123
Building a Chemical Informatics Grid Marlon Pierce Community Grids Laboratory Indiana University
| Date post: | 22-Dec-2015 |
| Category: |
Documents |
| View: | 216 times |
| Download: | 1 times |

Building a Chemical Informatics Grid
Marlon Pierce
Community Grids Laboratory
Indiana University

Acknowledgments
CICC researchers and developers who contributed to this presentation: Prof. Geoffrey Fox, Prof. David Wild, Prof. Mookie Baik,
Prof. Gary Wiggins, Dr. Jungkee Kim, Dr. Rajarshi Guha, Sima Patel, Smitha Ajay, Xiao Dong
Thanks also to Prof. Peter Murray Rust and the WWMM group at Cambridge University
More info: www.chembiogrid.org and www.chembiogrid.org/wiki.

Chemical Informatics and the Grid
An overview of the basic problem and solution

Chemical Informatics as a Grid Application Chemical Informatics is the application of information technology
to problems in chemistry. Example problems: managing data in large scale drug discovery
and molecular modeling Building Blocks: Chemical Informatics Resources:
Chemical databases maintained by various groups NIH PubChem, NIH DTP
Application codes (both commercial and open source) Data mining, clustering Quantum chemistry and molecular modeling
Visualization tools Web resources: journal articles, etc.
A Chemical Informatics Grid will need to integrate these into a common, loosely coupled, distributed computing environment.

Problem: Connecting It Together The problem is defining an architecture for tying all of
these pieces into a distributed computing system. A “Grid”
How can I combine application codes, web resources, and databases to solve a particular problem that interests me? Specifically, how do I build a runtime environment that can
connect the distributed services I need to solve an interesting problem?
For academic and government researchers, how can I do all of this in an open fashion? Data and services can come from anywhere That is, I must avoid proprietary infrastructure.

NIH Roadmap for Medical Researchhttp://nihroadmap.nih.gov/ The NIH recognizes chemical and biological
information management as critical to medical research.
Federally funded high throughput screening centers. 100-200 HTS assays per year on small molecules. 100,000’s of small molecules analyzed Data published, publicly available through NIH PubChem
online database. What do you do with all of this data?
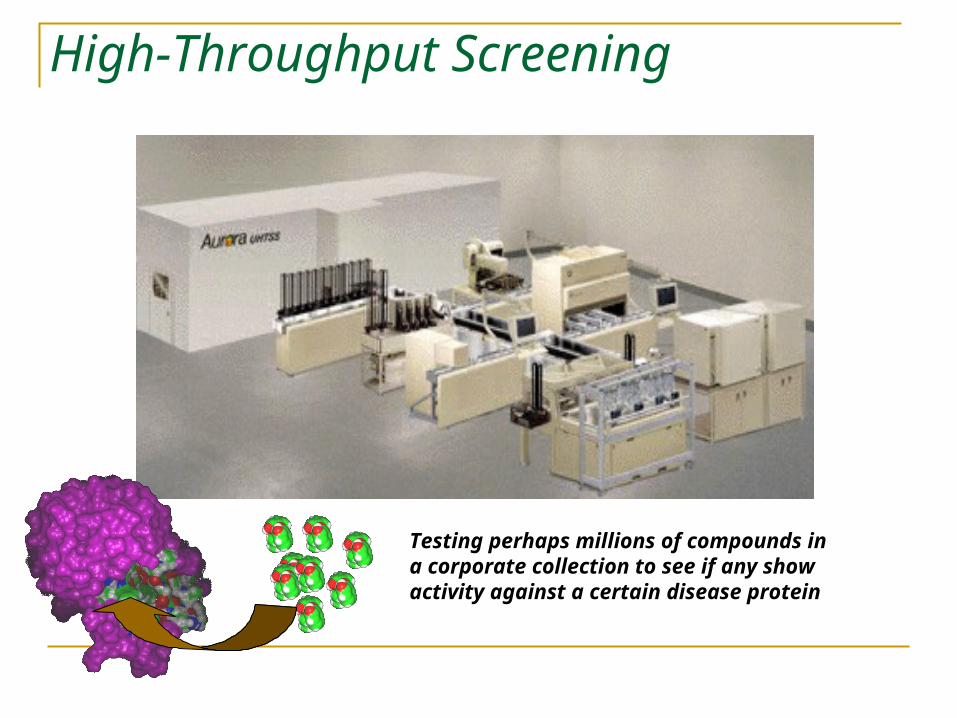
High-Throughput Screening
Testing perhaps millions of compounds in a corporate collection to see if any show activity against a certain disease protein

High-Throughput Screening
Traditionally, small numbers of compounds were tested for a particular project or therapeutic area
About 10 years ago, technology developed that enabled large numbers of compounds to be assayed quickly
High-throughput screening can now test 100,000 compounds a day for activity against a protein target
Maybe tens of thousands of these compounds will show some activity for the protein
The chemist needs to intelligently select the 2 - 3 classes of compounds that show the most promise for being drugs to follow-up

Informatics Implications
Need to be able to store chemical structure and biological data for millions of data points Computational representation of 2D structure
Need to be able to organize thousands of active compounds into meaningful groups Group similar structures together and relate to activity
Need to learn as much information as possible(data mining) Apply statistical methods to the structures and related
information Need to use molecular modeling to gain direct
chemical insight into reactions.

The Solution, Part I: Web Services Web Services provide the means for wrapping
databases, applications, web scavengers, etc, with programming interfaces. WSDL definitions define how to write clients to talk with
databases, applications, etc. Web Service messaging through SOAP Discovery services such as UDDI, MDS, and so on.
Many toolkits available Axis, .NET, gSOAP, SOAP::Lite, etc.
Web Services can be combined with each other into workflows Workflow==use case scenario More about this later.
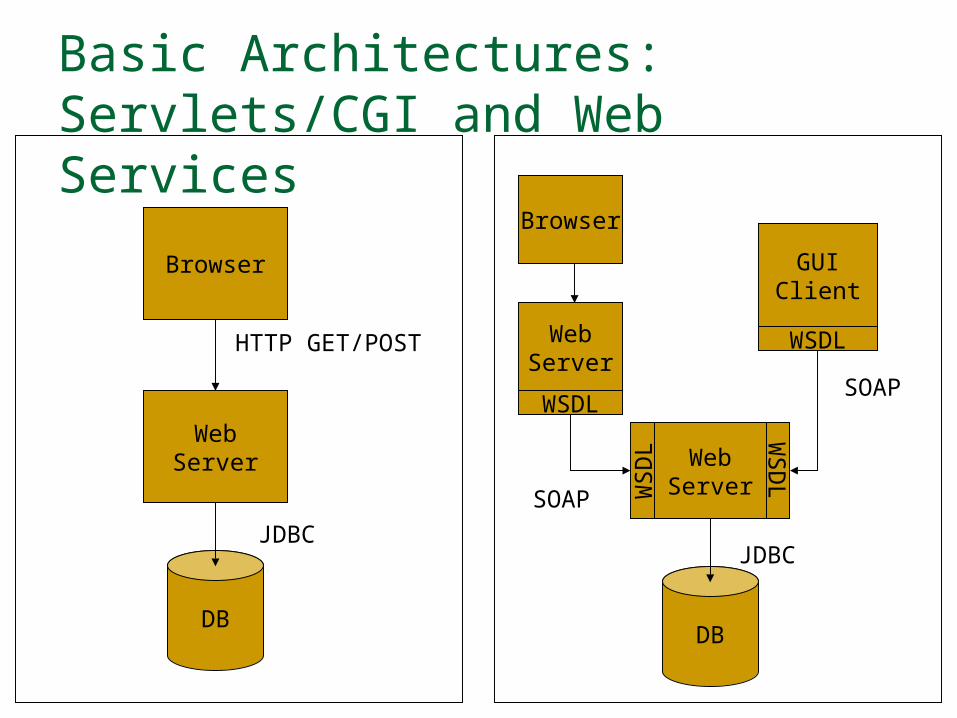
Basic Architectures: Servlets/CGI and Web Services
Browser
WebServer
HTTP GET/POST
DB
JDBC
WebServer
DB
JDBC
Browser
WebServer
SOAP
GUIClient
SOAPWSDL
WSDL
WSD
LWSD
L

Solution Part II: Grid Resources Many Grid tools provide powerful backend services
Globus: uniform, secure access to computing resources (like TeraGrid) File management, resource allocation management, etc.
Condor: job scheduling on computer clusters and collections
SRB: data grid access OGSA-DAI: uniform Grid interface to databases.
These have Web Service as well as other interfaces (or equivalently, protocols).

Solution, Part III: Domain Specific Tools and Standards -->More Services For Chemical Informatics, we have a number of tools and
standards. Chemical string representations
SMILES, InChI Chemistry Markup Language
XML language for describing, exchanging data. JUMBO 5: a CML parser and library
Glue Tools and Applications Chemistry Development Kit (CDK) OpenBabel
These are the basis for building interoperable Chemical Informatics Web Services
Analogous situations exist for other domains Astronomy, Geosciences, Biology/Bioinformatics

Solution Part IV: Workflows
Workflow engines allow you to connect services together into interesting composite applications.
This allows you to directly encode your scientific use case scenario as a graph of interacting services.
There are many workflow tools We’ll briefly cover these later. General guidance is to build web services first and then
use workflow tools on top of these services. Don’t get married to a particular workflow technology yet,
unless someone pays you.

Solution Part V: User Interfaces Web Services allow you to cleanly separate user
interfaces from backend services. Model-view-controller pattern for web applications
Client environments include Grid and web service scripting environments Desktop tools like Taverna and Kepler Portlet-based Web portal systems
Typically, desktop tools like Taverna are used by power users to define interesting workflows.
Portals are for running canned workflows.

Next steps
Next we will review the online data base resources that are available to us.
Databases come in two varieties Journal databases Data databases
As we will discuss, it is useful to build services and workflows for automatically interacting with both types.

Online Chemical Journal and Data Resources

MEDLINE: Online Journal Database MEDLINE (Medical Literature Analysis and Retrieval
System Online) is an international literature database of life sciences and biomedical information.
It covers the fields of medicine, nursing, dentistry, veterinary medicine, and health care.
MEDLINE covers much of the literature in biology and biochemistry, and fields with no direct medical connection, such as molecular evolution.
It is accessed via PubMed.
http://en.wikipedia.org/wiki/Medline

PubMed: Journal Search Engine PubMed is a free search engine offered by the United States National Library of Medicine as part of the Entrez information retrieval system.
The PubMed service allows searching the MEDLINE database. MEDLINE covers over 4,800 journals published in the United States
and more than 70 other countries primarily from 1966 to the present.
In addition to MEDLINE, PubMed also offers access to: OLDMEDLINE for pre-1966 citations. Citations to articles that are out-of-scope (e.g., general science and
chemistry) from certain MEDLINE journals In-process citations which provide a record for an article before it is
indexed with MeSH and added to MEDLINE Citations that precede the date that a journal was selected for
MEDLINE indexing Some life science journals
http://www.ncbi.nlm.nih.gov/entrez/query/static/overview.html

PubChem: Chemical Database PubChem is a database of chemical molecules. The system is maintained by the
National Center for Biotechnology Information (NCBI) which belongs to the United States National Institutes of Health (NIH).
PubChem can be accessed for free through a web user interface. And Web Services for programmatic access
PubChem contains mostly small molecules with a molecular mass below 500.
Anyone can contribute The database is free to use, but it is not curated, so value of a
specific compound information could be questionable. NIH funded HTS results are (intended to be) available through
pubchem.
http://pubchem.ncbi.nlm.nih.gov/

NIH DTP Database
Part of NIH’s Developmental Therapeutics Program.
Screens up to 3,000 compounds per year for potential anticancer activity.
Utilizes 59 different human tumor cell lines, representing leukemia, melanoma and cancers of the lung, colon, brain, ovary, breast, prostate, and kidney.
DTP screening results are part of PubChem and also available as a separate database.
http://dtp.nci.nih.gov/
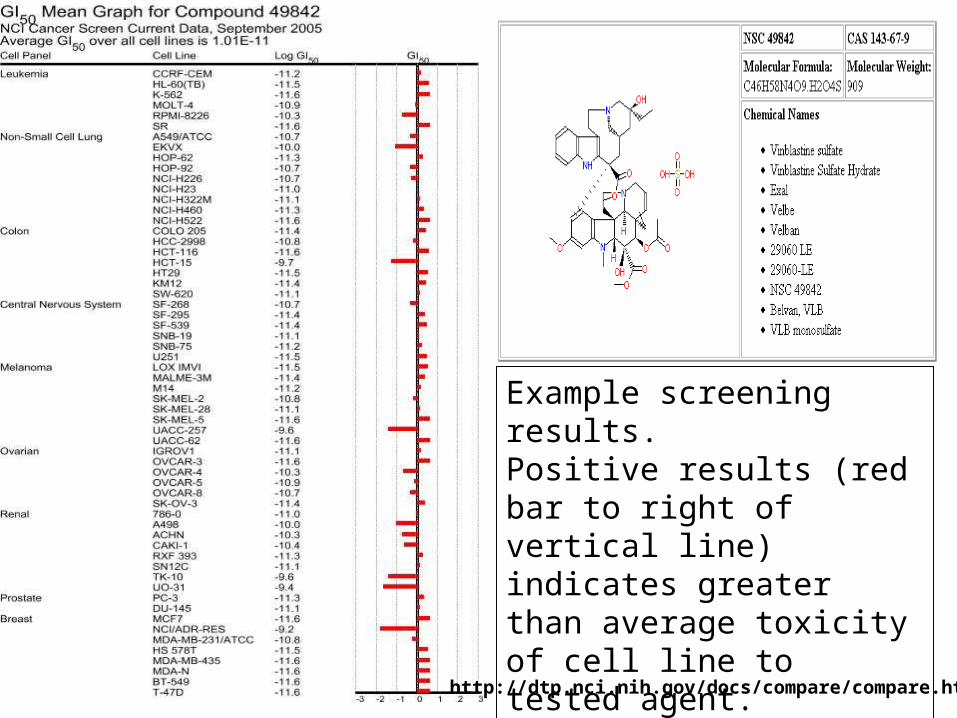
Example screening results. Positive results (red bar to right of vertical line) indicates greater than average toxicity of cell line to tested agent.
http://dtp.nci.nih.gov/docs/compare/compare.html

DTP and COMPARE
COMPARE is an algorithm for mining DTP result data to find and rank order compounds with similar DTP screening results.
Why COMPARE? Discovered compounds may be less toxic to
humans but just as effective against cancer cell lines.
May be much easier/safer to manufacture. May be a guide to deeper understanding of
experiments
http://dtp.nci.nih.gov/docs/compare/compare_methodology.html

Many Other Online Databases Complementary protein information
Indiana University: Varuna project Discussed in this presentation
University of Michigan: Binding MOAD “Mother of All Databases” Largest curated database of protein-ligand complexes Subset of protein databank Prof. Heather Carlson
University of Michigan: PDBBind Provides a collection of experimentally measured binding affinity
data (Kd, Ki, and IC50) exclusively for the protein-ligand complexes available in the Protein Data Bank (PDB)
Dr. Shaomeng Wang

The Point Is… All of these databases can be accessed on line with
human-usable interfaces. But that’s not so important for our purposes
More importantly, many of them are beginning to define Web Service interfaces that let other programs interact with them. Plenty of tools and libraries can simulate browsers, so you
can also build your own service. This allows us to remotely analyze databases with
clustering and other applications without modifying the databases themselves.
Can be combined with text mining tools and web robots to find out who else is working in the area.

Encoding chemistry

Chemical Machine Languages Interestingly, chemistry has defined three simple
languages for encoding chemical information. InChI, SMILES, CML Can generate these by hand or automatically
InChIs and SMILES can represent molecules as a single string/character array. Useful as keys for databases and for search queries in
Google. You can convert between SMILES and InChIs
OpenBabel, OELib, JOELib CML is an XML format, and more verbose, but
benefits from XML community tools

SMILES: Simplified Molecular Input Line Entry Specification Language for describing the structure of
chemical molecules using ASCII strings.
http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html

InChI: International Chemical Identifier IUPAC and NIST Standard similar to SMILES Encodes structural information about compounds Based on open an standard and algorithms.
http://wwmm.ch.cam.ac.uk/inchifaq/

InChI in Public Chemistry Databases US National Institute of Standards and Technology (NIST) - 150,000
structures NIH/NCBI/PubChem project - >3.2 million structures Thomson ISI - 2+ million structures US National Cancer Institute(NCI) Database - 23+ million structures US Environmental Protection Agency(EPA)-DSSToX Database - 1450
structures Kyoto Encyclopaedia of Genes and Genomes (KEGG) database - 9584
structures University of California at San Francisco ZINC - >3.3 million structures BRENDA enzyme information system (University of Cologne) - 36,000
structures Chemical Entities of Biological Interest (ChEBI) database of the European
Bioinformatics Institute - 5000 structures University of California Carcinogenic Potency Project - 1447 structures Compendium of Pesticide Common Names - 1437 (2005-03-03) structures

Journals and Software Using InChI Journals
Nature Chemical Biology. Beilstein Journal of Organic Chemistry
Software ACD/Labs ACD/ChemSketch. ChemAxon Marvin. SciTegic Pipeline Pilot. CACTVS Chemoinformatics Toolkit by Xemistry, GmbH.
http://wwmm.ch.cam.ac.uk/inchifaq/

Chemistry Markup Language
CML is an XML markup language for encoding chemical information. Developed by Peter Murray Rust, Henry Rzepa and others. Actually dates from the SGML days before XML
More verbose than InChI and SMILES But inherits XML schema, namespaces, parsers, XPATH,
language binding tools like XML Beans, etc. Not limited to structural information Has OpenBabel support.
http://cml.sourceforge.net/, http://cml.sourceforge.net/wiki/index.php/Main_Page

InChI Compared to SMILES SMILES is proprietary and
different algorithms can give different results.
Seven different unique SMILES for caffeine on Web sites: [c]1([n+]([CH3])[c]([c]2([c]([n+]1[CH3])
[n][cH][n+]2[CH3]))[O-])[O-] CN1C(=O)N(C)C(=O)C(N(C)C=N2)=C1
2 Cn1cnc2n(C)c(=O)n(C)c(=O)c12 Cn1cnc2c1c(=O)n(C)c(=O)n2C N1(C)C(=O)N(C)C2=C(C1=O)N(C)C=N
2 O=C1C2=C(N=CN2C)N(C(=O)N1C)C CN1C=NC2=C1C(=O)N(C)C(=O)N2C
On the other hand, some claimSMILES are more intuitive for human readers.
http://wwmm.ch.cam.ac.uk/inchifaq/
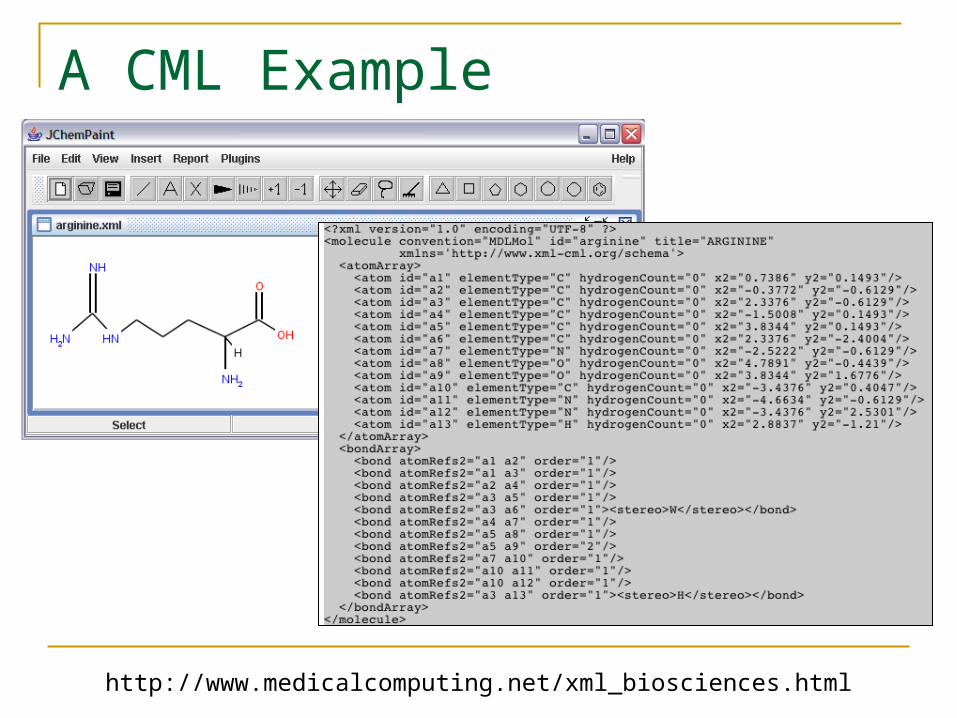
A CML Example
http://www.medicalcomputing.net/xml_biosciences.html

Clustering Techniques, Computing Requirements, and Clustering Services
Computational techniques for organizing data

The Story So Far
We’ve discussed managing screening assay output as the key problem we face Must sift through mountains of data in PubChem
and DTP to find interesting compounds. NIH funded High Throughput Screening will make
this very important in the near future. Need now a way to organize and analyze the
data.

Clustering and Data Analysis Clustering is a technique that can be applied to large data sets to
find similarities Popular technique in chemical informatics
Data sets are segmented into groups (clusters) in which members of the same cluster are similar to each other.
Clustering is distinct from classification, There are no pre-determined characteristics used to define the
membership of a cluster, Although items in the same cluster are likely to have many
characteristics in common. Clustering can be applied to chemical structures, for example, in
the screening of combinatorial or Markush compound libraries in the quest for new active pharmaceuticals.
We also note that these techniques are fairly primitive More interesting clustering techniques exist but apparently are not
well known by the chemical informatics community.

Non-Hierarchical Clustering Clusters form around centroids. The number of which can be specified by the user. All clusters rank equally and there is no particular
relationship between them.
http://www.digitalchemistry.co.uk/prod_clustering.html

Hierarchical Clustering Clusters are arranged in hierarchies
Smaller clusters are contained within larger ones; the bottom of the hierarchy consists of individual objects in "singleton" clusters, while the top of it consists of one cluster containing all the objects in the dataset.
Such hierarchies can be built either from the bottom up (agglomerative) or the top downwards (divisive)
http://www.digitalchemistry.co.uk/prod_clustering.html

Fingerprinting and Dictionaries--What Is Your Parameter Space? Clustering algorithms require a parameter space
Clusters defined along coordinate axes. Coordinate axes defined by a dictionary of chemical
structures. Use binary on/off for fingerprinting a particular compound
against a dictionary.
http://www.digitalchemistry.co.uk/prod_fingerprint.html

Cluster Analysis and Chemical Informatics Used for organizing datasets into chemical series, to build
predictive models, or to select representative compounds Clustering Methods
Jarvis-Patrick and variants O(N2), single partition
Ward’s method Hierarchical, regarded as best, but at least O(N2)
K-means < O(N2), requires set no of clusters, a little “messy”
Sphere-exclusion (Butina) Fast, simple, similar to JP
Kohonen network Clusters arranged in 2D grid, ideal for visualization

Limitations of Ward’s method forlarge datasets (>1m) Best algorithms have O(N2) time requirement (RNN) Requires random access to fingerprints
hence substantial memory requirements (O(N)) Problem of selection of best partition
can select desired number of clusters Easily hit 4GB memory addressing limit on 32 bit
machines Approximately 2m compounds

Scaling up clustering methods Parallelization
Clustering algorithms can be adapted for multiple processors
Some algorithms more appropriate than others for particular architectures
Ward’s has been parallelized for shared memory machines, but overhead considerable
New methods and algorithms Divisive (“bisecting”) K-means method Hierarchical Divisive Approx. O(NlogN)

Divisive K-means Clustering
New hierarchical divisive method Hierarchy built from top down, instead of bottom up Divide complete dataset into two clusters Continue dividing until all items are singletons Each binary division done using K-means method Originally proposed for document clustering
“Bisecting K-means” Steinbach, Karypis and Kumar (Univ. Minnesota)
http://www-users.cs.umn.edu/~karypis/publications/Papers/PDF/doccluster.pdf
Found to be more effective than agglomerative methods Forms more uniformly-sized clusters at given level

BCI Divkmeans
Several options for detailed operation Selection of next cluster for division size, variance, diameter affects selection of partitions from hierarchy, not shape of hierarchy
Options within each K-means division step distance measure choice of seeds batch-mode or continuous update of centroids termination criterion
Have developed parallel version for Linux clusters / grids in conjunction with BCI
For more information, see Barnard and Engels talks at: http://cisrg.shef.ac.uk/shef2004/conference.htm
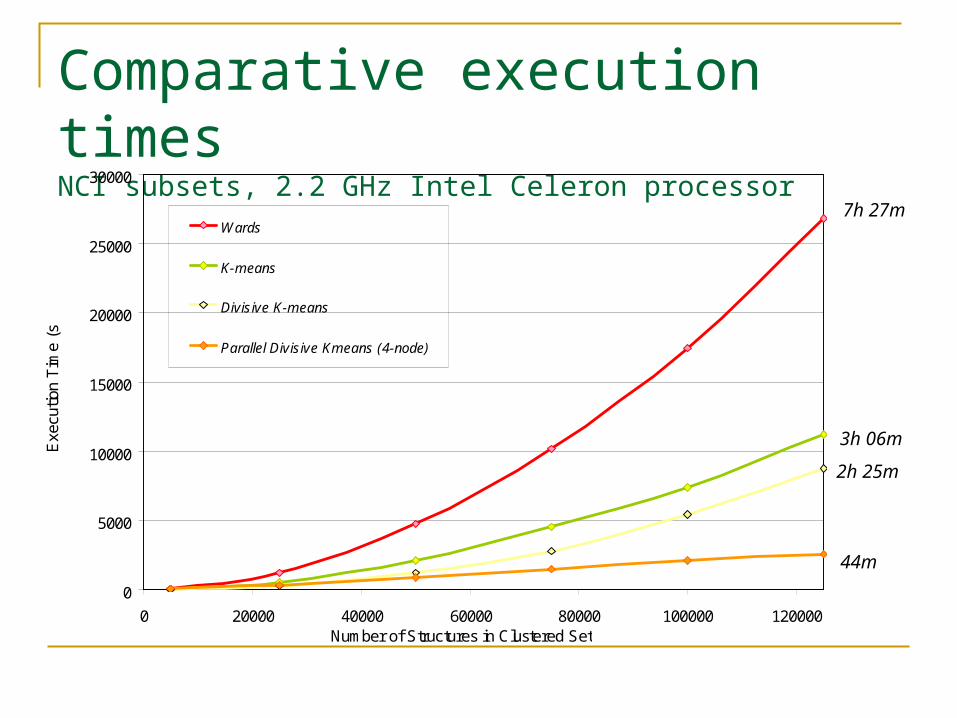
Comparative execution timesNCI subsets, 2.2 GHz Intel Celeron processor
7h 27m
3h 06m
2h 25m
44m
0
5000
10000
15000
20000
25000
30000
0 20000 40000 60000 80000 100000 120000Number of Structures in Clustered Set
Exe
cutio
n T
ime
(s)
Wards
K-means
Divisive K-means
Parallel Divisive Kmeans (4-node)

Divisive K-means: Conclusions Much faster than Ward’s, speed comparable to K-means,
suitable for very large datasets (millions) Time requirements approximately O(N log N) Current implementation can cluster 1m compounds in under
a week on a low-power desktop PC Cluster 1m compounds in a few hours with a 4-node parallel
Linux cluster Better balance of cluster sizes than Wards or Kmeans Visual inspection of clusters suggests better assembly of
compound series than other methods Better clustering of actives together than previously-
studied methods Memory requirements minimal Experiments using AVIDD cluster and Teragrid
forthcoming(50+ nodes)

Conclusions Effective exploitation of large volumes and diverse sources of
chemical information is a critical problem to solve, with a potential huge impact on the drug discovery process
Most information needs of chemists and drug discovery scientists are conceptually straightforward, but complex to implement
All of the technology is now in place to implement may of these information need “use-cases”: the four level model using service-oriented architectures together with smart clients look like a neat way of doing this
In conjunction with grid computing, rapid and effective organization and visualization of large chemical datasets is feasible in a web service environment
Some pieces are missing: Chemical structure search of journals (wait for InChI) Automated patent searching Effective dataset organization Effective interfaces, especially visualization of large numbers of 2D structures

Divisive K-Means as a Web Service The previous exercise was intended to show
that Divisive K-Means is a classic example of Grid application. Needs to be parallelized Should run on TeraGrid
How do you make this into a service? We’ll go on a small tour before getting back
to our problem.

Wrapping Science Applications as Services Science Grid services typically must wrap legacy
applications written in C or Fortran. You must handle such problems as
Specifying several input and output files These may need to be staged in
Launching executables and monitoring their progress. Specifying environment variables
Often these have also shell scripts to do some miscellaneous tasks.
How do you convert this to WSDL? Or (equivalently) how do you automatically generate the
XML job description for WS-GRAM?

Generic Service Toolkit (GFAC)(G. Kandaswamy, IU and RENCI) The Generic Service Toolkit can "wrap" any command-line
application as an application service. Given a set of input parameters, it runs the application, monitors
the application and returns the results. Requires no modification to program code. Also has web user interface generating tools.
When a user accesses an application service, the user is presented with a graphical user interface (GUI) to that service.
The GUI contains a list of operations that the user is allowed to invoke on that service.
After choosing an operation, the user is presented with a GUI for that operation, which allows the user to specify all the input parameters to that operation. The user can then invoke the operation on the service and get the output
results.
www.extreme.indiana.edu/gfac/

OPAL (S. Krishan, SDSC)
Features include scheduling (using Globus and Condor/SGE) and security (using GSI-based certificates), and persistent state management.
The WSDL defines operations to do the following: getAppMetadata: includes usage information, arbitrary application-specific
metadata specified as an array of other elements, e.g. description of the various options that are passed to the application
binary. launchJob: runs job with specified input and returns a Job ID. queryStatus: returns status code, message, and URL of the working
directory getOutputs: returns the outputs from a job that is identified by a Job ID.
URLs for the standard output and error Array of structures representing the output file names and URLs
getOutputAsBase64ByName: This operation returns the contents of an output file as Base64 binary.
destroy: This operation destroys a running job identified by a Job ID. launchJobBlocking: This operation requires the list of arguments as a string,
and an array of structures representing the input files.
http://grid-devel.sdsc.edu/gridsphere/gridsphere?cid=nbcrws

Our Solution: Apache Ant Services We’ve found using Apache Ant to be very useful for
wrapping services. Can call executables, set environment variables. Lots of useful built-in shell-like tasks. Extensible (write your own tasks). Develop build scripts to run your application
You can easily call Ant from other Java programs. So just write a wrapper service We use both blocking (hold connection until return) and
non-blocking version (suitable for long running codes). In non-blocking case, “Context” web service is used for
callbacks.
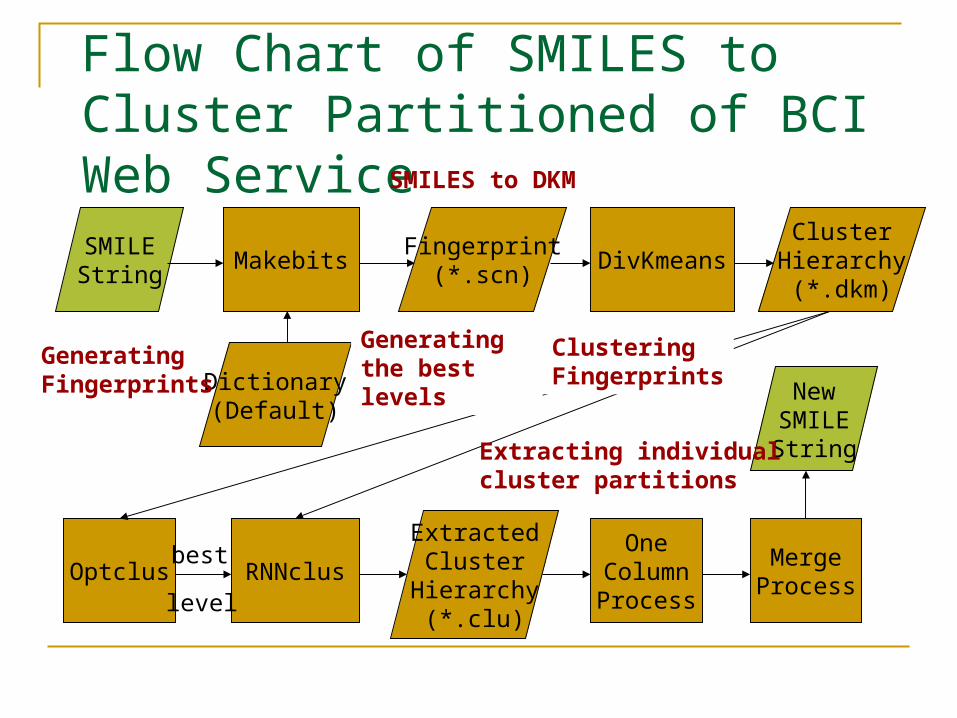
Flow Chart of SMILES to Cluster Partitioned of BCI Web ServiceSMILEString
Makebits
Dictionary(Default)
Fingerprint(*.scn)
DivKmeansCluster
Hierarchy(*.dkm)
Optclus RNNclusOne
ColumnProcess
MergeProcess
ExtractedCluster
Hierarchy(*.clu)
NewSMILEString
GeneratingFingerprints
ClusteringFingerprints
Generatingthe best levels
SMILES to DKM
Extracting individualcluster partitions
best
level
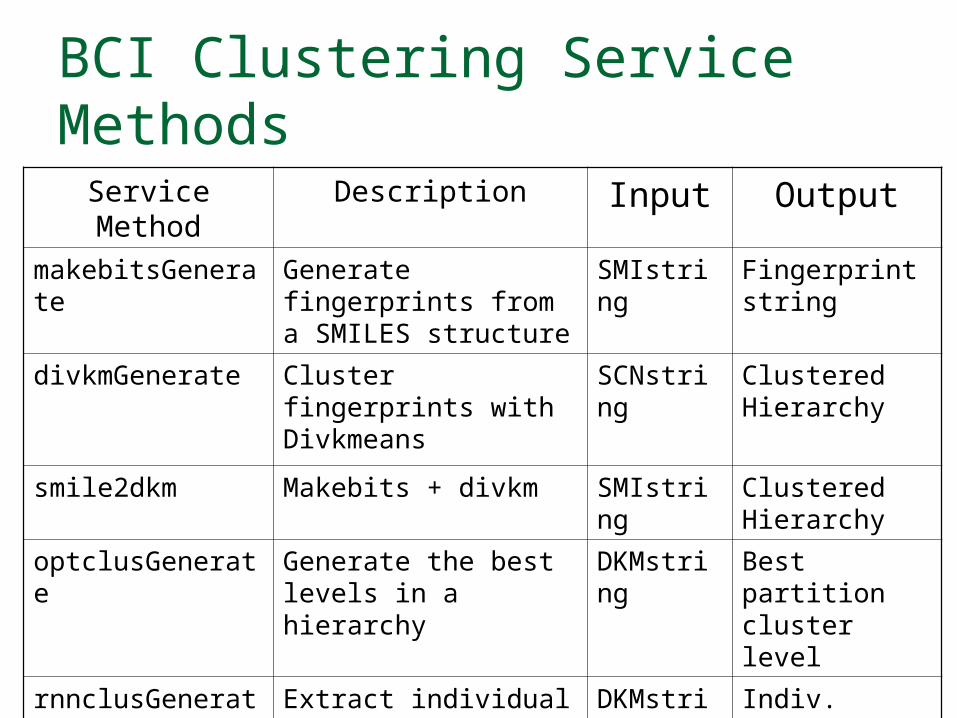
BCI Clustering Service Methods
Service Method Description Input Output
makebitsGenerate Generate fingerprints from a SMILES structure
SMIstring Fingerprint string
divkmGenerate Cluster fingerprints with Divkmeans
SCNstring Clustered Hierarchy
smile2dkm Makebits + divkm SMIstring Clustered Hierarchy
optclusGenerate Generate the best levels in a hierarchy
DKMstring Best partition cluster level
rnnclusGenerate Extract individual cluster partitions
DKMstring Indiv. cluster partitions
smile2ClusterPartitioned
Generate a new SMILES structure w/ extra col.
SMIstring New SMILES structure

A Library of Chemical Informatics Web Services

All Services Great and Small
Like most Grids, a Chemical Informatics Grid will have the classic styles: Data Grid Services: these provide access to data sources
like PubChem, etc. Execution Grid Services: used for running cluster analysis
programs, molecular modeling codes, etc, on TeraGrid and similar places.
But we also need many additional services Handling format conversions (InChI<->SMILES) Shipping and manipulating tabular data Determining toxicity of compounds Generating batch 2D images
So one of our core activities is “build lots of services”

VOTables: Handling Tabular Data Developed by the Virtual Observatory community for encoding
astronomy data. The VOTable format is an XML representation of the tabular
data (data coming from BCI, NIH DTP databases, and so on). VOTables-compatible tools have been built
We just inherit them. SAVOT and JAVOT JAVA Parser APIs for VOTable allow us
to easily build VOTable-based applications Web Services Spread sheet Plotting applications.
VOPlot and TopCat are two

Document Structure of VOTable
Compound
Name
Cluster
Number
Acemetacin 1
Candesartan 1
Acenocoumarol 2
Dicumarol 2
Phenprocoumon 2
Trioxsalen 2
Warfarin 2
<?xml version="1.0"?>
<VOTABLE version="1.1“ xmlns:xsi=http://www.w3.org/2001/XMLSchema-instance xsi:noNamespaceSchemaLocation="http://www.ivoa.net/xml/VOTable/VOTable/v1.1">
<RESOURCE >
<TABLE name="results">
<FIELD name=“CompoundName" ID="col1" datatype=“char" arraysize=“*”/>
<FIELD name=“ClustureNumber” ID="col2“ datatype=“int”/>
<DATA>
<TABLEDATA>
<TR><TD>Acemetacin</TD><TD>1</TD</TR>
<TR><TD>Candesartan</TD><TD>1</TD></TR>
<TR><TD>Acenocoumarol</TD><TD>2</TD></TR>
<TR><TD>Dicumarol</TD><TD>2</TD></TR>
<TR><TD>Phenprocoumon</TD><TD>2</TD></TR>
<TR><TD>Trioxsaken</TD><TD>2</TD></TR>
<TR><TD>warfarin</TD><TD>2</TD></TR>
</TABLEDATA>
</DATA>
</TABLE>
</RESOURCE>
</VOTABLE>
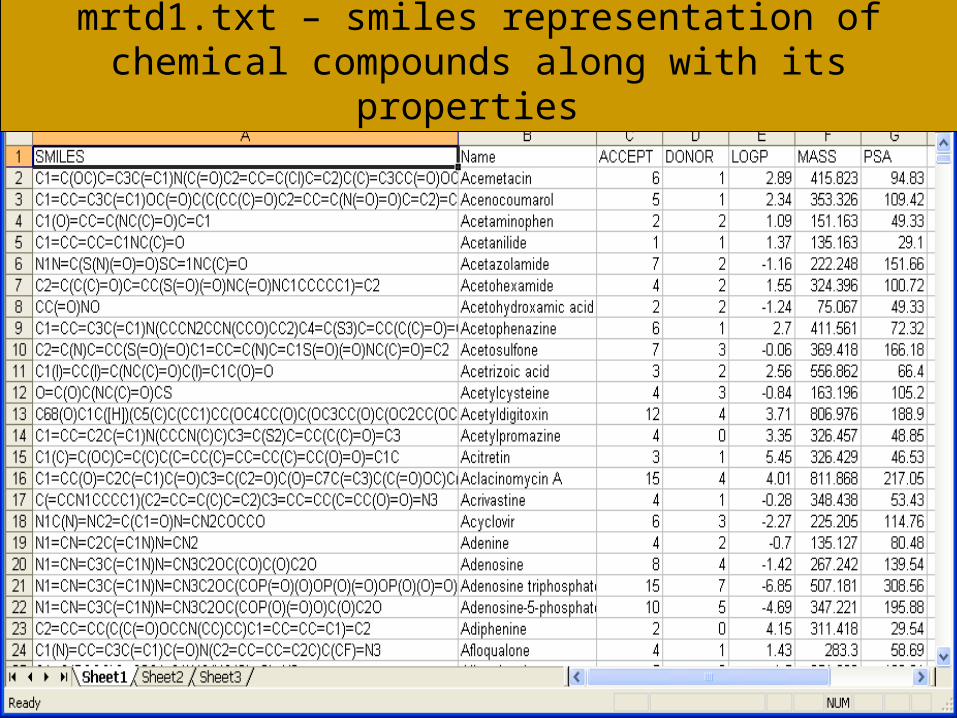
mrtd1.txt – smiles representation of chemical compounds along with its properties
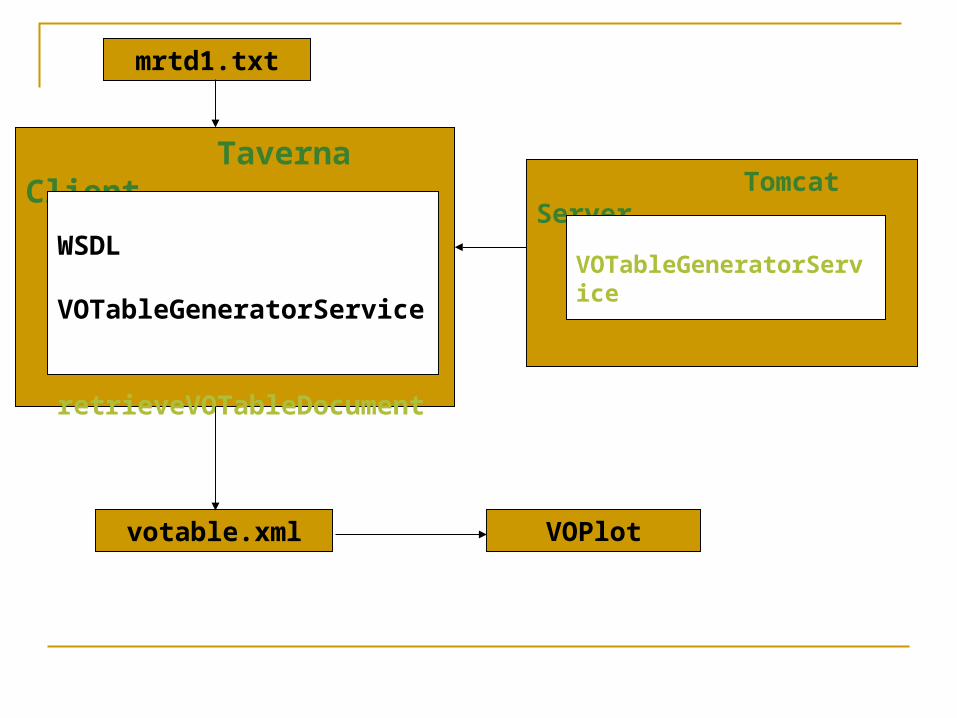
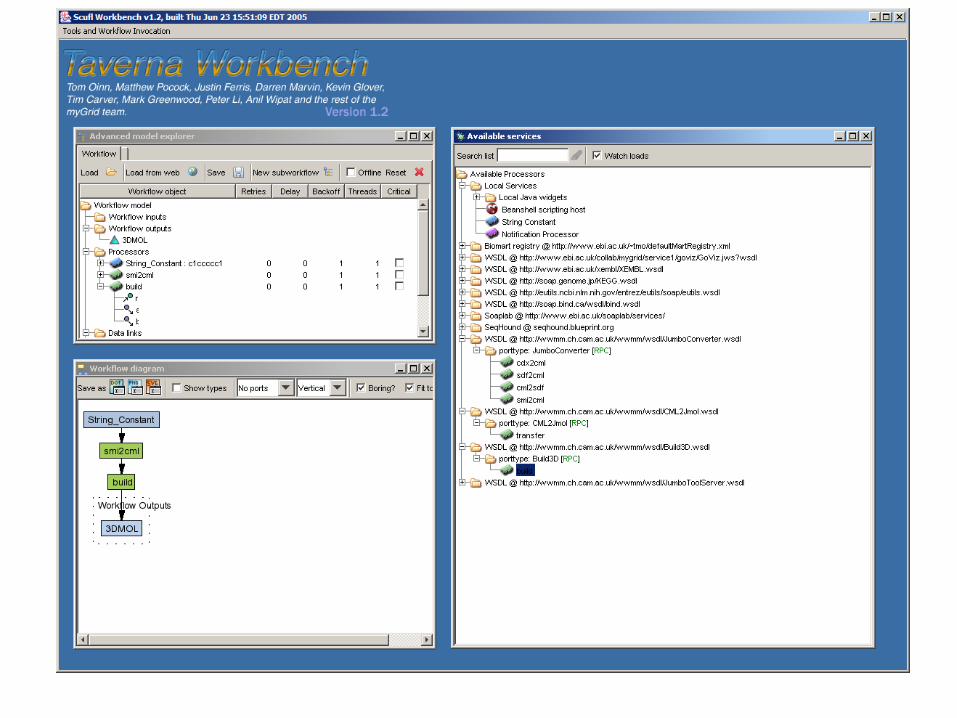
Taverna Client
WSDL VOTableGeneratorService retrieveVOTableDocument
Tomcat Server
VOTableGeneratorService
mrtd1.txt
votable.xml VOPlot
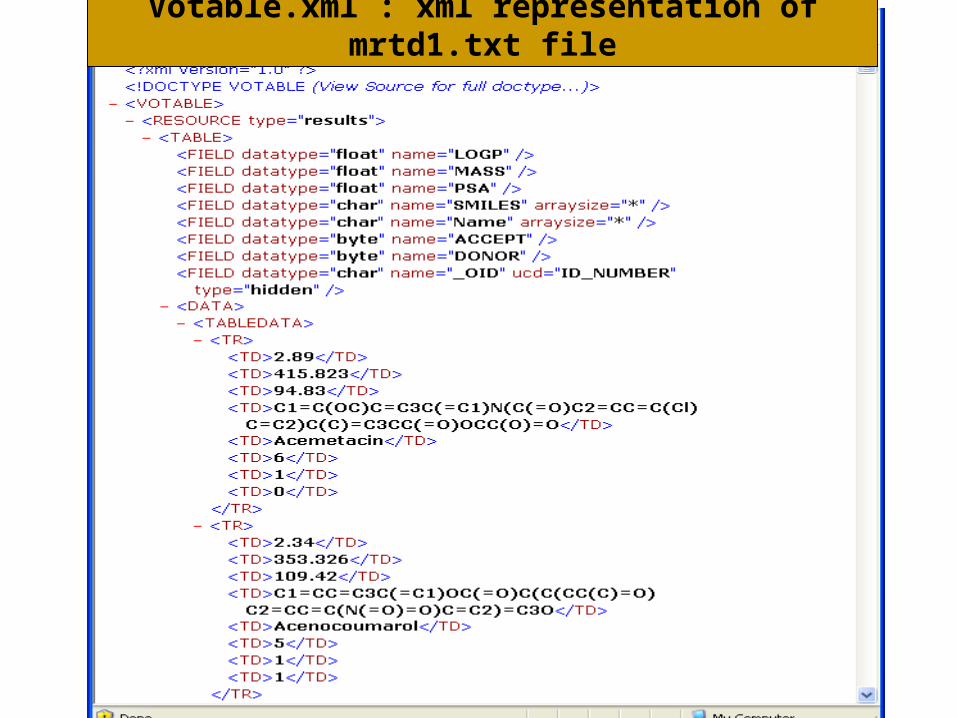
Votable.xml : xml representation of mrtd1.txt file
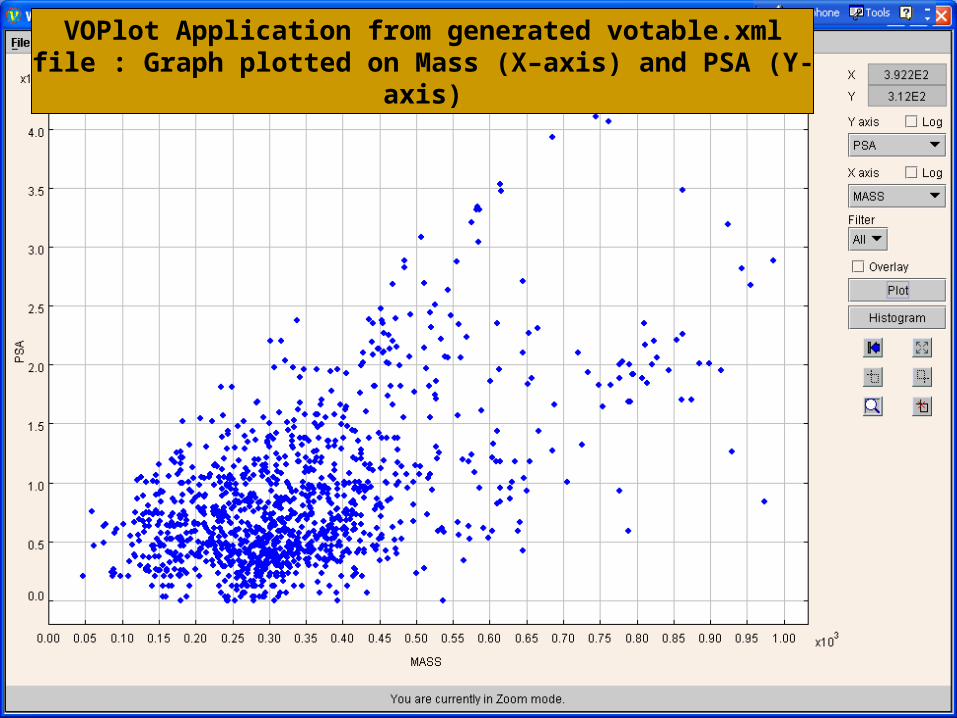
VOPlot Application from generated votable.xml file : Graph plotted on Mass (X–axis) and PSA (Y-axis)

Other Uses for VOTables
VOTables is a useful intermediate format for exchanging data between data bases.
Simple example: exchange data between VARUNA databases. Each student in the Baik group maintains his/her on copy
(sandbox purposes). Often need to import/export individual data sets.
It is also good for storing intermediate results in workflows. Value is not the format, but the fact that the XML can be
manipulated programmatically. Unions, subset, intersection operations

More Services: WWMM ServicesServices Descriptions Input Output
InChIGoogle Search an InChI structure through Google
inchiBasic
type
Search result in HTML format
InChIServer Generate InChI version
format
An InChI structure
OpenBabelServer
Transform a chemical format to another using Open Babel
format
inputData
outputData
options
Converted chemical structure string
CMLRSSServer
Generate CMLRSS feed from CML data
mol, title description link, source
Converted CMLRSS feed of CML data

CDK-Based Services
Common Substructure
Calculates the common substructure between two molecules.
CDKsim Takes two SMILES and evaluates the Tanimoto coefficient (ratio of intersection to union of their fingerprints).
CDKdesc Calculates a variety of molecular and atomic descriptors for QSAR modeling
CDKws Fingerprint generation
CDKsdg Creates a jpeg of the compound’s 2D structure
CDKStruct3D Generates 3D coordinates of a molecule from its SMILE
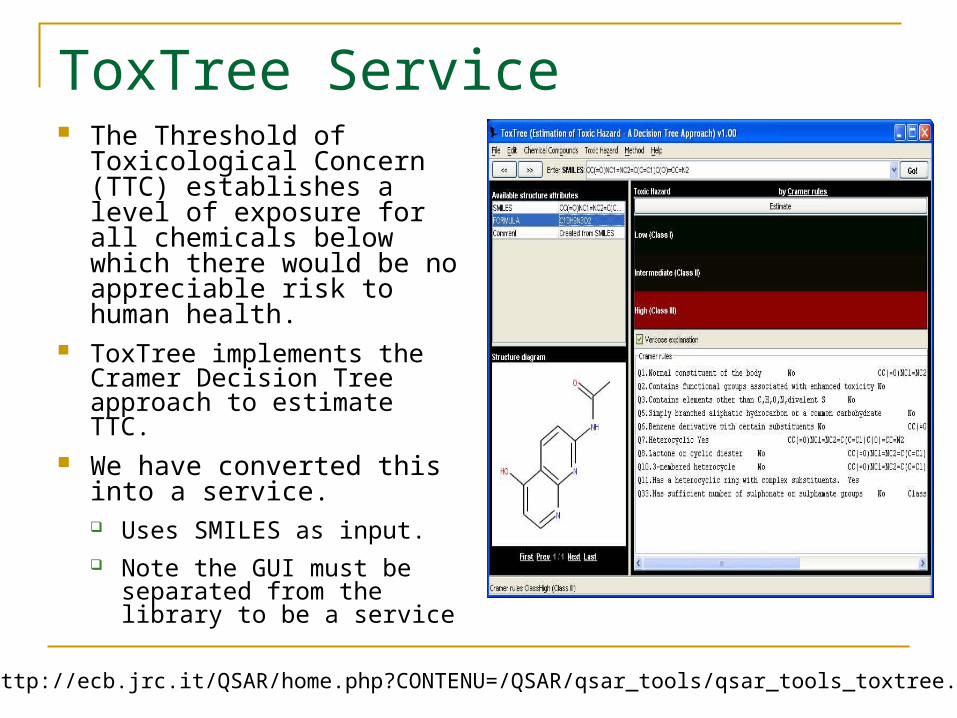
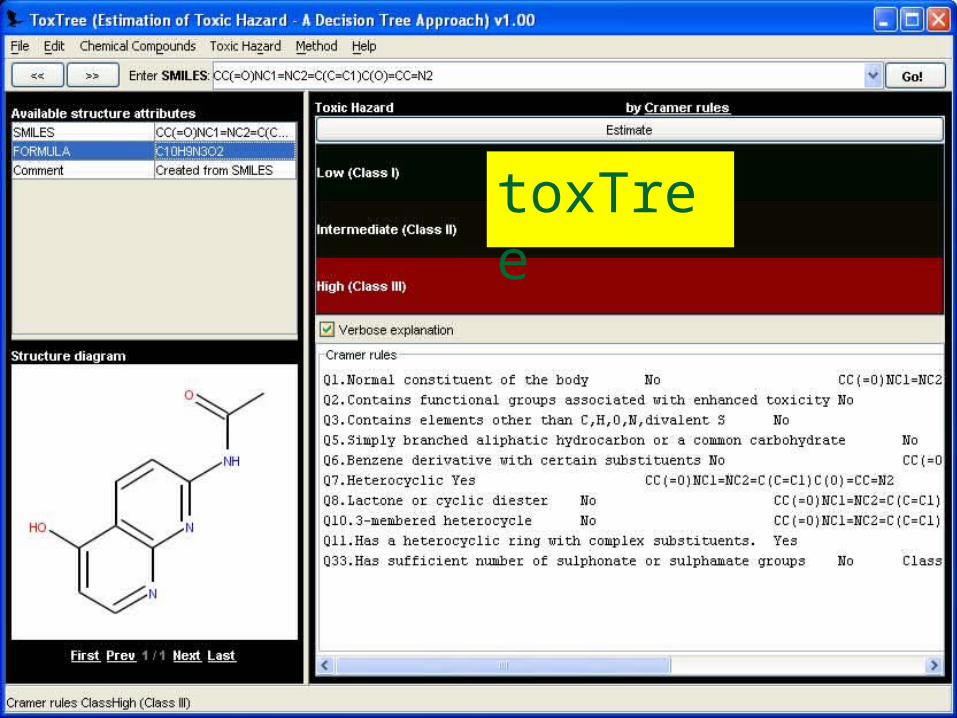
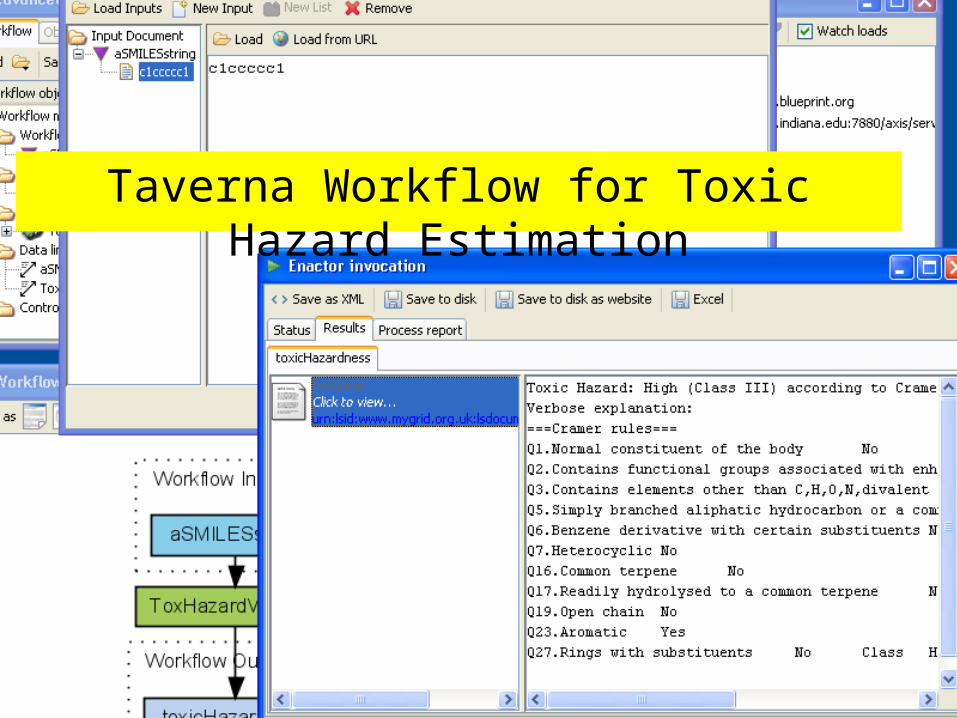
ToxTree Service The Threshold of
Toxicological Concern (TTC) establishes a level of exposure for all chemicals below which there would be no appreciable risk to human health.
ToxTree implements the Cramer Decision Tree approach to estimate TTC.
We have converted this into a service. Uses SMILES as input. Note the GUI must be
separated from the library to be a service
http://ecb.jrc.it/QSAR/home.php?CONTENU=/QSAR/qsar_tools/qsar_tools_toxtree.php
toxTree
Taverna Workflow for Toxic Hazard Estimation

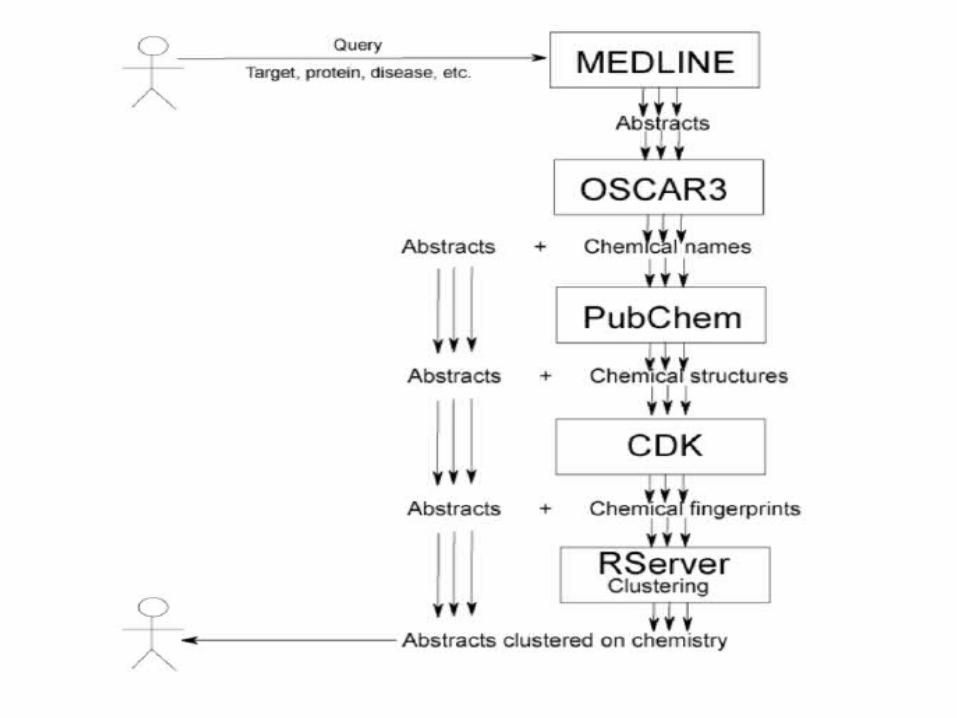
OSCAR3 Service Oscar3 is a tool for shallow, chemistry-specific
natural language parsing of chemical documents (i.e. journal articles).
It identifies (or attempts to identify): Chemical names: singular nouns, plurals, verbs etc., also
formulae and acronyms. Chemical data: Spectra, melting/boiling point, yield etc. in
experimental sections. Other entities: Things like N(5)-C(3) and so on.
There is a larger effort, SciBorg, in this area http://www.cl.cam.ac.uk/~aac10/escience/sciborg.html
This (like ToxTree) is potentially productively pleasingly parallelized.
It also has potentially very interesting Workflows
http://wwmm.ch.cam.ac.uk/wikis/wwmm/index.php/Oscar3

Use Cases and Workflows
Putting data and clustering together in a distributed environment.

Chemical Informatics as a Grid Problem NIH-Funded experimental screening
NIH DTP and HTS projects are generating a wealth of raw data on small compounds.
Available in PubChem Journal and chemical data sources often have public Web clients
and GUIs. But we need Web Service interfaces, not just Web interfaces. These provide a programming interfaces for building both human
and machine clients. These need to be connected to computing resources for running
clustering, data mining, and molecular modeling applications. Excellent candidates for running on the TeraGrid
We can formulate scientific problems that map to inter-connections of Grid services. This is generally called “Grid workflow” or “Service Orchestration”
Oracle Database (HTS)
Compounds were tested against related assays and showed activity, including
selectivity within target families
Oracle Database (Genomics)
? None of these compounds have been tested in a
microarray assay
Computation
The information in the structures and known activity data is good enough to create
a QSAR model with a confidence of 75%
External Database (Patent)
Some structures with a similarity > 0.75 to these
appear to be covered by a patent held by a competitor
Computation
All the compounds pass the Lipinksi Rule of Five and
toxicity filters
Excel Spreadsheet (Toxicity)
One of the compounds was previously tested for
toxicology and was found to have no liver toxicity
Word Document (Chemistry)
Several of the compounds had been followed up in a
previous project, and solubility problems prevented further
development
Journal Article
A recent journal article reported the effectiveness of some compounds in a related series against a target in the same family
Word Document (Marketing)
A report by a team in Marketing casts doubt on
whether the market for this target is big enough to make development cost-effective
SCIENTIST
“These compounds look promising from their HTS results. Should I commit some
chemistry resources to following them up?”
?

Workflow, Services, and Science Web Services work best as simple stateless
services. No implicit input, output, or interdependency of
methods. Services must be composed into interesting
applications. This is called workflow. A good workflow ...
Is composed of independent services Completely specifies an interesting science
problem.

Some Open Source Grid Workflow Projects UK e-Science Project’s Taverna
Scufl.xml scripting, GUI interface, works with Web Services. Kepler
Works with Web services and the Globus Toolkit. Condor DAGMan
www.cs.wisc.edu/condor Works over the top of Condor’s scheduler. Extended by the GriPhyN Virtual Data System
Java CoGKit’s Karajan XML workflow specification for scripting COG clients. Works with GT 2 and 4.
Community Grids Lab’s HPSearch www.hpsearch.org JavaScript scripting, works with Web services.
Indiana Extreme Lab’s Workflow Composer www.extreme.indiana.edu/xgws/xwf/index.html Jython, BPEL (soon) scripting
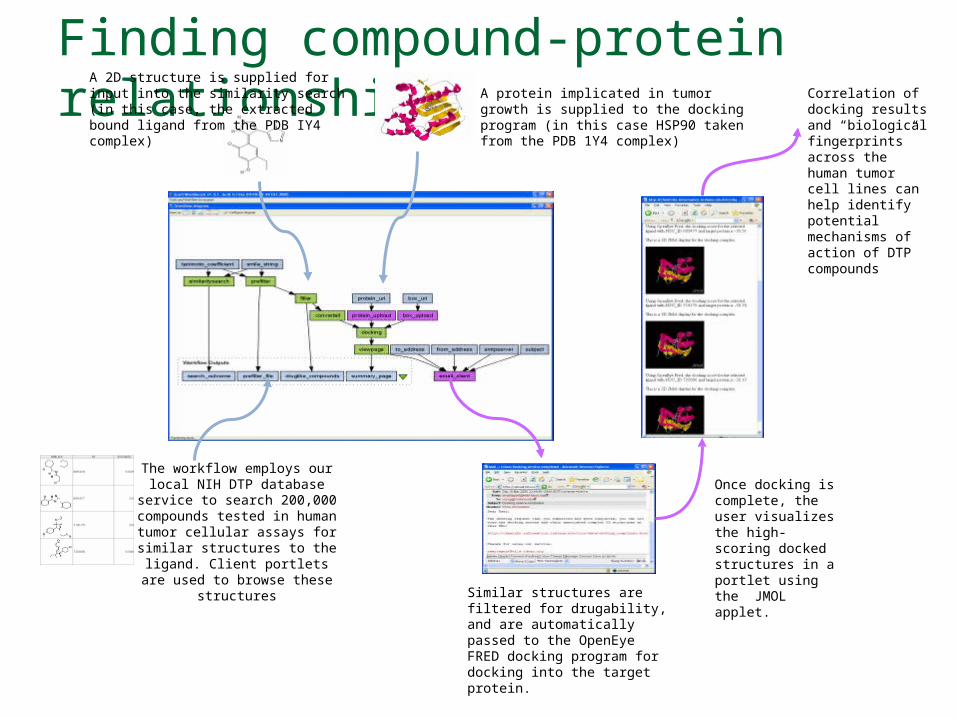
Finding compound-protein relationships A protein implicated in tumor growth is
supplied to the docking program (in this case HSP90 taken from the PDB 1Y4 complex)
The workflow employs our local NIH DTP database
service to search 200,000 compounds tested in human
tumor cellular assays for similar structures to the
ligand. Client portlets are used to browse these
structures
Once docking is complete, the user visualizes the high-scoring docked structures in a portlet using the JMOL applet.
Similar structures are filtered for drugability, and are automatically passed to the OpenEye FRED docking program for docking into the target protein.
A 2D structure is supplied for input into the similarity search (in this case, the extracted bound ligand from the PDB IY4 complex)
Correlation of docking results and “biological fingerprints” across the human tumor cell lines can help identify potential mechanisms of action of DTP compounds
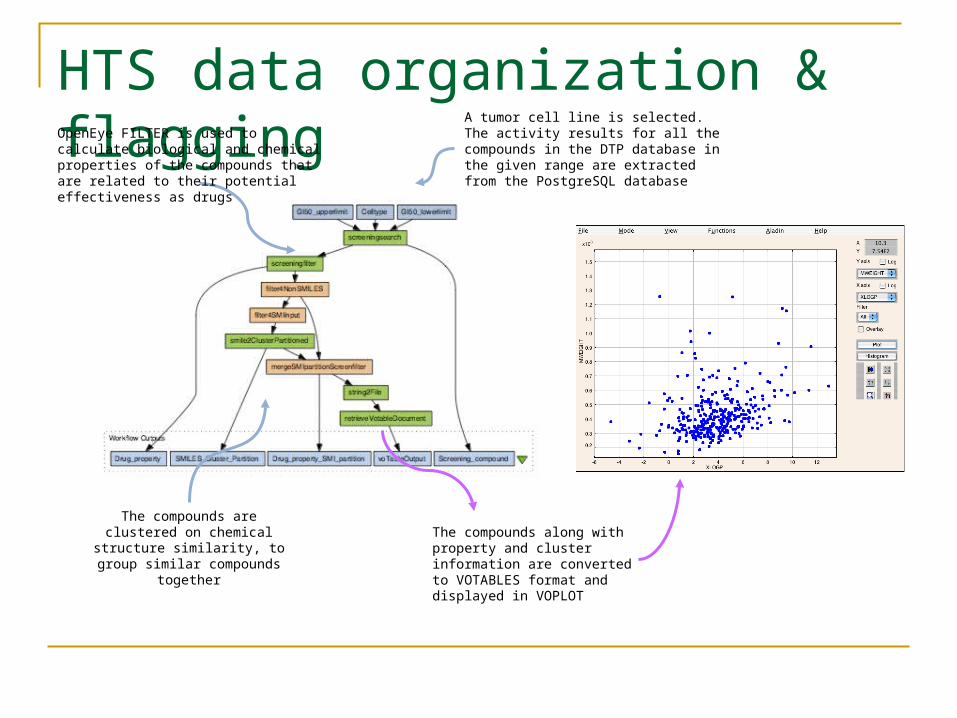
HTS data organization & flagging
A tumor cell line is selected. The activity results for all the compounds in the DTP database in the given range are extracted from the PostgreSQL database
The compounds are clustered on chemical structure
similarity, to group similar compounds together
The compounds along with property and cluster information are converted to VOTABLES format and displayed in VOPLOT
OpenEye FILTER is used to calculate biological and chemical properties of the compounds that are related to their potential effectiveness as drugs

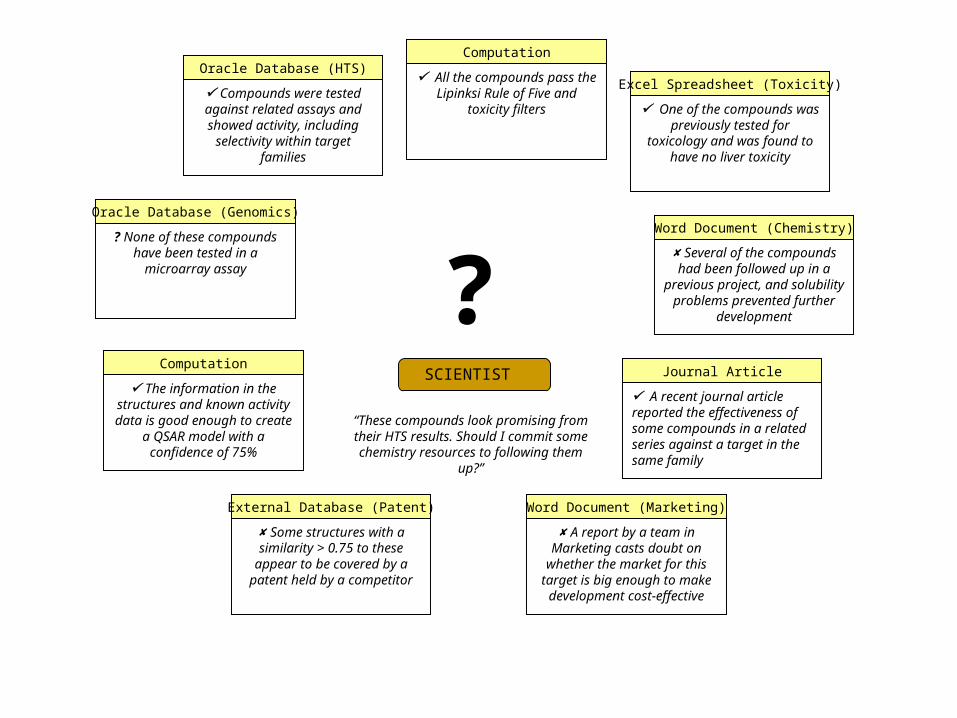
Use Case: Which of these hits should I follow up? An HTS experiment has produced 10,000 possible hits out of a
screening set of 2m compounds. A chemist on the project wants to know what the most promising series of compounds for follow-up are, based on: Series selection cluster analysis Structure-activity relationships modal fingerprints/stigmata Chemical and pharmacokinetic properties mitools, chemaxon Compound history gNova / PostgreSQL Patentability BCI Markush handling software Toxicity Synthetic feasibility + requires visualization tools!

A Workflow Scenario: HTS Data Organization and Flagging This workflow demonstrates how screening data can be flagged
and organized for human analysis. The compounds and data values for a particular screen are
retrieved from the NIH DTP database and then are filtered to remove compounds with reactive groups, etc. A tumor cell line is selected. The activity results for all the
compounds in the DTP database in the given range are extracted from the PostgreSQL database
OpenEye FILTER is used to calculate biological and chemical properties of the compounds that are related to their potential effectiveness as drugs
ToxTree is used to flag the potential toxicities of compounds. Divkmeans is used to add a column of cluster numbers. Finally, the results are visualized using VOPlot and the 2D
viewer applet.
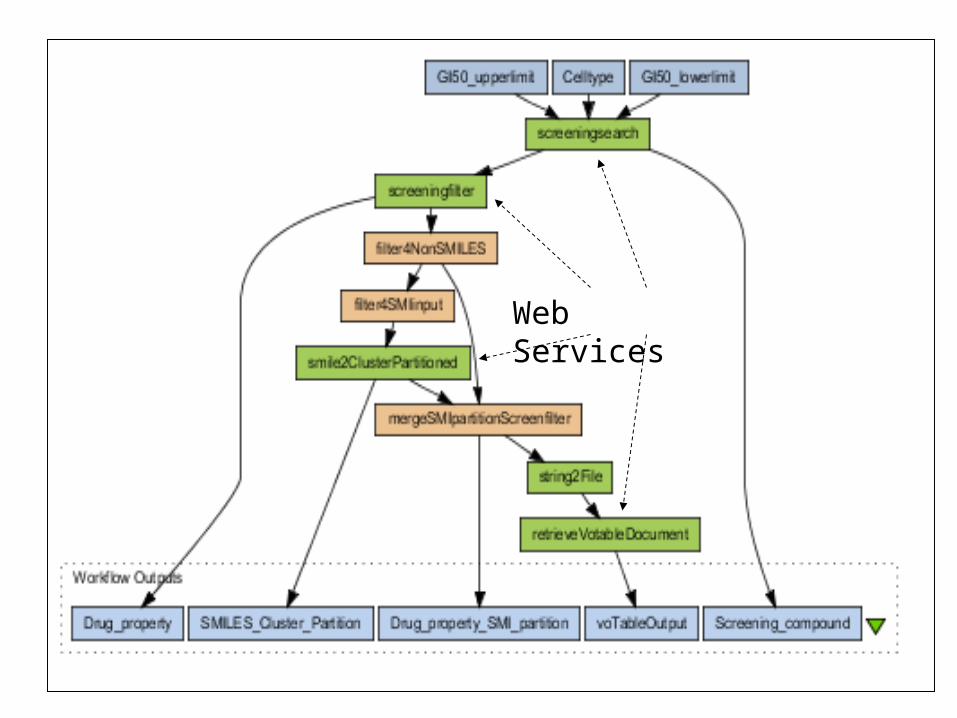
Web Services
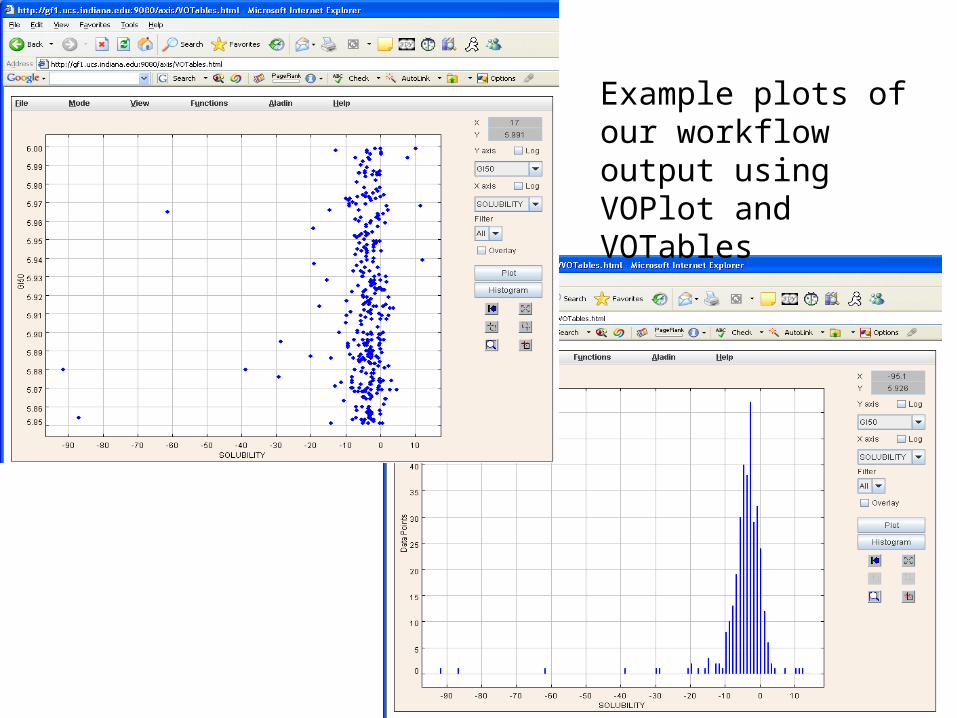
Example plots of our workflow output using VOPlot and VOTables
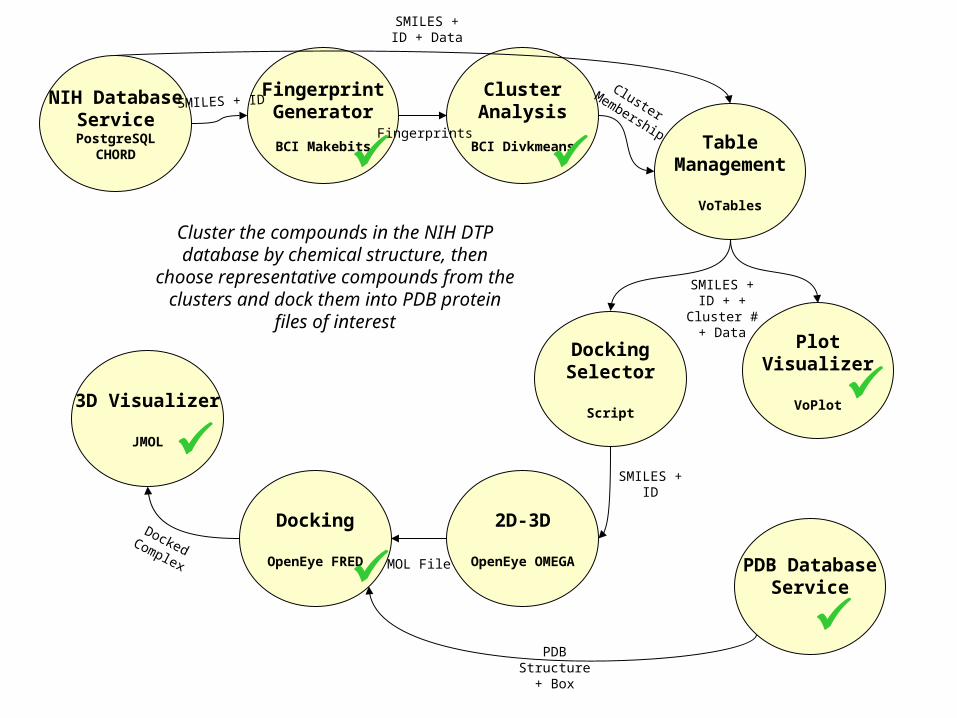
NIH DatabaseService
PostgreSQLCHORD
FingerprintGenerator
BCI Makebits
ClusterAnalysis
BCI Divkmeans TableManagement
VoTables
PlotVisualizer
VoPlot
DockingSelector
Script
2D-3D
OpenEye OMEGA
Docking
OpenEye FRED
3D Visualizer
JMOL
Cluster the compounds in the NIH DTP database by chemical structure, then
choose representative compounds from the clusters and dock them into
PDB protein files of interest
SMILES + ID
Fingerprints
PDB DatabaseService
SMILES + ID + Data
ClusterMembership
SMILES + ID + + Cluster # + Data
SMILES + ID
MOL File
PDB Structure +
Box
Docked Complex

Use Case: Are there any good ligands for my target? A chemist is working on a project involving a
particular protein target, and wants to know: Any newly published compounds which might fit the protein
receptor site gNova / PostgreSQL, PubChem search, FRED Docking
Any published 3D structures of the protein or of protein-ligand complexes PDB search
Any interactions of compounds with other proteins gNova / PostgreSQL, PubChem search
Any information published on the protein target Journal text search

Use Case: Who else is working on these structures? A chemist is working on a chemical series for a particular
project and wants to know: If anyone publishes anything using the same or related
compounds ~ PubChem search Any new compounds added to the corporate collection which are
similar or related gNova CHORD / PostgreSQL If any patents are submitted that might overlap the compounds
he is working on ~ BCI Markush handling software Any pharmacological or toxicological results for those or related
compounds gNova CHORD / PostgreSQL, MiToolkit The results for any other projects for which those compounds
were screened gNova CHORD / PostgreSQL, PubChem search

VARUNA – Towards a Grid-based Molecular Modeling Environment
A brief overview of Prof. Mookie Baik’s VARUNA project.

Chemical Informatics in Academic Research? Industrial Research: Target
Oriented Not bound to a specific
molecular system Not bound to a method Not concerned with
generality Aware of Efficiency Aware of Overall Cost Aware of Toxicity Concerned about
Formulations Cares about active
MOLECULES
Academic Research: Concept Oriented Specialized on few
molecular families Method Development is
important Obsessed with generality Does not care much about
efficiency Cost is unimportant Often can’t even assess for
Toxicity Formulation is a minor issue Cares mostly about
REACTIONS, i.e.ways to GET to a molecule

AutoGeFF, Varuna and Workflows Metalloproteins are extremely important in
biochemical processes Understanding their chemistry is difficult To add value to the small molecule DB’s
(PubChem, etc.), we must somehow connect them to PDB’s, BindMOAD, etc.
By extending Varuna’s functionality to handling, storing Metalloproteins, we could provide a connection

Automatic Generator of ForceFields (AutoGeFF) Developing a service that can take ANY
drug-like molecule (from PubChem, for example) metal complexes metalloenzymes (from PDB, for example) unnatural or functionalized amino acids, nucleobases (from in-
house db)for which molecular mechanics force fields are not available andautomatically generate FF’s based on High level Quantum Simulations (using Varuna as a Web
service)for Sophisticated Molecular Mechanics Simulations
First Step: Coding of a specialized Prototype that can reproduce our manually derived novel force fields for Cu-A Alzheimer’s Disease as a Proof-Of-Principles Study.

Automatic Quantum Mechanical Curation of Structure Data Chemical Research logic is often driven by molecular
structure Large-scale, small molecule DB’s (such as PubChem)
have low-resolution structure data Often key properties are not consistently available:
e.g.: Rotation-barriers, Redox Potentials, Polarizabilities, IR frequencies, reactivity towards nucleophiles
QM web-services will provide tools for generating high-resolution data that will curate the results of traditional ChemInfo studies allow for combinatorial computational chemistry access a database of modeling data
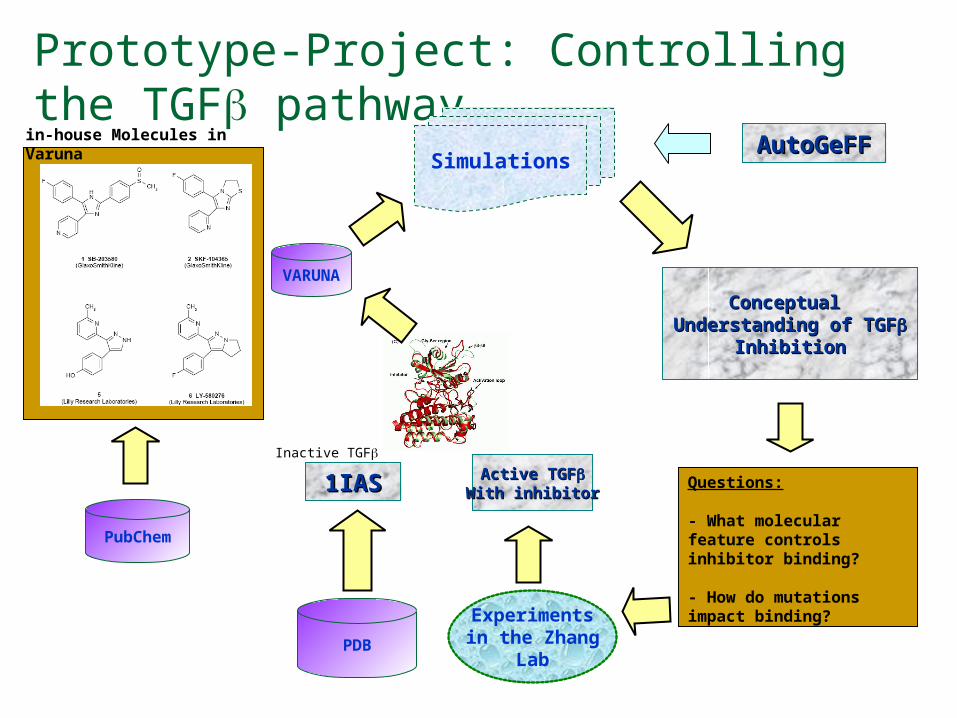
Prototype-Project: Controlling the TGF pathway
PDB
1IAS1IASInactive TGF
VARUNA
Experimentsin the Zhang
Lab
Active TGFActive TGFWith inhibitorWith inhibitor
PubChem
in-house Molecules in Varuna
Conceptual Conceptual Understanding of Understanding of TGFTGF
InhibitionInhibition
SimulationsAutoGeFFAutoGeFF
Questions:
- What molecular feature controls inhibitor binding?
- How do mutations impact binding?

Consequences for ChemInfo Design for Academia TWO Strategies are needed: Making traditional ChemInfo tools that are often available in
commercial research available to Academia is in principle straightforward.
New ChemInfo Tools that are CONCEPT centered and include REACTIONS in addition to MOLECULES must be developed.
Our approach: Development of
(a) Quantum Chemical Database(b) Molecular Modeling Database
Harness the power of recent advances in Molecular Modeling (QM, QM/MM, MM, MD) through information management.
Data-depository for Quantum Chemical Data including both Properties & Mechanisms
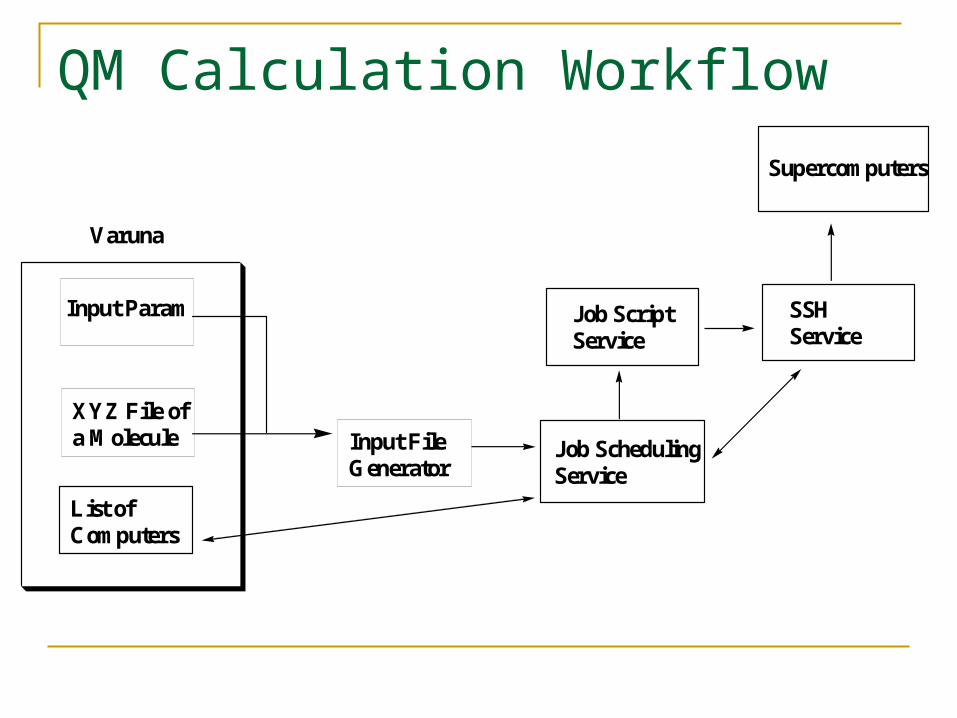
QM Calculation Workflow
XYZ File ofa Molecule Input File
Generator
Input Param
Varuna
Job SchedulingService
List ofComputers
Job ScriptService
SSHService
Supercomputers

More Information
Contact me: [email protected] Most of this was taken from our CICC project. See
www.chembiogrid.org/wiki. Note we’ve found wikis to be extremely useful and fun to
use for maintaining collaborative web sites. See also www.crisisgrid.org and www.gorerle.com/vlab-wiki
for other examples using Media Wiki. Many elements of our approach are based on Prof.
Peter Murray Rust’s group’s approach. WWMM Wiki: wwmm.ch.cam.ac.uk/wikis/wwmm/index.php
SourceForge Project Site http://sourceforge.net/projects/cicc-grid

Additional Slides

Use Case - CICCWhich of these hits should I follow up? An MLI HTS experiment has produced 10,000 possible hits out
of a screening set of 2m compounds. A chemist at another laboratory wants to know if there are any interesting active series she might want to pursue, based on: Structure-activity relationships Chemical and pharmacokinetic properties Compound history Patentability Toxicity Synthetic feasibility

CICC Web Services I
BCI Clustering Provides Bernard Chemical Information (BCI) clustering packages A module of the workflow for HTS data organization and flagging Status:
Added URL output support to the previous solid prototype (Multi-user durable) Taverna Beanshell Scripting for data format adjusting (e.g. Filtering out the head part listing
column names) To do: Evaluating the URI(URL) based workflow design
ToxTree Estimates toxic hazard by applying a decision tree approach A module of the workflow for HTS data organization and flagging Status: A test prototype producing the level of toxicity in a brief or verbose explanation
against a SMILE structure To do:
Refining the Web service for cluster input and external property support The Taverna Beanshell scripting for data merging not used in some modules

CICC Web Services II
Workflow for HTS data organization and flagging Demonstrates how screening data can be flagged and organized for human
analysis Status: Individual modules except the visualization are in prototype To do:
Defining at least XML schema or DTD for the workflow data (at most the Ontology) Redefining current workflow model to reflect the new feature of Taverna 1.4
supporting complex data structures and the provenance plugin Other Planed Web Services
Open Source Chemistry Analysis Routines (OSCAR) Extracts chemical information from text and produces an XML instance highlighting
the chemical information A module of the PMR workflow Status: OSCAR3 is available and works fine as a Java application To do: Studying XML instances for extracting chemical names
InfoChem’s SPRESI Web Service Provides access to the SPRESI molecule database Status: Perl scripts for accessing SPRESI Web Service To do: Developing a Web service wrapper to utilize InfoChem’s SPRESI Web
Service

BCI Clustering URL Service Methods
Service Method Description Input URLOutput
makebitsURLGenerate
Generate fingerprints from a SMILES structure
SMIstring Fingerprint and program output
divkmURLGenerate
Cluster fingerprints with Divkmeans
SCNstring DKM data and program output
smile2dkmURL Makebits + divkm SMIstring All SMI, DKM and std. outputs
optclusURLGenerate
Generate the best levels in a hierarchy
SMIstring
DKMstring
Best data and program output
rnnclusURLGenerate
Extract individual cluster partitions
SMIstring
DKMstring
New partition and std. output
smile2ClusterPartitionedURL
Generate a new SMILES structure w/ extra col.
SMIstring All intermediate data and output
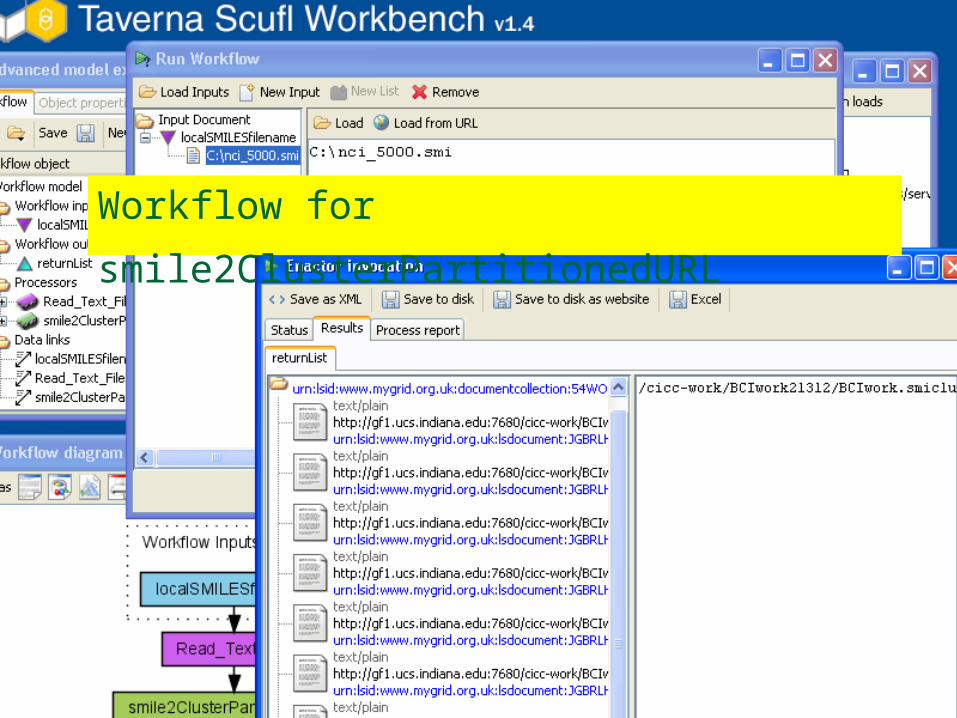
Workflow for
smile2ClusterPartitionedURL
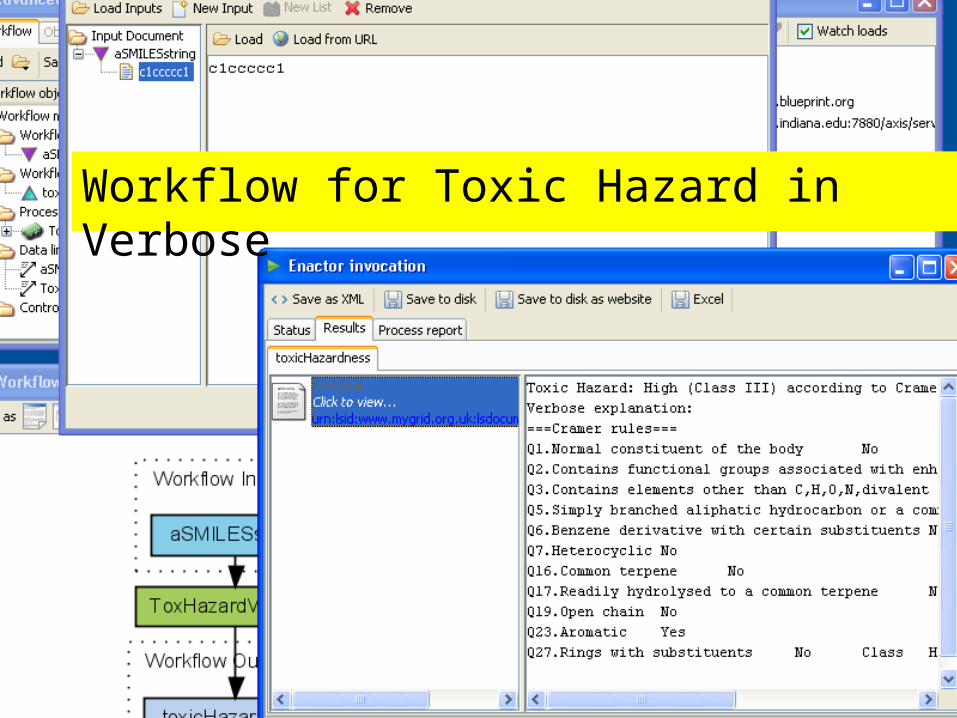
Workflow for Toxic Hazard in Verbose
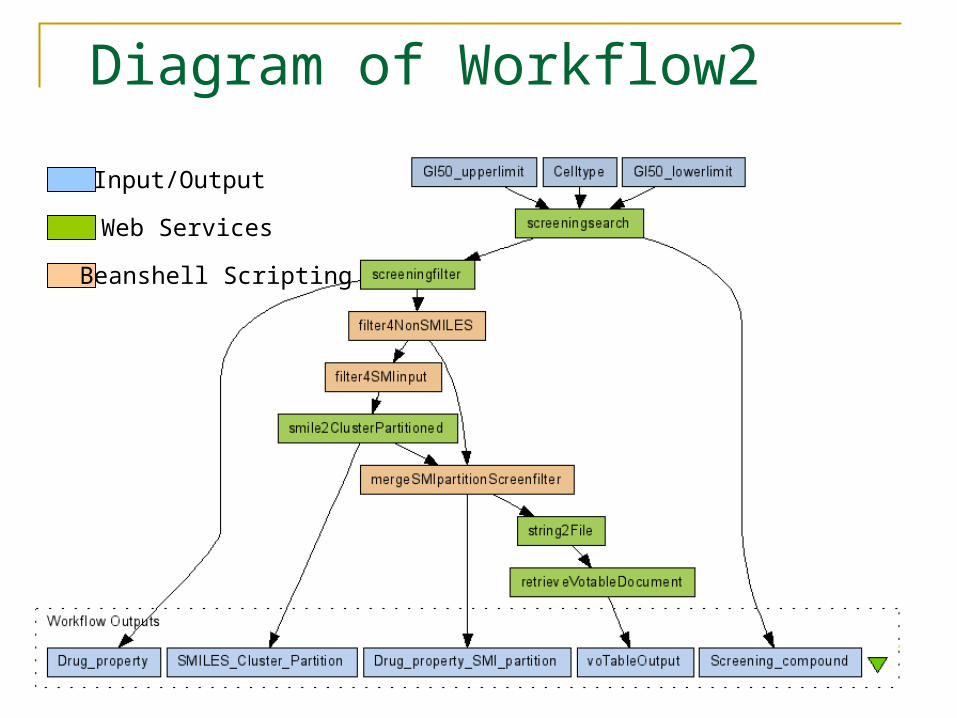
Diagram of Workflow2
Input/Output
Web Services
Beanshell Scripting

Informatics
Informatics is the discipline of science which investigates the structure and properties (not specific content) of scientific information, as well as the regularities of scientific information activity, its theory, history, methodology and organization. The purpose of informatics consists in developing optimal methods and means of presentation (recording), collection, analytical-synthetic processing, storage, retrieval and dissemination of scientific information.
A. I. Mikhailov, A. I. Chernyi, R. S. Gilyarevskii (1967) “Informatics -- New Name of the Theory of Scientific Information”

Chemical informatics is …
More usually know as chemoinformatics or cheminformatics
Very differently defined, reflecting its cross-disciplinary nature Librarian Chemist (synthetic, medicinal, theoretical) Biologist / Bioinformatician Molecular modeler Pharmaceutical or Chemical Engineer Computer Scientist / Informatician

More definitions
Computational Chemistry – The application of mathematical and computational methods to particularly to theoretical chemistry
Molecular Modeling – Using 3D graphics and optimization techniques to help understand the nature and action of compounds and proteins
Computer-Aided Drug Design – The discipline of using computational techniques (including chemical informatics) to assist in the discovery and design of drugs.

Traditional areas of application
Pharmaceutical & life science industry particularly in early stage drug design
Databases of available chemicals Electronic publishing
including searchable chemical structure information in journals, etc.
Government and patent databases

The –ics so far (1960’s to present) … How do you represent 2D and 3D chemical structures?
Not just a pretty picture How do you search databases of chemical structures?
Google doesn’t help (much, but it might do soon…) How do you organize large amounts of chemical information? How do you visualize chemical structures & proteins? Can computers predict how chemicals are going to behave
… in the test tube? … in the body?

Current trends & hot topics
The decorporatization of chemical informatics (PubChem, MLI, eScience, open source)
Service-oriented architectures Packaging & processing large volumes of complex
information for human consumption Integration with other –ics (bioinformatics,
genomics, proteomics, systems biology)

Main players (Commercial)
MDL www.mdl.com Tripos, inc. www.tripos.com Accelrys www.accelrys.com Daylight CIS, inc. www.daylight.com

Main players (Academia)
“Pure” Chemoinformatics University of Sheffield, UK (Willett / Gillet)
http://www.shef.ac.uk/uni/academic/I-M/is/research/cirg.html Erlangen, Germany (Gasteiger)
http://www2.chemie.uni-erlangen.de/ Cambridge Unilever Center
http://www-ucc.ch.cam.ac.uk/ Indiana University School of Informatics
http://www.informatics.indiana.edu/ Related (computational chemistry, etc.)
UCSF (Kuntz) http://mdi.ucsf.edu/
University of Texas (Pearlman) http://www.utexas.edu/pharmacy/divisions/pharmaceutics/faculty/pearlman.html
Yale (Jorgensen) http://zarbi.chem.yale.edu/
University of Michigan (Crippen) http://www.umich.edu/~pharmacy/MedChem/faculty/crippen/

“Traditional” Journals
Journal of Chemical Information & Modeling (formerly JCICS) http://pubs.acs.org/journals/jcisd8/index.html
Journal of Computer-Aided Molecular Design http://www.kluweronline.com/issn/0920-654X
Journal of Molecular Graphics and Modeling http://www.elsevier.com/inca/publications/store/5/2/5/0/1/2/
Journal of Computational Chemistry http://www3.interscience.wiley.com/cgi-bin/jhome/33822
Journal of Chemical Theory and Computation http://pubs.acs.org/journals/jctcce/
Journal of Medicinal Chemistry http://pubs.acs.org/journals/jmcmar/

“Informal” publications
Network Science (online) http://www.netsci.org/Science/index.html
Chemical & Engineering News http://pubs.acs.org/cen/
Drug Discovery Today http://www.drugdiscoverytoday.com/
Scientific Computing World http://www.scientific-computing.com/
Bio-IT World http://www.bio-itworld.com/
CINF-L Distribution List
Chemical Information Sources Discussion List
Created by Gary Wiggins at IUB http://www.indiana.edu/~cheminfo/
network.html
Yahoo! Chemoinformatics Discussion List For
Job postings Ideas exchange Questions Industry – Student
connections All students encouraged to
join Open to others
To join, go to http://groups.yahoo.com/group/chemoinf
Or send an email to [email protected]

Open Source / Free Software
Blue Obelisk - http://wiki.cubic.uni-koeln.de/dokuwiki/doku.php InChI - http://www.iupac.org/inchi/ JMOL – http://jmol.sourceforge.net FROWNS - http://frowns.sourceforge.net/ OpenBabel - http://openbabel.sourceforge.net/ CML - http://cml.sourceforge.net/ CDK - http://almost.cubic.uni-koeln.de/cdk/ MMTK - http://starship.python.net/crew/hinsen/MMTK/

Example 23D Visualization & Docking
3D Visualization of interactions between compounds and proteins
“Docking” compounds into proteins computationally

3D Visualization
X-ray crystallography and NMR Spectroscopy can reveal 3D structure of protein and bound compounds
Visualization of these “complexes” of proteins and potential drugs can help scientists understand the mechanism of action of the drug and to improve the design of a drug
Visualization uses computational “ball and stick” model of atoms and bonds, as well as surfaces
Stereoscopic visualization available

Docking algorithms
Require 3D atomic structure for protein, and 3D structure for compound (“ligand”)
May require initial rough positioning for the ligand Will use an optimization method to try and find the
best rotation and translation of the ligand in the protein, for optimal binding affinity

Genetic Algorithms
Create a “population” of possible solutions, encoded as “chromosomes”
Use “fitness function” to score solutions Good solutions are combined together
(“crossover”) and altered (“mutation”) to provide new solutions
The process repeats until the population “converges” on a solution
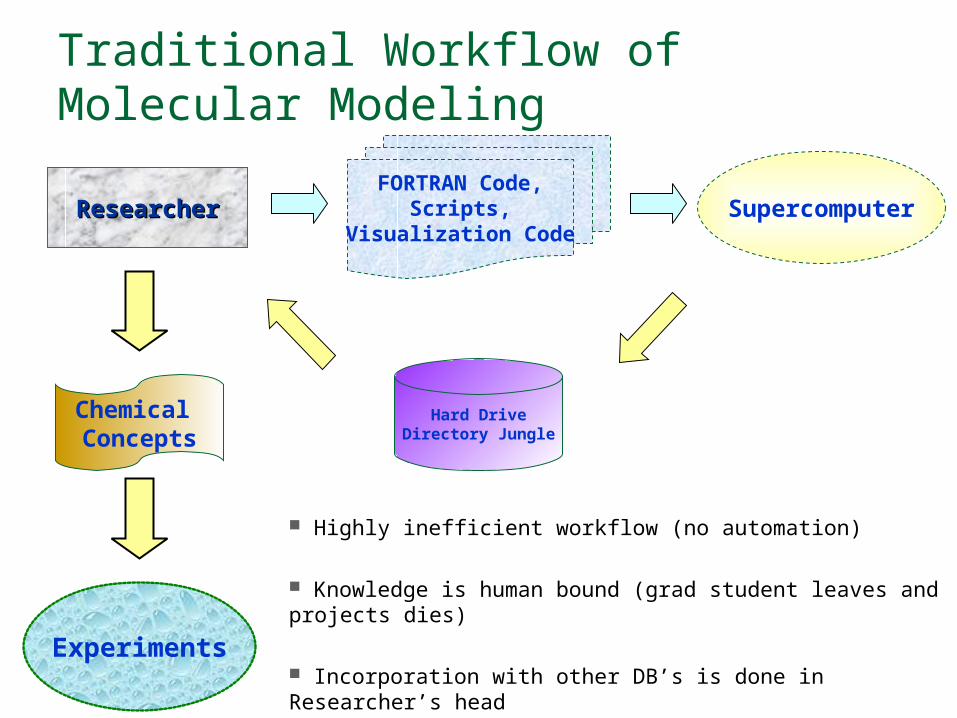
Traditional Workflow of Molecular Modeling
SupercomputerResearcherResearcherFORTRAN Code,
Scripts,Visualization Code
Hard DriveDirectory Jungle
Chemical Concepts
Experiments
Highly inefficient workflow (no automation)
Knowledge is human bound (grad student leaves and projects dies)
Incorporation with other DB’s is done in Researcher’s head
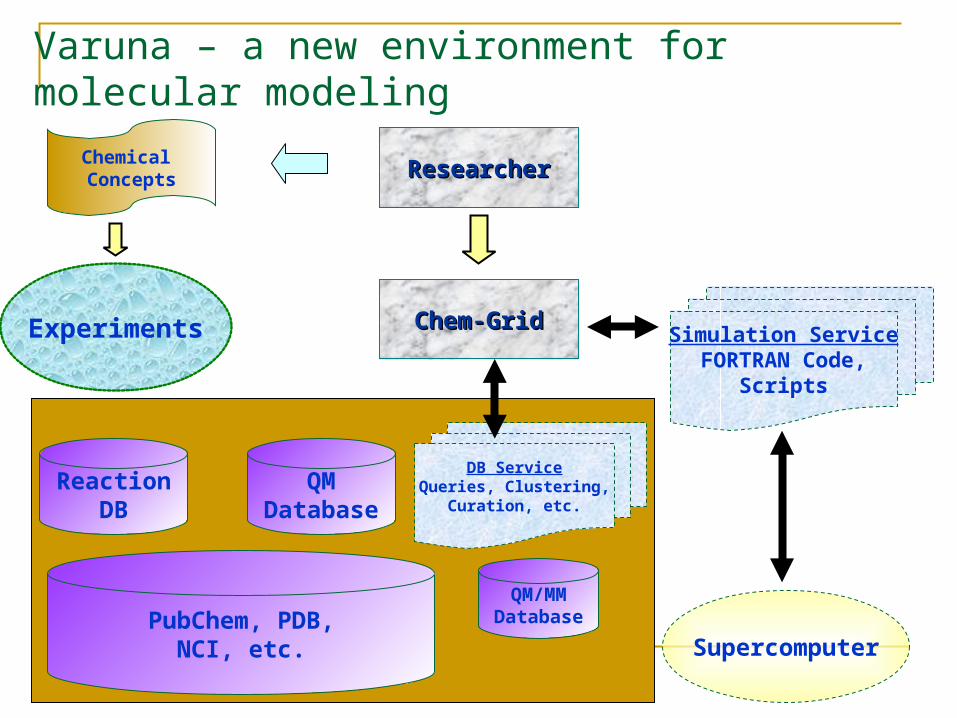
Varuna – a new environment for molecular modeling
QMDatabase
Supercomputer
ResearcherResearcher
Simulation ServiceFORTRAN Code,
Scripts
Chemical Concepts
Experiments
QM/MMDatabasePubChem, PDB,
NCI, etc.
Chem-GridChem-Grid
ReactionDB
DB ServiceQueries, Clustering,
Curation, etc.
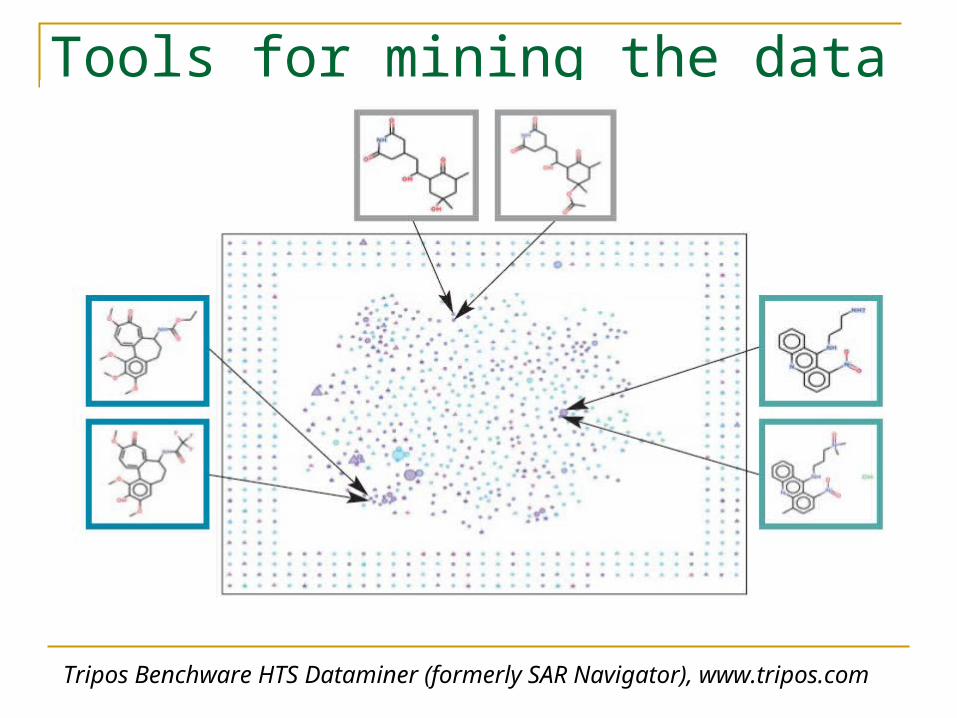
Tools for mining the data
Tripos Benchware HTS Dataminer (formerly SAR Navigator), www.tripos.com

















