Aging and excitotoxic stress exacerbate neural circuit reorganizationin amyloid precursor protein intracellular domain transgenic mice
Kaushik Ghosal*, Sanjay W. PimplikarDepartment of Neurosciences, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA
Received 4 January, 2010; received in revised form 6 April 2010; accepted 20 April 2010
bstract
The cleavage of amyloid precursor protein (APP) by presenilins simultaneously generates amyloid-� (A�) and APP intracellular DomainAICD) peptides. A� plays a pivotal role in Alzheimer’s disease (AD) pathology and recently AICD was also shown to contribute to AD.ransgenic mice overexpressing AICD show age-dependent tau phosphorylation and aggregation, memory deficits, and neurodegeneration.oreover, these mice show aberrant electrical activity and silent seizures beginning at 3–4 months of age. Here we show that AICD mice
lso displayed abnormal mossy fiber sprouting beginning about the same time and that this sprouting intensified as the animals aged.xpression of neuropeptide Y was increased in mossy fiber terminals in aged but not young AICD mice. Importantly, young AICD mice
njected with kainic acid showed similar pathology to that observed in aged AICD mice. These data show that elevated levels of AICDender neurons hypersensitive to stress and induce hippocampal circuit reorganization, which can further exacerbate hyperexcitability. Theseesults further demonstrate that AICD, in addition to A�, can play a significant role in AD pathogenesis.
Alzheimer’s disease (AD) is a progressive neurodegen-rative disease characterized by the histopathological hall-arks of neurofibrillary tangles and senile plaques made up
f amyloid-� (A�) peptides (Price and Sisodia, 1998). Theeurological symptoms of AD include loss of short-termemory, confusion, disorientation to time and place, and
oss of initiative, which finally lead to complete loss ofognition (Hardy and Selkoe, 2002; Zheng and Koo, 2006).
significant number of AD patients also show alterations inlectroencephalograms (EEG) and the presence of silenteizures (Amatniek et al., 2006; Cabrejo et al., 2006; Snidert al., 2005). Although the exact cause of AD is not known,large number of studies show that A� peptides play a
ivotal role in disease pathology. However, it has becomencreasingly clear that AD etiology is highly complex and
* Corresponding author at: Department of Neurosciences, Lerner Re-earch Institute, Cleveland Clinic, Cleveland, OH 44195, USA. Tel: (216)44 9513; fax: (216) 444 7900.
hat other factors also contribute to disease pathogenesisPimplikar, 2009; Small and Duff, 2008).
A� peptides are produced by proteolytic processing ofmyloid precursor protein (APP). The presenilin-mediatedleavage of APP that causes extracellular release of A� alsoesults in intracellular generation of APP intracellular Do-ain (AICD). We and others have shown that AICD regu-
ates gene expression, alters cell signaling pathways, andauses deleterious effects in tissue culture cells (Giliberto etl., 2008; Muller et al., 2008). We also showed that AICD-verexpresing transgenic mice recapitulate AD-like featuresuch as hyperphosphorylation of tau, impaired memory, andeurodegeneration (Ghosal et al., 2009). Interestingly,ICD transgenic mice also exhibit nonconvulsive seizures
nd aberrant EEGs (Vogt et al., 2009), which are also seenn human AD and mouse models of AD (Del Vecchio et al.,004; Minkeviciene et al., 2009; Palop et al., 2007). Worky Mucke and colleagues (Palop et al., 2007) highlightedhe occurrence of seizures as a significant pathological fea-ure of AD and characterized the role of A� peptides in
ausing aberrant EEGs and the histopathological changes in
2 K. Ghosal, S.W. Pimplikar / Neurobiology of Aging xx (2010) xxx
ARTICLE IN PRESS
ippocampal circuits associated with neural hyperactivity.ere we show that elevated levels of AICD in transgenic mice
lso cause seizure-associated histopathological changes thatre similar, but not identical to, those seen in other mouseodels of AD. Aging or exposure to excitotoxic stress in
oung transgenic mice exacerbates these pathologicaleatures, suggesting that elevated levels of AICD rendereurons hypersensitive to environmental insults and ag-ng.
. Methods
.1. Animals
Transgenic mice coexpressing the AICD and Fe-65FeC�25) or Fe-65 alone (Fe27) under the control of theAMKII� promoter were previously described (Ghosal etl., 2009; Ryan and Pimplikar, 2005). Wild-type littermatesere used as controls. For all experiments mice were used
t 3–5 or � 18 months of age. All experiments were ap-roved by the Institutional Animal Use and Care Committeef The Cleveland Clinic.
.2. Kainic acid injections
Kainic acid (Sigma) was injected intraperitoneally (i.p.)t a dose of, 20 mg kg�1 (Vogt et al., 2009). Mice givenublethal doses were sacrificed after 3 days and transcardi-lly perfused with ice cold PBS followed by 4% parafor-aldehyde and processed as described below.
.3. Immunohistochemistry
Hemi brains were fixed in PBS containing 4% parafor-aldehyde overnight, sunk in 30% sucrose and embedded
n OCT. Sagittal sections (30 �m) were cut and every 12thection was used for immunohistochemistry. Sections wereashed in PBS, treated with 3% H2O2 in PBS, and incu-ated in blocking solution (5% normal goat serum in PBSontaining 0.01% Triton-X) for 1 hour at room temperature.ections were incubated with primary antibody against neu-opeptide Y (NPY) (1 : 1,000) or calbindin-D28k (1 : 500)vernight at 4 °C followed by respective secondary anti-odies (1 : 250) and developed using the ABC kit (Vectoraboratories). Microscopy was performed using a LeicaMR microscope equipped with a CCD camera for brighteld imaging.
.4. Quantitative analysis of immunoreactivity
Digital images of brain sections were obtained from 5 tosagittal sections per mouse with a Magnafire-Firewire
igital camera (Optronics, Goleta, CA) on a Leica DMRicroscope (Leica, Germany). Integrated optical density
IOD) was measured using ImageJ software (National In-titutes of Health (NIH)) from a defined area of 0.51 � 0.44m (0.223 mm2) in the mossy fiber terminal region, theolecular layer of the dentate gyrus and the stratum radia-
um of the CA1 region. No changes were detected in the s
tratum radiatum. The IOD of the stratum radiatum wasubtracted from the IOD of the mossy fiber region to get aormalized IOD value for the mossy fiber region relative tohe background intensity. The mean IOD for each animalas calculated from the normalized IOD for each section
5–7 sections per animal) and used for statistical tests.mage analysis was done blind to the treatment group.
.5. Timm staining and scoring
Timm staining was performed as described previouslyor other mouse models of AD (Palop et al., 2007)Minkeviciene et al., 2009). Briefly, mice were administeredn i.p. injection of sodium selenite (Sigma) in 0.9% normalaline at a dose of, 20 mg kg�1. After 30 min, mice wereeeply anesthetized with Avertin and transcardially per-used with ice cold PBS. Brains were harvested and post-xed in 4% paraformaldehyde in PBS. Every 12th sagittalection was used for Timm staining. Briefly, sections wereashed in PBS and incubated in dark conditions for 90 min
n Timm developing solution (30% acacia, 0.11% silveractate, 0.85% hydroquinone, 2.35% sodium citrate, and.55% citric acid). The developing reaction was terminatedy repeated washes with water. Sections were counter-tained with cresyl violet and mounted with glycerol. Mossyber sprouting was scored according to conventional tech-iques (Cavazos et al., 1991; Nissinen et al., 2001) that werereviously reported for other AD mouse models (Minkevicienet al., 2009; Palop et al., 2007), where the following scale wassed: (0) no granules; (1) sparse granules in the supragranu-ar region and the inner molecular layer; (2) evenly dis-ributed granules in the supragranular region and thenner molecular layer; (3) almost continuous layer ofimm-positive staining in the supragranular region and the
nner molecular layer; (4) continuous band of Timm-posi-ive staining in the supragranular region and the inner mo-ecular layer; and (5) dense and thick band of Timm stainingovering the inner molecular layer. The experimenter waslind to the treatment groups during scoring.
.6. Statistical analysis
Statistics were performed using GraphPad InStat soft-are (version 4). One way ANOVA was employed and
ignificance was set at p � 0.05.
. Results
We previously showed that AICD transgenic mice ex-ibit abnormal EEG patterns and seizure-like hyperactivityVogt et al., 2009). Seizures can induce reorganization ofossy fibers in the dentate gyrus (Buckmaster and Dudek,
997) which can lead to further overexcitation resulting inrolonged seizure-like symptoms (Santhakumar et al., 2005).ecause FeC�25 mice show aberrant EEG activity, we exam-
ned the integrity of hippocampal neural circuits by Timm
taining, which stains mossy fiber terminals. 4-Months-old
FsasmEim(F(ao1F
btmi
meebntlmm
F(s(aattn
3K. Ghosal, S.W. Pimplikar / Neurobiology of Aging xx (2010) xxx
ARTICLE IN PRESS
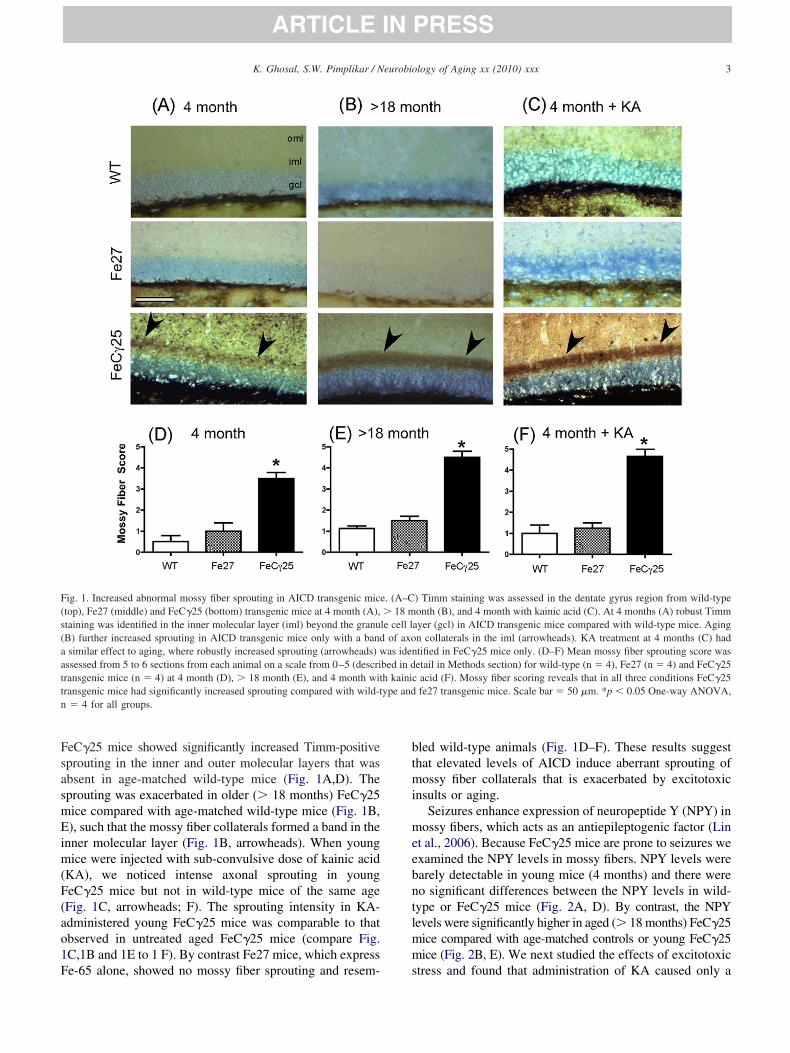
eC�25 mice showed significantly increased Timm-positiveprouting in the inner and outer molecular layers that wasbsent in age-matched wild-type mice (Fig. 1A,D). Theprouting was exacerbated in older (� 18 months) FeC�25ice compared with age-matched wild-type mice (Fig. 1B,), such that the mossy fiber collaterals formed a band in the
nner molecular layer (Fig. 1B, arrowheads). When youngice were injected with sub-convulsive dose of kainic acid
KA), we noticed intense axonal sprouting in youngeC�25 mice but not in wild-type mice of the same ageFig. 1C, arrowheads; F). The sprouting intensity in KA-dministered young FeC�25 mice was comparable to thatbserved in untreated aged FeC�25 mice (compare Fig.C,1B and 1E to 1 F). By contrast Fe27 mice, which express
ig. 1. Increased abnormal mossy fiber sprouting in AICD transgenic mictop), Fe27 (middle) and FeC�25 (bottom) transgenic mice at 4 month (A),taining was identified in the inner molecular layer (iml) beyond the granuB) further increased sprouting in AICD transgenic mice only with a bandsimilar effect to aging, where robustly increased sprouting (arrowheads) w
ssessed from 5 to 6 sections from each animal on a scale from 0–5 (descriransgenic mice (n � 4) at 4 month (D), � 18 month (E), and 4 month witransgenic mice had significantly increased sprouting compared with wild-t� 4 for all groups.
e-65 alone, showed no mossy fiber sprouting and resem- s
led wild-type animals (Fig. 1D–F). These results suggesthat elevated levels of AICD induce aberrant sprouting ofossy fiber collaterals that is exacerbated by excitotoxic
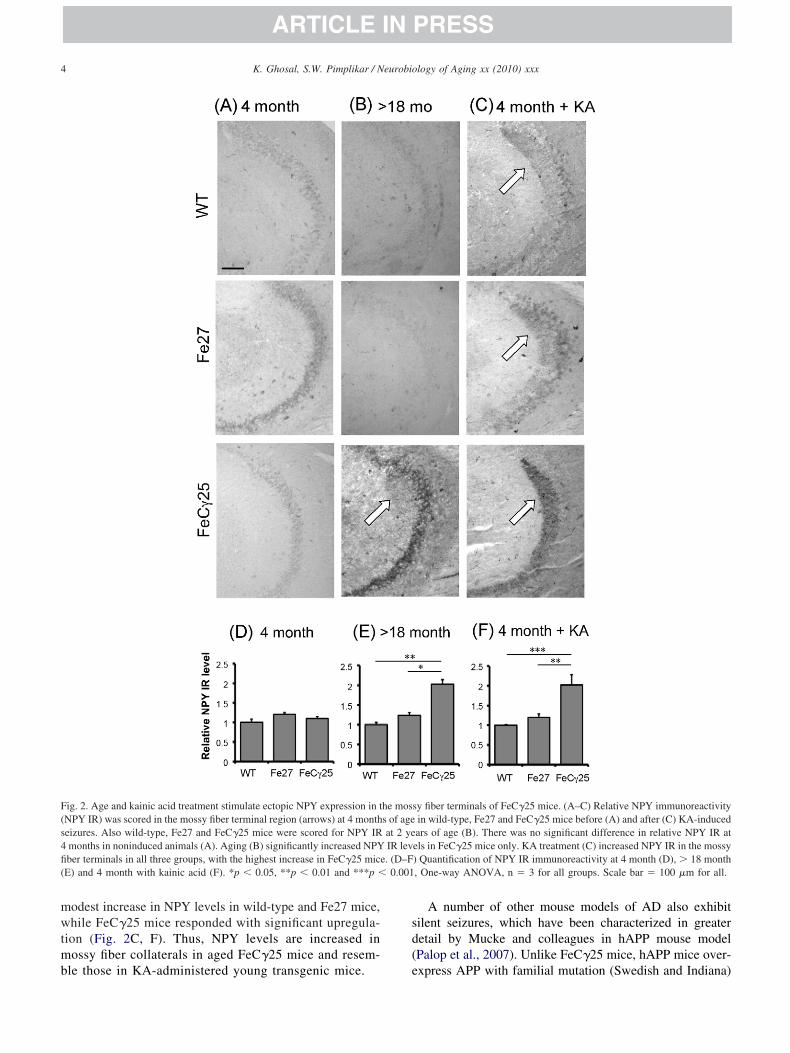
nsults or aging.Seizures enhance expression of neuropeptide Y (NPY) in
ossy fibers, which acts as an antiepileptogenic factor (Lint al., 2006). Because FeC�25 mice are prone to seizures wexamined the NPY levels in mossy fibers. NPY levels werearely detectable in young mice (4 months) and there wereo significant differences between the NPY levels in wild-ype or FeC�25 mice (Fig. 2A, D). By contrast, the NPYevels were significantly higher in aged (� 18 months) FeC�25ice compared with age-matched controls or young FeC�25ice (Fig. 2B, E). We next studied the effects of excitotoxic
) Timm staining was assessed in the dentate gyrus region from wild-typeonth (B), and 4 month with kainic acid (C). At 4 months (A) robust Timm
ayer (gcl) in AICD transgenic mice compared with wild-type mice. Agingn collaterals in the iml (arrowheads). KA treatment at 4 months (C) hadtified in FeC�25 mice only. (D–F) Mean mossy fiber sprouting score wasetail in Methods section) for wild-type (n � 4), Fe27 (n � 4) and FeC�25acid (F). Mossy fiber scoring reveals that in all three conditions FeC�25fe27 transgenic mice. Scale bar � 50 �m. *p � 0.05 One-way ANOVA,
e. (A–C� 18 mle cell l
of axoas iden
bed in dh kainicype and
tress and found that administration of KA caused only a
mwtmb
sd(
F(s4fi( 0.001
4 K. Ghosal, S.W. Pimplikar / Neurobiology of Aging xx (2010) xxx
ARTICLE IN PRESS
odest increase in NPY levels in wild-type and Fe27 mice,hile FeC�25 mice responded with significant upregula-
ion (Fig. 2C, F). Thus, NPY levels are increased inossy fiber collaterals in aged FeC�25 mice and resem-
ig. 2. Age and kainic acid treatment stimulate ectopic NPY expression in tNPY IR) was scored in the mossy fiber terminal region (arrows) at 4 montheizures. Also wild-type, Fe27 and FeC�25 mice were scored for NPY IRmonths in noninduced animals (A). Aging (B) significantly increased NPYber terminals in all three groups, with the highest increase in FeC�25 micE) and 4 month with kainic acid (F). *p � 0.05, **p � 0.01 and ***p �
le those in KA-administered young transgenic mice. e
A number of other mouse models of AD also exhibitilent seizures, which have been characterized in greateretail by Mucke and colleagues in hAPP mouse modelPalop et al., 2007). Unlike FeC�25 mice, hAPP mice over-
sy fiber terminals of FeC�25 mice. (A–C) Relative NPY immunoreactivityin wild-type, Fe27 and FeC�25 mice before (A) and after (C) KA-inducedars of age (B). There was no significant difference in relative NPY IR atels in FeC�25 mice only. KA treatment (C) increased NPY IR in the mossy) Quantification of NPY IR immunoreactivity at 4 month (D), � 18 month, One-way ANOVA, n � 3 for all groups. Scale bar � 100 �m for all.
he moss of ageat 2 yeIR lev
e. (D–F
xpress APP with familial mutation (Swedish and Indiana)
aptotfi3
rldFlmp
F(aNS
5K. Ghosal, S.W. Pimplikar / Neurobiology of Aging xx (2010) xxx
ARTICLE IN PRESS
nd show elevated levels of A� and exacerbated plaqueathology. These mice also show increased levels of NPY inhe mossy fiber terminals as well as in the molecular layerf the dentate gyrus. We next measured NPY immunoreac-ivity in the molecular layer in AICD transgenic mice andound no detectable presence of NPY in the molecular layern wild-type, Fe27 or FeC�25 mice at 4 months of age (Fig.
ig. 3. Ectopic NPY expression is absent in the dentate gyrus of FeC�25 mm) of dentate gyrus above the granule cell layer (g) in wild-type, Fe27 andcid (C). There was no significant difference in relative NPY IR at 4 monPY expression in FeC�25 mice. (D–F) Quantification of NPY IR at 4 mocale bar � 100 �m for all.
A,D). Surprisingly, in contrast to the mossy fiber terminal F
egion, there was no increase in NPY levels in the molecularayer of aged FeC�25 mice and there were no significantifferences in the NPY levels in the molecular layer ofeC�25 mice, wild-type or Fe27 mice (Fig. 3B, E). Simi-
arly, KA-induced excitotoxicity did not increase NPY im-unoreactivity in the molecular layer of FeC�25 mice com-
ared with age-matched wild-type or Fe27 mice (Fig. 3C,
C) Relative NPY immunoreactivity (IR) was scored in the molecular layertransgenic mice at 4 month (A), � 18 month (B) and 4 month with kainic
oninduced animals (A). Aging (B) and KA treatment (C) did not induce, � 18 month (E) and 4 month with kainic acid (F). n � 3 for all groups.
ice. (A–FeC�25ths in nnth (D)
). These results suggest that NPY levels are increased in
mFsni
tc2i(wirpncisn4cwmtsatlimr
3
rot2hrecemoTsmtahAa
ilceci
ersgrmmghMot2KsapnnAcAmg(haDtnict
oePaafHplc(A(
6 K. Ghosal, S.W. Pimplikar / Neurobiology of Aging xx (2010) xxx
ARTICLE IN PRESS
ossy fiber collaterals but not in the molecular layer in agedeC�25 mice or young AICD mice exposed to excitotoxictress. Thus, unlike hAPP mice, AICD transgenic mice doot show compensatory inhibitory projections that terminaten the molecular layer of the dentate gyrus.
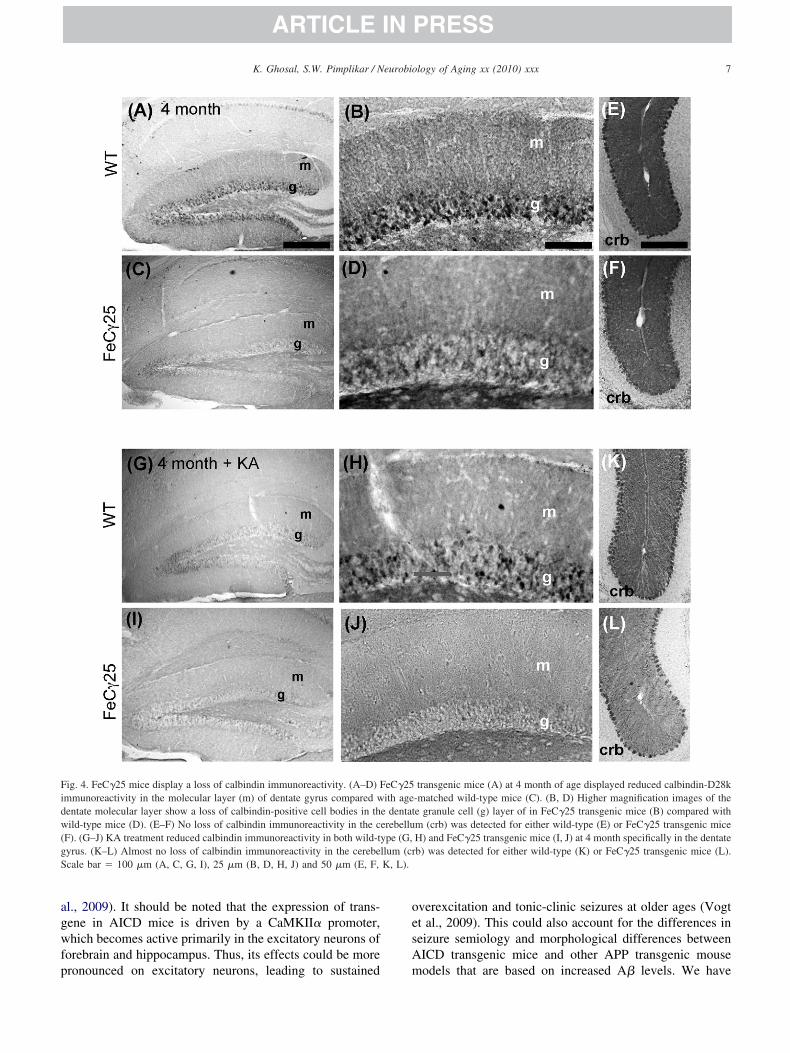
Increased hyperexcitability observed in different APPransgenic mouse models of AD is associated with a loss ofalbindin D28k immunoreactivity (Minkeviciene et al.,009; Palop et al., 2007). More recently, loss of calbindinmmunoreactivity has been reported in human epilepsyMagloczky, 2010). Therefore, we stained brain sectionsith anti-calbindin antibodies and found reduced calbindin
mmunoreactivity in the molecular layer of the dentate gy-us in FeC�25 mice compared with wild-type mice (com-are Fig 4A–C). At a higher magnification, the reducedumber of calbindin-positive neurons in the dentate granuleell layer was visible (Fig 4B v. 4 D). The loss of calbindinmmunoreactivity seemed restricted to the hippocampus,ince there were no significant changes in calbindin immu-oreactivity in the cerebellum of AICD transgenic mice (FigE, F). We also found that administration of KA reducedalbindin immunoreactivity in the hippocampus of bothild-type mice and FeC�25 mice (Fig. 4G, I). At higheragnification, the loss of calbindin-positive cells in wild-
ype mice was apparent (Fig. 4H), whereas it was moreeverely reduced in AICD mice (Fig. 4J) compared withge-matched wild-type controls (Fig. 4H). By comparison,he loss of calbindin in cerebellum in KA-treated mice wasess severe (Fig. 4K, L). Together with our previous find-ngs, these observations show that seizure activity in AICDice is accompanied by a loss of calbindin-D28k immuno-
eactivity.
. Discussion
A large body of literature shows that A� plays a pivotalole in AD pathogenesis. Nonetheless, an increasing numberf studies indicate that factors other than A� also contributeo AD pathogenesis (Holmes et al., 2008; Simon et al.,008). Our earlier findings that AICD transgenic mice ex-ibit several AD-like pathological features including aber-ant electrical activity, silent seizures, and sensitivity toxcitotoxic stress (Vogt et al., 2009) suggest that AICDould be one such important factor. In the present study, wextended these observations and demonstrated that AICDice show mossy fiber sprouting beginning at 3–4 months
f age and increased expression of NPY in aged animals.hese pathologies were exacerbated after excitotoxic in-ults, which resulted in the neural circuits from young AICDice administered with KA resembling those from old un-
reated AICD mice. Aberrant axonal sprouting (Cotman etl., 1990; Masliah et al., 1993) and elevated levels of NPYave been observed in AD patients and mouse models ofD (Diez et al., 2003; Minkeviciene et al., 2009; Palop et
l., 2007; Ramos et al., 2006) and have been attributed to s
ncreased A� levels (Palop et al., 2007). Because the patho-ogical features observed in AICD mice occur without in-reased A� levels or alterations in APP processing (Ghosalt al., 2009), our studies suggest that both AICD and A� canontribute to axonal sprouting and can increase NPY levelsn AD brains.
Although AICD transgenic mice and other mouse mod-ls of AD (such as hAPP or APdE9) show sprouting andeorganization of hippocampal neural circuits, there areubtle but significant differences in the observed patholo-ies. The axonal sprouting in AICD transgenic animalsesembles that in temporal lobe epilepsy; in both cases,ossy fiber collaterals terminate as a band in the innerolecular layer and cause further excitation of adjacent
ranule cells. By contrast, sprouting of mossy fibers inAPP transgenic mice (which overexpress Swedish K670N/67 1L and Indiana V717F mutations in APP) terminates
n basket cells and results in proper maintenance of exci-atory-inhibitory balance (Cavazos et al., 1991; Palop et al.,007). By contrast, APdE9 mice, expressing both APP695
595N/M59 6L and PS1- � exon nine mutations, showeizures but exhibit no axonal sprouting (Minkeviciene etl., 2009). These results suggest that seizures and hippocam-al circuit reorganization are regulated by complex mecha-isms and that A� and AICD affect these processes in aonidentical manner. Lack of compensatory mechanisms inICD mice, as opposed to that observed in hAPP mice,
ould explain the occurrence of convulsive seizures in agedICD transgenic mice (Vogt et al., 2009) but not in hAPPice. This difference could also explain why AICD trans-
enic mice (Ghosal et al., 2009) but not the hAPP or APdE9Minkeviciene et al., 2009; Palop et al., 2007) mice showippocampal neuronal loss during aging. Neuronal loss isssociated with an epileptic phenotype (Buckmaster andudek, 1997; Cavazos and Sutula, 1990) and the cumula-
ive effect of long-term overexcitation may contribute toeurodegeneration in AICD transgenic mice. However, sim-lar to hAPP and APdE9 models, we observed a loss ofalbindin immunoreactivity in the hippocampus of AICDransgenic mice.
Our findings that aged AICD mice show elevated levelsf NPY agree with reports in AD patients and mouse mod-ls of AD (Diez et al., 2003; Minkeviciene et al., 2009;alop et al., 2007; Ramos et al., 2006). NPY is known to ben anticonvulsant and antiepileptic (Baraban et al., 1997)nd its levels in mossy fibers are elevated because of aeedback response to hyperactivity (Nadler et al., 2007).owever, unlike hAPP-J20 mice, the increase in NPY ex-ression in AICD mice was not observed in the molecularayer of the dentate gyrus where NPY counteracts overex-itability and protects mice from generalized seizuresPalop et al., 2007). Instead, the NPY expression pattern inICD mice resembled that of normal temporal lobe epilepsy
Vezzani et al., 1999) and other mouse models of AD with
eizure-like phenotypes (Diez et al., 2003; Minkeviciene et
agwfp
oesA
Fidw(gS K, L).
7K. Ghosal, S.W. Pimplikar / Neurobiology of Aging xx (2010) xxx
ARTICLE IN PRESS
l., 2009). It should be noted that the expression of trans-ene in AICD mice is driven by a CaMKII� promoter,hich becomes active primarily in the excitatory neurons of
orebrain and hippocampus. Thus, its effects could be more
ig. 4. FeC�25 mice display a loss of calbindin immunoreactivity. (A–D) Fmmunoreactivity in the molecular layer (m) of dentate gyrus compared wentate molecular layer show a loss of calbindin-positive cell bodies in thild-type mice (D). (E–F) No loss of calbindin immunoreactivity in the c
F). (G–J) KA treatment reduced calbindin immunoreactivity in both wild-tyrus. (K–L) Almost no loss of calbindin immunoreactivity in the cerebecale bar � 100 �m (A, C, G, I), 25 �m (B, D, H, J) and 50 �m (E, F,
ronounced on excitatory neurons, leading to sustained m
verexcitation and tonic-clinic seizures at older ages (Vogtt al., 2009). This could also account for the differences ineizure semiology and morphological differences betweenICD transgenic mice and other APP transgenic mouse
transgenic mice (A) at 4 month of age displayed reduced calbindin-D28k-matched wild-type mice (C). (B, D) Higher magnification images of thee granule cell (g) layer of in FeC�25 transgenic mice (B) compared withm (crb) was detected for either wild-type (E) or FeC�25 transgenic miceH) and FeC�25 transgenic mice (I, J) at 4 month specifically in the dentateb) was detected for either wild-type (K) or FeC�25 transgenic mice (L).
eC�25ith age
e dentaterebelluype (G,llum (cr
odels that are based on increased A� levels. We have
a(sdsm
tsvvelwmyesnApCtttf
htsnttt
D
i
A
mpRtmt
R
A
B
B
C
C
C
C
D
D
G
G
H
H
L
M
M
M
M
N
N
8 K. Ghosal, S.W. Pimplikar / Neurobiology of Aging xx (2010) xxx
ARTICLE IN PRESS
nalyzed EEG patterns in the R1.40 mouse model of ADexpressing APPsweK670N/M67 1L fAD mutation) thathows similar levels of AICD as the FeC�25 mice butisplays higher levels of A�. We found that R1.40 mice alsohowed a seizure phenotype, similar to the AICD transgenicodel (Ryan and Pimplikar, 2005; Vogt et al., 2009).Another important observation of the present study is
hat the elevated levels of AICD render neurons hypersen-itive to stress associated with aging or excitotoxicity inivo. AICD was shown to increase sensitivity in neurons initro in cell culture (Giliberto et al., 2008) and our studyxtends these findings in vivo. We previously showed that aow dose of KA produced moderate convulsive seizures inild-type mice but caused lethality in AICD transgenicice (Vogt et al., 2009). Consistent with these findings,
oung AICD mice subjected to excitotoxic insult developedxacerbated histopathological features that resembled thoseeen during aging. Similarly, young AICD mice showedormal levels of NPY and no neurodegeneration, while agedICD mice or young AICD mice injected with KA dis-layed higher NPY levels (this study) and a severe loss ofA3 neurons (Ghosal et al., 2009). Thus, it could be argued
hat the effects of acute excitotoxic stress are equivalent tohose seen during aging. These findings are consistent withhe observations that aging is the most significant risk factoror ad.
In summary, we report that AICD transgenic mice showistopathological changes in the hippocampus similar tohose seen in AD patients and mouse models of AD. Thistudy further confirms that elevated levels of AICD rendereurons vulnerable to stress associated with aging or exci-otoxicity. Uncovering the precise mechanisms underlyinghe effects of AICD will be important since it may offer newargets for therapeutic interventions.
isclosure statement
The authors declare no actual or potential conflicts ofnterest.
cknowledgements
The authors express sincere thanks to the two anony-ous reviewers whose comments made significant im-
rovement in our studies. This work was supported by01AG026146, Alzheimer’s Association and CART-Ro-
ary funds to SWP. We thank Chris Nelson for his com-ents and Daniel Vogt for performing kainic acid injec-
ions.
eferences
matniek, J.C., Hauser, W.A., delCastillo-Castaneda, C., Jacobs, D.M.,Marder, K., Bell, K., Albert, M., Brandt, J., Stern, Y., 2006. Incidenceand predictors of seizures in patients with Alzheimer’s disease. Epi-
uckmaster, P.S., Dudek, F.E., 1997. Neuron loss, granule cell axonreorganization, and functional changes in the dentate gyrus of epileptickainate-treated rats. J Comp Neurol 385, 385–404.
abrejo, L., Guyant-Marechal, L., Laquerriere, A., Vercelletto, M., De laFourniere, F., Thomas-Anterion, C., Verny, C., Letournel, F., Pasquier,F., Vital, A., Checler, F., Frebourg, T., Campion, D., Hannequin, D.,2006. Phenotype associated with APP duplication in five families.Brain 129, 2966–76.
avazos, J.E., Sutula, T.P., 1990. Progressive neuronal loss induced bykindling: a possible mechanism for mossy fiber synaptic reorganizationand hippocampal sclerosis. Brain Res 527, 1–6.
avazos, J.E., Golarai, G., Sutula, T.P., 1991. Mossy fiber synaptic reor-ganization induced by kindling: time course of development, progres-sion, and permanence. J Neurosci 11, 2795–803.
otman, C.W., Geddes, J.W., Kahle, J.S., 1990. Axon sprouting in therodent and Alzheimer’s disease brain: a reactivation of developmentalmechanisms? Prog Brain Res 83, 427–34.
el Vecchio, R.A., Gold, L.H., Novick, S.J., Wong, G., Hyde, L.A., 2004.Increased seizure threshold and severity in young transgenic CRND8mice. Neurosci Lett 367, 164–7.
iez, M., Danner, S., Frey, P., Sommer, B., Staufenbiel, M., Wiederhold,K.H., Hokfelt, T., 2003. Neuropeptide alterations in the hippocampalformation and cortex of transgenic mice overexpressing beta-amyloidprecursor protein (APP) with the Swedish double mutation (APP23).Neurobiol Dis 14, 579–94.
hosal, K., Vogt, D.L., Liang, M., Shen, Y., Lamb, B.T., Pimplikar, S.W.,2009. Alzheimer’s disease-like pathological features in transgenic miceexpressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA106, 18367–72.
iliberto, L., Zhou, D., Weldon, R., Tamagno, E., De Luca, P., Tabaton,M., D’Adamio, L., 2008. Evidence that the Amyloid beta PrecursorProtein-intracellular domain lowers the stress threshold of neurons andhas a “regulated” transcriptional role. Mol Neurodegener 3, 12.
ardy, J., Selkoe, D.J., 2002. The amyloid hypothesis of Alzheimer’sdisease: progress and problems on the road to therapeutics. Science297, 353–6.
olmes, C., Boche, D., Wilkinson, D., Yadegarfar, G., Hopkins, V., Bayer,A., Jones, R.W., Bullock, R., Love, S., Neal, J.W., Zotova, E., Nicoll,J.A., 2008. Long-term effects of Abeta42 immunisation in Alzheimer’sdisease: follow-up of a randomised, placebo-controlled phase I trial.Lancet 372, 216–23.
in, E.J., Young, D., Baer, K., Herzog, H., During, M.J., 2006. Differentialactions of NPY on seizure modulation via Y1 and Y2 receptors:evidence from receptor knockout mice. Epilepsia 47, 773–80.
agloczky, Z., 2010. Sprouting in human temporal lobe epilepsy: Excita-tory pathways and axons of interneurons. Epilepsy Res 89, Nos. 1,52–9.
asliah, E., Miller, A., Terry, R.D., 1993. The synaptic organization of theneocortex in Alzheimer’s disease. Med Hypotheses 41, 334–40.
uller, T., Meyer, H.E., Egensperger, R., Marcus, K., 2008. The amyloidprecursor protein intracellular domain (AICD) as modulator of geneexpression, apoptosis, and cytoskeletal dynamics-relevance for Alzhei-mer’s disease. Prog Neurobiol 85, 393–406.
adler, J.V., Tu, B., Timofeeva, O., Jiao, Y., Herzog, H., 2007. Neuropep-tide Y in the recurrent mossy fiber pathway. Peptides 28, 357–364.
issinen, J., Lukasiuk, K., Pitkanen, A., 2001. Is mossy fiber sproutingpresent at the time of the first spontaneous seizures in rat experimental
temporal lobe epilepsy? Hippocampus 11, 299–310.
P
P
P
R
R
S
S
S
S
V
V
Z
9K. Ghosal, S.W. Pimplikar / Neurobiology of Aging xx (2010) xxx
implikar, S.W., 2009. Reassessing the amyloid cascade hypothesis ofAlzheimer’s disease. Int J Biochem Cell Biol 41, 1261–1268.
rice, D.L., Sisodia, S.S., 1998. Mutant genes in familial Alzheimer’sdisease and transgenic models. Annu Rev Neurosci 21, 479–505.
amos, B., Baglietto-Vargas, D., del Rio, J.C., Moreno-Gonzalez, I., Santa-Maria, C., Jimenez, S., Caballero, C., Lopez-Tellez, J.F., Khan, Z.U., Ruano,D., Gutierrez, A., Vitorica, J., 2006. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenicmodel of Alzheimer’s disease. Neurobiol Aging 27, 1658–1672.
yan, K.A., Pimplikar, S.W., 2005. Activation of GSK-3 and phosphory-lation of CRMP2 in transgenic mice expressing APP intracellulardomain. J Cell Biol 171, 327–335.
anthakumar, V., Aradi, I., Soltesz, I., 2005. Role of mossy fiber sprouting andmossy cell loss in hyperexcitability: a network model of the dentate gyrus
incorporating cell types and axonal topography. J Neurophysiol 93, 437–453.
imon, A.M., Schiapparelli, L., Salazar-Colocho, P., Cuadrado-Tejedor,M., Escribano, L., Lopez de Maturana, R., Del Rio, J., Perez-Media-villa, A., Frechilla, D., 2008. Overexpression of wild-type human APPin mice causes cognitive deficits and pathological features unrelated toAbeta levels. Neurobiol Dis 33, Nos. 3, 369–378.
mall, S.A., Duff, K., 2008. Linking Abeta and tau in late-onset Alzhei-mer’s disease: a dual pathway hypothesis. Neuron 60, 534–42.
nider, B.J., Norton, J., Coats, M.A., Chakraverty, S., Hou, C.E., Jervis, R.,Lendon, C.L., Goate, A.M., McKeel, D.W., Jr, Morris, J.C., 2005.Novel presenilin 1 mutation (S17 0F) causing Alzheimer disease withLewy bodies in the third decade of life. Arch Neurol 62, 1821–30.
ezzani, A., Sperk, G., Colmers, W.F., 1999. Neuropeptide Y: emergingevidence for a functional role in seizure modulation. Trends Neurosci22, 25–30.
ogt, D.L., Thomas, D., Galvan, V., Bredesen, D.E., Lamb, B.T., Pimp-likar, S.W., 2009. Abnormal neuronal networks and seizure suscepti-bility in mice overexpressing the APP intracellular domain. NeurobiolAging Oct 12 [Epub ahead of print].