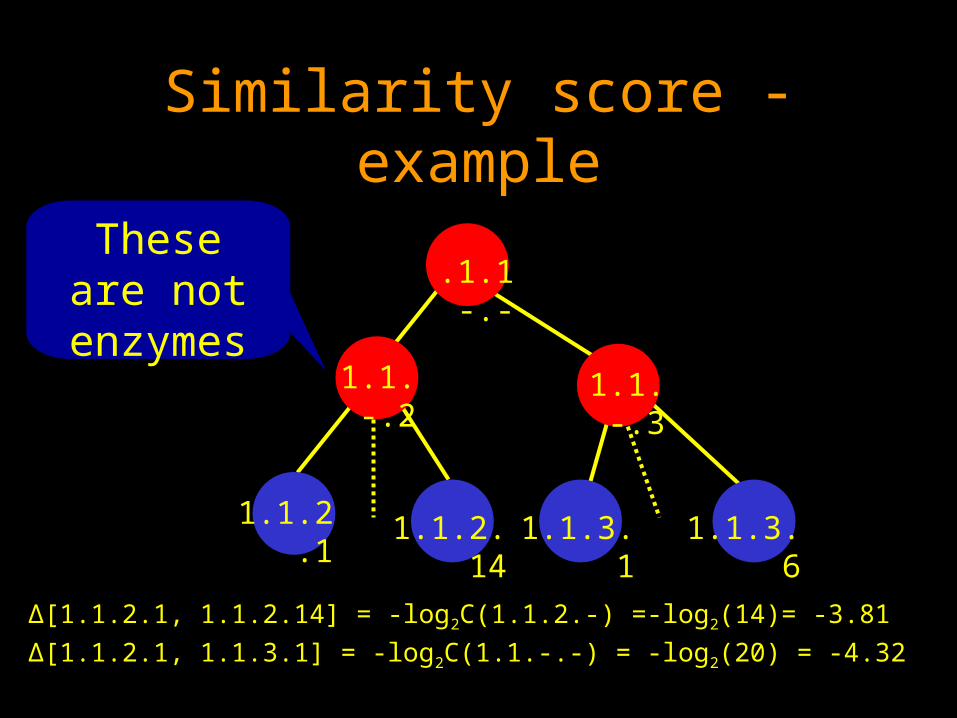
About the similarity score
MetaPathwayHunter uses the EC (Enzyme Commission) classification.
Four sets of numbers that categorize the type of the catalyzed chemical reaction. (e.g 1.2.5.23).
For an enzyme class h, C(h) denotes the number of enzymes whoose classes are included under h.
For two enzymes ei and ej, if their lowest common upper class is hij, then the similarity between then is –log2C(h).
Statistical significance is base on p-value. The p-value of an alignment (scored s) is
calculated by aligning the same query against 100 random pathway graphs, and counting the fraction of graphs containing an alignment that receive score s or higher.
A random pathway is a graph containing the same set of nodes and the same number of edges for each node, with random switch of the nodes.
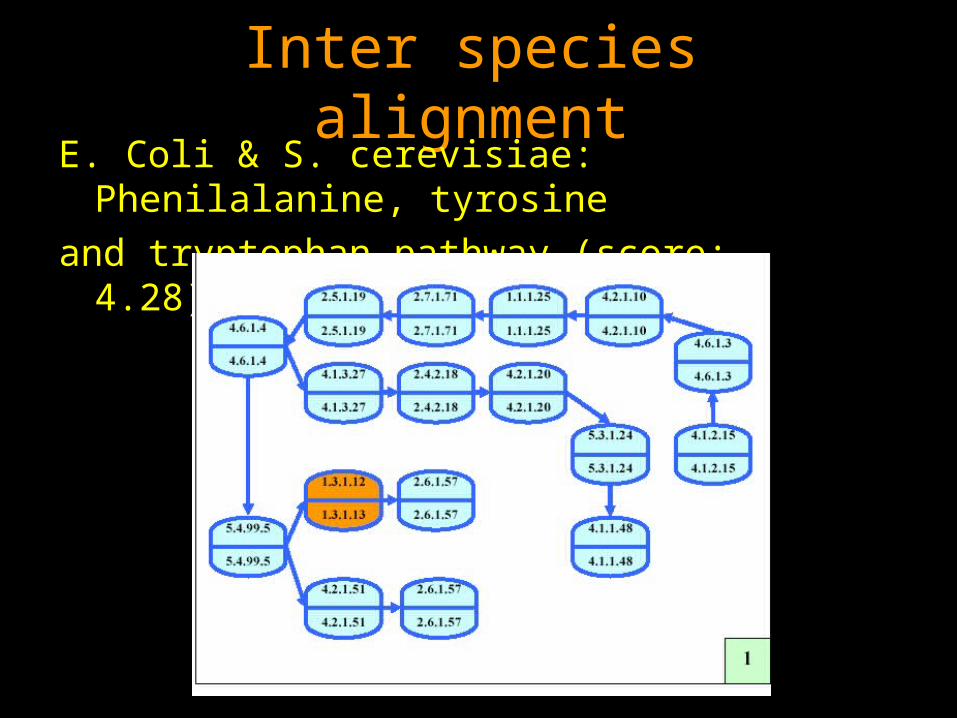
Inter species alignment
113 E. coli pathways and 151 S. cerevisiae
pathways. 610 pathway pairs had at least one
statistically significant alignment between them.
63% of the E. coli and 66% of S.
cerevisiae had at least one statistically
significantly aligned pair-mate from the other species
Inter species alignmentE. Coli & S. cerevisiae: Phenilalanine, tyrosine
and tryptophan pathway (score: -4.28) from [1]
Inter species alignment
What is the single mismatch? In E. coli: the enzyme uses NAD+ In S. cerevisia: the enzyme uses
NADP+ These two enzyme doesn’t have a
significant sequence similarity. == Two functional orthologs.
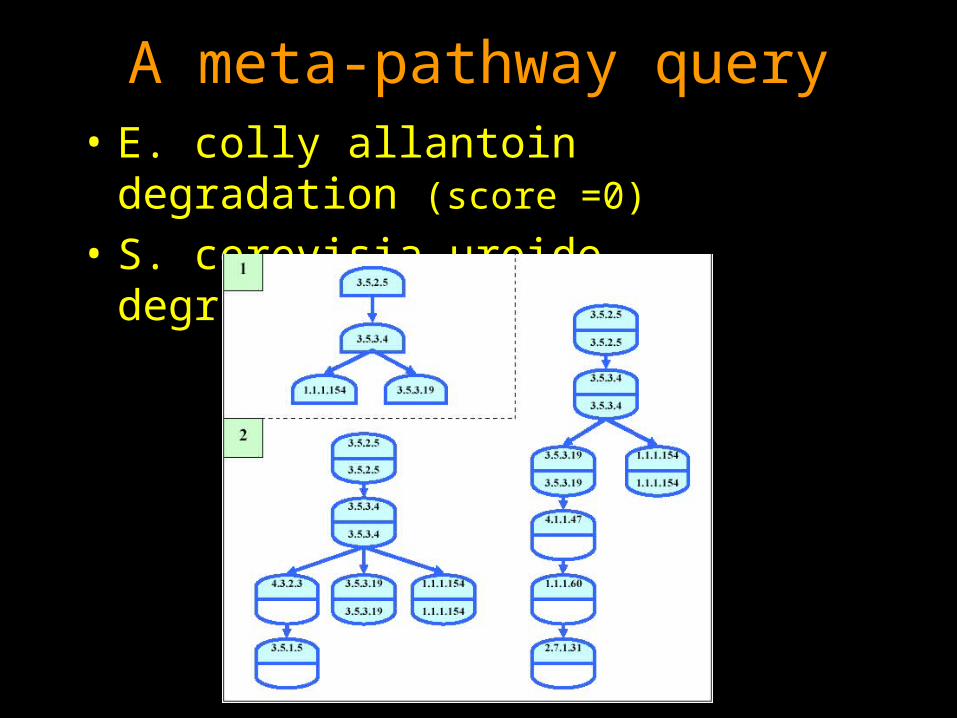
A meta-pathway query• E. colly allantoin degradation (score =0)
• S. cerevisia ureide degradation (score=0)
summary
• Biological motivation• Homeomorphism• Scoring and deleting• From assignment to matching• The algorithm for rooted unordered





















































![DS 09 Heaps.ppt [相容模式]Symmetric Min-Max Heap, SMMH Double-Ended Priority Queue, DEPQ Properties (old version) The root is empty The Left subtree is a min-heap The Right subtree](https://static.documents.pub/doc/80x56/60c2d421c957244ad70462db/ds-09-heapsppt-c-symmetric-min-max-heap-smmh-double-ended-priority.jpg)















