Augmented Protein Kinase C-�–Induced MyofilamentProtein Phosphorylation Contributes to Myofilament
Dysfunction in Experimental Congestive Heart FailureRashad J. Belin, Marius P. Sumandea, Edward J. Allen, Kelly Schoenfelt, Helen Wang,
R. John Solaro, Pieter P. de Tombe
Abstract—It is becoming clear that upregulated protein kinase C (PKC) signaling plays a role in reduced ventricularmyofilament contractility observed in congestive heart failure. However, data are scant regarding which PKC isozymesare involved. There is evidence that PKC-� may be of particular importance. Here, we examined PKC-� quantity,activity, and signaling to myofilaments in chronically remodeled myocytes obtained from rats in either early heart failureor end-stage congestive heart failure. Immunoblotting revealed that PKC-� expression and activation was unaltered inearly heart failure but increased in end-stage congestive heart failure. Left ventricular myocytes were isolated bymechanical homogenization, Triton-skinned, and attached to micropipettes that projected from a force transducer andmotor. Myofilament function was characterized by an active force–[Ca2�] relation to obtain Ca2�-saturated maximalforce (Fmax) and myofilament Ca2� sensitivity (indexed by EC50) before and after incubation with PKC-�, proteinphosphatase type 1 (PP1), or PP2a. PKC-� treatment induced a 30% decline in Fmax and 55% increase in the EC50 incontrol cells but had no impact on myofilament function in failing cells. PP1-mediated dephosphorylation increased Fmax
(15%) and decreased EC50 (�20%) in failing myofilaments but had no effect in control cells. PP2a-dependentdephosphorylation had no effect on myofilament function in either group. Lastly, PP1 dephosphorylation restoredmyofilament function in control cells hyperphosphorylated with PKC-�. Collectively, our results suggest that inend-stage congestive heart failure, the myofilament proteins exist in a hyperphosphorylated state attributable, in part, toincreased activity and signaling of PKC-�. (Circ Res. 2007;101:000-000.)
Key Words: heart failure � protein kinase C-� � myofilament proteins � Ca2� sensitivity� protein phosphatase type 1 � phosphorylation � troponin � myocyte
It has been predicted that the global incidence and preva-lence of the clinical syndrome of congestive heart failure
(CHF) will continue to rise.1 The “road” to CHF usuallybegins with some inciting event (eg, myocardial infarction),which imposes a heightened mechanical strain on the myo-cardium. Ventricular dysfunction ensues resulting in a declinein cardiac output. In turn, key regulatory neurohormonalsignals are recruited, which, in the acute phase, maintaincardiac output and “mask” the underlying ventricular con-tractile deficit. However, prolonged exposure of the heart tothese signals coupled with the prevailing mechanical over-load proves deleterious resulting in contractile dysfunction,myocyte hypertrophy, and death, heralding a downwardspiral wherein ventricular dysfunction becomes manifest andthe clinical features of CHF overt. Not surprisingly, consid-erable attention is now being focused on unraveling themolecular and cellular complexities that conspire to promotecontractile dysfunction of the failing cardiac myocyte, withthe underlying aim being identification of novel molecules
that may be potential foci for therapeutic intervention. Onepromising therapeutic target is the multifunctional proteinkinase C (PKC) signaling system. PKC comprises a family ofserine/threonine kinases activated by mechanical and neuro-hormonal signals that are involved in the management of amyriad of cellular processes including: transcription, cellgrowth, cell death, Ca2�-handling, and myofilament function.2–5
Recent studies have suggested that PKC-mediated, site-specific phosphorylation of cardiac troponin (cTn)I and cTnTdepresses myofilament contractility, intrinsic myocyte func-tion, and ventricular pump function.4–10 Moreover, we andothers have observed that, in various animal models ofend-stage cardiac failure (eg, myocardial infarction, pressureoverload) and in different cardiac muscle preparations (eg,cells and trabeculae) isolated from different regions of themyocardium (right versus left ventricle), myofilament func-tion is severely depressed.11–17 Furthermore, in a recent reportusing 2 distinct rat models of ventricular failure, we observeddepressed myofilament function secondary to dysfunction of
Original received January 15, 2007; revision received May 3, 2007; accepted May 24, 2007.From the Department of Physiology and Biophysics, Center for Cardiovascular Research, University of Illinois at Chicago.Correspondence to Dr Pieter P. de Tombe, PhD, University of Illinois at Chicago, Department of Physiology and Biophysics, 835 S Wolcott (M/C 901),
the regulatory cTn complex and increased phosphorylation ofcTnI.11 Our central tenet is that prolonged excess mechanicalstress/strain on the myocyte activates several PKC isozymes,a process that, in addition to promoting cell growth and death,also triggers functionally important phosphorylations of sev-eral myofilament proteins causing depressed myofilamentand, consequently, myocyte function. Indeed, several studieshave shown that activity and expression of numerous PKCisozymes is upregulated in experimental and human CHF.18–20
Once activated, there are several distinct PKC isozymesthat would affect the cardiac sarcomere to elicit myofilamentdysfunction in failing hearts.7,21,22 Recently, PKC-� hasemerged as a key player in contractile dysfunction, develop-ment of heart failure (HF), and control of myofilamentactivity. Studies from Braz et al indicate that depletion ofmyocardial PKC-� results in increased myocardial contrac-tility, whereas transgenic overexpression of the moleculeleads to marked ventricular dysfunction.23 These investiga-tors proposed that myocyte dysfunction in the PKC-� trans-genic mouse is caused by alterations in Ca2� homeostasis.23
Both cTnI and cTnT are viable substrates for PKC-�–dependent phosphorylation,4,5,24 and studies by Sumandea etal5 demonstrate that PKC-� depresses myofilament contrac-tility through site-specific phosphorylation of cTnT at thethreonine-206 residue. Thus, it is plausible that augmentedsignaling through PKC-� elicits myofilament dysfunction ofthe failing myocyte through phosphorylation of cTnI and/orcTnT. However, studies examining the functional link be-tween PKC-� and myofilament dysfunction in failing ventri-cles are lacking. Accordingly, we examined PKC-� expres-sion, activation, and impact on myofilament function inskinned left ventricular myocytes isolated from rats subjectedto chronic (8 to 9 months) pressure overload and myocardialinfarction; experimental CHF models in which myofilamentfunction is severely depressed.11 We also examined PKC-�expression and activity in rats at an early stage of HF toelucidate the role of PKC-� in mediating the transition toend-stage CHF. To assess whether myofilament proteinhyperphosphorylation causes the depressed myofilament phe-notype in experimental CHF, we determined whether dephos-phorylation of failing myofilaments with protein phosphatasetype 1 (PP1) or PP2a could rescue myofilament force devel-opment and Ca2�sensitivity. Here, we report that PKC-�expression and activity was unaltered in early HF but wassimilarly upregulated in 2 distinct rat models of end-stageCHF. In skinned myocytes, PKC-�–dependent phosphoryla-tion resulted in marked depression of control myofilamentfunction but had no impact on failing myofilaments. Also,PP1-elicited dephosphorylation increased myofilament func-tion in failing myocytes and control cells hyperphosphorylat-ed with PKC-� but had no effect in untreated control cells.Our data indicate that myofilament dysfunction in end-stageCHF arises, in part, from increased myofilament proteinphosphorylation through PKC-�.
Materials and MethodsAnimal Models of Ventricular Hypertrophyand CHFAscending aortic banding and myocardial infarction (MI) wereperformed on 4-week female Sprague–Dawley rats as described
previously, with slight modifications.11,15 Animals in the early HFcohort were followed for a period of 12 weeks,25 whereas animals inthe end-stage CHF group were followed for 32 to 36 weeks until theytransitioned to end-stage CHF. Unoperated age-matched animalsserved as controls. Previously, we found no difference between shamoperated and age-matched control animals.11,14
Force–[Ca2�] Measurements in SkinnedVentricular MyocytesMyocytes were isolated from the interventricular septum and leftventricular free wall of CHF, left ventricular hypertrophy (LVH),and age-matched control hearts by mechanical homogenizationand chemically permeabilized (skinned) with 0.3% TritonX-100.11,14 We observed no difference in myofilament functionbetween septal and left ventricular myocytes.11 Details of thesolutions for cell isolation and experimentation have been previ-ously described in detail.11,14,26 Cells were stored on ice and usedwithin 20 hours of isolation. All experiments were performed on thestage of an inverted microscope (Figure 3).11 Sarcomere length wasset to 2.10 �m by video micrometry.11
PKC-�, PP1, and PP2a ExpressionApproximately 30 to 40 mg of left ventricular early HF, LVH, CHF,and age-matched control myocardium was homogenized in homog-enization buffer (20 mmol/L HEPES, 150 mmol/L NaCl, 15%[vol/vol] glycerol, 5 mmol/L MgCl2, 1 mmol/L EGTA, 1 mmol/LEDTA, 1 mmol/L Na3VO4, 100 mmol/L NaF, 10 mmol/L Na-pyrophosphate, 1% [vol/vol] Triton X-100, 1% [vol/vol] Na-deoxycholate, 1 mmol/L dithiothreitol, 0.1% [vol/vol], 1 mmol/L4-[2-aminoethyl]benzenesulfonyl fluoride, 50 �g/mL aprotinin,5 mmol/L pepstatin A, and 50 �g/mL leupeptin) on ice. The homoge-nate was centrifuged at 4°C and 100 000g for 1 hour. The proteinconcentration of the supernatant was determined and the samples werestored at �80°C. A total of 150 �g of protein was loaded onto 10%SDS-PAGE gels. The electrophoresed proteins were transferred topoly(vinylidene difluoride) membranes. Membranes were incubatedwith primary antibodies against PKC-�, phospho-specific PKC-�,PKC-�, PKC-�, PKC-�, PP1, and PP2a (Upstate Biotechnology).Secondary anti-mouse and anti-rabbit IgG peroxidase conjugates(Sigma-Aldrich) were used. The relative abundance of single proteinswas detected using enhanced chemiluminescence (Amersham Bio-sciences). Suitable films were scanned and PKC-�, phospho-specificPKC-�, PKC-�, PKC-�, PKC-�, PP1, PP2a, and actin band density wasquantified using commercially available software (Image J; NIH).Scanning units were normalized to actin units.
Expression and Purification of Recombinant PKC-�Expression and purification of recombinant human PKC-� wasexecuted as described previously in detail.5
PKC-�–Mediated Phosphorylation and PP1- andPP2a-Mediated Dephosphorylation of MyofilamentProteins in Ventricular MyocytesTo assess the impact of PKC-� mediated phosphorylation onmyofilament function an attached cell was washed with PKC-�buffer less the enzyme for 2 minutes. The cell was then incubated inPKC-� buffer (1 mmol/L NaF, 1 mmol/L Na3VO4, 0.5 mmol/L MgCl2,100 mmol/L leupeptin, 100 mmol/L pepstatin, 150 mmol/LPMSF�EtOH, 1 mmol/L dithiothreitol, 0.8 �mol/L Ca2�, 20 �mol/Ldiacylglycerol [DAG], 0.3 mmol/L phosphatidylserine [PS], and 0.1�g/mL recombinant PKC-�) at 22°C to 25°C for 60 minutes. For PP1-and PP2a-induced dephosphorylation, myocytes were incubated inrelaxing solution containing the catalytic subunit of PP1 (0.15 U/mL;Upstate Biotechnology) or PP2a (0.15 U/mL; Upstate Biotechnol-ogy) along with 1 mmol/L dithiothreitol at 22°C to 25°C for 60minutes. Following the incubation, the attached cell was washed andexposed to 4 submaximal Ca2� concentrations.
2 Circulation Research July 20, 2007
by guest on July 12, 2018http://circres.ahajournals.org/
Data and Statistical AnalysesCell data were analyzed as described earlier.11,14,26 Data are ex-pressed as means�SEM. Statistical differences in PKC-�, PP1, andPP2a expression between early HF, end-stage CHF, and age-matchedcontrol ventricles were determined using unpaired Student’s t test.The impact of lipids, PKC-�, PP1, and PP2a on myofilamentfunction was determined by a paired Student’s t test with a P�0.05considered statistically significant.
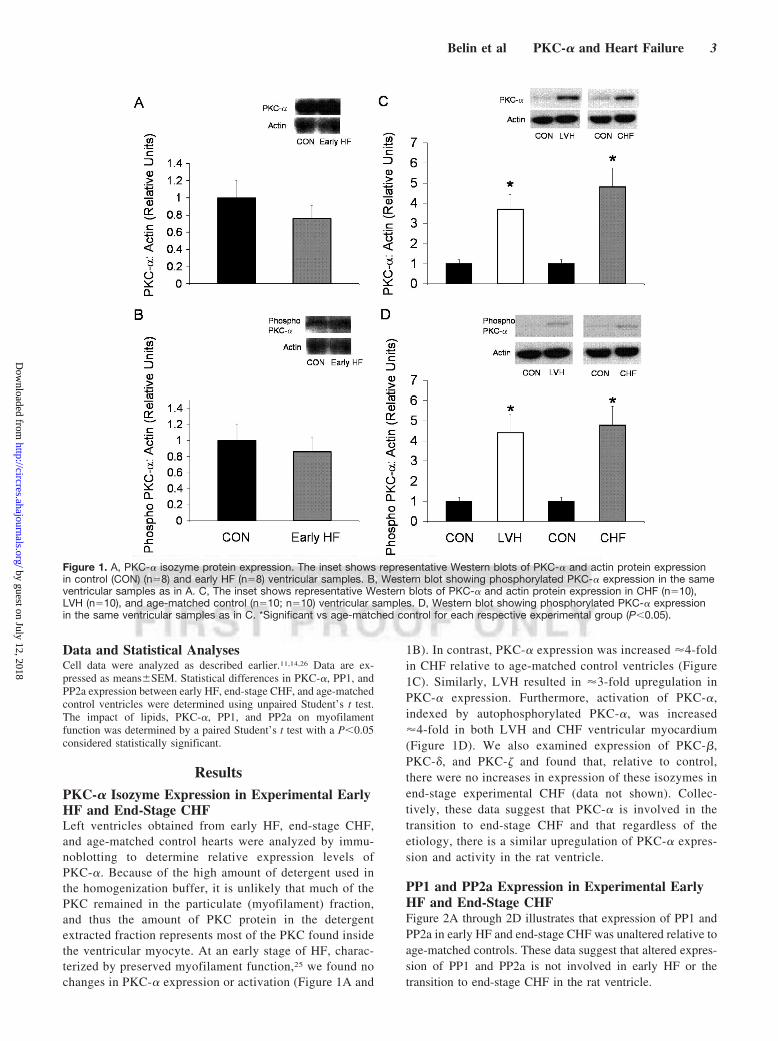
ResultsPKC-� Isozyme Expression in Experimental EarlyHF and End-Stage CHFLeft ventricles obtained from early HF, end-stage CHF,and age-matched control hearts were analyzed by immu-noblotting to determine relative expression levels ofPKC-�. Because of the high amount of detergent used inthe homogenization buffer, it is unlikely that much of thePKC remained in the particulate (myofilament) fraction,and thus the amount of PKC protein in the detergentextracted fraction represents most of the PKC found insidethe ventricular myocyte. At an early stage of HF, charac-terized by preserved myofilament function,25 we found nochanges in PKC-� expression or activation (Figure 1A and
1B). In contrast, PKC-� expression was increased �4-foldin CHF relative to age-matched control ventricles (Figure1C). Similarly, LVH resulted in �3-fold upregulation inPKC-� expression. Furthermore, activation of PKC-�,indexed by autophosphorylated PKC-�, was increased�4-fold in both LVH and CHF ventricular myocardium(Figure 1D). We also examined expression of PKC-�,PKC-�, and PKC-� and found that, relative to control,there were no increases in expression of these isozymes inend-stage experimental CHF (data not shown). Collec-tively, these data suggest that PKC-� is involved in thetransition to end-stage CHF and that regardless of theetiology, there is a similar upregulation of PKC-� expres-sion and activity in the rat ventricle.
PP1 and PP2a Expression in Experimental EarlyHF and End-Stage CHFFigure 2A through 2D illustrates that expression of PP1 andPP2a in early HF and end-stage CHF was unaltered relative toage-matched controls. These data suggest that altered expres-sion of PP1 and PP2a is not involved in early HF or thetransition to end-stage CHF in the rat ventricle.
Figure 1. A, PKC-� isozyme protein expression. The inset shows representative Western blots of PKC-� and actin protein expressionin control (CON) (n�8) and early HF (n�8) ventricular samples. B, Western blot showing phosphorylated PKC-� expression in the sameventricular samples as in A. C, The inset shows representative Western blots of PKC-� and actin protein expression in CHF (n�10),LVH (n�10), and age-matched control (n�10; n�10) ventricular samples. D, Western blot showing phosphorylated PKC-� expressionin the same ventricular samples as in C. *Significant vs age-matched control for each respective experimental group (P�0.05).
Belin et al PKC-� and Heart Failure 3
by guest on July 12, 2018http://circres.ahajournals.org/
Effect of PKC-�–Mediated Phosphorylation onMyofilament FunctionFigure 4 illustrates the effect of DAG and PS on myofila-ment function in control cells. Treatment of cardiac cellswith the lipids had no appreciable effect on Ca2�-saturatedmaximal force (Fmax) and the myofilament Ca2�-sensitivityindex (EC50) (P�0.05). The effect of PKC-� mediatedphosphorylation on myofilament function of control cellsis delineated in Figure 5A through 5C. PKC-�– dependentphosphorylation resulted in a �30% reduction in the Fmax
parameter and �50% increase in the EC50 (decreasedmyofilament Ca2� sensitivity). In contrast, the Fmax andEC50 in failing cells were not significantly affected byPKC-� treatment (Figure 5D through 5F). Furthermore,there was no statistical difference between Fmax and EC50 incontrol cells treated with PKC-� and untreated CHF cells(P�0.05). Taken together, these results suggest that in-creased PKC-� mediated myofilament phosphorylationcontributes to myofilament dysfunction in end-stage ex-perimental CHF.
Impact of PP1- and PP2a-MediatedDephosphorylation on Myofilament FunctionTo confirm that augmented phosphorylation contributes tothe depressed myofilament phenotype in experimental CHF,
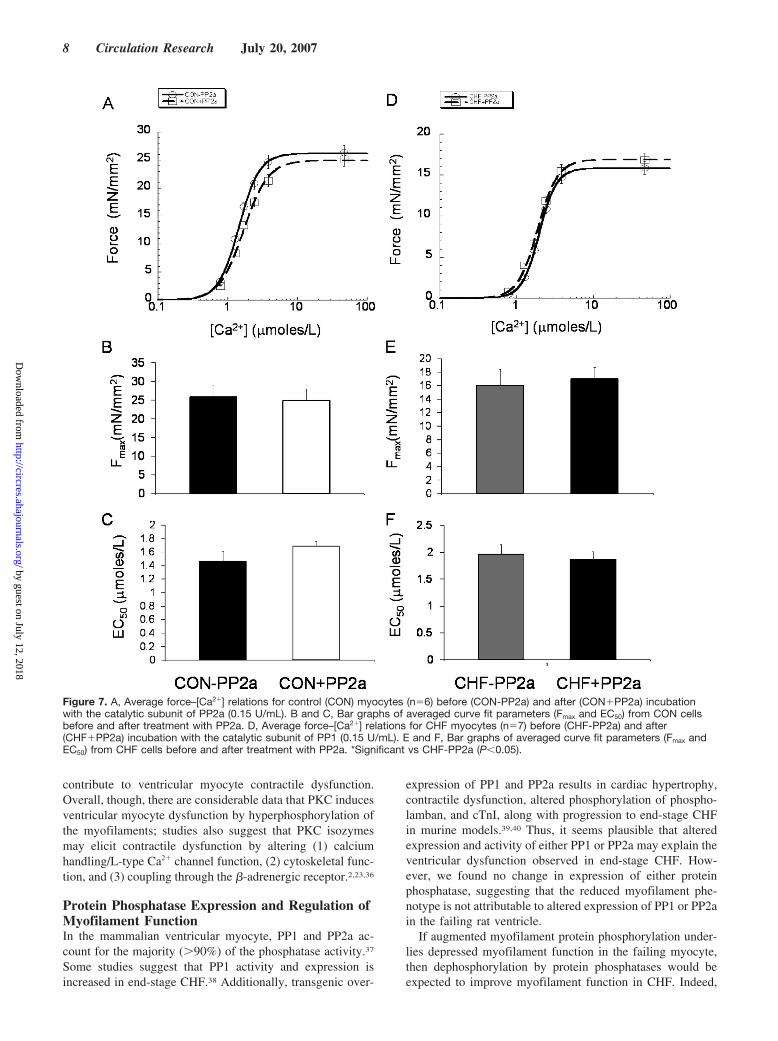
we determined the impact of PP1- and PP2a-dependentdephosphorylation on myofilament function in skinned myo-cytes isolated from control and failing left ventricles (Figures6 and 7). Incubation of control myofilaments with thecatalytic subunit of PP1 had no effect on Fmax or EC50 (Figure6A through 6C). In contrast, PP1-mediated dephosphoryla-tion of failing myofilament proteins resulted in a significant�15% increase in the Fmax and �20% decrease in the EC50
parameter (increased myofilament Ca2� sensitivity; Figure6D through 6F). PP2a-dependent dephosphorylation had noeffect on Fmax or EC50 in control or CHF cells (Figure 7).Finally, to confirm that increased PKC-�–dependent phos-phorylation reduces myofilament function, we treated non-failing cells with PKC-� followed by PP1. PKC-� treatmentresulted in a marked depression of myofilament function(decreased Fmax by �30%; increased EC50 by �35%). Impor-tantly, PP1-mediated dephosphorylation normalized myofil-ament activity and resulted in a �25% increase in Fmax and30% decrease in the EC50 (Figure 8). Overall, our findingsstrongly suggest that increased PKC-�–elicited phosphoryla-tion is partially responsible for myofilament dysfunction inend-stage experimental CHF and that this dysfunctionalphenotype can be partially reversed by dephosphorylationwith PP1.
Figure 2. A, PP1 protein expression. The inset shows representative Western blots of PP1 and actin protein expression in control(CON) (n�8) and early HF (n�8) ventricular samples. B, Western blot showing PP2a expression in the same ventricular samples as inA. C, The inset shows representative Western blots of PP1 and actin protein expression in CHF (n�10), LVH (n�10), and age-matchedcontrol (n�10) ventricular samples. D, Western blot showing PP2a expression in the same ventricular samples as in C.
4 Circulation Research July 20, 2007
by guest on July 12, 2018http://circres.ahajournals.org/
DiscussionThese results are the first to show that PKC-� expression andactivity is upregulated to a similar degree in two divergent ratmodels of end-stage CHF. Our observation that in nonfailingcells, PKC-� induces significant depression of myofilamentfunction, whereas in failing cells it is without a functional effect,suggests a role for PKC-� in the modulation of myofilamentfunction in both health and disease. Our finding that PP1treatment can recover myofilament force development andCa2� sensitivity in failing cells and nonfailing cells treatedwith PKC-� confirms that augmented phosphorylation ofmyofilament targets, attributable, in part, to increased PKC-�signaling, plays a key role in the contractile dysfunctionobserved in failing ventricular myocytes. Therefore, phos-phorylation of myofilament proteins mediated by PKC-� maybe a worthy signaling mechanism to target to restore contrac-tile function of the ventricular myocyte in end-stage CHF.
PKC-� Expression and Activation inExperimental CHFSeveral PKC isozymes have been linked to ventricularremodeling secondary to myocardial infarction and pressureoverload.2 However, previous studies have found increased orunaltered activation and expression of PKC-� in experimen-tal models of LVH and CHF.18,27,28 These divergent findingsmay relate to the duration (acute/compensated versus chronic/end-stage) and/or etiology (myocardial infarction, pressureoverload, volume overload, genetic hypertension) of heartdisease. For these reasons, we examined PKC-� expressionand activation in left ventricles obtained from rats in early HFand end-stage CHF. At the early stage, PKC-� concentrationsand activity were unchanged. In contrast, at end-stage CHF,we found comparable increases in PKC-� content and activ-ity in both experimental models, which is in agreement withprevious work.18,20,23 Indeed, our data suggest that PKC-�plays a key role in mediating the transition to end-stage CHFand is involved in myofilament dysfunction in the rat ventri-
cle. The role of PKC-� in impaired ventricular myocytecontractility has thus far been primarily attributed to alter-ations in Ca2� homeostasis.23 Our results suggest that PKC-�–dependent blunting of ventricular myocyte contractility ismore complex and also involves direct hyperphosphorylationof myofilament proteins.
PKC-�–Dependent Myofilament ProteinPhosphorylation in End-Stage CHFMyofilament dysfunction as a basis for reduced ventricularmyocyte contractility has emerged as an important molecularmechanism in the failing heart.3,29 That is, we and others haveshown both in experimental and human CHF a significantblunting of myofilament force development, Ca2� sensitivity,and cross-bridge cycling kinetics.3,11–14,16,17,25,26,29,30 In trans-genic mouse models of HF, reports indicate that increased
Figure 3. A, Photomicrograph of an attached ventricular myo-cyte in relaxing solution at a sarcomere length of 2.10 �m. B,Side view illustrating direct measurement of myocyte height inrelaxing solution at a sarcomere length of 2.10 �m.
Figure 4. A, Average force–[Ca2�] relations for control (CON)myocytes (n�5) before (CON-DAG/PS) and after (CON�DAG/PS) incubation with DAG and PS in the PKC myocyte incubationbuffer. B and C, Bar graphs of averaged curve fit parameters(Fmax and EC50) from control cells before and after treatment withphospholipids.
Belin et al PKC-� and Heart Failure 5
by guest on July 12, 2018http://circres.ahajournals.org/
myocardial concentrations of PKC-� and PKC-� elicit aug-mented phosphorylation of cTnI and cTnT coincident withmyofilament and ventricular dysfunction.7,21,22 Moreover,several studies clearly indicate that phosphorylation of cTnIand cTnT by numerous PKC isozymes are central in thecontrol of ventricular myofilament force development, Ca2�
sensitivity, and contractility.4–6,9,24 For example, using recon-stituted myofilament preparations, Jideama et al have shownthat PKC-�, PKC-�, PKC-�, and PKC-� phosphorylate cTnIand cTnT at multiple sites.24 Moreover, because of theirpropensity for cross phosphorylation of cTnI and cTnT sites,the number of phosphorylation sites, and the myriad PKCisozymes, clearly defining the relative contribution of eachspecific PKC isozyme in control of myofilament force devel-opment, and Ca2� sensitivity is rather difficult to discern.4,24
In addition to PKC-�, we also examined protein expression ofother PKC isozymes known to (1) phosphorylate cTnI andcTnT; (2) be altered in ventricular hypertrophy and failure;and (3) be predominately expressed in the nonfailing ratventricle, namely PKC-�, PKC-�, and PKC-�.31 We found no
changes in expression of these enzymes in end-stage CHF(unpublished observations). Thus, we chose to focus onPKC-� for several reasons. First, as mentioned, it was theonly PKC isozyme that, in our hands, appears involved inboth reduced myofilament contractility and in mediating thetransition to end-stage CHF. Secondly, PKC-� expressionand activity are increased in end-stage human CHF of diverseetiology, thus making it a clinically relevant moleculartarget.19,30 Lastly, PKC-� translocates to the cardiac sarco-mere following heightened neurohormonal stimulation32 andblunts myofilament function.4,5,24
Until now, data were scant regarding PKC-�–dependentsignaling to myofilaments in chronically failing myocardium.Importantly, we have recently demonstrated that alterations incardiac troponin, most likely phosphorylation, cause myofil-ament dysfunction in our CHF models. In skinned ventricularmyocytes, we found that PKC-�–induced phosphorylationhad no impact on myofilament mechanics in failing cells butresulted in a significant depression of Ca2�-saturated forceand myofilament Ca2� sensitivity in control cells. Further-
Figure 5. A, Average force–[Ca2�] relations for control (CON) myocytes (n�10) before (CON-PKC�) and after (CON�PKC�) incubationwith recombinant PKC-�. B and C, Histograms of averaged curve fit parameters (Fmax and EC50) from control cells before and aftertreatment with bacterially expressed PKC-�. D, Average force–[Ca2�] relations for CHF myocytes (n�7) before (CHF-PKC�) and after(CHF�PKC�) incubation with recombinant PKC-�. E and F, Histograms of averaged curve fit parameters (Fmax and EC50) from CHFcells before and after treatment with bacterially expressed PKC-�.*Significantly different vs CON-PKC� (P�0.05).
6 Circulation Research July 20, 2007
by guest on July 12, 2018http://circres.ahajournals.org/
more, we also found that PP1-dependent dephosphorylationcould rescue the depressed myofilament phenotype in controlmyofilaments hyperphosphorylated with PKC-�. Taken to-gether, these findings confirm that heightened PKC-� signal-ing contributes to myofilament dysfunction in end-stageCHF. In agreement with our findings, Noguchi et al alsofound increased activation of PKC-� in parallel with reducedthin-filament function in reconstituted myofilaments preparedfrom failing human hearts.30 Moreover, in that study, normal-ization of PKC-� activity by mechanical unloading prompteda restoration of maximal force development in failing myo-filaments. Hence, our findings in animal CHF may also beapplicable to human end-stage CHF. In contrast, recentreports show a decline in myofilament Ca2� sensitivity aftertreatment with the catalytic subunit of PKC in diseased ornondiseased human and porcine skinned myocytes but to agreater extent in failing myocardium.17,33 Those results sug-gest a reduction in PKC-mediated phosphorylation of failingmyofilaments. These investigators also found no effect ofPKC treatment on maximal Ca2�-saturated force develop-ment. The inconsistencies may relate, in part, to the use of
human tissue where significant catecholamine signaling indonor myocardium may have occurred.34,35 Furthermore,because those studies relied on the use of the catalyticsubfragment of PKC, it is plausible that reagent inducedpromiscuous phosphorylation of other myofilament proteins(eg, cTnI, cTnT, myosin light chain-2, and myosin-bindingprotein C) occurred, making these data difficult to interpret.Here, we avoided this potentially confounding factor byexamining the direct functional impact of PKC-�; the specificisoform that we found upregulated in CHF. In reconstitutedsystems and multicellular preparations, PKC-� has beenshown to phosphorylate serines 43 and 45 on cTnI and cTnT,which diminishes myofilament contractility.24 Moreover,Sumandea et al showed that PKC-�–dependent phosphory-lation of cTnT at the threonine 206 residue was bothnecessary and sufficient to induce depressed myofilamentfunction.5 Collectively, these data suggest a specific role forPKC-� in both failing and nonfailing myofilament function.However, further studies are required to identify which siteson cTnI and cTnT undergo PKC-�–dependent phosphoryla-tion in failing myofilaments and are the specific sites that
Figure 6. A, Average force–[Ca2�] relations for control (CON) myocytes (n�9) before (CON-PP1) and after (CON�PP1) incubation withthe catalytic subunit of protein PP1 (0.15 U/mL). B and C, Bar graphs of averaged curve fit parameters (Fmax and EC50) from controlcells before and after treatment with PP1. D, Average force–[Ca2�] relations for CHF myocytes (n�8) before (CHF-PP1) and after(CHF�PP1) incubation with the catalytic subunit of protein PP1 (0.15 U/mL). E and F, Bar graphs of averaged curve fit parameters (Fmax
and EC50) from CHF cells before and after treatment with PP1. *Significant vs CHF (P�0.05).
Belin et al PKC-� and Heart Failure 7
by guest on July 12, 2018http://circres.ahajournals.org/
contribute to ventricular myocyte contractile dysfunction.Overall, though, there are considerable data that PKC inducesventricular myocyte dysfunction by hyperphosphorylation ofthe myofilaments; studies also suggest that PKC isozymesmay elicit contractile dysfunction by altering (1) calciumhandling/L-type Ca2� channel function, (2) cytoskeletal func-tion, and (3) coupling through the �-adrenergic receptor.2,23,36
Protein Phosphatase Expression and Regulation ofMyofilament FunctionIn the mammalian ventricular myocyte, PP1 and PP2a ac-count for the majority (�90%) of the phosphatase activity.37
Some studies suggest that PP1 activity and expression isincreased in end-stage CHF.38 Additionally, transgenic over-
expression of PP1 and PP2a results in cardiac hypertrophy,contractile dysfunction, altered phosphorylation of phospho-lamban, and cTnI, along with progression to end-stage CHFin murine models.39,40 Thus, it seems plausible that alteredexpression and activity of either PP1 or PP2a may explain theventricular dysfunction observed in end-stage CHF. How-ever, we found no change in expression of either proteinphosphatase, suggesting that the reduced myofilament phe-notype is not attributable to altered expression of PP1 or PP2ain the failing rat ventricle.
If augmented myofilament protein phosphorylation under-lies depressed myofilament function in the failing myocyte,then dephosphorylation by protein phosphatases would beexpected to improve myofilament function in CHF. Indeed,
Figure 7. A, Average force–[Ca2�] relations for control (CON) myocytes (n�6) before (CON-PP2a) and after (CON�PP2a) incubationwith the catalytic subunit of PP2a (0.15 U/mL). B and C, Bar graphs of averaged curve fit parameters (Fmax and EC50) from CON cellsbefore and after treatment with PP2a. D, Average force–[Ca2�] relations for CHF myocytes (n�7) before (CHF-PP2a) and after(CHF�PP2a) incubation with the catalytic subunit of PP1 (0.15 U/mL). E and F, Bar graphs of averaged curve fit parameters (Fmax andEC50) from CHF cells before and after treatment with PP2a. *Significant vs CHF-PP2a (P�0.05).
8 Circulation Research July 20, 2007
by guest on July 12, 2018http://circres.ahajournals.org/
we observed that PP1-dependent dephosphorylation of failingmyofilament proteins improved myofilament function infailing cells but not in control cells. This result is consistentwith that of Knott and coworkers who observed thin filamentdysfunction in human CHF in parallel with altered myofila-ment protein phosphorylation. Depressed thin-filament func-tion in that study was normalized by nonspecific dephosphor-ylation with alkaline phosphatase. Likewise, a recent studyusing human skinned myocytes also found improved functionfollowing PP1 treatment, although these investigators attrib-uted this to dephosphorylation of myosin light chain-2. It has
been suggested that PP1 predominantly modulates Ca2�
homeostasis by dephosphorylating phospholamban. How-ever, PP1 is also docked at the myofilament and thus maymodulate myofilament protein phosphorylation.41 PP2a hasbeen suggested to be specific for dephosphorylation of cTnIat Ser23 and -24 sites. However, we found that PP2atreatment in either skinned failing or control myocytes waswithout a significant effect on myofilament function. Recentwork by Chen et al shows that PP2a dephosphorylatesMLC-2 in skinned ventricular myocytes. Thus, it is possiblethat in addition to dephosphorylating cTnI, PP2a also dephos-phorylates MLC-2. It may be that the cumulative impact ofthese functionally opposing dephosphorylation events leavesmyofilament function relatively unaltered. On the other hand,in reconstituted failing myofilaments from human ventricles,Noguchi et al, found that PP2a treatment caused an increasein maximal force development and a decrease in maximalsliding velocity. It is conceivable, therefore, that PP2a is moreeffective in human cardiac muscle; such a finding wouldimply important species differences; further investigation isrequired to resolve this issue. Our observation that PP1-induced dephosphorylation restores myofilament function infailing myocytes and control myocytes treated with PKC-�clearly suggests that augmented phosphorylation of myofila-ment constituents plays an important role in depressed myo-filament function in end-stage CHF.
In summary, we have identified PKC-� as an importantsignaling molecule that converges onto the myofilamentlattice to induce hyperphosphorylation and consequent de-pression of ventricular myofilament force development, Ca2�
sensitivity, and contractility in different models of end-stageCHF. Inhibition of PKC-� activity may be a novel therapeuticroute through which ventricular myofilament contractilitycould be restored in the setting of CHF.
AcknowledgmentsWe thank David L. Geenen, Dalia Urbonini, and Milana Yuzhakovafor assistance in generating the experimental models.
Sources of FundingThis work was supported by NIH grants HL64035, HL77195,HL62426, and T32-007692 and American Heart Association Grants0335199N and 0230038N. R.J.B. was supported by a United NegroCollege Fund–MERCK Predoctoral Fellowship and an AmericanPhysiological Society Porter Physiology Fellowship.
DisclosuresNone.
References1. Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T,
Zheng ZJ, Flegal K, O’Donnell C, Kittner S, Lloyd-Jones D, Goff DC Jr,Hong Y, Adams R, Friday G, Furie K, Gorelick P, Kissela B, Marler J,Meigs J, Roger V, Sidney S, Sorlie P, Steinberger J, Wasserthiel-SmollerS, Wilson M, Wolf P. Heart disease and stroke statistics–2006 update: areport from the American Heart Association Statistics Committee andStroke Statistics Subcommittee. Circulation. 2006;113:e85–e151.
2. Vlahos CJ, McDowell SA, Clerk A. Kinases as therapeutic targets forheart failure. Nat Rev Drug Discov. 2003;2:99–113.
3. de Tombe PP, Solaro RJ. Integration of cardiac myofilament activity andregulation with pathways signaling hypertrophy and failure. Ann BiomedEng. 2000;28:991–1001.
Figure 8. Average force–[Ca2�] relations for control (CON) myo-cytes (n�6) before treatment (CON-Baseline), after treatmentwith PKC-� (CON�PKC-�), and after treatment with PP1(CON�PP1). *Significant vs CON-Baseline (P�0.05); #significantvs CON-Baseline and CON�PP1 (P�0.05).
Belin et al PKC-� and Heart Failure 9
by guest on July 12, 2018http://circres.ahajournals.org/
4. Sumandea MP, Burkart EM, Kobayashi T, De Tombe PP, Solaro RJ.Molecular and integrated biology of thin filament protein phosphorylationin heart muscle. Ann N Y Acad Sci. 2004;1015:39–52.
5. Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. Iden-tification of a functionally critical protein kinase C phosphorylationresidue of cardiac troponin T. J Biol Chem. 2003;278:35135–35144.
6. Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, HomsherE, Solaro RJ. Phosphorylation or glutamic acid substitution at proteinkinase C sites on cardiac troponin I differentially depress myofilamenttension and shortening velocity. J Biol Chem. 2003;278:11265–11272.
7. Montgomery DE, Rundell VL, Goldspink PH, Urboniene D, Geenen DL,de Tombe PP, Buttrick PM. Protein kinase C epsilon induces systoliccardiac failure marked by exhausted inotropic reserve and intact Frank-Starling mechanism. Am J Physiol Heart Circ Physiol. 2005;289:H1881–H1888.
8. Montgomery DE, Wolska BM, Pyle WG, Roman BB, Dowell JC,Buttrick PM, Koretsky AP, Del Nido P, Solaro RJ. alpha-Adrenergicresponse and myofilament activity in mouse hearts lacking PKC phos-phorylation sites on cardiac TnI. Am J Physiol Heart Circ Physiol.2002;282:H2397–H2405.
9. Pyle WG, Sumandea MP, Solaro RJ, De Tombe PP. Troponin I serines43/45 and regulation of cardiac myofilament function. Am J PhysiolHeart Circ Physiol. 2002;283:H1215–H1224.
10. Roman BB, Goldspink PH, Spaite E, Urboniene D, McKinney R, GeenenDL, Solaro RJ, Buttrick PM. Inhibition of PKC phosphorylation of cTnIimproves cardiac performance in vivo. Am J Physiol Heart Circ Physiol.2004;286:H2089–H2095.
11. Belin RJ, Sumandea MP, Kobayashi T, Walker LA, Rundell VL,Urboniene D, Yuzhakova M, Ruch SH, Geenen DL, Solaro RJ, de TombePP. Left ventricular myofilament dysfunction in rat experimental hyper-trophy and congestive heart failure. Am J Physiol Heart Circ Physiol.2006;291:H2344–H23453.
12. Perez NG, Hashimoto K, McCune S, Altschuld RA, Marban E. Origin ofcontractile dysfunction in heart failure: calcium cycling versus myo-filaments. Circulation. 1999;99:1077–1083.
13. De Tombe PP, Wannenburg T, Fan D, Little WC. Right ventricularcontractile protein function in rats with left ventricular myocardialinfarction. Am J Physiol. 1996;271:H73–H79.
14. Fan D, Wannenburg T, de Tombe PP. Decreased myocyte tension devel-opment and calcium responsiveness in rat right ventricular pressureoverload. Circulation. 1997;95:2312–2317.
15. Kagaya Y, Hajjar RJ, Gwathmey JK, Barry WH, Lorell BH. Long-termangiotensin-converting enzyme inhibition with fosinopril improvesdepressed responsiveness to Ca2� in myocytes from aortic-banded rats.Circulation. 1996;94:2915–2922.
16. Noguchi T, Kihara Y, Begin KJ, Gorga JA, Palmiter KA, LeWinter MM,VanBuren P. Altered myocardial thin-filament function in the failing Dahlsalt-sensitive rat heart: amelioration by endothelin blockade. Circulation.2003;107:630–635.
17. van der Velden J, Merkus D, Klarenbeek BR, James AT, Boontje NM,Dekkers DH, Stienen GJ, Lamers JM, Duncker DJ. Alterations in myo-filament function contribute to left ventricular dysfunction in pigs earlyafter myocardial infarction. Circ Res. 2004;95:e85–e95.
18. Bayer AL, Heidkamp MC, Patel N, Porter M, Engman S, Samarel AM.Alterations in protein kinase C isoenzyme expression and autophosphor-ylation during the progression of pressure overload-induced left ventric-ular hypertrophy. Mol Cell Biochem. 2003;242:145–152.
19. Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, MintzeK, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G,King GL, Vlahos CJ. Increased protein kinase C activity and expression ofCa2�-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391.
20. Wang J, Liu X, Sentex E, Takeda N, Dhalla NS. Increased expression ofprotein kinase C isoforms in heart failure due to myocardial infarction.Am J Physiol Heart Circ Physiol. 2003;284:H2277–H2287.
21. Goldspink PH, Montgomery DE, Walker LA, Urboniene D, McKinneyRD, Geenen DL, Solaro RJ, Buttrick PM. Protein kinase Cepsilon over-expression alters myofilament properties and composition during theprogression of heart failure. Circ Res. 2004;95:424–432.
22. Takeishi Y, Chu G, Kirkpatrick DM, Li Z, Wakasaki H, Kranias EG,King GL, Walsh RA. In vivo phosphorylation of cardiac troponin I byprotein kinase Cbeta2 decreases cardiomyocyte calcium responsiveness
and contractility in transgenic mouse hearts. J Clin Invest. 1998;102:72–78.
24. Jideama NM, Noland TA Jr, Raynor RL, Blobe GC, Fabbro D, KazanietzMG, Blumberg PM, Hannun YA, Kuo JF. Phosphorylation specificitiesof protein kinase C isozymes for bovine cardiac troponin I and troponinT and sites within these proteins and regulation of myofilament prop-erties. J Biol Chem. 1996;271:23277–23283.
25. Daniels MC, Naya T, Rundell VL, de Tombe PP. Development ofcontractile dysfunction in rat heart failure: hierarchy of cellular events.Am J Physiol Regul Integr Comp Physiol. 2007. In press.
26. Jweied EE, McKinney RD, Walker LA, Brodsky I, Geha AS, MassadMG, Buttrick PM, de Tombe PP. Depressed cardiac myofilament functionin human diabetes mellitus. Am J Physiol Heart Circ Physiol. 2005;289:H2478–H2483.
27. Braun MU, LaRosee P, Schon S, Borst MM, Strasser RH. Differentialregulation of cardiac protein kinase C isozyme expression after aorticbanding in rat. Cardiovasc Res. 2002;56:52–63.
28. Johnsen DD, Kacimi R, Anderson BE, Thomas TA, Said S, Gerdes AM.Protein kinase C isozymes in hypertension and hypertrophy: insight fromSHHF rat hearts. Mol Cell Biochem. 2005;270:63–69.
29. de Tombe PP. Altered contractile function in heart failure. CardiovascRes. 1998;37:367–380.
30. Noguchi T, Hunlich M, Camp PC, Begin KJ, El-Zaru M, Patten R, LeavittBJ, Ittleman FP, Alpert NR, LeWinter MM, VanBuren P. Thin-filament-based modulation of contractile performance in human heartfailure. Circulation. 2004;110:982–987.
31. Dorn GW 2nd, Force T. Protein kinase cascades in the regulation ofcardiac hypertrophy. J Clin Invest. 2005;115:527–537.
32. Huang X, Walker JW. Myofilament anchoring of protein kinaseC-epsilon in cardiac myocytes. J Cell Sci. 2004;117:1971–1978.
33. van der Velden J, Narolska NA, Lamberts RR, Boontje NM, Borbely A,Zaremba R, Bronzwaer JG, Papp Z, Jaquet K, Paulus WJ, Stienen GJ.Functional effects of protein kinase C-mediated myofilament phosphor-ylation in human myocardium. Cardiovasc Res. 2006;69:876–887.
34. van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, OwenVJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2�-sensitivity of the contractile apparatus in end-stage human heart failureresults from altered phosphorylation of contractile proteins. CardiovascRes. 2003;57:37–47.
35. Jweied E, Detombe P, Buttrick PM. The use of human cardiac tissue inbiophysical research: the risks of translation. J Mol Cell Cardiol. 2007;42:722–726.
36. Hahn HS, Marreez Y, Odley A, Sterbling A, Yussman MG, Hilty KC,Bodi I, Liggett SB, Schwartz A, Dorn GW 2nd. Protein kinase Calphanegatively regulates systolic and diastolic function in pathological hyper-trophy. Circ Res. 2003;93:1111–1119.
37. Luss H, Klein-Wiele O, Boknik P, Herzig S, Knapp J, Linck B, MullerFU, Scheld HH, Schmid C, Schmitz W, Neumann J. Regional expressionof protein phosphatase type 1 and 2A catalytic subunit isoforms in thehuman heart. J Mol Cell Cardiol. 2000;32:2349–2359.
38. Gupta RC, Mishra S, Rastogi S, Imai M, Habib O, Sabbah HN. CardiacSR-coupled PP1 activity and expression are increased and inhibitor 1protein expression is decreased in failing hearts. Am J Physiol Heart CircPhysiol. 2003;285:H2373–H2381.
39. Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C,Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, GruppIL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, anegative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–4135.
40. Gergs U, Boknik P, Buchwalow I, Fabritz L, Matus M, Justus I, HanskeG, Schmitz W, Neumann J. Overexpression of the catalytic subunit ofprotein phosphatase 2A impairs cardiac function. J Biol Chem. 2004;279:40827–40834.
41. Chu Y, Wilson SE, Schlender KK. A latent form of protein phosphatase1 alpha associated with bovine heart myofibrils. Biochim Biophys Acta.1994;1208:45–54.
10 Circulation Research July 20, 2007
by guest on July 12, 2018http://circres.ahajournals.org/
Solaro and Pieter P. de TombeRashad J. Belin, Marius P. Sumandea, Edward J. Allen, Kelly Schoenfelt, Helen Wang, R. John
Contributes to Myofilament Dysfunction in Experimental Congestive Heart Failure-Induced Myofilament Protein PhosphorylationαAugmented Protein Kinase C-
http://circres.ahajournals.org/content/early/2007/06/07/CIRCRESAHA.107.148288.citationWorld Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on July 12, 2018http://circres.ahajournals.org/