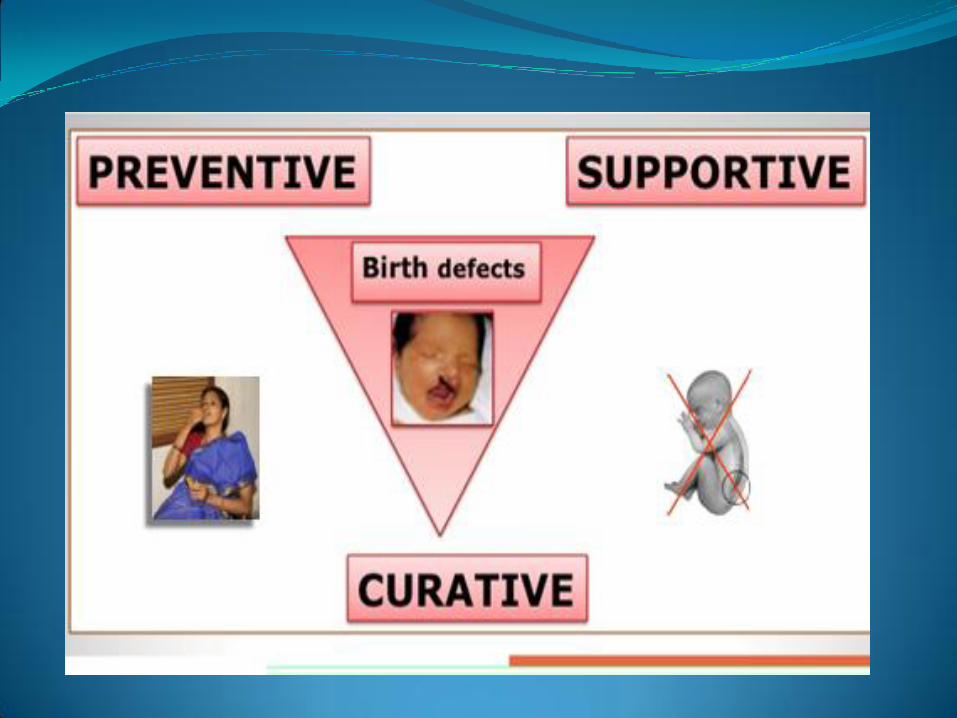
316
| Date post: | 07-May-2015 |
| Category: |
Health & Medicine |
| Upload: | rajasthan-govt |
| View: | 1,649 times |
| Download: | 0 times |


PRE-EMBYRONIC STAG <4 WEEKS
First cell division 30 hours
Zygote reaches uterine cavity 4 days
Implantation 5-6 days
Bilminar disc 12 days
Lynozation of female 16days
Fromation of trilminar disc 19 days
and primitive streak

Embryonic stage 4-12 weeks
ORGANOGENESIS 4-8 WEEKS
BRAIN & SPINAL CORD ARE FORMING 4 WKEEKS
FIRST SING OF HEART BEAT & LIMB BUDS 6 WEEKS
BRAIN,EYES, HEART & LIMBS – DEVELOP RAPIDLY
BOWEL AND LUNG BEGINNING TO DEVELOP
DIGITS APPEARED, EARS,KIDNEYS,LIVER & 8 WEEKS
MUSCLES ARE DEVELOPING.
PALATE CLOSES AND JOINT FORM 10 WEEKS
SEXUAL DIFFERENTIATION ALMOST COMPLETE 12 WEEKS

Deformation
External force causing distortion of an otherwise normal structure is called
deformation
Because of intrauterine crowding - as with multiple fetal pregnancy
Amniotic fluid leakage
Ex – Hip dislocation, Talipes equinovarus.
Deformation carry good prognosis

Disruption
Damage or dissolution of a part following normal development of body part
Ex - Amniotic band, thromboembolic episode.

Dysplasia
Morphological defects due to abnormal maturation and organization of cells into
Tissue is known as dysplasia.
Ectodermal dysplasia – abnormal skin, hair, nail and teeth.
Skeletal dysplasia - spondyloepipyseal dysplasia

Malformation Morphological defects occur due to error in the normal development
And differentiation of embryo is known as malformation.
Type of malformation
Sequence
Syndrome
Association
14% minor
3% two or >2 malformation
3% single major malformation
0.7% multiple malformation
Minor malformation : they do not cause any function defect
Major malformation : If malformation left uncorrected leads to
Significant Impairment of body function

Sequence The chain of events resulting in multiple defect –
Chronic leakage of amniotic fluid or less production of amniotic fluid leads
To fetus compression –
Potter sequence -squashed face, dislocation of hip
talipes equinovarus, pulmonary hypoplasia.
Pierre- Robin sequence- Microganthia, Tongue fall back and prevent closure
palate, leading to cleft palate.

Syndrome Co-occurrence of several distinct abnormalities(group of features)
definitely or presumably if
caused by same etiological factor in all affected individuals
Is known as syndrome.

Association
Co-occurrence of group of malformations, more frequently
Than expected by chance, without definite cause is called association.
VATER - VERTEBRAL, anal, tracheal, oesophageal and renal
CHARGE - coloboma, heart, atresia choanae, retarded growth, genital
and ear malformation

FETAL STAGE >12 – 38 WEEKS FETAL MOVEMENT 16-18 WEEKS
EYELIDS OPEN 24-26 WEEKS
FETUS VIABLE WITH SPECIAL CARE 24-26 WEEKS
RAPID WEIGHT GAIN 28-28 WEEKS

BIRTH DEFECT
A BIRTH DEFCT IS DEFIND AS THE MARCH OF DIMES IS “ FUNCTIONAL OR STRUCTURAL”, THAT PRESENT IN INFANCY OR LATER IN LIFE AND IS CAUSED BY EVENTS PRECEDING BIRTH WHETHER INHERITED OR ACQURIED.

BIRTH DEFECTS
DEFINED
AS AN ABNORMALITY OF THE BODY’ STRUCTURE
OR INHERENT FUNCTION IN
LIVE BORN
FETUS, INTRAUTERINE FETAL DEMISES,
STILL BIRTH
AND
IN MEDICALLY TERMINATED
PREGNANCIES.
WHICH IS PRESENT AT BIRTH , WHETHER SUCH
ABNORMALITY IS MANIFEST AT THE
DELIVERY OR BECOME APPARENT
LATER IN LIFE.

BIRTH DEFECT
1. Congenital malformation :- It is a primary structural defect
arising from a localised error in morphogenesis,resulting in
the abnormal formation of a tissue or organ.
2. Disruption : :- It is a structural defect resulting from the
destruction of a structure that had formed normally before
the insult such as - ischemia, infection & trauma.
3. Deformation : is a defect resulting from an abnormal
mechanical forces that distorts an otherwise normal
structure.
4. Dysplasia: is an abnormal orgination of cells into tissue.
Most dysplasia are cause by sinle gene defects & are
associated with high recurrence risk for sibling &/or
offspring

It is estimated that 1 in 40 or 2.5% of new born have a
recongisable malformations at birth.
In a about half of case a single isolated malformations and
other half display multiple malformations.
It is estimated that 10% of paediatric hospital admission
have a non-genetic condition, 18% have congenital defect
of unknown etiology and 40% have surgical admission are
patient with congenital malformations.
20-30% of infant death and 30-50% death after the
neonatal period are due to congenital abnormalities.
When several malformation occurred in a single
individual they are classified a syndrome, sequences or
association.

3. Deformation :- It is an alteration in shape or structural of a structure or organ that
has differentiated normally.
Uterine compression
Intrinsic
Oligohydramnios
Uterine hypertonia
Multiple foetuses
Large fetus
Uterine deformities
(biocornate)
Extrinsic
Small pelvis
Bony lumbar spines
Increased abdominal tone
Abnormal fetal posture
(including breech)
Abnormal fetal
Muscular toneIncreased mechanical forces
Fetal constraint
Deformations
Craniofacial Extremity Other
Scaphocephaly Dislocated hips Torticollis
Plagiocephaly Metatarsus adductus Lung hypoplasia
Mandibular asymmetry Equinovarus foot Scoliosis
Flattened facies Calcaneovalgus foot
Deviated nasal septum Tibial bowing
Crumpled ear Hyperflexed hips
Craniostenosis Hyperextended knees
Contractures
Internal tibial torsion

DEATHS DUE TO BIRTH DEFECTS
• CHROMOSOMAL
• CNS
• RESPIRATORY
• CHD
28% 15%
15%12%

What causes birth defects?Birth defects have a variety of causes, such as:
Genetic problems -caused when one or moregenes doesn't work properly or part of a gene ismissing
Problems with chromosomes- such as having anextra chromosome or missing part of achromosome
Environmental factors- that a woman is exposedto during pregnancy, such as rubella or Germanmeasles while pregnant, or using drugs or alcoholduring pregnancy.

• GENETIC –CHROMOSOMAL ANOMALY
• MATERNAL ILLNESS ,DRUGS AND INFECTION
• MULTIFACTORIAL• SPORADIC
40-60% 20-25%
12-25%10-13%

MOLECULAR MECHANISMS OF MALFORMATIONS:
Inborn Errors of Development
The gene mutation in malformation syndrome are key factor for
development events.
Gene mutation
Environmental agent Teratogenes
Transduction pathway Transcription pathway
Regulatory Protein
Development Events

Genetic factors A gene is a tiny, invisible unit containinginformation (DNA) that guides how the body forms and functions.
Each child gets half of its genes from each parent, arranged on 46chromosomes. Genes control all aspects of the body, how it works,and all its unique characteristics, including eye color and body size.
Genes are influenced by chemicals and radiation, but sometimeschanges in the genes are unexplained accidents.
In each pair of genes, one will take precedence (dominant) over theother (recessive) in determining each trait, or characteristic.
Birth defects caused by dominant inheritance include a form ofdwarfism called achondroplasia; high cholesterol; Huntington'sdisease, a progressive nervous system disorder; Marfan syndrome,which affects connective tissue; some forms of glaucoma, andpolydactyly (extra fingers or toes).

If both parents carry the same recessive gene, they have aone-in-four chance that the child will inherit the disease.Recessive diseases are severe and may lead to an earlydeath. They include sickle cell anemia, a blood disorderthat affects blacks, and Tay-Sachs disease, which causesmental retardation in people of eastern European Jewishheritage. Two recessive disorders that affect mostlyare: cystic fibrosis, a lung and digestive disorder,and phenylketonuria (PKU), a metabolic disorder.
If only one parent passes along the genes for the disorder,the normal gene received from the other parent willprevent the disease, but the child will be a carrier. Havingthe gene is not harmful to the carrier, but there is the 25%chance of the genetic disease showing up in the child oftwo carriers.

Some disorders are linked to the sex-determiningchromosomes passed along by parents.Hemophilia, a condition that prevents blood fromclotting, and Duchenne muscular dystrophy,which causes muscle weakness, are carried on theX chromosome.
Genetic defects can also take place when the eggor sperm are forming if the mother or fatherpasses along some faulty gene material. This ismore common in older mothers. The mostcommon defect of this kind is Down syndrome, apattern of mental retardation and physicalabnormalities, often including heart defects,caused by inheriting three copies of achromosome rather than the normal pair.

A less understood cause of birth defects results from the interactionof genes from one or both parents plus environmental influences.
These defects are thought to include:
Cleft lip and palate, which are malformations of the mouth
Clubfoot, ankle or foot deformities.
Spina bifida, an open spine caused when the tube that forms thebrain and spinal chord does not close properly.
Water on the brain (hydrocephalus), which causes brain damage.
Diabetes mellitus, an abnormality in sugar metabolism thatappears later in life.
Congenital Heart defects

DRUGS (TERATOGENS)
Only a few drugs are known to cause birth defects, but all have the potential to cause harm. Thalidomide is known to cause defects of the arms and legs.
Steroid cleft lip & palate
Lithium Ebstein’s anomaly
Retinoic acid Conotruncal anomaly
Valproic acid Coarctation of forta,HLHS,PA
Carbamazepin,valporic acid Spina bifida
radiation Microcephaly, spina bifida, blindness,cleft palate
Drugs Birth defects
Hyperthermia Spina bifida
Warfarin Hypoplastic nasal bone,skeletal dysplasia
Vitamin D Supravuvular aortic stenosis
D penacillamine Cutis laxa syndrome

The enzyme 5,10-methylenetetrahydrofolate reductase (MTHFR) isresponsible for converting folic acid to 5-methyltetrahydrofolate. 5-methyltetrahydrofolate serves as a methyl group donor in theconversion of homocysteine to methionine.
This methylation is important in providing carbon units to rapidlydividing cells and the synthesis of nucleotide bases. Thus, folic aciddeficiency would result in a neural tube defect.
Moreover, if there is a mutation in the gene regulating MTHFR,homocysteine will not get converted to methionine and affectedindividuals will have neural tube defect.
Increase folic acid intake can overcome the neural tube defectdue to genetically mediated enzyme deficiency.

Now with control of infections in neonates,BIRTH DEFECTS
are becoming an important cause of perinatal mortality in india.
If perinatal mortility is to be reduced further one should reducebirth defects at an early stage that is when fetus is in the age ofnonsurvival or by planning birth of such babies at tertiary carecentres dedicated for care of such babies.
THIS IS POSSIBLE BY FETAL MEDICINE.

E.g. Sonic Hedgehog as model :- The SHH pathway is developmentally important during
embryogeneous to induce controlled proliferation in a tissue specific manner, disruption of specific
steps in this pathway results in a variety of related developmental disorder and malformation.
1Sonic Hedgehog of
2Cleavage
N-Shin
Cholesterol
N-Shin-Chol Holoprosencephaly1
Microcephaly
Mental retardation
Failure of CNS
lateralization
Hypotelorism ti
Smith-Lemil-Opitz Syndrome2
Microcephaly
Mental retardation
Short, Upturned nose
Hypospadias
Post axial prolydactyly
Pallister-Hall
Syndrome3
Hypothalamic
hamartoma
Short, upturned nose
Central and postaxial
polydactyly
Bild epiglottis
Greig Greig
Cephalopolysyndactyly
syndrome4
Macrocephaly
Hypertelorism
Pre and postaxial polydactyly
Rubinstein-Taybi
Syndrome5
Microcephaly
Mental retardation
Prominent beaked nose
Broad thumbs
Hirsutism
GLI-1 GLI-2
4GLI-3.5CBP GLI1
PTC1 Twist
HNF3β

The birth defects are groups according to ICD-10 classification:
(Q00-Q07) nervous system,
(Q10-Q18) eye, ear, face and neck,
(Q20-Q28) circulatory system,
(Q30-Q34) respiratory system,
(Q35-Q37) cleft lip and cleft palate,
(Q38-Q45) digestive system,
(Q50-Q56) genital organs,
(Q60-Q64) urinary system,
(Q65-Q79) musculoskeletal system,
(Q80-Q89) other defects and
(Q90-Q99) chromosomal abnormalities, not elsewhere classified.

Nervous system:
(740) Anencephalus and similar anomalies
(740.0) Anencephalus
(741) Spina bifida
(742) Other congenital anomalies of nervous system
(742.1) Microcephalus
(742.3) Hydrocephalus

TYPES OF CONGENITAL MALFORMATIONS
1. Central Nervous System
• Neural Tube defects
• Spina bifida
• Meningocele
• Meningomyelocele
• Encephalocele
• Anencephaly
• Hydrocephalus and ventriculomegaly
• Holoprosencephaly
• Agenesis of the corpus collosum
• Dandy-Walker complex
• Microcephaly
• Megalencephaly
• Destructive cerebral lesions
• Arachnoid cysts
• Choroid plexus cysts
• Vein of Galen aneurysm
2. Face
• Orbital defects
• Facial cleft
• Micrognathia
• Ear defects

3. Cardiovascular system• Atrial septal defects
• Ventricular septal defects
• Atrioventricular septal defects
• Univentricular heart
• Aortic stenosis
• Coarctation and tubular hypoplasia of the aorta
• Interrupted aortic arch
• Hypoplastic left heart syndrome
• Pulmonary stenosis and pulmonary atresia
• Ebstein’s anomaly and tricuspid valve dysplasia
• Conotruncal malformations
• Transposition of the great arteries
• Tetralogy of Fallot
• Double-outlet right ventricle
• Truncus arteriosus communis
• Cardiosplenic syndromes
• Echogenic foci

4. Pulmonary abnormalities
• Cystic adenomatoid malformation
• Diaphragmatic hernia
• Pleural effusions
• Sequestration of the lungs
5. Anterior abdominal wall
• Exomphalos
• Gastroschisis
• Body stalk anaomaly
• Bladder exstrophy and cloacal exstrophy
6. Gastrointestinal tract
• Esopageal atresia
• Duodenal atresia
• Intestinal obstruction
• Hirschsprung’s disease
• Meconium peritonitis
• Hepatosplenomegaly
• stenosis and imperforate
• Hepatic calcifications
• Abdominal cysts

7. Kidneys and urinary tract• Renal agenesis
• Infantile polycystic Kidney disease (Potter type I)
• Multicystic dysplastic kidney disease (Potter type II)
• Potter type III renal dysplasia
• Obstructive uropathies
8. Skeleton• Skeletal anomalies
• Osteochondrodysplasias
• Limb deficiency or congenital amputations
• Split hand and foot syndrome
• Clubhands
• Polydactyly
• Fetal akinesia deformation sequence (FADS)
9. Hydrops fetalis

How we should approach for detection of
Congenital Malformations ?
CLINICAL EVALUATION
1.By History :-
(i) Pedigree analysis
(ii) Parental ages at the time of conception
(iii) Parental consanguinity
(iv) History of abortions
(v) Still birth and exposure to the drug teratogens
(vi) Maternal disorders and infections

2. By Examinations :-
A good observation is essential to recognize the
malformations.
(i) The defects produced due to an abnormality of a
development of a body part early in the prenatal life eg.
Cleft lip and palate and polydactyly and holoprosencephaly.
(ii) Anthropometry is important as is the measurement of
any other relevant dysmorphic feature eg.
Hypo/hypertelorism, low set ears etc.
(iii) Look for the presence of abnormal genitalia or delayed
puberty e.g. Smith Lemi Optiz syndrome, Tumer syndrome
etc.
(iv) Look for the presence of abnormal genitalia or delayed
puberty, e.g. Smith Lemi Optiz syndrome, Tumer syndrome
etc.

INVESTIGATIONS:-
1. Chromosomal analysis :-
• Karyotype analysis e.g. Down syndrome
• Fluorescent in situ hybridization (F.I.S.H.) e.g.
William syndrome, Prader Willi syndrome,
Angelman syndrome, Velocardiofacial syndrome.
• PCR studies
• Micro-array technology
2. Imaging studies (CT, MRI) e.g. CNS malformations
3. Echo done in all cases of Down syndrome and
velocardio facial syndrome.
4. Metabolic study particularly in (amino acids and
organic acids) e.g. like Mucopolysaccharidosis,
Zellweger syndrome, Smith Lemli Opitz syndrome

(v) Psychomotor delay, speech delay or mental retardation
are common feature many syndrome e.g. down syndrome,
Fragile-X syndrome.
(vi) Examination of presence of hearing loss and
abnormalities of the eye are essential in dysmorphogical
examination. It provides diagnostic clues for some
syndromes like chorioretinal lacunae in Aicardi syndrome,
Brusfield spots in Down syndrome.
(vii) Some clinical features suggest a specific diagnosis. These
features have been termed as

“Pearls of Dysmorphology” by Hall?
Pursed up lips – Whistling face syndrome
Broad thumbs/great toes – Rubinstein Taybi syndrome, Pfeiffer
syndrome.
Radial ray defects – Holt Oram syndrome, Thrombocytopenia absent
radius syndrome, Fanconi anemia.
Absent clavicles – Cleidocranial dysostosis.
Heterochromia iridis – Waardenburg syndrome
Mitten hands – Apert syndrome
Inverted nipples – Congenital disorder of glycosylation.
Webbing of the neck – Turner and Noonan syndromes.
Eversion of the lateral third of the lower eyelid – Kabuki Make-up
syndrome.
Hyper extensibility of skin and joins – Ehlers Danlos syndrome.

CNS
AT 7 WEEKS FLUID FILLED
VESICLE SEEN –
ROMBENCEPHALIC
VESICLE
AT 9 WEEKSCONVULATED
PATTERN OF THE
THREE PRIMARY
CEREBRAL VESICLE
IS VISIBLE.
AT 11 WEEKSBRIGHT ECHOGENIC
CHOROID PLEXUS
FILL LARGE LATERAL
VENTRICLES.





A simple classification of anomalies of brain & spine is as follows:
Failure of dorsal induction:
1. Anomaly of cranial development failure:Anaencephaly, cephalocele
Chiari malformation
Dysraphism
2.Anomaly of ventral induction failure:Holoprosencephalies
Facial abnormalities
Posterior fossa malformation: Dany –Walker malformation,Joubertsyndrome , Rhomboenvephalosynapsis.
3.Failure of histogenesis, neuronal proliferatio, migration &organizationDisorder of sulcation & cell migration: lissencephalic &nonlissencephalic dysplasia.
Gray mater hetrotropia
Cortical dysplasia- schizencephaly
Abnormalities of corpus callosum
Phakomatosis -

• RECURRENCE RISK IF ONE PARENT OR PREVIOUS SIB HAVE NTD
• RECURRENCE RISK IN NEXT SIB
• ENCEPHALOCELE• ANENCEPHALY
• SPINA BIFIDA
95% 5%
5-10%2-4/1000

NEURAL TUBE DEFECTSRACHICHISIS
SEVERE FORM OF SPINA BIFIDA.
INCOMPATIBLE WITH
LIFE
MENINGOMYELOSIS
COMMON TYPE
FEW SEGMENT BIFID
SPINA BIFIDA OCCULTA
ONLY BONE BIFID
TELL-TALE SIGN MAY BE SEEN

CNS malformation
1. NEURAL TUBE DEFECTS
Classification
A.Primary NTD -95 % failure of closure of neural tube at
17 to 28 days of gestation
-Meningomyelocele
-Encephalocele
-Anencephaly
B.Secondary NTD -5% occurs after neural tube closure due
to defect in mesoderm.
-meningocele
-Lipomeningocele
-Diastometomyelia
-Dorsal sermal sinus
- Tethered cord

Etiology of NTD
1. Multifactorial inheritance
2. Maternal risk factor alcohol, radiation,
valproate, methotraxate
3. Genetic MTHFR, gene defect
4. Chromosomal abnormality Trisomy 13 & 18
PREVENTION Folate supplementation 0.4mg/day to all mothers 1
month before to 3 months of pregnancy.
If there is any previous affected child than give
5mg/day.- prenatal diagnosis in subsequent pregnancy by MSAFP estimation
and fetal ultrasound at 12 week and 16-20 week of gestation.

SPINA BIFIDA
ASSOCITED SIGN
LEMON SIGN
BANANA SIGN
FETAL THERAPY: IN UTERO CLOSURE
OF SPINA BIFIDA REDUCES
RISK OF HANDICAP; BECAUSE AMNIOTIC
FLUID IN THIRD TRIMESTER IS NEUROTOXIC

Spina Bifida Occulta :- This is a midline defect of vertebral bodies without protrusion of the spinal
cord or meninges.
Most individuals are asymptomatic and lack neurologic sign.
In some cases, patch of hair, lipoma, discoloration of skin, dermal sinus in
the midline of lower back suggest a more significance malformations of
spinal cord.
A Spine X-ray shows a defect in closure of posterior vertebral arches and
laminae, typically involve in L5 and S1.
It is occasionally associated with more significant developmental
abnormalities of the spinal cord, including syringomyelia,
Diastematomyelia and tethered cord.
These are the best identified with MRI.
A dermoid sinus usually forms a small skin opening, which lead to narrow
duct, some time indicated by protruding hairs, hairy patch or vascular
nervus.
Demoid sinus occur in the midline at the sight of meningocele or
enencephalocele may occur.
Demoid sinus tracts may pass through the dura, acting age conduit for the
spread of infection.

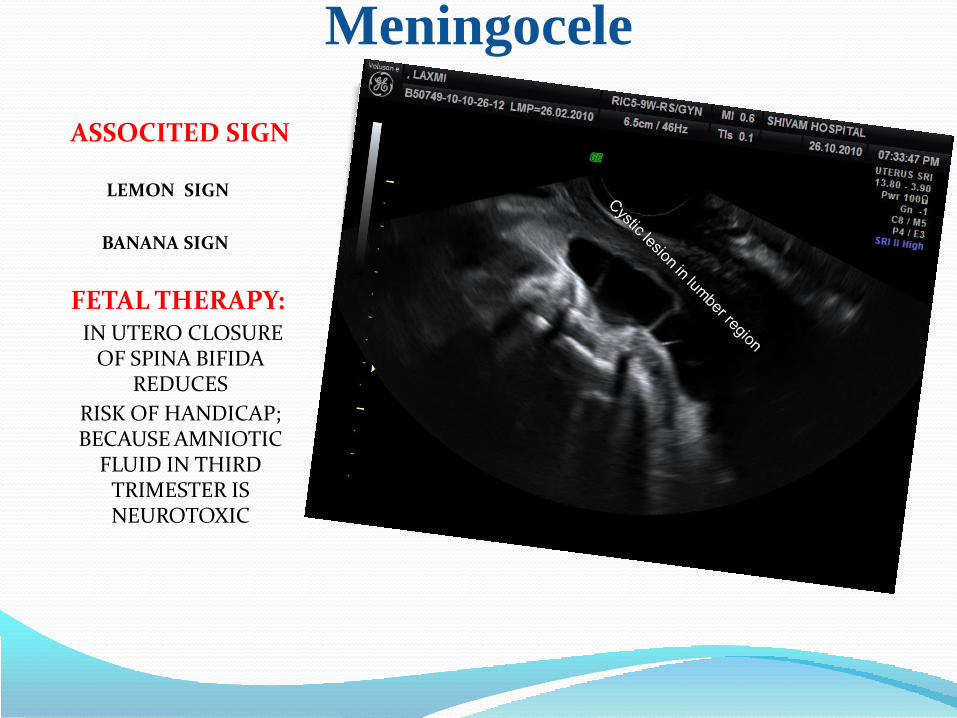
Meningocele
ASSOCITED SIGN
LEMON SIGN
BANANA SIGN
FETAL THERAPY: IN UTERO CLOSURE
OF SPINA BIFIDA REDUCES
RISK OF HANDICAP; BECAUSE AMNIOTIC
FLUID IN THIRD TRIMESTER IS NEUROTOXIC
Meningocele
ASSOCITED SIGN
LEMON SIGN
BANANA SIGN
FETAL THERAPY: IN UTERO CLOSURE
OF SPINA BIFIDA REDUCES
RISK OF HANDICAP; BECAUSE AMNIOTIC
FLUID IN THIRD TRIMESTER IS NEUROTOXIC
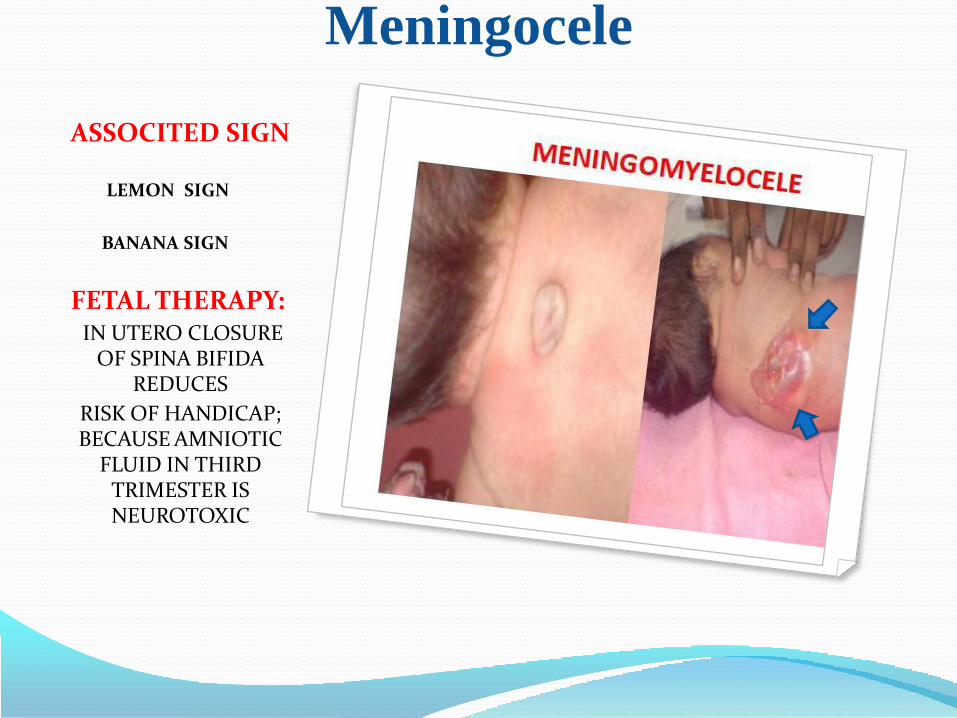
Meningocele
ASSOCITED SIGN
LEMON SIGN
BANANA SIGN
FETAL THERAPY: IN UTERO CLOSURE
OF SPINA BIFIDA REDUCES
RISK OF HANDICAP; BECAUSE AMNIOTIC
FLUID IN THIRD TRIMESTER IS NEUROTOXIC

Meningocele
ASSOCITED SIGN
LEMON SIGN
BANANA SIGN
FETAL THERAPY: IN UTERO CLOSURE
OF SPINA BIFIDA REDUCES
RISK OF HANDICAP; BECAUSE AMNIOTIC
FLUID IN THIRD TRIMESTER IS NEUROTOXIC

.Meningocele :- It is formed when the meninges
herminate through a defect in the posterior vertebral
arches.
A functuant midline mass that may transilluminate
occurs along the vertebral column, usually in the
lower back.
Asymptomatic children with normal neurological
finding and fullthickness skin covering may have
surgery delayed.
Those patients with leaking CSF should under go
immediate surgery.

MYELOMENINGOCELEIt is the most severe form of dysraphism involving the
vertebral column.
Incidence = 1/4000 live births.
Treatment :-Management and supervision of a child and family
myelomeningocele require a multidisciplinary team including
surgeon, physician, therapisis.
Surgery is often done within a day or so of birth but can be
delay for several days when there is a CSF leak.
Prognosis :-For a child who is born with a myelomeningocele and who is
treated aggressively mortality 10-15%.

ENENCEPHALOCELE :- Two major forms of dysraphism affect the skull, resulting in protrusion
of tissue through a bony midline defect, called cranium bifidum.
A cranial meningocele consists of CSF filled meningeal sac only, and a
cranial encephalocele contain the sac + cereberal cortex, cerebellium,
portions of the brainstem.
This defects occur most commonly in the ocipital region but in certain
part of world, frontal or nasofrontal enencephalocele are more
prominent.
Meckel-Gruber syndrome is a rare autosomal recessive condition that is
characterized by occipital enencephalocele, cleft lip or palate, microcephaly,
microphthalamia, abnormal genitalia, polycystic kidney and polydactyly.
Diagnosis MRI & CT Scan :-
Maternal serum alpha fetoprotein level and ultrasound measurement of BPD as
well as identification of enencephalocele in utero .

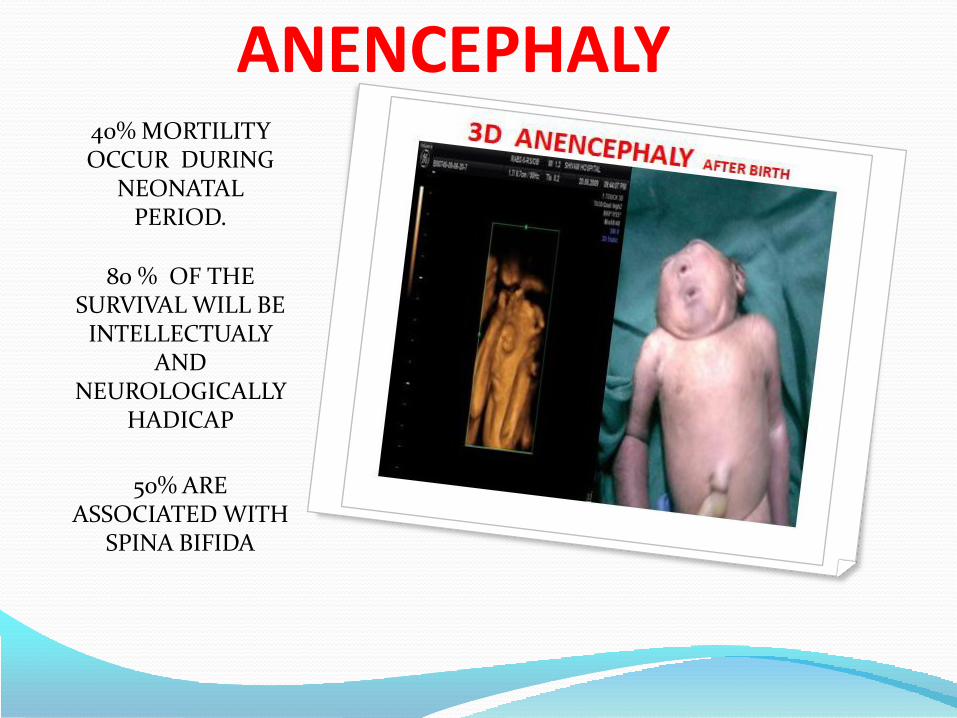
ANENCEPHALY40% MORTILITY OCCUR DURING
NEONATAL PERIOD.
80 % OF THE SURVIVAL WILL BE
INTELLECTUALY AND
NEUROLOGICALLY HADICAP
50% ARE ASSOCIATED WITH
SPINA BIFIDA
ANENCEPHALY40% MORTILITY OCCUR DURING
NEONATAL PERIOD.
80 % OF THE SURVIVAL WILL BE
INTELLECTUALY AND
NEUROLOGICALLY HADICAP
50% ARE ASSOCIATED WITH
SPINA BIFIDA


ANENCEPHALY :-
It is distinctive appearance with a large defect of calvarium, meninges
and scalp associated with a rudimentary brain, which results from failure
of closure of the rostral neuropore the opening of the anterior neural
tube.
The incidence – 1/1000 live births. The most anenecephalic infants die
within several days of birth.
The recurrence risk is 4% and increase to 10% if a couple has had two
previously affected pregnancies.
Approximately 50% of cases of anencephaly have associated
polyhydraminos.
Diagnosis :-
Couples who have had an anencephalic infant
should have successive pregnancy monitored,
including amniocentesis, determination of
AFP levels and ultrasound examination
between the 14th and 16th week of gestation.

HYDROCEPHALUS AND VENTRICULOMEGALY
In hydrocephalus there is pathological increase in the size of the cerebral
ventricles.
PrevalenceHydrocephalus is found in about 2 per 1,000 births. Ventriculomegaly
(lateral ventricle diameter of 10 mm or more) is found in 1% of
pregnancies at the 18-23 week scan. Therefore the majority of fetuses
with ventriculomegaly do not develop hydrocephalus.
EtiologyThis may result from chromosomal and genetic abnormalities,
intrauterine hemorrhage or congenital infection, although many cases
have as yet no clear-cut etiology.
DiagnosisFetal hydrocephalus is diagnosed sonographically, by the
demonstration of abnormally dilated lateral cerebral ventricles.

PrognosisFetal or perinatal death and
neurodevelopment in survivors are strongly
related to the presence of other
malformations and chromosomal defects.
Although mild, also referred to as borderline,
ventriculomegaly is generally associated with a
good prognosis,

HOLOPROSENCEPHALYPREVALENCE : 1/10,000BIRTHand occurs with arate of 1 in 250 duringembryogenesis
There are threeclassifications ofholoprosencephaly. Alobar,in which the brain has notdivided at all, is usuallyassociated with severe facialdeformities. Semilobar, inwhich the brain'shemispheres havesomewhat divided, causesan intermediate form of thedisorder. Lobar, in whichthere is considerableevidence of separate brainhemispheres, is the leastsevere form. In some casesof lobar holoprosencephalythe baby's brain may be
nearly normal.

HOLOPROCENCEPHALYCAUSES: A variety ofteratogens, chromosomalabnormalities (in 25-50% ofcases), and single genemutations can result inholoprosencephaly. Trisomy 13(in about 40% of cases) andtrisomy 18 are the mostfrequently identifiedchromosomal anomalies. Manysingle-gene disorders (18-25%)can result in syndromes with avariable incidence ofholoprosencephaly. Examplesinclude Pallister-Hall,Rubinstein-Taybi, Kallmann,Smith-Lemli-Opitz, Meckel,hydrolethalus, pseudotrisomy
13, and microtia -anotia
syndromes. Maternal diabeteshas been implicated in about1% of cases.
RECURRENCE RISK 6%
ALOBAR &SEMILOBAR
-LETHAL
LOBAR : MR

ABSENT SEPTUM PELLUCIDUMAbsence of the septumpellucidum is reported to be anunusual anomaly that occurs inan estimated 2 to 3 individualsper 1 00,000 people in thegeneral
population Absence of the SPalone is not a disorder but isinstead a characteristic noted inchildren with septo-opticdysplasia.
The prognosis for individualswith septo-opticdysplasia varies according tothe presence and severity ofsymptoms
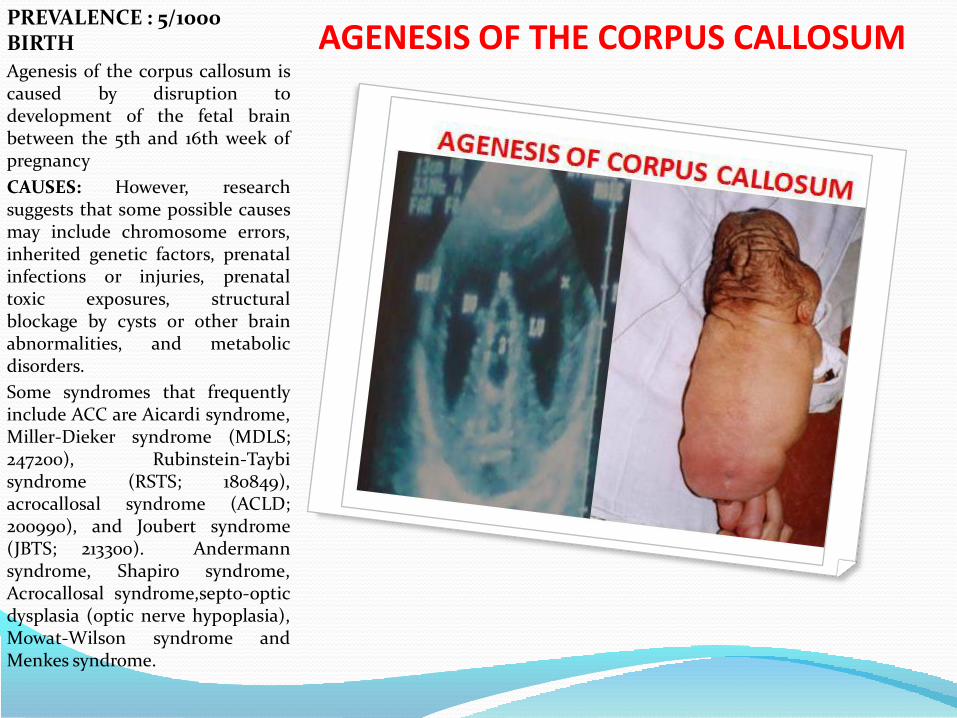
AGENESIS OF THE CORPUS CALLOSUM PREVALENCE : 5/1000 BIRTH
Agenesis of the corpus callosum iscaused by disruption todevelopment of the fetal brainbetween the 5th and 16th week ofpregnancy
CAUSES: However, researchsuggests that some possible causesmay include chromosome errors,inherited genetic factors, prenatalinfections or injuries, prenataltoxic exposures, structuralblockage by cysts or other brainabnormalities, and metabolicdisorders.
Some syndromes that frequentlyinclude ACC are Aicardi syndrome,Miller-Dieker syndrome (MDLS;247200), Rubinstein-Taybisyndrome (RSTS; 180849),acrocallosal syndrome (ACLD;200990), and Joubert syndrome(JBTS; 213300). Andermannsyndrome, Shapiro syndrome,Acrocallosal syndrome,septo-opticdysplasia (optic nerve hypoplasia),Mowat-Wilson syndrome andMenkes syndrome.

DANDY WALKER SYNDROME
PREVALENCE:
1/30,000
CAUSES:
LOW RECURRENCE RISK 1-5%
13 & 18 TRISOMIES
TRIPLOIDY
50 GENETIC SYNDROME
CONGENITAL INFECTION
WARFARIN
20 NEONATAL MORTILITY
50% INTELLECTUAL AND NEUROLOGICAL
HANDICAP.
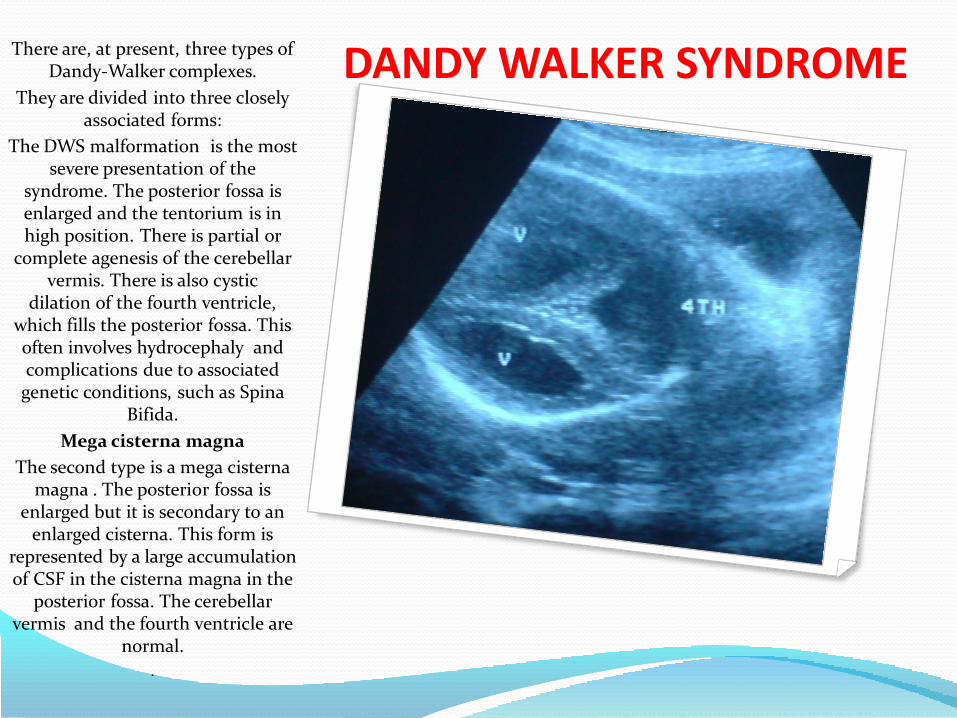
DANDY WALKER SYNDROME There are, at present, three types of Dandy-Walker complexes.
They are divided into three closely associated forms:
The DWS malformation is the most severe presentation of the
syndrome. The posterior fossa is enlarged and the tentorium is in high position. There is partial or
complete agenesis of the cerebellar vermis. There is also cystic
dilation of the fourth ventricle, which fills the posterior fossa. This often involves hydrocephaly and complications due to associated
genetic conditions, such as Spina Bifida.
Mega cisterna magna
The second type is a mega cisterna magna . The posterior fossa is
enlarged but it is secondary to an enlarged cisterna. This form is
represented by a large accumulation of CSF in the cisterna magna in the
posterior fossa. The cerebellar vermis and the fourth ventricle are
normal.
.

DANDY WALKER SYNDROMEThe fourth ventricle is only mildly enlarged and there is
mild enlargement of the posterior fossa. The cerebellar vermis is hypoplastic and has a variably sized cyst space. This is caused by open communication
of the posteroinferior fourth ventricle and the cisterna
magna through the enlarged vallecula. Patients
exhibit hydrocephalus in 25% of cases and supratentorial CNS variances are uncommon, only present in 20% of cases. There
is notorcular-lambdoid inversion, as usually
seen in patients with the malformation. The third and
lateral ventricles as well as the brain stem are normal.

ARNOLD CHIARI MALFORMATIONIncidence : The Chiarimalformation, defined astonsilar herniations of 3 to5 mm or greater
The incidence is approximately1 in 1,200.The incidence ofsymptomatic Chiari is less butunknown.
A prevalence of approximatelyin 1000 has been described.
The Austrian pathologist HansChiari in the late 1800sdescribed seemingly relatedanomalies of the hindbrain, theso called Chiari malformationsI, II and III. Later, otherinvestigators added a fourth(Chiari IV) malformation. Thescale of severity is rated I -IV, with IV being the mostsevere. Types III and IV arevery rare

Type Presentation Other notes
IIs generally asymptomatic during childhood, but often manifests with headaches and cerebellarsymptoms. Herniation of cerebellar tonsils.
The most common form.
II
Usually accompanied by a myelomeningocele leading to partial or complete paralysis below the spinal defect. Abnormal development of the cerebellarvermis and medulla oblongata occur, and they both descend into the foramen magnum. Hydrocephalus is frequently present.
IIICauses severe neurological defects. It is associated with an encephalocele
IV Characterized by a lack of cerebellar development.[
The brainstem, cranial nerves, and the lower portion of the cerebellum may be stretched
or compressed. Therefore, any of the functions controlled by these areas may be
affected. The blockage of Cerebro-Spinal Fluid (CSF) flow may also cause a syrinx to
form, eventually leading to syringomyelia. Chiari is often associated with major
headaches, sometimes mistaken for migraines. Chiari headaches usually include
intense pressure in the back of the head, aggravated by Valsalva maneuvers, such as
yawning, laughing, crying, coughing, sneezing or straining. Chiari also includes muscle
weakness, facial pain, hearing problems, and extreme fatigue. It also can cause
insomnia cycles of sleep deprivation followed by inabilities to remain awake cycling
between them. 15% of patients with adult Chiari malformation are asymptomatic

TreatmentOnce symptomatic onset occurs, a common treatment is decompression surgery,[14] in
which a neurosurgeon usually removes the lamina of the first and sometimes the second
or even third cervical vertebrae and part of the occipital bone of the skull to relieve
pressure. The flow of spinal fluid may be accompanied by a shunt. Since this surgery
usually involves the opening of the dura mater and the expansion of the space beneath, a
dural graft is usually applied to cover the expanded posterior fossa.
A small number of neurological surgeons believe that detethering the spinal cord as an
alternate approach relieves the compression of the brain against the skull opening
(foramen magnum), obviating the need for decompression surgery and associated
trauma. However, this approach is significantly less documented in the medical literature,
with reports on only a handful of patients. It should be noted that the alternative spinal
surgery is also not without risk.
PrognosisThe prognosis differs dependent on the type of malformation (i.e., type I, II, III, or IV). Type
I is generally adult-onset and, while not curable, treatable and non-fatal. Types I and II
sufferers may also develop syringomyelia. Type II is typically diagnosed at birth or
prenatally. Approximately 33% of individuals with Chiari II malformation develop
symptoms of brainstem damage within five years; a 1996 study found a mortality rate of
33% or more among symptomatic patients, with death frequently occurring due to
respiratory failure. 15% of individuals with Chiari II malformation die within two years of
birth. Among children under two who also have myelomeningocele, it is the leading cause
of death. Prognosis among children with Chiari II malformation who do not have spina
bifida is linked to specific symptoms; the condition may be fatal among symptomatic
children when it leads to neurological deterioration, but surgical intervention has shown
promise. Types III and IV are extremely rare and patients generally do not survive past the
age of two or three

ARNOLD CHIARI MALFORMATIONArnold Chiari Malformation:-
Type I –It is usually not associatedwith hydrocephalus patientcomplain of headache,neckpain,urinary frequency andprogressive lower extremityspasticity.The deformity consist ofdisplacement of cerebellar tonsilinto cervical canal. Althoughpathogenesis is unknown, a theorysuggest that obstruction of caudalportion of the IV ventrical duringfetal development is responsible.
Type II :-It is charactarised byprogressive hydrocephalus with amyelomeningocele, pointing offrontal horn & colpocephaly(dialted occipital horn) .This lesionrepresent and anomaly of hindbrainprobably due to failure of pontineflexure during embriyogenesis,andresult in elongation of the IVventrical and kinking of thebrainstem with displacement ofinferior vermis,pons,medulla intocervical canal.This anomaly istreated by surgical decompression.

ARNOLD CHIERI MALFORMATION
Type III - in this there is highcervical encephalo-meningocele: in which themedulla, 4TH ventricle, andentire cerebellum reside.

AQUEDUCTAL STENOSISCongenital: Some patients are bornwith a congenitally narrow orcompletely obstructed aqueduct. Incomplete, this usually presents aspediatric hydrocephalus. However,if the obstruction is more minor, thepatient may be asymptomatic ormay not present until older age. Theobstruction can appear as a generalnarrowing of the aqueduct or canappear as small webs or rings oftissue across the channel.
Post-Infectious or Post-Hemorrhage:Infection in the cerebrospinal fluidor hemorrhage into the ventriclesfrom other causes can occasionallylead to scarring that creates webs orrings that cause aqueductal stenosisand block flow through theaqueduct.
Idiopathic Acquired: Some patientspresent in adulthood with the newonset or gradual onset ofhydrocephalus. In many cases it isunclear what the underlying causeof the stenosis was and isconsidered idiopathic

ARACHNOID CYSTArachnoid cysts are fluid-filled cystscontained within the arachnoidspace.
Prevalence : Arachnoid cysts areextremely rare.
Etiology :Unknown; infectiousprocess has been hypothesized butit is unlikely that this may explainthe congenital cysts.
Diagnosis : Arachnoid cysts appearon antenatal ultrasound assonolucent lesions with a thinregular outline, that do not containblood flow, do not communicatewith the lateral ventricles andanyhow are not associated with lossof brain tissue. They occur mostfrequently in the area of the cerebralfissure and in the midline. .Prognosis : Large cysts may causeintracranial hypertension andrequire neurosurgical treatment.However, a normal intellectualdevelopment in the range of 80-90% is reported by most series.Spontaneous remission has beendescribed both in the postnatal aswell as in the antenatal period.

CHOROID PLEXUS CYSTPrevalence : Choroid plexus cystsare found in about 2% of fetuses at20 weeks of gestation but in morethan 90% of cases they resolve by 26weeks.
Etiology : Choroid plexus cystscontain cerebrospinal fluid andcellular debris.
Diagnosis :The diagnosis is madeby the presence of single or multiplecystic areas (greater than 2 mm indiameter) in one or both choroidplexuses.
Prognosis :They are usually of nopathological significance, but theyare associated with an increasedrisk for trisomy 18 if maternal age>35years, serum beta hCG >0.3MoM, nuchal fold >6mm,echogenic bowel, hydronephrosisand cyst > 10 mm and possiblytrisomy 21. In the absence of othermarkers of trisomy 18 the maternalage-related risk is increased by afactor of 1.5. The choroid plexuscyst < 10 mm sometime disappearspontaneously.

VEIN OF GALEN MALFORMATIONVein of Galen aneurysm is avery rare abnormality.
Prevalence : Vein of Galenaneurysm is a sporadicabnormality.
Diagnosis : The diagnosis ismade by the demonstration of asupratentorial mid-linetranslucent elongated cyst.
Prognosis : In the neonatalperiod about 50% of the infantspresent with heart failure andthe rest are asymptomatic. Inlater life hydrocephalus andintracranial hemorrhage maydevelop.
Good results can beachieved bycatheterization andembolization of themalformation.

LISSENCEPHALYLissencephaly, whichliterally means smoothbrain, is a rare brainformation disorder causedby defective neuronalmigration during the 12th to24th weeks of gestation,resulting in a lack ofdevelopment of brain folds(gyri) and grooves (sulci). Itis a form of cephalicdisorder. Terms such as'agyria' (no gyri) or'pachygyria' (broad gyri) areused to describe theappearance of the surface ofthe brain. Affected childrendisplay severe psychomotorretardation, failure tothrive, seizures, andmuscle spasticity or
hypotonia.[
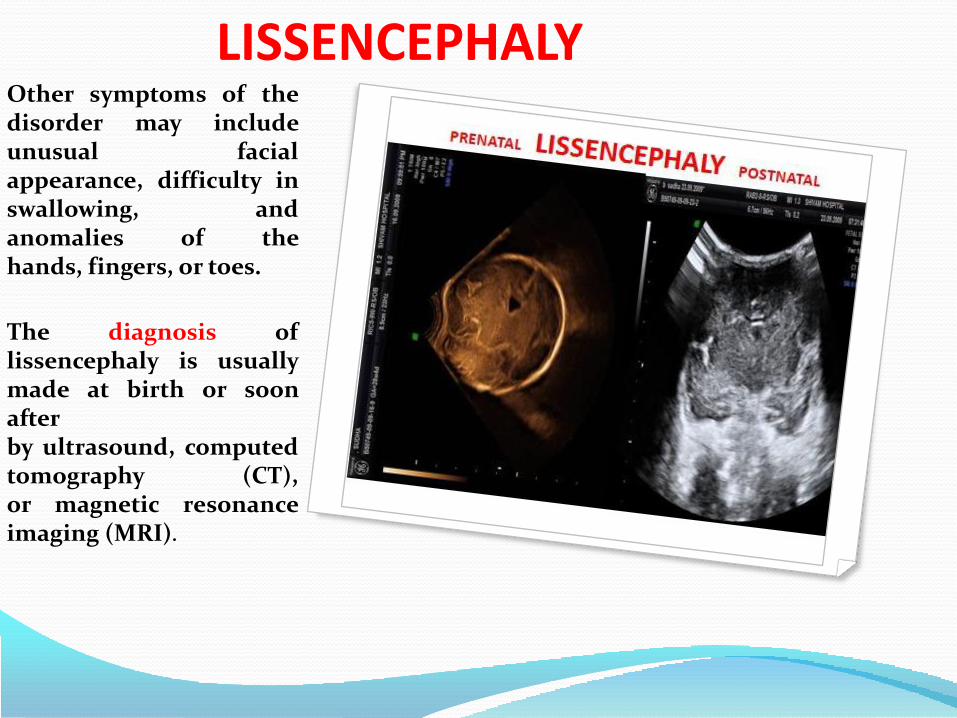
LISSENCEPHALYOther symptoms of thedisorder may includeunusual facialappearance, difficulty inswallowing, andanomalies of thehands, fingers, or toes.
The diagnosis oflissencephaly is usuallymade at birth or soonafterby ultrasound, computedtomography (CT),or magnetic resonanceimaging (MRI).

category type
Classical (type 1) LIS: Lissencephaly due to PAFAH1B1 gene mutation
Type I isolated lissencephaly (601545Miller –dicker syndrome(247200)
LI SX : lissencephaly due to double cortin(DCX) gene mutation(330121)Lissencephaly type I without genetic disorder
Cobblestone (type 2) Walker –Warburg syndrome(236670)Fukuyama syndrome (253800)Muscle Eye Brain disease (MEB)253280
other LIS2: Norman –Robert syndrome 253280LIS3: TUBA1A, 611603LISX2 : ARX, 300215MICRO-LISSENCEPHALY

DESTRUCTIVE CEREBRAL LESIONS
Prevalence : Destructive cerebral lesions are found in
about 1 per 10,000 births. These lesions
include hydranencephaly porencephaly and schizencephaly .

HYDRANENCEPHALY
hydranencephaly thereis absence of the cerebralhemispheres withpreservation of themid-brain andcerebellum.
Complete absence ofechoes from theanterior and middlefossae distinguisheshydranencephaly fromsevere hydrocep’
PrognosisHydranencephaly isusually incompatiblewith survival beyondearly infancyhalus .

PORENCEPHALYIn porencephaly there arecystic cavities within the brainthat usually communicatewith the ventricular system,the subarachnoid space orboth.
Etiology : Porencephaly may becaused by infarction of thecerebral arteries orhemorrhage into the brainparenchyma.
Diagnosis : In trueporencephaly there is one ormore cystic areas in thecerebral cortex, whichcommunicates with theventricle while in pseudoporencephalic cyst cavity donotcommunictes with ventricle.
Prognosis : The prognosis inporencephaly is related to thesize and location of the lesionand although there isincreased risk of impairedneurodevelopment in somecases development is normal

SCHIZENCEPHALYSchizencephaly is associatedwith clefts in the fetal brainconnecting the lateralventricles with thesubarachnoid space.
Etiology : Schizencephaly maybe a primary disorder of braindevelopment or it may be due tobilateral occlusion of themiddle cerebral arteries.
Dignosis : In schizencephalythere are bilateral cleftsextending from the lateralventricles to the subarachnoidspace, and is usually associatedwith absence of the cavumseptum pellucidum.
Prognosis : Schizencephaly isassociated with severeneurodevelopmental delay andseizures.

ENCEPHALOMALACIACystic encephalomalaciaan irregular cystic area inthe brain parenchymawhich is characterised bythe presence of multipleglial septations surroundedby astrocytic proliferation.This may be caused byinfarction, infection ortrauma. They may be focalor diffuse and theirdistribution will depend onthe cause and severity ofthe injury and the postconceptual age of thepatient. Encephalomalaciacaused by infarction maybe in the distribution of amajor cerebral artery.

ENCEPHALOMALACIAIf the injury is caused bymild to moderatehypotension the areas ofencephalomalacia may liein the boundary zonesbetween the major cerebralarteries, whereas severehypotension may result inwidespread cysticencephalomalacia withsparing of the deepperiventricular whitematter only. The presenceof reactive astrocytosis andglial septationsdistinguishes cysticencephalomalacia from anarea of porencephaly andindicates that the injuryoccurred late in gestation,in the perinatal period, orafter birth

CHOROID PLEXUS PAPILLOMAGuerard described the first CPP ina 3-year-old girl in 1832, andPerthes described the firstsuccessful surgical removal in 1910.The male-to-female incidence ratioof CPP is 2.8 : 1.
CPPs are rare, comprising less than1% of brain tumors in patients ofall ages. However, CPPs most oftenoccur in children and constitute upto 3% of childhood intracranialneoplasms with a predilection foryounger ages. CPPs comprise 4-6%of the intracranial neoplasms inchildren younger than 2 years and12-13% of intracranial neoplasms inchildren younger than 1 year.

Circulatory system(745) Bulbus cordis anomalies and anomalies of cardiac septal closure
(745.4) Ventricular septal defect
(745.5) Atrial septal defect
(746) Other congenital anomalies of heart
(747) Other congenital anomalies of circulatory system
(747.1) Coarctation of aorta
(747.11) Interruption of aortic arch
(747.2) Other congenital anomalies of aorta
(747.3) Congenital anomalies of pulmonary artery
(747.4) Congenital anomalies of great veins
(747.5) Absence or hypoplasia of umbilical artery
(747.6) Arteriovenous malformation, unspec.
(747.8) Other specified anomalies of circulatory system
(747.81) Arteriovenous malformation of brain
(746.82) Cor triatriatum
(746.83) Infundibular pulmonic stenosis congenital
(746.84) Congenital obstructive anomalies of heart not elsewhere classified
(746.85) Coronary artery anomaly congenital
(746.86) Congenital heart block
(746.87) Malposition of heart and cardiac apex
(747.89) Other specified congenital anomalies of heart
Brugada syndrome
(747.9) Unspecified congenital anomaly of circulatory system

CONGENITAL HEART DISEASES
80-90% CHD’S
LOW RISK PREGNANCIES
MJORITY OF THEM ARE
PRIMI
9% INFANT MORTILITY IN
U.K.
DUE – C.H.D.
SIX TIME MORE COMMON THAN
TRISOMIES 21,18,13
FOUR TIMES MORE COMMON THAN
NEURAL TUBE DEFECTS
PREVALENCE
8/1000 LIVE BIRTH
30/1000
STILL BIRTH

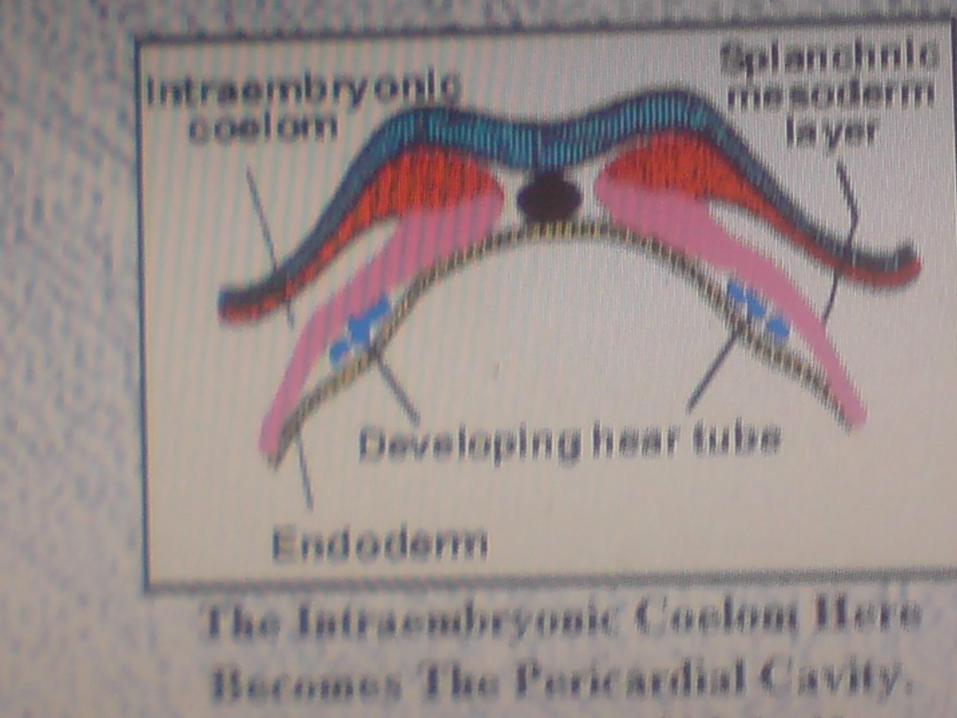
FIRST ORGAN TO BE FUNCTIONALIN HUMAN BEING - HEART
CARDIOGENESIS
TWO POOL OF CARDIAC
PRECURSORS
SECOND FIELD
DEVELOP INTO
RV, OFT, SINUS
VENOSUS
FIRST FIELD DVELOPS
INTO
RA,LA, LV


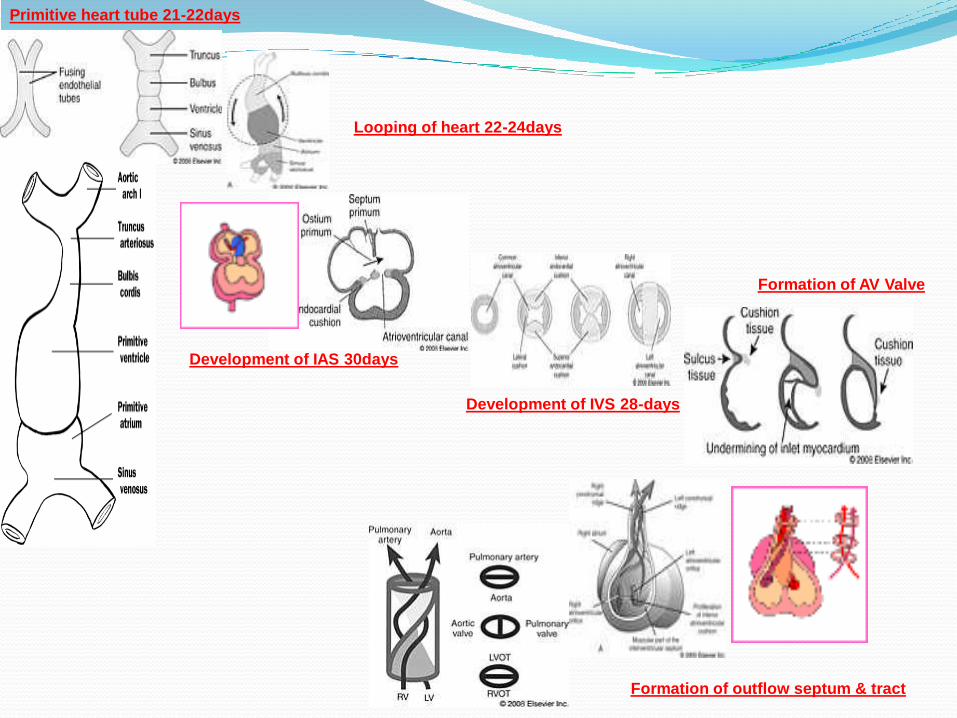
Primitive heart tube 21-22days
Looping of heart 22-24days
Development of IAS 30days
Development of IVS 28-days
Formation of AV Valve
Formation of outflow septum & tract

STREETER’S HORIZONS STAGES (CVS DEVELOPMENT)
22 24 26 28 30 32 34 36 38DAYSOF LIFE
HEART
PULSAIONSINO –ATRIALFORAMEN
*CIRCUL ATION*AV CUSHION*D.V.*3ARCHES
•P.VS.•PA -6•AORTA 4 ARCH
PVS->LA
*RV,LV*AV NODE
O.P.>CL*O.SEC.*RV-6A*LV-4A
TA-SEPTATION
S.CUSP
TVMVIVSNERVE
IVS

GENETIC ASPECT OF THE CHDs
• 40%• 100%
• 80%• 40-50%
21 T 13 T
OX18 T

VENRICULAR SEPTAL DEFECT 2/1000
ATRIAL SEPTAL DEFECT 1/3000
AORTIS STENOSIS 1/7000
PULMONARY STENOSIS 1/1000
PULMONARY ATRESIA 1/10,000
d – TRANSPOSITION OF GREAT ARTERIES
1/5000
TETRALOGY OF FALLOT’S 1/3000
DOUBLE OUTLET RIGHT VENTRCLE
1/10,000
TRUNCUS ARTERIOSUS 1/10,000
CARDIO SPLENIC SYBDROME 1/10,000
CHD
30%
10%
3%
0.1%
0.01%
2%

OVERALL RECURRENCE RISK IN CHDs
%
GENERAL POPULATION 1
SIBS OF ISOLATED CASE 2
OFFSPRING OF ISOLATED CASE 3
TWO AFFECTED SIBS ( SIB +PARENT) 10
> TWO AFFECTED FIRST DEGREE RELATIVE
50
MOTHER WITH CHD 10
FATHER WITH CHD 2

VENTRICULAR SEPTAL DEFECT
30% OF CHD’S
2/1000 BIRTH
50% V.S.D. ARE ISOLATED
PERIMEMBRANOUS 80%
INLET V.S.D.
MUSCULAR V.S.D.
OUTLET V.S.D.
90 SMALL V.S.D. CLOSE SPONTANEOUSLY.
SURGICAL OUT COME IS GOOD

ATRIAL SEPTAL DEFECT1/3000 BIRTH
10% OF ALL CHD’S
F.OVALE 3%
A.S.D. SECONDUM (ABOVE F.OVALE)
A.S.D. PRIMUM
(BELOW F. OVALE)
A.S.D. SINUS VENOSUS
CORONARY SINUS A.S.D.;
A rare type A.S.D. in whichcoronary sinu and left atrium openpartially or completely unroofed ,leading to left to right shunt.
50% with A.S.D. Have otherassociated other cardiac defects.
? DIFICULT TO DIAGNOSED ANTENATALLY
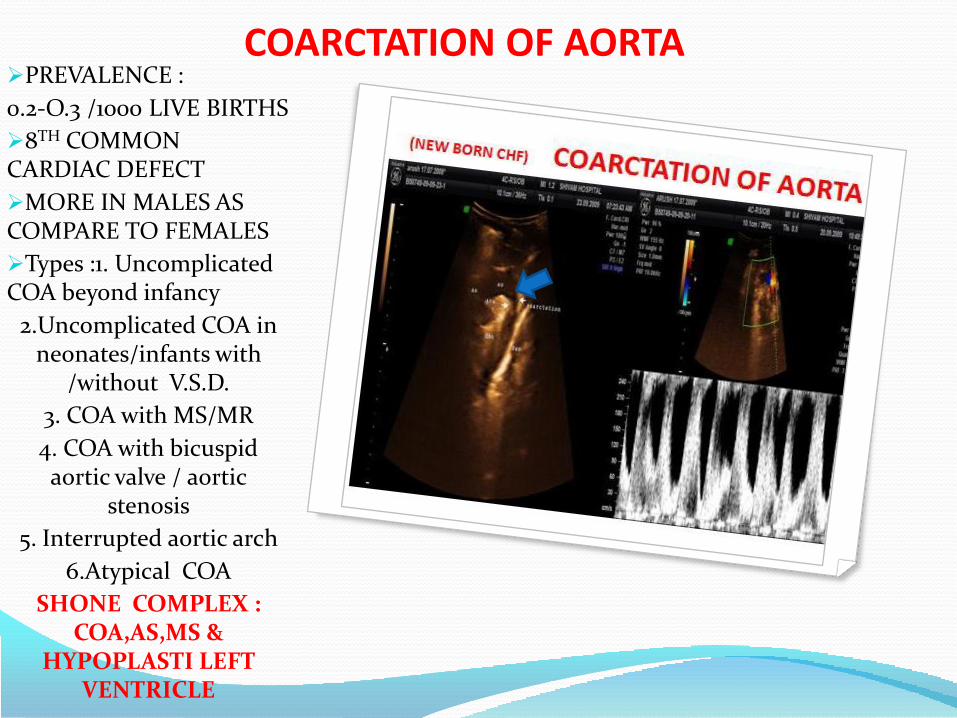
COARCTATION OF AORTAPREVALENCE :
0.2-O.3 /1000 LIVE BIRTHS
8TH COMMON CARDIAC DEFECT
MORE IN MALES AS COMPARE TO FEMALES
Types :1. Uncomplicated COA beyond infancy
2.Uncomplicated COA in neonates/infants with
/without V.S.D.
3. COA with MS/MR
4. COA with bicuspid aortic valve / aortic
stenosis
5. Interrupted aortic arch
6.Atypical COA
SHONE COMPLEX : COA,AS,MS &
HYPOPLASTI LEFT VENTRICLE
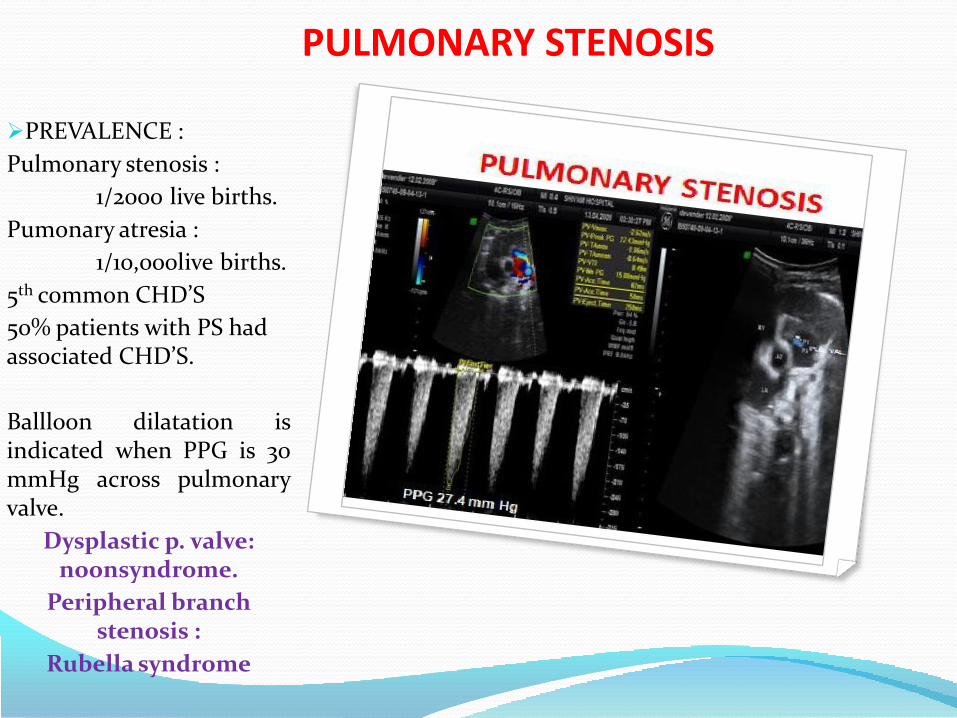
PULMONARY STENOSIS
PREVALENCE :
Pulmonary stenosis :
1/2000 live births.
Pumonary atresia :
1/10,000live births.
5th common CHD’S
50% patients with PS had associated CHD’S.
Ballloon dilatation isindicated when PPG is 30mmHg across pulmonaryvalve.
Dysplastic p. valve: noonsyndrome.
Peripheral branch stenosis :
Rubella syndrome

FETAL AORTIC STENOSIS
3% OF ALL CHD’S
1/7000 BIRTH
TYPE
SUPRA VALVULAR :
MEMBRANE
LOCALIZED NARROWING
DIFFUSE NARROWING
VALVULAR
BICUSPID AORTIC VALVE
DYSPLASTIC AORTIC VALVE.
SUBVALVULAR
FETAL VALVUAL AORTIC =STENOSIS

PATENT DUCTUS ATERIOSUSPREVALENCE :
O.138-O.8/1000 LIVE BIRTHSEighty percent (80%) of the DA interm infants close by 48 hours andnearly 100% by 96 hours.
Failure of the ductus arteriosus to close within 48-96 hours of postnatal age results in a left to right shunt across the ductus and overloading of the pulmonary circulation.
A hemodynamically significant shuntdue to PDA has been reported in 40%of infants less
than 1000 grams and 20% of infants between 1000-1500 grams Initial Indomethacin.
0.2 mg/kg stat followed by age adjusted doses:
Subsequent dose
< 2 day- 0.1 mg/kg/dose 12 hourly for 2 doses
2-7 day- 0.2 mg/kg/dose 12 hourly for 2 doses
7 day- 0.25 mg/kg/dose 12 hourly for 2 doses.
Ibuprofen : 10 mg/kg stat followed by 5 mg/kg/dose 24 hourly for 2 doses
PULMONARY ARTERY
DAO

SINGLE VENTRICLE1.5 % OF ALL CHD’S
Univentricular heart includesboth those cases in which twoatrial chambers are connected, byeither two distinct atrioventricularvalves or by a common one, to amain ventricular chamber(double-inlet single ventricle) aswell as those cases in which,because of the absence of oneatrioventricular connection(tricuspid or mitral atresia), oneof the ventricular chambers iseither rudimentary or absent.
Surgical treatment (the Fontanprocedure) involves separation ofthe systemic circulations byanastomosing the superior andinferior vena cava directly to thepulmonary artery.
GALEN SHUNT
FONATAN PROCEDURE
COMPLICATIONS :
ARRHYTHMIA
THROMBUS FORMATION
PROTEIN LOOSING ENTERO PATHY

CONGENITAL MITRAL STENOSISCongenital MS is rare,occurring in 0.5% ofpatients with congenitalheart disease (CHD)Congenital MS, a rare entity,takes several forms. Theseinclude hypoplasia of themitral valve annulus, mitralvalve commissural fusion,double orifice mitral valve,shortened or thickenedchordae tendinae, andparachute mitral valve, inwhich all chordae attach toa single papillary muscle.The most commonassociated malformationsarecoarctation of theaorta, aortic valve stenosis,and subvalvular aorticstenosis.
LA
LA

CONGENITAL MITRAL ATRESIAThe association ofmultiple levels of left-sided inflow and outflowtract obstruction istermed the Shonecomplex.
Severe hypoplasia, oratresia, of the mitralvalve results in ahypoplastic LV cavity sizethat is not capable ofsustaining the systemiccardiac output.
This situation isconsidered part of thespectrum ofthe hypoplastic left heartsyndrome

ATRIO – VENTRICULAR SEPTAL DEFECTPREVALENCE : 7% OF ALL CHD’S
1/3000 LIVE BIRTHS
50% of cases are associatedwith aneuploidy, 60%being trisomy 21, 25%trisomy 18 ( associated withextra-cardiac anomalies)or in fetuses withcardiosplenic syndromesassociated with multiplecardiac anomalies andabnormal disposition ofthe abdominal organs arealmost the rule.
Diagnosis :Antenatal echocardiographicdiagnosis of completeatrioventricular septal defects isusually easy. The incompleteforms are more difficult torecognize.
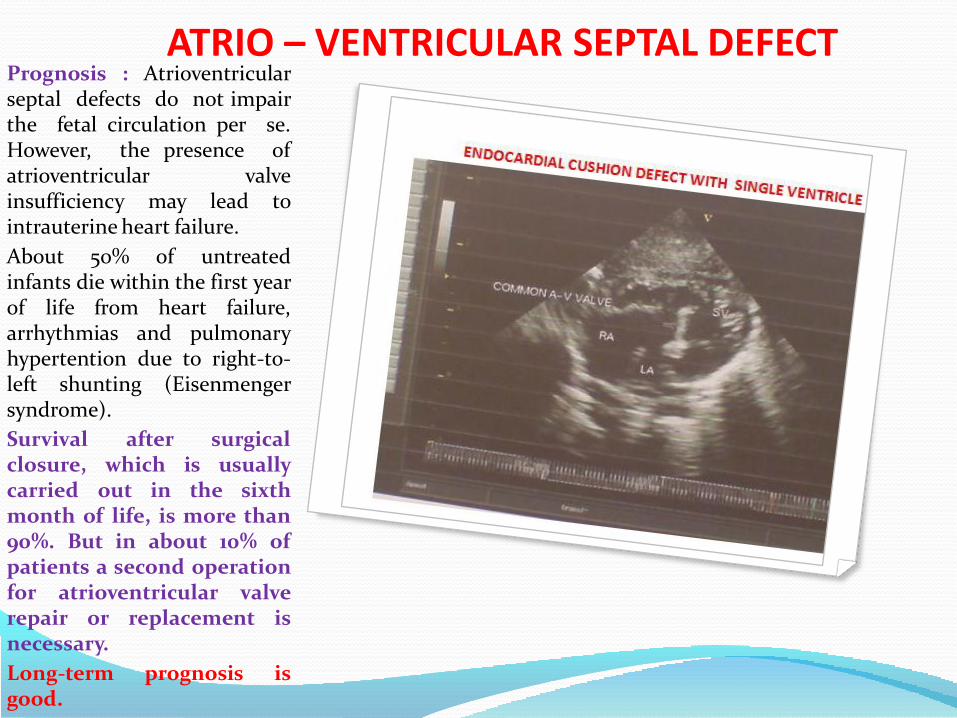
ATRIO – VENTRICULAR SEPTAL DEFECTPrognosis : Atrioventricularseptal defects do not impairthe fetal circulation per se.However, the presence ofatrioventricular valveinsufficiency may lead tointrauterine heart failure.
About 50% of untreatedinfants die within the first yearof life from heart failure,arrhythmias and pulmonaryhypertention due to right-to-left shunting (Eisenmengersyndrome).
Survival after surgicalclosure, which is usuallycarried out in the sixthmonth of life, is more than90%. But in about 10% ofpatients a second operationfor atrioventricular valverepair or replacement isnecessary.
Long-term prognosis isgood.

d - TRANSPOSITION OF GREAT ARTERIESPREVALENCE : 0.24/1000 LIVE BIRTHS(1/5000)
2ND MOST COMMON CHD’S ENCOUNTERED IN INFANCY &
REQUIRE TRANSFER TO TERTIARY CARE CENTER WITHIN FIRST TWO
WEEK OF LIFE.
TYPE OF TGA
Those with intact ventricular septum with or without
pulmonary stenosis,
Those with ventricular septal defects and
Those with ventricular septaldefect and pulmonary stenosis.Diagnosis : Completetransposition is probably one ofthe most difficult cardiaclesions to recognize in utero. Inmost cases the four-chamberview is normal, and the cardiaccavities and the vessels havenormal
PROGNOSIS :Surgery whichinvolves arterial switch toestablish anatomic andphysiological correction, isusually carried out within thefirst two weeks of life..
.
P.A.
LA

TETRALOGY OF FALLOT’SPrevalence : 3-26/10.000 live
births.
Mutation : NKX2,5 for 4% TOF
Deletion of human TBX1 ; chromosome 22q11.2, for 15% TOF
Trisomy 21 ,18,13 for 10% TOF.
Thus in 70% TOF is genetic etiology remains to be determine.
Anatomical lesion:Underdevelopment of
pulmonary infundibulum, subaortic V.S.D.,
overriding of aorta and
right ventricular hypertrophy
61% simple TOF
33% pulmonary atresia
3% absent pulmonary valve
3% common atrio-ventricular canal
Tetralogy of Fallot It is the mostcommon cyanotic heart defect,representing 55-70%, and the mostcommon cause of blue babysyndrome. It was described in 1672by Niels Stensen, in 1773 by EdwardSandifort, and in 1888 by theFrench physician Étienn-LouisArthur Fallot, for whom it isnamed

TETRALOGY OF FALLOT’SWhen severe pulmonic stenosisis present, cyanosis tends todevelop immediately after birth.With lesser degrees ofobstruction to pulmonaryblood flow the onset ofcyanosis may not appear untillater in the first year of life.
Diagnosis : Echocardiographicdiagnosis of tetralogy of Fallotrelies on the demonstration of aventricular septal defect in theoutlet portion of the septumand an overriding aorta. Thereis an inverse relationshipbetween the size of theascending aorta and pulmonaryartery, with a disproportion that isoften striking. A large aortic rootis indeed an important diagnosticclue.
Prognosis : Cardiac failure isnever seen in fetal life as well aspostnatally.

TETRALOGY OF FALLOT’S
Even in cases of tight pulmonarystenosis or atresia, the wideventricular septal defectprovides adequate combinedventricular output, while thepulmonary vascular bed issupplied in a retrograde mannerby the ductus. . When there ispulmonary atresia, rapid andsevere deterioration followsductal constriction. Survivalafter complete surgical repair(which is usually carried out inthe third month of life) is morethan 90% and about 80% ofsurvivors have normal exercisetolerance.

TRUNCUS ARTERIOSUS
7% OF ALL CHD’S
1/10,000 BIRTH
30% HAVE EXTRACARDIAC MALFORMATION
TRUNCUS IS CONNECTED TO :
40% RGIHT VENTRCLE
20% LEFT VENTRICLE
40% TO BOTH VENTRICLE
TYPE :
I : MAIN PA CONNECTED TO TA
II : PA BRANCH FROM LATERAL ASPECT OF TRUNCUS
III: PA BRANCH FROM POSTERIOR ASPECT OF TA.
IV : NO PA; LUNG GETS BLOOD SUPLLY FROM – AORTIC COLATERALS


DOUBLE OULET RIGHT VENTRICLE7% OF ALL CHD’S
PREVALENCE : 1/10,000 BIRTH
In double-outlet right ventricle(DORV) most of the aorta andpulmonary valve arisecompletely or almostcompletely from the rightventricle. The relation betweenthe two vessels may vary,ranging from a Fallot-like to aTGA-like situation (the Taussig-Bing anomaly). Pulmonarystenosis is very common in alltypes of DORV, but left outflowobstructions, from subaorticstenosis to coarctation andinterruption of the aortic arch,can also be seen.
Diagnosis : Prenatal diagnosis ofDORV can be reliably made in thefetus but differentiation from otherconotruncal anomalies can be verydifficult.
PROGNOSIS: Since the fetalheart works as a commonchamber where the blood ismixed and pumped, DORV isnot associated with intrauterineheart failure.
RV
PA
AO
LA
IVS
LA
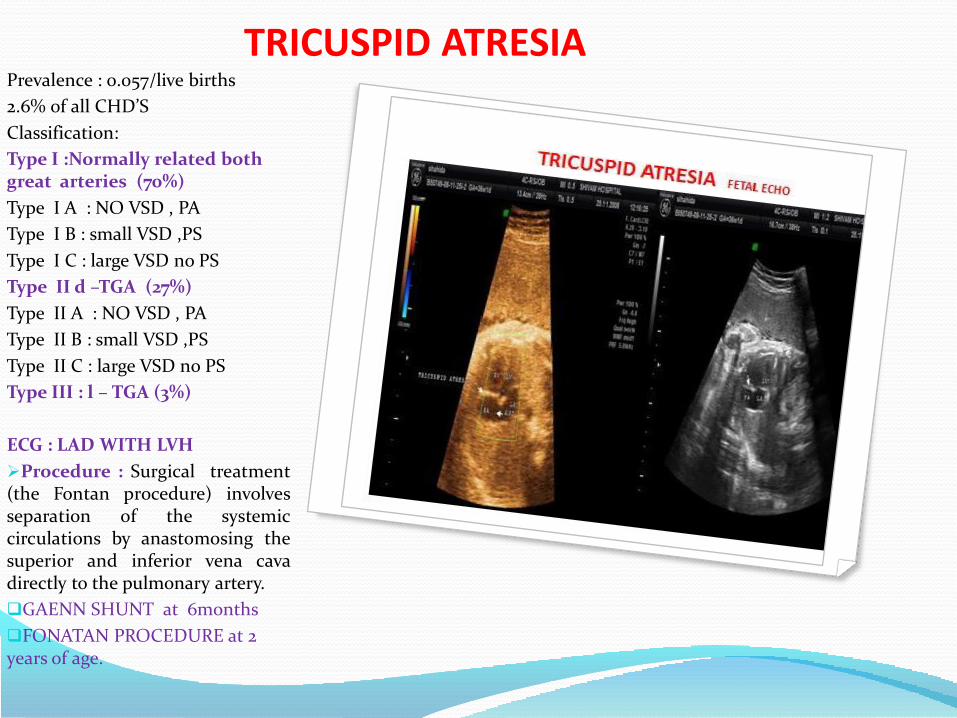
TRICUSPID ATRESIAPrevalence : 0.057/live births
2.6% 0f all CHD’S
Classification:
Type I :Normally related both great arteries (70%)
Type I A : NO VSD , PA
Type I B : small VSD ,PS
Type I C : large VSD no PS
Type II d –TGA (27%)
Type II A : NO VSD , PA
Type II B : small VSD ,PS
Type II C : large VSD no PS
Type III : l – TGA (3%)
ECG : LAD WITH LVH
Procedure : Surgical treatment(the Fontan procedure) involvesseparation of the systemiccirculations by anastomosing thesuperior and inferior vena cavadirectly to the pulmonary artery.
GAENN SHUNT at 6months
FONATAN PROCEDURE at 2 years of age.

TRICUSPID ATRESIAPrevalence : 0.057/live births
2.6% 0f all CHD’S
Classification:
Type I :Normally related both great arteries (70%)
Type I A : NO VSD , PA
Type I B : small VSD ,PS
Type I C : large VSD no PS
Type II d –TGA (27%)
Type II A : NO VSD , PA
Type II B : small VSD ,PS
Type II C : large VSD no PS
Type III : l – TGA (3%)
ECG : LAD WITH LVH
Procedure : Surgical treatment(the Fontan procedure) involvesseparation of the systemiccirculations by anastomosing thesuperior and inferior vena cavadirectly to the pulmonary artery.
GAENN SHUNT at 6months
FONATAN PROCEDURE at 2 years of age.

TRICUSPID ATRESIAPrevalence : 0.057/live births
2.6% 0f all CHD’S
Classification:
Type I :Normally related both great arteries (70%)
Type I A : NO VSD , PA
Type I B : small VSD ,PS
Type I C : large VSD no PS
Type II d –TGA (27%)
Type II A : NO VSD , PA
Type II B : small VSD ,PS
Type II C : large VSD no PS
Type III : l – TGA (3%)
ECG : LAD WITH LVH
Procedure : Surgical treatment(the Fontan procedure) involvesseparation of the systemiccirculations by anastomosing thesuperior and inferior vena cavadirectly to the pulmonary artery.
GAENN SHUNT at 6months
FONATAN PROCEDURE at 2 years of age.
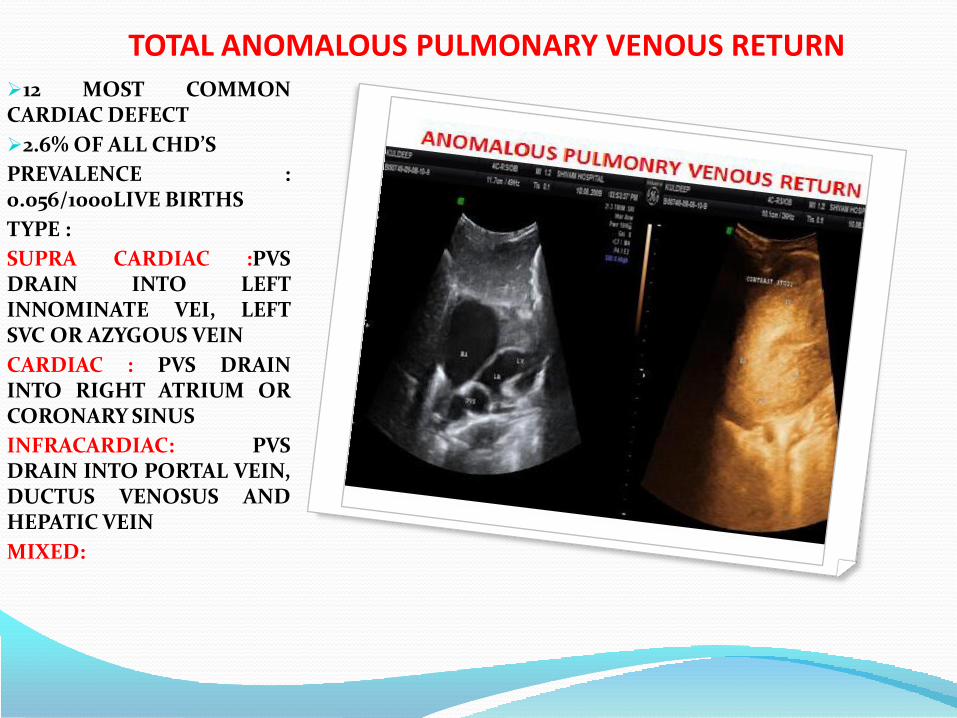
TOTAL ANOMALOUS PULMONARY VENOUS RETURN12 MOST COMMONCARDIAC DEFECT
2.6% OF ALL CHD’S
PREVALENCE :0.056/1000LIVE BIRTHS
TYPE :
SUPRA CARDIAC :PVSDRAIN INTO LEFTINNOMINATE VEI, LEFTSVC OR AZYGOUS VEIN
CARDIAC : PVS DRAININTO RIGHT ATRIUM ORCORONARY SINUS
INFRACARDIAC: PVSDRAIN INTO PORTAL VEIN,DUCTUS VENOSUS ANDHEPATIC VEIN
MIXED:

RUPTURE OF SINUS OF VALSALVASinus of Valsalva aneurysmcomprises approximately 0.1-3.5% of all congenital cardiacanomalies. Discovery in thepediatric age group isunusual.Congenital sinus ofValsalva aneurysm was firstdescribed by Hope. The 3 sinusesof Valsalva are located in the mostproximal portion of the aorta, justabove the cusps of the aortic valve.The sinuses correspond to theindividual cusps of the aortic valve.Aneurysm of a sinus of Valsalva is arare congenital cardiac defect thatcan rupture, causing heart failureor other catastrophic cardiacevents. If the aneurysm remainsunruptured, it occasionally causesobstruction of cardiac flowresulting from compression ofnormal structures. Aneurysmstypically develop as a discreteflaw in the aortic media withinone of the sinuses of Valsalva.Aneurysms most often involve theright aortic sinus (67-85% ofpatients, often associated with asupracristal ventricular septaldefect), followed by thenoncoronary sinus, whereas ananeurysm of the left sinus is rare.

RUPTURE OF SINUS OF VALSALVADistortion and prolapse of thesinus and aortic valve tissue canlead to progressive aortic valveinsufficiency. Unrupturedaneurysm may cause distortionand obstruction in the rightventricular outflow tract.Distortion and compression mayalso cause myocardial ischemia (bycoronary artery compression) and,possibly, heart block (bycompressing the conductionsystem).
Rupture may occur into anychamber, although rupture mostcommonly occurs into the aorticright ventricular communication.Rupture into the right atrium is thesecond most common, inassociation with a noncoronarycusp aneurysm. Rupture may occurless commonly into the left-sidedchambers, the pulmonaryartery, and rarely extends into thepericardium.
RSOVs are commonly "wind-sock"-like, with a broader aorticend, ADO is best suited for thisdefect, although otherAmplatzer devices

ALCPAALCAPA or
Blannd-Garland-Whitesyndrome is a rarecongenital anomaly withincidence of 1 in 3 lac livebirths, accounting for0.25% of congenital heartdisease.
Wesselhoeft et al.
classified the clinicalspectrum of
ALCAPA as follows:
1. Infantile Syndrome :This is the most commonform. Patient developsacute episode ofrespiratory insufficiency,cyanosis, irritability andprofuse sweating. Mostof them die within twoyears.
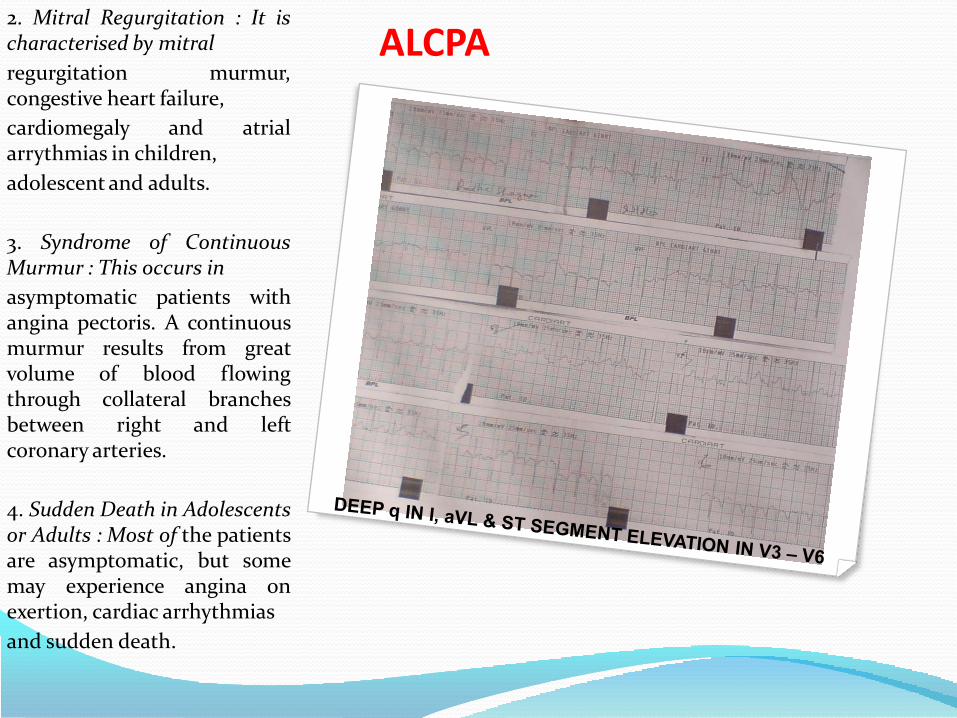
ALCPA2. Mitral Regurgitation : It ischaracterised by mitral
regurgitation murmur,congestive heart failure,
cardiomegaly and atrialarrythmias in children,
adolescent and adults.
3. Syndrome of ContinuousMurmur : This occurs in
asymptomatic patients withangina pectoris. A continuousmurmur results from greatvolume of blood flowingthrough collateral branchesbetween right and leftcoronary arteries.
4. Sudden Death in Adolescentsor Adults : Most of the patientsare asymptomatic, but somemay experience angina onexertion, cardiac arrhythmias
and sudden death.

EBSTEIN’S ANOMALY TRICUSPID VALVE
Ebstein disease
Prevalence : 0.012-0.06/1000live births Ebstein's may beassociated with trisomy 13, 21,Turner, Cornelia de Lange andMarfan syndromes. Maternalingestion of lithium has alsobeen incriminated as a causalfactor
Ebstein's anomaly resultsfrom a faulty implantation ofthe tricuspid valve. Theposterior and septal leafletsare elongated and tetheredbelow their normal level ofattachment on the annulus ordisplaced apically, away fromthe annulus, down to thejunction between the inlet andtrabecular portion of the rightventricle. . Associatedanomalies include atrial septaldefect, pulmonary atresia,ventricular septal defect, andsupraventricular tachycardia.

EBSTEIN’S ANOMALY TRICUSPID VALVE
Ebstein disease
Prevalence : 0.012-0.06/1000live births Ebstein's may beassociated with trisomy 13, 21,Turner, Cornelia de Lange andMarfan syndromes. Maternalingestion of lithium has alsobeen incriminated as a causalfactor
Ebstein's anomaly resultsfrom a faulty implantation ofthe tricuspid valve. Theposterior and septal leafletsare elongated and tetheredbelow their normal level ofattachment on the annulus ordisplaced apically, away fromthe annulus, down to thejunction between the inlet andtrabecular portion of the rightventricle. . Associatedanomalies include atrial septaldefect, pulmonary atresia,ventricular septal defect, andsupraventricular tachycardia.

EBSTEIN’S ANOMALY Ebstein disease
Prevalence : 0.012-0.06/1000live births Ebstein's may beassociated with trisomy 13, 21,Turner, Cornelia de Lange andMarfan syndromes. Maternalingestion of lithium has alsobeen incriminated as a causalfactor
Ebstein's anomaly resultsfrom a faulty implantation ofthe tricuspid valve. Theposterior and septal leafletsare elongated and tetheredbelow their normal level ofattachment on the annulus ordisplaced apically, away fromthe annulus, down to thejunction between the inlet andtrabecular portion of the rightventricle. . Associatedanomalies include atrial septaldefect, pulmonary atresia,ventricular septal defect, andsupraventricular tachycardia.
LA

CORTRIATRIATUMThe incidence of cor triatriatumis less than 1 in 10,000.
First reported in 1868, cortriatriatum, that is, a heart with3 atria (triatrial heart), is acongenital anomaly in whichthe left atrium (cor triatriatumsinistrum) or right atrium (cortriatriatum dextrum) is dividedinto 2 parts by a fold of tissue, amembrane, or a fibromuscularband. Classically, the proximal(upper or superior) portion ofthe corresponding atriumreceives venous blood, whereasthe distal (lower or inferior)portion is in contact with theatrioventricular valve andcontains the atrial appendageand the true atrial septum thatbears the fossa ovalis. Themembrane that separates theatrium into 2 parts variessignificantly in size and shape.

CORTRIATRIATUMIt may appear similar to adiaphragm or be funnel-shaped,bandlike, entirely intact(imperforate) or contain one ormore openings (fenestrations)ranging from small, restrictive-type to large and widely open. Cortriatriatum dexter is a rare cardiacabnormality in which the rightatrium is subdivided into twodistinct chambers. This anomalyis generally attributed to thepersistence of the right sinusvenosus valve and it is frequentlyassociated with severemalformations of other rightheart structures. Cor triatriatumdexter results from persistence ofthe entire right sinus venosusvalve, which forms a large,obstructive flap or septum acrossthe right atrium and divides itinto 2 separate chambers. Theupstream chamber receives superiorand inferior vena caval flow, while thedownstream chamber incorporates theright atrial appendage.
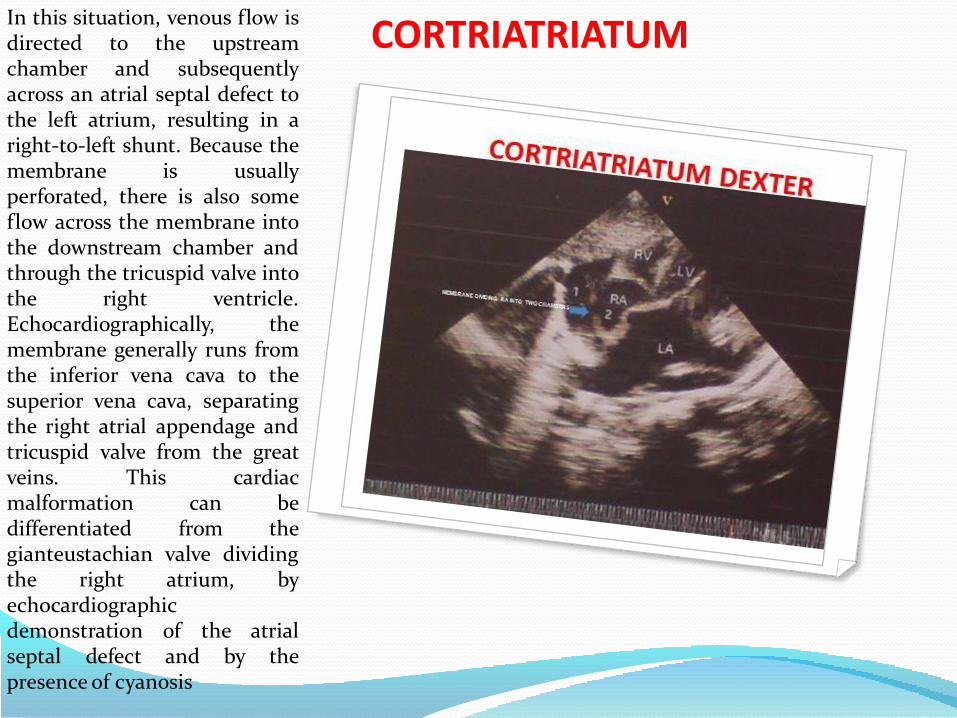
CORTRIATRIATUMIn this situation, venous flow isdirected to the upstreamchamber and subsequentlyacross an atrial septal defect tothe left atrium, resulting in aright-to-left shunt. Because themembrane is usuallyperforated, there is also someflow across the membrane intothe downstream chamber andthrough the tricuspid valve intothe right ventricle.Echocardiographically, themembrane generally runs fromthe inferior vena cava to thesuperior vena cava, separatingthe right atrial appendage andtricuspid valve from the greatveins. This cardiacmalformation can bedifferentiated from thegianteustachian valve dividingthe right atrium, byechocardiographicdemonstration of the atrialseptal defect and by thepresence of cyanosis

ENLARGE CORONARY SINUS The coronary sinus is enlarge
1. If left superior vena cava orpulmonary vein open into it.
PREVALENCE : 0.5% in generalpopulation & 3-10 %among childernwith CHD.
2. In condition associated withraise right atrial pressure like –tricuspid atresia, severe pulmonaryarterial hypertension.
3. Increased left main coronaryartery flow and increasedcoronary sinus return.
Dilated coronary sinus is aprompt to look for further cardiacabnormalities such as intracardiacshunts or thoracic venousabnormalities.
The complex of an unroofedcoronary sinus (UCS) and apersistent left superior vena cava(PLSVC) is a rare congenital heartdisease first described by Raghibet al. in 1965.1 A normal coronarysinus drains the cardiac veins intothe right atrium. A UCS, in additionto draining the cardiac veins, alsocommunicates abnormally with theleft atrium.

ENLARGE CORONARY SINUS This abnormal communication isthought to be due to impaireddevelopment of the partitionbetween the left atrium and thecoronary sinus – an alternativeexplanation is subsequentdissolution of this partition.
A PLSVC, abnormally draining theleft internal jugular and subclavianveins into the coronary sinus, isdue to impaired degeneration of theembryonic left counterpart of thenormal right superior vena cava.A UCS or a PLSVC may be furtherassociated with other cardiacabnormalities. UCS and PLSVCmay cause no symptoms or maycause right ventricular failure,paradoxical cerebral embolism andcerebral abscess, or cyanosis thatmay vary with neck position. UCSand PLSVC may be furtherassociated with other cardiacabnormalities such asatrioventricular septal defect, atrialappendage anomalies andcoronary sinus ostial atresia .
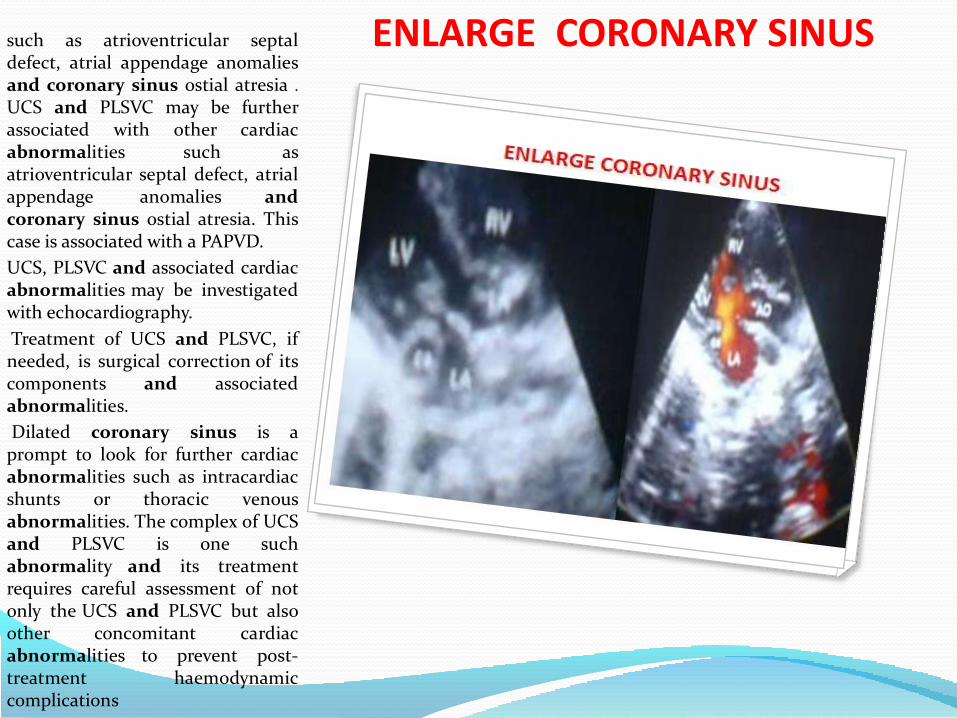
ENLARGE CORONARY SINUSsuch as atrioventricular septaldefect, atrial appendage anomaliesand coronary sinus ostial atresia .
UCS and PLSVC may be furtherassociated with other cardiacabnormalities such asatrioventricular septal defect, atrialappendage anomalies andcoronary sinus ostial atresia. Thiscase is associated with a PAPVD.
UCS, PLSVC and associated cardiacabnormalities may be investigatedwith echocardiography.
Treatment of UCS and PLSVC, ifneeded, is surgical correction of itscomponents and associatedabnormalities.
Dilated coronary sinus is aprompt to look for further cardiacabnormalities such as intracardiacshunts or thoracic venousabnormalities. The complex of UCSand PLSVC is one suchabnormality and its treatmentrequires careful assessment of notonly the UCS and PLSVC but alsoother concomitant cardiacabnormalities to prevent post-treatment haemodynamiccomplications

CARDIAC MALPOSITIONPREVALENCE : 0.103/1000LIVE BIRTH
1% OF ALL CHD’S
TYPE :
DEXTROCARDIA
ECTOPIA CORDIS -PENTALOGY OF CANTRELL
ASPLENIA
POLYSPLENIA

RHABDOMYOMAPrevalence: Any cardiactumor 1-2/10,000; over 90%are benign. Rhabdomyomais the most commonbenign congenital tumor..
occurring in the fetus and neonate,with most identified within thefirst year of life
Recurrence risk: Frequent inpatients with tuberous sclerosis.
Associated anomalies: Tuberoussclerosis (50-86%), cardiacdysrhythmia, non-immunehydrops.
Intracavitary growth of the tumorsmay cause disruption ofintracardiac blood flow leading tocongestive heart failure andhydrops. Cardiac dysrhythmias,caused by compression of theconducting system, are alsofrequently identified.Rhabdomyomas grow slowly inutero but tend to regressspontaneously after birth.

FETAL P S V T
Adenosine :Per umbilical
0.05 to 0.2mg
Flecanide : oral 200-300mg
Digoxin : Oral, parenteral
Transplacental, 0.5- 1 mg
Amiodarone : parenteral
600-800mg
Sotatlol : oral; 80-320 mg
FETAL PSVT ========

FETAL COMPLETE A-V BLOCK1901 MORQUIO gave first
description of CCAVB.
1908 Van den heuvel – ECG
1929 Yater IN UTERO
diagnosis of CCAVB.
Can be diagnosed as early as 16th week of
gestation.
1976 McCue & Chameides - association
between CCAVB & connective tissue
disorder .
75% anti –Ro positive
Prevalence : 1/22,000 live births
1/3 to ¼ have -structural heart defects
- L – TGA
ECD

Eye, ear, face and neck(743) Congenital anomalies of eye
(743.0) Anophthalmos
(743.1) Microphthalmos
(743.2) Buphthalmos
(743.3) Congenital cataract and lens anomalies
(743.4) Coloboma and other anomalies of anterior segment
(743.45) Aniridia
(743.5) Congenital anomalies of posterior segment
(743.6) Congenital anomalies of eyelids, lacrimal system, and orbit
(744) Congenital anomalies of ear, face, and neck
(744.0) Anomalies of ear causing impairment of hearing
(744.1) Accessory auricle
(744.2) Other specified congenital anomalie of ea
(744.22) Macrotia
(744.23) Microtia
(744.3) Unspecified congenital anomaly of ear
(744.4) Branchial cleft cyst or fistula; preauricular sinus
(744.5) Webbing of neck
(744.8) Other specified congenital anomalies of face and neck
(744.81) Macrocheilia
(744.82) Microcheilia
(744.83) Macrostomia
(744.84) Microstomia

OPHTHALAMIC BIRTH DEFECTS CONGENITAL CORNEAL OPACITY
Most ocular abnormalitieshave occurred in patients withchromosomal defects. Majorocular abnormalities, such asanophthalmia, cyclopia,retinoblastoma,microphthalmia, cornealopacities, coloboma,cataracts, intraocularcartilage, retinal dysplasiaand absent optic nerves;and, minor abnormalities,such as ptosis, abnormaleyelid fissures, andBrushfield spots are presentin individuals with abnormalchromosomes. Thechromosome errors are usuallypresent in all somatic tissues.Consequently, multiple tissueabnormalities would beexpected in most patients withchromosome abnormalities.

CONGENITAL PTOSISMental retardation is verycommon in those patientswith abnormalities ofautosomes. Therefore, it isunlikely that an isolated singleclinical or histopathologicalocular abnormality will be theresult of a chromosome error.However, if the individual hasmultiple systemicabnormalities, then achromosome error can beconsidered reasonably. Anychromosome disorder can beidentified correctly by anappropriate bandingchromosome determinationon the affected individuals.With the possible exception ofthe association of 13ql4- andretinoblastoma, there does notappear to be anypathognomonic ocularabnormalities that occur inindividuals with chromosomeerrors.

Mental retardation is verycommon in those patientswith abnormalities ofautosomes. Therefore, it isunlikely that an isolated singleclinical or histopathologicalocular abnormality will be theresult of a chromosome error.However, if the individual hasmultiple systemicabnormalities, then achromosome error can beconsidered reasonably. Anychromosome disorder can beidentified correctly by anappropriate bandingchromosome determinationon the affected individuals.With the possible exception ofthe association of 13ql4- andretinoblastoma, there does notappear to be anypathognomonic ocularabnormalities that occur inindividuals with chromosomeerrors.

CONGENITAL GLUCOMABuphthalmos is defined as a"large eye" [bu (Greek) = ox orcow]. It is most often presentin both eyes in children due tocongenital open-angleglaucoma of the eye, noted byunusually large corneas andincreased overall size of theeyeball. An abnormally narrowangle between the cornea andiris blocks the outflow ofaqueous humor, which leads toan increased intraocularpressure and a characteristicbulging enlargement of theeyeball. Patient symptoms mayinclude excessive tearing andlight sensitivity("photophobia"). Cupping ofthe optic disk, which may bethe first sign to be seen ondilated examination by an eyecare professional. Congenitalglaucoma untreated usuallyleads to blindness.

CONGENITAL GLUCOMAAniridia is a rare congenital
condition characterized by theunderdevelopment of the eye's iris.This usually occurs in both eyes. Itis associated with poordevelopment of the retina at theback of the eye preventing normalvision development. Aniridia doesnot always cause lack of vision, butusually leads to a number ofcomplications with the eye TheAN2 region of the short arm ofchromosome 11 (11p13) includes thePAX6 gene (named for its PAiredboX status), whose gene producthelps regulate a cascade of othergenetic processes involved in thedevelopment of the eye Aniridia isa heterozygotic disease, meaningthat only one of the twochromosome 11 copies is affected.When both copies are altered(homozygous condition), the resultis a uniformly fatal condition withnear complete failure of entire eyeformation
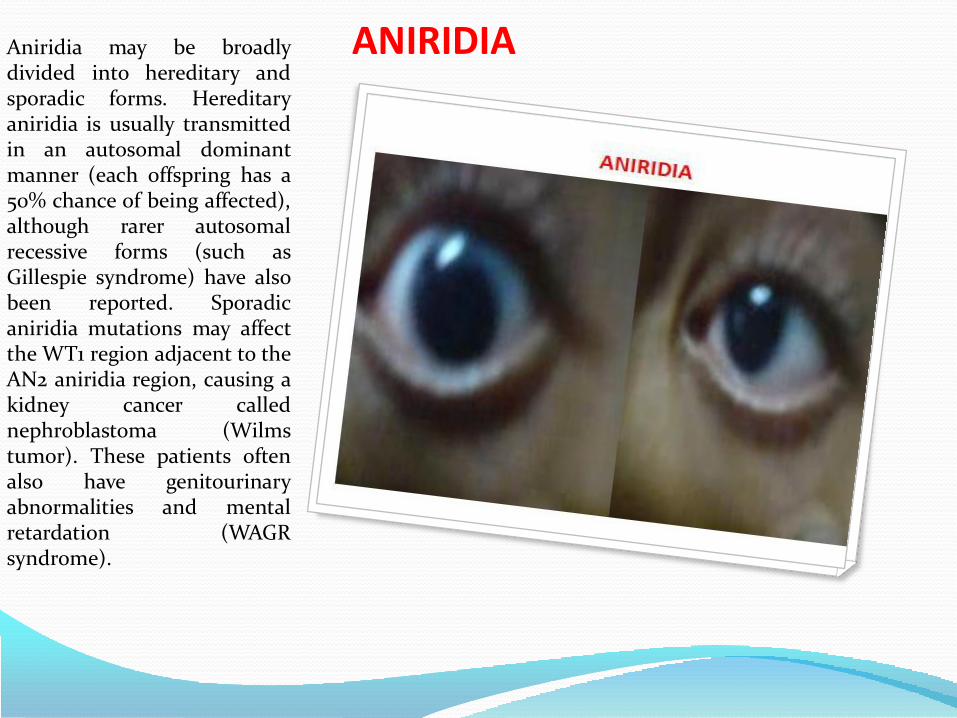
ANIRIDIAAniridia may be broadlydivided into hereditary andsporadic forms. Hereditaryaniridia is usually transmittedin an autosomal dominantmanner (each offspring has a50% chance of being affected),although rarer autosomalrecessive forms (such asGillespie syndrome) have alsobeen reported. Sporadicaniridia mutations may affectthe WT1 region adjacent to theAN2 aniridia region, causing akidney cancer callednephroblastoma (Wilmstumor). These patients oftenalso have genitourinaryabnormalities and mentalretardation (WAGRsyndrome).

ANIRIDIAAniridia is a rare congenital
condition characterized by theunderdevelopment of the eye’s iris. Thisusually occurs in both eyes. It isassociated with poor development ofthe retina at the back of the eyepreventing normal visiondevelopment. Aniridia does not alwayscause lack of vision, but usually leads to anumber of complications with the eyeThe AN2 region of the short arm ofchromosome 11 (11p13) includes the PAX6gene (named for its PAired boX status),whose gene product helps regulate acascade of other genetic processesinvolved in the development of the eye(as well as other nonocular structures).[
Aniridia is a heterozygotic disease,meaning that only one of the twochromosome 11 copies is affected. Whenboth copies are altered (homozygouscondition), the result is a uniformly fatalcondition with near complete failure ofentire eye formation. In 2001, two casesof homozygous An iridia patients werereported; the fetuses died prior to birthand had severe brain damage. In mice,homozygous Small eye defect (mousePax-6) led to loss of eyes, nose and thefetuses suffered severe brain damage.[Aniridia may be broadly divided intohereditary and sporadic forms.

ANIRIDIAHereditary aniridia is usually transmittedin an autosomal dominant manner (eachoffspring has a 50% chance of beingaffected), although rarer autosomalrecessive forms (such as Gillespiesyndrome) have also been reported.Sporadic aniridia mutations may affectthe WT1 region adjacent to the AN2aniridia region, causing a kidney cancercalled nephroblastoma (Wilms tumor).These patients often also havegenitourinary abnormalities and mentalretardation (WAGR syndrome). Severaldifferent mutations may affect the PAX6gene. Some mutations appear to inhibitgene function more than others, withsubsequent variability in the severity ofthe disease. Thus, some aniridicindividuals are only missing a relativelysmall amount of iris, do not have fovealhypoplasia, and retain relatively normalvision. Presumably, the genetic defect inthese individuals causes less"heterozygous insufficiency," meaningthey retain enough gene function to yielda milder phenotype
(OMIM) 106210 AN
(OMIM) 106220 Aniridia and absentpatella
(OMIM) 106230 Aniridia, microcornea,and spontaneously reabsorbed cataract
(OMIM) 206700 Aniridia, cerebellarataxia, and mental deficiency (Gillespiesyndrome)

CONGENITAL ANTERIOR STAPHYLOMAStaphyloma : It is theprotrusion of the sclera orcornea, usually lined withuveal tissue. In 1827 F.A. VonAmmon describe it.
Anterior staphylomastaphyloma is in the anteriorpart of the eye.
corneal staphyloma 1.bulging of the cornea withadherent uveal tissue.
2. One formed by protrusionof the iris through a cornealwound.
posterior staphyloma,staphyloma posticumbackward bulging of sclera atposterior pole of eye.
scleral staphylomaprotrusion of the contents ofthe eyeball where the sclerahas become thinned

CONGENITAL ANTERIOR STAPHYLOMA
Probabilities are that twotypes of such congenitalanterior staphyloma exist –one is of inflammatoryorigin and the other is dueto developmental defect.The latter is, however, allthe more rare.

FACE - CLEFT
MICRO OPHTHALMIA
ANOPHTHALMIA
HYPOTELORISM
(STENOPIA)
HYPERTELORISM

FACE - CLEFT
• ONLY PALATE
• ONLY LIP
• BOTH LIP & PALATE ARE CLEFT
• PREVALENCE
1/800
BIRTH50%
25%25%

CLEFT LIP & PALATE
INHERITANCE
AD, AR, XR & XD
1-2 % WITH 13 & 18 TRISOMY
5% TERATOGEN –ANTIEPILEPTICS
WITH 100 GENETIC DISODERS
.
80% CLEFT S LIP WITH /WITHOUT
PALATE
ISOLATED
20 % CLEFT ASSOCIATED
WITH SYNDROME
PREVALENCE
1/800 BIRTH
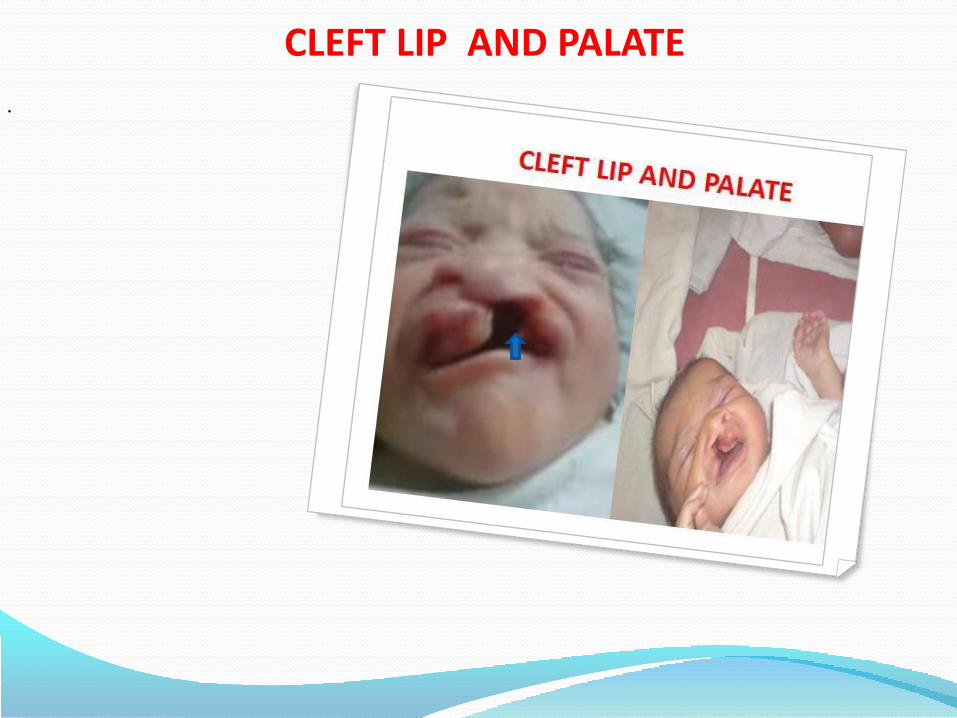
CLEFT LIP AND PALATE
.

CLEFT LIP
MEDIAN CLEFT LIP PREVALENCE IS 0.5 %
ASSOCIATE WITH HOLOPROSENCEPHALY
&
ORAL – FACIAL DIGITAL SYNDROME
.
MEDIAN
&
LATERAL
CLEFT
LIP
TYPE OF CLEFT LIP

CLEFT LIP
.

FACIAL CLEFT This term refers to a wide spectrum of clefting defects (unilateral, bilateral
and less commonly mid-line) usually involving the upper lip, the palate, or both.
Cleft palate without cleft lip is a distinct disorder. Facial clefts encompass a broad
spectrum of severity, ranging from minimal defects, such as a bifid uvula, linear
indentation of the lip, or submucous cleft of the soft palate, to large deep defects
of the facial bones and soft tissues.
The typical cleft lip will appear as a linear defect extending from one side of the lip
into the nostril.
Cleft palate associated with cleft lip may extend through the alveolar ridge and hard
palate, reaching the floor of the nasal cavity or even the floor of the orbit. Isolated
cleft palate may include defects of the hard palate, the soft palate, or both.
Both cleft lip and palate are unilateral in about 75% of cases and the left side is
more often involved than the right side.
PrevalenceThe incidence of cleft lip with or without cleft palate 1/750, the incidence of cleft
palate alone is 1/2500.
TreatmentSurgical closer of cleft lip is usually performed by three months of the age when the
infant has satisfactory weight gain and free from respiratory and systemic infection.
Closer of palate is usually done before 1 year of age to enhance normal speech
development.

ANGULAR CLEFT LIP
.

ABSENT DEPRESSOR ANGULARIS ORISThe depressor angularis orismuscle (DAOM) originates fromthe oblique line of the mandibleand extends upward andmedially to the orbi-cularis oris.It attaches to the skin and themucous membrane of the lowerlip. The DAOM draws the lowercorner of the mouth downwardand everts the lower lip. Thecause for agenesis of the muscleis unknown. The absence orhypoplasia of the DAOMproduces characteristic findings.The lower lip on the affected sidelooks thinner because of the lackof eversion and feels thinnerbecause of the muscle agenesis.When crying, the corner of themouth on the affected side isdisplaced toward the normalside and the lower lip on thenormal side moves downwardand outward

ABSENT ORBICULARIS ORIS .
These patients havesymmetrical foreheadwrinkling, eye closure, andnasolabial fold depth. Thediagnosis may be confirmed byelectrophysiologic studies. Thefacial nerve conduction timeto the mentalis and orbicularisoris muscle are normal. Thereis no fibrillation in the areanormally occupied by theDAOM. Motor units aredecreased or absent in thesame area.Agenesis of the DAOM canoccur as an isolated anomalybut it has also been reported inassociation withcardiovascular,musculoskeletal,genitourinary, and respiratorydefects.
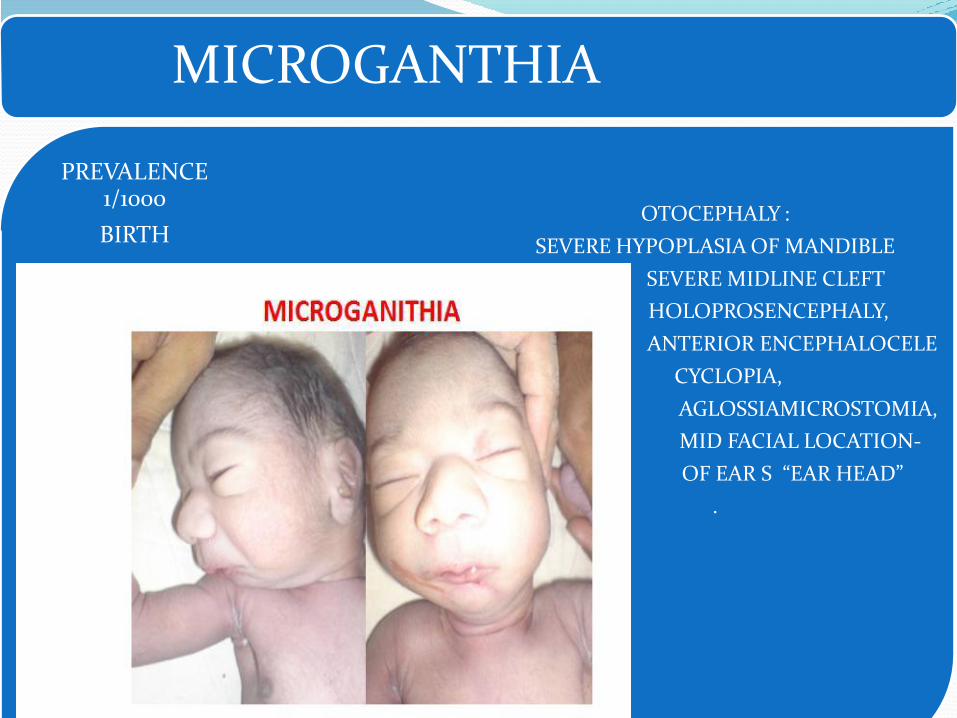
MICROGANTHIA
OTOCEPHALY :
SEVERE HYPOPLASIA OF MANDIBLE
SEVERE MIDLINE CLEFT
HOLOPROSENCEPHALY,
ANTERIOR ENCEPHALOCELE
CYCLOPIA,
AGLOSSIAMICROSTOMIA,
MID FACIAL LOCATION-
OF EAR S “EAR HEAD”
.
PREVALENCE 1/1000
BIRTH

MICROGANTHIA
• GENETIC SYNDROME
• CHROMOSOMAL
• TERATOGENIC DRUG
• ROBIN ANOMALAD
• (SPORADIC)
40% METHOTREXATE
TREACHER-COLLINS, ROBIN & ROBERT
SYNDROME
18 T
TRIPLOIDY

NECK
TURNER SYNDROME
45 0X.
CYSTIC HYGROMA

Respiratory system(748) Congenital anomalies of respiratory system
(748.0) Choanal atresia

RESPIRATORY SYSTEM
CONGENITAL
DIAPHRAGMATIC
HERNIA
PLEURAL
EFFUSION
SEQUESTRATION OF LUNG
CYSTIC ADENOMATOID
MALFORMATION
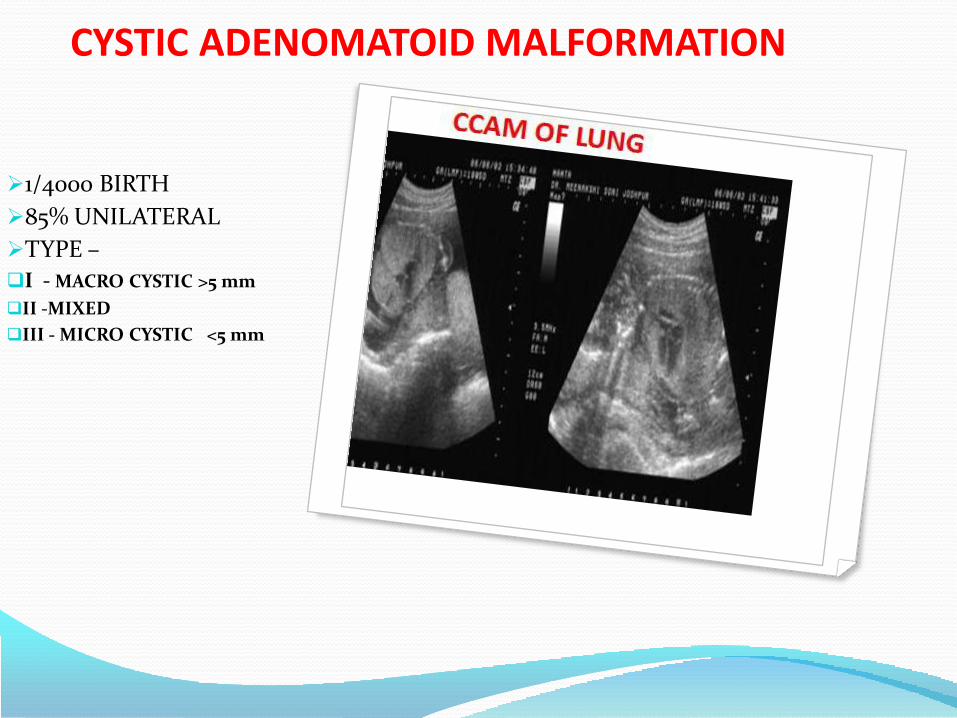
CYSTIC ADENOMATOID MALFORMATION
1/4000 BIRTH
85% UNILATERAL
TYPE –
I - MACRO CYSTIC >5 mm
II -MIXED
III - MICRO CYSTIC <5 mm

PULMONARY ABNORMALITIES
CYSTIC ADENOMATOID MALFORMATION (CAM)
Cystic adenomatoid malformation of the lung is a developmental
abnormality arising from an overgrowth of the terminal respiratory
bronchioles.
The condition may be bilateral involving all lung tissue, but in the
majority of cases it is confined to a single lung or lobe. The lesions are
either macrocystic (cysts of at least 5mm in diameter) or microcystic
(cysts less than 5 mm in diameter).
In 85% of cases, the lesion is unilateral with equal frequency in the right
and left lungs and equal frequency in the microcystic and macrocystic
types.
Prevalence: CCAM is common 1-4/100000 birth.
Clinical Features :
New born present with respiratory distress recurrent respiratory infection
and pneumothorax.
The lesion may be confused with a diaphragmatic hernia.

Treatment : Antenatal treatment is
controversial it may include
excision of affected lobe,
aspiration of macrocystic
lesion.
In the postnatal period surgery
is indicated for all symptomatic
patients.

CONGENITAL DIAPHRAGMATIC HERNIA
• GENETIC SYNDROME
• CHROMOSOMAL
• OTHER
• SPORADIC
50%
CARNIO SPINAL DEFECT
INIENCEPHALY
CARDIAC DEFECT
FRYNS SYNDROME
MARFAN
DE LANG
SYNDROME
18 & 13T
MOSAIC
TETRASOMY12 p
PALLISTER-KILLIAN SYN

CONGENITAL DIAPHRAGMATIC HERNIA
1/4000 BIRTHS
TYPE :
RIGHT CDH LIVER HERNIATING
INTO CHEST
HEART ON LEFT SIDE OF CHEST
FETAL THERAPY:TRACHEAL OCCLUSION
EITHER BY FETSCOPE OR
BY BALLON OCCLUSION
HEART RIGHT HEMITHORAX LEFT
LIVER IN RIGHT HEMITHORAX

CONGENITAL DIAPHRAGMATIC HERNIA
1/4000 BIRTHS
TYPE :
LEFT CDH STOMACH,SPLEEN & BOWEL HERNIATING
INTO CHEST
FETAL THERAPY:Initial approach was
tracheal occlusion by clips on the trachea.
It is now performed with intra-tracheal inflatable balloon. The balloon is
inserted at 26 to 28 weeks and removed at 34 weeks.
HEART RIGHT HEMITHORAX

DIAPHRAGMATIC HERNIA
Development of the diaphragm is usually completed by the 9th
week of gestation.
In the presence of a defective diaphragm, there is herniation of the
abdominal viscera into the thorax at about 10–12 weeks, when the
intestines return to the abdominal cavity from the umbilical cord.
However, at least in some cases, intrathoracic herniation of viscera
may be delayed until the second or third trimester of pregnancy.
Prevalence:Diaphragmatic hernia is found in about 1 per 4000 births.
Clinical Features:Respiratory distress grunting use of accessory muscle and cyanosis
and child have scaphoid abdomen.

Etiology Diaphragmatic hernia is usually a sporadic abnormality.
However, in about 50% of affected fetuses there are associated
chromosomal abnormalities (mainly trisomy 18, trisomy 13 and
Pallister–Killian syndrome – mosaicism for tetrasomy 12p), other
defects (mainly craniospinal defects, including spina bifida,
hydrocephaly and the otherwise rare iniencephaly, and cardiac
abnormalities) and genetic syndromes (such as Fryns syndrome, de
Lange syndrome and Marfan syndrome).
TreatmentAggressive respiratory support it include rapid endotracheal
intubation, sedation, possibly paralysis.
Surfactant is commonly used but no study has proven.
High frequency oscillation ventilation (HFOV).
Extracorporeal membrane oxygeneration (ECMO).
Nitric oxide is used has selective vasodilator.
Surgery : Ideal time to repair after stabilization.
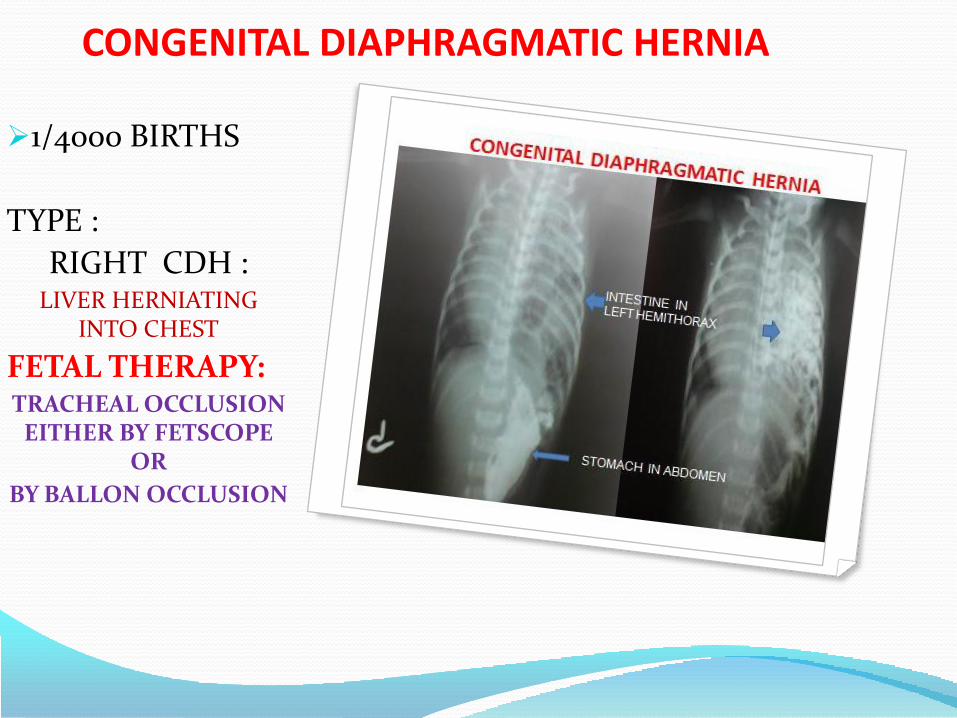
CONGENITAL DIAPHRAGMATIC HERNIA
1/4000 BIRTHS
TYPE :
RIGHT CDH :LIVER HERNIATING
INTO CHEST
FETAL THERAPY:TRACHEAL OCCLUSION
EITHER BY FETSCOPE OR
BY BALLON OCCLUSION

PLEURAL EFFUSIONUnilateral BilateralPrimarymost often chylous; often on the right
SecondaryClear; as part of non-immune hydrops
Isolatedusually associated with an underlying structural anomaly: pulmonary
lymphangiectasia cystic
adenomatoidmalformation of the lung
bronchopulmonary sequestration
diaphragmatic hernia
chest wall hamartoma
pulmonary vein atres
incidence of about one per 1,000 pregnancies.
Associated with other manifestations of hydrops subcutaneo
us skin oedema
pericardial effusion
ascites

PLEURAL EFFUSIONOne option in the management offetuses with pleural effusion isthoracocentesis and drainage ofthe effusions. However, in themajority of cases the fluidreaccumulates within 24-48 hoursrequiring repeated procedures andit is therefore preferable to achievechronic drainage by the insertionof pleural-amniotic shunts. Theclinical course of primary fetalhydrothorax is unpredictable.Whereas smaller unilateraleffusions might remain stable oreven regress, this is rarely the casewith larger collections. Bilateral
effusions, hydrops, pretermdelivery and the lack of antenataltherapy are all associated with pooroutcome. Once structural andchromosomal anomalies have beenexcluded, optimal managementdepends on gestational age, rate ofprogression, the development of
hydrops and associated maternalsymptoms. For very large effusionswith mediastinal shift, hydropsand/or hydramnios, or when there
is rapid enlargement of theeffusion, fetal intervention iswarranted

CHEST WALL LYMPHATIC HYGROMACongenital lymphangioma is amalformation of the lymphaticsystem. Althoughhistologically it is a benigndisorder, it has a propensity forrapid growth and localinvasion into the muscle,bone, and underlying tissue,and it may lead to a decreasedquality of life. Thislymphangioma can occur in
various anatomic locations,such as the axilla, the anteriorabdominal wall, and theextremities.
Chest wall lymphangioma,however, seems to be acompletely different disease,and prenatal diagnosis of thiscondition is rare.

CHEST WALL LYMPHATIC HYGROMA & LEG HAEMNGIOMA
The findings may be unilocular ormultilocular, and the lesions rangein size from several millimeters tomuch larger and contain a clear orcloudy lymphoid fluid.Lymphangiomas are believed to becaused by the anomalousdevelopment of the lymphaticsystem; the etiology is variable,probably multigenic.Lymphangiomas are made up oflymphatic vessels supported byconnective tissue. Nocommunication exists between thenormal lymphatic system and thelymphangioma. Lymphangiomashave a predilection for localinfiltration of the dermis,subcutaneous tissue, and softtissue and occasionally arewidespread.
In contrast to cystic hygroma, chestlymphangioma may be a differentcongenital anomaly

CHEST WALL LYMPHATIC HYGROMAWe suggest that chest walllymphangioma should beincluded in the entity offindings of low incidence forchromosomal abnormalities.These lesions are usually notassociated with othercongenital abnormalities orgeneralized lymphedema. Theprenatal finding of chest walllymphangioma is relativelysimple and easy to diagnosesonographically, and thetreatment of choice is surgicalexcision. The outcome isrelatively favorable, with arecurrence rate of 10% to 15%,depending on the technicalpossibilities of completeremoval of the pathologictissue

digestive system
(749) Cleft palate(749.0) Cleft palate, unspec.
(749.2) Cleft palate w/ cleft lip
(750) Other congenital anomalies of upper alimentary tract(750.0) Tongue tie
(750.5) Pyloric stenosis
(751) Other congenital anomalies of digestive system(751.0) Meckel's diverticulum
(751.2) Imperforate anus
(751.3) Hirschsprung's disease

ABDOMEN
EXOMPHALOS 1/4000
GASTROCHESIS 1/4000
BODY STALK ANOMALY 1/10,000
BADDER EXTROPHY 1/30,000
CLOACAL EXTORPHY 1/20,000

BODY STALK COMPLEX. . Limb body wall complex was
described for the first time by VanAllen et al. in 1987. Two of the threefollowing anomalies must be present toestablish the diagnosis:
1. Thoracic and/or abdominal celosomia.2. Exencephaly or encephalocele with afacial cleft.3. Anomalies of the extremities.
The anomaly consists of a poly-malformation syndrome with a thoraco-and/or abdomino-schisis associated withan eventration of the internal organs andanomalies of the extremities. Russo et al.in 1993 and later Cusi et al. in 1996distinguished two different phenotypesaccording to the fetoplacentalrelationships.
In the phenotype with the “cranio-placental attachment” a neural tubeclosure defect is associated with one ormore complex facial clefts and ananterior coelosomy, whereas amnioticbands are inconstant and anomalies ofthe extremities, if any, touch primarilythe upper limbs .
In the phenotype with the “abdomino-placental attachment” the authorsdescribe:
A persistence of the cavity of theextraembryonic coelom containing theexteriorized abdominal organs.

BODY STALK COMPLEX. . The umbilical cord is always
localized on the wall of this bag; itis short, non-free and isincompletely covered by theamnion.
Urogenital anomalies and thepersistence of the primitive cloaca.
Rachidian anomalies
Prevalence : is estimated tobe 0.7 in 10,000 births; 1/14000BIRTHS
IT IS A LETHAL ANOMALY, IN THEFIRST 12 WEEKS. THE ABDOMINALWALL WITH HERNIATED ORGANS IS
FUSED TO THE PLACENTA. Bodystalk anomaly is a severeabdominal wall defect causedby the failure to form a bodystalk and is characterized bythe absence of an umbilicalcord , naval & failure of fusionof the four flod of abdominalwall.THE ASSOCIATED MALFORMATIONSARE – NEURAL TUBEDEFECTS,GASTROINTESTINAL,
GENTOURINARY SYSTEM, HEART,
LIVER & LUNGS.
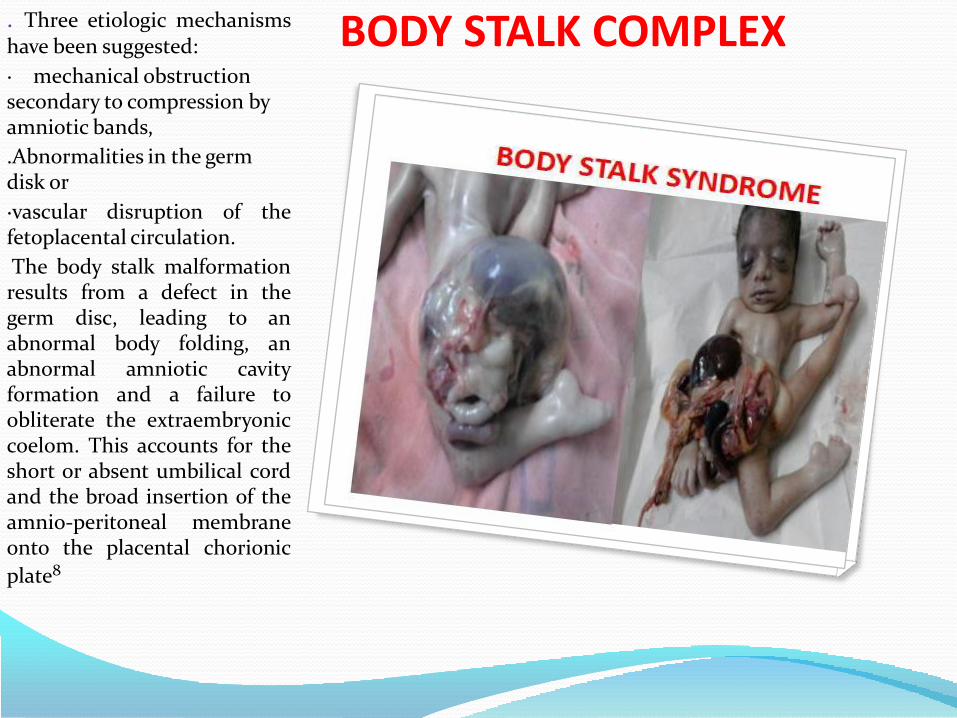
BODY STALK COMPLEX. Three etiologic mechanismshave been suggested:
· mechanical obstruction secondary to compression by amniotic bands,
.Abnormalities in the germ disk or
·vascular disruption of thefetoplacental circulation.
The body stalk malformationresults from a defect in thegerm disc, leading to anabnormal body folding, anabnormal amniotic cavityformation and a failure toobliterate the extraembryoniccoelom. This accounts for theshort or absent umbilical cordand the broad insertion of theamnio-peritoneal membraneonto the placental chorionic
plate8

BODY STALK COMPLEXClosing failure of theCephalic body fold defectslead to an anteriordiaphragmatic hernia, ectopiacordis, sternal cleft, cardiacdefects and an upper midlineomphalocele as observed inthe Pentalogy of Cantrell.
Closing failure of the caudalbody fold results in exstrophyof the bladder, imperforateanus, partial colonic agenesisand agenesis of one umbilicalartery together with ahypogastric omphalocele.
Aplasia or hypoplasia of theparaspinous or thoracolumbarmusculature is responsible forthe severe scoliosis.Insufficiency in both cephalicand caudal body folding leadsto a combination of the above-mentioned features.

EXOMPHALOS
1/4000 BIRTHS
RECURRENCE RISK 1%
50% WITH EXOMPHALOS HAVE 18/13 TRISOMY; AT 12
WEEKS OF GESTATION,30% AT MID
GESTATION & IN 15% NEONATES .
BECKWITH WIEDEMANN SYNDROME-SPORADIC, AD,AR, X LINKED AND
POLYGENIC INHERITANCE
ABDOMEN
OMPHALOCELE
OMPHALOCELE

EXOMPHALOS
LESS OFTEN ASSOCIATED WITH FAILURE IN THE CEPHALIC EMBYRONIC
FOLD-
PENTALOGY OF CANTRAL
ACRANIA
STERNAL DEFECT
ECTOPIA CORDIS
DIAPHRAGMATIC HERNIA
EXOMPHALOS
FILURE OF CAUDAL FOLD WITH EXTROPHY OF BLADDER & CLOACA,
IMPERFORATED ANUS, COLONIC ATRESIA AND
SACRAL VERTEBRAL DEFECTS
ABDOMEN
OMPHALOCELE
OMPHALOCELE

EXOMPHALOS
1/4000 BIRTHS
RECURRENCE RISK 1%
50% WITH EXOMPHALOS HAVE 18/13 TRISOMY; AT 12
WEEKS OF GESTATION,30% AT MID
GESTATION & IN 15% NEONATES .
BECKWITH WIEDEMANN SYNDROME-SPORADIC, AD,AR, X LINKED AND
POLYGENIC INHERITANCE
ABDOMEN
OMPHALOCELE
OMPHALOCELE

GASTROSCHISIS
1/4000 BIRTH
UMBILICUS IS NORMAL
INTESTINE HERNIATE THROUGH DEFECT IN
ABDOMINAL WALL JUST LATERAL & USUALLY ON
RIGHT SIDE OF THE UMBILICUS
SPORADIC ABNORMALITY
CHILD REQUIRE TOTAL PARENTRAL NUTRITION
DURING IMMEDIATE POSTNATAL PERIOD
DEATH IS AROUD 4 YEARS OF AGE DUE TO
LIVER FAILURE
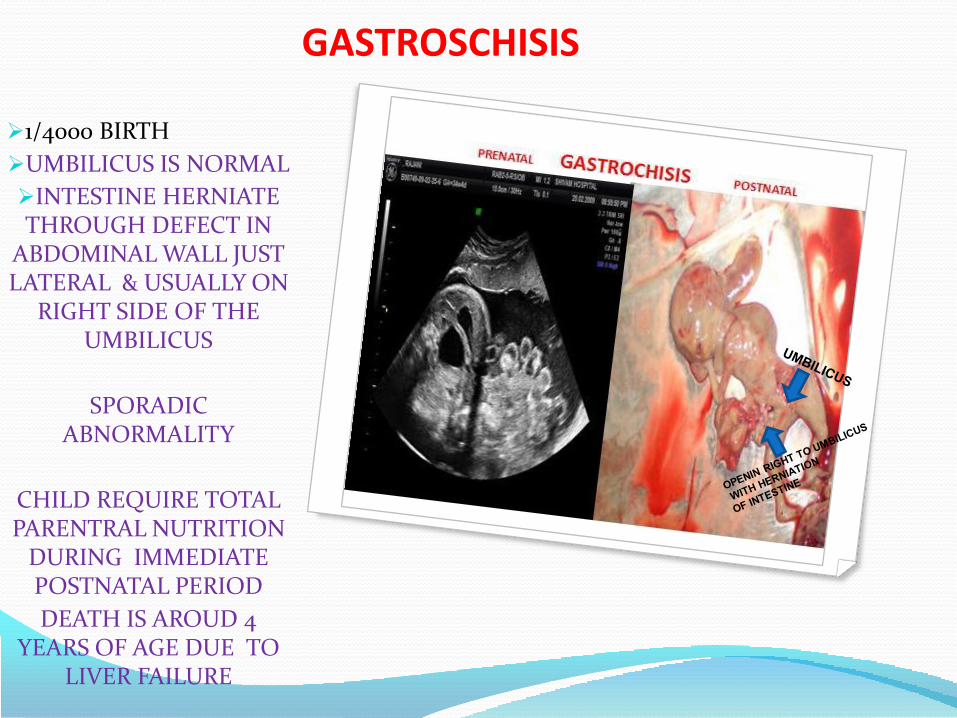
GASTROSCHISIS
1/4000 BIRTH
UMBILICUS IS NORMAL
INTESTINE HERNIATE THROUGH DEFECT IN
ABDOMINAL WALL JUST LATERAL & USUALLY ON
RIGHT SIDE OF THE UMBILICUS
SPORADIC ABNORMALITY
CHILD REQUIRE TOTAL PARENTRAL NUTRITION
DURING IMMEDIATE POSTNATAL PERIOD
DEATH IS AROUD 4 YEARS OF AGE DUE TO
LIVER FAILURE

BADDER EXTROPHY
1/3000 0BIRTH
SPORADIC ABNORMALITY
DIAGNOSIS:
NORMAL AMNIOTIC FLUID
URINARY BLADDER NOT VISUALIZED
ANECHOGENIC MASS
PROTUDING FROM LOWER ABDOMINAL WALL

CLOACAL EXTROPHY
½0,000BIRTH
SPORADIC ABNORMALITY
DIAGNOSIS:
NORMAL AMNIOTIC FLUID
URINARY BLADDER NOT VISUALIZED
ANECHOGENIC MASS
PROTUDING FROM LOWER ABDOMINAL
WALL .
POSTERIOR ANOMALOUS COMPONENT-
HERNIATED BOWEL &/OR
MENINGOMYELOCELE

Digestive system(749) Cleft palate
(749.0) Cleft palate, unspec.
(749.2) Cleft palate w/ cleft lip
(750) Other congenital anomalies of upper alimentary tract
(750.0) Tongue tie
(750.5) Pyloric stenosis
(751) Other congenital anomalies of digestive system
(751.0) Meckel's diverticulum
(751.2) Imperforate anus
(751.3) Hirschsprung's disease

GASTROINTESTINAL DEFECTS
EOSOPHAGEAL ATRESIA 1/3000 BIRTHS
DUODENAL ATRESIA 1/5000 BIRTHS
INTESTINAL OBSTRUCTION 1/2,000 BIRTHS
HIRSCHPRUNG DISEASE 1/3,000 BIRTHS
MECONIUM PERITONITIS 1/3,000 BIRTHS
HEPATIC CALCIFICATION 1/2000 BIRTHS
ANORECTAL MALFORMATION 1/4000 BIRTHS
ESOPHAGEAL WEB OR RINGEsophageal webs are thin (2-3mm) membranes of normalesophageal tissue consisting ofmucosa and submucosa. Theycan be congenital or acquired.Congenital webs commonlyappear in the middle andinferior third of the esophagus,and they are more likely to becircumferential with a centralor eccentric orifice. esophagealwebs and rings are estimatedto occur in 1 in 25,000 to 1 in50,000 live births. They aremainly observed in thePlummer-Vinson syndrome,which is associated withchronic iron deficiencyanemia. Esophageal webs areassociated with bullousdiseases (such asepidermolysis bullosa,pemphigus, and bullouspemphigoid), with graft versushost disease involving theesophagus, and with celiacdisease.

TRACHEO- ESOPHAGEAL FISTLA
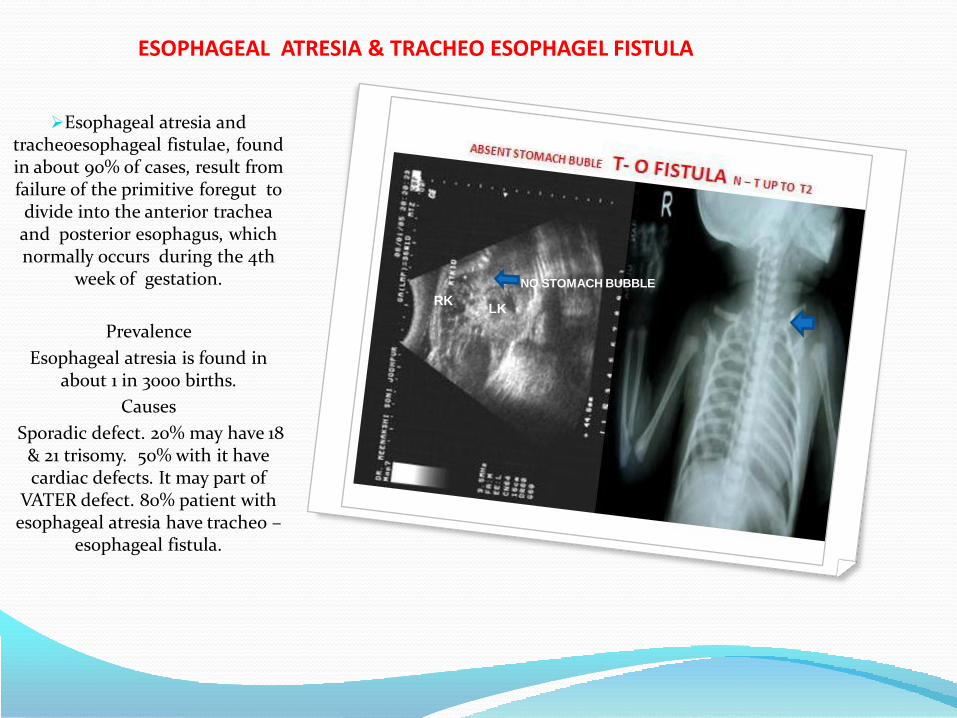
ESOPHAGEAL ATRESIA & TRACHEO ESOPHAGEL FISTULA
Esophageal atresia and tracheoesophageal fistulae, found in about 90% of cases, result from failure of the primitive foregut to divide into the anterior trachea and posterior esophagus, which normally occurs during the 4th
week of gestation.
Prevalence
Esophageal atresia is found in about 1 in 3000 births.
Causes
Sporadic defect. 20% may have 18 & 21 trisomy. 50% with it have cardiac defects. It may part of
VATER defect. 80% patient with esophageal atresia have tracheo –
esophageal fistula.
RKLK
NO STOMACH BUBBLE

ESOPHAGEAL ATRESIA & TRACHEO ESOPHAGEL FISTULA
Diagnosis
Prenatally, the diagnosis of esophageal atresia is suspected when, in the presence of polyhydramnios (usually after 25 weeks), repeated ultrasonographic examinations fail to demonstrate the fetal stomach.
Prognosis
Survival is primarily dependent on gestation at delivery and the
presence of other anomalies. Thus, for babies with an isolated
tracheoesophageal fistula, born after 32 weeks, when an early
diagnosis is made, avoiding reflux and aspiration pneumonitis, postoperative survival is more than
95%.
RKLK
NO STOMACH BUBBLE
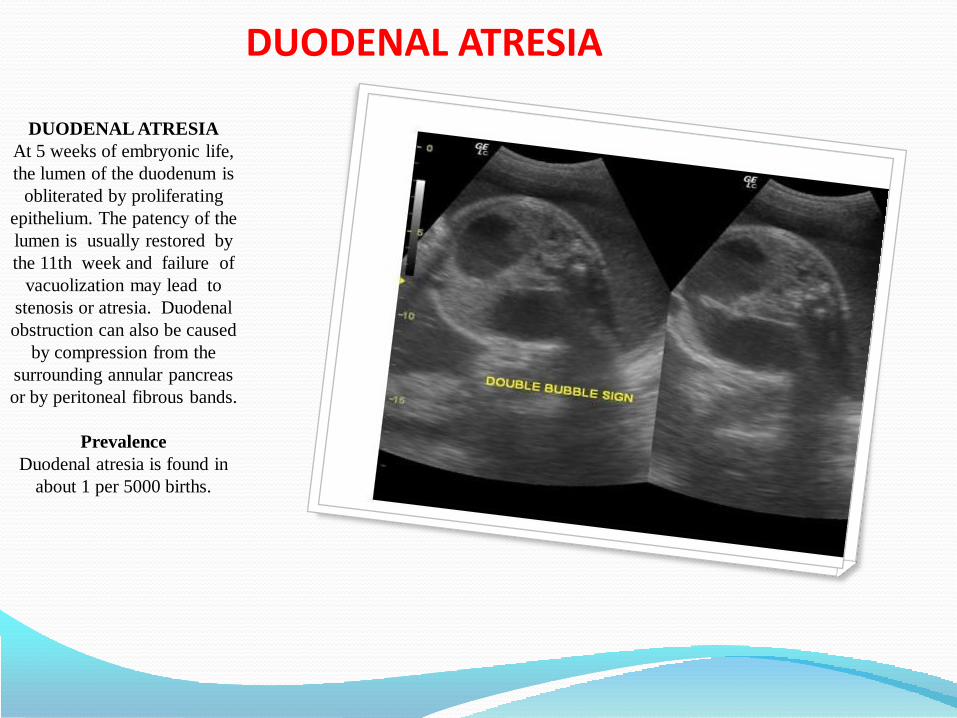
DUODENAL ATRESIA
DUODENAL ATRESIA
At 5 weeks of embryonic life,
the lumen of the duodenum is
obliterated by proliferating
epithelium. The patency of the
lumen is usually restored by
the 11th week and failure of
vacuolization may lead to
stenosis or atresia. Duodenal
obstruction can also be caused
by compression from the
surrounding annular pancreas
or by peritoneal fibrous bands.
Prevalence
Duodenal atresia is found in
about 1 per 5000 births.
DOUBLE BUBBLE

DUODENAL ATRESIAEtiology
Duodenal atresia is a sporadic abnormality, although, in some
cases, there is an autosomal recessive pattern of inheritance.
Approximately half of fetuses with duodenal atresia have associated abnormalities, including trisomy 21 (in about 40% of fetuses) and
skeletal defects (vertebral and rib anomalies, sacral agenesis, radial
abnormalities and talipes), gastrointestinal abnormalities
(esophageal atresia/tracheoesophageal fistula, intestinal malrotation, Meckel’s
diverticulum and anorectal atresia), cardiac and renal defects.
Diagnosis
Prenatal diagnosis is based on the demonstration of the
characteristic ‘double bubble’ appearance of the dilated
stomach and proximal duodenum, commonly associated
with polyhydramnios.
Prognosis
Survival after surgery in cases with isolated duodenal atresia is more
than 95%.
DOUBLE BUBBLE

INTESTINAL OBSTRUCTIONIntrinsic lesions result from
absent (atresia) or partial (stenosis) recanalization of the intestine. In cases of atresia, the two segments of the gut may be either completely separated or connected by a fibrous cord. In
cases of stenosis, the lumen of the gut is narrowed or the two
intestinal segments are separated by a septum with a central
diaphragm.. The most frequent site of small bowel obstruction is distal ileus (35%), followed by proximal jejunum (30%), distal jejunum (20%), proximal ileus (15%). In about 5% of cases, obstructions
occur in multiple sites. Anorectal atresia results from abnormal
division of the cloaca during the 9th week of development.
Prevalence
Intestinal obstruction is found in about 1 per 2000 births; in about half of the cases, there is small
bowel obstruction and in the other half anorectal atresia.
Small intestine 1/750 BIRTHS
DIALTED BOWEL
LOOP
Dx ILEAL ATRESIA
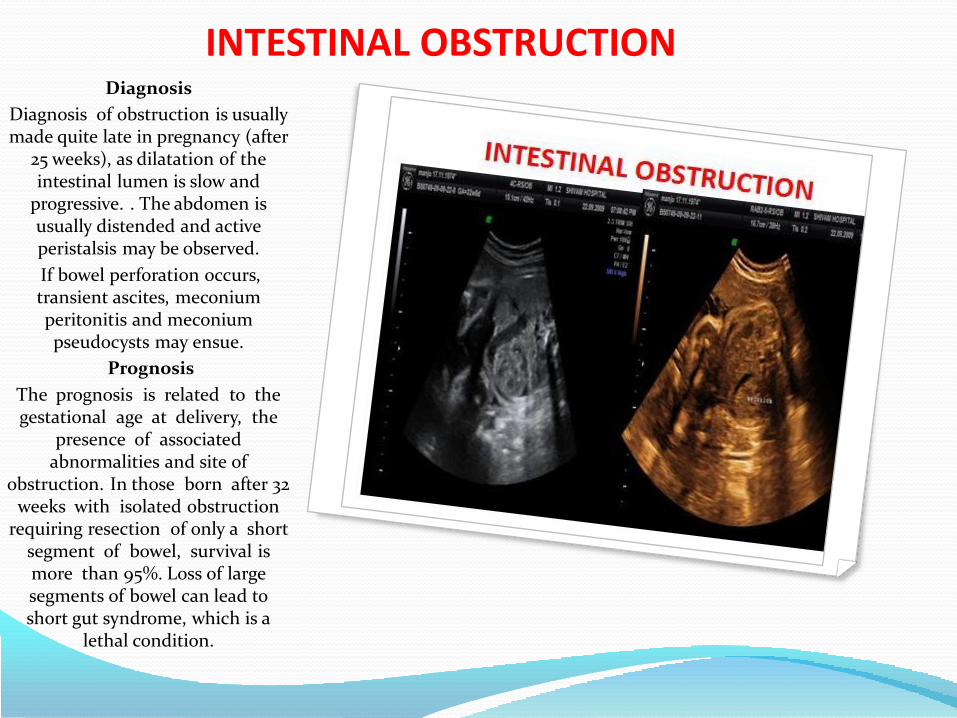
INTESTINAL OBSTRUCTIONDiagnosis
Diagnosis of obstruction is usually made quite late in pregnancy (after
25 weeks), as dilatation of the intestinal lumen is slow and
progressive. . The abdomen is usually distended and active peristalsis may be observed.
If bowel perforation occurs, transient ascites, meconium peritonitis and meconium pseudocysts may ensue.
Prognosis
The prognosis is related to the gestational age at delivery, the
presence of associated abnormalities and site of
obstruction. In those born after 32 weeks with isolated obstruction
requiring resection of only a short segment of bowel, survival is more than 95%. Loss of large segments of bowel can lead to short gut syndrome, which is a
lethal condition.

HIRSCHSPRUNG’S DISEASEHirschsprung’s disease is
characterized by congenital absence of intramural
parasympathetic nerve ganglia in a segment of the colon. It derives
from failure of migration of neuroblasts from the neural crest
to the bowel segments, which generally occurs between the 6th
and 12th weeks of gestation. Another theory suggests that the
disease is caused by degeneration of normally
migrated neuroblasts during either pre- or postnatal life.
Prevalence
1 in 3000 births.
Etiology
It is considered to be a sporadic disease, although in about 5% of
cases there is a familial inheritance. In a small number of cases, Hirschsprung’s disease is
associated with trisomy 21.
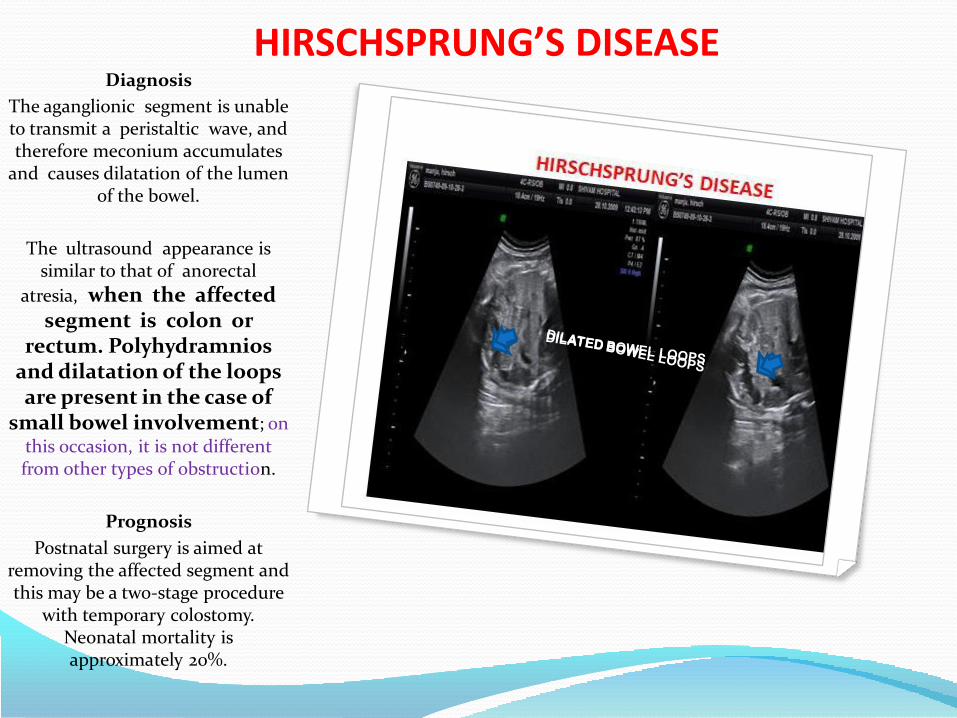
HIRSCHSPRUNG’S DISEASEDiagnosis
The aganglionic segment is unable to transmit a peristaltic wave, and therefore meconium accumulates
and causes dilatation of the lumen of the bowel.
The ultrasound appearance is similar to that of anorectal
atresia, when the affected segment is colon or
rectum. Polyhydramnios and dilatation of the loops are present in the case of
small bowel involvement; on this occasion, it is not different from other types of obstruction.
Prognosis
Postnatal surgery is aimed at removing the affected segment and this may be a two-stage procedure
with temporary colostomy. Neonatal mortality is approximately 20%.
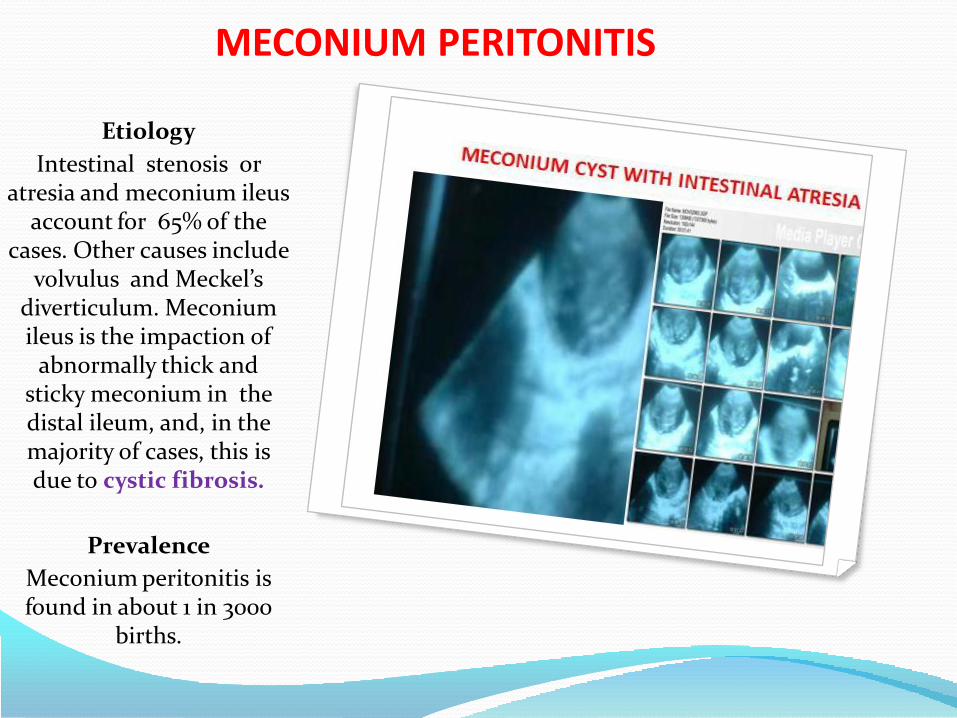
MECONIUM PERITONITIS
Etiology
Intestinal stenosis or atresia and meconium ileus
account for 65% of the cases. Other causes include
volvulus and Meckel’s diverticulum. Meconium ileus is the impaction of
abnormally thick and sticky meconium in the distal ileum, and, in the majority of cases, this is due to cystic fibrosis.
Prevalence
Meconium peritonitis is found in about 1 in 3000
births.
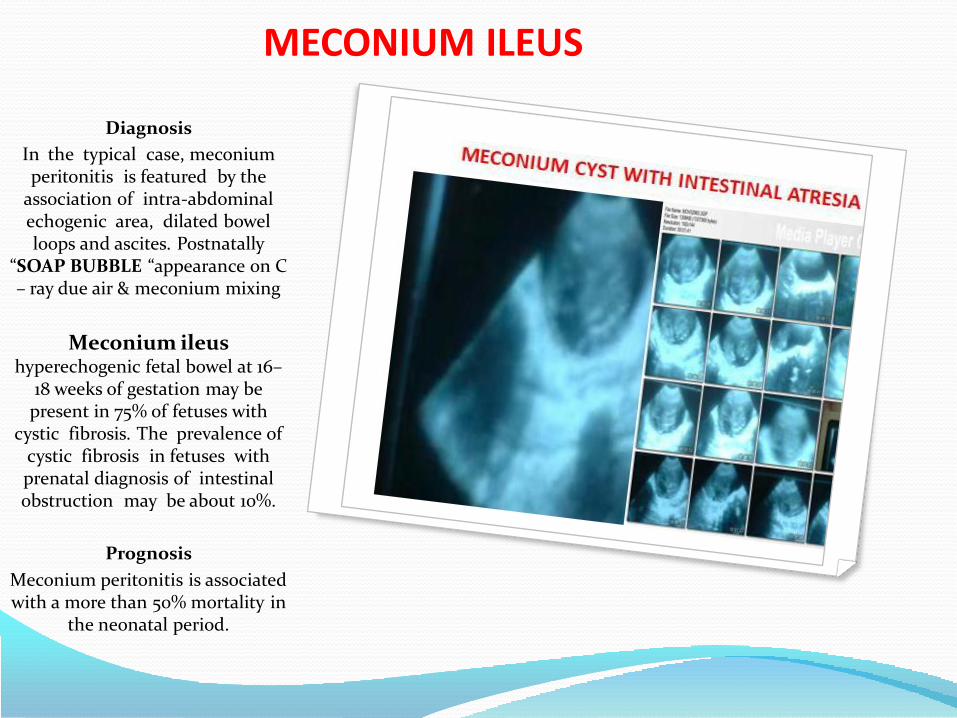
MECONIUM ILEUS
Diagnosis
In the typical case, meconium peritonitis is featured by the
association of intra-abdominal echogenic area, dilated bowel loops and ascites. Postnatally
“SOAP BUBBLE “appearance on C – ray due air & meconium mixing
Meconium ileus hyperechogenic fetal bowel at 16–
18 weeks of gestation may be present in 75% of fetuses with
cystic fibrosis. The prevalence of cystic fibrosis in fetuses with prenatal diagnosis of intestinal obstruction may be about 10%.
Prognosis
Meconium peritonitis is associated with a more than 50% mortality in
the neonatal period.

ANORECTAL MALFORMATION
.Incidence 1 in 4000.
Imperforated anus
It can be devided into low lesion where the rectum has descended through the
sphincter comlex,and high lesion where it has not.
Manifestation and diagnosis
:Low anal atresia
anal opening is covered with skin, after 24 hrs meconium bulding may be seen.
In these cases immediate perineal procedure performed followed by
dilation programm.
-High lesion
Perineum appear flat,or there may be air or meconium passed via the penis or
urethra, in this condition entering the bulbur,prostatic urethra and even the
bladder.They have good prognosis with a normal sacrum and anal dimple and
intact sphincter function.
UB

HEPATO-BILIARY SYSTEM
Fetal cholelithiasis was firstdiagnosed in 1983 and since thenthere have been only few reportsabout the presence of gallstonesin the fetus. Maternal conditions,fetal or obstetrical predisposingrisk factors have been proposedto have a causative role, but thepathogenesis of fetal gallstonesremains unknown. fetalcholelithiasis confirmed to be aself-limiting disease withoutcomplications and did notrequire any form of therapy.However, a close follow-up isindicated in these patients untilspontaneous resolution isdemonstrated by US.

HEPATO-BILIARY SYSTEMFetal cholelithiasis was firstdiagnosed in 1983 and sincethen there have been only fewreports about the presence ofgallstones in the fetus.Maternal conditions, fetal orobstetrical predisposing riskfactors have been proposed tohave a causative role, but thepathogenesis of fetal gallstonesremains unknown. fetalcholelithiasis confirmed to be aself-limiting disease withoutcomplications and did notrequire any form of therapy.However, a close follow-up isindicated in these patients untilspontaneous resolution isdemonstrated by US.

ABDOMINAL CYSTCholedochal,uncommoncongenital disordercharacterized by a globular orfusiform dilatation of thecommon bile duct just belowthe site of entry of the cysticduct. It is detected with anincidence of 0.2 - 0.5% per1.000.000 population. There isa 3:1 female predominance.Type I choledocal cyst refers toaneurysmal dilatation of thecommon bile duct oftenaccompanied by distalnarrowing. It accounts for80% - 90% of cases.
Types II choledochocoelewhich involves only theintraduodenal portion of themain bile duct and accountsfor 1 - 5% of cases. thecommon bile duct terminatesinto the choledochocoelewhich drains into theduodenum via an aperture inits wall. The main pancreaticduct may also empty into thecholedochocoele.

ABDOMINAL CYSTType III occurs when the
common bile duct entersnormally into the papilla andthe choledococoele fills andempties directly into thecommon bile duct. In thisvariant the pancreatic ductusually enters into the mainbile duct proximally to thecholedochocoele.
Type IV refers to multiplecysts. Type IVa refers tomultiple intrahepatic cysts andan extrahepatic cyst and TypeIVb refers to multipleextrahepatic cysts.
Type V refers to Carolis diseasecomplete biliary tree isinvolved.
CHOLEDOCAL CYST
MESENTRIC CYST
OMENTAL CYST
INTTESTINAL
DUPLICATION CYST
OVARIAN CYST
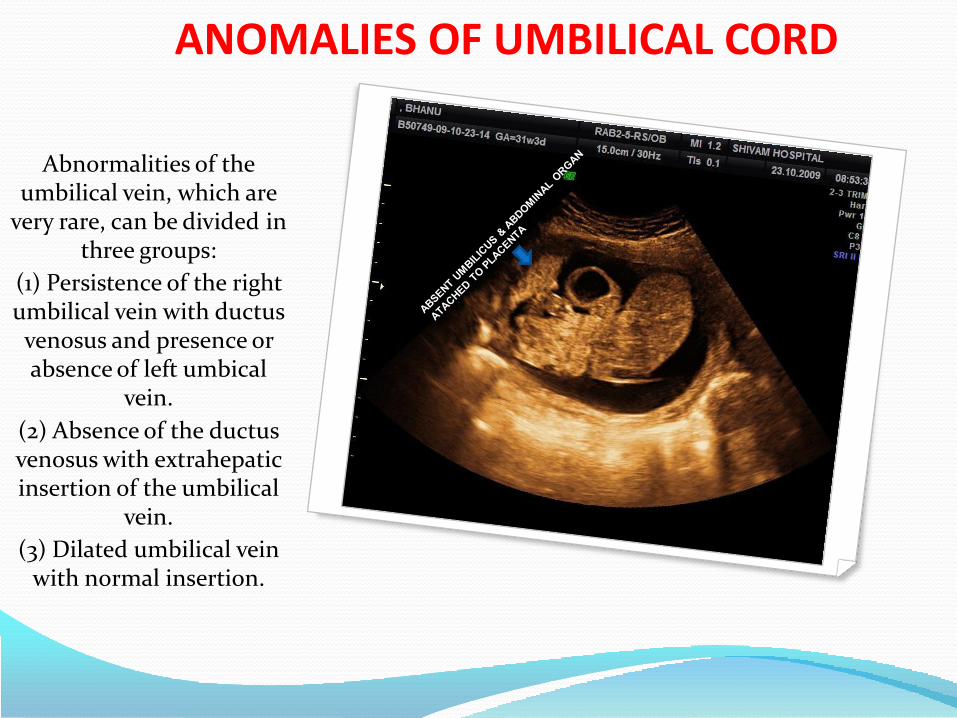
ANOMALIES OF UMBILICAL CORD
Abnormalities of the umbilical vein, which are
very rare, can be divided in three groups:
(1) Persistence of the right umbilical vein with ductus venosus and presence or absence of left umbical
vein.
(2) Absence of the ductus venosus with extrahepatic insertion of the umbilical
vein.
(3) Dilated umbilical vein with normal insertion.

PENTALOGY OF CANTERLLPentalogy of Cantrell is a
condition in which a persontypically has two or three of thefollowing birth defects, with veryfew people having all five findings:(1) a deficiency of the front part ofthe diaphragm (the thin layerof muscle underneath the lungsinvolved in breathing); (2) a defectof the middle part of the abdomenabove the belly button; (3) a defectin the pericardium (the outerlayer) of the diaphragm; (4) variouscongenital (present at birth) heartabnormalities; and (5) a defect ofthe lower part of the sternum(breastbone). The condition isbelieved to be caused by a failure in
development that occurs when the
fetus is about 14-18 days old.Treatment is based on thesymptoms present in thepersonCephalic body fold defectslead to an anterior diaphragmatichernia, ectopia cordis, sternal cleft,cardiac defects and an uppermidline omphalocele as observedin the Pentalogy of Cantrell

Urinary system(753) Congenital anomalies of urinary system
(753.0) Renal agenesis and dysgenesis
(753.1) Cystic kidney disease
(753.2) Obstructive defects of renal pelvis and ureter
(753.3) Other specified anomalies of kidneyRenal ectopia
Horseshoe kidney
(753.4) Other specified anomalies of ureter
Ectopic ureter
(753.5) Exstrophy of urinary bladder
(753.6) Atresia and stenosis of urethra and bladder neck
(753.7) Anomalies of urachus
Urachal cyst
(753.8) Other specified anomalies of bladder and urethra
(753.9) Unspecified anomaly of urinary system

KIDNEY AND URINARY TRACT
RENAL AGENESIS BILATERAL
1/5000 BIRTHS
RENAL AGENESIS UNILATERAL
1/2000 BIRTHS
INFANTILE POLCYSTIC KIDNEY 1/30,000 BIRTHS
MULTICYSTIC DYSPLASTIC KIDNEY DISEASE
1/1,000 BIRTHS
ADULTPOLCYSTIC KIDNEY DISEASE
1/1,000 BIRTHS
URETERO –PELVIC JUNCTION OBSTRUCTION
VESICO URETERIC REFLUX
POSTERIOR URETHRAL VALVE 1/3000 BIRTH
RECRRENCE RISK 3%
AR ; RECURRENCE RISK 25%

RENAL AGENESIS
Renal agenesis
is the consequence of failure of differentiation of the metanephric blastema during the 25–28th day of development and both ureters and
kidneys and renal arteries are absent.
Prevalence
Bilateral renal agenesis is found in 1 per 5000 births, while unilateral disease is
found in 1 per 2000 births.
Etiology
Renal agenesis is usually an isolated sporadic abnormality but, in a few
cases, it may be secondary to a chromosomal abnormality or part of a
genetic syndrome (such as Fraser syndrome), or a developmental defect
(such as VACTERL association). In non-syndromic cases, the risk of
recurrence is approximately 3%.However, in about 15% of cases, one of
the parents has unilateral renal agenesis and in these families the risk of
recurrence is increased.
SPS

RENAL AGENESIS
Diagnosis
Antenatally, the condition is suspected by the combination of anhydramnios
(from 17 weeks) and empty fetal bladder (from as early as 14 weeks).
Examination of the renal areas is often hampered by the
oligohydramnios and the ‘crumpled’ position adopted by these fetuses, and
care should be taken to avoid the mistaken diagnosis of perirenal fat and
large fetal adrenals for the absent kidneys..
Prognosis
Bilateral renal agenesis is a lethal condition, usually in the neonatal
period due to pulmonary hypoplasia.
The prognosis with unilateral agenesis is normal.
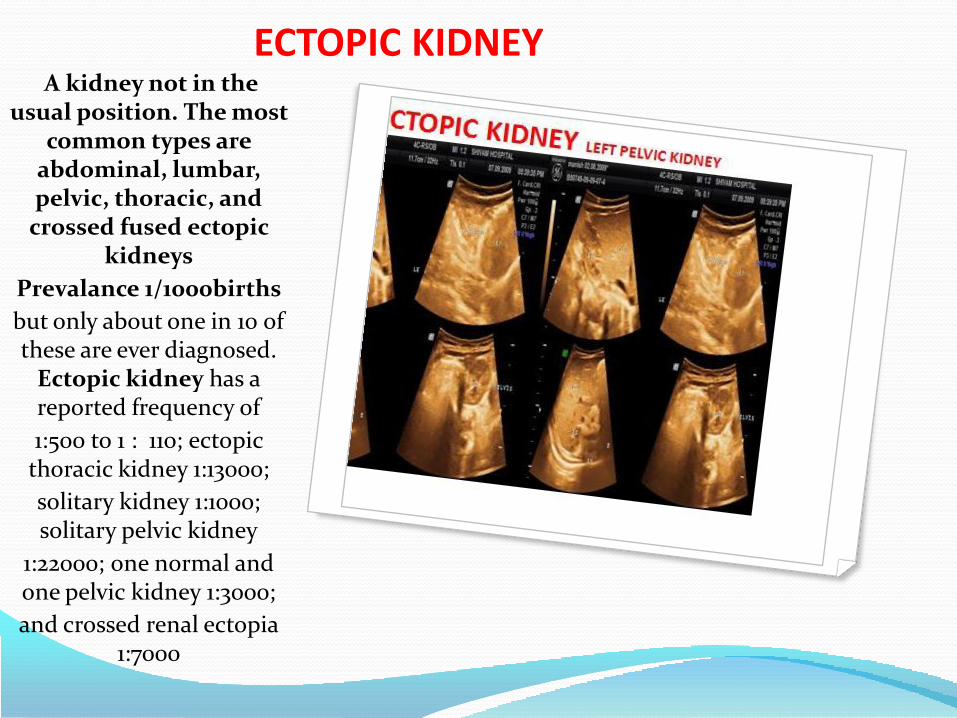
ECTOPIC KIDNEYA kidney not in the
usual position. The most common types are
abdominal, lumbar, pelvic, thoracic, and
crossed fused ectopic kidneys
Prevalance 1/1000births
but only about one in 10 of these are ever diagnosed.
Ectopic kidney has a reported frequency of
1:500 to 1 : 110; ectopic thoracic kidney 1:13000;
solitary kidney 1:1000; solitary pelvic kidney
1:22000; one normal and one pelvic kidney 1:3000;
and crossed renal ectopia 1:7000

ECTOPIC KIDNEYSometimes the kidney will not
form at all, and never develops, and is called renal agenesis.
Sometimes the kidney tissue forms, but forms poorly and forms a scarred and poorly functioning
kidney, known as renal dysplasia or as a multi-cystic dysplastic
kidney (MCDK).
Sometimes these kidneys will stay fairly big and can be fairly easily
seen and followed.
Other times they will shrink down and atrophy and involute and may more or less completely disappear.
Sometimes the kidney tissue will form normally, but the kidney will
be small in size, known as a hypoplastic kidney.
Sometimes the kidney tissue will form normally, and the kidneys will be normal size, but the two
kidneys may migrate abnormally or may be fused together known as
ectopic kidneys or as a horseshoe kidney.
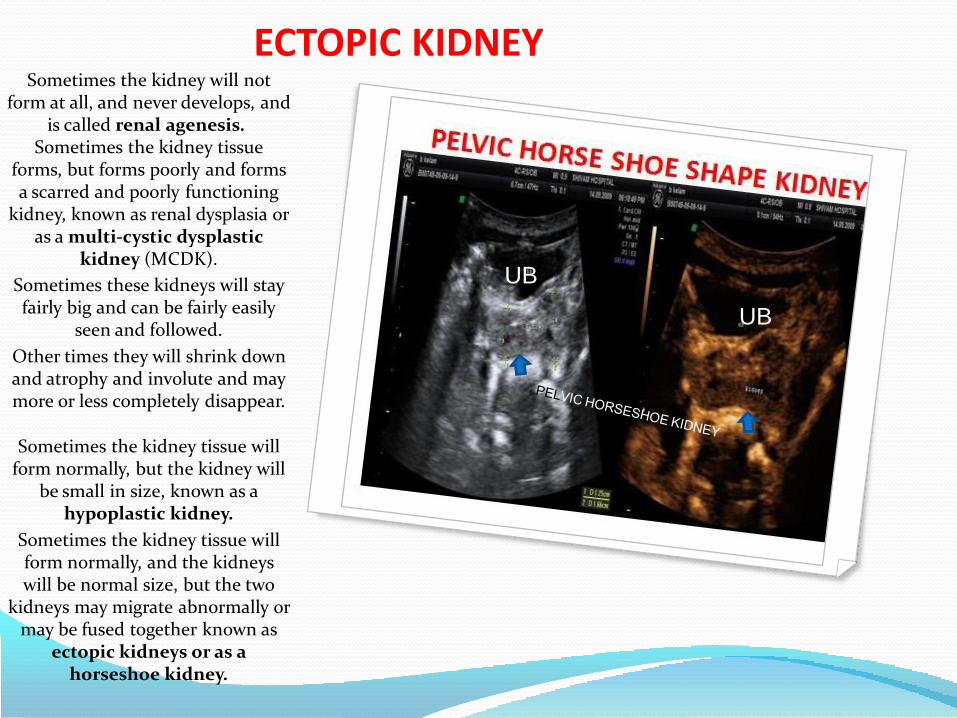
ECTOPIC KIDNEYSometimes the kidney will not
form at all, and never develops, and is called renal agenesis.
Sometimes the kidney tissue forms, but forms poorly and forms a scarred and poorly functioning
kidney, known as renal dysplasia or as a multi-cystic dysplastic
kidney (MCDK).
Sometimes these kidneys will stay fairly big and can be fairly easily
seen and followed.
Other times they will shrink down and atrophy and involute and may more or less completely disappear.
Sometimes the kidney tissue will form normally, but the kidney will
be small in size, known as a hypoplastic kidney.
Sometimes the kidney tissue will form normally, and the kidneys will be normal size, but the two
kidneys may migrate abnormally or may be fused together known as
ectopic kidneys or as a horseshoe kidney.
UB
UB

INFANTILE POLYCYSTIC DISEASE (POTTER TYPE I)
Prevalence
Infantile polycystic kidney disease is found
in about 1 per 30 000 births.
Etiology
This is an autosomal recessive condition. The
responsible gene is in the short arm of
chromosome 6 and prenatal diagnosis in families at risk can be
carried out by first-trimester chorion villous
sampling.

INFANTILE POLYCYSTIC DISEASE (POTTER TYPE I)
Diagnosis
Prenatal diagnosis is based on the demonstration of bilaterally
enlarged and homogeneously hyperechogenic kidneys. There is often associated oligohydramnios, but this is not invariably so. These
sonographic appearances, however, may not become apparent before
24 weeks of gestation and, therefore, serial scans should be performed for exclusion of the
diagnosis.

INFANTILE POLYCYSTIC DISEASE (POTTER TYPE I)
Prognosis
The perinatal type is lethal either in utero or in the neonatal period
due to pulmonary hypoplasia.
The neonatal type results in death due to renal failure within the 1st
year of life.
The infantile and juvenile types result in chronic renal failure, hepatic fibrosis and portal
hypertension; many cases survive into their teens and require
renal transplantation.

MULTICYSTIC DYSPLASTIC KIDNEY DISEASE (POTTER TYPE II)
Multicystic dysplastic kidney disease is thought to be a
consequence of either developmental failure of the mesonephric blastema to form
nephrons or early obstruction due to urethral or ureteric atresia. The collecting tubules become cystic
and the diameter of the cysts determines the size of the
kidneys, which may be enlarged or small.
Prevalence
Multicystic dysplastic kidney disease is found in about 1 per 1000
births.
unilateral ½,500-4,500 children
Bilateral 25,000 children
Etiology
In the majority of cases, this is a sporadic abnormality but
chromosomal abnormalities (mainly trisomy 18), genetic syndromes and other defects
(mainly cardiac) are present in about 50% of the cases.
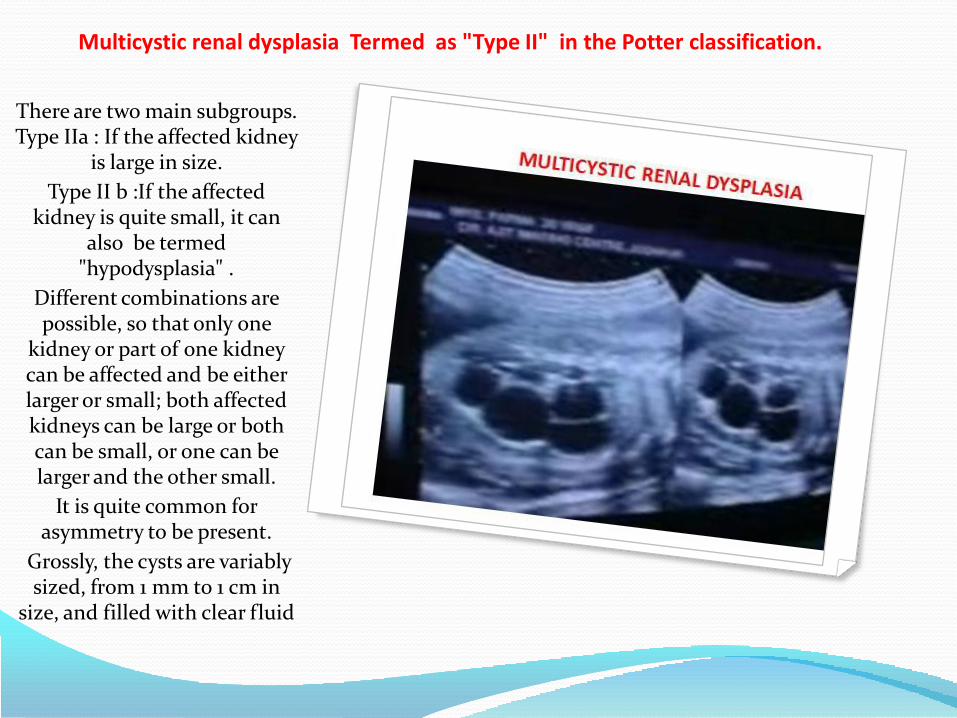
Multicystic renal dysplasia Termed as "Type II" in the Potter classification.
There are two main subgroups. Type IIa : If the affected kidney
is large in size.
Type II b :If the affected kidney is quite small, it can
also be termed "hypodysplasia" .
Different combinations are possible, so that only one
kidney or part of one kidney can be affected and be either larger or small; both affected kidneys can be large or both can be small, or one can be larger and the other small.
It is quite common for asymmetry to be present.
Grossly, the cysts are variably sized, from 1 mm to 1 cm in
size, and filled with clear fluid

MULTICYSTIC DYSPLASTIC KIDNEY DISEASE (POTTER TYPE II)There are few recognizable
glomeruli and tubules microscopically, and the remaining
glomeruli are not affected by the cystic change.
The hallmark of renal dysplasia is the presence of "primitive ducts" lined by cuboidal to columnar epithelium and
surrounded by a collagenous stroma.
This increased stroma may contain small islands of cartilage. The liver will not show congenital hepatic
fibrosis.
Multicystic renal dysplasia is often the only finding, but it may occur in combination with other anomalies
and be part of a syndrome (e.g., Meckel-Gruber syndrome), in which
case the recurrence risk will be defined by the syndrome.
If this disease is bilateral, the problems associated with
oligohydramnios are present, with pulmonary hypoplasia .

Dominant Polycystic Kidney Disease (DPKD)
This condition is inherited in an autosomal dominant pattern, so the recurrence risk in affected families is 50%. However,
this disease rarely manifests itself before
middle age.
If DPKD is manifested in fetuses and infants, the cysts may involve the gomeruli (so-called "glomerular cysts").
It is possible in some cases for the liver to be
more severely affected, so that hepatic failure results.
Patients with DPKD are also prone to have berry
aneurysms of the cerebral arteries.

Cystic Change with ObstructionWhen the obstruction is complete and occurs early in fetal life, renal
hypoplasia (deficiency in total nephron population) and dysplasia (Potter type II; formation of abnormal
nephrons and mesenchymal stroma)
ensue.
On the other hand, where intermittent obstruction allows for normal renal
development, or when it occurs in the second half of pregnancy, hydronephrosis will result and the severity of the renal damage will depend on the degree and duration of the
obstruction.

Cystic Change with ObstructionGrossly, this form of
cystic disease may not be apparent. The cysts may be no more than 1 mm in size.
The cysts may be more than 1 mm in size.
Microscopically, the cysts form in association with
the more sensitive developing glomeruli in the nephrogenic zone so
that the cysts tend to be in a cortical location.
Thus, "cortical microcysts" are the
hallmark of this form of cystic disease.
if the obstruction is at the bladder outlet,
oligohydramnios with pulmonary hypoplasia can
result.

Medullary sponge kidney (MSK) It is a congenital
condition that most often occurs sporadically, without a defined
inheritance pattern.
It is often bilateral, but incidental and found only
on radiologic imaging studies, with an incidence
of 0.5 to 1% in adults.
MSK may become symptomatic in young adults, with onset of recurrent hematuria and/or urinary tract
infection as a consequence of formation of calculi,
which develop in 60% of cases. Renal failure is
unlikely to occur, but may result from severe
pyelonephritis.

HYDRONEPHROSISVarying degrees of
pelvicalyceal dilatation are found in about 1% of
fetuses.
Transient hydronephrosis may be due to relaxation of smooth muscle of the urinary tract by the high
levels of circulating maternal hormones, or
maternal–fetal overhydration. In the majority of cases, the
condition remains stable or resolves in the neonatal period.

HYDRONEPHROSIS. Mild hydronephrosis or
pyelectasia
is defined by the presence of an anteroposterior
diameter of the pelvis is
4 mm at 15–19 weeks,
> 5 mm at 20–29 weeks and
> 7 mm at 30–40 weeks.
Moderate hydronephrosis
Is characterized by an anteroposterior pelvic diameter of more than
10 mm and pelvicalyceal dilatation, is usually
progressive and in more than 50% of cases surgery
is necessary during the first 2 years of life.

URETEROPELVIC JUNCTION OBSTRUCTIO
In about 20% of cases underlying cause may be ureteropelvic junction
obstruction or vesicoureteric reflux.
This is usually sporadic.
Although in some cases there is an anatomic
cause, such as ureteral valves.
In 80% of cases, the condition is unilateral.

URETEROPELVIC JUNCTION OBSTRUCTIOPrenatal diagnosis is based
on the demonstration of hydronephrosis in the
absence of dilated ureters and bladder.
The degree of pelvicalyceal dilatation is variable and, occasionally, perinephric
urinomas and urinary ascites may be present.
Spotaneous resolution of it occur in 50-70% of all fetuses during antenatal period or in postnatal life upto three years.
Postnatally, renal function is assessed by serial isotope
imaging studies and, if there is deterioration, pyeloplasty is
performed. About 20% of cases, requires postnatal follow-up and possible
surgery.
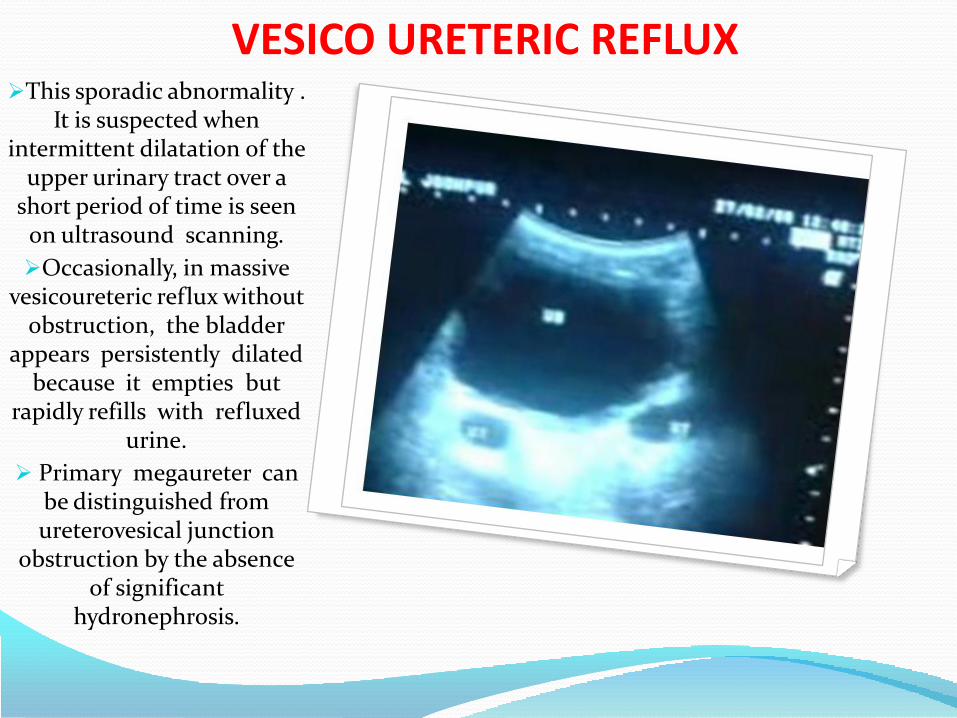
VESICO URETERIC REFLUXThis sporadic abnormality .
It is suspected when intermittent dilatation of the
upper urinary tract over a short period of time is seen
on ultrasound scanning.
Occasionally, in massive vesicoureteric reflux without
obstruction, the bladder appears persistently dilated
because it empties but rapidly refills with refluxed
urine.
Primary megaureter can be distinguished from ureterovesical junction
obstruction by the absence of significant
hydronephrosis.

URETERO VESICLE JUNCTION OBSTRUCTIONThis is a sporadic
abnormality characterized by hydronephrosis and
hydroureter in the presence of a normal bladder.
The etiology is diverse, including ureteric stricture or
atresia, retrocaval ureter, vascular obstruction, valves,
diverticulum, ureterocele, and vesicoureteral reflux.
Ureteroceles visible as a thin-walled and fluid-filled small
circular area inside the bladder; are usually found in association with duplication of the collecting system. In
ureteral duplication, the upper pole moiety
characteristically obstructs and the lower one refluxes. The dilated upper pole may enlarge to displace the non-dilated lower pole inferiorly
and laterally.

URETHRAL OBSTRUCTIONUrethral obstruction can be caused by urethral agenesis,
persistence of the cloaca, urethral stricture or posterior
urethral valves.
Posterior urethral valves occur only in males and are
the commonest cause of bladder outlet obstruction. The condition is sporadic.
Prevalence is about 1 in 3000 male fetuses. With posterior
urethral valves, there is usually incomplete or
intermittent obstruction of the urethra, resulting in an enlarged and hypertrophied
bladder with varying degrees of hydroureters,
hydronephrosis, a spectrum of renal hypoplasia and
dysplasia, oligohydramnios and pulmonary hypoplasia.
RK LK RKLK
UB
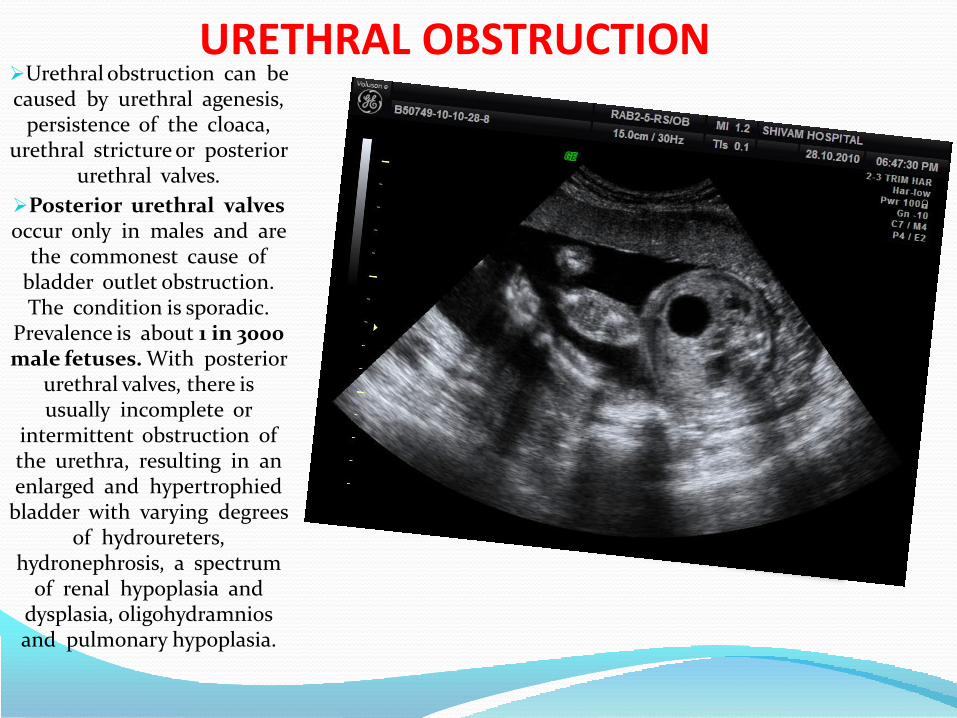
URETHRAL OBSTRUCTIONUrethral obstruction can be caused by urethral agenesis,
persistence of the cloaca, urethral stricture or posterior
urethral valves.
Posterior urethral valves occur only in males and are
the commonest cause of bladder outlet obstruction. The condition is sporadic.
Prevalence is about 1 in 3000 male fetuses. With posterior
urethral valves, there is usually incomplete or
intermittent obstruction of the urethra, resulting in an enlarged and hypertrophied
bladder with varying degrees of hydroureters,
hydronephrosis, a spectrum of renal hypoplasia and
dysplasia, oligohydramnios and pulmonary hypoplasia.
RK LK RKLK
UB

URETHRAL OBSTRUCTIONUrethral obstruction can be caused by urethral agenesis,
persistence of the cloaca, urethral stricture or posterior
urethral valves.
Posterior urethral valves occur only in males and are
the commonest cause of bladder outlet obstruction. The condition is sporadic.
Prevalence is about 1 in 3000 male fetuses. With posterior
urethral valves, there is usually incomplete or
intermittent obstruction of the urethra, resulting in an enlarged and hypertrophied
bladder with varying degrees of hydroureters,
hydronephrosis, a spectrum of renal hypoplasia and
dysplasia, oligohydramnios and pulmonary hypoplasia.
RK LK RKLK
UB

FETAL TUMORSThe most common site oforigin of fetal tumors areheart, face and neck andabdomen.
The most common site oforigin of tumor is the heart(20/84, 23.8%), followed bythe face and neck region(19/84, 22.6%) and theabdomen (16/84, 19%).
Lymphangiomas (21/84, 25%)and rhabdomyomas (19/84,22.6%) comprised half of thetumor histologically. Lessfrequently, teratomas (14/84,16.6%) and hemangiomas(12/84, 14.2%) were seen.
Heart tumor isRhabdomyoma. The birthprevalence is 1 per 10,000.In 50% tumor is associatedwith Tuberous sclerosis(AD).
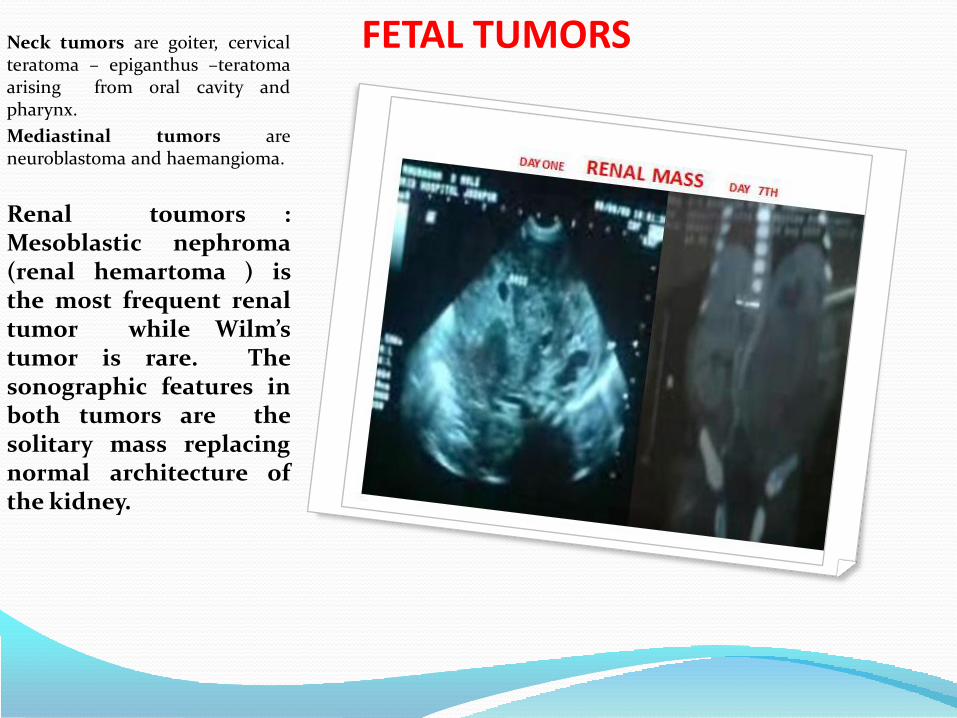
FETAL TUMORSNeck tumors are goiter, cervicalteratoma – epiganthus –teratomaarising from oral cavity andpharynx.
Mediastinal tumors areneuroblastoma and haemangioma.
Renal toumors :Mesoblastic nephroma(renal hemartoma ) isthe most frequent renaltumor while Wilm’stumor is rare. Thesonographic features inboth tumors are thesolitary mass replacingnormal architecture ofthe kidney.

FETAL TUMORSNeck tumors are goiter, cervicalteratoma – epiganthus –teratomaarising from oral cavity andpharynx.
Mediastinal tumors areneuroblastoma and haemangioma.
Renal toumors :Mesoblastic nephroma(renal hemartoma ) isthe most frequent renaltumor while Wilm’stumor is rare. Thesonographic features inboth tumors are thesolitary mass replacingnormal architecture ofthe kidney.
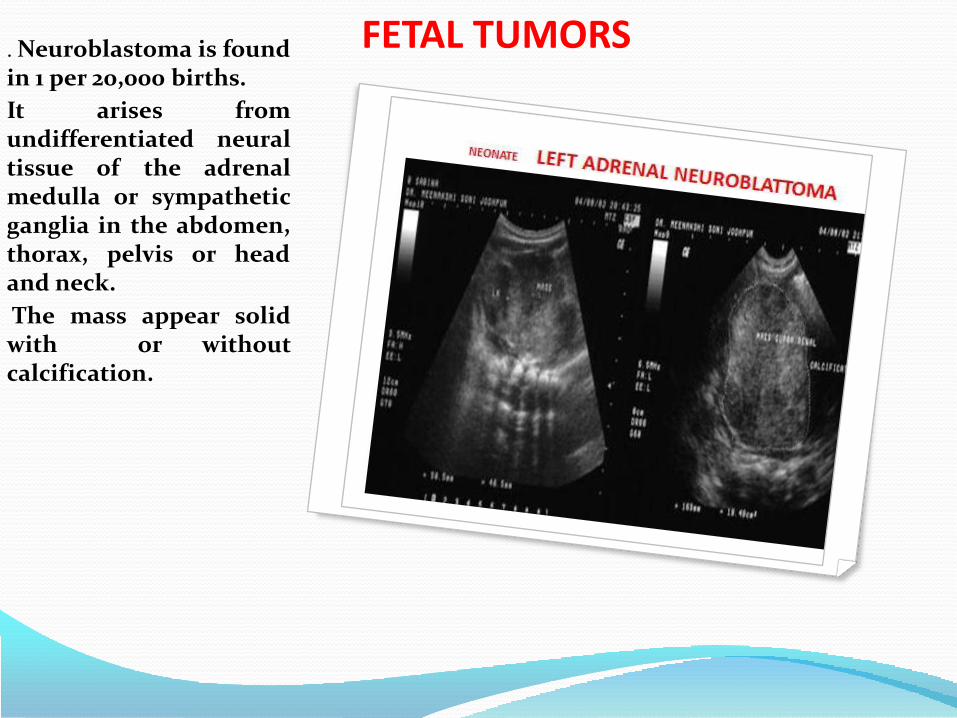
FETAL TUMORS. Neuroblastoma is foundin 1 per 20,000 births.
It arises fromundifferentiated neuraltissue of the adrenalmedulla or sympatheticganglia in the abdomen,thorax, pelvis or headand neck.
The mass appear solidwith or withoutcalcification.

CHOROID PLEXUS PAPILLOMAIntracranial tumorsare teratomas,epidermoid, dermoid,meduloblastoma,lipoma of corpuscallosum, choroidplexus papilloma etc.

SACROCOCCYGEAL TERATOMA
Tumors of the skin :
Sacrococcygeal teratoma –
It is found in about 1 per40.000 births. Female arefour times morecommonly affected thanmales, but malignantchanges is more commonin males.

LYMPHANGIOMA
Tumors of theextrimities: arevascular hamartoma,hemangioma,lymphangioma andsarcoma.

.
.

Genital organs(752) Congenital anomalies of genital organs
(752.4) Anomalies of cervix, vagina, and external female genitalia
(752.42) Imperforate hymen
(752.5) Undescended testicle
(752.6) Hypospadias and epispadias
(752.61) Hypospadias
(752.62) Epispadias
(752.63) Congenital chordee
(752.64) Micropenis

BICORNUATE UTERUSMüllerian duct anomalies in thefemale patient include a lack ofdevelopment (hypoplasia,aplasia, or the unicornuateuterus), lack of fusion at themidline (didelphys orbicornuate uterus), or lack ofresorption of midline tissueafter fusion (septate and arcuateuteri). Those anomalies thatresult in the appearance of 2symmetric endometrial cavities(septate, bicornuate, anddidelphys uteri) may be difficultto distinguish. This distinctionis important if infertilitywarrants surgical repair. Nosurgery is undertaken indidelphys uteri, whereas theapproach in bicornuate uteri islaparoscopic and hysteroscopicin septate uteri.
Uterine didelphys is completenonfusion of the müllerianducts with 2 uteri, 2 cervices,and duplication of the upperthird of the vagina.

BICORNUATE UTERUSThe bicornuate uterus resultsfrom partial nonfusion with 1cervix and vagina. In a septateuterus, there is complete fusionwith a single uterus, but lack ofresorption of the midlineseptum. Septal resorptionoccurs from caudal to cranialand may be arrested at anypoint, resulting in variablelength of the septum. In themost severe case, the septumextends to the level of thecervix, or even perhaps theupper vagina, resulting in asimilar appearance to adidelphys uterus.
Characterization of theintervening uterine tissue andpresence or absence of cervicalduplication are not reliabledistinguishing features. The keyto diagnosis is the contour ofthe fundus.
The fundal contour isnormal in the septateuterus, with a deepconcavity (>1 cm) indidelphys and bicornuateuteri.

BICORNUATE UTERUSCongenital malformations of ovaries, fallopian tubes and broad ligaments
(Q51.) Congenital malformations of uterus and cervix
(Q51.0) Agenesis and aplasia of uterus
(Q51.1) Doubling of uterus with doubling of cervix and vagina
(Q51.2) Other doubling of uterus
(Q51.3) Bicornate uterus
(Q51.4) Unicornate uterus
(Q51.5) Agenesis and aplasia of cervix
(Q51.6) Embryonic cyst of cervix
(Q51.7) Congenital fistulae between uterusand digestive and urinary tracts
(Q51.8) Other congenital malformations of uterusand cervix
(Q51.9) Congenital malformation of uterusand cervix, unspecified

HYPOAPADIASPrevalence : 1 in 4000 to as high as 1 in 125 boys.
Sporadic occurrence
The urethral meatus opens onthe glans penis in about 50–75% ofcases; these are categorized as firstdegree hypospadias. Seconddegree (when the urethra opens onthe shaft), and third degree (whenthe urethra opens onthe perineum) occur in up to 20and 30% of cases respectively. Themore severe degrees are morelikely to be associatedwith chordee, in which the phallusis incompletely separated from theperineum or is still tethereddownwards by connective tissue,or with undescended testes(cryptorchidism). Up to 10% ofboys with hypospadias have atleast one undescended testis, anda similar number have an inguinalhernia. An enlarged prostaticutricle is common when thehypospadias is severe (scrotal orperineal), and can predisposeto urinary tract infections, pseudo-incontinence, or even stoneformation

Musculoskeletal system(754) Certain congenital musculoskeletal deformities
(754.1) Torticollis, sternomastoid
(754.3) Dislocation of hip, unilateral
(754.5) Varus deformities of feet
(754.51) Talipes equinovarus(754.6) Valgus deformities of feet
(754.8) Other specified nonteratogenic anomalies
(754.81) Pectus excavatum(755) Other congenital anomalies of limbs
(755.0) Polydactyly
(755.5) Other congenital anomalies of upper limb including shoulder girdle
(755.55) AcrocephalosyndactylyApert syndrome
(755.9) Limb anomaly, unspec.
(756) Other congenital musculoskeletal anomalies
(756.1) Anomalies of spine
(756.12) Spondylolisthesis
(756.17) Spina bifida occulta(756.5) Osteodystrophies
(756.51) Osteogenesis imperfecta

SKELETAL ANOMALIES
• 1: 4000
PREVALENCE
• 25%
STILL BIRTH
• 30%
NEONATAL DEATH

CLASIFICTION OF SKELETAL DYSPLASIA
I- The Osteochondrodysplasias, in which there is, generalized
abnormality in bone or cartilage. This group is subdivided into three
main categories:
Defects of the growth of tubular bones and or spine
(chondrodysplasias).
Abnormalities of density or cortical diaphyseal structure and or
metaphyseal modeling.
Disorganized development of cartilage and fibrous components of
the skeleton.
II- Dysostoses: This group refers to malformations or absence of individual
bones singly or in combination. They are mostly static and their malformations
occur during blastogenesis (1st 8 weeks of embryonic life). This is in contrast to
osteochondrodysplasias, which often present after this stage, has a more general skeletal involvement and
continue to evolve as a result of active gene involvement throughout life .
The dysostoses group can be sub-classified into three main categories:
Those primarily concerned with craniofacial involvement and includes in
various craniosynostosis.
Those with predominant axial involvement including the various
segmentation defect disorders.
Those affecting only the limbs.

SKELETAL ANOMALY.
According to the International
Nomenclature for Skeletal Dysplasias, the diseases are
subdivided into three different groups:
(1) Osteochondrodysplasias (abnormalities of cartilage and / or bone growth and development)
(2) Disorganized development of cartilaginous and fibrous components of the skeleton;
and
(3) Idiopathic osteolyses.

SKELETAL ANOMALY
ACHONDROPLASIA 1 : 26.000 FGFR3
THANATOPHORIC DYSPLASIA 1 : 10,000 FGFR3
ACHONDROGENESIS 1 : 40,000 COL2A1
OSTEOGENESIS IMPERFECTA TYPE I
1 : 30,000 COL1A1, COL1A2
ASPHYXIATING THORACIC DYRTROPHY (JENUE SYNDROME)
1 : 70,000
HYPOPHOSPHATASIA 1 : 100,000
CAMPOMELIC DYSPLASIA 1 : 200,000
SPLIT HAND & FOOT SYNDROME TYPICAL 1:90,000: ATYPICAL +1 : 15,0000
FGFR3
AD
AD/AR
AD
AR
AD
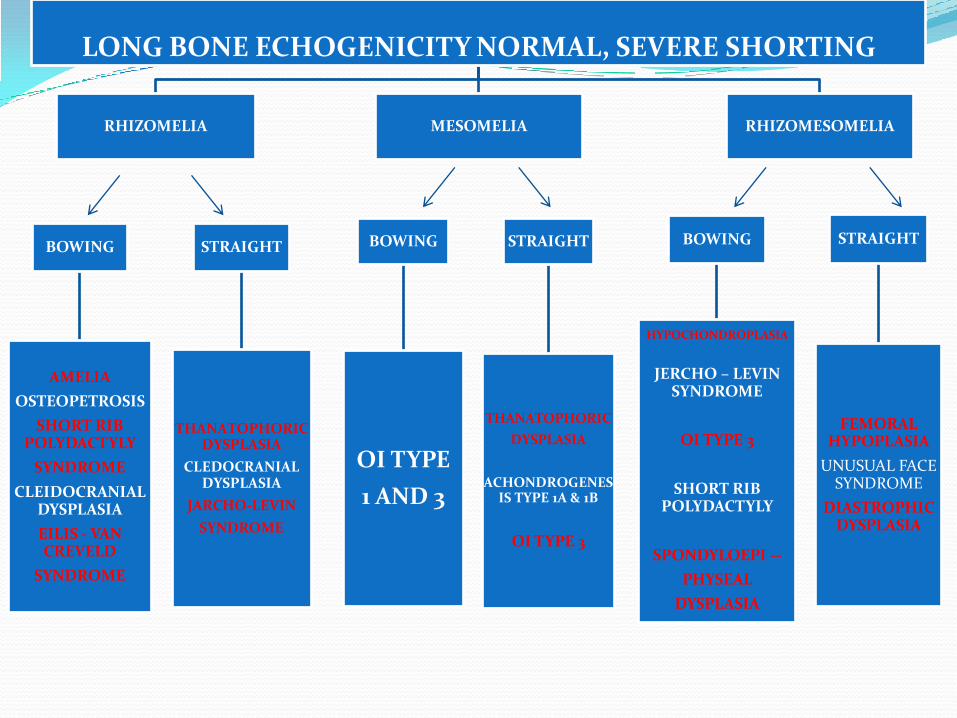
BOWING
AMELIA
OSTEOPETROSIS
SHORT RIB POLYDACTYLY
SYNDROME
CLEIDOCRANIAL DYSPLASIA
EILIS - VAN CREVELD
SYNDROME
STRAIGHT
THANATOPHORIC DYSPLASIA
CLEDOCRANIAL DYSPLASIA
JARCHO-LEVIN
SYNDROME
LONG BONE ECHOGENICITY NORMAL, SEVERE SHORTING
RHIZOMELIA MESOMELIA RHIZOMESOMELIA
BOWING
OI TYPE
1 AND 3
STRAIGHT
THANATOPHORIC
DYSPLASIA
ACHONDROGENESIS TYPE 1A & 1B
OI TYPE 3
BOWING
HYPOCHONDROPLASIA
JERCHO – LEVIN SYNDROME
OI TYPE 3
SHORT RIB POLYDACTYLY
SPONDYLOEPI --
PHYSEAL
DYSPLASIA
STRAIGHT
FEMORAL HYPOPLASIA
UNUSUAL FACE SYNDROME
DIASTROPHIC DYSPLASIA

BOWING
EILIS - VAN CREVELD
SYNDROME
STRAIGHT
EILIS – VAN CREVELD
SYNDROME
LONG BONE ECHOGENICITY NORMAL, MODERATE SHORTING
RHIZOMELIA MESOMELIA RHIZOMESOMELIA
MESOMELIC SYNDROME
SPONDYLOEPIPHYSEAL
DYSPLASIA CONGENITA
BOWING
OI TYPE 1
SHORT RIB POLYDACTYLY
SPONDYLOEPI --
PHYSEAL
DYSPLASIA
CONGENITA
STRAIGHT
OI TYPE 1
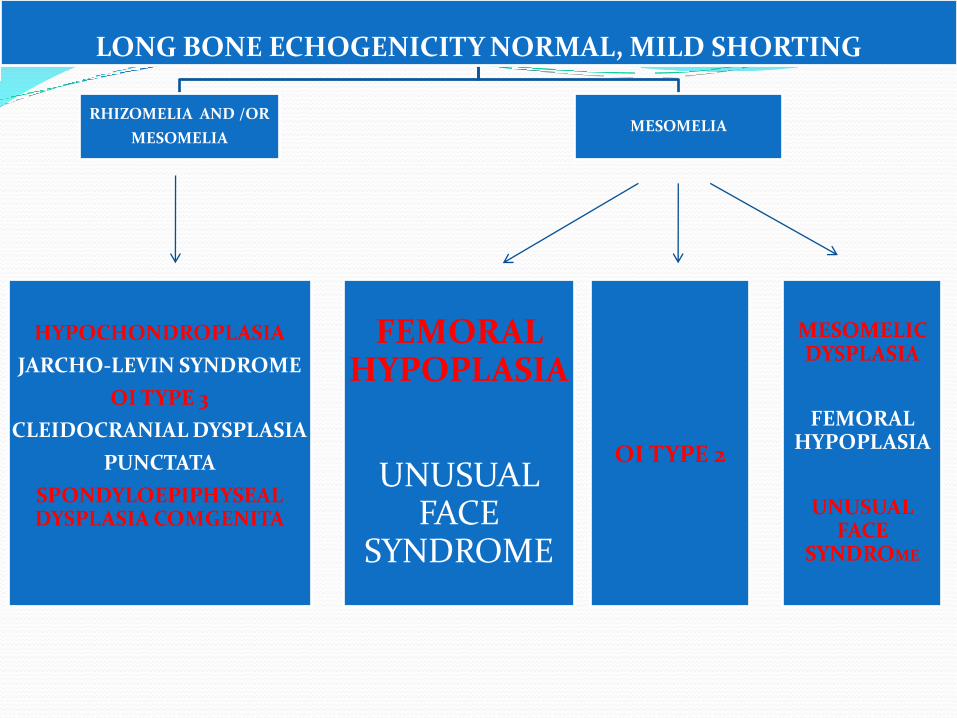
HYPOCHONDROPLASIA
JARCHO-LEVIN SYNDROME
OI TYPE 3
CLEIDOCRANIAL DYSPLASIA
PUNCTATA
SPONDYLOEPIPHYSEAL DYSPLASIA COMGENITA
LONG BONE ECHOGENICITY NORMAL, MILD SHORTING
RHIZOMELIA AND /OR
MESOMELIAMESOMELIA
FEMORAL HYPOPLASIA
UNUSUAL FACE
SYNDROME
OI TYPE 2
MESOMELIC DYSPLASIA
FEMORAL HYPOPLASIA
UNUSUAL FACE
SYNDROME

Polydactyly and syndactyly are dueto errors in the process of fetaldevelopment. For example,syndactyly results from the failureof the programmed cell death thatnormally occurs between digits.Most often these errors are due togenetic defects.
Polydactyly and syndactyly canboth occur by themselves asisolated conditions or inconjunction with other symptomsas one aspect of a multi-symptomdisease. There are several forms ofisolated syndactyly and severalforms of isolated polydactyly; eachof these, where the genetics isunderstood, is caused by anautosomal dominant gene. Thismeans that since the gene isautosomal (not sex-linked), malesand females are equally likely toinherit the trait. This also meansthat since the gene is dominant,children who have only oneparent with the trait have a 50%chance of inheriting it. However,people in the same family carryingthe same gene can have differentdegrees of polydactyly orsyndactyly

Polydactyly and syndactyly are alsopossible outcomes of a large numberof rare inherited and developmentaldisorders. One or both of them canbe present in
over 100 different disorders wherethey are minor features compared toother characteristics of thesediseases.
For example, polydactyly is acharacteristic of Meckel syndromeand Laurence-Moon-Biedlsyndrome. Polydactyly may also bepresent in Patau's syndrome,asphyxiating thoracic dystrophy,hereditary spherocytic hemolyticanemia, Moebius syndrome,VACTERL association, and Klippel-Trenaunay syndrome.
Syndactyly is a characteristic ofApert syndrome, Poland syndrome,Jarcho-Levin syndrome, oral-facial-digital syndrome, Pfeiffersyndrome, and Edwards syndrome.Syndactyly may also occur withGordon syndrome, Frasersyndrome, Greigcephalopolysyndactyly,phenylketonuria, Saethre-Chotzensyndrome, Russell-Silver syndrome,and triploidy.

In some isolated cases ofpolydactyly or syndactyly, it is notpossible to determine the cause.Some of these cases mightnevertheless be due to geneticdefects; sometimes there is toolittle information to demonstrate agenetic cause. Some cases might bedue to external factors likeexposure to toxins or wombanomalies.
Postaxial polydactyly (80% of cases)
Extra digit on ulnar or fibular side oflimb
Preaxial polydactyly Extra digit onradial or tibial side of limb

TALIPES EQUINOVAROUSThe incidence (i.e., how often itoccurs) of the condition varies withrace, sex, and familial incidence:
Boys are twice as likely to get thedisease as girls
The incidence among Caucasians isaround 1 per 1,000 live births
The incidence among children inJapan is 0.5 per 1,000 live births
The incidence among natives ofthe South Pacific is nearly 7 per1,000 live births
The incidence for children whohave a sibling with clubfoot isapproximately 3%
The incidence for children whohave 1 parent that had clubfoot is3-4%; if both parents had it, theincidence is 15%
Children born with clubfoot have ahigher-than-normal incidence(around 14%) of other geneticconditions, including Edward'sSyndrome , Larsen's syndrome,spina bifida, neural tube defects,and congenital heart defects
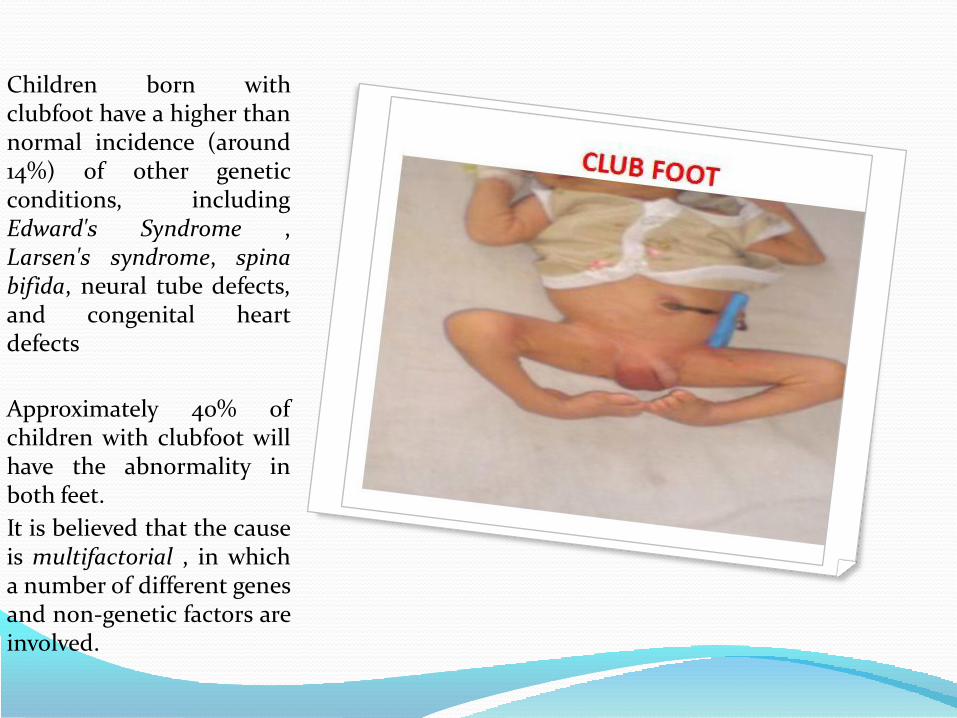
Children born withclubfoot have a higher thannormal incidence (around14%) of other geneticconditions, includingEdward's Syndrome ,Larsen's syndrome, spinabifida, neural tube defects,and congenital heartdefects
Approximately 40% ofchildren with clubfoot willhave the abnormality inboth feet.
It is believed that the causeis multifactorial , in whicha number of different genesand non-genetic factors areinvolved.

TALIPES EQUINOVAROUSAbout one in every 1,000 babies isborn with clubfoot.
Clubfoot does not have anythingto do with the baby’s position inthe womb.
It is mostly a problem passed fromparents to children (genetic), andit may run in families.
If you have one baby with clubfoot,the chances of having a secondchild with the condition are aboutone in 40.
About half of children with thecondition have two clubfeet.Children with certain neurological
and chromosome conditions aremore likely to have clubfoot.
Most times children who haveclubfoot are otherwise completelyhealthy.
Clubfoot is the common nameused to describe the conditiontalipes equinovarus, in which thereis a deformity of the foot or feetthat affects children at birth.
Clubfoot may be classified ascongenital (i.e. existing at birth)or teratologic (in which it isassociated with a neuromusculardisorder). .

.

LIMB DEFICIENCY
0.49/10,000 BIRTHS
AMELIA : COMPLETE ABSENCE OF LIMB
MEROMELIA : PARTIAL ABSENCE OF LIMB.
TERMINAL DEFICIENCY- ALL SKELETAL ELEMENTS ALONG A LONGITUDINAL RAY BEYOND A GIVEN POINT ARE ABSENT.
INERCALARY : ABSENCE OF PROXIMAL OR MIDDLE SEGMENT OF A LIMB WITH ALL THE DISTAL SEGMENT PRESENT.
FURTHER SUBGROUPING IS BASED ON THE AXIS OF DEFICIENCY – TRANSVERSE OR LONGITUDINAL
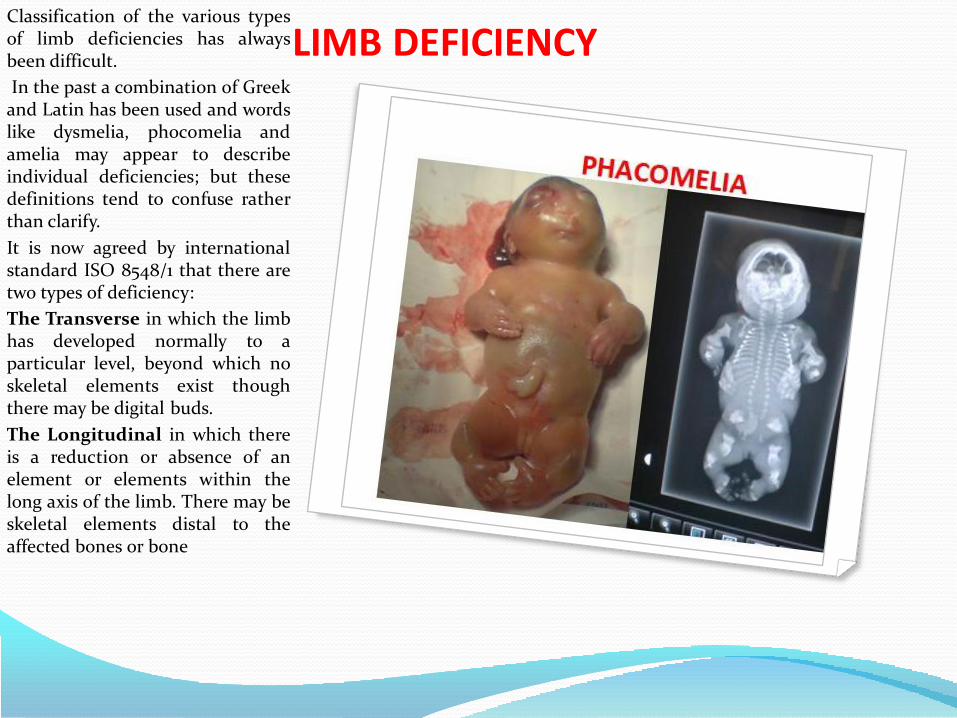
LIMB DEFICIENCYClassification of the various typesof limb deficiencies has alwaysbeen difficult.
In the past a combination of Greekand Latin has been used and wordslike dysmelia, phocomelia andamelia may appear to describeindividual deficiencies; but thesedefinitions tend to confuse ratherthan clarify.
It is now agreed by internationalstandard ISO 8548/1 that there aretwo types of deficiency:
The Transverse in which the limbhas developed normally to aparticular level, beyond which noskeletal elements exist thoughthere may be digital buds.
The Longitudinal in which thereis a reduction or absence of anelement or elements within thelong axis of the limb. There may beskeletal elements distal to theaffected bones or bone

ACHONDROPLASIAPrevalence : 1 in 26 000
It is inherited as autosomaldominant disorder, majorityof cases represent newmutations. It is the mostcommon nonlethal skelataldysplasia.The characteristicfeatures of heterozygousachondroplasia include shortlimbs, lumbar lordosis, shorthands and fingers,macrocephaly with frontalbossing and depressed nasalbridge.
Intelligence and lifeexpectancy are normal.
Prenatally, limb shorteningand typical facis usuallybecome apparent only after 22weeks of gestation. In thehomozygous state, which is alethal condition, short limbsare associated with a narrowthorax.

ACHONDROPLASIA
Achondroplasia is due to aspecific mutation withinthe fibroblast growth factorreceptor type 3 gene(FGFR3) located on 4p16.3.and can now be diagnosed byDNA analysis of fetal blood oramniotic fluid obtained incases of suspicioussonographic findings.
In cases where bothparents haveachondroplasia, there is a25% chance that the fetusis affected by the lethal typeand the diagnosis can be madeby first-trimester chorionvillous sampling

OSTEOGENESIS OF IMPERFECTAOsteogenesis imperfecta is agenetically heterogeneous group ofdisorders .The underlying defect is a
dominant negative mutationaffecting COL1A1 or COL1A2alleles, which encode theproA1(I) and proa2(I) chains oftype I collagen, a protein ofparamount importance fornormal skin and bonedevelopment. The mutations resultin the production of abnormalquantity (O I type I) or quality (typesII, III and IV) of collagen.
There are four clinical subtypes.Type I : which is an autosomaldominant condition with a birthprevalence of about 1 in 30 000.The affected individuals havefragile bones, blue sclerae, loosejoints, growth deficiency andprogressive deafness, but lifeexpectancy is normal. Prenataldiagnosis is available by DNAanalysis. Ultrasonography in thesecond and third trimesters maydemonstrate fractures of long bones.

OSTEOGENESIS OF IMPERFECTA
Type II: which is a lethal disorder .Prevalence 1 in 60 000 births.
Most cases represent newdominant mutations(recurrence is about 6%). Thedisorder is characterized byearly prenatal onset of severebone shortening and bowingdue to multiple fracturesaffecting all long bones andribs, and poor mineralizationof the skull.

OSTEOGENESIS IMPERFECTA
Type III is a progressivelydeforming condition characterizedby multiple fractures, usuallypresent at birth, resulting inscoliosis and very short stature.Bowing of the femur has beendescribed in these cases in utero.Both autosomal dominant andrecessive modes of inheritancehave been reported.
Type IV is an autosomal dominantcondition with variableexpressivity. Severely affectedindividuals may have deformitiesof the long bones due to fractures.
Prenatal diagnosis of types IIIand IV can be made by chorionvillous sampling and DNAanalysis, or by demonstration ofabnormal collagen productionin cultured fibroblasts.

CAMPOMELIC DYSPLASIAPrevalence: 1 in 20,000births.
It lethal , autosomaldominant syndrome dueto mutation in the SOX9gene at,an SRY – relatedgene at 17q23-qter. ischaracterized byshortening and bowing ofthe long bones of the legs,narrow chest, hypoplasticscapulae, and largecalvarium withdisproportionately smallface.Some of the affectedgenetically maleindividuals show a femalephenotype.
Patients usually die in theneonatal period frompulmonary hypoplasia.

CAMPOMELIC DYSPLASIAPrevalence: 1 in 20,000births.
It lethal , autosomaldominant syndrome dueto mutation in the SOX9gene at,an SRY – relatedgene at 17q23-qter. ischaracterized byshortening and bowing ofthe long bones of the legs,narrow chest, hypoplasticscapulae, and largecalvarium withdisproportionately smallface.Some of the affectedgenetically maleindividuals show a femalephenotype.
Patients usually die in theneonatal period frompulmonary hypoplasia.

ASPHXIATED THORACIC DYSPLASIA(JEUNE SYNDROME)
Prevalence : 1 in 70 000 births.
It is the autosomalrecessive disorder.
It ‘s characteristic featuresare narrow chest andrhizomelic limbshortening, postaxialpolydactyly andintracerebral anomalies.There is a variablephenotypic expressionand, consequently, theprognosis varies fromneonatal death, due topulmonary hypoplasia(70%), to normal survival.Limb shortening is mildto moderate and this maynot become apparent untilafter 24 weeks of gestation.

ELLIS – VAN CREVELD SYNDROME
This rare,autosomal recessivecondition ischaracterized byacromelic andmesomelicshortness of limbs,postaxialpolydactyly, smallchest, ectodermaldysplasia, midlinecleft or notchedupper lip andcongenital heartdefects in more than50% of cases.

SHORT LIMB POLYDACTYLY SYNDROMEThis group of lethal disorders ischaracterized by short limbs,narrow thorax and postaxialpolydactyly. Associated anomaliesare frequently found, includingcongenital heart disease, polycystickidneys, and intestinal atresia.
Four different types have beenrecognized.
Type I (Saldino–Noonan) hasnarrow metaphyses;
Type II (Majewski) has cleft lipand palate and disproportionallyshortened tibiae;
Type III (Naumoff) has widemetaphyses with spurs;
Type IV (Beemer–Langer) ischaracterized by median cleft lip,small chest with extremely shortribs, protuberant abdomen withumbilical hernia and ambiguousgenitalia in some 46,XYindividuals.

JARCHO- LEVIN SYNDROME
This is a heterogeneousdisorder, characterized byvertebral and ribabnormalities(misalignment of thecervical spine and ribs). Anautosomal recessive type ischaracterized by aconstricted short thoraxand respiratory death ininfancy. Anotherautosomal recessive and anautosomal dominant typeare associated with a shortstature and are compatiblewith survival to adult lifebut with some degree ofphysical disability.

DIASTROPHIC DYSPLASIA This autosomal recessivecondition due to mutation in thediastrophic dysplasia sulfatasetransporter gene located atchromosome 5q32-q33.1 ; resultngin undersulfated proteoglycans inthe cartilage matrix. It ischaracterized by severeshortening and bowing of alllong bones, talipes equinovarus,hand deformities with abductedposition of the thumbs(‘hitchhiker thumb’), multiplejoint flexion contractures andscoliosis.
The bones are characterized bycrescent – shaped flattenedepiphyses , a short and broadfemoral neck, and shorting ofthe metaphyseal widening oftubular bones. There is a widespectrum in phenotypicexpression and some cases may
not be diagnosable in utero. This
disease is not lethal andneurodevelopment is normal.

HYPOPHOSPHATASIAPrevalence : 1 in 10000births.
It autosomal recessivedisoder & is characterizedby severe shortening of thelong bones, small thorax,hypomineralization of theskull and long bones.There is absence of liverand bone isoenzymes ofalkaline phosphatase, andfirst-trimester diagnosis ismade by measurement ofalkaline phosphataseisoenzymes in chorionvillous samples.
The diagnosis can also bemade by DNA studies.

FETAL AKINESIA DEFORMATION SEQUENCE
Prevalence : 1 in 3000 Births.Neurological, muscular,connective tissue, and skeletalabnormalities result in multiplejoint contractures, includingbilateral talipes and fixedflexion or extension deformitiesof the hips, knees, elbows andwrists.
This sequence includescongenital lethal arthrogryposis,multiple pterygium and Pena–Shokeir syndromes.
The deformities are usuallysymmetric and, in most cases, allfour limbs are involved. Theseverity of the deformitiesincreases distally in the involvedlimb, with the hands and feettypically being the most severelyaffected. The condition iscommonly associated withpolyhydramnios (usually after 25weeks), narrow chest,micrognathia and nuchal edema(or increased nuchal translucencyat 10–13 +6 weeks).

POLYDACTYLY SYNDROMEPolydactyly is the presence of anadditional digit, which may rangefrom a fleshy nubbin to acomplete digit with controlledflexion and extension. Postaxialpolydactyly (the most commonform) occurs on the ulnar side ofthe hand and fibular side of thefoot. Preaxial polydactyly ispresent on the radial side of thehand and the tibial side of thefoot. The majority of conditionsare isolated with an autosomaldominant mode of inheritance.Some of them are part of asyndrome, usually an autosomalrecessive one. Preaxialpolydactyly, especiallytriphalangeal thumb, is mostlikely to be part of amultisystem syndrome. Centralpolydactyly, which consists of anextra digit (usually hiddenbetween the long and the ringfinger), is often bilateral and isassociated with other hand andfoot malformations; it is inheritedwith an autosomal mode ofinheritance.

POLYDACTYLY SYNDROMEPolydactyly is the presence of anadditional digit, which may rangefrom a fleshy nubbin to acomplete digit with controlledflexion and extension. Postaxialpolydactyly (the most commonform) occurs on the ulnar side ofthe hand and fibular side of thefoot. Preaxial polydactyly ispresent on the radial side of thehand and the tibial side of thefoot. The majority of conditionsare isolated with an autosomaldominant mode of inheritance.Some of them are part of asyndrome, usually an autosomalrecessive one. Preaxialpolydactyly, especiallytriphalangeal thumb, is mostlikely to be part of amultisystem syndrome. Centralpolydactyly, which consists of anextra digit (usually hiddenbetween the long and the ringfinger), is often bilateral and isassociated with other hand andfoot malformations; it is inheritedwith an autosomal mode ofinheritance.

CLUB HAND DEFORMITYClubhand deformities areclassified into two main categories:radial and ulnar.
Radial clubhand includes a widespectrum of disorders thatencompass absent thumb, thumbhypoplasia, thin first metacarpaland absent radius.
Ulnar clubhand, which is lesscommon, ranges from mild deviationsof the hand on the ulnar side of theforearm to complete absence of theulna. While radial clubhand isfrequently syndromatic, ulnarclubhand is usually an isolatedanomaly.
Clubhand deformities are oftenfound in association withchromosomal abnormalities (suchas trisomy 18), hematologicalabnormalities (such as Fanconi’spancytopenia, TAR syndrome andAase syndrome), or geneticsyndromes with cardiac defects (suchas Holt–Oram syndrome, or theLewis upper limb–cardiovascularsyndrome).
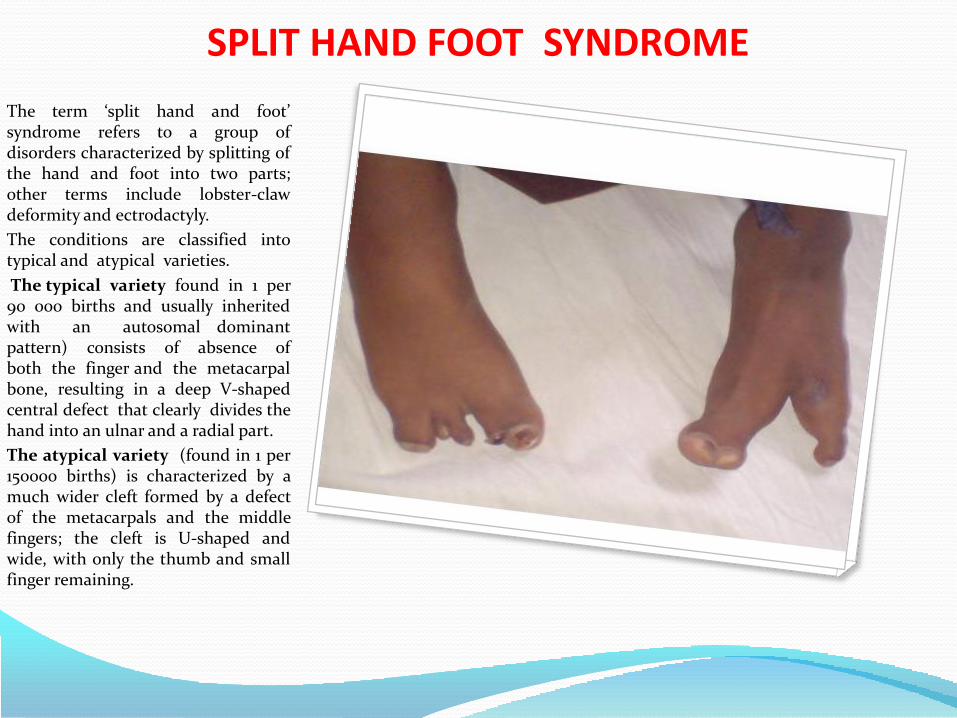
SPLIT HAND FOOT SYNDROME
The term ‘split hand and foot’syndrome refers to a group ofdisorders characterized by splitting ofthe hand and foot into two parts;other terms include lobster-clawdeformity and ectrodactyly.
The conditions are classified intotypical and atypical varieties.
The typical variety found in 1 per90 000 births and usually inheritedwith an autosomal dominantpattern) consists of absence ofboth the finger and the metacarpalbone, resulting in a deep V-shapedcentral defect that clearly divides thehand into an ulnar and a radial part.
The atypical variety (found in 1 per150000 births) is characterized by amuch wider cleft formed by a defectof the metacarpals and the middlefingers; the cleft is U-shaped andwide, with only the thumb and smallfinger remaining.

LIMBS AMNIOTIC BAND SYNDROMEg constriction rings of limbs or digits
g amputation of limbs or digits
g lymphedemag pseudosyndactyly (pseudosyndactyly involves only the distalportion of the digits,whereas syndactyly includes the base of the digits)
g abnormal dermal ridge patterns
g simian creaseg clubfeet
Cranium
g multiple and asymmetric cephaloceles
g anencephalyg acrania
Face
g cleft-lip, -palate & -faceg nasal deformitiesg asymmetric microphthalmos
g incomplete or absent calcifications of the skull
Thorax
g rib cleftingSpine
g scoliosisAbdominal wall
g gastroschisisg omphaloceleg bladder Exstrophy
Perineum
g ambiguous Genitaliag imperforate Anus

AMNIOTIC BAND SYNDROMEAmniotic band syndrome is a set ofcongenital malformations rangingfrom minor constriction rings andlymph edema of the digits to complex,bizarre multiple congenital anomaliesthat are attributed to amniotic bandsthat stick, entangle and disrupt fetalparts.
Prevalence: The prevalence for livebirths is 7.7:10,0001; for spontaneousabortions it can be as high as178:10.0003. M1:F1
Amniotic banding affectsapproximately 1 in 1,200 live births. Itis also believed to be the cause of 178in 10,000 miscarriages.
Up to 50% of cases have othercongenital anomalies including cleftlip, cleft palate, and clubfootdeformity. Hand and finger anomaliesoccur in up to 80%.
Pathogenesis: Rupture of theamnion in early pregnancy leads toentrapment of fetal structures by“sticky” mesodermic bands thatoriginate from the chorionic side ofthe amnion, followed by disruption.
This theory has been contestedrecently based on clinical andexperimental data

AMNIOTIC BAND SYNDROMEAmniotic band syndrome is a set ofcongenital malformations rangingfrom minor constriction rings andlymph edema of the digits to complex,bizarre multiple congenital anomaliesthat are attributed to amniotic bandsthat stick, entangle and disrupt fetalparts.
Prevalence: The prevalence for livebirths is 7.7:10,0001; for spontaneousabortions it can be as high as178:10.0003. M1:F1
Amniotic banding affectsapproximately 1 in 1,200 live births. Itis also believed to be the cause of 178in 10,000 miscarriages.
Up to 50% of cases have othercongenital anomalies including cleftlip, cleft palate, and clubfootdeformity. Hand and finger anomaliesoccur in up to 80%.
Pathogenesis: Rupture of theamnion in early pregnancy leads toentrapment of fetal structures by“sticky” mesodermic bands thatoriginate from the chorionic side ofthe amnion, followed by disruption.
This theory has been contestedrecently based on clinical andexperimental data

AMNIOTIC BAND SYNDROMEAmniotic band syndrome is a set ofcongenital malformations rangingfrom minor constriction rings andlymph edema of the digits to complex,bizarre multiple congenital anomaliesthat are attributed to amniotic bandsthat stick, entangle and disrupt fetalparts.
Prevalence: The prevalence for livebirths is 7.7:10,0001; for spontaneousabortions it can be as high as178:10.0003. M1:F1
Amniotic banding affectsapproximately 1 in 1,200 live births. Itis also believed to be the cause of 178in 10,000 miscarriages.
Up to 50% of cases have othercongenital anomalies including cleftlip, cleft palate, and clubfootdeformity. Hand and finger anomaliesoccur in up to 80%.
Pathogenesis: Rupture of theamnion in early pregnancy leads toentrapment of fetal structures by“sticky” mesodermic bands thatoriginate from the chorionic side ofthe amnion, followed by disruption.
This theory has been contestedrecently based on clinical andexperimental data

AMNIOTIC BAND SYNDROMEAmniotic band syndrome is a set ofcongenital malformations rangingfrom minor constriction rings andlymph edema of the digits to complex,bizarre multiple congenital anomaliesthat are attributed to amniotic bandsthat stick, entangle and disrupt fetalparts.
Prevalence: The prevalence for livebirths is 7.7:10,0001; for spontaneousabortions it can be as high as178:10.0003. M1:F1
Amniotic banding affectsapproximately 1 in 1,200 live births. Itis also believed to be the cause of 178in 10,000 miscarriages.
Up to 50% of cases have othercongenital anomalies including cleftlip, cleft palate, and clubfootdeformity. Hand and finger anomaliesoccur in up to 80%.
Pathogenesis: Rupture of theamnion in early pregnancy leads toentrapment of fetal structures by“sticky” mesodermic bands thatoriginate from the chorionic side ofthe amnion, followed by disruption.
This theory has been contestedrecently based on clinical andexperimental data

AMNIOTIC BAND SYNDROMEAmniotic band syndrome is a set ofcongenital malformations rangingfrom minor constriction rings andlymph edema of the digits to complex,bizarre multiple congenital anomaliesthat are attributed to amniotic bandsthat stick, entangle and disrupt fetalparts.
Prevalence: The prevalence for livebirths is 7.7:10,0001; for spontaneousabortions it can be as high as178:10.0003. M1:F1
Amniotic banding affectsapproximately 1 in 1,200 live births. Itis also believed to be the cause of 178in 10,000 miscarriages.
Up to 50% of cases have othercongenital anomalies including cleftlip, cleft palate, and clubfootdeformity. Hand and finger anomaliesoccur in up to 80%.
Pathogenesis: Rupture of theamnion in early pregnancy leads toentrapment of fetal structures by“sticky” mesodermic bands thatoriginate from the chorionic side ofthe amnion, followed by disruption.
This theory has been contestedrecently based on clinical andexperimental data

AMNIOTIC BAND SYNDROMEAmniotic band syndrome is a set ofcongenital malformations rangingfrom minor constriction rings andlymph edema of the digits to complex,bizarre multiple congenital anomaliesthat are attributed to amniotic bandsthat stick, entangle and disrupt fetalparts.
Prevalence: The prevalence for livebirths is 7.7:10,0001; for spontaneousabortions it can be as high as178:10.0003. M1:F1
Amniotic banding affectsapproximately 1 in 1,200 live births. Itis also believed to be the cause of 178in 10,000 miscarriages.
Up to 50% of cases have othercongenital anomalies including cleftlip, cleft palate, and clubfootdeformity. Hand and finger anomaliesoccur in up to 80%.
Pathogenesis: Rupture of theamnion in early pregnancy leads toentrapment of fetal structures by“sticky” mesodermic bands thatoriginate from the chorionic side ofthe amnion, followed by disruption.
This theory has been contestedrecently based on clinical andexperimental data

In medicine, the term collodion babyapplies to newborns who appear tohave an extra layer of skin (known asa collodion membrane) that has acollodion-like quality. It is adescriptive term, not a specificdiagnosis or disorder . Ichthyosislammellaris (lamellar ichthyosis) andnonbullous congenital ichthyosis, is arare inherited skin disorder, affectingaround 1 in 600,000 people. Theappearance is often described as ashiny film looking like a layer ofvaseline. The eyelids and mouth mayhave the appearance of being forcedopen due to the tightness of the skin.There can be associated eversion ofthe eyelids (ectropion). Most cases(approximately 75%) of collodionbaby will go on to develop a type ofautosomal recessive congenitalichthyosis (either lamellar ichthyosisor congenital ichthyosiformerythrodema) This condition is anautosomal recessive genetic disorder,which means the defective gene islocated on an autosome, and bothparents must carry one copy of thedefective gene in order to have a childborn with the disorder. Carriers of arecessive gene usually do not showany signs or symptoms of thedisorder. Ichthyosis lamellaris isassociated with a deficiency of theenzyme keratinocytetransglutaminase. Genes involvedinclude TGM1, ABCA12, and CYP4F22.[

The appearance can be caused by severalskin diseases, and it is most often notassociated with other birth defects. Inmost cases, the baby develops anichthyosis or ichthyosis-like condition orother rare skin disorder.
Most cases (approximately 75%) ofcollodion baby will go on to develop atype of autosomal recessive congenitalichthyosis (either lamellar ichthyosis orcongenital ichthyosiform erythrodema).
In around 10% of cases the baby shedsthis layer of skin and has normal skinfor the rest of its life.. This is knownas self-healing collodion baby.
The remaining 15% of cases are caused bya variety of diseases involvingkeratinization disorders.[ Known causesof collodion baby include ichthyosisvulgaris and trichothiodystrophy. Lesswell documented causes include Sjögren-Larsson syndrome, Netherton syndrome,Gaucher disease type 2, congenitalhypothyroidism, Conradi syndrome,Dorfman-Chanarin syndrome,ketoadipiaciduria, koraxitrachiticsyndrome, ichthyosis variegata andpalmoplantar keratoderma withanogenital leukokeratosis. Since many ofthese conditions have an autosomalrecessive inheritance pattern, they arerare and can be associated withconsanguinity.
Tests that can be used to find the causeof collodion baby include examination ofthe hairs, blood tests and a skin biopsy

Ectodermal dysplasia is the term used todescribe a group of rare congenitalanomalies characterized by abnormaldevelopment of 1 or several ectoderm-derived tissues. At least 154 differenttypes, divided into 11 clinical subgroups,have been recognized.1 Among them, thehypohidrotic type is the most commonform, withan incidence of 1 per 10,000 to1 per 100,000 live births.2–4 Thiscondition, originally known as anhidroticectodermal dysplasia because of thenotable reduction of sweat glandfunction, is clinically characterized byhypohidrosis, hypotrichosis, andhypodontia.2–4 Most cases are inheritedas an X-linked recessive trait, with thegene responsible being mapped to Xq12-q13.1.5 The autosomal recessive anddominant patterns of inheritance havealso been documented.6,7
Prenatal diagnosis of this condition hasbeen reported previously in high-riskpregnancies on the basis of histologicanalysisof fetal skin obtained by second-trimester fetoscopy-guided skin biopsy.8,9
DNA-based linkage analysis has alsomade thediagnosis possible with the useof chorionic villi in the first trimester.10 Inthis report, we describe noninvasiveprenatal diagnosis of hypohidroticectodermal dysplasia in a pregnancy atrisk for this condition.

Thrombocytopenia-absent radius (TAR)syndrome is a rare condition in whichthrombocytopenia is associated withbilateral radial aplasia. TAR syndromewas first described in 1951. An autosomalrecessive inheritance pattern wasproposed because TAR affected morethan one member of some families. In1969, TAR was defined as a syndrome andfurther classified as the association ofhypomegakaryocytic thrombocytopeniaand absent radii. TAR syndrome iscongenital, and patients usually presentwith symptomatic thrombocytopenia inthe first week of life.

Epidermolysis bullosa (EB) isan inherited connectivetissuedisease causing blisters inthe skin and mucosalmembranes, with anincidence of 1/50,000. Itsseverity ranges from mild tolethal. It is caused by amutation in the keratinor collagen gene. Of thesecases, approximately 92%are EBS, 5% are DEB, 1% areJEB, and 2% are unclassified.Carrier frequency rangesfrom 1 in 333 for Junctional,to 1 in 450 for Dystrophic.Carrier frequency for Simplexis not indicated in this article,but is presumed to be muchhigher than JEB or DEB.
Epidermolysis bullosa simplex
Generalized epidermolysis bullosa simplex(Koebner variant of generalized epidermolysis bullosa simplex)
Localized epidermolysis bullosa simplex(Weber-Cockayne variant of generalized epidermolysis bullosa simplex)
Epidermolysis bullosa herpetiformis (Dowling-Meara epidermolysis bullosa simplex)
Epidermolysis bullosa simplex of Ogna
Epidermolysis bullosa simplex with muscular dystrophy
Epidermolysis bullosa simplex with mottled pigmentation

OMIM Name Locus Gene
609352epidermolysis bullosa simplex with migratory circinate erythema
12q13 KRT5
131960epidermolysis bullosasimplex with mottled pigmentation; EBS-MP
12q13 KRT5
601001epidermolysis bullosa simplex, autosomal recessive
17q12-q21 KRT14
131900epidermolysis bullosa simplex, Koebner type; EBS2
17q12-q21, 12q13 KRT5,KRT14
131800epidermolysis bullosa simplex, Weber-Cockayne type
17q12-q21, 17q11-qter, 12q13 KRT5,KRT14
131760epidermolysis bullosa herpetiformis, Dowling-Meara type
17q12-q21, 12q13 KRT5,KRT14
226670epidermolysis bullosa simplex with muscular dystrophy
8q24 PLEC1
612138epidermolysis bullosa simplex with pyloric atresia
8q24 PLEC1
131950epidermolysis bullosasimplex, Ogna type
8q24 PLEC1
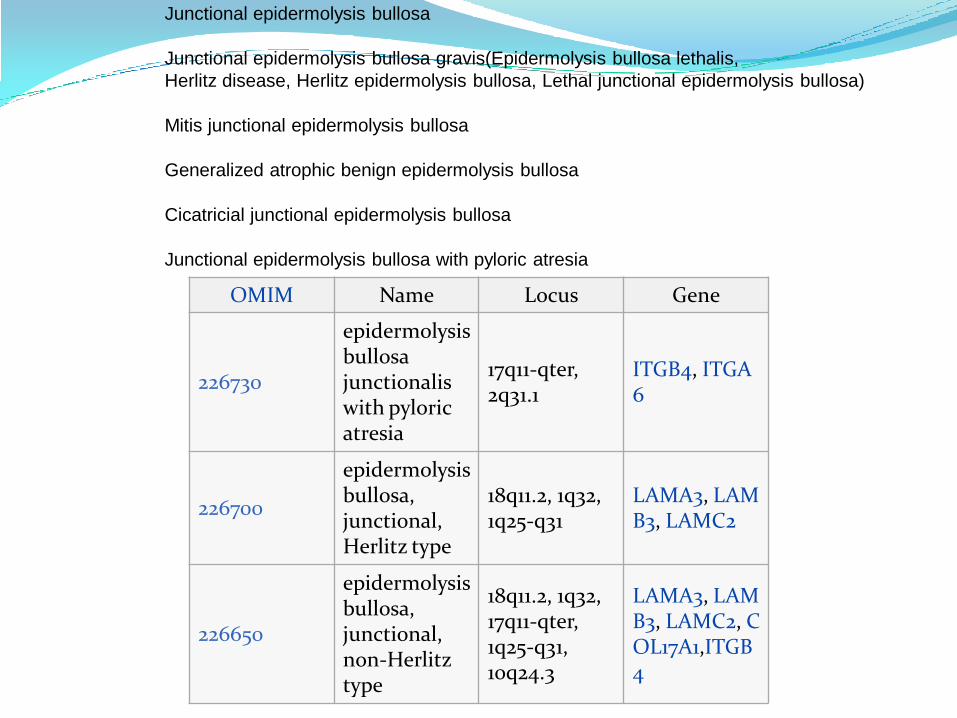
OMIM Name Locus Gene
226730
epidermolysis bullosa junctionalis with pyloric atresia
17q11-qter, 2q31.1
ITGB4, ITGA6
226700
epidermolysis bullosa, junctional, Herlitz type
18q11.2, 1q32, 1q25-q31
LAMA3, LAMB3, LAMC2
226650
epidermolysis bullosa, junctional, non-Herlitz type
18q11.2, 1q32, 17q11-qter, 1q25-q31, 10q24.3
LAMA3, LAMB3, LAMC2, COL17A1,ITGB4
Junctional epidermolysis bullosa
Junctional epidermolysis bullosa gravis(Epidermolysis bullosa lethalis,
Herlitz disease, Herlitz epidermolysis bullosa, Lethal junctional epidermolysis bullosa)
Mitis junctional epidermolysis bullosa
Generalized atrophic benign epidermolysis bullosa
Cicatricial junctional epidermolysis bullosa
Junctional epidermolysis bullosa with pyloric atresia

OMIM Name Locus Gene
131750
epidermolysis bullosa dystrophica, autosomal dominant; DDEB
3p21.3 COL7A1
226600
epidermolysis bullosa dystrophica, autosomal recessive; RDEB
11q22-q23, 3p21.3 COL7A1, MMP1
131850epidermolysis bullosa dystrophica, pretibial
3p21.3 COL7A1
604129epidermolysis bullosa pruriginosa
3p21.3 COL7A1
132000
epidermolysis bullosa with congenital localized absence of skin and deformity of nails
3p21.3 COL7A1
131705transient bullous dermolysis of the newborn; TBDN
3p21.3 COL7A1
• Dystrophic epidermolysis bullosa
• Dominant dystrophic epidermolysis bullosa(Cockayne-Touraine disease)
• Recessive dystrophic epidermolysis bullosa (Hallopeau-Siemens variant of
epidermolysis bullosa)

Integument(757) Congenital anomalies of the integument
(757.3) Other specified anomalies of skin
(757.32) Birthmarks
(757.39) Other specified congenital anomalies of skin
Bloom syndrome
Epidermolysis bullosa
Pseudoxanthoma elasticum
(757.6) Supernumerary nipple

Deafness
Recent studies estimate that over 50% of non-syndromicdeafness is due to genetic factors with mutations in GJB2, thegene for Connexin 26, causing approximately 30% of cases ofsporadic hearing loss and over 50% of cases of congenitalsevere-to-profound deafness in which there is an affectedsibling. It is anticipated that genetic testing will provide anumber of advantages by facilitating diagnosis andultimately impacting prevention and intervention. Thepurpose of this is to review the clinical process of genetictesting including genetic counselling for deaf and hard ofhearing patients and their families.
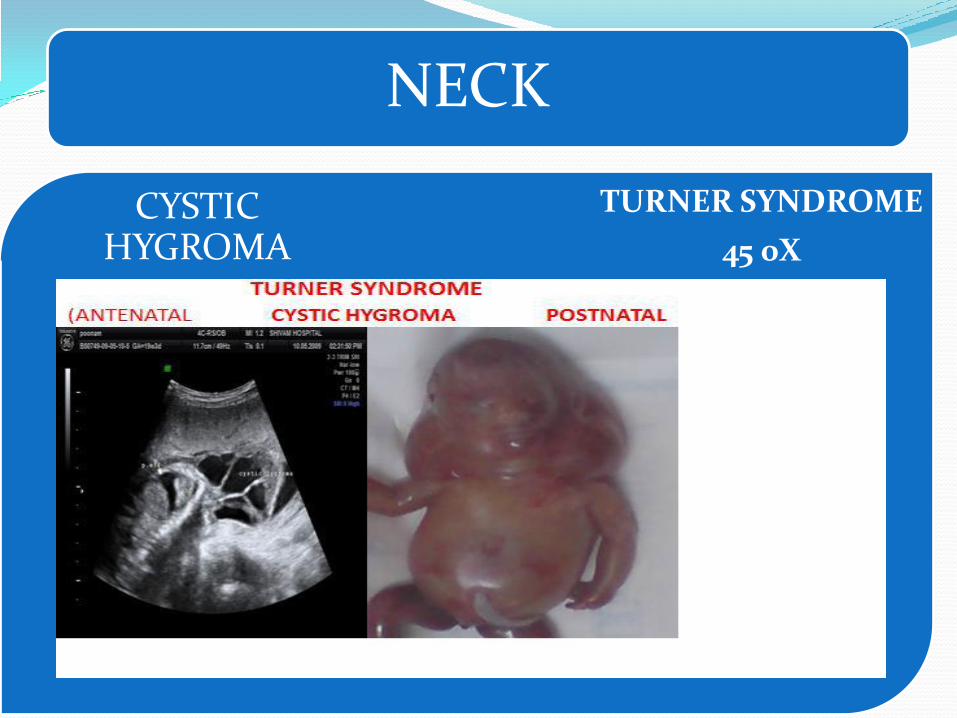
NECK
TURNER SYNDROME
45 0X.
CYSTIC HYGROMA
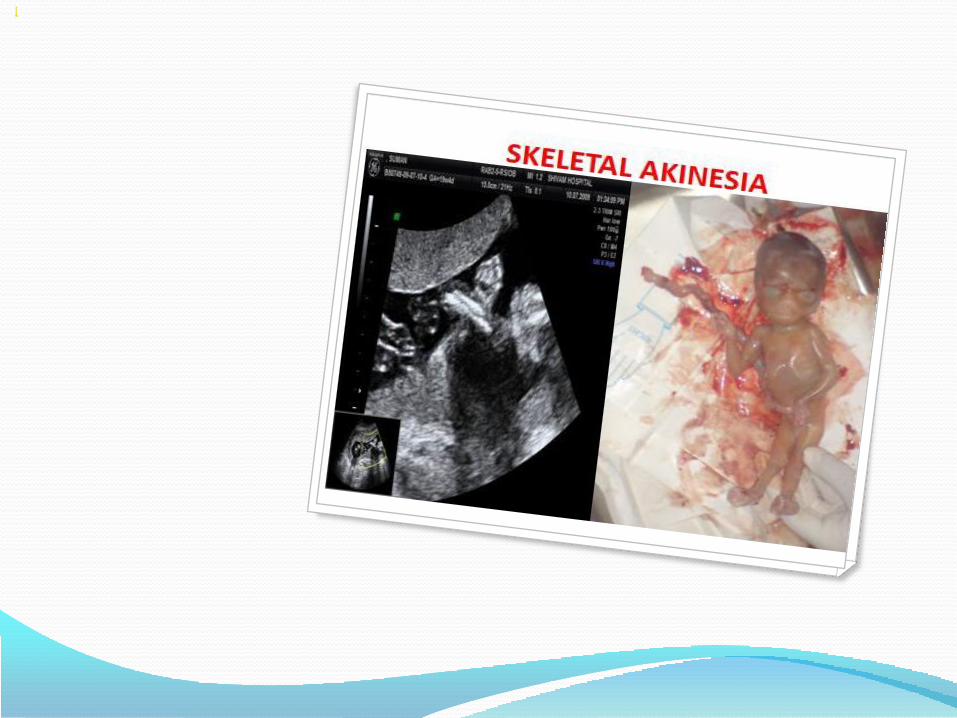
[

BARTSOCAS – PAPAS SYNDROMEBartsocas – papas syndrome (BPS)MIM263650 is a severe and rareautosomal recessive syndromecharacterized by popliteal pterigium/webbing, oligo-syndactyly, genitalanomalies and typical face with shortpalpebral fissures, ankyloblepharon ,hypoplastic nose, oro-facial clefts andsmall mouth It is inherited as autosomalrecessive disorder and X - linkedrecessive disorder. In autosomalrecessive variant recurrence risk is 25%.This syndrome is characterized by themultiple pterygium - chinto-sternum,cervical, axillary, antecubital, poplitealand crural; flexion contracture; skeletalabnormalities – absences of metacarpal,metatarsal, phalangeal bones andscapulae; hypoplastic heart and lung;facial anomalies - ectropion, medialcanthal web, blephrophimosis,hypoplasia of nose, oral andnasopharyngeal cavities, vocal cord,tongue, microganithia, orolabialsynechia; sad and expression less face;short neck; asymmetric nipples; stenosisof rectum; hypoplastic labia majora;complete syndactyly of all fingers andtoes; pes equinovarus and absence nailsand phalangeal creases. This syndromeshould be differentiated form anautosomal recessive - Pena – Shokeirsyndrome. It is characterized byneurogenic arthogryphosis, facialanomalies, pulmonary hypo plasia anddismorphic features, resulting from fetalakinesia
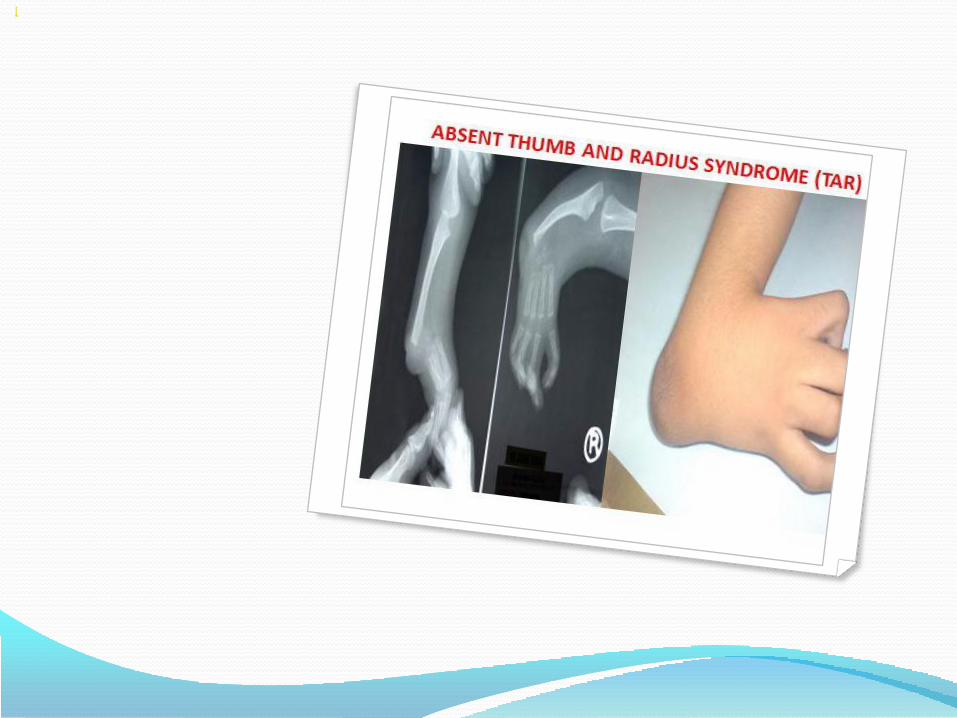
[

[

[

References
1. Nelson Text book of Pediatrics 18th edition.
2. Ghai Essential Pediatrics 2nd edition.
3. Manuals of Neonatal care cloherty 6th edition.
4. Rudolf Pediatrics
5. Pediatrics Cardiology Robert Anderson, ellot M. Tyran.
6. Centers for Disease Control and Prevention.
7. The National Library of Medicine or MEDLINE/PubMed Mesh
(medical subject heading) term
8. American College of Medical Genetics Centre New York.
9. Essentials of Embryology and Birth Defects, 6th ed. 2003.
10. Cohen MM Jr. Child with Multiple Birth Defects. New York.
11. Buyse ML. Birth Defects Encyclopedia. Oxford, 1990.
12. Hall BD. The state of the art of dysmorphology. Am J; 1993.