27
Aylwyn Scally, Wellcome Trust Sanger Institute March 2012
| Date post: | 02-Dec-2018 |
| Category: |
Documents |
| Upload: | hoangtuong |
| View: | 214 times |
| Download: | 0 times |

Aylwyn Scally, Wellcome Trust Sanger Institute March 2012

Key tasks in sequence analysis
• Data handling • Alignment to a reference sequence • Alignment file handling • Variant calling
• SNPs, genotypes • structural variation
• Sequence assembly

Data handling
• Important to have a data hierarchy corresponding to experimental factors
lane/run
library
sequencing technology
individual
strain/subspecies/population
species hsa
YRI
NA12878
SLX
NA12878-WG
297_1 297_2
454
CEU
NA19240
SLX
NA19240-WG
505_7 505_8

Raw sequence data
• FASTQ format • original Sanger standard for capillary data • derived from FASTA format • sequence and an associated per base quality
score • PHRED quality scores encoded as ASCII
printable characters (ASCII 33–126) • standard offset 33 but older Solexa/Illumina variants
used 64 @title
sequence +optional_text
quality
@SRR010930.8436795/1!ACCCCAGGATCAACACTTCACATGCATTAGCAGAGAGAGATAAATCAA!+!=>=??A?<@B@A:?B?D;AC@@CAAAD<AAA:99?:@=?@B@77C><4!

PHRED quality scores
• Encodes the probability of an erroneous call • quality score Q = –10 log10 P • error probability P = 10–Q/10
• example: call with Q = 30 has error probability P = 10-3 = 1 in 1000
• ASCII encoding ! ” # $ % & ’ ( ) * + , - . / 0 1 2 3 4 !0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19!
encoding Q score

DATA PROCESSING
ALIGNMENT
SAM FILE PROCESSING
Alignment pipeline
• Get data • Prepare and index reference
• sequence names; alternate haplotypes etc • Align data
• by lane or smaller unit – optimise throughput • Sort by position • Merge alignments • Improve alignments • Merge libraries • Index final alignment

Alignment pipeline
BAM BAM BAM Library merge Library
… Alignment
Fastq
BAM
BAM
Fastq
BAM
BAM
Fastq
BAM
BAM
Fastq
BAM
BAM
Fastq
BAM
BAM BAM Improvement
Lane/plex
BAM BAM Sample/Platform Sample merge
…
…

bwa • bwa index [-a bwtsw|div|is] [-c] <in.fasta>
• Burroughs-Wheeler transform construction algorithm • ‘bwtsw’ for vertebrate sized genomes, ‘is’ for smaller genomes
• bwa aln [options] <prefix> <in.fq> • align each single-ended fastq file individually • <prefix> is name of reference file • options control alignment parameters, scoring matrix, seed length
• bwa sampe [options] <prefix> <in1.sai> <in2.sai> <in1.fq> <in2.fq> • generates pairwise alignment from sai files produced by bwa aln • produces SAM output
• bwa bwasw [options] <prefix> <query.fa> • alignment of long reads in query.fa • produces SAM output

bwa usage notes
• bwa finds matches up to a finite edit distance • by default for 100-bp reads allows 5 edits
• Important to quality-clip reads • -q in bwa aln, e.g. set to 20
• Non-ACGT bases on reads are treated as mismatches
• Parallelise for speed • split data into 1 Gbp blocks • bwa takes ~8 hrs per block
• Check for truncated BAM files • e.g. with samtools flagstats

Alignment improvement
• Library duplicate removal • samtools, Picard
• Realignment around indels • GATK
• Base quality recalibration • GATK

Library duplicate removal • PCR amplification step in library preparation can result
in duplicate DNA fragments • PCR-free protocols exist but require larger volumes of
input DNA • Generally a low number of duplicates in good libraries;
increases with depth of sequencing • Duplicates can result in false SNP calls
• manifest as high read depth support • Removal method
• Identify read-pairs where outer ends map to the same position on the genome and remove all but one copy
• samtools rmdup • Picard/GATK MarkDuplicates

Realignment • Short indels in the sample relative to reference pose
difficulties for alignment • Indels occurring near the ends of reads often not aligned
correctly • Aligners prefer to introduce SNPs rather than an indel
• Realignment algorithm • Input set of known indel sites and a BAM file
• Previously published indel sites, dbSNP, 1000 Genomes, or estimate from alignment
• At each site, model the indel and reference haplotypes and select best fit with data
• New BAM file produced, modified where indels have been introduced by realignment
• Implemented in GATK (IndelRealigner function)

Additional alignment issues
• Separate chromosomal BAMs • easier to process in parallel
• Realign/assemble unmapped reads • recover sequence missed due to reference
incompatibility or incompleteness

SAM/BAM
• Sequence Alignment/Map format • unified format for storing read alignments to a
reference genome • BAM (Binary Alignment/Map) format
• binary equivalent of SAM • Features
• stores alignments from most alignment programs • supports multiple sequencing technologies • supports indexing for quick retrieval • reads can be classed into logical groups
• e.g. lanes, libraries, individuals

SAM file format • Header • Alignment lines (one per read)
• 11 mandatory fields • several optional fields (format TAG:TYPE:VALUE)
Col Field Type Description 1 QNAME str query name of the read or the read pair 2 FLAG int bitwise flag (pairing, mapped, mate mapped, etc.) 3 RNAME str reference sequence name 4 POS int 1-based leftmost position of clipped alignment 5 MAPQ int mapping quality (Phred scaled) 6 CIGAR str extended CIGAR string (details of alignment) 7 RNEXT str mate reference name (‘=’ if same as RNAME) 8 PNEXT int position of mate/next segment 9 TLEN int observed template length 10 SEQ str segment sequence 11 QUAL str ASCII of Phred-scaled base quality

SAM format
• Example
• http://picard.sourceforge.net/explain-flags.html
IL4_315:7:105:408:43!177!X!1741!0!1S35M!X!56845228!0!ATTTGGCTCTCTGCTTGTTTATTATTGGTGTATNGG!+1,1+16;>;166>;>;;>>;>>>>>>,>>>>>+>>!
QNAME FLAG
RNAME POS
MAPQ CIGAR RNEXT PNEXT
TLEN SEQ
QUAL

SAM/BAM file processing tools • samtools
• C program and library • http://samtools.sourceforge.net
• view: SAM-BAM conversion • sort, index, merge multiple BAM files • flagstat: summary counts of mapping flags
• Picard • Java program suite
http://picard.sourceforge.net • MarkDuplicates, CollectAlignmentSummaryMetrics,
CreateSequenceDictionary, SamToFastq, MeanQualityByCycle • Pysam
• Python interface to samtools API • http://code.google.com/p/pysam/

Variant calling • Call SNPs with genotypes (heterozygous and
homozygous), indels and structural variants • Tools
• samtools, bcftools • GATK, SOAPsnp, Dindel • SVMerge
• File formats: • VCF, pileup
• Filters and calling protocols • depth, quality, strand bias, multiple samples
• Indels harder to call accurately than SNPs • structural variation harder still

Variant Call Format (VCF)
• Stores polymorphism data with annotation • SNPs, insertions, deletions and structural variants
• Can be indexed for fast data retrieval • Variant calls across many samples • Metadata
• e.g. dbSNP accession, filter status, validation status • Arbitrary tags can be used to describe new types
of variant • Note: binary BCF produced by samtools
• get vcf with samtools mpileup | bcftools view

VCF Format • Header
• Arbitrary number of INFO definition lines starting with ‘##’ • Column definition line starts with single ‘#’
• Mandatory columns • Chromosome (CHROM) • Position of the start of the variant (POS) • Unique identifiers of the variant (ID) • Reference allele (REF) • Comma separated list of alternate non-reference alleles
(ALT) • Phred-scaled quality score (QUAL) • Site filtering information (FILTER) • User extensible annotation (INFO)

VCF format
• Example 3!74393!.!G!T!999!. DP=31;AF1=0.7002;AC1=4;DP4=4,0,22,2;… GT:PL:DP:GQ!1/1:181,57,0:19:57!1/1:90,15,0:5:16!0/0:0,12,85:4:7!
CHROM POS
ID REF ALT
QUAL FILTER
INFO FORMAT sample1 sample2 sample3
see H. Li, Bioinformatics 27(21): 2987–2993 (2011) for details of likelihood and population genetic calculations

More information • SNP calling and genotyping
• Samtools • http://bioinformatics.oxfordjournals.org/content/25/16/2078.long • http://samtools.sourceforge.net
• GATK • http://www.broadinstitute.org/gsa/wiki/index.php/
The_Genome_Analysis_Toolkit • VCF
• VCFtools • http://vcftools.sourceforge.net • Danacek et al. Bioinformatics 27(15): 2156-2158 (2011)
• http://www.1000genomes.org/wiki/Analysis/Variant%20Call%20Format/vcf-variant-call-format-version-41
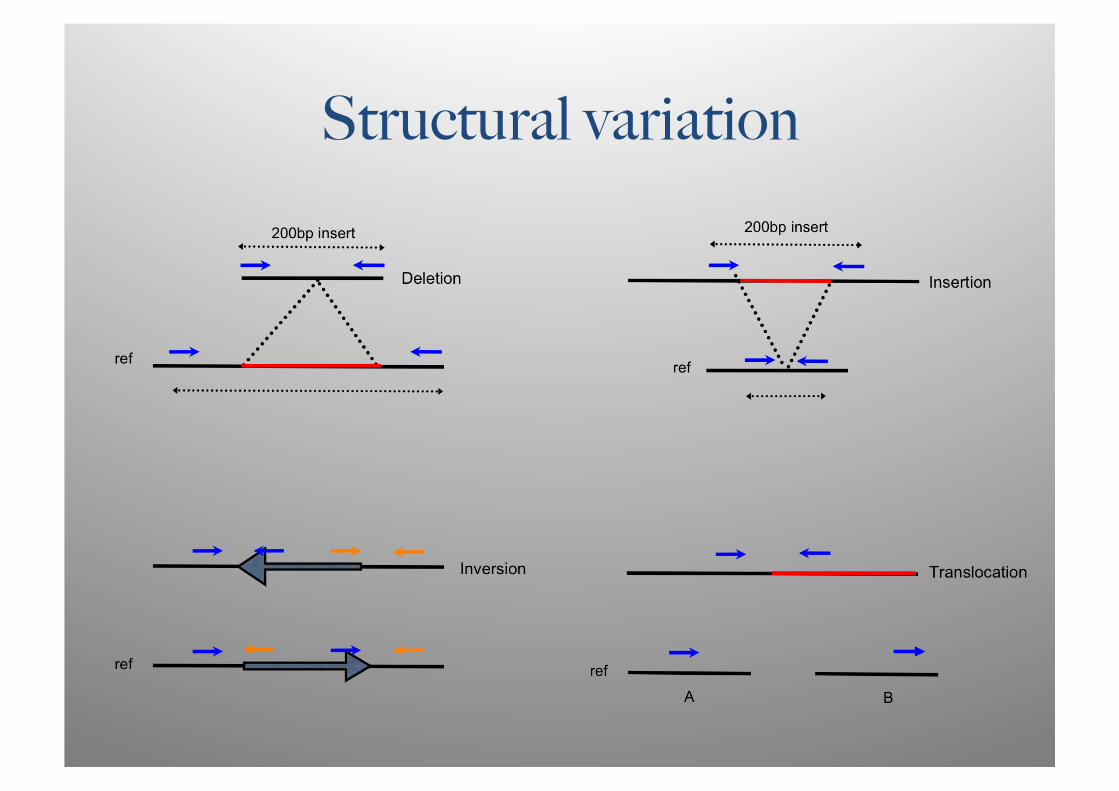
Structural variation

Structural variation • Read depth and pairing information used to detect events
• deviations from the expected fragment size • presence/absence of mate pairs • excessive/reduced read depth (CNV)
• Several methods/tools released • SVMerge pipeline
• makes SV predictions using a collection of callers • Input is one BAM file per sample • callers run individually & outputs converted into standard BED
format • calls merged • computationally validated using local de novo assembly • http://svmerge.sourceforge.net/

Assembly
• Tools • Abyss
• http://www.bcgsc.ca/platform/bioinfo/software/abyss • SGA
• https://github.com/jts/sga • SOAPdenovo
• http://soap.genomics.org.cn/soapdenovo.html • ALLPATHS-LG
• http://www.broadinstitute.org/software/allpaths-lg/blog • Cortex
• http://cortexassembler.sourceforge.net/ • Velvet
• http://www.ebi.ac.uk/~zerbino/velvet/

Assembly metrics
• N50, N10, N90 etc • x % of assembly is in fragments larger than
Nx • Number of contigs, mean/max contig
length • Realignment
• fraction of read pairs mapped correctly • correct homozygous SNPs • identify breakpoints

Thanks to Thomas Keane and the Vertebrate Resequencing team at WTSI for several slides

















