13586 J. Phys. Chem. 1993, 97, 13586-13597 Butadiene. 3. Charge Distribution in Electronically Excited States Kenneth B. Wiberg,' Christopher M. Hadad,$ G. Barney Ellison,$ and James B. Foresman Department of Chemistry, Yale University, New Haven, Connecticut 0651 1 Received: September 30, 1993' The vertical transition energies for butadiene have been calculated using the CIS/6-3 1 1 (2+)G* theoretical model. The observed energies were satisfactorily reproduced. The charge distribution for each of the excited states was calculated so that the change from the ground-state distribution could be examined. The nature of the Rydberg states are discussed. Quantitative information on the degree of charge-transfer and bond-order changes was obtained for the excited states. The adiabatic geometries of the valence and Rydberg states were examined, and vibrational frequencies for the excited states were calculated, providing agreement with experiment. The triplet states were also studied for both the vertical and adiabatic surfaces. Analysis of the charge density gave information on the charge reorganization that occurs in the excited states. It showed the remarkable similarity of the atomic orbital-like Rydberg states to the corresponding radical cation and, moreover, showed the unique nature of the 1'B, A - A* valence state. The vertical 2lA, state was examined with the MCSCF method. For a comparison, the 1 'B", 13B,, and 13A, states were studied in the same fashion. The relationship between these states is discussed. Introduction As the simplest of the chemically important polyenes, the electronicallyexcited states of butadiene have received extensive experimental' and theoretical study.* The ground state of butadiene has A, symmetry, and the electronicallyexcited states may have A,, A,, B,, or B, symmetry. For a one-photon process the transition moment is zero for a g - g* transition, and only transitions to an A, or B, states are allowed. The B, states are in-plane (x,y) polarized and the A,, states are out-of-plane (2) polarized. The B, states are expected to be relatively intense since the transition dipole length can approach half the length of the *-system. The A, transitions are expected to be relatively weak since the transition moment length in the z direction must of necessity be small. The electronic spectrum of butadiene is shown in Figure 1. It is now generally agreed that the first allowed excited state corresponds to the B, A - A* transition. The 0,O transition is at 46 258 cm-1 (5.74 eV) and the vertical transition is at 47 678 cm-1 (5.91 eV). This is followed by some weaker bands and then by a set of A, Rydberg transitions starting at 56 970 cm-l (7.06 eV Table I). In the simple *-electron approximation, the A* excited state is formed by taking an electron from an MO which has a node between C2 and C3 and placing it in an MO which has nodes between C1 and C2 and between C3 and Cd. Thus, one would expect increased double-bond character between C2 and C3 and decreased double-bond character between the other C-C bonds. We were interested in seeing how large a charge shift actually occurs. Rydberg states are important in the electronic spectrum of butadiene and of other h y d r ~ a r b o n s . ~ In the case of atoms Rydberg states are formed by transitions from occupied atomic orbitals to virtual atomic orbitals, and with approximately spherical molecules, one could visualizetransitions from occupied orbitals to diffuse atomic-orbital-like configurations. However, with an extended planar systems such as butadiene, it is not as clear as to what is meant by a Rydberg state. Here again, a study of the charge distribution in the excited states should be. useful. The geometries of the excited states of butadiene have been extensively disc~ssed.~ While octatetraene and longer polyenes fluoresce, butadiene does not. The lack of fluorescence of the A - A* state has been attributed to nonradiative energy transfer f Department of Chemistry, University of Colorado, Boulder, CO 80309. @ Abstract published in Advance ACS Abstracfs, December I, 1993. 0022-365419312097-13586$04.00/0 80.0 . 70.0. - 60.0. 5 50.0. 40.0. z 6 0 30.0. VI FREQUENCY [CM-1 X 10-31 Figure 1. Electronic spectrum of 1,3-butadiene along with a comparison of theexperimentalandcalculated (CIS/6-311(2+)G*) vertical transition energies for the singlet excited states. The average error of the CIS energies (0.2 eV) corresponds to 1600 cm-I. to a different excited state which is of a similar geometry.1° The low-energy region of the absorption spectrum is quite broad for butadiene, and the bandwidth becomes significantly narrower for hexatriene and octatetraene.Ij The bandwidth of butadiene also is relatively unaffected by temperature. The cause of the large bandwidth is not known. Also, while some vibrational frequencies for the A - A* state are known, thedegreeof twisting of the carbon framework has been debated.1j~0~2j,k.q The recent implementation of the configuration interaction with singles (CIS) excitation method,5and its success in thestudy of the excited states of bicyclo[ 1.1 .O]b~tane,~ ethylene,' form- aldehyde, acetaldehyde,8 and pyridine5 has prompted our study of butadiene. We have now examined its vertical and adiabatic excited states. Other procedures such as MRD-C12m have the potential of giving somewhat more accurate transition energies. However, our main interestis in learning more about the properties of excited states, and the CIS formalism is particularly well suited for studiesof adiabatic geometries and of excited-statevibrational frequencies. Calculated Transition Energies and Intensities The effect of basis set on the calculated transition energies was examined, and as with the other molecules we have studied, it was necessary to include a double set of diffuse functions in order 0 1993 American Chemical Society
Transcript
13586 J . Phys. Chem. 1993, 97, 13586-13597
Butadiene. 3. Charge Distribution in Electronically Excited States
Kenneth B. Wiberg,' Christopher M. Hadad,$ G. Barney Ellison,$ and James B. Foresman Department of Chemistry, Yale University, New Haven, Connecticut 0651 1
Received: September 30, 1993'
The vertical transition energies for butadiene have been calculated using the CIS/6-3 1 1 (2+)G* theoretical model. The observed energies were satisfactorily reproduced. The charge distribution for each of the excited states was calculated so that the change from the ground-state distribution could be examined. The nature of the Rydberg states are discussed. Quantitative information on the degree of charge-transfer and bond-order changes was obtained for the excited states. The adiabatic geometries of the valence and Rydberg states were examined, and vibrational frequencies for the excited states were calculated, providing agreement with experiment. The triplet states were also studied for both the vertical and adiabatic surfaces. Analysis of the charge density gave information on the charge reorganization that occurs in the excited states. It showed the remarkable similarity of the atomic orbital-like Rydberg states to the corresponding radical cation and, moreover, showed the unique nature of the 1 'B, A - A* valence state. The vertical 2lA, state was examined with the MCSCF method. For a comparison, the 1 'B", 13B,, and 13A, states were studied in the same fashion. The relationship between these states is discussed.
Introduction
As the simplest of the chemically important polyenes, the electronically excited states of butadiene have received extensive experimental' and theoretical study.* The ground state of butadiene has A, symmetry, and the electronically excited states may have A,, A,, B,, or B, symmetry. For a one-photon process the transition moment is zero for a g - g* transition, and only transitions to an A, or B, states are allowed. The B, states are in-plane (x,y) polarized and the A,, states are out-of-plane (2 )
polarized. The B, states are expected to be relatively intense since the transition dipole length can approach half the length of the *-system. The A, transitions are expected to be relatively weak since the transition moment length in the z direction must of necessity be small.
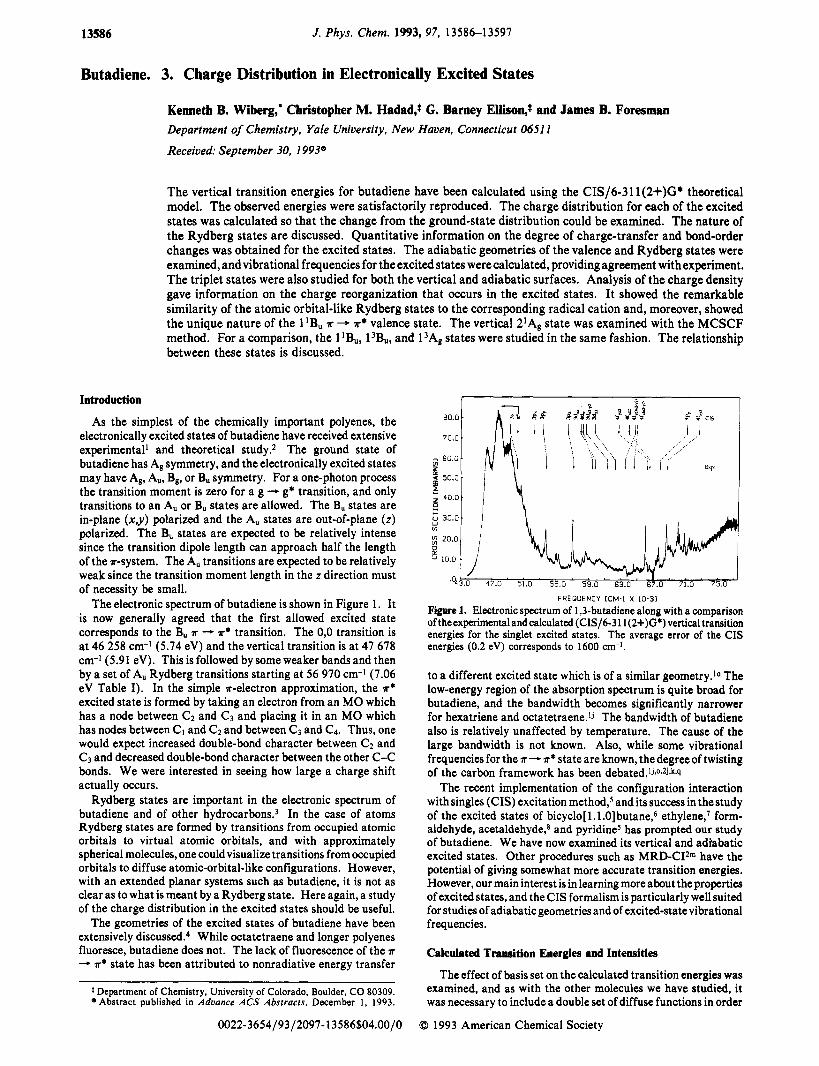
The electronic spectrum of butadiene is shown in Figure 1 . It is now generally agreed that the first allowed excited state corresponds to the B, A - A* transition. The 0,O transition is at 46 258 cm-1 (5.74 eV) and the vertical transition is at 47 678 cm-1 (5.91 eV). This is followed by some weaker bands and then by a set of A, Rydberg transitions starting at 56 970 cm-l (7.06 eV Table I). In the simple *-electron approximation, the A*
excited state is formed by taking an electron from an MO which has a node between C2 and C3 and placing it in an MO which has nodes between C1 and C2 and between C3 and Cd. Thus, one would expect increased double-bond character between C2 and C3 and decreased double-bond character between the other C-C bonds. We were interested in seeing how large a charge shift actually occurs.
Rydberg states are important in the electronic spectrum of butadiene and of other hydr~arbons .~ In the case of atoms Rydberg states are formed by transitions from occupied atomic orbitals to virtual atomic orbitals, and with approximately spherical molecules, one could visualize transitions from occupied orbitals to diffuse atomic-orbital-like configurations. However, with an extended planar systems such as butadiene, it is not as clear as to what is meant by a Rydberg state. Here again, a study of the charge distribution in the excited states should be. useful.
The geometries of the excited states of butadiene have been extensively disc~ssed.~ While octatetraene and longer polyenes fluoresce, butadiene does not. The lack of fluorescence of the A - A* state has been attributed to nonradiative energy transfer
f Department of Chemistry, University of Colorado, Boulder, CO 80309. @ Abstract published in Advance ACS Abstracfs, December I , 1993.
0022-365419312097-13586$04.00/0
80.0 .
70.0.
- 60.0.
5 50.0.
40.0.
z
6 0 30.0.
VI
FREQUENCY [CM-1 X 10-31
Figure 1. Electronic spectrum of 1,3-butadiene along with a comparison of theexperimentalandcalculated (CIS/6-3 11(2+)G*) vertical transition energies for the singlet excited states. The average error of the CIS energies (0.2 eV) corresponds to 1600 cm-I.
to a different excited state which is of a similar geometry.1° The low-energy region of the absorption spectrum is quite broad for butadiene, and the bandwidth becomes significantly narrower for hexatriene and octatetraene.Ij The bandwidth of butadiene also is relatively unaffected by temperature. The cause of the large bandwidth is not known. Also, while some vibrational frequencies for the A - A* state are known, thedegreeof twisting of the carbon framework has been debated.1j~0~2j,k.q
The recent implementation of the configuration interaction with singles (CIS) excitation method,5 and its success in thestudy of the excited states of bicyclo[ 1.1 .O]b~tane,~ ethylene,' form- aldehyde, acetaldehyde,8 and pyridine5 has prompted our study of butadiene. We have now examined its vertical and adiabatic excited states. Other procedures such as MRD-C12m have the potential of giving somewhat more accurate transition energies. However, our main interest is in learning more about the properties of excited states, and the CIS formalism is particularly well suited for studies of adiabatic geometries and of excited-state vibrational frequencies.
Calculated Transition Energies and Intensities
The effect of basis set on the calculated transition energies was examined, and as with the other molecules we have studied, it was necessary to include a double set of diffuse functions in order
0 1993 American Chemical Society
Charge Distribution in Butadiene
tmts-l,1Butadiene (CIS/6311(2+)Cf)'
The Journal of Physical Chemistry, Vol. 97, No. 51, 1993 13587
TABLE I: Calculated Vertical Transition Energies of trans-Butadiene Singlet n + n* State Projection Density Differences
Figure 2. Projection density difference plots for the vertical * - r* state of trans-butadiene with respect to the ground state at the CIS/6-311- (2+)G* level. The upper plots are from the generalized (relaxed) density matrix, and the lower ones are using the one-particle (unrelaxed) density matrix. The outer contour is 0.0001 e/B3, and the contours increase by a factor of 2.
bond. As a result, the (22-9 bond order should increase, whereas the C,-C2 and C 3 4 bond orders should decrease on going to the excited state.
a Using the MP2/6-31G* geometry. CIS calculations allowed cor- The density matrix that was derived from the CIS/6-311+G*
density was derived from the latter. One of the convenient ways relation of all electrons. The total energies of the ground state are RHF, -154.950 806, and MPZ(ful1). -155.566 212 hartrees.
calculation was transformed into natural orbitals, and the charge
to obtain satisfactory results. The basis set used in our calculations, 6-31 1 (2+)G(d) corresponds to a valence triple4 basis set with two diffuse functions on each carbon with exponents of 0.0438 and 0.013 193, along with a set of d functions on the carbons. The transition energies calculated using the MP2/6- 3 lGf geometry, which is close to the experimental geometry, are summarized in Table I. A comparison with the experimental data (Table 11) shows that the CIS energies give a good representation of the experimental data.
Singlet Excited States
in which to examine the charge density for a planar moleculeis to make a projection density plot in which the charge density above and below a point on the molecular plane is summed and assigned to that point.9 The projection density difference plots for the vertical T-T* excited state and the ground state is shown in Figure 2. Here, solid contours indicate regions in which charge density has increased on going to the excited state, and dashed contours indicate regions in which the charge density has been depleted. The shift in s-electrons on going to the excited state is expected, and a corresponding plot for the u electrons showed no significant shift in charge density.
The small u wlarization paralleling the shift in T-charaedensitv One of our major interests was in the change in charge
distribution that occurs during an electronic transition. The T - T* transition of butadiene appeared to be a good starting point because as indicated above it was expected to lead to a shift in *-electron population from the C-C double bonds to the single
TABLE II: Comparison of Calculated Vertical Transition Energies for trsos-1,3-Butadiene
seemed surpri'sing and might be an artifact of the use of thk one-particle density matrix (1PDM) as is commonly done. It has been found that a more accurate density matrix can be derived from the derivative of the CIS energy.5 This generalized density matrix (GDM)l0 allows the electronic distribution to relax after
state assignment MRD-CIb QDVFT SAC-CI~ EVSHC RPM C I S expth
Figure 4. Charge density depletion regions (negative contours) for the first eight vertical singlet excited states and the radical cation of trans- butadiene (0.0001 e/B3).
TABLE IIk Atomic Orbitals under C2) Symmetry symmetry atomic orbitals
3'8, n2 + 3d, (10) 3'4, n2 + 4fF2 ( 1 1 )
density for the other excited states of trans-butadiene. While projection density plots are useful to analyze the in-plane polarized states such as the 1 B, state, it is not as clear for the out-of-plane polarized states. We have, instead, utilized 3-dimensional contour plots. For these, cubes of charge density (40 au on each side with 80 points/side) were generated for each excited state and for the ground state. Subtraction of the ground-state distribution from the excited-state density was followed by construction of 3D plots corresponding to the 1 X la" e/au3 surface.
These plots for states 1-1 8 are depicted in Figure 3 where solid (positive) contours show where charge has accumulated in the excited state relative to the ground state and dashed (negative) contours show charge depletion. The plots provide a simple method for visualizing the character of the different excited states and clearly reveals the nature of the excitation. The character of the s, p, d, and f atomic orbitals under C2h symmetry are listed in Table 111. From Figure 3 one can easily see atomic-orbital- like Rydberg s, p, d, and f states and also valence states, as in the lBu state.
In Figure 4, we have shown just the depletion zones (dashed, negativecontours) for states 1-8 and thevertical 7r2 radical cation obtained at the UHF level with the same basis set. It can be seen that all of the states are similar to the radical cation except for the lB, state which has some valence character.
A similar analysis of each of the other excited states allowed the assignments listed in Table I. A comparison of the CIS predictions with those of other studies and with the available experimental data for the vertical excitations of trans-butadiene is given in Table 11. While the ?r - u* vertical excitation is known (5.91 eV), most of the other transition energies are only known as adiabatic or adiabatic-vertical (advert). Nonetheless, we find satisfactory agreement between the CIS transition energies and the experimental values. Agreement with all of the states, including high-lying d and f Rydberg states,' is better than 0.5 eV. In some cases, the agreement is as good as 0.1 eV.
It is also possible to obtain transition energies with a further inclusion of electron correlation via Maller-Plesset perturbation
Figure 3. Charge density difference plots for the first 18 vertical singlet excited states of trans-butadiene at the CIS/6-3 1 1 (2+)G* level with respect to theground state. The GDM was used for this and the following plots. The contour level is 0.0001 e/B3.
the gross reorganization caused by promotion into the CI space. With the use of the latter the ?r-projection density difference was contracted slightly (Figure 2), and now a u polarization opposite the u shift was found. This observation helps resolve the fact that the calculated oscillator strength (based on 1PDM) is 0.8 whereas the observed value is only 0.4.
To determine the magnitude of the charge reorganization, we summed the number of electrons that shift on going to the ?r - ?r* state for the density difference plots. Using the generalized density matrix, 0.34 e are shifted for the total density distribution in the u - u* state as compared to the ground state. There is a larger reorganization in the ?r system (0.43 e) than in the u system (0.16 e). Since the a-system opposes the ?r-system, the total change is smaller than just the ?r-shift. The one-particle density matrix is quite different. The total, ?r, and u charge shifts are 0.87, 0.87, and 0.02 e, respectively, and this confirms the overestimation of the oscillator strength via the IPDM.
Since the generalized density matrix is the better representation of the charge density, we have also obtained the corresponding
Charge Distribution in Butadiene The Journal of Physical Chemistry, Vol. 97, No. 51, 1993 13589
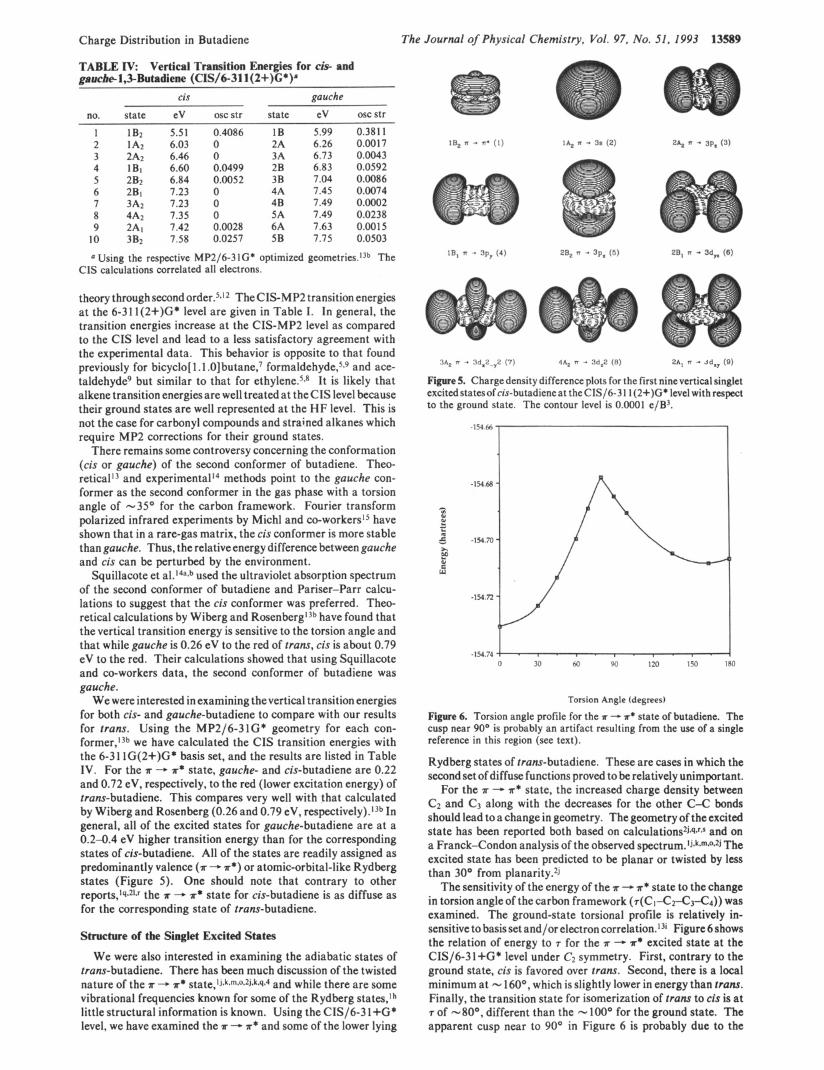
TABLE I V Vertical Transition Energies for cis- and gaucbe-1,3-Butadiene (CIS/6-311(2+)C*)a
cis gauche
no. state eV oscstr state eV oscstr 1 1B2 5.51 2 1A2 6.03 3 2A2 6.46 4 1Bl 6.60 5 2B2 6.84 6 2B1 7.23 7 3A2 7.23 8 4A2 7.35 9 2Al 7.42
Using the respective MP2/6-3 lG* optimized geometries.13b The CIS calculations correlated all electrons.
theory through second ~ r d e r . ~ J ~ The CIS-MP2 transition energies at the 6-31 1(2+)G* level are given in Table I. In general, the transition energies increase at the CIS-MP2 level as compared to the CIS level and lead to a less satisfactory agreement with the experimental data. This behavior is opposite to that found previously for bicyclo[ 1.1 .O] butane,' f~rmaldehyde,~.~ and ace- taldehyde9 but similar to that for e th~ lene .~ .~ It is likely that alkene transition energies are well treated at the CIS level because their ground states are well represented at the HF level. This is not the case for carbonyl compounds and strained alkanes which require MP2 corrections for their ground states.
There remains some controversy concerning the conformation (cis or gauche) of the second conformer of butadiene. Theo- reticalI3 and experimental14 methods point to the gauche con- former as the second conformer in the gas phase with a torsion angle of -35' for the carbon framework. Fourier transform polarized infrared experiments by Michl and co-worker~~~ have shown that in a rare-gas matrix, the cis conformer is more stable than gauche. Thus, the relative energy difference between gauche and cis can be perturbed by the environment.
Squillacote et al. 14a*b used the ultraviolet absorption spectrum of the second conformer of butadiene and Pariser-Parr calcu- lations to suggest that the cis conformer was preferred. Theo- retical calculations by Wiberg and R ~ s e n b e r g l ~ ~ have found that the vertical transition energy is sensitive to the torsion angle and that while gauche is 0.26 eV to the red of trans, cis is about 0.79 eV to the red. Their calculations showed that using Squillacote and co-workers data, the second conformer of butadiene was gauche.
We were interested in examining thevertical transition energies for both cis- and gauche-butadiene to compare with our results for trans. Using the MP2/6-31G* geometry for each con- former,13b we have calculated the CIS transition energies with the 6-3 1 lG(2+)G* basis set, and the results are listed in Table IV. For the ?r -. ?r* state, gauche- and cis-butadiene are 0.22 and 0.72 eV, respectively, to the red (lower excitation energy) of trans-butadiene. This compares very well with that calculated by Wiberg and Rosenberg (0.26 and 0.79 eV, re~pectively).~~b In general, all of the excited states for gauche-butadiene are at a 0.2-0.4 eV higher transition energy than for the corresponding states of cis-butadiene. All of the states are readily assigned as predominantly valence (?r - T * ) or atomic-orbital-like Rydberg states (Figure 5 ) . One should note that contrary to other reports,1q*21.r the ?r - 'A* state for cis-butadiene is as diffuse as for the corresponding state of trans-butadiene.
Structure of the Singlet Excited States We were also interested in examining the adiabatic states of
trans-butadiene. There has been much discussion of the twisted nature of the ?r - T* ~ t a t e , ~ j ~ ~ ~ ~ ~ ~ ~ ~ J ~ ~ ~ q ~ ~ and while there are some vibrational frequencies known for some of the Rydberg states,Ih little structural information is known. Using the CIS/6-3 1 +G* level, we have examined the ?r - ?r* and some of the lower lying
18, n + no (1) 1% n - 3s (2) 2% n - 3P, (3)
3A, n - 3d,2-,2 (7) 4A2 n -. 3d,2 (8) ZA, n - Jd,, (9)
Figure 5. Charge density difference plots for the first nine vertical singlet excitedstatesof cis-butadieneat theCIS/b311(2+)G* level withrespect to the ground state. The contour level is 0.0001 e/B3.
0 120 150 180 30 60 90
Torsion Angle (degrees) Figure 6. Torsion angle profile for the II - II* state of butadiene. The cusp near 90° is probably an artifact resulting from the use of a single reference in this region (see text).
Rydberg states of trans-butadiene. These are cases in which the second set of diffuse functions proved to be relatively unimportant.
For the ?r + ?r* state, the increased charge density between C2 and C3 along with the decreases for the other C-C bonds should lead to a change in geometry. The geometry of the excited state has been reported both based on calculations2jvq*r*s and on a Franck-Condon analysis of the observed spectrum.1j-k*m*o*2j The excited state has been predicted to be planar or twisted by less than 30' from planarity.2j
The sensitivity of the energy of the ?r - ?r* state to the change in torsion angle of the carbon framework ( ~ ( C I - C A & S ) ) was examined. The ground-state torsional profile is relatively in- sensitive to basis set and/or electron ~orre1ation.I~~ Figure 6 shows the relation of energy to T for the ?r - ?r* excited state at the CIS/6-31+G* level under C2 symmetry. First, contrary to the ground state, cis is favored over trans. Second, there is a local minimum at - 160°, which is slightly lower in energy than trans. Finally, the transition state for isomerization of trans to cis is at T of -80°, different than the - 100' for the ground state. The apparent cusp near to 90' in Figure 6 is probably due to the
13590 The Journal of Physical Chemistry, Vol. 97, No. 51, 1993 Wiberg et al.
TABLE V Geometrical Parameters of the w -* W* Singlet States of Butadiene (CIS/6-31+G*)n
Bond distances in angles and angles indegrees. Number of imaginary vibrational frequencies. E Unable to obtain the vibrational frequencies since the geometry is not converged. Lowest real or imaginary vibrational frequencyin cm-1 (unscaled). In unitsofhartrees. IConverged to0.0001 hartree; see ref 16. 8 In electronvolts.
known deficiency of an RHF reference in dealing with a region of the potential energy surface where two determinants are mixing. In this region at -90°, a better reference for the configuration interaction is needed beyond a single RHF determinant.
To fully characterize the conformational preferences of the x - x* state, we havecarried out completegeometry optimizations, andthestructuraldataat theCIS/6-31+G* levelaresummarized in Table V. For each conformer, the C-C bond lengths become very similar (- 1.40 and - 1.39 A), and the structural read- justment is due to the charge reorganization that occurs upon excitation. Starting from trans-butadiene, the C2h-optimized geometry (7 180°, planar) is a transition state based on thederived harmonic vibrational frequencies. The vibrational mode corre- sponding to the imaginary frequency was a conrotatory twisting motion of the ethylenic units with respect to each other. Optimizationunder Czsymmetryalloweda 17Otwistofthecarbon framework (T 163'). This structure also was a transition state. Optimization under CI symmetry revealed a structure composed of a near-planar allyl radical and a perpendicular methylene radical.16 It should be noted that the CI structure has a near- planar methylene radical center, and this does not support the 'sudden-polarization" arguments (Le., pyramidalization of one of the methylene groups with the generation of a large dipole moment) as mentioned in the literature for the excited states of 01efins.~q.'~ This lack of sudden polarization was also seen in our study of ethylene.' For all of these geometries, the initial bond length adjustments from the C2h structure are relatively constant, and only the degree of twisting changes. The C1 structure, as well as the ones described below, are shown in Figure 7. The charge polarized excited states may play a more important role in condensed media where the energetic cost of charge separation is much smaller.
Starting from cis-butadiene, optimization of the x - x* state under C2" symmetry provided a structure where the bond lengths are very similar to the C2h and the C2 geometries above. Vibrational frequency analysis confirmed that this C2" geometry
n
I Figrw7. (a) Geometryofther--.r* stateofcis-butadiene;(b) geometry of the r - A* state of trans-butadiene.
was a transition state, and optimization under Cs symmetry (where both methylenes must twist in a disrotatory sense) yielded a stable stationary point (zero imaginary frequencies). For this geometry, the carbon framework is planar, but the terminal methylenes are twisted by -15' (Figure 7). Optimization of the gauche- butadiene u - x* state under C2 symmetry yielded the CzV geometry that was obtained from the cis-butadiene starting geometry, consistent with the energy-torsional angle profile (cf. Figure 6). The adiabatic transition energy for both the C, and CI structures of the x - x* state is -5.3 and is - 1 eV lower in energy than the vertical transition energy to the lB, x - x* state.
The adiabatic geometries of the Rydberg excited states of butadiene and their relationship to the x - x* state also was of interest. The geometries of the x - 3s (llBg), x - 3p, (llAu), x - 3py (2IAu), and x - 3p, (2IBu) excited states as well as the 2B, ( x ) radical cation were optimized. The CIS/6-31+G* level was used for the excited states and the UHF and UMP2/6- 3 1+G* levels were used for the radical cation. The corresponding geometrical data are listed in Table VI.
The Rydberg and radical cation states were found to have a very similar geometry. Under C2h symmetry all of these states are minima and have zero imaginary vibrational frequencies. The derived geometries have similar C-C bond lengths (- 1.38 and - 1.40 A), and the c2<3 bond length is longer than that for the ( 2 1 x 2 bond. This is contrary to the x - x* state and emphasizes the larger charge reorganization that occurs in the x - A* state. The bond distances and bond angles are very similar to the radical cation geometries and again shows the close similarity between the Rydberg excited states and the corre- sponding radical cation. Our calculated geometrical parameters for the radical cation are in reasonable agreement with the recent theoretical values of Cave and Perrott.18 Also, the calculated CZh symmetry for the adiabatic radical cation is supported by experiment.
The calculated adiabatic transition energies for the Rydberg and radical cation states as well as the experimental data are given in Table VI. For the radical cation, the inclusion of electron correlation at the UMP2 level provides an adiabatic ionization potential within 0.2 eV of experiment. The CIS/6-31+G* adiabatic energies for the Rydberg states are high by -0.3 eV, except for the x - 3p,, Rydberg state (the out-of-plane p orbital)
Charge Distribution in Butadiene
TABLE VI: Geometrical Parameters of Rydberg and Radical Cation States of ttaasButadiene (6-31+6*).
The Journal of Physical Chemistry, Vol. 97, No. 51, 1993 13591
1.3835 1.4077 1.0852 1.0865 1.0883 120.80 121.32 121.19 117.49 119.64 119.56 180 180 0 0 C c -1 55.12968 8.81
9.06'
Bond distances in angstroms and angles in degrees. Number of imaginary vibrational frequencies. Vibrational frequencies were not calculated. d Lowest real or imaginary vibrational frequency, cm-I. In units of hartrees. f In electronvolts. 8 Transition energy at the CIS/6-311(2+)G*//CIS/ 6-31+G* Level. The ground state RHF/6-31 I(2+)GS//RHF/6-31+G* energy is -154.952 115 hartrees. Reference lh. Brundle, C. R.; Robin, M. R. J. Am. Chem. Soc. 1970, 92, 5550.
TABLE VIk Vibrational Frequencies of hos-Butadiene 'A, Ground and r - r* Excited States (6-31+6*). cis (T -L x * ) 1B2 IA, ground state (&) tram ( x - T * ) lB,
0 The data for the other symmetry blocks are available as supplementary material. Frequencies are given in cm-l. Reference 4, p 33, and ref 13a. reference 13b. Ratio of experimental to calculated frequencies. e There was an imaginary frequency in the Bg block. /Reference lm.
which deviates by 0.9 eV. The 6-31+G* basis set may be inadequate for describing the diffuse charge distribution above the molecular plane for the A - 3p, state (cf. Figure 3) and thereby leads to an underestimation of the adiabatic transition energy. Indeed, single point energy calculations at the CIS/6- 31 1(2+)G*//CIS/6-31+G* level for eachofthe Rydbergstates reveals a significant decrease in the calculated adiabatic transition energies, and the values are closer to the experimental results. In particular, the transition energy for the A - 3p, state is within 0.2 eV of the experimental adiabatic value at the CIS/6-311- (2+)G* level.
In agreement with previous work for the r - A* state, it can be seen that electronic excitation results in a significant decrease in the central C-C bond length and an increase in the other C-C lengths. This change in geometry would require an excitation of both C=C and C-C vibrational modes on going to the vertical excited state. The vibrational modes of the excited states are commonly related to thoseof thegroundstate with theassumption that they will have somewhat weakened force constants. However, the reversal of single and double-bond lengths makes one wonder if such an approach is appropriate for butadiene. Thevibrational frequencies at the CIS/6-31+G* level are compared with the corresponding calculation for thegroundstate (RHF/6-3 lG+G*) in Table VII.
Hartree-Fockvibrational frequencies are known to be too high by about 10% since the corresponding bond lengths are too short due to the neglect of electron correlation.20 The vibrational
frequencies obtained for the 1 B, (C2h) structure are very similar to the RHF frequencies for the ground state, and it would suggest that the scaling factors typically used to scale Hartree-Fock frequencies would be appropriate for the CIS-derived frequencies. This method of scaling has worked relatively well for our studies of bicyclo[ 1.1 .O]butane,6 ethylene,' and carbonyl compounds.8 It also has been used by Elston et al. in their study of pdifluo- robenzene.21 In Table VI1 we have used the experimental vibrational frequencies'3b for trans-butadiene to obtain the corresponding scale factors between the theoretical frequencies and those obtained experimentally. The scale factors are typically in the range 0.85-0.9 1, with a few of the lower energy frequencies having larger scale factors.
Let us first consider the ?r - A* state. Using the normal-mode displacements for the ground state, we have determined the vibrational frequencies for the A - A* excited states which correspond to the ground-state frequencies in Table VII. The frequencies for the trans and cis geometries are quite similar, except for some of the twisting modes. We have applied the ground-state scaling factors to the C, (cisoid) geometry, and these scaled frequencies are also listed. There are only two vibrational frequencies observed for the butadiene A - r* state, and they wereassigned as the totally symmetric C = C and C-C stretches." We find reasonable agreement with the scaled frequencies from the C, structure and the experimental values. Unfortunately, we are unable to obtain vibrational frequencies for the C1 structure since it was not fully optimized.16
13592 The Journal of Physical Chemistry, Vol. 97, No. 51, I993
TABLE MI: Vibrational Frequencies' of Rydberg and Radical Cptim States of bnrr9-Butadiene (631+G*)
a Frequencies in cm-I. The ratio bctween the experimental and RHF frquencies for the ground state are used as scaling factors to obtain the scaled frequencies for each excited state (see text). The mode assignments correspond to those of the ground state. Reference 1 h. Reference lh, 3pl series (strong intensity). Reference lh, 3p2 series (very strong intensity). f Reference 22. Reference l h assigned this frequency to UT. * References l a and 1 b. ' This frequency could not be obtained because of a numerical problem.
For the Rydberg and radical cation states, we have used the ground-state scale factors to obtain some scaled theoretical vibrational frequencies to compare with experiment. Table VI11 lists the calculated and experimental frequencies for the s- and p-Rydberg states of trans-butadiene. One can see good agreement with our theoretical (scaled) vibrational frequencies and those obtained experimentally for butadiene at room temperature. Typically, we obtain agreement within 90 cm-I for all of the states examined, and the qualitative trends between excited states are also reproduced. Agreement for the radical cation2* is excellent. Only vg for the r - 3py (2IAU) state is calculated to bevery different (557 calculated vs 375 cm-' experimentIh). Since the experimental 375 cm-I frequency is very different than v9 for any of the other states (despite very similar geometries), we are unsure of the cause for the discrepancy. Finally, we should note that the UHF calculation for the radical cation leads to slight spin contamination ( ( S 2 ) is 0.89 and should be 0.75 for a pure doublet). The corresponding geometry and derived vibrational frequencies may be affected by this spin contamination. We do note, however, that agreement with the experimental frequencies for the radical cation is quite good. We are not aware of a proven method for obtaining the geometry and vibrational frequencies for the radical cation while avoiding the problem of the spin contamination.
Triplet Excited States
We were interested in determining the difference in charge density distribution between the singlet excited states and the corresponding triplet states of tram-butadiene. Using the CIS method, the vertical transition energies were calculated with the 6-31G+G*, 6-311+GS, and 6-311(2+)G* basis sets. The transition energies for the 13Bu and 13Ag valence transition^^^ were not significantly affected by the change in basis set. The best agreement with experiment for the '(-3s) state was found with the larger basis set, emphasizing the diffuse character of this excited state. The excitation energies for the higher triplet
a The MP2/6-31G* geometry of the ground state was used. The CIS calculations correlated all electrons. Masher, 0. A.; Flicker, W. M.; Kupperman, A. Chem. Phys. Lett. 1973,19,332. Reference 3, p 319.
Rydberg state are greatly affected by the addition of diffuse functions. The results are summarized in Table IX.
The projection density difference plots for the 13Bu and 13A, valence transitions are shown in Figure 8. As with the singlet r - ?r* state, the 13B, state has charge depletion for the C1-C2 and C 3 4 4 bonds, and a concentration of charge in the C 2 4 3 bond. The shifts in the r and u systems oppose each other as was seen with the singlet state. The 13Ag state, on the other hand, has charge accumulation at each of the carbons and depletion at each of the C-C bonds. Instead, all of the charge increase is concentrated in the periphery of the molecule. For both the 13B, and 13A, states, the projection density plots demonstrate the greater diffuse character of the singlet states as compared with their triplet counterparts. This was also seen in our analysis of the excited states of bicyclo[ 1.1 .O] butane6 and eth~lene.~
The total charge reorganization that occurs in forming these vertical excited states was calculated using the GDM. The 13Bu state has a 0.30 e shift for the total density, and 0.30 and 0.06 e for the r and u systems, respectively. This is somewhat smaller than found for the corresponding singlet state. The 13Ag state
Charge Distribution in Butadiene The Journal of Physical Chemistry, Vol. 97, No. 51, 1993 13593
trans-Butadiene Triplet Excited States Projection Density Differences TABLE X Geometrical Parameters of the 3B,, and jAe States of tnms-l,3-Butadiene (6-316+6*).
1%, (1) total density 1%" (1) m density 1'8, (1) u density
1'4 (2) total density 13A, (2) m density 13A, (2) u density
Figure 8. Projection density difference plots for the vertical triplet 13Bu (upper) and 13Ag (lower) states of trans-butadiene with respect to the ground state. The outer contour is 0.0001 e/B3 and the contours increase by a factor of 2.
z3Al - 3 4 (9)
Figure9. Charge density difference plots for the first nine vertical triplet excited states of trans-butadiene at the CIS/6-311(2+)G* level with respect to the ground state. The contour level is 0.0001 e/B3.
has 0.24 e total shift, and 0.27 and 0.04 e respectively for the ?r
and u systems. The three-dimensional density difference plots with respect to
the ground state also were obtained and are shown in Figure 9. The first two states are significantly less diffuse than the others. The 13A, state is a mixture of two predominant configurations: 3 ( ~ 1 - TI*) and 3 ( ~ 2 - Q*). Here, TI and 7r2 are the occupied orbitals and ?rl* and ?r2* are the virtual orbitals. In the simple ?r-electron picture, each of these excitations would lead to loss of bonding character for the C1-C2 and C3-C4 bonds, as well as for the C&3 bond. This is what is found.
To see how a change in geometry might occur on going to the triplet valence states, geometry optimizations were carried out both at the UHF/6-3 1 +G* and UMP2/6-3 1+G* levels as well as the CIS/6-3 1+G* level. The results are summarized in Table X.
With the 3Bu state, the adiabatic structure had C2h symmetry at both CIS and UHF levels as verified by a vibrational frequency calculation. As for the singlet state, there is a reversal of the C-C bond lengths as compared to the ground-state geometry.
Bond distances in angstroms and angles in degrees. Number of imaginary vibrational frequencies. e Vibrational frequencies were not calculated. Lowest real or imaginary vibrational frequency in cm-1 (unscaled). In units of hartrees. J In electronvolts.
However, the central C-C bond is significantly shorter than for the singlet state. The CIS optimized geometry is qualitatively similar to that obtained at the UHFand UMP2 levels. Differences in geometry and transition energy may be due to the fact that spin contamination has been eliminated in the CIS calculations.
The adiabatic geometry of the 3A, state was examined at the CIS/6-3 1 +G* level. Optimization under C2h symmetry provided elongated C I - C ~ and CrC3 bonds, as suggested by the charge density difference plots. The geometry was, however, a transition state, and the vibrational mode corresponding to the imaginary frequency was a conrotatory twisting of the molecular framework. The resulting C2 symmetric geometry with a 94O twist between the plane of the ethylenic units was a true minimum. The C&3 bond length decreased to 1.479 A while the C 4 2 bond increased to 1.416 A. Each ethylenic unit, though, remains quite planar.
Atomic Populations and Covalent Bond Orders
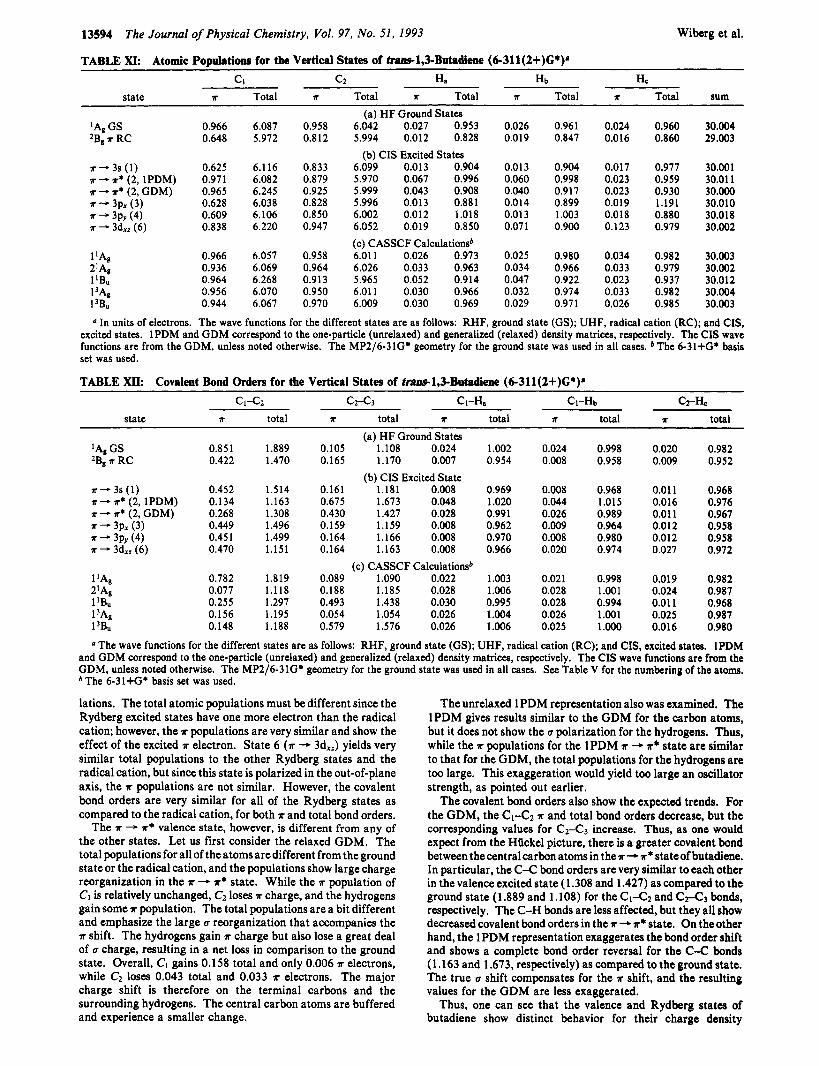
We were interested in obtaining information on the changes in electron population and bond orders that occur on electronic excitation. Bader's theory of atoms in molecules24 was used to obtain the populations, and the procedure of Cioslowski and M i ~ o n ~ ~ was used to derive the bond orders from the atomic overlap matrices. The ?r and total atomic populations for the ground state and some of the excited states of butadiene, as well as for the 2B, (4 radical cation are given in Table XI. The results for the radical cation shows that the unit positive charge is shared by all of the atoms in butadiene, with the hydrogens losing 0.68 e. The central carbon atoms, C2 and C3, suffer loss charge loss (0.05 e each) than the terminal carbon atoms (0.12 e each). The carbon atoms lose a good deal of ?r-charge on going to the radical cation, but the total charge is compensated by an increase in the a-contribution to the carbon atomic basins at the expense of the hydrogens.
Similar effects are also seen for the covalent bond orders (Table XII). There is a stronger covalent bond for the central carbon atoms (C2-C3) on going to the vertical radical cation as both the ?rand total bond orders increase. The C1-Q bond order, though, decreases by about a factor of 2 for the ?r bond strength with a loss of -0.43. The total bond order is decreased by a similar amount, emphasizing little a reorganization. The C-H bond orders also decrease by -0.04 for the vertical radical cation, with a loss of -0.01 5 in the ?r contribution. Thecentral hydrogen atoms (H,) change the least.
The Rydberg excited states show a similarity to their corre- sponding radical cation, especially with respect to the ?r-popu-
13594 The Journal of Physical Chemistry, Vol. 97, No. 51, 1993
TABLE XI: Atomic Populations for the Vertical Stntes of brUrs-l,lButadiene (6-311(2+)6*).
Wiberg et al.
CI c2 H. Hb HC state 7r Total A Total 7r Total U Total 7r Total sum
In units of electrons. The wave functions for the different states are as follows: RHF, ground state (GS); UHF, radical cation (RC); and CIS, excited states. lPDM and GDM correspond to the one-particle (unrelaxed) and generalized (relaxed) density matrices, respectively. The CIS wave functions are from the GDM, unless noted otherwise. The MP2/6-31GS geometry for the ground state was used in all cases. The 6-31+G* basis set was used.
TABLE XII: Covalent Bond Orders for the Vertical States of trans-l$-Butadiene (6-3l1(2+)Gr)* ~~
CI42 crc3 CI-H~ C1-b CtHc state T total 7r total 7r total x total U total
The wave functions for the different states are as follows: RHF, ground state (GS); UHF, radical cation (RC); and CIS, excited states. lPDM and GDM correspond to the one-particle (unrelaxed) and generalized (relaxed) density matrices, respectively. The CIS wave functions are from the GDM, unless noted otherwise. The MP2/6-31GS geometry for the ground state was used in all cases. See Table V for the numbering of the atoms. The 6-31+G* basis set was used.
lations. The total atomic populations must be different since the Rydberg excited states have one more electron than the radical cation; however, the T populations are very similar and show the effect of the excited x electron. State 6 ( a - 3d,,) yields very similar total populations to the other Rydberg states and the radical cation, but since this state is polarized in the out-of-plane axis, the a populations are not similar. However, the covalent bond orders are very similar for all of the Rydberg states as compared to the radical cation, for both T and total bond orders.
The T - a* valence state, however, is different from any of the other states. Let us first consider the relaxed GDM. The total populations for all of the atoms are different from the ground state or the radical cation, and the populations show large charge reorganization in the a - x* state. While the a population of Cl is relatively unchanged, C2 loses T charge, and the hydrogens gain some r population. The total populations are a bit different and emphasize the large u reorganization that accompanies the a shift. The hydrogens gain a charge but also lose a great deal of u charge, resulting in a net loss in comparison to the ground state. Overall, CI gains 0.158 total and only 0.006 a electrons, while C2 loses 0.043 total and 0.033 T electrons. The major charge shift is therefore on the terminal carbons and the surrounding hydrogens. The central carbon atoms are buffered and experience a smaller change.
The unrelaxed lPDM representation also was examined. The lPDM gives results similar to the GDM for the carbon atoms, but it does not show the u polarization for the hydrogens. Thus, while the a populations for the lPDM a + ?r* state are similar to that for the GDM, the total populations for the hydrogens are too large. This exaggeration would yield too large an oscillator strength, as pointed out earlier.
The covalent bond orders also show the expected trends. For the GDM, the CI-CZ T and total bond orders decrease, but the corresponding values for c 2 4 3 increase. Thus, as one would expect from the Hiickel picture, there is a greater covalent bond between thecentralcarbonatomsin the*-# stateofbutadiene. In particular, the C-C bond orders are very similar to each other in the valence excited state (1.308 and 1.427) as compared to the ground state (1 3 8 9 and 1.108) for the c1-C~ and C A a bonds, respectively. The C-H bonds are less affected, but they all show decreased covalent bond orders in the *+T* state. On the other hand, the lPDM representation exaggerates the bond order shift and shows a complete bond order reversal for the C-C bonds (1.163 and 1.673, respectively) as compared to the ground state. The true u shift compensates for the a shift, and the resulting values for the GDM are less exaggerated.
Thus, one can see that the valence and Rydberg states of butadiene show distinct behavior for their charge density
Charge Distribution in Butadiene
TABLE XIII: MCSCF Calculations for Vertical Excited States of bmrt41,3-Butadlene (Cz#
The Journal of Physical Chemistry, Vol. 97, No. 51, 1993 135%
a Using the 6-31+G* basis set at the MP2/6-31G* ground-state geometry. In hartrees relative to -154.0. In electronvolts. In electrons.
properties. The Rydberg states, in particular, are very similar to the corresponding radical cation. The valence states show different behavior from any of the other states. These states can easily be distinguished by their atomic populations and covalent bond orders.
The Mysterious 2lA, State The 2IA, state has been extensively d isc~ssed . l~~*~ Doering
and McDiarmid have assigned the vertical transition energy for a ]A, state at 7.4 eV.lg Our CIS calculations and charge density difference plots found a 'A, Rydberg state (17 - 3d,J at 7.19 eV which is in good agreement. However, it is believed that there is a lower energy valence 'A, state. Extensive searches by multiphoton ionization,Ic thermal lensing,Id and fluorescence spectroscopy5 have provided no assistance on the location of this state. Recent resonance Raman scattering experiments by Chadwick et al. have suggested that the (0-0) energy for the 2IA, state is about 0.25 eV lower in energy than the llB, (0-0) energyelp
The 2IA, state has been the subject of many theoretical calculations,Zf-gJ-n,q-t,v-x and it has been concluded that the multiconfigurational self-consistent field (MCSCF) procedure with the inclusion of doubly excited configurations is needed in order to correctly reproduce this state. We wished to examine the relationship of this state to the others that were found using the CIS formalism. We have made use of the complete active space (CASSCF) formalism26 including all four ?r electrons, and the four 7 orbitals (CASSCF(4,4)) using the 6-3 1+G* basis set. The results using even larger CAS spaces (8,8) did not differ significantly from the (4,4) results presented here. The energies are given in Table XIII. For a comparison, the first two triplet states also were calculated at this level of theory. It is interesting to compare the singlet A, and B, states with the corresponding triplet states (Figure 10).
The triplet states have charge distributions that agree with expectations from simple Hiickel *-wave functions. In the ground state the C 4 2 and C S 3 bond orders are 0.89 and 0.44, respectively. The T - ** state has bond orders of 0.45 and 0.72, respectively, and it can be seen that this corresponds to the shift in charge density found for the 3Bu state. The next excited state would be formed by the promotion of 71 - T I * and of u2 - a2*, and a linear combination of the two was found for the 3A, state in the CIS calculation. Here, the Hiickel bond orders would be C1-G = 0.45 and c2<3 = 0.22. Thus, both the double bonds and the central bond should lose charge density. This is found to be the case. The simplicity of the triplet states arises because of the spin orthogonality with respect to the ground state, eliminating the need for spatial orthogonality. It should be noted that physically more significant bond orders may be obtained from the charge density distribution and are given in Table XII.
The IB, state is similar to the 3B, state, except that it is much more diffuse. This probably arises in order to achieve spatial orthogonality to the ground state. The singlet 2 'A, state found in the CASSCF calculation is similar to the triplet A, state both in charge distribution and in spatial extent.
In the CASSCF calculation, the ground state is formed as a linear combination of the conventional ?r configurations, and a
Butadiene CAS(4,4)/6-31+C'//MP2/6-3lG* Vertical Projectlon Density Dllferencca
1'Bu - 1'Ag (gr) Z'Ag - 1'Ag (gs)
Figure 10. Charge density different plots for the first two vertical singlet and triplet excited states of trans-butadiene at the CAS(4,4)/6-31+G* level with respect to the ground state. The outermost contour is O.OOO1 e/B3.
set of doubly excited r configurations. The 2 'A, state is formed from the same configurations, but with different coefficients. The calculation forces it to be orthogonal to the ground state, without the necessity of becoming diffuse. It is interesting to note that the excited state also has contributions from the single excitations u1 - ul* and ?r2 - a** that are important for the corresponding triplet state. The electron populations and bond orders for this state are given in Tables XI and XII. There is little change in electron populations on excitation, but there are large changes in bond orders.
Contrary to the 2 (A, state, the transition energy for the IB, state is poorly calculated via CASSCF. Serrano-Andres et al. have shown that it is possible to improve the calculation of this state by a perturbation theory correction.2w The origin of the difference between the CASSCF and CIS results was examined by calculating the CIS transition energies using only a selected set of configurations. When it was restricted to MOs 14-21 which includes the r* MOs, the calculated transition energy was 6.94 eV, close to that found in the CASSCF calculation. When the two highest occupied MOs and all the virtual MOs were included (1673) the energy dropped to 6.78 eV. Incressing the number of occupied orbitals to 8 (8-73) gave a transition energy of 6.40 eV, and when all of the MOs were included, the transition energy was 6.1 1 eV. Therefore, correlating all of the electrons of butadiene is essential when examining this state. It is sufficient, however, to accomplish this with CIS and Hartree-Fock orbitals without the need to employ complete active space or perturbative corrections.
Conclusions The CIS method has been found to reproduce the experimental
vertical singlet transition energies with an average error of only 0.2 eV. The inclusion of a double set of diffuse functions was found to be important for describing the Rydberg states. An
13596 The Journal of Physical Chemistry, Vol. 97, No. 51, 1993 Wiberg et al.
analysis of the charge density distribution in the excited states has proven to be useful both in assigning the states and in providing quantitative information on the degree of charge reorganization. Here, the one particle density matrix was found to be inadequate, and only the generalized density matrix showed that charge reorganization occurred in both the r and u systems of the r - r* excited state.
The CIS method also provided optimized geometries for both the valence and Rydberg excited states of butadiene. The 1 IB, r - r* state showed large C-C bond length changes that agreed with expectations from Huckel theory. However, the planar form was a transition state, and the equilibrium geometry had a twisted structure. The corresponding state for cis-butadiene was ex- amined and showed similar changes in bond lengths. The equilibrium geometry also had a twisted structure.
The transition energies for the triplet states also were reasonably reproduced by the CIS calculations. The 1 3Bu state was much less diffuse that the corresponding singlet state. The 1 3A, state was a mixture of two predominant configurations: 3(rl - TI*) and 3(r2 - rz*), again in accord with simple Hiickel theory. The 1 3Bu state was calculated to remain planar, but the 1 3Ag state underwent rotation about the central C-C bond giving two relatively independent ethylenic units rotated 90° to each other. The triplet valence states can be relatively compact because of their spin orthogonality with respect to the ground state, whereas the singlet states must achieve spatial orthogonality, leading to more diffuse character.
The 2 'A, state was examined using a CASSCF(4,4) calcu- lation. It was found to have a charge distribution similar to that of the corresponding triplet state. The 1 IB, transition energy is not well calculated at this level of theory, and it was found that both correlation with higher virtual orbitals and with the u-occupied orbitals was important for describing this state.
Partitioning the charge densities into atomic contributions was useful. It provided quantitative data on the degree of charge transfer and bond-order reorganization that occurs in the excited states. The expected changes in bond orders were found for the valence states, and the Rydberg states were found to be quite similar to the corresponding radical cation.
Calculations
All calculations were performed with the GAUSSIAN 9227 series of programs. Standard basis sets were used in all cases, except for 6-31 1 (2+)G* which is the 6-31 1+G* basis set with an additional sp shell on carbon (0.013 192 8 exponent). Six d Cartesian functions (the 6D option) was used for all basis sets. All CIS and MP2 calculations correlated all electrons (full), except where noted. All optimizations utilized the analytical gradient method. CIS frequency analyses were performed via numerical differentiation of the analytic derivative. The relative energies listed in the text do not include the correction for the zero-point energy so as to render easy comparison of vertical and adiabatic energies. The density matrices were abstracted and converted to natural orbitals with PSICHK.28 Charge density difference plots were obtained by calculating a cube of charge density 40 au on a side with 80 points in each direction. Contouring and plotting were done using locally developed programs. Atomic properties werecalculated with the Atoms in Molecules Package (AIMPAC) from McMaster University and, in particular, a modified version of PROAIM.29 Covalent bond orders were calculated with BONDER.2S
Experimental Methods The room-temperature absorption spectrum of 1,3-butadiene
was taken on a McPherson 225 vacuum UV spectrometer with a 1-m focal length.30 Theinstrumental resolution was -25 cm-I. A rare-gas discharge lamp, driven by a microwave generator, was used as the light source. The relative intensity was detected by two photomultipliers which were monitored by phase-sensitive
lock-in amplifiers. The amplified signal was then transferred to a computer for data storage and analysis. The sample pressure of 1,3-butadiene was - 1 Torr, and the samplecell was a stainless steel tube (10 cm long with 1.905-cm i.d.) with LiF windows.
Acknowledgment. This investigation was supported by a grant from theNationa1 Science Foundation. C.M.H. thanks theFannie and John Hertz Foundation for a predoctoral fellowship. We are indebted to the Pittsburgh Supercomputing Center for a generous grant of computer time. We would also like to thank Todd Keith (McMaster University) for his help in obtaining some of the integrated properties for the excited states. The comments of one of the reviewers concerning the MCSCF procedure were particularly helpful and appreciated.
Supplemenclvy Material Available: Tables giving the butadiene ground state geometry, the effect of basis set on calculated transition energies, the calculated vibrational frequencies for excited states, and the expectation values of zz for some excited states ( 5 pages). Ordering information is given on any current masthead page.
References and Notes (1) (a) McDiarmid, R. Chem. Phys. Lett. 1975,34,130. (b) McDiarmid,
R. J . Chem. Phys. 1976,64,514. (c) Johnson, P. M. J. Chem. Phys. 1976, 64,4638. (d) Neiman, G. C.; Colson, S. D. J . Chem. Phys. 1978,68,2994. (e ) Vaida, V.; Turner, R. E.; Casey, J. L.; Colson, S. D. Chem. Phys. Len. 1978,54,25. (f) Doering, J. P. J. Chem. Phys. 1979,70, 3902. (g) Doering, J. P.; McDiarmid, R. J . Chem. Phys. 1980, 73, 3617. (h) Rothberg, L. J.; Gerrity, D. P.; Vaida, V. J . Chem. Phys. 1980, 73, 5508. (i) Doering, J. P.; McDiarmid, R. J. Chem. Phys. 1981,75,2477. u) Granville, M. F.; Kohler, B. E.;Snow, J. B. J . Chem. Phys. 1981,75,3765. (k) Hemley,R. J.; Dawson, J. I.; Vaida, V. J . Chem. Phys. 1983, 78, 2915. (1) Mallard, W. 0.; Miller, J. H.; Smyth, K. C. J . Chem. Phys. 1983, 79, 5900. (m) Leopold, D. G.; Pendley, R. D.; Roebber, J. L.; Hemley, R. J.; Vaida, V. J . Chem. Phys. 1984, 81,4218. (n) Chadwick, R. R.; Gerrity, D. P.; Hudson, B. S. Chem. Phys. Lerr. 1985, 115, 24. (0) McDarmid, R.; Sheybani, A,-H. J. Chem. Phys. 1988,89,1255. (p) Chadwick, R. R.; Zgierski, M. Z.; Hudson, B. S. J. Chem. Phys. 1991,95,7204. (4) McDiarmid, R. Chem. Phys. Lerr. 1992,188,423. (r) Phillips, D. L.; Zgierski, M. Z.; Myers, A. B. J. Phys. Chem. 1993, 97, 1800. (s) Trulson, M. 0.; Mathies, R. A. J . Phys. Chem. 1990, 91, 5741.
(2) (a) Buenker, R. J.; Whitten, J. L. J. Chem. Phys. 1968, 49, 5381. (b) Shih, S.; Buenker, R. J.; Peyerimhoff, S. Chem. Phys. Lerr. 1972,16,244. (c) Hosteny. R. P.; Dunning, T. H.; Gilman, R. R.; Pipano, A,; Shavitt, I. J. Chem. Phys. 1975, 62,4764. (d) Buenker, R. J.; Shih, S.; Peyerimhoff, S. D. Chem. Phys. Lett. 1976,44385. ( e ) Bonacic-Koutecky, V.; Buenker, R. J.; Peyerimhoff, S. D. J . Am. Chem. SOC. 1979,101,5917. (f) Nascimento, M. A. C.; Goddard, W. A., I11 Chem. Phys. 1979,36,147. (9) Nascimento, M. A. C.; Goddard, W. A., I11 Chem. Phys. 1980.53,215. (h) Lasaga, A. C.; Aerni, R. J.; Karplus, M. J . Chem. Phys. 1980, 73, 5230. (i) Bonacic- Koutecky, V.; Persico, M.; Dohnert, D.; Sevin, A. J. Am. Chem. Soc. 1982, 104,6900. Q) Dinur, U.; Hemley, R. J.; Karplus, M. J. Phys. Chem. 1983, 87,924. (k) Aoyagi, M.; Osamura, Y.; Iwata, S. J . Chem. Phys. 1985,83, 1140. (1) Cave, R. J.; Davidson, E. R. J . Phys. Chem. 1987,91,4481. (m) Cave, R. J.; Davidson, E. R. Chem. Phys. Left. 1988,143, 190. (n) Kitao, 0.; Nakatsuji, H. Chem. Phys. Lerr. 1988,143,528. (0 ) Galasso, V. J. Chem. Phys. 1988, 89, 4529. (p) Szalay, P. G.; Karpfen, A.; Lischka, H. Chem. Phys. 1989,130,219. (9) Aoyagi, M.; Osamura, Y. J . Am. Chem.Sof. 1989, 11 1,470. (r) Szalay, P. G.; Karpfen, A.; Lischka, H. Chem. Phys. 1989,130, 219. (s) Szalay, P. G.; Karpfen, A.; Lischka, H. Chem. Phys. 1990,141,355. (t) Graham, R. L.; Freed, K. F. J . Chem. Phys. 1992,96, 1304. (u) Cave, R. J. J . Chem. Phys. 1990,92,2450. (v) Serrano-Andres, L.; Sanchez-Marin, J.; Nebot-Gil, I. J. Chem. Phys. 1992, 97, 7499. (w) Serrano-Andrw, L.; Merchan, M.; Nebot-Gil, I.; Lindh, R.; Roos, B. 0. J. Chem. Phys. 1993,98, 3151. (x) Olivucci, M.; Ragazos, I. N.; Bernardi, F.; Robb, M. A. J. Am. Chem. Soc. 1993, 115, 3710.
(3) Robin, M. B. Higher Excited Sfafes of Polyatomic Molecules; Academic Press: New York, 1985; Vol. 111.
(4) Hudson, B. S.; Kohler, B. E.; Schulten, K. Linear Polyene Electronic Structureand Potential Surfaces. In ExcitedStares; Lim, E. C., Ed.; Academic Press: New York, 1982; Vol. VI, pp 2-95.
( 5 ) Foresman, J. B.; Head-Gordon, M.; Pople, J. A.; Frisch, M. J. J . Phys. Chem. 1992, 96, 135.
B.; Johnson, P. M.; Foresman, J. B. J . Am. Chem. Soc. 1991,113,4782. (6) Walters, V. A.; Hadad, C. M.; Thiel, Y.; Colson, S. D.; Wiberg, K.
(7) Wiberg, K. B.; Hadad, C. M.; Foresman, J. B.; Chupka, W. A. J . Phys. Chem. 1992, 96, 10756.
(8) Hadad, C. M.; Foresman, J. B.; Wiberg, K. B. J. Phys. Chem. 1993, 97, 4293.
(9) Streitwieser, A., Jr.; Collins, J. B.; McKelvey, J. M.;Grier, D.;Sender, J.; Toczko, A. G. Prof. Narl. Acad. Sci. U.S.A. 1979, 76,2499.
(10) Handy, N.C.;Schaefer,H. F.J. Chem.Phys. 1984,81,5031. Wiberg, K. B.; Hadad, C. M.; LePager, T. J.; Breneman, C. M.; Frisch, M. J. J . Phys. Chem. 1992, 96, 671.
Charge Distribution in Butadiene The Journal of Physical Chemistry, Vol. 97, No. 51, 1993 13597
(1 1) One should note that some assignments by Vaida and co-workerslb were for 4p and 4f Rydberg states. Since these configurations will have the same number of (radial and angular) nodes, the final states can be a mixture of both configurations. In Table 11, we have listed the experimental values for p and f Rydberg states based on the experimental assignments and the relative intensities.
(12) (a) Msller, C.; Plesset, M. S . Phys. Rev. 1934,46,618. (b) Binkley, J. S.; Pople, J. A. Inr. J. Quontum Chem. 1975, 9, 229. (c) Pople, J. A.; Binkley, J. S.; Seger, R. Inr. J. Quontum Chem. Symp. 1976, 10, 1.
(13) (a) Guo, H.; Karplus, M.J. Chem. Phys. 1991,94,3679. (b) Wiberg, K. B.; Rosenberg, R. E. J . Am. Chem.Soc. 1990,112, 1509. (c) Breulet, J.; Lee,T. J.;Schaefer, H. F., I11 J. Am. Chem. Soc. 1984,106,6250. (d) Feller, D.; Davidson, E. R. Theor. Chim. Acto 1985,68,57. (e) Bock, C. W.; George, P.; Trachtman, M. Theor. Chim. Acro 1984,64,293. (f) Kavana-Saebo, K.; Saebo, S.; Boggs, J. E. J . Mol. Srrucr. 1984,106,259. (8) Tai, J. C.; Allinger, N. L. J. Am. Chem. Soc. 1976,98,7928. (h) Radom, L.; Pople, J. A. J . Am. Chem. Soc. 1970, 92,4786. (i) Wiberg, K. B.; Rablen, P. R.; Marquez, M. J. Am. Chem. Soc. 1992,114,8654.
(14) (a) Squillacote, M. E.; Sheridan, R. S.; Chapman, 0. L.; Anet, F. A. J. Am. Chem. Soc. 1979, 101, 3657. (b) Squillacote, M. E.; Semple, T. C.; Mui, P. W. J . Am. Chem. Soc. 1985, 107, 6842. (c) Furakawa, Y.; Takenichi, H.; Harada, I.; Tasumi, M. Bull. Chem. Soc. Jpn. 1983,56,392. (d) Fisher, J. J.; Michl, J. J. Am. Chem. Soc. 1987,109, 1056. (e) Mui, P. W.;Gnmwald,E. J.Am. Chem.Soc. 1982,104,6562. (f)Engeln,R.;Consalvo, D.; Reuss, J. Chem. Phys. 1992, 160,427.
(15) Arnold, B. R.; Balaji, V.; Michl, J. J . Am. Chem. Soc. 1990, 112, 1808.
(16) Unfortunately we were unable to fully optimize the CI structure as it is near C, symmetry, and there is a RHF to UHF instability in the wave function. Attempts at using the UHF wave function as a reference for the CIS (UCIS) similarly failed. Our best geometry (converged to better than O.OOO1 hartrees, with small coordinate forces) is listed in Table V.
(17) Salem, L. Electrons in Chemical Reocrions: Firsr Principles; John Wiley: New York, 1982.
(18) Cave, R. J.; Perrott, M. G. J. Chem. Phys. 1992, 96, 3745. (19) Gerson. F.; Qin, X.-Z. Helv. Chim. Acro 1988, 71, 1065. (20) (a) DeFrecs, D. J.; Ragavachari, K.; Schlegel, H. B.; Pople, J. A. J.
Am. Chem. Soc. 1982,104, 5576. (b) Wiberg, K. B. J. Org. Chrm. 1985, 50, 5285. (c) h l a y , P.; Fogarasi, G.; Pongor, G.; Boggs, J.; Vorgha, A. J . Am. Chem. Soc. 1983,105,7037. (d) Hess, B. J., Jr.; Schaad, L. J.; Carsky, P.; Zahradnik, R. Chem. Rev. 1986, 86, 709.
(21) Elston, H. J.; Davidson, E. R.; Todd, F. G.; Parmenter, C. S. J . Phys. Chem. 1993,97, 5506.
(22) Tang, W.; Zhang, X.-L.; Bally, T. J . Phys. Chem. 1993, 97,4373. (23) A tableshowing the effect of basis set on calculated transition energies
may be found in the supplementary material. (24) Bader, R. F. W. Chem. Rev. 1991,91,893. Bader. R. F. W. A t o m
in Molecules: A Quantum Theory; Clarendon Press: Oxford, 1990. (25) Cioslowski, J.; Mixon, S . T. J. Am. Chem. Soc. 1991, 113, 4142.
Wiberg, K. B.; Hadad, C. M.; Rablen, P. R.; Cioslowski, J. J. Am. Chem. Soc. 1992, 14, 8644.
(26) Roos, B. 0.; Taylor, P. R.; Siegbahn, P. E. M. Chem. Phys. 1980, 48, 157. Ruedenberg, K.; Schmidt, M. W.; Gilbert, M. M.; Elbert, S. T. Chem. Phys. 1982,71,65. Ruedenberg, K.; Schmidt, M. W.; Gilbert, M. M. Chem. Phys. 1982, 71, 51. R m , B. 0. Adv. Chem. Phys. 1987, 69, 399.
(27) Frisch, M. J.; Trucks, G. W.; Head-Gordon, M.; Gill, P. M. W.; Wong, M. W.; Foresman, J. B.; Johnson, B. G.; Schlegel, H. B.; Robb, M. A,; Replogle, E. S.; Gomperts, R.; Andres, J. L.; Raghavachari, K.; Binkley, J. S.; Gonzalez, C.; Martin, R. L.; Fox, D. J.; DeFrees, D. J.; Baker, J.; Stewart, J. J. P.; Pople, J. A. GAUSSIAN 92, Revision A; Gaussian, Inc.: Pittsburgh, PA, 1992.
(28) .LePage, T. J.; Laidig, K. E.; Hadad, C. M.; Cheeseman, J. R. Yale University (program available upon request).