Bocavirus Infection Induces a DNA Damage Response That FacilitatesViral DNA Replication and Mediates Cell Death�
Yong Luo, Aaron Yun Chen, and Jianming Qiu*Department of Microbiology, Molecular Genetics and Immunology, University of Kansas Medical Center, Kansas City, Kansas
Received 22 July 2010/Accepted 23 October 2010
Minute virus of canines (MVC) is an autonomous parvovirus that replicates efficiently without helper virusesin Walter Reed/3873D (WRD) canine cells. We previously showed that MVC infection induces mitochondrion-mediated apoptosis and G2/M-phase arrest in infected WRD cells. However, the mechanism responsible forthese effects has not been established. Here, we report that MVC infection triggers a DNA damage response ininfected cells, as evident from phosphorylation of H2AX and RPA32. We discovered that both ATM (ataxiatelangiectasia-mutated kinase) and ATR (ATM- and Rad3-related kinase) were phosphorylated in MVC-infected WRD cells and confirmed that ATM activation was responsible for the phosphorylation of H2AX,whereas ATR activation was required for the phosphorylation of RPA32. Both pharmacological inhibition ofATM activation and knockdown of ATM in MVC-infected cells led to a significant reduction in cell death, amoderate correction of cell cycle arrest, and most importantly, a reduction in MVC DNA replication andprogeny virus production. Parallel experiments with an ATR-targeted small interfering RNA (siRNA) had noeffect. Moreover, we identified that this ATM-mediated cell death is p53 dependent. In addition, we localizedthe Mre11-Rad50-Nbs1 (MRN) complex, the major mediator as well as a substrate of the ATM-mediated DNAdamage response pathway to MVC replication centers during infection, and show that Mre11 knockdown ledto a reduction in MVC DNA replication. Our findings are the first to support the notion that an autonomousparvovirus is able to hijack the host DNA damage machinery for its own replication and for the induction ofcell death.
Bocavirus is a newly classified genus of the family Parvoviri-nae and includes human bocavirus (HBoV), minute virus ofcanines (MVC), and bovine parvovirus (BPV). HBoV was re-cently associated with acute respiratory wheezing and pneu-monia (3, 44, 72) and is commonly detected in association withother respiratory viruses (44, 72). In addition to being linked torespiratory illnesses, HBoV has been associated with gastro-enteritic diseases (2, 4, 50, 53, 85). Within their respectivehosts, two closely related animal bocaviruses share these char-acteristics (12, 17, 42, 58, 66, 76). Although differentiated hu-man airway epithelial cells were recently shown to supportHBoV replication, the fact that this was at an extremely lowlevel (31) makes this system a difficult one to study HBoVbiology. MVC infection of Walter Reed/3873D (WRD) cells,however, has been proven much more efficient (11, 79). Usingthis system, we have shown that MVC infection induces mito-chondrion-mediated apoptosis, that this effect is dependent onreplication of the viral genome, and that the MVC genome perse is able to arrest the cell cycle at the G2/M phase (19).
Infection by many DNA viruses has been found to induce acellular DNA damage response (DDR), which can either blockor enhance viral DNA replication, as well as cell cycle arrest (inresponse to mild damage) or apoptosis (in response to irrep-arable damage), in infected cells (56). DNA damage rapidly
activates conserved DDR pathways (41, 75) that involve threephosphatidylinositol 3-kinase-like kinases (PI3Ks): ATM(ataxia telangiectasia-mutated kinase), ATR (ATM- andRad3-related kinase), and DNA-PK (DNA-dependent proteinkinase) (7, 54, 65). ATM is activated primarily as a result ofDNA double-strand breaks (DSBs) and is recruited to DSBs bythe Mre11-Rad50-Nbs1 (MRN) complex. ATR, on the otherhand, responds to the detection of single-stranded DNA(ssDNA) breaks and stalled DNA replication forks and is re-cruited to RPA-coated ssDNA by an ATR-interacting protein(ATRIP) (15, 41). Like ATM, DNA-PK is activated in re-sponse to DSBs, but it is recruited to the damage site incomplex with Ku70 and Ku80. Once recruited to a site ofdamage, ATM, ATR, and DNA-PK phosphorylate a numberof substrates (including H2AX, RPA, CHK1 and CHK2, p53,SMC1, Nbs1, and BRCA1) that in turn target other proteins,with the ultimate outcome being the silencing of cyclin-depen-dent kinases (CDKs) and an arrest of cell cycle progression topromote DNA repair or elimination of the potential hazardouscells by apoptosis (6, 41, 45).
Parvovirus contains a linear ssDNA genome with terminalrepeat structures at both ends (24). Adeno-associated virus 2(AAV2), a member of the genus Dependovirus of the familyParvovirinae, in the case of infection by (UV-inactivated)AAV2 alone, provokes a DDR that mimics stalled replicationforks, with both ATM and ATR being activated, resulting inthe phosphorylation of CHK1 and H2AX and G2-phase arrest(32, 43, 67). It is the p5 promoter sequence, rather than theAAV2 terminal repeats, that triggers the DDR (32). However,when AAV2 undergoes a productive infection in the presenceof adenovirus, AAV2 DNA replication activates a DDR that is
* Corresponding author. Mailing address: Department of Microbi-ology, Molecular Genetics and Immunology, University of KansasMedical Center, Mail Stop 3029, 3901 Rainbow Blvd., Kansas City, KS66160. Phone: (913) 588-4329. Fax: (913) 588-7295. E-mail: [email protected].
� Published ahead of print on 3 November 2010.
133
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
mediated primarily through the DNA-PK pathway and leads tophosphorylation of the downstream targets H2AX, RPA32,Nbs1, CHK1, CHK2, and SMC1 (22, 73) in the absence of theMRN complex (73). Replication of AAV2 requires degrada-tion of the MRN complex, an upstream regulator essential foractivation of the ATM pathway (74). For this, AAV2 requiresthe help of another virus, such as adenovirus. Adenovirus per secan induce a DDR and cell death (26). Therefore, a simplemodel for studying the relationships among parvovirus DNAreplication, DDR, and induced cell death has not been estab-lished.
In the current study, we provide the first evidence that in-fection by MVC, an autonomous parvovirus, triggers a DDRthat is represented by phosphorylation of both H2AX andRPA32. We show that both ATM- and ATR-mediated path-ways are involved in the MVC infection-induced DDR but thatonly the ATM-mediated pathway, which is sensed by the MRNcomplex, is critical for replication of the MVC genome andMVC infection-induced cell death.
MATERIALS AND METHODS
Cell and virus. WRD canine cells were maintained in Dulbecco’s modifiedEagle’s medium with 10% fetal calf serum in 5% CO2 at 37°C. The MVC strainused in this study is the original strain, GA3, which was isolated at the Collegeof Veterinary Science, Cornell University. MVC was cultured and quantified aspreviously described, and the virus titer was determined as the number of fluo-rescence focus-forming units (FFU) per ml (19). The WRD cell line and theMVC strain were obtained as gifts from Colin Parrish at Cornell University.WRD cells were infected with MVC at a multiplicity of infection (MOI) of 5.
Chemicals and treatment. Hydroxyurea (HU) (Calbiochem) was diluted todeionized water as a stock solution at 250 mM. Inhibitors CGK733, KU55933,NU7441, and wortmannin were bought from Calbiochem and were diluted indimethyl sulfoxide (DMSO) as stock solutions at 10 mM. Bromodeoxyuridine(BrdU) was purchased from Sigma and diluted in deionized water as a stocksolution at 5 mM.
WRD cells were seeded on 60-mm dishes 1 day prior to chemical treatment.KU55933, CGK733, NU7441, and wortmannin were applied to cells at finalconcentrations of 20 �M, 2.5 �M, 10 �M, and 10 �M, respectively, 3 h prior toinfection. DMSO (0.25%) was used as a control. HU was added to cells at a finalconcentration of 2.5 mM in parallel with MVC infection.
siRNA, plasmids, and transfection. Small interfering RNA (siRNA) oligonu-cleotides were synthesized as dicer substrate RNA interference (RNAi) at Inte-grated DNA Technologies (IDT, Coralville, IA). The following siRNA se-quences were chosen for targeting the genes of interest: siRNA specific to ATM(siATM), 5�-GUACUAGUUGCUUGUGUAACUGUA-3�; siRNA specific toATR (siATR), 5�-AGAAAGGAUUGUAGGCUAAUGGAA-3�; siRNA spe-cific to the DNA-PK catalytic subunit (siDNA-PKcs), 5�-CUAGGAAAUCCAUCGGUAUCAUUAA-3�; siRNA specific to Mre11 (siMre11), 5�-GGUCUUCUACUCUUAGGGUUGUUCCUU-3�; and siRNA specific to p53 (sip53), 5�-CCACCAUCCCUAAACUAAUGTG-3�. The following scrambled RNA(scrambled) was used as a siRNA control: 5�-CUUCCUCUCUUUCUCUCCCUUGUGA-3�. Transfection of all siRNAs was performed using Trifectin reagent(IDT) following the manufacturer’s instructions. At 48 h posttransfection, thecells were fed with fresh medium and infected with MVC.
MVC plasmids pIMVC, pIMVCNS1(�), pIMVCNP1(�), pIMVCVP1/2(�),and pMVCNSCap and the method for transfection have been described previ-ously (19, 79).
Antibodies. Anti-MVC NS1 and anti-MVC NP1 antisera were producedpreviously (19, 79). Anti-phosphorylated H2AX (anti-�H2AX) (MilliporeCorporation), anti-phosphorylated RAP32 (anti-p-RAP32) (Ser33) (BethylLaboratories, Inc.), anti-p-ATM (Ser1981) (Rockland Immunochemicals, Inc.),anti-Rad50 (GeneTex, Inc.), anti-p-SMC1 (Ser957) (Genscript USA, Inc.), andanti-ATM, anti-ATR, and anti-DNA-PKcs (Calbiochem, EMD Chemicals, Inc.)were used in this study. Both a monoclonal anti-Mre11 antibody (clone 12D7;GeneTex), which was generated by immunizing a truncated Mre11 from aminoacids (aa) 182 to 582, and a polyclonal anti-Mre11 antibody (C-16; Santa CruzBiotechnology, Inc.), which was raised against a peptide mapping near the Cterminus of human Mre11, were used to detect Mre11. Anti-p-ATR (Ser428),
anti-p-Nbs1 (Ser343), and anti-p-p53 (Ser15) were obtained from Cell Signaling,Inc. Antibody dilutions used for Western blotting and immunofluorescence anal-ysis were those suggested in the manufacturers’ instructions.
Western blotting and immunofluorescence. Western blotting and immunoflu-orescence assays were performed as previously described (19). Confocal imageswere taken at a magnification of �100 (objective lens) with an Eclipse C1 Plusconfocal microscope (Nikon) controlled by Nikon EZ-C1 software.
For BrdU incorporation, WRD cells were seeded on a chamber slide andinfected with MVC at an MOI of 5. At 18 h postinfection (p.i.), BrdU was addedinto the cell culture medium at a final concentration of 5 �M. At 24 h p.i., cellswere fixed and coimmunostained with rat anti-MVC NS1 and mouse anti-BrdUto mark the MVC replication centers.
Southern blotting. Low-molecular-weight DNA (Hirt DNA) was extractedfrom WRD cells, and DpnI digestion and Southern blotting were performedusing an MVC NSCap probe as described previously (79).
Virus titration assay. WRD cells were transfected with siRNAs for 48 h ortreated with inhibitors for 3 h prior to MVC infection (MOI of 5). At 48 h p.i.,both the cells and the medium were collected and lysed by repeated freezing andthawing. After lysis, the samples were briefly centrifuged and the supernatantswere collected for the virus titration assay.
WRD cells were seeded on 4-well chamber slides (Lab-Tek) 24 h prior toinfection. Virus samples were serially diluted 10-fold and added to each well. At24 h p.i., cells were fixed in 100% acetone, stained with anti-MVC NS1, and thenprocessed for the immunofluorescence assays. The number of fluorescence-positive cells in each chamber was counted. The number of focus-forming unitsin each well was calculated by multiplying the number of fluorescence-positivecells per chamber by the dilution of the virus-containing supernatant. The viraltiter is expressed as the average number of focus-forming units per ml of super-natant (FFU/ml).
Flow cytometry analysis. Live/Dead Violet staining for detection of cell deathand DAPI (4�,6-diamidino-2-phenylindole) staining for analysis of cell cycle wereperformed as described previously (19). All of the processed samples wereanalyzed on a three-laser flow cytometer (LSR II; BD Biosciences) at the FlowCytometry Core of the University of Kansas Medical Center. All flow cytometrydata were analyzed using FACSDiva software (BD Biosciences).
RESULTS
MVC infection causes a DDR in infected cells. To examinewhether a DNA damage response (DDR) is induced duringMVC infection, we evaluated the phosphorylation status ofH2AX and RPA32 in MVC-infected cells. First, we used BrdUincorporation to identify the MVC DNA replication centersand inspected whether anti-BrdU staining colocalizes with theMVC NS1 protein. As shown in Fig. 1A, NS1 was present atactive replication foci, as indicated by anti-BrdU staining. Insubsequent experiments, we used anti-NS1 staining as amarker for the MVC DNA replication centers.
MVC-infected cells were coimmunostained with anti-NS1and anti-phosphorylated H2AX (�H2AX) or with anti-NS1and anti-RPA32 phosphorylated at serine 33 (p-RPA32). Inparallel, we treated cells with hydroxyurea (HU), an agentknown to induce a DDR (5, 86), as a positive control. At 48 hp.i., we found that MVC infection led to significant increases inthe levels of both �H2AX and p-RPA32 in NS1-expressingcells (Fig. 1B and C, MVC-infected), with most of the NS1-expressing cells (red) also positive for anti-�H2AX or anti-p-RPA (green), respectively. Interestingly, p-RPA32 colocalizedwith NS1 at replication foci, but �H2AX did not (Fig. 1B andC, MVC-infected). In the HU-treated positive-control cells,�H2AX and p-RPA32 were also expressed in the nuclei (Fig.1B and C, HU-treated). Thus, MVC infection specifically in-duces the phosphorylation of H2AX and RPA32. Western blotanalysis revealed that H2AX and RPA32 were increasinglyphosphorylated over time (Fig. 1D). Both H2AX and RPA32were phosphorylated starting at 18 h p.i. and were maximally
134 LUO ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
phosphorylated at 36 h p.i.; this correlated with the level ofMVC replication as assessed by NS1 expression (Fig. 1D).Collectively, these findings show that MVC infection induces asignificant DDR that is correlated with MVC replication.
Both ATM and ATR are activated in the MVC infection-induced DDR. We examined which kinase pathway underliesthe DDR induced during MVC infection. First, we used phar-macological inhibitors to ATM, ATR, and DNA-PK to blockthe phosphorylation of these kinases. Both the ATM-specificinhibitor KU55933 (38) and the ATR/ATM-specific inhibitorCGK733 (1, 10, 25, 35, 89, 92) reduced �H2AX levels approx-imately 5-fold compared with those in DMSO control cells(Fig. 2A, lanes 3 and 4 versus lane 2); this did not hold true forthe DNA-PK inhibitor NU7441 (51) (Fig. 2A, lane 5).CGK733 also significantly interfered with the phosphoryla-tion of RPA32, whereas KU55933 and NU7441 did not (Fig.2B). The pan-PI3K inhibitor wortmannin (61) also inhibitedthe phosphorylation of RPA32 but not as potently as didCGK733 (Fig. 2B). Dephosphorylation of ATM by theinhibitors KU55933 and CGK733 was confirmed by anti-p-ATM staining (Fig. 2C, lanes 3 and 4), and ATR dephosphor-ylation by the inhibitor CGK733 was confirmed by anti-p-ATRstaining (Fig. 2D, lane 4).
We next used siRNA molecules to specifically knock downATM, ATR, and the DNA-PK catalytic subunit (DNA-PKcs).As shown in Fig. 3A, the ATM-specific siRNA inhibited ATMexpression approximately 4-fold compared to that seen withthe scrambled siRNA control (Fig. 3A). Consistent with aninhibition of ATM expression, the ATM siRNA reduced�H2AX more than 5-fold, to the background level (Fig. 3D,lane 3), but did not affect phosphorylation of RPA32 (Fig. 3E,lane 3). Similarly, the ATR-specific siRNA reduced ATR ex-pression (Fig. 3B) and accordingly decreased the level of phos-phorylated RPA32 to the background level in the mock controlbut not that of �H2AX (Fig. 3D and E, lane 4 versus lane 1).However, neither phosphorylation event was diminished inMVC-infected cells treated with the DNA-PKcs-specificsiRNA (Fig. 3D and E, lane 5), in spite of the fact that the
FIG. 1. MVC infection induces a DDR. (A) Detection of the MVCDNA replication foci using BrdU incorporation. Punctate staining byanti-NS1 (red) or anti-BrdU (green) antibody indicates the replicationfoci. (B and C) Immunofluorescence analysis of MVC infection-in-duced DDR. At 48 h p.i., MVC-infected cells were coimmunostainedwith anti-MVC NS1 (red) and anti-�H2AX (green) (B) or anti-MVCNS1 (red) and anti-p-RPA (Ser33) (green) (C). Cells treated withhydroxyurea (HU) were used as a positive control for an induced DDR(5, 86). Nuclei were marked by DAPI staining. (D) Western blotting ofMVC infection-induced DDR. MVC-infected cells were harvested atthe indicated time points and analyzed by Western blotting. In additionto the levels of the MVC NS1 protein, phosphorylation of the DDRmarkers H2AX and RPA32 was assessed using the appropriate anti-bodies. The same membrane was reprobed with each antibody. HU-treated cells served a positive control, and anti-�-actin was used as aloading control. The arrowhead shows phosphorylated RPA32 atserine 33; two bands of phosphorylated RPA32 were detected in HU-treated cells.
FIG. 2. Treatment with pharmacological inhibitors of ATM andATR reduces MVC infection-induced phosphorylation of H2AX andRPA32, respectively. WRD cells were treated with DMSO (0.25%)and inhibitors and then infected with MVC at 3 h posttreatment.MVC-infected cells were harvested at 48 h p.i. and were lysed forimmunoblotting using anti-�H2AX (A), anti-p-RPA (Ser33) (B), anti-p-ATM (Ser1981) (C), and anti-p-ATR (Ser428) (D). In all blots,anti-�-actin was used to control for loading. Mock-infected and HU-treated cells were used as a negative and a positive control, respec-tively.
VOL. 85, 2011 BOCAVIRUS DDR ROLE IN DNA REPLICATION AND CELL DEATH 135
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
DNA-PKcs-specific siRNA inhibited nearly 90% of the DNA-PKcs (Fig. 3C, lane 3).
Together, these results show that the increase in �H2AXduring MVC infection is mediated by ATM phosphorylation,whereas the increase in RPA32 phosphorylation is a conse-quence of ATR activation. Thus, the MVC infection-inducedDDR appears to involve activation of both the ATM and theATR pathway. Due to the lack of an antibody to detect phos-phorylated canine DNA-PK, we were not able to test whetherDNA-PK is phosphorylated during MVC infection. However,based on results using the DNA-PKcs-specific siRNA, we be-lieve that DNA-PK is less likely to be involved in the DDRinduced by MVC infection.
The ATM-mediated DDR plays an important role in induc-ing the cytopathic effects that occur during MVC infection. Weused the above-described pharmacological inhibitors andsiRNAs to examine the effects of inhibiting ATM, ATR, andDNA-PK on the cell death that is triggered in WRD cells byMVC infection (19). At 24 and 48 h p.i., the cells were har-vested for flow cytometry analysis with the cell death markerdye Live/Dead Violet and an anti-MVC NS1 antibody. NS1-expressing cells were selectively gated to determine the per-centage of dead cells. We found that when either an ATMinhibitor (KU55933 or CGK733) or an ATM-specific siRNAwas applied to MVC-infected cells, cell death at 48 h p.i. wassignificantly inhibited (Fig. 4). More specifically, treatmentwith KU55933 and CGK733 reduced cell death by 55% and
FIG. 3. Treatment with siRNAs targeting ATM and ATR reducesMVC infection-induced phosphorylation of H2AX and RPA32, re-spectively. WRD cells were transfected with the indicated siRNA tosilence ATM, ATR, or DNA-PK. The cells were infected with MVC at48 h posttransfection. (A to C) To confirm the efficiency of knock-down, at 48 h p.i., cells were collected and lysed for immunoblottingusing antibodies against ATM (A), ATR (B), and DNA-PKcs (C). Ascrambled siRNA served as a negative control. (D and E) To assess theeffect of the knockdown on the DDR, at 48 h p.i., cells were collectedand lysed for immunoblotting using anti-�H2AX (D) and anti-p-RPA(Ser33) (E). Both blots were reprobed with anti-�-actin antibody.Mock-infected cells were used as a background control.
FIG. 4. Both inhibition of ATM phosphorylation and knockdown of ATM expression significantly reduce MVC infection-induced cell death.WRD cells were treated with inhibitors for 3 h prior to MVC infection or transfected with the indicated siRNAs 48 h prior to MVC infection.(A) At 24 or 48 h p.i., cells were collected and analyzed using Live/Dead Violet and anti-NS1 costaining by flow cytometry. The result shown isone representative from three independent experiments. Percentages of dead cells in NS1-expressing cells are shown in each histogram.(B) Statistical analysis of the percentages of dead cells in NS1-expressing cells from three independent experiments. Averages (numerical values)and standard deviations (error bars) are shown for each treatment group.
136 LUO ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
84%, respectively, over that seen in the DMSO control, andapplication of the ATM-specific siRNA reduced cell death by70% compared to that achieved when the scrambled siRNAcontrol was applied. Treatment of the cells with the ATR-specific siRNA resulted in only a slight inhibition of cell death,by approximately 11% (Fig. 4). In contrast, application of nei-ther the DNA-PK inhibitor NU7441 nor DNA-PKcs-specificsiRNA resulted in significant inhibition of the cell death in-duced by MVC infection at 48 h p.i. (Fig. 4). These resultssuggest that ATM activation is important to the cell deathinduced by MVC infection, whereas the ATR pathway con-tributes minimally. At 24 h p.i., cells in the DMSO controlgroup did not undergo cell death at a significant level; there-fore, inhibitory effects of the ATM inhibitor or of the siRNAcould not easily be evaluated (Fig. 4). Moreover, application ofthe ATM-specific siRNA inhibited phosphorylation of p53 atserine 15 (Fig. 5A). Knockdown of p53 by a p53-specific siRNAreduced cell death by approximately 61% compared to thatachieved with the treatment with the scrambled siRNA (Fig.5B and C) but did not correct the cell cycle arrest (Fig. 5D).
Thus, our results suggest that MVC infection induces an ATM-mediated and p53-dependent cell death.
We reported previously that a G2/M cell cycle arrest occursduring late MVC infection (19). When KU55933 and CGK733were applied to WRD cells prior to infection, the G2/M arrest,which normally occurs at 48 h p.i., was inhibited to someextent; only approximately 56% (CGK733) and 67%(KU55933) were in the G2/M phase, compared to 76% in theDMSO control sample (Fig. 6). In contrast, the DNA-PK in-hibitor had no effect (Fig. 6). Consistent with these results, onlythe ATM siRNA reduced the MVC infection-induced G2/Marrest, by approximately 16% compared to that seen in thecells treated with the control scrambled siRNA (Fig. 6); nei-ther the ATR- nor the DNA-PKcs-specific siRNA had an ef-fect (Fig. 6). These results suggest that ATM activation is likelyinvolved in the G2/M cell cycle arrest during MVC infection.
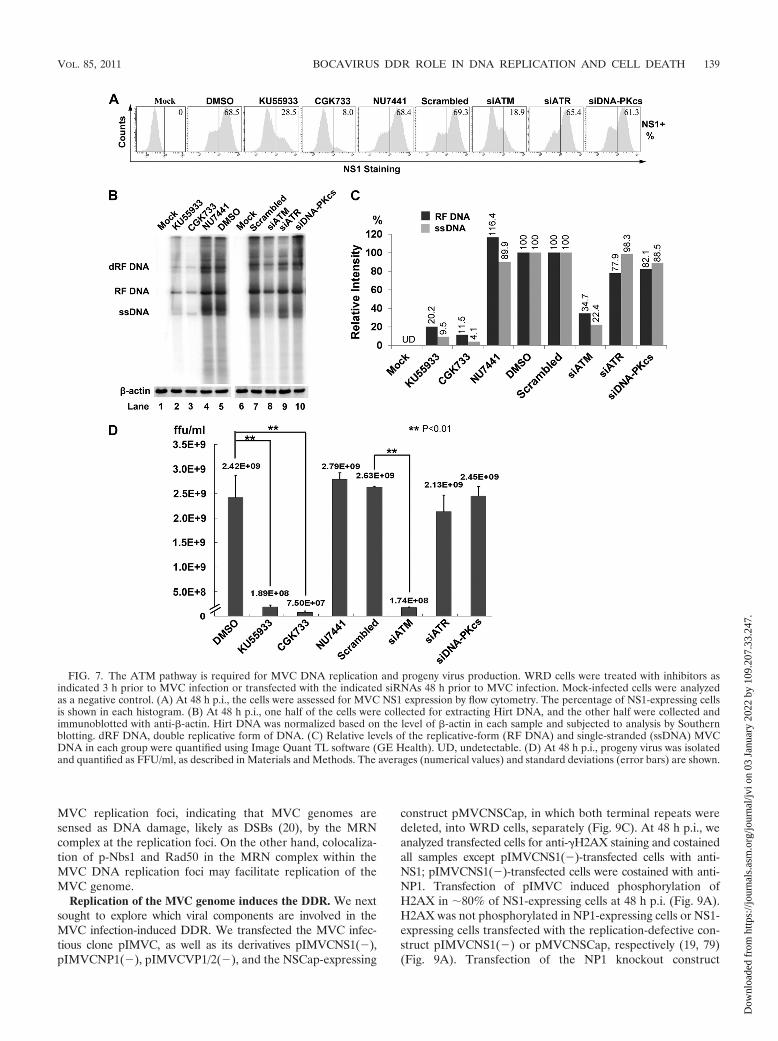
The ATM-mediated DDR is required for replication of theMVC genome. To test whether MVC replication was impairedby deactivation of any of the three DDR pathways, we evalu-ated the percentage of MVC-infected cells by intracellularstaining using anti-NS1 antiserum. A typical experiment isshown in Fig. 7A, where, at 48 h p.i., approximately 70% ofWRD cells were infected with MVC (Fig. 7A, DMSO andScrambled). In cells treated with KU55933 or CGK733, NS1-expressing cells were decreased to 41.6% or 11.6%, respec-tively, of the numbers seen in the control, indicating that ATMinactivation reduced MVC infection. Furthermore, knockdownof ATM using an ATM-specific siRNA led to a 72% decreasein NS1-expressing cells compared to the number in the scram-bled siRNA control group (Fig. 7A). However, treatment witheither an ATR- or a DNA-PKcs-specific siRNA failed to re-duce the number of NS1-expressing cells significantly (Fig.7A). These results indicate that ATM activation may facilitateMVC replication.
To confirm the role of ATM activation in MVC DNA rep-lication, we treated WRD cells with our panel of kinase inhib-itors and siRNAs and then analyzed MVC DNA replication bySouthern blotting. As shown in Fig. 7B, treatment of cells witheither KU55933 or CGK733 reduced the level of the replica-tive form (RF DNA) of the MVC DNA approximately 5-fold(Fig. 7B, lanes 2 and 3 versus lane 5), whereas treatment withthe DNA-PK inhibitor NU7441 did not (Fig. 7B, lane 4). No-tably, both KU55933 and CGK733 significantly blocked syn-thesis of the MVC ssDNA, approximately 10-fold (Fig. 7B,lanes 2 and 3 versus lane 5); this effect of ATM inhibition wasmore pronounced when the ATM-specific siRNA was applied(Fig. 7B, lane 8 versus lane 7). In contrast, ATR- and DNA-PKcs-specific siRNAs failed to inhibit synthesis of both the RFDNA and the ssDNA of MVC (Fig. 7B, lanes 9 and 10). Theinhibition of ssDNA synthesis was confirmed by measuringprogeny virus production from MVC-infected cells subjectedto each treatment. As expected, the virus titers in the ATM-inhibited groups (KU55933, CGK733, and siATM treated)were reduced more than 12-fold compared with those in theirrespective control groups (Fig. 7D, Mock and Scrambled).Consistent with results from Southern blotting, CGK733 treat-ment reduced progeny virus production 32-fold; however, theinhibition of ATR alone using an ATR-specific siRNA did notsignificantly decrease the production of progeny virus (Fig. 7D,siATR). Likewise, treatment of cells with a DNA-PK-specific
FIG. 5. MVC infection-induced cell death is dependent on phos-phorylation of p53. WRD cells were transfected with the indicatedsiRNAs 48 h prior to MVC infection. (A) At 48 h p.i., cells werecollected and lysed for immunoblotting using antibodies against phos-phorylated p53 at serine 15. The blot was reprobed with anti-�-actin.(B and C) Cells were collected and analyzed using Live/Dead Violetand anti-NS1 costaining by flow cytometry. (B) Results from a repre-sentative experiment are shown, with percentages of dead cells inNS1-expressing cells given in each histogram. (C) Averages (numericalvalues) and standard deviations (error bars) are indicated for eachtreatment group. (D) Cells were collected and analyzed for cell cycleusing DAPI and anti-NS1 costaining by flow cytometry. A representa-tive experiment is shown, with percentages of cells in each cell cycle.
VOL. 85, 2011 BOCAVIRUS DDR ROLE IN DNA REPLICATION AND CELL DEATH 137
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
inhibitor or siRNA did not affect the production of progenyvirus compared to that seen in the respective controls (Fig. 7D,NU7441 and siDNA-PKcs).
Taken together, these results show that the ATM-mediatedDDR is essential to MVC DNA replication and that this par-ticular DDR is the most important with respect to synthesis ofthe MVC ssDNA genome during infection. This is the firsttime to demonstrate that an autonomous parvovirus is able tohijack the cellular DNA damage response machinery for itsproductive replication.
The MRN complex facilitates MVC DNA replication. Toexamine whether the MRN complex is involved in MVC DNAreplication, we first evaluated whether this complex forms inearly infection. At 24 h p.i., Mre11 in infected cells colocalizedwith the replication foci (punctate patterns) as well as withRad50 and phosphorylated Nbs1 (p-Nbs1) (Fig. 8A); in unin-fected cells, Mre11, Rad50, and p-Nbs1 were broadly distrib-uted throughout the nuclei without forming bright foci (datanot shown). As MVC infection proceeded, Mre11 was de-graded, as evident from a decrease in Mre11 in the MVCreplication foci at 48 h p.i. (Fig. 8A). Importantly, Rad50 andp-Nbs1 remained at the same level in these locations as at 48 hp.i. (Fig. 8A). Immunoblotting revealed a clear transition ofMre11 expression between 24 and 36 h p.i. (Fig. 8B, Mre11);this period corresponds to the time point that is critical forreplication of the MVC DNA (Fig. 8B, NS1). Notably, startingat 18 h p.i., we observed a smaller band of approximately 70kDa (Fig. 8B, Mre11a). This small Mre11 band reached amaximal level at 24 h p.i. and decreased in late infection; thiscorrelates with the timing of MVC DNA replication, as indi-cated by anti-NS1 staining (Fig. 8B, NS1). It was not expressed
in HU-treated cells (Fig. 8B) and was confirmed not to be aviral protein (data not shown). We speculate that it is an activeform of Mre11, which may play the important role of sensingDSBs and recruiting p-Nbs1 and Rad50 to the MVC replica-tion foci. We used another anti-Mre11 antibody to confirm thatMre11 is degraded. Indeed, a slight reduction of the Mre11band was observed, and only in infected cells, at 24 and 36 h p.i.(Fig. 8B, Mre11b). In contrast, the level of Rad50 remainedconstant throughout MVC infection (Fig. 8B, Rad50).
To further explore the role of Mre11 in MVC DNA repli-cation, we knocked down Mre11 and then measured MVCDNA replication. Unlike the loss of MRN complex function,which occurs during replication of AAV2 when it is coinfectedwith adenovirus (74), Mre11 knockdown led to a significantdecrease, approximately 3-fold, in MVC DNA replication (Fig.8C and D). We speculated that this was caused by the failureto activate ATM, since the MRN complex acts as an upstreamregulator of the ATM pathway (52, 63, 81). Indeed, we foundthat silencing of Mre11 reduced ATM phosphorylation signif-icantly, to a level similar to that produced by ATM knockdown(Fig. 8E). Moreover, phosphorylation of the ATM substrateSMC1 (47, 71, 93) was reduced in this context (Fig. 8E). Inaddition, we found that in contrast to the results we obtainedby silencing Mre11, knockdown of ATM reduced Nbs1 phos-phorylation only slightly, indicating that a low level of activatedATM is sufficient to phosphorylate Nbs1. As Nbs1 is essentialfor DSB repair and genome stability (29), the persistent pres-ence of p-Nbs1 in the MVC replication foci (Fig. 8A) suggeststhat this activated form may play a role in replication of theMVC genome.
These results show that the MRN complex localizes to the
FIG. 6. Both inhibition of ATM phosphorylation and knockdown of ATM attenuate MVC infection-induced G2/M arrest. (A) WRD cells weretreated with inhibitors for 3 h prior to MVC infection or transfected with siRNAs 48 h prior to MVC infection. At 48 h p.i., the cells were subjectedto flow cytometry for an assessment of G2/M arrest using DAPI and anti-NS1 costaining. The percentage of cells in each phase of the cell cyclewas quantified and is shown at the bottom. Significant changes in the numbers of cells in the G2/M phase are shown. Mock-infected cells wereanalyzed as a normal cell cycle control. (B) Statistical analysis of the percentage of cells in the G2/M phase in NS1-expressing cells from threeindependent experiments. Averages (numerical values) and standard deviations (error bars) are shown for each treatment group.
138 LUO ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
MVC replication foci, indicating that MVC genomes aresensed as DNA damage, likely as DSBs (20), by the MRNcomplex at the replication foci. On the other hand, colocaliza-tion of p-Nbs1 and Rad50 in the MRN complex within theMVC DNA replication foci may facilitate replication of theMVC genome.
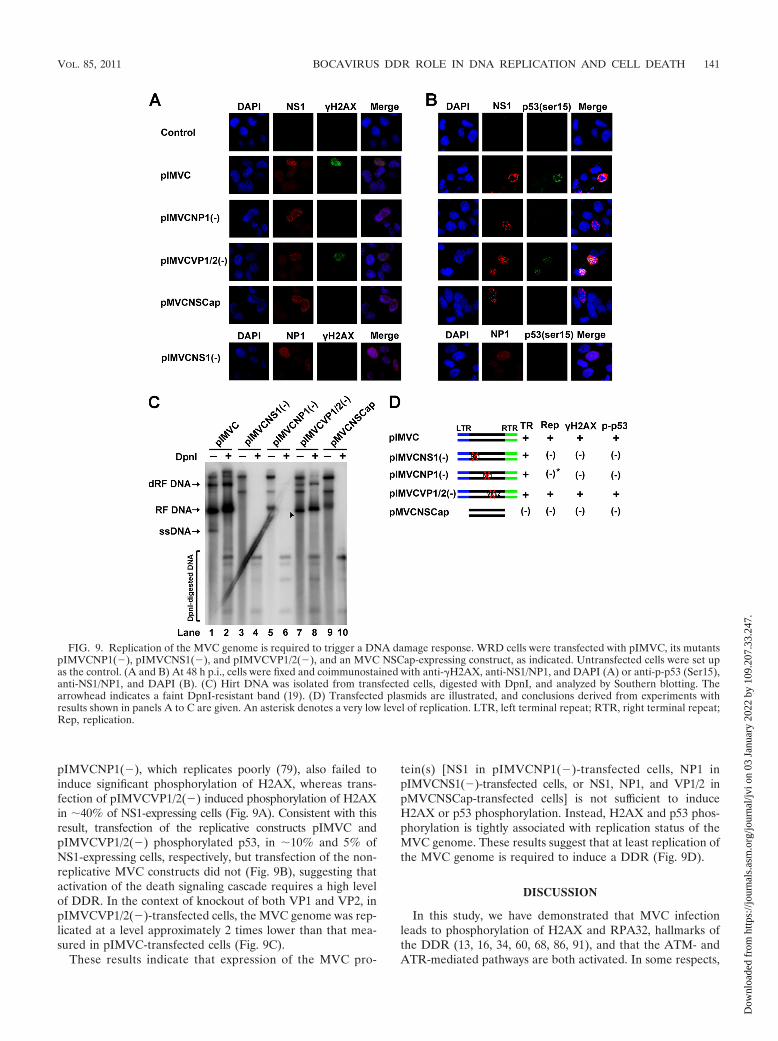
Replication of the MVC genome induces the DDR. We nextsought to explore which viral components are involved in theMVC infection-induced DDR. We transfected the MVC infec-tious clone pIMVC, as well as its derivatives pIMVCNS1(�),pIMVCNP1(�), pIMVCVP1/2(�), and the NSCap-expressing
construct pMVCNSCap, in which both terminal repeats weredeleted, into WRD cells, separately (Fig. 9C). At 48 h p.i., weanalyzed transfected cells for anti-�H2AX staining and costainedall samples except pIMVCNS1(�)-transfected cells with anti-NS1; pIMVCNS1(�)-transfected cells were costained with anti-NP1. Transfection of pIMVC induced phosphorylation ofH2AX in �80% of NS1-expressing cells at 48 h p.i. (Fig. 9A).H2AX was not phosphorylated in NP1-expressing cells or NS1-expressing cells transfected with the replication-defective con-struct pIMVCNS1(�) or pMVCNSCap, respectively (19, 79)(Fig. 9A). Transfection of the NP1 knockout construct
FIG. 7. The ATM pathway is required for MVC DNA replication and progeny virus production. WRD cells were treated with inhibitors asindicated 3 h prior to MVC infection or transfected with the indicated siRNAs 48 h prior to MVC infection. Mock-infected cells were analyzedas a negative control. (A) At 48 h p.i., the cells were assessed for MVC NS1 expression by flow cytometry. The percentage of NS1-expressing cellsis shown in each histogram. (B) At 48 h p.i., one half of the cells were collected for extracting Hirt DNA, and the other half were collected andimmunoblotted with anti-�-actin. Hirt DNA was normalized based on the level of �-actin in each sample and subjected to analysis by Southernblotting. dRF DNA, double replicative form of DNA. (C) Relative levels of the replicative-form (RF DNA) and single-stranded (ssDNA) MVCDNA in each group were quantified using Image Quant TL software (GE Health). UD, undetectable. (D) At 48 h p.i., progeny virus was isolatedand quantified as FFU/ml, as described in Materials and Methods. The averages (numerical values) and standard deviations (error bars) are shown.
VOL. 85, 2011 BOCAVIRUS DDR ROLE IN DNA REPLICATION AND CELL DEATH 139
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
FIG. 8. The MRN complex is an upstream regulator of the ATM pathway and facilitates replication of the MVC genome. (A) Immu-nofluorescence analysis of colocalization of the MRN complex in the MVC replication center. At 24 or 48 h p.i. as indicated, Mre11,phosphorylated Nbs1 at serine 343 (p-Nbs1), and Rad50 were examined for colocalization with MVC NS1 by use of their respectiveantibodies. A monoclonal anti-Mre11 antibody (clone 12D7) was used to detect Mre11. (B) Western blot analysis of MVC-infected cells.MVC-infected cells were collected at various times postinfection as indicated and were analyzed by immunoblotting for the level of Mre11.Mre11 was examined using an anti-Mre11 monoclonal antibody (Mre11a) and a polyclonal antibody (Mre11b). The blot was reprobed usinganti-Rad50 (Rad50), anti-NS1 (NS1), and anti-�-actin (�-actin) antibodies, sequentially. (C and D) WRD cells were transfected with theMre11-specific siRNA (siMre11) and a scrambled siRNA as a control 48 h prior to MVC infection. Mock-infected cells were used as anegative control. At 48 h p.i., cells were collected and immunoblotted with anti-Mre11 and reprobed using anti-�-actin (C); Hirt DNA wasprepared from infected cells, normalized based on the level of �-actin in each sample, and analyzed by Southern blotting (D). (E) WRD cellswere transfected with siRNAs as indicated 48 h prior to MVC infection. At 48 h p.i., the cells were collected and immunoblotted withanti-Mre11 (C-16; Santa Cruz) and reprobed sequentially with anti-p-ATM (Ser1918), anti-p-SMC1 (Ser957), anti-p-Nbs1 (Ser343), andanti-�-actin. Mock-infected cells were used as a background control.
140 LUO ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
pIMVCNP1(�), which replicates poorly (79), also failed toinduce significant phosphorylation of H2AX, whereas trans-fection of pIMVCVP1/2(�) induced phosphorylation of H2AXin �40% of NS1-expressing cells (Fig. 9A). Consistent with thisresult, transfection of the replicative constructs pIMVC andpIMVCVP1/2(�) phosphorylated p53, in �10% and 5% ofNS1-expressing cells, respectively, but transfection of the non-replicative MVC constructs did not (Fig. 9B), suggesting thatactivation of the death signaling cascade requires a high levelof DDR. In the context of knockout of both VP1 and VP2, inpIMVCVP1/2(�)-transfected cells, the MVC genome was rep-licated at a level approximately 2 times lower than that mea-sured in pIMVC-transfected cells (Fig. 9C).
These results indicate that expression of the MVC pro-
tein(s) [NS1 in pIMVCNP1(�)-transfected cells, NP1 inpIMVCNS1(�)-transfected cells, or NS1, NP1, and VP1/2 inpMVCNSCap-transfected cells] is not sufficient to induceH2AX or p53 phosphorylation. Instead, H2AX and p53 phos-phorylation is tightly associated with replication status of theMVC genome. These results suggest that at least replication ofthe MVC genome is required to induce a DDR (Fig. 9D).
DISCUSSION
In this study, we have demonstrated that MVC infectionleads to phosphorylation of H2AX and RPA32, hallmarks ofthe DDR (13, 16, 34, 60, 68, 86, 91), and that the ATM- andATR-mediated pathways are both activated. In some respects,
FIG. 9. Replication of the MVC genome is required to trigger a DNA damage response. WRD cells were transfected with pIMVC, its mutantspIMVCNP1(�), pIMVCNS1(�), and pIMVCVP1/2(�), and an MVC NSCap-expressing construct, as indicated. Untransfected cells were set upas the control. (A and B) At 48 h p.i., cells were fixed and coimmunostained with anti-�H2AX, anti-NS1/NP1, and DAPI (A) or anti-p-p53 (Ser15),anti-NS1/NP1, and DAPI (B). (C) Hirt DNA was isolated from transfected cells, digested with DpnI, and analyzed by Southern blotting. Thearrowhead indicates a faint DpnI-resistant band (19). (D) Transfected plasmids are illustrated, and conclusions derived from experiments withresults shown in panels A to C are given. An asterisk denotes a very low level of replication. LTR, left terminal repeat; RTR, right terminal repeat;Rep, replication.
VOL. 85, 2011 BOCAVIRUS DDR ROLE IN DNA REPLICATION AND CELL DEATH 141
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
the DDR induced by MVC infection is beneficial to virusinfection, i.e., facilitating replication of viral DNA, especiallysynthesis of ssDNA, as well as inducing cell death, which isessential for virus egress. On the other hand, the MVC infec-tion-induced DDR is detrimental to the host in that it leads toactivation of cell cycle checkpoints and apoptosis of infectedcells. Notably, the DDR was not triggered by expression ofviral proteins and the delivery of plasmids containing the non-replicative MVC DNA but rather by replication of the MVCDNA. Thus, we provide convincing evidence that a DDR in-duced by autonomous parvovirus plays critical roles in the viruslife cycle and virus infection-induced cytopathic effects.
MVC infection induced a DDR mediated by both the ATMand the ATR pathway. ATR and its downstream effectorRPA32 were phosphorylated during MVC infection, and phos-phorylated RPA32 colocalized with MVC NS1 in the replica-tion centers (Fig. 1B). Interestingly, RPA32 is an ssDNA bind-ing protein that is essential for replication of both the minutevirus of mice (MVM) (21) and the AAV2 (62) genome. Acti-vation of ATR and subsequent phosphorylation of RPA32have been shown to play a pivotal role in the DDR induced byinfection with UV-inactivated AAV2 (32, 43). Moreover,AAV2 DNA replication activates DNA-PK, which then phos-phorylates RPA32 at multiple sites (73). RPA32 phosphoryla-tion also has been shown to increase as infection by Epstein-Barr virus (EBV) progresses (48). However, in our study, wefound that inhibition of RPA32 phosphorylation by silencingATR did not affect MVC DNA replication. In fact, it has beenreported that RPA32 phosphorylation appears to occur out-side the cellular replication sites (33, 84). CGK733 inhibitsboth ATM and ATR activation (1, 10, 25, 35, 89, 92) and wasthe most effective inhibitor of DDR-mediated cell death andcell cycle arrest in MVC-infected cells. We believe that thesepotent inhibitory effects are due to the fact that CGK733 is amore potent inhibitor of ATM than is the ATM-specific inhib-itor KU55933 in WRD cells, rather than due to its additionalinhibition of ATR phosphorylation.
The DDR that is induced during simian virus 40 (SV40)infection has been suggested to be activated by the large Tantigen via Bub1 binding (37). The large T antigen also inter-acts with the MRN complex (30, 49, 90). The human papillo-mavirus (HPV) E7 protein directly binds to ATM, and thisinduces an ATM-mediated DDR upon HPV infection of dif-ferentiated epithelia (59). In parvoviruses, both AAV2 Rep78and parvovirus H-1 NS1 have been implicated in the phosphor-ylation of H2AX (9, 39), which was hypothesized to occur as aresponse by either nonspecific nicking of the cellular DNA byRep78 (9) or NS1-induced reactive oxygen species (ROS) as aDNA damage agent (39). However, Rep78 accounts for only asmall portion of the DDR that is induced during AAV2 rep-lication (73). Notably, our results suggest that neither NS1 nora stalled replication fork (Fig. 9), which would potentially beassembled in the region of the MVC replication origin toactivate ATR (32, 43), is responsible for the DDR inducedduring MVC infection. The low level of DNA replicationachieved by transfection of the NP1-deficient infectious clone[pIMVCNP1(�)] in WRD cells failed to induce a clear DDR.Given that a moderate level of genome replication is absolutelyrequired for the MVC-induced DDR, we hypothesize thatspecific nicking of the replicative form (RF) of the MVC ge-
nome by the helicase activity of NS1 may create lesions thatmimic DSBs (Fig. 10, c) and that a DDR is triggered when thissignal accumulates to a certain level. In fact, ATM is a primecandidate for mediating the cellular damage response to DSBs(52), as DSBs are sensed by the MRN complex, which triggersATM-mediated H2AX phosphorylation (52, 64). How ATM isactivated during virus infection is not clearly understood.Based on studies of herpes simplex virus type 1 (HSV-1), it wasproposed that DSBs may arise as a consequence of replicationfork collapse at sites of oxidative damage (57, 82), possibly dueto cleavage of the viral � sequences by endonuclease G duringgenome isomerization (40, 88). Parvovirus NS1 nicks only thepositive strand of the RF DNA at the terminal resolution site(23), and it has been shown that opening of the DNA helix isrequired for MRN complex stimulation of functional ATM(52, 63). Self-complementary recombinant AAV2 (scAAV), ofwhich the genome is an RF DNA, contains palindromic hair-pin-structured terminal repeats, which resemble a repair inter-mediate of DSBs (20). Thus, NS1-nicked RF DNA and un-paired (replication) intermediates (Fig. 10, d and e) might beperfectly opened DNA helices that function as DSBs and trig-ger ATM activation.
In addition, delivery of the AAV2 genome by UV-inacti-vated AAV2 has been proven to induce an ATM/ATR-medi-ated DDR (32, 43, 67), which differs from the DNA-PK-me-diated DDR induced by AAV2 DNA replication (22, 73). Inour study, we observed that the extent of DDR induced byMVC DNA replication somehow correlated with the replica-tion efficiency and the accumulation of the ssDNA genomeduring infection. Interestingly, H2AX was significantly phos-phorylated when cells were inoculated with UV-inactivatedMVC at a high MOI of 40 but not at a low MOI of 5 (data not
FIG. 10. Proposed DNA damage response pathways induced dur-ing MVC infection. The proposed pathways are described in detail inthe Discussion. The model of MVC DNA replication refers to DNAreplication of the minute virus of mice (23). Bax translocalization andcaspase activation have been shown previously (19) to induce apoptoticcell death during MVC infection, and upregulation of cyclin B/CDK1was confirmed to be responsible for the G2/M arrest that is inducedduring MVC infection (19). L, left terminal repeat; R, right terminalrepeat.
142 LUO ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
shown). Thus, we speculate that the accumulated ssDNA ge-nome of MVC may also contribute to the DDR induced duringinfection (Fig. 10, g), which warrants further investigation.
Based on the information summarized above, we hypothe-size that during the virus life cycle, replication of the MVCgenome leads to an accumulation of strand breaks and thatthese are registered as DSBs and thus trigger ATM activation.RPA-coated ssDNA breaks, on the other hand, could po-tentially trigger ATR activation (Fig. 10, a and f) duringreplication (95). Notably, MVM infection also induced anATM-activated DDR that helps MVM DNA replication inMVM-permissive cells (David Pintel, personal communica-tion). Both MVC and MVM are autonomous parvoviruses,meaning that replication of their genomes does not require thefunction from a helper virus. Thus, the MVC and MVM in-fection systems both provide simple models in which to studythe DDR induced by the ssDNA genome of parvovirus. It isnow clear that, like other DNA viruses (27, 56), the autono-mous parvovirus hijacks the cellular DDR machinery to facil-itate replication of its genome.
Only the ATM activation-mediated DDR facilitates MVCDNA replication and elicits cell death in MVC-infected cells.Although MVC infection induces activation of both ATM andATR, we found that only the ATM-mediated DDR contributesto MVC DNA replication and cell death. In response to DNAdamage, cells activate a complex network of factors (6, 45) thatsilence CDKs and thereby arrest the cell cycle, promotingDNA repair (8). Interestingly, MVC infection impaired cellproliferation and disturbed the cell cycle, allowing a transitionfrom the S-phase accumulation to the G2/M arrest as theinfection progresses (19). Thus, the MVC infection-inducedDDR supports replication of the viral DNA by first arrestingcell cycle progression at the S phase and then impairing the cellcycle at the G2/M phase to prevent mitosis, which would leadto apoptotic cell death. On the other hand, if the cell sustainsDNA damage that cannot be repaired, the DDR triggers acascade of apoptotic cell death, through either a p53-depen-dent or a p53-independent pathway (69). We found that p53was phosphorylated upon MVC infection and that it was de-phosphorylated in the context of ATM inactivation (Fig. 5).Furthermore, replication of transfected MVC RF DNA in-duced phosphorylation of p53, albeit at a low level (Fig. 9),which presumably is due to the low level of DNA replication bytransfection compared with that during MVC infection. Thesefindings suggest that phosphorylated ATM activates apoptosisin a p53-dependent manner (Fig. 10). Bax translocalizationand caspase activation have been shown to occur during celldeath triggered by MVC infection (19). We hypothesize thatphosphorylated p53 may activate the BH3-only molecules, e.g.,tBID, BIM, and PUMA, which further activate Bax/Bak (46).
We have demonstrated that the ATM-mediated DDR isinvolved to some extent in cell cycle arrest, which is p53 inde-pendent. We believe that CHK2 (checkpoint kinase 2) likelysignals to activate this ATM-mediated G2/M arrest (6). Nota-bly, we did not observe a clear DDR in cells transfected withnonreplicative and poorly replicating MVC constructs (Fig. 9).The cell cycle of these transfected cells, however, was arrestedat G2/M phase (19). We think that the DDR-induced replica-tion of the MVC genome may not fully account for the cellcycle arrest during MVC infection and that an unknown mech-
anism may contribute to the cell cycle arrest induced by theviral genome, specifically the terminal repeats (19). Actually,both inhibition of ATM activation and knockdown of ATMonly moderately rescued the cell cycle arrest. It could also bethat a low level of DDR induced by the MVC genome, whichis able to induce cell cycle arrest but not cell death (19), is notsufficient to induce a significant increase of �H2AX. There-fore, in the context of the DDR induced during MVC infec-tion, it may be easier to prevent cell death than to preventarrest of the cell cycle.
The MRN complex localizes to the MVC replication center.The MRN complex is involved in the initial processing of DSBsas a sensor and is required for ATM activation by DNA dam-age (52, 63, 81, 83). It is also critical to the repair of DNAdamage (28, 78). The MRN complex is required to signal DDRinduction during HSV-1 infection (55), mutant adenovirus in-fection (18), and HPV infection (59). In contrast, the MRNcomplex has to be destroyed during adenovirus (77), AAV2(74), and SV40 (94) infections. During MVC infection, theMRN complex was assembled at early stages of infection, butMre11 was slightly degraded at later stages, while the virus wasactively replicating. A loss of Mre11 at later times followingHSV-1 infection has also been reported (36). However, theMRN complex was colocalized to the MVC replication centerduring the course of MVC infection. Further evidence thatMre11 knockdown reduced MVC replication approximately3-fold (Fig. 8) strongly supports the notion that the MRNcomplex is required for replication of the MVC genome (Fig.10) and that its role may be to recruit DNA repair factors tothe replication center (14, 37), as p-Nbs1 is essential to DSBrepair (29). On the other hand, the MRN complex senses ATMactivation, which in turn may mediate proteasome-dependentdegradation of the MRN subunit (94) at later stages of infec-tion.
HSV-1, SV40, and HPV have all been shown to induce theATM-mediated DDR, whereby a number of repair factors arerecruited to the replication center (59, 80, 87, 94). Exactly howthe DDR microenvironment helps viral DNA replication islargely unknown. DSBs can be repaired by either of two dis-tinct repair pathways: nonhomologous end joining (NHEJ) orhomologous recombination (HR) (70). Components of theserepair machineries have been shown to support viral DNAreplication (56). For example, DDR-induced Rad51 facilitatesreplication of the EBV and SV40 genomes (14, 48). Studyingthe MVC replication-induced DDR and how this responsefeeds back to help MVC DNA replication will likely help us tounderstand the mechanism underlying the virus infection-in-duced DDR.
In conclusion, MVC infection-caused cytopathic effects areunique and are mediated by the DDR induced by replication ofthe viral genome. We believe that the DDR is induced duringparvovirus infection and that the ensured cell death and cellcycle arrest may be common and potentially synergistic mech-anisms underlying parvovirus infection-induced cytopathic ef-fects. Understanding the mechanism underlying the MVC-in-duced DDR and the DDR-induced cell death and cell cyclearrest pathways will potentially elucidate the molecular patho-genesis of Bocavirus infection, as well as unravel the mecha-nism underlying the regulatory DDR pathways.
VOL. 85, 2011 BOCAVIRUS DDR ROLE IN DNA REPLICATION AND CELL DEATH 143
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
ACKNOWLEDGMENTS
This work was supported by PHS grant 1R21AI085236 from NIAIDand grant P20 RR016443 from the NCRR COBRE program.
We thank Colin Parrish at the James A. Baker Institute, CornellUniversity, for valuable reagents. We are indebted to Mary AshleyRimmer for initiating the experiment identifying the MVC replicationcenter and to Fang Cheng for technical help.
REFERENCES
1. Alao, J. P., and P. Sunnerhagen. 2009. The ATM and ATR inhibitorsCGK733 and caffeine suppress cyclin D1 levels and inhibit cell proliferation.Radiat. Oncol. 4:51.
2. Albuquerque, M. C., L. N. Rocha, F. J. Benati, C. C. Soares, A. G. Maranhao,M. L. Ramirez, D. Erdman, and N. Santos. 2007. Human bocavirus infectionin children with gastroenteritis, Brazil. Emerg. Infect. Dis. 13:1756–1758.
3. Allander, T., T. Jartti, S. Gupta, H. G. Niesters, P. Lehtinen, R. Osterback,T. Vuorinen, M. Waris, A. Bjerkner, A. Tiveljung-Lindell, B. G. van denHoogen, T. Hyypia, and O. Ruuskanen. 2007. Human bocavirus and acutewheezing in children. Clin. Infect. Dis. 44:904–910.
4. Arnold, J. C., K. K. Singh, S. A. Spector, and M. H. Sawyer. 2006. Humanbocavirus: prevalence and clinical spectrum at a children’s hospital. Clin.Infect. Dis. 43:283–288.
5. Balajee, A. S., and C. R. Geard. 2004. Replication protein A and gamma-H2AX foci assembly is triggered by cellular response to DNA double-strandbreaks. Exp. Cell Res. 300:320–334.
6. Bartek, J., C. Lukas, and J. Lukas. 2004. Checking on DNA damage in Sphase. Nat. Rev. Mol. Cell Biol. 5:792–804.
7. Bartek, J., and J. Lukas. 2003. Chk1 and Chk2 kinases in checkpoint controland cancer. Cancer Cell 3:421–429.
8. Bartek, J., and J. Lukas. 2006. Cell biology. Balancing life-or-death deci-sions. Science 314:261–262.
9. Berthet, C., K. Raj, P. Saudan, and P. Beard. 2005. How adeno-associatedvirus Rep78 protein arrests cells completely in S phase. Proc. Natl. Acad.Sci.U. S. A. 102:13634–13639.
10. Bhattacharya, S., R. M. Ray, and L. R. Johnson. 2009. Role of polyaminesin p53-dependent apoptosis of intestinal epithelial cells. Cell. Signal. 21:509–522.
11. Binn, L. N., E. C. Lazar, G. A. Eddy, and M. Kajima. 1970. Recovery andcharacterization of a minute virus of canines. Infect. Immun. 1:503–508.
12. Binn, L. N., R. H. Marchwicki, E. H. Eckermann, and T. E. Fritz. 1981. Viralantibody studies of laboratory dogs with diarrheal disease. Am. J. Vet. Res.42:1665–1667.
13. Binz, S. K., A. M. Sheehan, and M. S. Wold. 2004. Replication protein Aphosphorylation and the cellular response to DNA damage. DNA Repair(Amst.) 3:1015–1024.
14. Boichuk, S., L. Hu, J. Hein, and O. V. Gjoerup. 2010. Multiple DNA damagesignaling and repair pathways deregulated by simian virus 40 large T antigen.J. Virol. 84:8007–8020.
15. Branzei, D., and M. Foiani. 2008. Regulation of DNA repair throughout thecell cycle. Nat. Rev. Mol. Cell Biol. 9:297–308.
16. Burma, S., B. P. Chen, M. Murphy, A. Kurimasa, and D. J. Chen. 2001.ATM phosphorylates histone H2AX in response to DNA double-strandbreaks. J. Biol. Chem. 276:42462–42467.
17. Carmichael, L. E., D. H. Schlafer, and A. Hashimoto. 1991. Pathogenicity ofminute virus of canines (MVC) for the canine fetus. Cornell Vet. 81:151–171.
18. Carson, C. T., R. A. Schwartz, T. H. Stracker, C. E. Lilley, D. V. Lee, andM. D. Weitzman. 2003. The Mre11 complex is required for ATM activationand the G2/M checkpoint. EMBO J. 22:6610–6620.
19. Chen, A. Y., Y. Luo, F. Cheng, Y. Sun, and J. Qiu. 2010. Bocavirus infectioninduces a mitochondrion-mediated apoptosis and cell cycle arrest at G2/Mphase. J. Virol. 84:5615–5626.
20. Choi, V. W., D. M. McCarty, and R. J. Samulski. 2006. Host cell DNA repairpathways in adeno-associated viral genome processing. J. Virol. 80:10346–10356.
21. Christensen, J., and P. Tattersall. 2002. Parvovirus initiator protein NS1 andRPA coordinate replication fork progression in a reconstituted DNA repli-cation system. J. Virol. 76:6518–6531.
22. Collaco, R. F., J. M. Bevington, V. Bhrigu, V. Kalman-Maltese, and J. P.Trempe. 2009. Adeno-associated virus and adenovirus coinfection induces acellular DNA damage and repair response via redundant phosphatidylino-sitol 3-like kinase pathways. Virology 392:24–33.
23. Cotmore, S. F., and P. Tattersall. 2005. A rolling-haipin strategy: basicmechanisms of DNA replication in the parvoviruses, p. 171–181. In J. Kerr,S. F. Cotmore, M. E. Bloom, R. M. Linden, and C. R. Parrish (ed.), Parvo-viruses. Hoddler Arond, London, United Kingdom.
24. Cotmore, S. F., and P. Tattersall. 2005. Structure and organization of theviral genome, p. 73–94. In J. Kerr, S. F. Cotmore, M. E. Bloom, R. M.Linden, and C. R. Parrish (ed.), Parvoviruses. Hodder Arnold, London,United Kingdom.
25. Cruet-Hennequart, S., M. T. Glynn, L. S. Murillo, S. Coyne, and M. P. Carty.
2008. Enhanced DNA-PK-mediated RPA2 hyperphosphorylation in DNApolymerase eta-deficient human cells treated with cisplatin and oxaliplatin.DNA Repair (Amst.) 7:582–596.
26. Cuconati, A., C. Mukherjee, D. Perez, and E. White. 2003. DNA damageresponse and MCL-1 destruction initiate apoptosis in adenovirus-infectedcells. Genes Dev. 17:2922–2932.
27. Dahl, J., J. You, and T. L. Benjamin. 2005. Induction and utilization of anATM signaling pathway by polyomavirus. J. Virol. 79:13007–13017.
28. D’Amours, D., and S. P. Jackson. 2002. The Mre11 complex: at the cross-roads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol.3:317–327.
29. Difilippantonio, S., and A. Nussenzweig. 2007. The NBS1-ATM connectionrevisited. Cell Cycle 6:2366–2370.
30. Digweed, M., I. Demuth, S. Rothe, R. Scholz, A. Jordan, C. Grotzinger, D.Schindler, M. Grompe, and K. Sperling. 2002. SV40 large T-antigen disturbsthe formation of nuclear DNA-repair foci containing MRE11. Oncogene21:4873–4878.
31. Dijkman, R., S. M. Koekkoek, R. Molenkamp, O. Schildgen, and L. van derHoek. 2009. Human bocavirus can be cultured in differentiated human air-way epithelial cells. J. Virol. 83:7739–7748.
32. Fragkos, M., M. Breuleux, N. Clement, and P. Beard. 2008. Recombinantadeno-associated viral vectors are deficient in provoking a DNA damageresponse. J. Virol. 82:7379–7387.
33. Francon, P., J. M. Lemaitre, C. Dreyer, D. Maiorano, O. Cuvier, and M.Mechali. 2004. A hypophosphorylated form of RPA34 is a specific compo-nent of pre-replication centers. J. Cell Sci. 117:4909–4920.
34. Furuta, T., H. Takemura, Z. Y. Liao, G. J. Aune, C. Redon, O. A. Sedelni-kova, D. R. Pilch, E. P. Rogakou, A. Celeste, H. T. Chen, A. Nussenzweig,M. I. Aladjem, W. M. Bonner, and Y. Pommier. 2003. Phosphorylation ofhistone H2AX and activation of Mre11, Rad50, and Nbs1 in response toreplication-dependent DNA double-strand breaks induced by mammalianDNA topoisomerase I cleavage complexes. J. Biol. Chem. 278:20303–20312.
35. Goldstein, M., W. P. Roos, and B. Kaina. 2008. Apoptotic death induced bythe cyclophosphamide analogue mafosfamide in human lymphoblastoidcells: contribution of DNA replication, transcription inhibition and Chk/p53signaling. Toxicol. Appl. Pharmacol. 229:20–32.
36. Gregory, D. A., and S. L. Bachenheimer. 2008. Characterization of mre11loss following HSV-1 infection. Virology 373:124–136.
37. Hein, J., S. Boichuk, J. Wu, Y. Cheng, R. Freire, P. S. Jat, T. M. Roberts, andO. V. Gjoerup. 2009. Simian virus 40 large T antigen disrupts genomeintegrity and activates a DNA damage response via Bub1 binding. J. Virol.83:117–127.
38. Hickson, I., Y. Zhao, C. J. Richardson, S. J. Green, N. M. Martin, A. I. Orr,P. M. Reaper, S. P. Jackson, N. J. Curtin, and G. C. Smith. 2004. Identifi-cation and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64:9152–9159.
39. Hristov, G., M. Kramer, J. Li, N. El-Andaloussi, R. Mora, L. Daeffler, H.Zentgraf, J. Rommelaere, and A. Marchini. 2010. Through its nonstructuralprotein NS1, parvovirus H-1 induces apoptosis via accumulation of reactiveoxygen species. J. Virol. 84:5909–5922.
40. Huang, K. J., B. V. Zemelman, and I. R. Lehman. 2002. Endonuclease G, acandidate human enzyme for the initiation of genomic inversion in herpessimplex type 1 virus. J. Biol. Chem. 277:21071–21079.
41. Jackson, S. P. 2009. The DNA-damage response: new molecular insights andnew approaches to cancer therapy. Biochem. Soc. Trans. 37:483–494.
42. Jarplid, B., H. Johansson, and L. E. Carmichael. 1996. A fatal case of pupinfection with minute virus of canines (MVC). J. Vet. Diagn. Invest. 8:484–487.
43. Jurvansuu, J., K. Raj, A. Stasiak, and P. Beard. 2005. Viral transport ofDNA damage that mimics a stalled replication fork. J. Virol. 79:569–580.
44. Kahn, J. 2008. Human bocavirus: clinical significance and implications. Curr.Opin. Pediatr. 20:62–66.
45. Kastan, M. B., and J. Bartek. 2004. Cell-cycle checkpoints and cancer.Nature 432:316–323.
46. Kim, H., H. C. Tu, D. Ren, O. Takeuchi, J. R. Jeffers, G. P. Zambetti, J. J.Hsieh, and E. H. Cheng. 2009. Stepwise activation of BAX and BAK bytBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 36:487–499.
47. Kitagawa, R., C. J. Bakkenist, P. J. McKinnon, and M. B. Kastan. 2004.Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 18:1423–1438.
48. Kudoh, A., S. Iwahori, Y. Sato, S. Nakayama, H. Isomura, T. Murata, and T.Tsurumi. 2009. Homologous recombinational repair factors are recruitedand loaded onto the viral DNA genome in Epstein-Barr virus replicationcompartments. J. Virol. 83:6641–6651.
49. Lanson, N. A., Jr., D. B. Egeland, B. A. Royals, and W. C. Claycomb. 2000.The MRE11-NBS1-RAD50 pathway is perturbed in SV40 large T antigen-immortalized AT-1, AT-2 and HL-1 cardiomyocytes. Nucleic Acids Res.28:2882–2892.
50. Lau, S. K., C. C. Yip, T. L. Que, R. A. Lee, R. K. Au-Yeung, B. Zhou, L. Y.So, Y. L. Lau, K. H. Chan, P. C. Woo, and K. Y. Yuen. 2007. Clinical and
144 LUO ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 03
Jan
uary
202
2 by
109
.207
.33.
247.
molecular epidemiology of human bocavirus in respiratory and fecal samplesfrom children in Hong Kong. J. Infect. Dis. 196:986–993.
51. Leahy, J. J., B. T. Golding, R. J. Griffin, I. R. Hardcastle, C. Richardson, L.Rigoreau, and G. C. Smith. 2004. Identification of a highly potent andselective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) byscreening of chromenone libraries. Bioorg. Med. Chem. Lett. 14:6083–6087.
52. Lee, J. H., and T. T. Paull. 2005. ATM activation by DNA double-strandbreaks through the Mre11-Rad50-Nbs1 complex. Science 308:551–554.
53. Lee, J. I., J. Y. Chung, T. H. Han, M. O. Song, and E. S. Hwang. 2007.Detection of human bocavirus in children hospitalized because of acutegastroenteritis. J. Infect. Dis. 196:994–997.
54. Li, J., and D. F. Stern. 2005. Regulation of CHK2 by DNA-dependentprotein kinase. J. Biol. Chem. 280:12041–12050.
55. Lilley, C. E., C. T. Carson, A. R. Muotri, F. H. Gage, and M. D. Weitzman.2005. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc.Natl. Acad. Sci.U. S. A. 102:5844–5849.
56. Lilley, C. E., R. A. Schwartz, and M. D. Weitzman. 2007. Using or abusing:viruses and the cellular DNA damage response. Trends Microbiol. 15:119–126.
57. Milatovic, D., Y. Zhang, S. J. Olson, K. S. Montine, L. J. Roberts, J. D.Morrow, T. J. Montine, T. S. Dermody, and T. Valyi-Nagy. 2002. Herpessimplex virus type 1 encephalitis is associated with elevated levels of F2-isoprostanes and F4-neuroprostanes. J. Neurovirol. 8:295–305.
58. Mochizuki, M., M. Hashimoto, T. Hajima, M. Takiguchi, A. Hashimoto, Y.Une, F. Roerink, T. Ohshima, C. R. Parrish, and L. E. Carmichael. 2002.Virologic and serologic identification of minute virus of canines (canineparvovirus type 1) from dogs in Japan. J. Clin. Microbiol. 40:3993–3998.
59. Moody, C. A., and L. A. Laimins. 2009. Human papillomaviruses activate theATM DNA damage pathway for viral genome amplification upon differen-tiation. PLoS Pathog. 5:e1000605.
60. Mukherjee, B., C. Kessinger, J. Kobayashi, B. P. Chen, D. J. Chen, A.Chatterjee, and S. Burma. 2006. DNA-PK phosphorylates histone H2AXduring apoptotic DNA fragmentation in mammalian cells. DNA Repair(Amst.) 5:575–590.
61. Nakanishi, S., S. Kakita, I. Takahashi, K. Kawahara, E. Tsukuda, T. Sano,K. Yamada, M. Yoshida, H. Kase, and Y. Matsuda. 1992. Wortmannin, amicrobial product inhibitor of myosin light chain kinase. J. Biol. Chem.267:2157–2163.
62. Ni, T. H., W. F. McDonald, I. Zolotukhin, T. Melendy, S. Waga, B. Stillman,and N. Muzyczka. 1998. Cellular proteins required for adeno-associatedvirus DNA replication in the absence of adenovirus coinfection. J. Virol.72:2777–2787.
63. Paull, T. T., and J. H. Lee. 2005. The Mre11/Rad50/Nbs1 complex and itsrole as a DNA double-strand break sensor for ATM. Cell Cycle 4:737–740.
64. Petrini, J. H., and T. H. Stracker. 2003. The cellular response to DNAdouble-strand breaks: defining the sensors and mediators. Trends Cell Biol.13:458–462.
65. Pommier, Y., J. N. Weinstein, M. I. Aladjem, and K. W. Kohn. 2006. Chk2molecular interaction map and rationale for Chk2 inhibitors. Clin. CancerRes. 12:2657–2661.
66. Pratelli, A., D. Buonavoglia, M. Tempesta, F. Guarda, L. Carmichael, and C.Buonavoglia. 1999. Fatal canine parvovirus type-1 infection in pups fromItaly. J. Vet. Diagn. Invest. 11:365–367.
67. Raj, K., P. Ogston, and P. Beard. 2001. Virus-mediated killing of cells thatlack p53 activity. Nature 412:914–917.
68. Rao, V. A., A. M. Fan, L. Meng, C. F. Doe, P. S. North, I. D. Hickson, andY. Pommier. 2005. Phosphorylation of BLM, dissociation from topoisomer-ase IIIalpha, and colocalization with gamma-H2AX after topoisomeraseI-induced replication damage. Mol. Cell. Biol. 25:8925–8937.
69. Roos, W. P., and B. Kaina. 2006. DNA damage-induced cell death byapoptosis. Trends Mol. Med. 12:440–450.
70. Sancar, A., L. A. Lindsey-Boltz, K. Unsal-Kacmaz, and S. Linn. 2004. Mo-lecular mechanisms of mammalian DNA repair and the DNA damage check-points. Annu. Rev. Biochem. 73:39–85.
71. Schar, P., M. Fasi, and R. Jessberger. 2004. SMC1 coordinates DNA double-strand break repair pathways. Nucleic Acids Res. 32:3921–3929.
72. Schildgen, O., A. Muller, T. Allander, I. M. Mackay, S. Volz, B. Kupfer, andA. Simon. 2008. Human bocavirus: passenger or pathogen in acute respira-tory tract infections? Clin. Microbiol. Rev. 21:291–304.
73. Schwartz, R. A., C. T. Carson, C. Schuberth, and M. D. Weitzman. 2009.
Adeno-associated virus replication induces a DNA damage response coor-dinated by DNA-dependent protein kinase. J. Virol. 83:6269–6278.
74. Schwartz, R. A., J. A. Palacios, G. D. Cassell, S. Adam, M. Giacca, and M. D.Weitzman. 2007. The Mre11/Rad50/Nbs1 complex limits adeno-associatedvirus transduction and replication. J. Virol. 81:12936–12945.
75. Shiloh, Y. 2003. ATM and related protein kinases: safeguarding genomeintegrity. Nat. Rev. Cancer 3:155–168.
76. Spahn, G. J., S. B. Mohanty, and F. M. Hetrick. 1966. Experimental infectionof calves with hemadsorbing enteric (HADEN) virus. Cornell Vet. 56:377–386.
77. Stracker, T. H., C. T. Carson, and M. D. Weitzman. 2002. Adenovirusoncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Na-ture 418:348–352.
78. Stracker, T. H., J. W. Theunissen, M. Morales, and J. H. Petrini. 2004. TheMre11 complex and the metabolism of chromosome breaks: the importanceof communicating and holding things together. DNA Repair (Amst.) 3:845–854.
79. Sun, Y., A. Y. Chen, F. Cheng, W. Guan, F. B. Johnson, and J. Qiu. 2009.Molecular characterization of infectious clones of the minute virus of caninesreveals unique features of bocaviruses. J. Virol. 83:3956–3967.
80. Taylor, T. J., and D. M. Knipe. 2004. Proteomics of herpes simplex virusreplication compartments: association of cellular DNA replication, repair,recombination, and chromatin remodeling proteins with ICP8. J. Virol. 78:5856–5866.
81. Uziel, T., Y. Lerenthal, L. Moyal, Y. Andegeko, L. Mittelman, and Y. Shiloh.2003. Requirement of the MRN complex for ATM activation by DNAdamage. EMBO J. 22:5612–5621.
82. Valyi-Nagy, T., S. J. Olson, K. Valyi-Nagy, T. J. Montine, and T. S. Dermody.2000. Herpes simplex virus type 1 latency in the murine nervous system isassociated with oxidative damage to neurons. Virology 278:309–321.
83. van den Bosch, M., R. T. Bree, and N. F. Lowndes. 2003. The MRN complex:coordinating and mediating the response to broken chromosomes. EMBORep. 4:844–849.
84. Vassin, V. M., M. S. Wold, and J. A. Borowiec. 2004. Replication protein A(RPA) phosphorylation prevents RPA association with replication centers.Mol. Cell. Biol. 24:1930–1943.
85. Vicente, D., G. Cilla, M. Montes, E. G. Perez-Yarza, and E. Perez-Trallero.2007. Human bocavirus, a respiratory and enteric virus. Emerg. Infect. Dis.13:636–637.
86. Ward, I. M., and J. Chen. 2001. Histone H2AX is phosphorylated in anATR-dependent manner in response to replicational stress. J. Biol. Chem.276:47759–47762.
87. Wilkinson, D. E., and S. K. Weller. 2004. Recruitment of cellular recombi-nation and repair proteins to sites of herpes simplex virus type 1 DNAreplication is dependent on the composition of viral proteins within prerep-licative sites and correlates with the induction of the DNA damage response.J. Virol. 78:4783–4796.
88. Wohlrab, F., S. Chatterjee, and R. D. Wells. 1991. The herpes simplex virus1 segment inversion site is specifically cleaved by a virus-induced nuclearendonuclease. Proc. Natl. Acad. Sci. U. S. A. 88:6432–6436.
89. Won, J., M. Kim, N. Kim, J. H. Ahn, W. G. Lee, S. S. Kim, K. Y. Chang, Y. W.Yi, and T. K. Kim. 2006. Small molecule-based reversible reprogramming ofcellular lifespan. Nat. Chem. Biol. 2:369–374.
90. Wu, X., D. Avni, T. Chiba, F. Yan, Q. Zhao, Y. Lin, H. Heng, and D.Livingston. 2004. SV40 T antigen interacts with Nbs1 to disrupt DNA rep-lication control. Genes Dev. 18:1305–1316.
91. Wu, X., S. M. Shell, and Y. Zou. 2005. Interaction and colocalization ofRad9/Rad1/Hus1 checkpoint complex with replication protein A in humancells. Oncogene 24:4728–4735.
92. Yang, X. H., B. Shiotani, M. Classon, and L. Zou. 2008. Chk1 and Claspinpotentiate PCNA ubiquitination. Genes Dev. 22:1147–1152.
93. Yazdi, P. T., Y. Wang, S. Zhao, N. Patel, E. Y. Lee, and J. Qin. 2002. SMC1is a downstream effector in the ATM/NBS1 branch of the human S-phasecheckpoint. Genes Dev. 16:571–582.
94. Zhao, X., R. J. Madden-Fuentes, B. X. Lou, J. M. Pipas, J. Gerhardt, C. J.Rigell, and E. Fanning. 2008. Ataxia telangiectasia-mutated damage-signal-ing kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1subunits in simian virus 40-infected primate cells. J. Virol. 82:5316–5328.
95. Zou, L., and S. J. Elledge. 2003. Sensing DNA damage through ATRIPrecognition of RPA-ssDNA complexes. Science 300:1542–1548.
VOL. 85, 2011 BOCAVIRUS DDR ROLE IN DNA REPLICATION AND CELL DEATH 145