Artificial Protein Block Polymer Libraries Bearing Two SADs: Effectsof Elastin Domain RepeatsMin Dai,† Jennifer Haghpanah,† Navjot Singh,† Eric W. Roth,‡ Alice Liang,‡ Raymond S. Tu,§
and Jin Kim Montclare*,†,∥
†Department of Chemical and Biological Sciences, Polytechnic Institute of NYU, Brooklyn, New York 11201, United States‡Skirball Institute Image Core Facility, New York University Medical Center, New York, New York 10016, United States§Department of Chemical Engineering, City College of New York, New York, New York 10031, United States∥Department of Biochemistry, SUNY Downstate Medical Center, Brooklyn, New York 11203, United States
*S Supporting Information
ABSTRACT: We have generated protein block polymer EnCand CEn libraries composed of two different self-assemblingdomains (SADs) derived from elastin (E) and the cartilageoligomeric matrix protein coiled-coil (C). As the E domain isshortened, the polymers exhibit an increase in inverse transitiontemperature (Tt); however, the range of temperature changediffers dramatically between the EnC and CEn library. Whereasall polymers assemble into nanoparticles, the bulk mechanical properties of the EnC are very different from CEn. The EnCmembers demonstrate viscolelastic behavior under ambient conditions and assemble into elastic soft gels above their Tt values.By contrast, the CEn members are predominantly viscous at all temperatures. All library members demonstrate binding tocurcumin. The differential thermoresponsive behaviors of the EnC and CEn libraries in addition to their small moleculerecognition abilities make them suitable for potential use in tissue engineering and drug delivery.
■ INTRODUCTION
The fabrication of stimuli-responsive, multifunctional nanoma-terials that can self-assemble into defined structures bears tre-mendous potential in drug delivery and regenerative medicine.1−4
Whereas there has been remarkable progress in synthetic self-assembling systems, nature provides a wealth of highly orderedstructures with defined features from the nano- to the mesoscalelevel. In fact, a large fraction of the structures in nature arecomposed of proteins. Proteins not only provide a diversity ofchemical functionality as the building blocks are composed of20 amino acids but also present critical 3D structures that pro-vide order on the nano- to meso-length scales. Inspired by natureand fueled by recent advances in molecular and syntheticbiology,5,6 we7,8 and others9−22 have generated “smart” proteinpolymers capable of self-assembling and responding to externalstimuli.Recently, we engineered three protein block polymers7,8
−EC, CE and ECE−composed of two distinct self-assemblingdomains (SADs) derived from elastin (E)23,24 and cartilageoligomeric matrix protein coiled-coil (C).25,26 Character-ization of the proteins revealed that the orientation andnumber of blocks influenced the overall secondary structure,stability, supramolecular assembly, and small-molecule bindingability. Whereas the protein polymers exhibited interestingphysicochemical properties, their thermoresponsive behaviorswere limited; all three proteins possessed inverse temperaturetransitions (Tt values) at room temperature or below. The Edomain length for the three constructs possessed a five repeat
elastin (n = 5). To expand the thermoresponsive range of thesematerials, a library of EnC and CEn diblock polymers wasconstructed in which the E domain was systematically truncated,where n ranged from 4 to 1 repeats (Figure 1). We hypothesizedthat as the E domain is shortened, the Tt would increase based on
Received: August 2, 2011Revised: September 22, 2011Published: October 26, 2011
Figure 1. (a) Illustration of the EnC and CEn protein block polymersand their sequences where n represents the number of E repeats.(b) 12% SDS-PAGE of purified protein polymers.
previous literature on elastin-based polypeptides.22 Whereas anincrease in Tt was observed upon decreasing the E domain lengthas expected, the orientation of the diblocks affected the secondarystructure, supramolecular assembly, mechanical properties, andsmall molecule recognition.
■ MATERIALS AND METHODSGeneral. BamHI, SacI, SalI, HindIII and dNTPs were obtained
from Roche Applied Science, whereas PicoMaxx high fidelity waspurchased from Agilent technologies. Yeast extract and curcumin wereobtained from Acros Organics. Tryptic soy agar was acquired from MPBiomedicals. Ampicillin, isopropyl β-D-1-thiogalactopyranoside (IPTG),imidazole, sodium monobasic phosphate, sodium dibasic phos-phate, sodium hydroxide, sodium chloride, sucrose, tryptone, and ureawere obtained from Fisher Scientific. 4-(2-Hydroxyethyl)-1-piperazinee-thanesulfonic acid (HEPES), magnesium sulfate, and nickel chloridewere purchased from Sigma Aldrich. Ethylenediaminetetraacetic acid(EDTA), hydrochloric acid was acquired from VWR. HPLC grademethanol was obtained from Ricca Chemical Company. Sinapinic acidwas purchased from Thermo Scientific. Protein standards used forMALDI MS were acquired from New England Biolabs.Construction of DNA Libraries. The following primers were used
to amplify the library of E domain length from pUC19ELP pentamer(gift from D. Tirrell) plasmid: BamHI:5′-ggaggccGGATCCaagcc-gattgcggctagcgcggtgccgg-3′, SacI:5′-gccccGAGCTCcgatccctcgagcggcacccc-gac-3′, SalI:5′-ggaggccGTCGACaagccgattgcggctagcgcggtgccgg-3′, and Hin-dIIII: 5′-ggccccAAGCTTcgcaccggtacccgatccctcgagcggcaccccgac-3′.Fragments of E of 150, 200, 300, and 350 bps were amplified by acombination of increasing the plasmid DNA to 300 ng/μL,decreasing the number of cycles and decreasing the annealingtemperature. The inserts were gel-purified and restricted withappropriate restriction enzymes. E inserts were cloned into thePCR-assembled pQE30/C between restriction sites BamHI and SacIto generate pQE30/EnC or SalI and HindIII yielding pQE30/CEn.The clones were verified by forward and reverse DNA sequencing(MWG operon).Expression and Purification. The resulting plasmids were
transformed into XL1-blue cells and expressed according to previouslypublished protocols.7,8 Transformed library members were cultured in1 L LB media bearing 0.57 mM ampicillin at 37 °C until the opticaldensity at 600 nm (OD600) reached ∼0.6. IPTG was added to 0.52mM final concentration to induce overexpression for overnight. Cellswere harvested and resuspended to 7 mL of lysis buffer (buffer A: 6 Murea, 50 mM Na2HPO4, 20 mM imidazole, pH 8.0), the cellsuspensions were stored at −80 °C. The suspension was thawed at4 °C, osmotically shocked27 by incubation with 40 mL of sucrosebuffer (50 mM HEPES, 20% sucrose, 1 mM EDTA pH 7.9), andpelleted. Supernatants were discarded, and pellets were resuspended in25 mL of 5 mM MgSO4, incubated on ice for 10 min, and harvested.Cell pellets were resuspended in 25 mL of buffer A and subjected tolysis via French press (Thermo Scientific). The proteins in the lysatewas purified via FPLC using a HiTrap IMAC FF column (5 mLvolume) that was charged with NiCl2 and equilibrated with lysis bufferusing an ATKA purifier system (GE Life Sciences). Protein was elutedby adjusting percentage of buffer B (same components as buffer A butwith 200 mM Imidazole) with 50 mL of 1%, 30 mL of 5%, and 50 mLof 100% at 5 mL/min flow rate. The elutions bearing purified proteinwere collected and dialyzed against 10 mM phosphate buffer pH 8.0 at4 °C. Purity was confirmed by SDS-PAGE and ImageQuant-TL 1D gelanalysis program prior to dialysis (GE Life Sciences). Concentrationsof protein were determined via micro-BCA analysis (ThermoScientific) by using SpectraMax M2 (Molecular Devices). Molecularweights were confirmed via matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (MALDI-TOF MS) on a BrukerOmniflex (Table 1).Circular Dichroism. Wavelength-dependent circular dichroism
(CD) spectra were collected on a Jasco J-815 CD spectrometerequipped with a PTC-423S single position Peltier temperature controlsystem and counter-cooled with an Isotemp 3016S (Fisher Scientific)
water bath. Samples were loaded in a Hellma 218 quartz cuvette (500 μL,1 mm path length). A far-UV temperature-dependent wavelength scanfrom 185 to 260 nm as a function of temperature was completed forEnC and CEn library members at 10 μM in 10 mM phosphate bufferpH 8.0 at scan rate of 50 nm/min at 1 °C/min temperature increasingrate. All scans were performed three times for accumulation. CD datawas converted into mean residue molar ellipticity ([θ]mrw) via anequation [θ]mrw = θ·MRW/(10·C·l), where θ is the data obtained inmdeg, MRW is mean residue weight, C is concentration in milligramsper milliliter, and l is path length in centimeters.28 Fitting andcalculation of protein secondary structure was processed withCDSSTR methods.29−32
UV/vis Spectrometry. The inverse temperature transition (Tt)was determined using UV−vis instrument Cary-50 (Varian) withTC125 temperature controller (Quantum Northwest) by observingthe change in turbidity at 320 nm from 15 to 80 °C at 1 °C/mintemperature increasing rate. Protein samples were prepared in 0.2 mg/mL in 10 mM phosphate buffer, 500 mM NaCl, pH 8. Samples wereloaded in type 21 quartz cuvette with 10 mm path length (BuckScience). Scans were performed on at least two different proteinsample preps to calculate the average Tt. Tt was determined at theinflection point of the absorbance curve.33
Transmission Electron Microscopy. Transmission electronmicroscopy (TEM) was used to identify the potential nanometer-sized structures that resulted from self-assembly at 4 °C. Samples wereprepared in water at 0.2 mg/mL concentrations in 10 mM phosphate,500 mM NaCl pH 8.0, and gently vortexed. The samples were appliedon a carbon-coated 400 mesh Cu/Rh grids and negatively stained with1% uranyl acetate. The images of the samples were collected on aPhillips CM12 Tungsten Emission TEM at 120 kV. The particle areaand size were measured using ImageJ.34 Sizes of all particles weredetermined from at least >25 particles from two separately purifiedprotein samples.Microrheology. Lypohilized protein samples were resuspended in
10 mM phosphate buffer, 500 mM NaCl, pH 8.0 at a final con-centration of 15 mg/mL. We added 2 μL of 2% (wt %) of fluorescentamidated polystyrene beads (1.0 μm) to 10 μL of the resuspendedprotein sample. Epifluorescence was monitored on an inverted Nikonmicroscope. A Linkam LST120 peltier cell was used to control temper-ature. All samples were analyzed in replicate both below and abovetheir Tt values (E1C = 45 °C, E2C = 42 °C, E3C = 40 °C, E4C = 30 °C,CE1 = 64 °C, CE2 = 53 °C, CE3 = 37 °C, and CE4 = 33 °C). Sampleswere equilibrated at their appropriate temperatures for ∼4 min on thetemperature stage prior to their run. Videos were recorded in triplicatefrom various locations on the slide with a QiCam (640 × 480 pixels at30 and 60 fps) and converted to 8-bit tiff file for IDL analysis. Particletrajectories were obtained from three videos with IDL image softwareanalysis, and the dynamic moduli were determined using the Stokes−Einstein relation.35−40
Fluorescence. Protein stock samples were prepared at 6 μM con-centration in 10 mM phosphate buffer, pH 8. Curcumin was freshlydissolved in HPLC grade methanol, followed by dilution into 10 mMphosphate buffer pH 8 to give stock standards ranging from 1.5 to 54 μM.Proteins were loaded in a Costar 96-well black plate (Corning LifeScience); subsequently, curcumin standards were added using Biomek
Table 1. Protein Polymer Yields and Molecular Weights
NXP Laboratory Automation Workstation (Beckman Coulter) to startall binding reactions at the same time. Final protein polymerconcentration was 4 μM, and curcumin concentrations ranged from0.5 to 18 μM.8 For each experiment, three replicates of curcuminstandards and two replicates of protein with curcumin were prepared.The average dissociation constant (Kd) was calculated from twoseparate batches of proteins from two different expressions.Fluorescence was recorded after 2 h of incubation by usingSpectraMax M2 (Molecular Devices) with 420 nm excitation, 495nm emission, and 30 readings per well. Fitting and determination of Kdwere carried with Sigmaplot using one site saturation withoutnonspecific binding equation: Fb = Fs[L]/(Kd + [L]) in which Fb isthe fluorescence signal of bound ligand, [L] is the ligand concentrationin micromolar, and Fs is the plateau signal at saturation.25
■ RESULTS
Biosynthesis of Protein Libraries. To create the EnC andCEn libraries in which the E domain was systematicallytruncated, we PCR amplified the E gene using primers bearingBamHI and SacI resitriction sites or SalI and HindIII restrictionsites. The E fragments ranging from 150 to 350 bps werecloned into the parent plasmid pQE30/C (bearing the Cdomain) to produce pQE30/EnC or pQE30/CEn.
7 Thisproduced library members in which one to four repeats ofthe E domain was expressed (Figure 1). The protein polymerlibrary members were overexpressed, purified, and character-ized. After affinity purification, 3.0−11.3 mg was recovered foreach protein polymer (Table 1). Whereas SDS-PAGE analysisdemonstrated slightly higher molecular weights for EnC andCEn (Figure 1B), the exact molar masses were confirmed byMALDI-TOF MS (Table 1).Inverse Transition Temperature. We hypothesized that
shortening the E domain length would cause an increase in theTt and overall thermoresponsive behavior of the protein blockpolymers. To test this, we monitored the UV/vis absorbance ofthe EnC and CEn library members as a function of temperature. Allprotein polymers exhibited incrementally elevated Tt values upontruncation of E at 0.2 mg/mL concentrations (Table 2, Figure 2).
Whereas E4C and CE4 illustrated similar Tt values of 28.5 and27.0 °C, respectively, the rest of the library members revealeddifferent values. The EnC library presented a narrow range ofTt values with a maximal difference of 9.5 °C when comparingE4C to E1C. By contrast, the CEn library presented a morebroad temperature range with a 32 °C maximal dif-ference. The degree by which the temperature changed wasdependent on the orientation of the diblocks as the EnC and
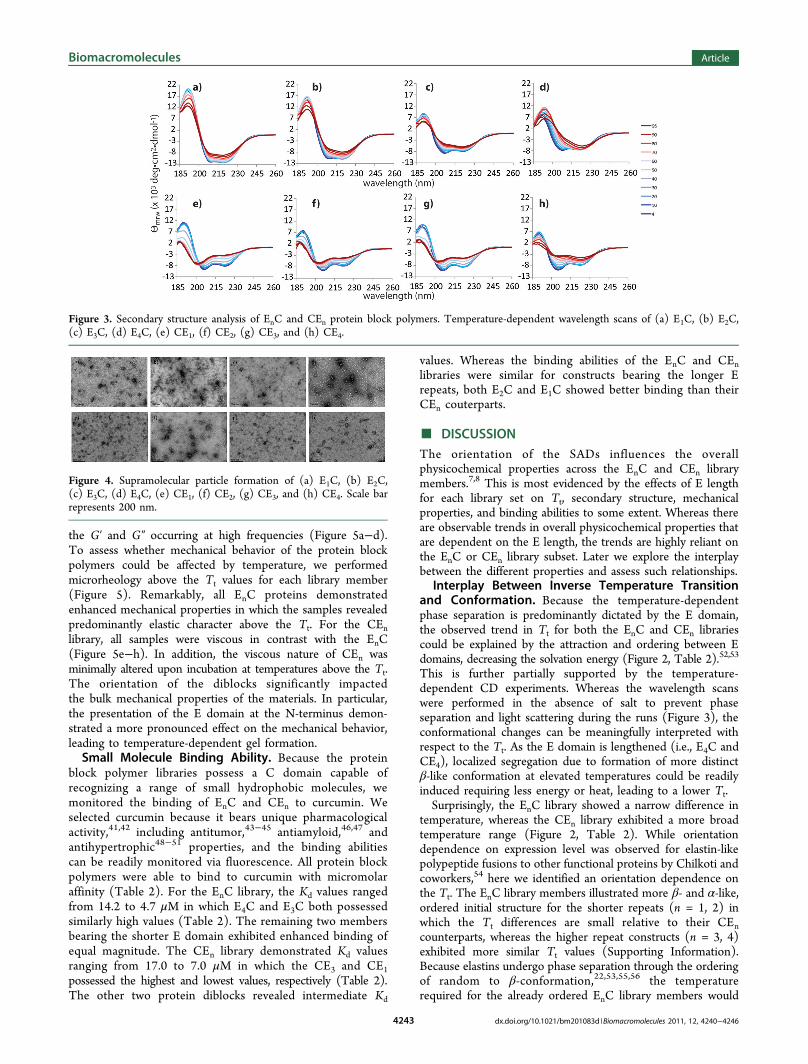
determine the effects of E domain truncation on the overallsecondary structure of the EnC and CEn libraries, far-UV wave-length scans were performed as a function of temperature(Figure 3). In our previous work, we demonstrated that thediblocks EC and CE were structurally different even thoughthey were nearly identical in composition.7 As expected, theEnC and CEn diblocks differed in secondary structure andexhibited different temperature-dependent conformationalchanges (Figure 3, Supporting Information). The E4C waspredominantly unordered or random-like at 4 °C, and uponincreasing temperature, it assumed more β-conformation. Asimilar trend was observed for E3C, in which the initialstructure was more random-like and the final structure was lessβ-like relative to E4C. Interestingly a shift in the trend wasobserved for E2C; at 4 °C, it was helical and random-like, and atelevated temperature it was predominantly β-like in con-formation. The E1C exhibited a helical signature and a finalstructure that was mostly β-structured with almost an equalamount of random conformation. For the CEn library, allproteins exhibited a significant random-like conformation at4 °C and became more β-like with a substantial unorderedpopulation. As the E domain was shortened, the startingstructure was less random with more helical contribution, andthe final structure at elevated temperatures was more β-like yetmaintained a predominantly unordered conformation for theCEn series. Essentially, the decrease in E length led to areduction in β-conformation and increase in helical contribu-tion from the C domain for both series at low temperatures.Supramolecular Particle Assemblies. Because previous
studies showed that such protein block polymers couldassemble into supramolecular structures, the library memberswere visualized via TEM to assess the assemblies formed. Allproteins revealed the formation of discrete nanometer-sizedparticles confirming the supramolecular assemblies at 0.2 mg/mLconcentrations (Figure 4, Supporting Information). In the caseof the EnC library members, the particles ranged from 26.4 ±3.8 to 28.5 ± 3.1 nm, whereas for CEn library members theparticles ranged from 26.0 ± 3.0 to 30.4 ± 6.1 nm (Table 2).The truncation of the E domain for both libraries did notsubstantially influence the size of the particles because they allwere within the same size range.Temperature-Dependent Mechanical Properties. To
determine the mechanical properties of the protein polymerlibrary, microrheology was performed at 12.5 mg/mL proteinconcentrations. In the case of the EnC library at 22 °C, allproteins exhibited viscoelastic behavior with a crossover of
Table 2. Supramolecular Assembly and Small MoleculeBinding of Protein Library
aValues are obtained from an average of at least three trials of twoindependent protein preps. bSizes determined from TEM usingImageJ. cCalculated values are determined and an average of at leasttwo trials of two independent protein preps.
Figure 2. Characterization of inverse thermal transitions. (a) Turbidityprofiles for E1C (circle), E2C (diamond), E3C (triangle), and E4C(square) of the EnC library. (b) Turbidity profiles for CE1 (circle), CE2(diamond), CE3 (triangle), and CE4 (square) of the CEn library.
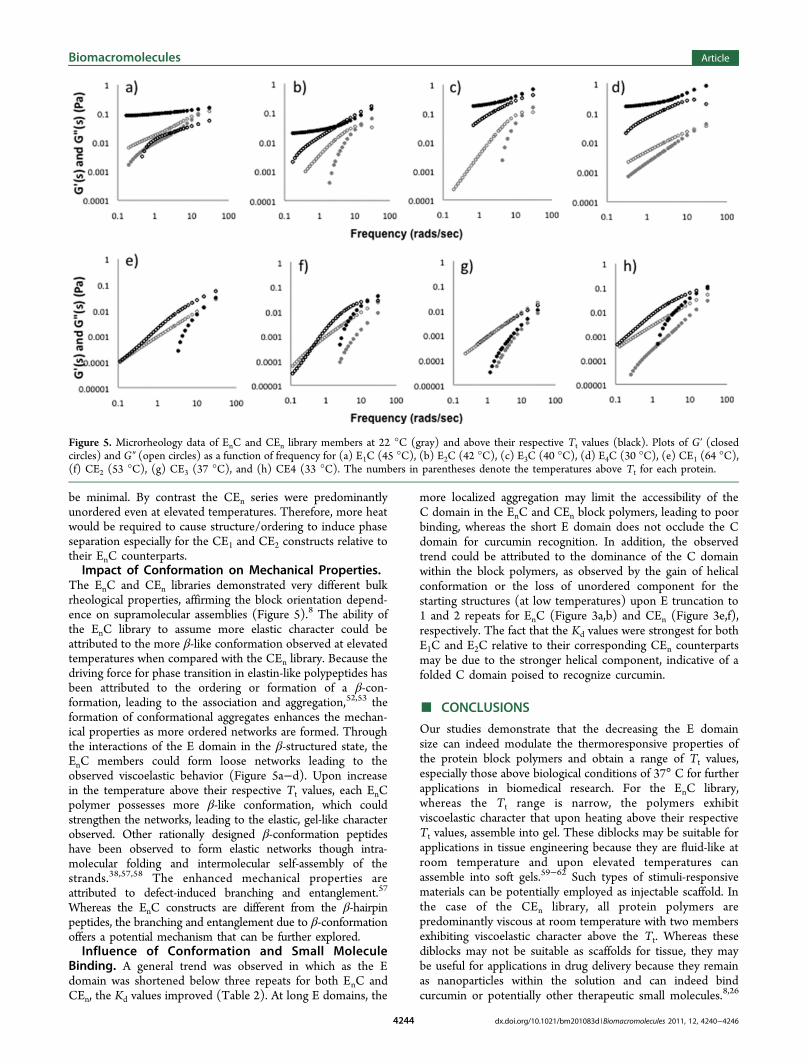
the G′ and G″ occurring at high frequencies (Figure 5a−d).To assess whether mechanical behavior of the protein blockpolymers could be affected by temperature, we performedmicrorheology above the Tt values for each library member(Figure 5). Remarkably, all EnC proteins demonstratedenhanced mechanical properties in which the samples revealedpredominantly elastic character above the Tt. For the CEnlibrary, all samples were viscous in contrast with the EnC(Figure 5e−h). In addition, the viscous nature of CEn wasminimally altered upon incubation at temperatures above the Tt.The orientation of the diblocks significantly impactedthe bulk mechanical properties of the materials. In particular,the presentation of the E domain at the N-terminus demon-strated a more pronounced effect on the mechanical behavior,leading to temperature-dependent gel formation.Small Molecule Binding Ability. Because the protein
block polymer libraries possess a C domain capable ofrecognizing a range of small hydrophobic molecules, wemonitored the binding of EnC and CEn to curcumin. Weselected curcumin because it bears unique pharmacologicalactivity,41,42 including antitumor,43−45 antiamyloid,46,47 andantihypertrophic48−51 properties, and the binding abilitiescan be readily monitored via fluorescence. All protein blockpolymers were able to bind to curcumin with micromolaraffinity (Table 2). For the EnC library, the Kd values rangedfrom 14.2 to 4.7 μM in which E4C and E3C both possessedsimilarly high values (Table 2). The remaining two membersbearing the shorter E domain exhibited enhanced binding ofequal magnitude. The CEn library demonstrated Kd valuesranging from 17.0 to 7.0 μM in which the CE3 and CE1possessed the highest and lowest values, respectively (Table 2).The other two protein diblocks revealed intermediate Kd
values. Whereas the binding abilities of the EnC and CEnlibraries were similar for constructs bearing the longer Erepeats, both E2C and E1C showed better binding than theirCEn couterparts.
■ DISCUSSIONThe orientation of the SADs influences the overallphysicochemical properties across the EnC and CEn librarymembers.7,8 This is most evidenced by the effects of E lengthfor each library set on Tt, secondary structure, mechanicalproperties, and binding abilities to some extent. Whereas thereare observable trends in overall physicochemical properties thatare dependent on the E length, the trends are highly reliant onthe EnC or CEn library subset. Later we explore the interplaybetween the different properties and assess such relationships.Interplay Between Inverse Temperature Transition
and Conformation. Because the temperature-dependentphase separation is predominantly dictated by the E domain,the observed trend in Tt for both the EnC and CEn librariescould be explained by the attraction and ordering between Edomains, decreasing the solvation energy (Figure 2, Table 2).52,53
This is further partially supported by the temperature-dependent CD experiments. Whereas the wavelength scanswere performed in the absence of salt to prevent phaseseparation and light scattering during the runs (Figure 3), theconformational changes can be meaningfully interpreted withrespect to the Tt. As the E domain is lengthened (i.e., E4C andCE4), localized segregation due to formation of more distinctβ-like conformation at elevated temperatures could be readilyinduced requiring less energy or heat, leading to a lower Tt.Surprisingly, the EnC library showed a narrow difference in
temperature, whereas the CEn library exhibited a more broadtemperature range (Figure 2, Table 2). While orientationdependence on expression level was observed for elastin-likepolypeptide fusions to other functional proteins by Chilkoti andcoworkers,54 here we identified an orientation dependence onthe Tt. The EnC library members illustrated more β- and α-like,ordered initial structure for the shorter repeats (n = 1, 2) inwhich the Tt differences are small relative to their CEncounterparts, whereas the higher repeat constructs (n = 3, 4)exhibited more similar Tt values (Supporting Information).Because elastins undergo phase separation through the orderingof random to β-conformation,22,53,55,56 the temperaturerequired for the already ordered EnC library members would
Figure 3. Secondary structure analysis of EnC and CEn protein block polymers. Temperature-dependent wavelength scans of (a) E1C, (b) E2C,(c) E3C, (d) E4C, (e) CE1, (f) CE2, (g) CE3, and (h) CE4.
be minimal. By contrast the CEn series were predominantlyunordered even at elevated temperatures. Therefore, more heatwould be required to cause structure/ordering to induce phaseseparation especially for the CE1 and CE2 constructs relative totheir EnC counterparts.Impact of Conformation on Mechanical Properties.
The EnC and CEn libraries demonstrated very different bulkrheological properties, affirming the block orientation depend-ence on supramolecular assemblies (Figure 5).8 The ability ofthe EnC library to assume more elastic character could beattributed to the more β-like conformation observed at elevatedtemperatures when compared with the CEn library. Because thedriving force for phase transition in elastin-like polypeptides hasbeen attributed to the ordering or formation of a β-con-formation, leading to the association and aggregation,52,53 theformation of conformational aggregates enhances the mechan-ical properties as more ordered networks are formed. Throughthe interactions of the E domain in the β-structured state, theEnC members could form loose networks leading to theobserved viscoelastic behavior (Figure 5a−d). Upon increasein the temperature above their respective Tt values, each EnCpolymer possesses more β-like conformation, which couldstrengthen the networks, leading to the elastic, gel-like characterobserved. Other rationally designed β-conformation peptideshave been observed to form elastic networks though intra-molecular folding and intermolecular self-assembly of thestrands.38,57,58 The enhanced mechanical properties areattributed to defect-induced branching and entanglement.57
Whereas the EnC constructs are different from the β-hairpinpeptides, the branching and entanglement due to β-conformationoffers a potential mechanism that can be further explored.Influence of Conformation and Small Molecule
Binding. A general trend was observed in which as the Edomain was shortened below three repeats for both EnC andCEn, the Kd values improved (Table 2). At long E domains, the
more localized aggregation may limit the accessibility of theC domain in the EnC and CEn block polymers, leading to poorbinding, whereas the short E domain does not occlude the Cdomain for curcumin recognition. In addition, the observedtrend could be attributed to the dominance of the C domainwithin the block polymers, as observed by the gain of helicalconformation or the loss of unordered component for thestarting structures (at low temperatures) upon E truncation to1 and 2 repeats for EnC (Figure 3a,b) and CEn (Figure 3e,f),respectively. The fact that the Kd values were strongest for bothE1C and E2C relative to their corresponding CEn counterpartsmay be due to the stronger helical component, indicative of afolded C domain poised to recognize curcumin.
■ CONCLUSIONS
Our studies demonstrate that the decreasing the E domainsize can indeed modulate the thermoresponsive properties ofthe protein block polymers and obtain a range of Tt values,especially those above biological conditions of 37° C for furtherapplications in biomedical research. For the EnC library,whereas the Tt range is narrow, the polymers exhibitviscoelastic character that upon heating above their respectiveTt values, assemble into gel. These diblocks may be suitable forapplications in tissue engineering because they are fluid-like atroom temperature and upon elevated temperatures canassemble into soft gels.59−62 Such types of stimuli-responsivematerials can be potentially employed as injectable scaffold. Inthe case of the CEn library, all protein polymers arepredominantly viscous at room temperature with two membersexhibiting viscoelastic character above the Tt. Whereas thesediblocks may not be suitable as scaffolds for tissue, they maybe useful for applications in drug delivery because they remainas nanoparticles within the solution and can indeed bindcurcumin or potentially other therapeutic small molecules.8,26
Figure 5. Microrheology data of EnC and CEn library members at 22 °C (gray) and above their respective Tt values (black). Plots of G′ (closedcircles) and G″ (open circles) as a function of frequency for (a) E1C (45 °C), (b) E2C (42 °C), (c) E3C (40 °C), (d) E4C (30 °C), (e) CE1 (64 °C),(f) CE2 (53 °C), (g) CE3 (37 °C), and (h) CE4 (33 °C). The numbers in parentheses denote the temperatures above Tt for each protein.
Furthermore, it can be possible to exploit the Tt for localizationof the nanoparticle delivery vehicle within a particular regionvia hyperthermia.13−16 Experiments are underway to explorethe applications of these protein polymers for such biomedicalapplications.
■ ASSOCIATED CONTENT*S Supporting InformationDNA gels, secondary structural analysis via CDSSTR, TEManalysis, DTT experiments, and fluorescence binding plots.This material is available free of charge via the Internet athttp://pubs.acs.org.
■ ACKNOWLEDGMENTSThis work was supported by AFOSR FA-9550-07-1-0060 and FA-9550-08-1-0266 (J.K.M.), partially by the NSF MRSEC Programunder award number DMR-0820341, Society of Plastic Engineers(J.S.H.), and GK-12 Fellows grant DGE-0741714 (J.S.H.).
■ REFERENCES(1) Channon, K. J.; Devlin, G. L.; MacPhee, C. E. J. Am. Chem. Soc.
2009, 131, 12520.(2) John, G.; Vemula, P. K. Soft Matter 2006, 2, 909.(3) Kopecek, J. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5929.(4) Shen, X. C.; Mo, X. Q.; Moore, R.; Frazier, S. J.; Iwamoto, T.;
Tomich, J. M.; Sun, X. Z. S. J. Nanosci. Nanotechnol. 2006, 6, 837.(5) Connor, R. E.; Tirrell, D. A. Polym. Rev. 2007, 47, 9.(6) Voloshchuk, N.; Montclare, J. K. Mol. BioSyst. 2010, 6, 65.(7) Haghpanah, J. S.; Yuvienco, C.; Civay, D. E.; Barra, H.; Baker, P. J.;
Khapli, S.; Voloshchuk, N.; Gunasekar, S. K.; Muthukumar, M.;Montclare, J. K. ChemBioChem 2009, 10, 2733.(8) Haghpanah, J. S.; Yuvienco, C.; Roth, E. W.; Liang, A.; Tu, R. S.;
Montclare, J. K. Mol. Biosyst. 2010, 6, 1662.(9) Krishnaji, S. T.; Huang, W.; Rabotyagova, O.; Kharlampieva, E.;
Choi, I.; Tsukruk, V. V.; Naik, R.; Cebe, P.; Kaplan, D. L. Langmuir2011, 27, 1000.(10) Rabotyagova, O. S.; Cebe, P.; Kaplan, D. L. Macromol. Biosci.
1739.(12) Xu, C.; Kopecek, J. Pharm. Res. 2008, 25, 674.(13) Furgeson, D. Y.; Dreher, M. R.; Chilkoti, A. J. Controlled Release
2006, 110, 362.(14) Mackay, J. A.; Chilkoti, A. Int. J. Hyperthermia 2008, 24, 483.(15) Dreher, M. R.; Simnick, A. J.; Fischer, K.; Smith, R. J.; Patel, A.;
Schmidt, M.; Chilkoti, A. J. Am. Chem. Soc. 2008, 130, 687.(16) Dreher, M. R.; Liu, W. G.; Michelich, C. R.; Dewhirst, M. W.;
Chilkoti, A. Cancer Res. 2007, 67, 4418.(17) Nagarsekar, A.; Crissman, J.; Crissman, M.; Ferrari, F.;
Cappello, J.; Ghandehari, H. J. Biomed. Mater. Res. 2002, 62, 195.(18) Nagarsekar, A.; Crissman, J.; Crissman, M.; Ferrari, F.;
Cappello, J.; Ghandehari, H. Biomacromolecules 2003, 4, 602.(19) Dandu, R.; Von Cresce, A.; Briber, R.; Dowell, P.; Cappello, J.;
Ghandehari, H. Polymer 2009, 50, 366.(20) Shen, W.; Kornfield, J. A.; Tirrell, D. A. Soft Matter 2007, 3, 99.(21) Shen, W.; Kornfield, J. A.; Tirrell, D. A. Macromolecules 2007,
40, 689.(22) Meyer, D. E.; Chilkoti, A. Biomacromolecules 2004, 5, 846.(23) Urry, D. W.; Mitchell, L. W.; Ohnishi, T. Biochim. Biophys. Acta,
Protein Struct. 1975, 393, 296.(24) Baker, P. J.; Haghpanah, J. S.; Montclare, J. K. Elastin Based
Protein Polymers. In Green Polymer Chemistry: Biocatalysis and
Biomaterials; Cheng, H. N., Gross, R. A., Eds.; Oxford UniversityPress: Oxford, U.K., 2008.(25) Guo, Y.; Bozic, D.; Malashkevich, V. N.; Kammerer, R. a.;
Schulthess, T.; Engel, J. EMBO J. 1998, 17, 5265.(26) Gunasekar, S. K.; Asnani, M.; Limbad, C.; Haghpanah, J. S.;
Hom, W.; Barra, H.; Nanda, S.; Lu, M.; Montclare, J. K. Biochemistry2009, 48, 8559.(27) Magnusdottir, A.; Johansson, I.; Dahlgren, L.-G.; Nordlund, P.;
Berglund, H. Nat. Methods 2009, 6, 477.(28) Lee, D. L.; Mant, C. T.; Hodges, R. S. J. Biol. Chem. 2003, 278,
22918.(29) Sreerama, N.; Venyaminov, S. Y.; Woody, R. W. Anal. Biochem.
2000, 287, 243.(30) Whitmore, L.; Wallace, B. A. Biopolymers 2008, 89, 392.(31) Whitmore, L.; Wallace, B. A. Nucleic Acids Res. 2004, 32, W668.(32) Sreerama, N.; Woody, R. W. Anal. Biochem. 2000, 287, 252.(33) Nuhn, H.; Klok, H.-A. Biomacromolecules 2008, 9, 2755.(34) Abramoff, M. D. M.; Paulo, J.; Ram, S. J. Biophotonics Int. 2004,
11, 6.(35) Veerman, C.; Rajagopal, K.; Palla, C. S.; Pochan, D. J.;
Schneider, J. P.; Furst, E. M. Macromolecules 2006, 39, 6608.(36) Gardel, M. L.; Valentine, M. T.; Weitz, D. A. Microscale
Diagnostic Techniques; Breuer, K. S., Ed.; Springer: Berlin, 2005.(37) Breedveld, V.; Pine, D. J. J. Mater. Sci. 2003, 38, 4461.(38) Schneider, J. P.; Pochan, D. J.; Ozbas, B.; Rajagopal, K.; Pakstis,
L.; Kretsinger, J. J. Am. Chem. Soc. 2002, 124, 15030.(39) Mason, T. G.; Ganesan, K.; van Zanten, J. H.; Wirtz, D.; Kuo, S.
C. Phys. Rev. Lett. 1997, 79, 3282.(40) Mason, T. G.; Weitz, D. A. Phys. Rev. Lett. 1995, 74, 1250.(41) Shishodia, S.; Sethi, G.; Aggarwal, B. B. Ann. N.Y. Acad. Sci.
2005, 1056, 206.(42) Shehzad, A.; Lee, Y. S. Drugs Future 2010, 35, 113.(43) Mehta, K.; Pantazis, P.; McQueen, T.; Agarwal, B. Anticancer
Drugs 1997, 8, 471.(44) Cheng, A. L.; Hsu, C. H.; Lin, J. K.; Hsu, M. M.; Ho, Y. F.;
Shen, T. S.; Ko, J. Y.; Lin, J. T.; Lin, B. R.; Wu, M. S.; Yu, H. S.; Jee, S.H.; Chen, G. S.; Chen, T. M.; Chen, C. A.; Lai, M. K.; Pu, Y. S.; Pan,M. H.; Wang, Y. J.; Tsai, C. C.; Hsieh, C. Y. Anticancer Res. 2001, 21,2895.(45) Bisht, S.; Feldmann, G.; Soni, S.; Ravi, R.; Karikar, C.; Maitra, A.
J. Nanobiotechnol. 2007, 5, 3.(46) DaSilva, K. A.; Shaw, J. E.; McLaurin, J. Exp. Neurol. 2010, 223,
311.(47) Darvesh, A. S.; Carroll, R. T.; Bishayee, A.; Geldenhuys, W. J.;
Van der Schyf, C. J. Expert Rev. Neurother. 2010, 10, 729.(48) Suzuki, H.; Morimoto, T.; Sunagawa, Y.; Kawamura, T.; Wada,
(59) Kretsinger, J. K.; Haines, L. A.; Ozbas, B.; Pochan, D. J.;Schneider, J. P. Biomaterials 2005, 26, 5177.(60) Morihara, Y.; Ogata, S.-I.; Kamitakahara, M.; Ohtsuki, C.;
Tanihara, M. J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 6048.(61) Collier, J. H.; Hu, B. H.; Ruberti, J. W.; Zhang, J.; Shum, P.;
Thompson, D. H.; Messersmith, P. B. J. Am. Chem. Soc. 2001, 123,9463.(62) Betre, H.; Liu, W.; Zalutsky, M. R.; Chilkoti, A.; Kraus, V. B.;
Setton, L. A. J. Controlled Release 2006, 115, 175.