Bonding energy analysis in cationic borylene complexes of palladium and platinum: A theoretical study Krishna K. Pandey ⇑ School of Chemical Sciences, Devi Ahilya University Indore, Indore 452017, India article info Article history: Available online 15 May 2012 Dedicated to Alfred Werner on the 100th anniversary of his Nobel prize in chemistry in 1913. Keywords: DFT Bonding Platinum Palladium Borylene: EDA abstract Quantum chemical calculations at DFT/BP86/TZ2P have been carried out for cationic metal borylene complexes of palladium and platinum trans-[X(PMe 3 ) 2 M(BR)] + (X = Cl, Br, I; M = Pd, Pt; R = Mesityl, Xylyl, Ph, NMe 2 ). The bonding analysis was carried out with charge decomposition and energy decomposition analysis. The calculated geometry of the platinum borylene complex trans-[Br(PMe 3 ) 2 Pt(BMes)] + is in excellent agreement with the experimental structural data for trans-[Br(PCy 3 ) 2 Pt(BMes)] + . Pauling bond order of the optimized structures shows that the M–B bonds in these complexes have multiple M–B bond character, which is also supported by the performed energy decomposition analysis. The orbital interac- tions between the metal and boron arise mainly from M BR r donation. In all complexes, the p bond- ing contribution to the orbital interactions is relatively small. The bonding analysis clearly shows that the platinum borylene complexes [X(PMe 3 ) 2 Pt(BR)] + are more strongly stabilized by Pt–BR interactions than the platinum borylene complexes [X(PMe 3 ) 2 Pd(BR)] + which is probably the reason the complex platinum borylene complexes [X(PCy 3 ) 2 Pt(BMes)] + could become isolated. The contributions of the electrostatic interaction terms DE elstat in the M@BR bonding are significantly larger in all borylene complexes than the covalent bonding DE orb term. Thus, the M@BR bond in the studied cationic borylene complexes of pal- ladium and platinum has a greater degree of ionic character. The larger gap between HOMOs of metal fragments [X(PMe 3 ) 2 M)] + and LUMO of BNMe 2 allow relatively weak [X(PMe 3 ) 2 M] + ? BNMe 2 p-back bonding. Ó 2012 Elsevier Ltd. All rights reserved. 1. Introduction Synthesis, structure, reactivity and bonding in transition metal borylene complexes have been a provocative subject since the first report of structurally characterized two coordinated transition metal borylene complexes in 1998 [1,2]. So far a number of struc- turally characterized terminal transition metal borylene complexes [1–24] has been reported (Table 1). A number of interesting review articles have been written during these periods by Braunschweig et al. [25–30] and Aldridge et al. [31–33]. Theoretical approaches have been proven to be an indispensable part of the studies of terminal metal borylene complexes [34–50]. Previously, the BLYP/LANL2DZ and B3LYP/LANL2DZ approaches have been applied to study the geometry and electronic structure of terminal cationic borylene complexes [(g 5 -C 5 H 5 )(CO) 2 Fe{B(g 5 - C 5 Me 5 )}] + , [(g 5 -C 5 H 5 )(CO) 2 Fe(BMes)] + , [(g 5 -C 5 H 5 )(CO) 2 Fe(BN Me 2 )] + and [(g 5 -C 5 H 5 )(CO) 2 Ru(BNMe 2 )] + [15,19,20,50]. Structure and bonding energy analysis of transition metal borylene com- plexes have been studied in detail [42,45,47–50]. To the best of our knowledge, the bonding analyses of terminal cationic borylene complexes of palladium and platinum have never been studied before. One should note that borylene complexes of palladium studied in this paper are not known so far. Only a single represen- tative example of cationic platinum borylene complex trans- [Br(PCy 3 ) 2 Pt{BMes] + was reported by Braunschweig and coworkers [18]. Structures of the other studied borylene complexes of palla- dium and platinum are hitherto unknown. However, a number of cationic boryl complexes of platinum [51] as well as neutral boryl complexes of palladium [52] have been synthesized and structur- ally characterized by Braunschweig and coworkers. Here, for the first time, we report the geometry, electronic structure and nature of M@BR bonds in borylene complexes of pal- ladium and platinum trans-[X(PMe 3 ) 2 M(BR)] + (M = Pd, Pt; X = Cl, Br, I; R = Ph, Xylyl, Mes, BNMe 2 ) at the DFT level using BP86/ TZ2P/ZORA. The main goals of the present study are (i) to investi- gate the geometries and to analyze the nature of M@B bonding in metal borylene complexes, (ii) to provide a quantitative differenti- ation of the M–BR bonds and (iii) to elucidate the role of the tran- sition metal atoms and substituent R of BR ligands in the stability of the M–BR bond. The bonding situation in the molecules was investigated with the energy decomposition analysis (EDA), which has previously been used in systematic studies of transition metal complexes [46–50,53–56]. The complexes I–IV, VI–XII, XIV–XVI 0277-5387/$ - see front matter Ó 2012 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.poly.2012.04.005 ⇑ Tel.: +91 731 2460208; fax: +91 731 2762342. E-mail address: [email protected]Polyhedron 52 (2013) 1431–1439 Contents lists available at SciVerse ScienceDirect Polyhedron journal homepage: www.elsevier.com/locate/poly
Transcript
Polyhedron 52 (2013) 1431–1439
Contents lists available at SciVerse ScienceDirect
Polyhedron
journal homepage: www.elsevier .com/locate /poly
Bonding energy analysis in cationic borylene complexes of palladium andplatinum: A theoretical study
Krishna K. Pandey ⇑School of Chemical Sciences, Devi Ahilya University Indore, Indore 452017, India
a r t i c l e i n f o a b s t r a c t
Article history:Available online 15 May 2012
Dedicated to Alfred Werner on the 100thanniversary of his Nobel prize in chemistryin 1913.
Keywords:DFTBondingPlatinumPalladiumBorylene: EDA
0277-5387/$ - see front matter � 2012 Elsevier Ltd. Ahttp://dx.doi.org/10.1016/j.poly.2012.04.005
Quantum chemical calculations at DFT/BP86/TZ2P have been carried out for cationic metal borylenecomplexes of palladium and platinum trans-[X(PMe3)2M(BR)]+ (X = Cl, Br, I; M = Pd, Pt; R = Mesityl, Xylyl,Ph, NMe2). The bonding analysis was carried out with charge decomposition and energy decompositionanalysis. The calculated geometry of the platinum borylene complex trans-[Br(PMe3)2Pt(BMes)]+ is inexcellent agreement with the experimental structural data for trans-[Br(PCy3)2Pt(BMes)]+. Pauling bondorder of the optimized structures shows that the M–B bonds in these complexes have multiple M–B bondcharacter, which is also supported by the performed energy decomposition analysis. The orbital interac-tions between the metal and boron arise mainly from M BR r donation. In all complexes, the p bond-ing contribution to the orbital interactions is relatively small. The bonding analysis clearly shows that theplatinum borylene complexes [X(PMe3)2Pt(BR)]+ are more strongly stabilized by Pt–BR interactions thanthe platinum borylene complexes [X(PMe3)2Pd(BR)]+ which is probably the reason the complex platinumborylene complexes [X(PCy3)2Pt(BMes)]+ could become isolated. The contributions of the electrostaticinteraction terms DEelstat in the M@BR bonding are significantly larger in all borylene complexes thanthe covalent bonding DEorb term. Thus, the M@BR bond in the studied cationic borylene complexes of pal-ladium and platinum has a greater degree of ionic character. The larger gap between HOMOs of metalfragments [X(PMe3)2M)]+ and LUMO of BNMe2 allow relatively weak [X(PMe3)2M]+ ? BNMe2 p-backbonding.
� 2012 Elsevier Ltd. All rights reserved.
1. Introduction
Synthesis, structure, reactivity and bonding in transition metalborylene complexes have been a provocative subject since the firstreport of structurally characterized two coordinated transitionmetal borylene complexes in 1998 [1,2]. So far a number of struc-turally characterized terminal transition metal borylene complexes[1–24] has been reported (Table 1). A number of interesting reviewarticles have been written during these periods by Braunschweig etal. [25–30] and Aldridge et al. [31–33].
Theoretical approaches have been proven to be an indispensablepart of the studies of terminal metal borylene complexes [34–50].Previously, the BLYP/LANL2DZ and B3LYP/LANL2DZ approacheshave been applied to study the geometry and electronic structureof terminal cationic borylene complexes [(g5-C5H5)(CO)2Fe{B(g5-C5Me5)}]+, [(g5-C5H5)(CO)2Fe(BMes)]+, [(g5-C5H5)(CO)2Fe(BNMe2)]+ and [(g5-C5H5)(CO)2Ru(BNMe2)]+ [15,19,20,50]. Structureand bonding energy analysis of transition metal borylene com-plexes have been studied in detail [42,45,47–50]. To the best ofour knowledge, the bonding analyses of terminal cationic borylene
ll rights reserved.
complexes of palladium and platinum have never been studiedbefore. One should note that borylene complexes of palladiumstudied in this paper are not known so far. Only a single represen-tative example of cationic platinum borylene complex trans-[Br(PCy3)2Pt{BMes]+ was reported by Braunschweig and coworkers[18]. Structures of the other studied borylene complexes of palla-dium and platinum are hitherto unknown. However, a number ofcationic boryl complexes of platinum [51] as well as neutral borylcomplexes of palladium [52] have been synthesized and structur-ally characterized by Braunschweig and coworkers.
Here, for the first time, we report the geometry, electronicstructure and nature of M@BR bonds in borylene complexes of pal-ladium and platinum trans-[X(PMe3)2M(BR)]+ (M = Pd, Pt; X = Cl,Br, I; R = Ph, Xylyl, Mes, BNMe2) at the DFT level using BP86/TZ2P/ZORA. The main goals of the present study are (i) to investi-gate the geometries and to analyze the nature of M@B bonding inmetal borylene complexes, (ii) to provide a quantitative differenti-ation of the M–BR bonds and (iii) to elucidate the role of the tran-sition metal atoms and substituent R of BR ligands in the stabilityof the M–BR bond. The bonding situation in the molecules wasinvestigated with the energy decomposition analysis (EDA), whichhas previously been used in systematic studies of transition metalcomplexes [46–50,53–56]. The complexes I–IV, VI–XII, XIV–XVI
Fig. 1. Schematic representation of the M–BR bonding.
1432 K.K. Pandey / Polyhedron 52 (2013) 1431–1439
and their fragments which are used for EDA have C2v symmetrywhich is very helpful because the contributions of the a1(r),a2(d), b1(p||) and b2(p\) orbitals can be distinguished. We will dis-cuss the degree of ionic and covalent character of the M@B bondsas well as the extent of the M BR r bonding and M ? BR p back-bonding contribution to the M–BR orbital interactions (Fig. 1).Understanding the nature of metal-borylene bonding in these spe-cies is of utmost importance and could lead to synthesis of newtransition metal-borylene complexes with interesting reactivity.
2. Computational methods
Calculations for the 16 cationic borylene complexes of palla-dium and platinum trans-[X(PMe3)2M(BR)]+ (M = Pd, X = Cl, I, R= Xylyl, II, R = NMe2; M = Pd, X = Br, III, R = Ph, IV, R = Xylyl, V,R = Mes, VI, R = NMe2; M = Pd, X = I, VII, R = Xylyl, XIII, R = NMe2;M = Pt, X = Cl, IX, R = Xylyl, X, R = NMe2; M = Pt, X = Br, XI, R = Ph,XII, R = Xylyl, XIII, R = Mes, XIV, R = NMe2; M = Pt, X = XV, R = Xy-lyl, XVI, R = NMe2) have been performed at the semi-local DFT levelof theory using the exchange functional of Becke [57] and the cor-relation functional of Perdew [58] (BP86). Scalar relativistic effectshave been considered using the ZORA formalism [59]. Uncon-tracted Slater-type orbitals (STOs) using triple-f basis sets
augmented by two sets of polarization functions were employedfor the SCF calculations [60]. The (1s)2 core electrons of boron,and carbon, (1s2s2p)10 core electrons of phosphorus and chlorine,(1s2s2p3s3p)18 core electrons of bromine, (1s2s2p3s3p3d)28 coreelectrons of palladium, (1s2s2p3s3p3d4s4p)36 core electrons ofiodine and (1s2s2p3s3p3d4s4p4d)46 core electrons of platinumwere treated by the frozen-core approximation [61]. An auxiliaryset of s, p, d, f and g STOs was used to fit the molecular densitiesand to represent the coulomb and exchange potentials accuratelyin each SCF cycle [62]. The calculations were performed utilizingthe program package ADF-2010.02 [63].
The binding between the metal fragments [X(PMe3)2M]+ andborylene fragments BR in complexes I–IV, VI–XII, XIV–XVI hasbeen analyzed with C2v symmetry and in complexes V, XIII withCs symmetry using the energy decomposition scheme of ADF pack-age [64] which is based on the Morokuma [65] and Ziegler andRauk [66] methods. Based on these studies, the bond energy DEbetween the fragments can be decomposed as:
DE ¼ DEint þ DEprep ð1Þ
where DEprep is the energy required to promote the free fragmentsfrom their equilibrium structure of the electronic ground state intothat which they take up in the molecule:
DEprep ¼ Etotalðdistorted fragmentsÞ� Etotalðfragments in the equilibrium structureÞ ð2Þ
The interaction energy, DEint is the difference between the energy ofthe molecule and energies of the two fragments of the molecule. Itcan be decomposed into three main components:
DEint ¼ DEelstat þ DEPauli þ DEorb ð3Þ
where DEelstat describes the classical Coulomb interaction betweenthe fragments. The size of electrostatic attraction depends on thetopology of the charge distribution. The DEPauli, which representsthe exchange repulsion or Pauli repulsion, takes into account thedestabilizing two-orbital three- or four-electron interactions be-tween the occupied orbitals of both fragments. The orbital interac-tion, DEorb is the interaction between the occupied and virtualorbitals of the two fragments. The orbital interaction, whichdepends on the energy values and the special distribution of theinteracting orbitals, is always attractive. It is possible to decomposethe orbital interaction DEorb into contributions from orbitalsbelonging to different irreducible representations of the point groupof the molecule.
The electronic structures of the studied complexes were exam-ined by NBO analysis [67]. All MO pictures were visualized byusing the MOLDEN program [68].
3. Results and discussion
3.1. Geometries
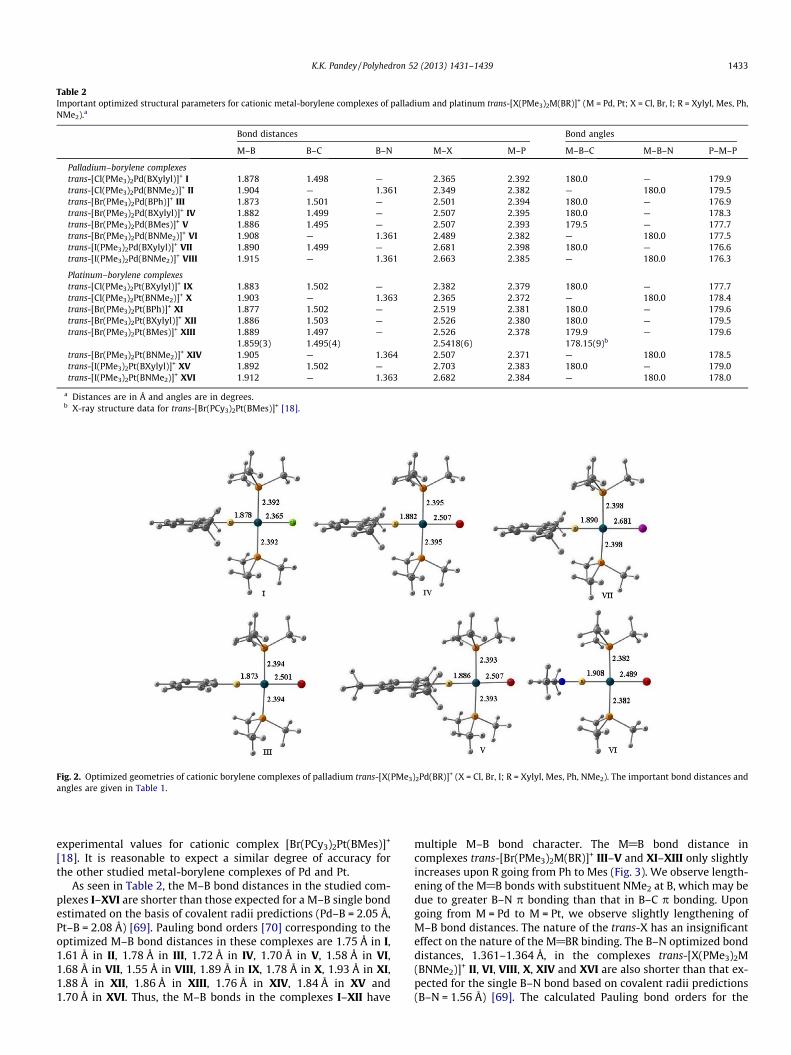
The important bond distances and angles of the complexesI–XVI calculated at the BP86/TZ2P/ZORA level of theory are pre-sented in Table 2. Their geometries are shown in Fig. 2 (only forthe case of M = Pd, i.e., for complexes I, III–VII; the structures ofthe platinum complexes are very similar to those presented in thisFig. and therefore are not included in the Fig. 2; the optimizedCartesian coordinates of all studied complexes are given in theSupporting Information). As mentioned above, the studied palla-dium borylene complexes are not known so far. Therefore, we can-not compare the calculated values for studied palladiumcomplexes with the experimental data. However, as seen in Table2, the calculated geometrical parameters of platinum borylenecomplex [Br(PMe3)2Pt(BMes)]+ are in excellent agreement with
Table 2Important optimized structural parameters for cationic metal-borylene complexes of palladium and platinum trans-[X(PMe3)2M(BR)]+ (M = Pd, Pt; X = Cl, Br, I; R = Xylyl, Mes, Ph,NMe2).a
Bond distances Bond angles
M–B B–C B–N M–X M–P M–B–C M–B–N P–M–P
Palladium–borylene complexestrans-[Cl(PMe3)2Pd(BXylyl)]+ I 1.878 1.498 — 2.365 2.392 180.0 — 179.9trans-[Cl(PMe3)2Pd(BNMe2)]+ II 1.904 — 1.361 2.349 2.382 — 180.0 179.5trans-[Br(PMe3)2Pd(BPh)]+ III 1.873 1.501 — 2.501 2.394 180.0 — 176.9trans-[Br(PMe3)2Pd(BXylyl)]+ IV 1.882 1.499 — 2.507 2.395 180.0 — 178.3trans-[Br(PMe3)2Pd(BMes)]+ V 1.886 1.495 — 2.507 2.393 179.5 — 177.7trans-[Br(PMe3)2Pd(BNMe2)]+ VI 1.908 — 1.361 2.489 2.382 — 180.0 177.5trans-[I(PMe3)2Pd(BXylyl)]+ VII 1.890 1.499 — 2.681 2.398 180.0 — 176.6trans-[I(PMe3)2Pd(BNMe2)]+ VIII 1.915 — 1.361 2.663 2.385 — 180.0 176.3
Platinum–borylene complexestrans-[Cl(PMe3)2Pt(BXylyl)]+ IX 1.883 1.502 — 2.382 2.379 180.0 — 177.7trans-[Cl(PMe3)2Pt(BNMe2)]+ X 1.903 — 1.363 2.365 2.372 — 180.0 178.4trans-[Br(PMe3)2Pt(BPh)]+ XI 1.877 1.502 — 2.519 2.381 180.0 — 179.6trans-[Br(PMe3)2Pt(BXylyl)]+ XII 1.886 1.503 — 2.526 2.380 180.0 — 179.5trans-[Br(PMe3)2Pt(BMes)]+ XIII 1.889 1.497 — 2.526 2.378 179.9 — 179.6
1.859(3) 1.495(4) 2.5418(6) 178.15(9)b
trans-[Br(PMe3)2Pt(BNMe2)]+ XIV 1.905 — 1.364 2.507 2.371 — 180.0 178.5trans-[I(PMe3)2Pt(BXylyl)]+ XV 1.892 1.502 — 2.703 2.383 180.0 — 179.0trans-[I(PMe3)2Pt(BNMe2)]+ XVI 1.912 — 1.363 2.682 2.384 — 180.0 178.0
a Distances are in Å and angles are in degrees.b X-ray structure data for trans-[Br(PCy3)2Pt(BMes)]+ [18].
Fig. 2. Optimized geometries of cationic borylene complexes of palladium trans-[X(PMe3)2Pd(BR)]+ (X = Cl, Br, I; R = Xylyl, Mes, Ph, NMe2). The important bond distances andangles are given in Table 1.
K.K. Pandey / Polyhedron 52 (2013) 1431–1439 1433
experimental values for cationic complex [Br(PCy3)2Pt(BMes)]+
[18]. It is reasonable to expect a similar degree of accuracy forthe other studied metal-borylene complexes of Pd and Pt.
As seen in Table 2, the M–B bond distances in the studied com-plexes I–XVI are shorter than those expected for a M–B single bondestimated on the basis of covalent radii predictions (Pd–B = 2.05 Å,Pt–B = 2.08 Å) [69]. Pauling bond orders [70] corresponding to theoptimized M–B bond distances in these complexes are 1.75 Å in I,1.61 Å in II, 1.78 Å in III, 1.72 Å in IV, 1.70 Å in V, 1.58 Å in VI,1.68 Å in VII, 1.55 Å in VIII, 1.89 Å in IX, 1.78 Å in X, 1.93 Å in XI,1.88 Å in XII, 1.86 Å in XIII, 1.76 Å in XIV, 1.84 Å in XV and1.70 Å in XVI. Thus, the M–B bonds in the complexes I–XII have
multiple M–B bond character. The M@B bond distance incomplexes trans-[Br(PMe3)2M(BR)]+ III–V and XI–XIII only slightlyincreases upon R going from Ph to Mes (Fig. 3). We observe length-ening of the M@B bonds with substituent NMe2 at B, which may bedue to greater B–N p bonding than that in B–C p bonding. Upongoing from M = Pd to M = Pt, we observe slightly lengthening ofM–B bond distances. The nature of the trans-X has an insignificanteffect on the nature of the M@BR binding. The B–N optimized bonddistances, 1.361–1.364 Å, in the complexes trans-[X(PMe3)2M(BNMe2)]+ II, VI, VIII, X, XIV and XVI are also shorter than that ex-pected for the single B–N bond based on covalent radii predictions(B–N = 1.56 Å) [69]. The calculated Pauling bond orders for the
Fig. 3. Variation of M–B bond distances with substituent at boron atom in trans-[Br(PMe3)2M(BR)]+.
1434 K.K. Pandey / Polyhedron 52 (2013) 1431–1439
optimized B–N bond distances (�1.90) in these complexes revealapproximately B@N double bonds. The M–B–N/C bond angles inthese complexes are deviated slightly from linearity.
Pd BXylylCl
PMe3
PMe30.53
0.53
(0.89)
-0.58 0.340.18
+
PdCl
PMe3
PMe30.52
0.52
(0
-0.56 0.18
Pd BXylylBr
PMe3
PMe30.53
0.53
(0.86)
-0.53 0.310.16
+
PdBr
PMe
PMe30.52
0.52
(0
-0.53 0.16
Pd BXylylI
PMe3
PMe30.51
0.51
(0.83)
-0.46 0.310.13
+
-0
Pt BXylylCl
PMe3
PMe30.56
0.56
(1.06)
-0.56 0.310.13
+
PtCl
PMe
PMe0.53
0.53
(1
-0.53 0.1
Pt BXylylBr
PMe3
PMe30.55
0.55
(1.02)
-0.49 0.300.09
+
PtBr
PMe
PMe0.55
0.55
(
-0.49 0.0
Pt BXylylI
PMe3
PMe30.54
0.54
(0.98)
-0.42 0.290.05
+
-
Fig. 4. M–B Wiberg bond indices (WBI) in parentheses and NPA charges in borylene c
3.2. Bonding analysis of the M@BR bonds of the complexes I–XVI
We start the analysis of the M@B bonding in the borylene com-plexes I–XVI with a discussion of bond orders and atomic charges.In Fig. 4, we presented the calculated Wiberg bond indices (WBI)[71], and Natural Population Analysis (NPA) charges. The resultsof the natural bond orbital (NBO) analysis are presented in Table3. As seen from Fig. 4, the WBI values of the M@B bonds of thecomplexes I–XVI are in the range 0.79–0.89 for palladiumcomplexes and in the range 0.93–1.06 for platinum complexes.Like the M@B bond distances, upon going from M = Pd to M = Pt,the WBI values of the M@B bonds increase. The lowest WBI valuesof the M@B bonds of the complexes [I(PMe3)2M(BNMe2)]+ VIII andXVI may be due to greater trans influence of iodine as well as smal-ler M@B p bonding. The calculated NPA charge distributionsindicate that the metal atom, BR and PMe3 ligands carry positivecharge in the studied cationic borylene complexes. The haloligands are negatively charged. Relatively larger positive chargeon BNMe2 ligand reveals weaker [X(PMe3)2M]+ ? [BNMe2] pback-bonding, which is confirmed by energy decompositionanalysis.
A more definitive picture of M@B bonding is obtained throughNBO analysis of the delocalized Kohn–Sham orbitals. The charac-teristics of the M@B r-orbitals are listed in Table 3. In most of
Table 3Results of the NBO analysis in cationic metal-borylene complexes of palladium and platinum trans-[X(PMe3)2M(BR)]+ (M = Pd, Pt; X = Cl, Br, I; R = Xylyl, Mes, Ph, NMe2) at BP86/TZP.
X = [X(PMe3)2Pd(BR)]+ [X(PMe3)2Pt(BR)]+
Cl Br I Cl Br I
R = Xylyl NMe2 Ph Xylyl Mes NMe2 Xylyl NMe2 Xylyl NMe2 Ph Xylyl Mes NMe2 Xylyl NMe2
the complexes I–XVI, the M–B r-bonding orbital is polarized to-ward the boron atom (i.e., the B-center contributes more to thebonding orbital). The B–C and B–N r-bonding orbitals are signifi-cantly polarized towards the carbon of Ph, Xylyl, Mes ligands andnitrogen of NMe2 ligand. The B–C p- bonding is small and is notobserved in NBO analysis. On the other hand, the B–N p-bondingis strong and like the B–N r-bonding orbitals, these molecularorbitals are significantly polarized towards the nitrogen atom ofNMe2 group. The contributions of boron to the r- and p-bondingare small (see Table 3). This conclusion is consistent with thenature of the bonding molecular orbitals given in Fig. 7 for[Br(PMe3)2Pt(BMes)]+ and Fig. 8 for the [Br(PMe3)2Pt(BNMe2)]+.
3.3. Energy decomposition analysis of the M@B bonding of complexesI–XVI
Besides the charge decomposition analysis at the NBO level, wealso carried out an energy decomposition analysis of the M@Bbonds in the calculated cationic borylene complexes of palladiumand platinum trans-[X(PMe3)2M(BR]+ (M = Pd, Pt; X = Cl, Br, I;R = Ph, Xylyl, Mes, NMe2) using the metal fragments [X(PMe3)2M]+
and borylene fragments [BR]:
½XðPMe3Þ2MðBRÞ�þ ! ½XðPMe3Þ2M�þ þ ½BR�
The tabulated bond dissociation energy in Table 4 reveals thatthe Pt@B bonds are stronger than the corresponding palladiumcomplexes. The interaction energies, DEint, show the same trendas the calculated BDEs, with the discrepancies between the two
values (DEprep). Fig. 5 shows schematically the variation in thebond dissociation energy DE (BDE), interaction energy DEint, orbi-tal interaction (covalent bonding) DEorb, electrostatic interaction(ionic bonding) DEelstat and Pauli repulsive interaction DEPauli inthe complexes I–XVI. The breakdown of the interaction energyDEint into the repulsive term DEPauli and the attractive terms DEorb
and DEelstat shows that the DEPauli repulsive interactions have thelarger absolute value for the studied complexes I–XVI and compar-atively larger (as expected) for platinum complexes (Table 4). Thelarger magnitude of the bonding energy components for platinumcomplexes than palladium complexes is consistent with the largersize and greater overlap potential of 5d than 4d orbitals.
The variation in M@B bond energy depends on three factors:metal atom, trans exerting ligands X and substituent at the boronatom. On going from palladium complexes to platinum complexes,the absolute values of DEPauli, DEelstat, DEorb increase. The values ofthe M@B bond dissociation energies decreases on going from trans-located ligand X = Cl to X = I. This weaken of M@B bonds is similarto the lengthening of M@B bond distances on going from X = Cl toX = I. However, it is important to point out that the value of bondenergy is not simply a function of interatomic distance. Marderand coworkers investigated in detail the trans effect in platinumboryl complexes [72]. The contributions of the electrostatic inter-action terms DEelstat in the M@BR bonding are significantly largerin all borylene complexes I–XVI than the covalent bonding DEorb
term. Thus, the M@BR bond in the studied cationic borylenecomplexes of palladium and platinum has a greater degree of ioniccharacter. Table 4 also gives a breakdown of the orbital interac-tions DEorb into the M BR r-donation and M ? BR p-back-dona-tion components. It is significant to note that the p-bonding
Table 4Energy decomposition analysisa of cationic metal-borylene complexes of palladium and platinum trans-[X(PMe3)2M(BR)]+ (M = Pd, Pt; X = Cl, Br, I; R = Xylyl, Mes, Ph, NMe2) atBP86/TZ2P.
[X(PMe3)2Pd(BR)]+ [X(PMe3)2Pt(BR)]+
X = Cl Br I Cl Br I
R = Xylyl NMe2 Ph Xylyl Mes NMe2 Xylyl NMe2 Xylyl NMe2 Ph Xylyl Mes NMe2 Xylyl NMe2
a Energy contributions in kcal/mol.b The values in parentheses are the percentage contribution to the total electrostatic interactions reflecting the ionic character of the bond.c Bond dissociation energy with negative sign.
Fig. 5. Values of the energy contributions of the bond dissociation energy,interaction energies, orbital interactions (covalent interactions) and electrostaticinteractions (ionic contribution) to the M–B bonding in terminal borylenecomplexes of palladium and platinum trans-[X(PMe3)2M(BR)]+ (X = Cl, Br, I;R = Xylyl, Mes, Ph, NMe2) (I–XVI).
Fig. 6. Energies of HOMOs and LUMOs of the metal fragments (a) [Cl(PMe3)2Pd]+,(b) [Br(PMe3)2Pd]+, (c) [I(PMe3)2Pd]+, (d) [Cl(PMe3)2Pt]+, (e) [Br(PMe3)2Pt]+, (f)[I(PMe3)2Pt]+ and the ligand fragments BR.
1436 K.K. Pandey / Polyhedron 52 (2013) 1431–1439
contribution in all studied palladium and platinum borylenecomplexes are larger than the other reported metal borylene com-plexes [46–50].
The nature of the substituent R of ligand BR (R = Ph, Xylyl, Mes)has an insignificant effect on the nature of the M@B bonding: theM@B bond energy in complexes [Br(PMe3)2M(BR)]+ only slightlyincreases upon R going from Ph to Mes. On going from thecomplexes [X(PMe3)2M(BR)]+ (R = Ph, Xylyl, Mes) to complexes[X(PMe3)2M(BNMe2)]+, the absolute values of, DEPauli, DEelstat,DEorb as well as of bond dissociation energy decrease. Therelatively larger M ? BR p-contribution is found for the complexes[X(PMe3)2M(BR)]+ (X = Cl, Br, I; R = Ph, Xylyl, Mes) than in the com-plexes [X(PMe3)2M(BNMe2)]+. This is due to greater B@N p-bond-ing than the B–C p-bonding (see Table 3). These observations aresimilar to those found in NBO analysis. The nature and propertiesof the HOMO and LUMO of the fragments BR and [X(PMe3)2M]+
(X = Cl, Br, I; M = Pd, Pt) play a role in explaining the orbital inter-action differences. The energy of the HOMOs and LUMOs of themetal fragments [X(PMe3)2M]+ and borylene ligand fragments BRare presented in Fig. 6. The larger gap between HOMOs of metalfragments [X(PMe3)2M)]+ and LUMO of BNMe2 allow relativelyweak [X(PMe3)2M]+ ? BNMe2 p-back bonding.
The EDA of the complexes [X(PMe3)2M(BR)]+ (R = Ph, Xylyl,NMe2) with C2v symmetry reveals the different contributions ofthe M ? BR p back-donation with respect to the p orbitals whichare in-plane DEp(b1) and out-of-plane DEp(b2). For example, inthe complexes [Br(PMe3)2M(BPh)]+ in-plane p back-donation isstronger than the out-of-plane contribution (Table 4). This isbecause the out-of-plane p(p) atomic orbital of boron is stabilizedby p-conjugation from the phenyl ring, whereas the in-plane p(p)orbital is empty. Furthermore, in the complexes [Br(PMe3)2M(BNMe2)]+, the values of DEp(b1) are significantly larger thanDEp(b2) due to strong out-of-plane B–N p-bonding.
It is instructive for the numerical analysis of the M@BR bondinganalysis to present and to discuss the molecular orbitals of themodel complexes [Br(PMe3)2Pt(BMes)]+ and [Br(PMe3)2Pt (BNMe2)]+. Figs. 7 and 8 display the envelope plots of relevantmolecular orbitals for complexes [Br(PMe3)2Pt(BMes)]+ and
Fig. 7. Plots of some relevant molecular orbitals of the platinum borylene complex trans-[Br(PMe3)2Pt(BMes)]+ (XIII).
K.K. Pandey / Polyhedron 52 (2013) 1431–1439 1437
[Br(PMe3)2Pt (BNMe2)]+ respectively. Figs. 7E and 8E contribute tothe Pt–B r bond. Figs. 7C and 8C (b1 symmetry) possess bondingcontribution to the Pt–B p|| bond, while Figs. 7D and 8D (b2 sym-metry) depicts a Pt–B p orbital in the P–Pt–P plane (p\). Figs. 7Fand 8F also show the lowest lying vacant orbital which is essen-tially a non-bonding p orbital centered at boron. Figs. 7B and 8Ahave only small contributions at the boron atom. Figs. 7A and 8Bgive pictorial description of the B–C p bonding and B–N p bonding,respectively.
The pictorial representations of the molecular orbitals can becomplemented with the numerical analysis of the electronicstructure and EDA of the complexes. From these representations,it can be concluded that: (i) the B–N p bonding is stronger thanthe B–C p bonding, (ii) thus, the Pt–BNMe2 p back-bonding isweaker than the Pt–BMes p back-bonding and (iii) the p back-bonding in the P–Pt–P plane (p\) is relatively weaker than thep back-bonding p||.
4. Conclusions
From the above-presented theoretical studies of the structureand bonding in 16 cationic borylene complexes of palladium andplatinum, one can draw the following conclusions:
1. Here, for the first time (except the Pt complex XIII), we reportedthe geometry and electronic structure of, as well as analyzedthe nature of, M@BR bonds in the terminal cationic metal bor-
ylene complexes of palladium and platinum [X(PMe3)2M(BR)]+
(where M = Pd and Pt, and R = Ph, Xylyl, Mes and BNMe2). Thecalculated geometry parameters of platinum borylene complex[Br(PMe3)2M(BMes)]+ XIII, are in excellent agreement withtheir available experimental values [Br(PCy3)2M(BMes)]+ [18].
2. The M–B bonds in the studied borylene complexes of palladiumand platinum I–XVI have multiple M–B bond character.
3. The lowest WBI values of the M@B bonds of the complexes[I(PMe3)2M(BNMe2)]+ VIII and XVI are due to greater transinfluence of I as well as weaker [X(PMe3)2M]+ ? [BNMe2] pback-bonding.
4. Relatively larger positive charge on BNMe2 ligand correlateswith weaker [X(PMe3)2M]+ ? [BNMe2] p back-bonding.
5. The bonding analysis clearly shows that the platinum borylenecomplexes [X(PMe3)2Pt(BR)]+ IX–XVI are more strongly stabi-lized by Pt–BR interactions than the palladium borylene com-plexes [X(PMe3)2Pd(BR)]+ I–VIII by Pd-BR interactions.
6. The contributions of the electrostatic interaction terms DEelstat
in the M@BR bonding are significantly larger in all borylenecomplexes I–XVI than the covalent bonding DEorb term. Thus,the M@BR bond in the studied cationic borylene complexes ofpalladium and platinum has a greater degree of ionic character.
We believe that a more detailed understanding of the bondingin metal-borylene complexes is a requisite, particularly for thesynthesis of terminal transition metal borylene complexes, as wellas the designing of efficient borylene transfer processes. In this
Fig. 8. Plots of some relevant molecular orbitals of the platinum borylene complex trans-[Br(PMe3)2Pt(BNMe2)]+ (XIV).
1438 K.K. Pandey / Polyhedron 52 (2013) 1431–1439
aspect, the above-presented findings are important contributionsto the fast developing metal-borylene chemistry.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.poly.2012.04.005.
References
[1] H. Braunschweig, C. Kollann, U. Englert, Angew. Chem., Int. Ed. 37 (1998) 3179.[2] A.H. Cowley, V. Lomeli, A. Voigt, J. Am. Chem. Soc. 120 (1998) 6401.[3] H. Braunschweig, M. Colling, C. Kollann, H.G. Stammler, B. Neumann, Angew.
Chem., Int. Ed. 40 (2001) 2298.[4] H. Braunschweig, M. Colling, C. Kollann, K. Merz, K. Radacki, Angew. Chem., Int.
Ed. 40 (2001) 4198.[5] H. Braunschweig, M. Colling, C. Hu, K. Radacki, Angew. Chem., Int. Ed. 43
(2003) 205.[6] H. Braunschweig, K. Radacki, D. Rais, K. Uttinger, Angew. Chem., Int. Ed. 45
(2006) 162.[7] H. Braunschweig, K. Radacki, D. Rais, K. Uttinger, Organometallics 25 (2006)
5159.[8] H. Braunschweig, K. Radacki, K. Uttinger, Angew. Chem., Int. Ed. 46 (2007)
3979.[9] B. Blank, M. Colling-Hendelkens, C. Kollann, K. Radacki, D. Rais, K. Uttinger, G.R.
Whittell, H. Braunschweig, Chem. Eur. J. 13 (2007) 4770.[10] H. Braunschweig, M. Burzler, T. Kupfer, K. Radacki, F. Seeler, Angew. Chem., Int.
Ed. 46 (2007) 7785.[11] H. Braunschweig, M. Forster, T. Kupfer, F. Seeler, Angew. Chem., Int. Ed. 47
(2008) 5981.[12] H. Braunschweig, T. Kupfer, K. Radacki, A. Schneider, F. Seeler, K. Uttinger, H.
Wu, J. Am. Chem. Soc. 130 (2008) 7974.[13] G. Alcaraz, U. Helmstedt, E. Clot, L. Vendier, S. Sabo-Etienne, J. Am. Chem. Soc.
130 (2008) 12878.[14] D.L. Coombs, S. Aldridge, C. Jones, D.J. Willock, J. Am. Chem. Soc. 125 (2003)
6356.[15] D.L. Coombs, S. Aldridge, A. Rossin, C. Jones, D.J. Willock, Organometallics 23
(2004) 2911.
[16] S. Aldridge, C. Jones, T. Gans-Eichler, A. Stasch, D.L. Kays, N.D. Coombs, D.J.Willock, Angew. Chem., Int. Ed. 45 (2006) 6118.
[17] D. Vidovic, M. Findlater, G. Reeske, A.H. Cowley, Chem. Commun. (2006) 3786.[18] (a) H. Braunschweig, K. Radacki, K. Uttinger, Angew. Chem., Int. Ed. 46 (2007)
3979;(b) N. Arnold, H. Braunschweig, P. Brenner, J. Oscar, C. Jimenez-Halla, T. Kuper,K. Organometallics (2012), http://dx.doi.org/10.1021/om2012248.
[19] G.A. Pierce, D. Vidovic, D.L. Kays, N.D. Coombs, A.L. Thompson, E.D. Jemmis, S.De, S. Aldridge, Organometallics 28 (2009) 2947.
[20] S. De, G.A. Pierce, D. Vidovic, D.L. Kays, N.D. Coombs, E.D. Jemmis, S. Aldridge,Organometallics 28 (2009) 2961.
[21] D.A. Addy, G.A. Pierce, D. Vidovic, D. Mallick, E.D. Jemmis, J.M. Goicoechea, S.Aldridge, J. Am. Chem. Soc. 132 (2010) 4586.
[22] D.A. Addy, N. Phillips, G.A. Pierce, D. Vidovic, T. Krämer, D. Mallick, E.D.Jemmis, G. Reid, S. Aldridge, Organometallics 31 (2012) 1092.
[23] M. O’Neill, D.A. Addy, I. Riddlestone, M. Kelly, N. Phillips, S. Aldridge, J. Am.Chem. Soc. 133 (2011) 11500.
[24] H. Braunschweig, Q. Ye, K. Radaski, Chem. Commun. 48 (2012) 2701.[25] H. Braunschweig, Adv. Organomet. Chem. 51 (2004) 163.[26] H. Braunschweig, C. Kollann, D. Rais, Angew. Chem., Int. Ed. 45 (2006) 5254.[27] H. Braunschweig, C. Kollann, F. Seeler, Struct. Bond. (Berlin) 130 (2008) 1.[28] H. Braunschweig, R.D. Dewhurst, Angew. Chem., Int. Ed. 48 (2009) 1893.[29] H. Braunschweig, R.D. Dewhurst, Angew. Chem., Int. Ed. 49 (2010) 3412.[30] H. Braunschweig, R.D. Dewhurst, A. Schneider, Chem. Rev. 110 (2010) 3924.[31] S. Aldridge, D.L. Coombs, Coord. Chem. Rev. 248 (2004) 535.[32] S. Aldridge, D.L. Kays, Main Group Chem. 5 (2006) 223.[33] D. Vidovic, G.A. Pierce, S. Aldridge, Chem. Commun. (2009) 1157.[34] A.W. Ehlers, E.J. Baerends, F.M. Bickelhaupt, U. Radius, Chem. Eur. J. 4 (1998)
210.[35] U. Radius, F.M. Bickelhaupt, A.W. Ehlers, N. Goldberg, R. Hoffmann, Inorg.
Chem. 37 (1998) 1080.[36] C.L.B. Macdonald, A.H. Cowley, J. Am. Chem. Soc. 121 (1999) 12113.[37] C. Boehme, G. Frenking, Chem. Eur. J. 5 (1999) 2184.[38] J. Uddin, C. Boehme, G. Frenking, Organometallics 19 (2000) 571.[39] Y. Chen, G. Frenking, Dalton Trans. (2001) 434.[40] T. Bollwein, P.J. Brothers, H.L. Hermann, P. Schwerdtfeger, Organometallics 21
(2002) 5236.[41] J. Uddin, G. Frenking, J. Am. Chem. Soc. 123 (2001) 1683.[42] C. Boehme, J. Uddin, G. Frenking, Coord. Chem. Rev. 197 (2000) 249.[43] G. Frenking, N. Fröhlich, Chem. Rev. 100 (2000) 717.[44] G. Frenking, K. Wichmann, N. Fröhlich, C. Loschen, M. Lein, J. Frunzke, V.M.
[45] S. Aldridge, A. Rossin, D.L. Coombs, D.J. Willock, Dalton Trans. (2004) 2649.[46] K.K. Pandey, A. Lledós, F. Maseras, Organometallics 28 (2009) 6442.[47] K.K. Pandey, D.G. Musaev, Organometallics 29 (2010) 142.[48] K.K. Pandey, H. Braunschweig, A. Lledós, Inorg. Chem. 50 (2011) 1402.[49] K.K. Pandey, H. Braunschweig, R.D. Dewhurst, Eur. J. Inorg. Chem. (2011)
2045.[50] K.K. Pandey, Organometallics 30 (2011) 5851.[51] H. Braunschweig, K. Radacki, K. Uttinger, Chem. Eur. J. 14 (2008) 7858.[52] H. Braunschweig, K. Gruss, K. Radacki, K. Uttinger, Eur. J. Inorg. Chem. (2008)
1462.[53] (a) A. Diefenbach, F.M. Bickelhaupt, G. Frenking, J. Am. Chem. Soc. 122 (2000)
6449;(b) G. Frenking, K. Wichmann, N. Froehlich, C. Loschen, M. Lein, J. Frunzke,V.M. Ravon, Coord. Chem. Rev. 238 (2003) 55;(c) M. von Hopffgerten, G. Frenking, WIREs Comput. Mol. Sci. 2 (2012) 43. andreferences therein.
[54] (a) H. Jacobsen, T. Zeigler, J. Am. Chem. Soc. 116 (1994) 3667;(b) H. Zhu, T. Zeigler, Organometallics 27 (2008) 1748;(c) M.P. Mitoraj, H. Zhu, A. Michalak, T. Zeigler, Organometallics 26 (2007)1627;(d) M.P. Mitoraj, A. Michalak, T. Ziegler, J. Chem. Theory Comput. 5 (2009) 962;(e) T. Zeigler, Struct. Bond. 47 (2011) 1.
[55] (a) A.W. Ehlers, E.J. Baerends, K. Lammertsma, J. Am. Chem. Soc. 124 (2002)2831;(b) A. Diefenbach, F.M. Bickelhaupt, J. Chem. Phys. 115 (2001) 4030.
[56] (a) K.K. Pandey, Coord. Chem. Rev. 253 (2009) 37;(b) K.K. Pandey, J. Phys. Chem. A 115 (2011) 8578.
[57] A.D. Becke, Phys. Rev. A 38 (1988) 3098.[58] J.P. Perdew, Phys. Rev. B 33 (1986) 8822.[59] (a) C. Chang, M. Pelissier, Ph. Durand, Phys. Scr. 34 (1986) 394;
(b) J.L. Heully, I. Lindgren, E. Lindroth, S. Lundquist, A.M. Martensson-Pendrill,J. Phys. B 19 (1986) 2799;(c) E. van Lenthe, E.J. Baerends, J.G. Snijders, J. Chem. Phys. 99 (1993) 4597;(d) E. van Lenthe, E.J. Baerends, J.G. Snijders, J. Chem. Phys. 105 (1996)6505;(e) E. van Lenthe, R. van Leeuwen, E.J. Baerends, J.G. Snijders, Int. J. Quantum
Chem. 57 (1996) 281;(f) E. van Lenthe, A.E. Ehlers, E.J. Baerends, J. Chem. Phys. 110 (1999) 8943.
[60] J.G. Snijders, E.J. Baerends, P. Vernooijs, At. Data Nucl. Data Tables (1982) 26.and 483.
[61] E.J. Baerends, D.E. Ellis, P. Ros, Chem. Phys. 2 (1973) 41.[62] G. te Velde, F.M. Bickelhaupt, S.J.A. van Gisbergen, C. Fonseca Guerra, E.J.
Baerends, J.G. Snijders, T. Ziegler, J. Comput. Chem. 22 (2001) 931.[63] E.J. Baerends, J. Autschbach, D. Bashford, A. Bérces, F.M. Bickelhaupt, C. Bo, P.M.
Boerrigter, L. Cavallo, D.P. Chong, L. Deng, R.M. Dickson, D.E. Ellis, M. vanFaassen, L. Fan, T.H. Fischer, C. Fonseca Guerra, A. Ghysels, A. Giammona, S.J.A.van Gisbergen, A.W. Götz, J.A. Groeneveld, O.V. Gritsenko, M. Grüning, F.E.Harris, P. van den Hoek, C.R. Jacob, H. Jacobsen, L. Jensen, G. van Kessel, F.Kootstra, M.V. Krykunov, E. van Lenthe, D.A. McCormack, A. Michalak, M.Mitoraj, J. Neugebauer, V.P. Nicu, L. Noodleman, V.P. Osinga, S. Patchkovskii,P.H.T. Philipsen, D. Post, C.C. Pye, W. Ravenek, J.I. Rodríguez, P. Ros, P.R.T.Schipper, G. Schreckenbach, M. Seth, J.G. Snijders, M. Solà, M. Swart, D.Swerhone, G. te Velde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang,T.A. Wesolowski, E.M. van Wezenbeek, G. Wiesenekker, S.K. Wolff, T.K. Woo,A.L. Yakovlev, T. Ziegler, ADF 2010.02, Scientific Computing & Modelling NV,The Netherlands.
[64] F.M. Bickelhaupt, E.J. Baerends, in: K.B. Lipkowitz, D.B. Boyd (Eds.), Reviews inComputational Chemistry, vol. 15, Wiley-VCH, New York, 2000, pp. 1–86.
[65] (a) K. Morokuma, J. Chem. Phys. 55 (1971) 1236;(b) K. Morokuma, Acc. Chem. Res. 10 (1977) 294.
[66] (a) T. Ziegler, A. Rauk, Theor. Chim. Acta 46 (1977) 1;(b) T. Ziegler, A. Rauk, Inorg. Chem. 18 (1979) 1558;(c) T. Ziegler, A. Rauk, Inorg. Chem. 18 (1979) 1755.
[67] A.E. Reed, L.A. Curtiss, F. Weinhold, Chem. Rev. 88 (1988) 899.[68] G. Schaftenaar, MOLDEN3.4, CAOSCAMM Center, The Netherlands, 1998.[69] P. Pyykkö, M. Atsumi, Chem. Eur. J. 15 (2009) 12770.[70] L. Pauling, The Nature of the Chemical Bond, third ed., Cornell University Press,
New York, 1960. The relationship of bond order to length is dn = d1–0.71 log(n)where n is the bond order, d1 and dn are the bond lengths with bond order 1and n, respectively..
[71] K.B. Wiberg, Tetrahedron 24 (1968) 1083.[72] J. Zhu, Z. Lin, T.B. Marder, Inorg. Chem. 44 (2005) 9384.