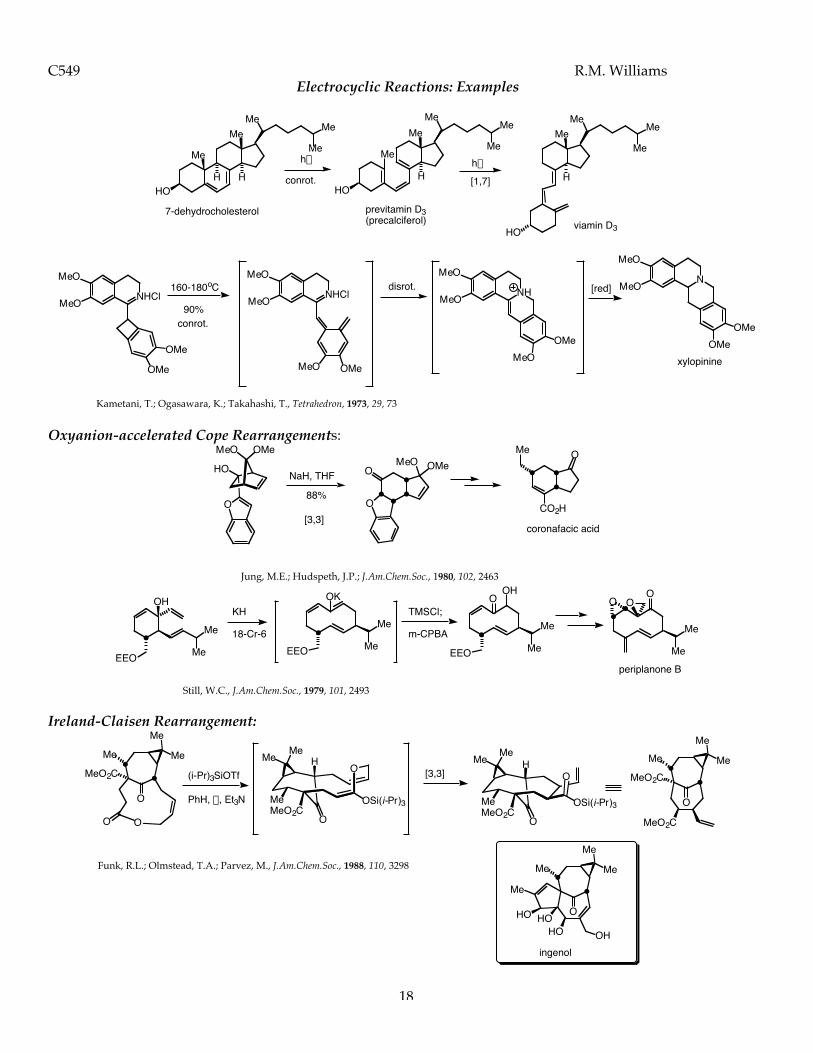
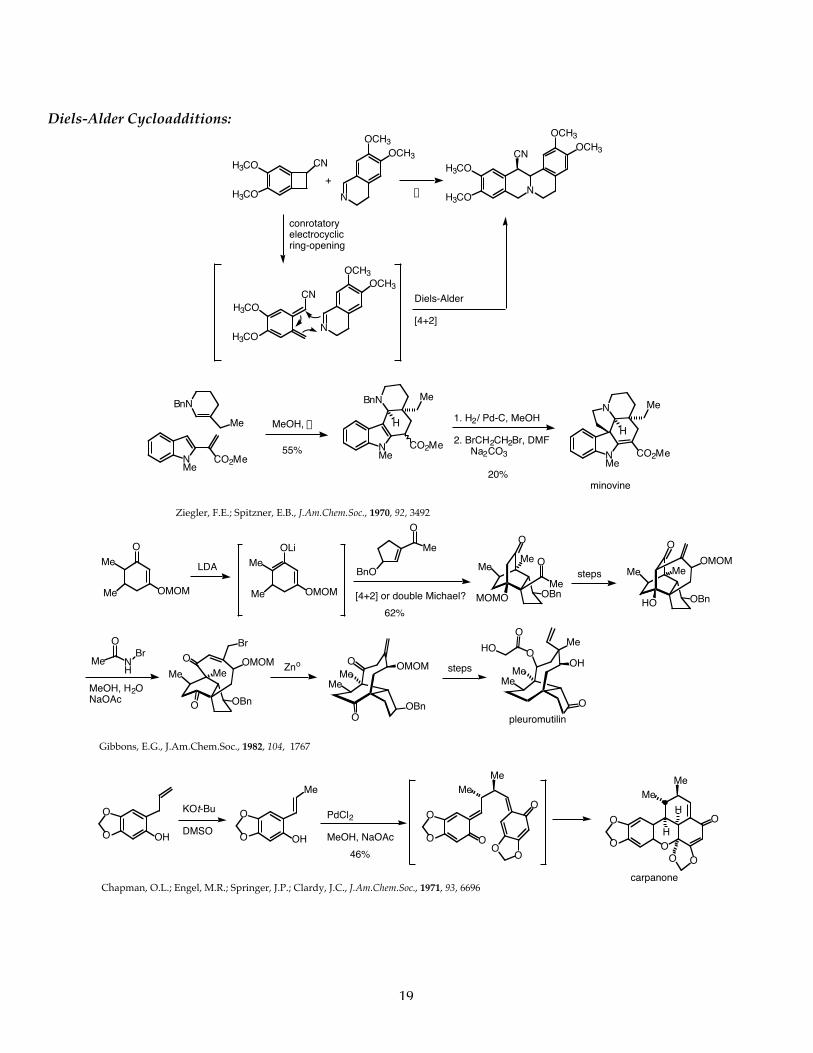
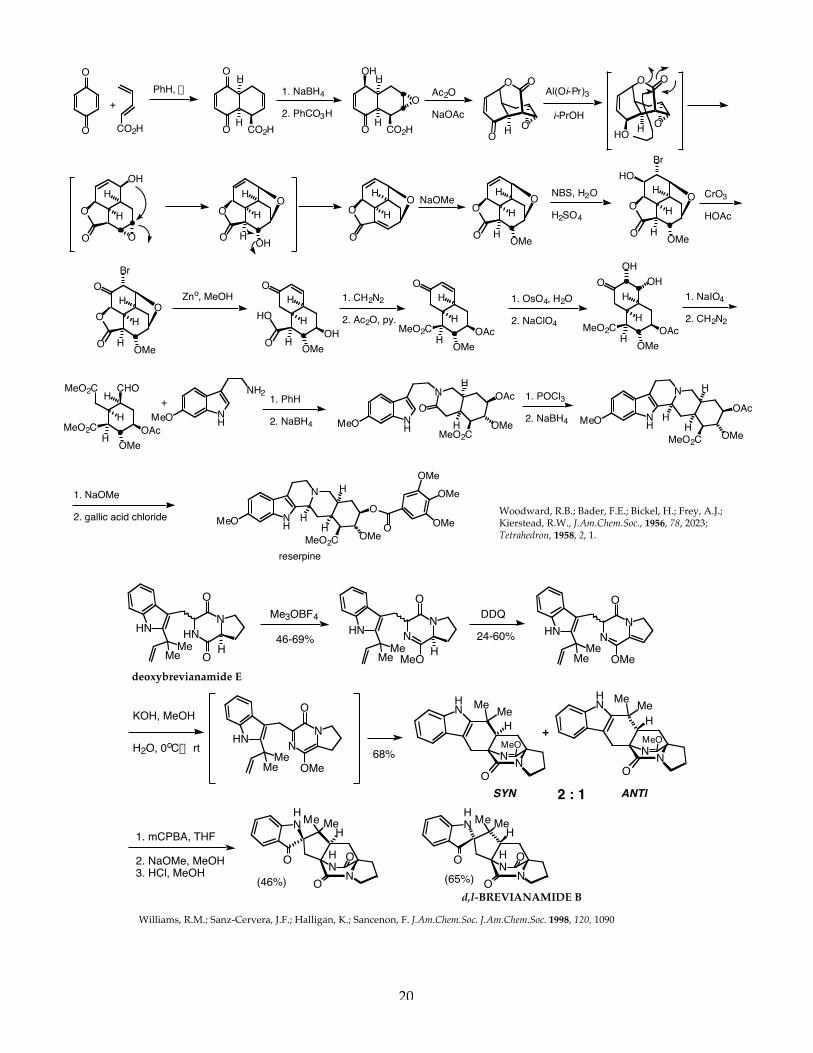
1 C549 Pericyclic Reactions Introduction Pericyclic reactions are a very special and important class of reactions that do not require the addition of any external chemical reagents. These reactions, called pericyclic reactions, only require the addition of energy in the form of heat or light to proceed. Chemists long ago recognized that certain molecules, when heated or exposed to light, would undergo chemical change. It wasn’t until the 1960’s, that a set of principles and mathematical theory to support these principles, were elucidated and published. By examining the orbitals that are interacting in the bond-breaking and bond-forming process, it is now possible to predict with a high degree of reliability and accuracy, the outcome of pericyclic reactions. The Conservation of Orbital Symmetry A theoretical basis for understanding pericyclic reactions was postulated in 1965 by Roald Hoffman of Cornell University and Robert B. Woodward of Harvard University. This theory, called the Conservation of Orbital Symmetry stated: “Orbital symmetry controls in an easily discernible manner the feasibility and stereochemical consequences of every concerted reaction”. This powerful theory relied on ..”the concepts of symmetry, overlap, interaction and the nodal structure of wave functions.” This theory was later elaborated by Fukui to introduce the concept of frontier molecular orbital theory. Basically, pericyclic reactions can be analyzed and rationalized by examining the nodal structure of the wave functions of the highest (energy) occupied (filled) molecular orbital (also abbreviated the HOMO) of one reactant and the lowest (energy) unoccupied molecular orbital (also abbreviated the LUMO) of the other reactant. The conservation of orbital symmetry theory predicted that the nodal symmetry of the interacting orbitals would be preserved throughout the reaction. Woodward and Hoffman state in one of their important early papers in 1968: “..it is possible to transform continuously the molecular orbitals of reactants into those of the product in such a way to preserve the bonding character of all occupied molecular orbitals at all stages of the reaction.” All three chemists, Woodward, Hoffman and Fukui won the Nobel Prize in Chemistry. Woodward won the Nobel prize in 1965 for the “art of complex molecule synthesis”, work that empirically contributed to developing the Woodward-Hoffman rules. Hoffman and Fukui shared the Nobel prize in 1981 for their work on frontier molecular orbital theory which was developed directly from the work on the Woodward-Hoffman rules for pericyclic reactions. Orbital Symmetry-Controlled Reactions have the following characteristics: 1. These reactions require only heat or light (no external reagents). 2. These reactions do NOT involve ionic intermediates or free radicals. 3. Bonds are made and broken simultaneously in a CYCLIC TRANSITION STATE. 4. These reactions are highly STEREOSPECIFIC. Electrocyclic Reactions: Molecular Orbitals of Conjugated Dienes Many orbital symmetry-controlled reactions involve the reactions of conjugated polyenes. Conjugated polyenes will contain either 4n or 4n+2 p-electrons (where n= a whole integer). For butadiene, the simplest conjugated diene, there are 4n p-electrons, where n=1. For ethylene, there are 4n+2 p-electrons where n=0. The p-molecular orbitals of these systems are comprised of atomic p-orbitals. According to the principle of the linear combination of atomic orbitals (LCAO) there must be the same number of p-molecular orbitals as there were atomic p-orbitals from which they (the p-system - a molecular system) were constructed. Similarly, for ethylene, there must be two p-molecular orbitals corresponding to the two atomic p-orbitals that were used to construct the p-bond. Butadiene has 4n p-electrons (where n=1). The molecular orbitals for butadiene and ethylene are shown below.
Transcript
1
C549Pericyclic Reactions
Introduction
Pericyclic reactions are a very special and important class of reactions that do not require the additionof any external chemical reagents. These reactions, called pericyclic reactions, only require the addition ofenergy in the form of heat or light to proceed. Chemists long ago recognized that certain molecules, whenheated or exposed to light, would undergo chemical change. It wasn’t until the 1960’s, that a set of principlesand mathematical theory to support these principles, were elucidated and published. By examining theorbitals that are interacting in the bond-breaking and bond-forming process, it is now possible to predict witha high degree of reliability and accuracy, the outcome of pericyclic reactions.
The Conservation of Orbital Symmetry
A theoretical basis for understanding pericyclic reactions was postulated in 1965 by Roald Hoffman ofCornell University and Robert B. Woodward of Harvard University. This theory, called the Conservation ofOrbital Symmetry stated: “Orbital symmetry controls in an easily discernible manner the feasibility andstereochemical consequences of every concerted reaction”. This powerful theory relied on ..”the concepts ofsymmetry, overlap, interaction and the nodal structure of wave functions.” This theory was later elaborated byFukui to introduce the concept of frontier molecular orbital theory. Basically, pericyclic reactions can beanalyzed and rationalized by examining the nodal structure of the wave functions of the highest (energy)occupied (filled) molecular orbital (also abbreviated the HOMO) of one reactant and the lowest (energy)unoccupied molecular orbital (also abbreviated the LUMO) of the other reactant. The conservation of orbitalsymmetry theory predicted that the nodal symmetry of the interacting orbitals would be preserved throughoutthe reaction.
Woodward and Hoffman state in one of their important early papers in 1968: “..it is possible to transformcontinuously the molecular orbitals of reactants into those of the product in such a way to preserve the bonding characterof all occupied molecular orbitals at all stages of the reaction.”
All three chemists, Woodward, Hoffman and Fukui won the Nobel Prize in Chemistry. Woodwardwon the Nobel prize in 1965 for the “art of complex molecule synthesis”, work that empirically contributed todeveloping the Woodward-Hoffman rules. Hoffman and Fukui shared the Nobel prize in 1981 for their workon frontier molecular orbital theory which was developed directly from the work on the Woodward-Hoffmanrules for pericyclic reactions.Orbital Symmetry-Controlled Reactions have the following characteristics:
1. These reactions require only heat or light (no external reagents).2. These reactions do NOT involve ionic intermediates or free radicals.3. Bonds are made and broken simultaneously in a CYCLIC TRANSITION STATE.4. These reactions are highly STEREOSPECIFIC.
Electrocyclic Reactions: Molecular Orbitals of Conjugated Dienes
Many orbital symmetry-controlled reactions involve the reactions of conjugated polyenes. Conjugatedpolyenes will contain either 4n or 4n+2 p-electrons (where n= a whole integer). For butadiene, the simplestconjugated diene, there are 4n p-electrons, where n=1. For ethylene, there are 4n+2 p-electrons where n=0.The p-molecular orbitals of these systems are comprised of atomic p-orbitals. According to the principle of thelinear combination of atomic orbitals (LCAO) there must be the same number of p-molecular orbitals as therewere atomic p-orbitals from which they (the p-system - a molecular system) were constructed. Similarly, forethylene, there must be two p-molecular orbitals corresponding to the two atomic p-orbitals that were used toconstruct the p-bond. Butadiene has 4n p-electrons (where n=1). The molecular orbitals for butadiene andethylene are shown below.
2
Antibonding
Bonding
LUMO *
LOMO
**
*
HUMO
LUMO*
*
*
HOMO
LOMO
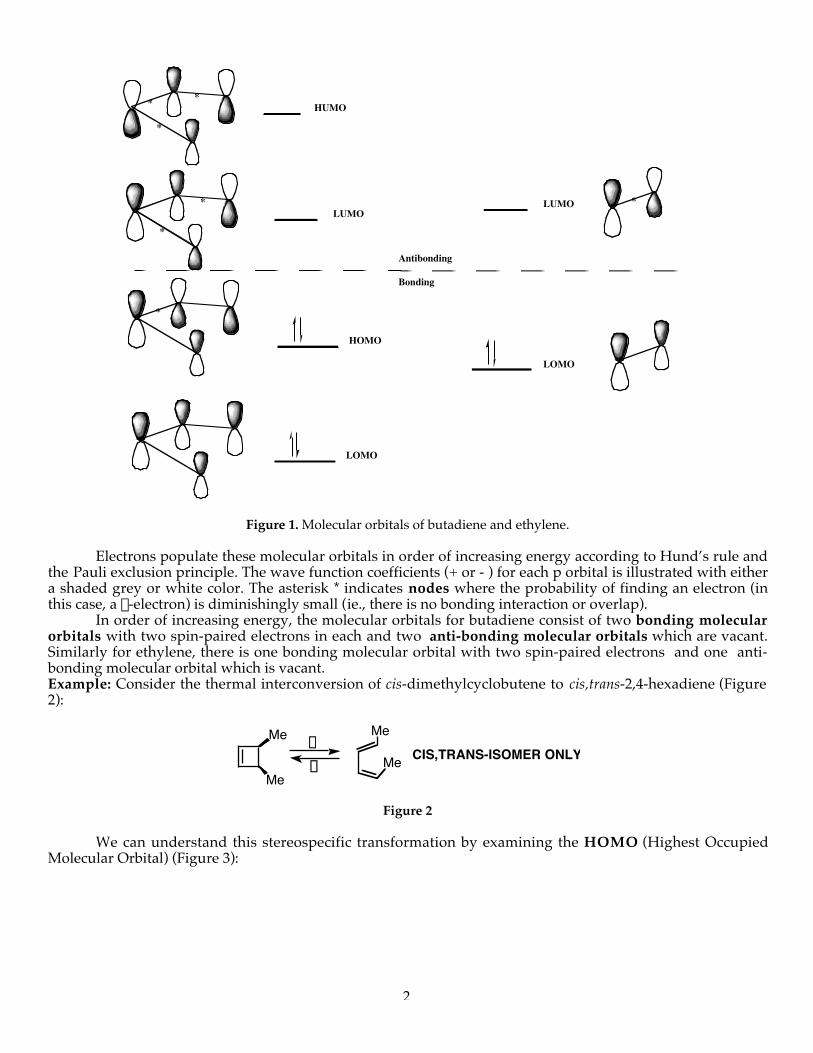
Figure 1. Molecular orbitals of butadiene and ethylene.
Electrons populate these molecular orbitals in order of increasing energy according to Hund’s rule andthe Pauli exclusion principle. The wave function coefficients (+ or - ) for each p orbital is illustrated with eithera shaded grey or white color. The asterisk * indicates nodes where the probability of finding an electron (inthis case, a p-electron) is diminishingly small (ie., there is no bonding interaction or overlap).
In order of increasing energy, the molecular orbitals for butadiene consist of two bonding molecularorbitals with two spin-paired electrons in each and two anti-bonding molecular orbitals which are vacant.Similarly for ethylene, there is one bonding molecular orbital with two spin-paired electrons and one anti-bonding molecular orbital which is vacant.Example: Consider the thermal interconversion of cis-dimethylcyclobutene to cis,trans-2,4-hexadiene (Figure2):
Me
Me
D
DCIS,TRANS-ISOMER ONLY
Me
Me
Figure 2
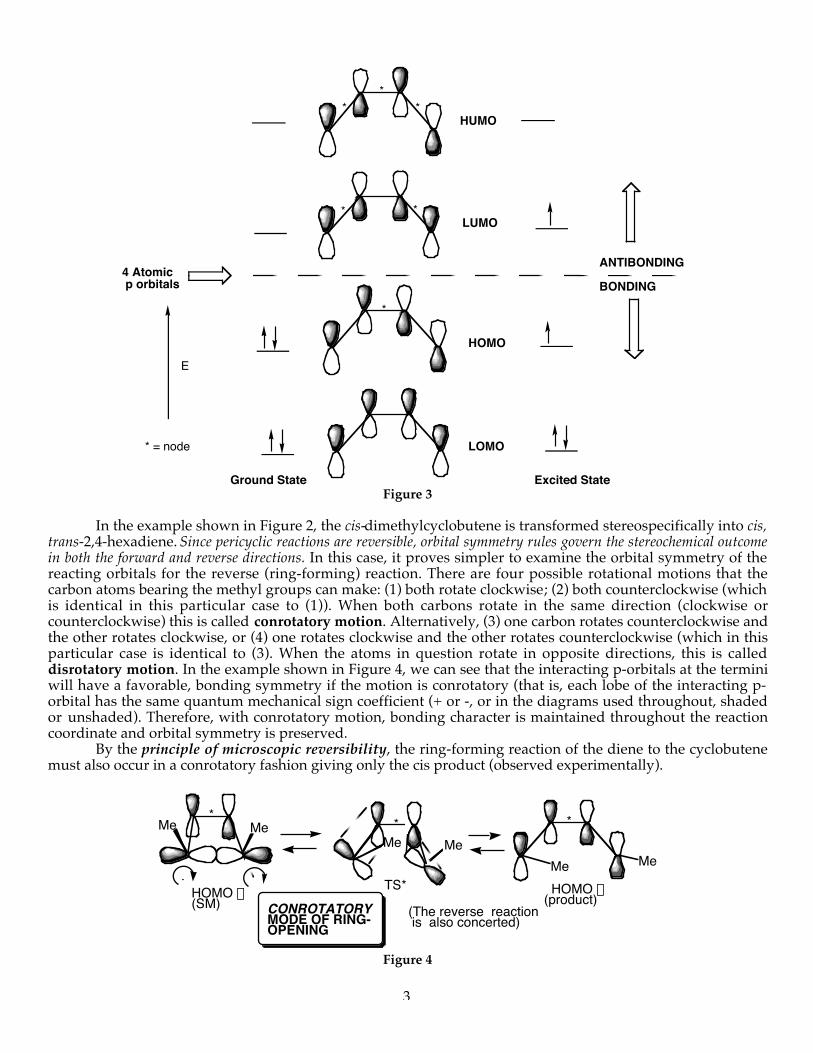
We can understand this stereospecific transformation by examining the HOMO (Highest OccupiedMolecular Orbital) (Figure 3):
3
ANTIBONDING
BONDING
Excited StateGround State
LOMO
HOMO
LUMO
HUMO*
**
* *
*
4 Atomic p orbitals
* = node
E
Figure 3
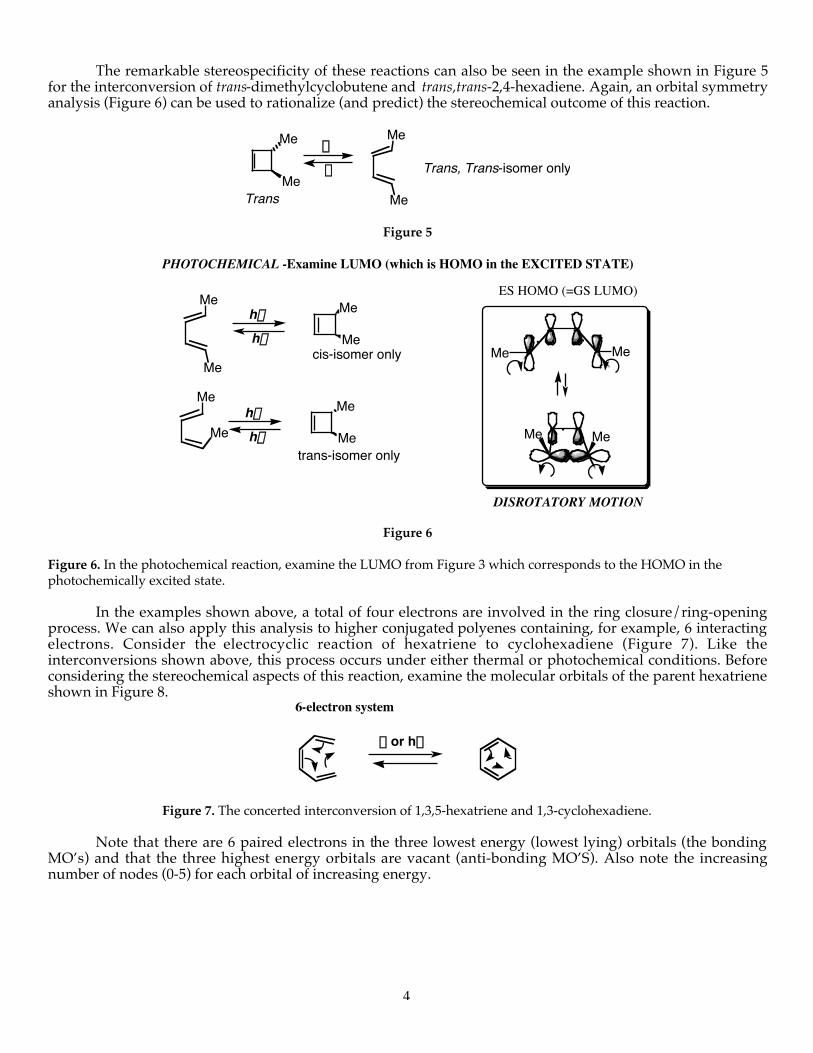
In the example shown in Figure 2, the cis-dimethylcyclobutene is transformed stereospecifically into cis,trans-2,4-hexadiene. Since pericyclic reactions are reversible, orbital symmetry rules govern the stereochemical outcomein both the forward and reverse directions. In this case, it proves simpler to examine the orbital symmetry of thereacting orbitals for the reverse (ring-forming) reaction. There are four possible rotational motions that thecarbon atoms bearing the methyl groups can make: (1) both rotate clockwise; (2) both counterclockwise (whichis identical in this particular case to (1)). When both carbons rotate in the same direction (clockwise orcounterclockwise) this is called conrotatory motion. Alternatively, (3) one carbon rotates counterclockwise andthe other rotates clockwise, or (4) one rotates clockwise and the other rotates counterclockwise (which in thisparticular case is identical to (3). When the atoms in question rotate in opposite directions, this is calleddisrotatory motion. In the example shown in Figure 4, we can see that the interacting p-orbitals at the terminiwill have a favorable, bonding symmetry if the motion is conrotatory (that is, each lobe of the interacting p-orbital has the same quantum mechanical sign coefficient (+ or -, or in the diagrams used throughout, shadedor unshaded). Therefore, with conrotatory motion, bonding character is maintained throughout the reactioncoordinate and orbital symmetry is preserved.
By the principle of microscopic reversibility, the ring-forming reaction of the diene to the cyclobutenemust also occur in a conrotatory fashion giving only the cis product (observed experimentally).
Me Me
Me MeMe Me
CONROTATORYMODE OF RING-OPENING
(The reverse reaction is also concerted)
HOMO p(product)
TS*HOMO s(SM)
* * *
Figure 4
4
The remarkable stereospecificity of these reactions can also be seen in the example shown in Figure 5for the interconversion of trans-dimethylcyclobutene and trans,trans-2,4-hexadiene. Again, an orbital symmetryanalysis (Figure 6) can be used to rationalize (and predict) the stereochemical outcome of this reaction.
Me
Me
Trans, Trans-isomer only
Trans
D
D
Me
Me
Figure 5
Me
Me
Me
Me
Me
MeMe
Me
MeMe
Me Me
ES HOMO (=GS LUMO)
DISROTATORY MOTION
.
. .
trans-isomer only
cis-isomer only
hn
hn
hn
hn
PHOTOCHEMICAL -Examine LUMO (which is HOMO in the EXCITED STATE)
Figure 6
Figure 6. In the photochemical reaction, examine the LUMO from Figure 3 which corresponds to the HOMO in thephotochemically excited state.
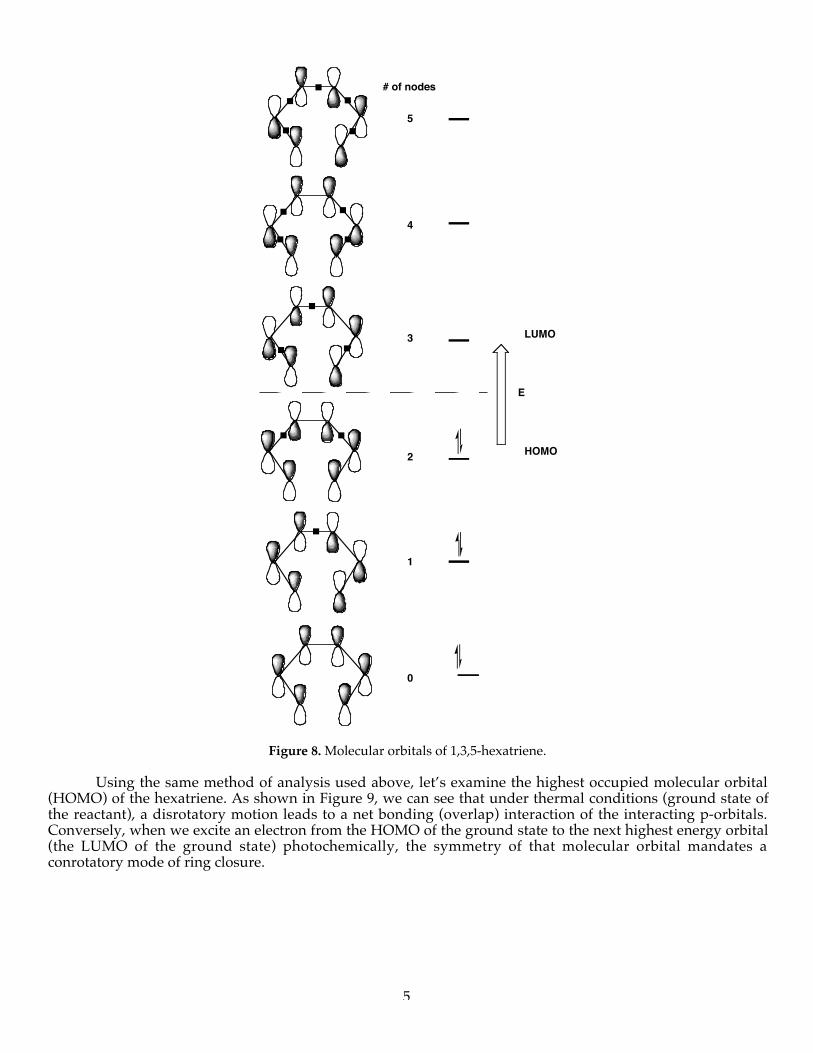
In the examples shown above, a total of four electrons are involved in the ring closure/ring-openingprocess. We can also apply this analysis to higher conjugated polyenes containing, for example, 6 interactingelectrons. Consider the electrocyclic reaction of hexatriene to cyclohexadiene (Figure 7). Like theinterconversions shown above, this process occurs under either thermal or photochemical conditions. Beforeconsidering the stereochemical aspects of this reaction, examine the molecular orbitals of the parent hexatrieneshown in Figure 8.
6-electron system
D or hn
Figure 7. The concerted interconversion of 1,3,5-hexatriene and 1,3-cyclohexadiene.
Note that there are 6 paired electrons in the three lowest energy (lowest lying) orbitals (the bondingMO’s) and that the three highest energy orbitals are vacant (anti-bonding MO’S). Also note the increasingnumber of nodes (0-5) for each orbital of increasing energy.
5
..
..
..
.
.. ...
.. .
5
4
3
2
1
0
E
# of nodes
LUMO
HOMO
Figure 8. Molecular orbitals of 1,3,5-hexatriene.
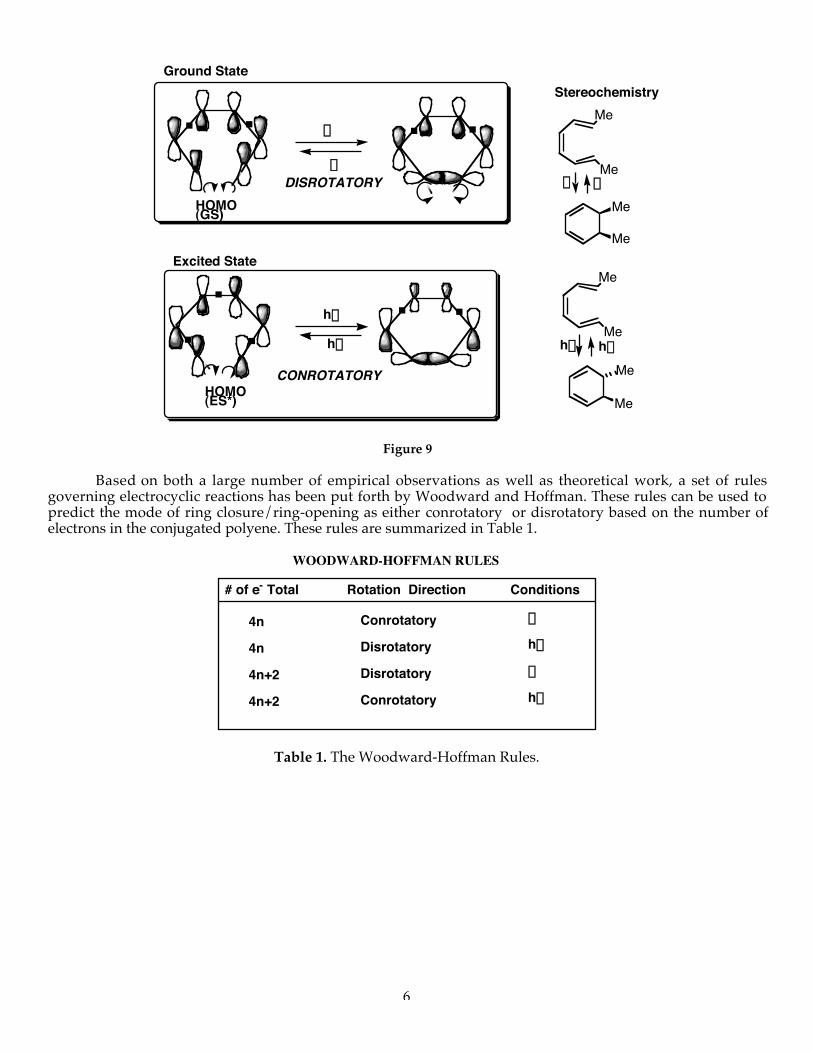
Using the same method of analysis used above, let’s examine the highest occupied molecular orbital(HOMO) of the hexatriene. As shown in Figure 9, we can see that under thermal conditions (ground state ofthe reactant), a disrotatory motion leads to a net bonding (overlap) interaction of the interacting p-orbitals.Conversely, when we excite an electron from the HOMO of the ground state to the next highest energy orbital(the LUMO of the ground state) photochemically, the symmetry of that molecular orbital mandates aconrotatory mode of ring closure.
6
Me
Me
Me
Me
..
hnhn
DD
Stereochemistry
hn...
HOMO(ES*)
hn .CONROTATORY
Excited State
Ground State
DISROTATORYD
D
HOMO(GS)
.
Me
Me
Me
Me
. .
Figure 9
Based on both a large number of empirical observations as well as theoretical work, a set of rulesgoverning electrocyclic reactions has been put forth by Woodward and Hoffman. These rules can be used topredict the mode of ring closure/ring-opening as either conrotatory or disrotatory based on the number ofelectrons in the conjugated polyene. These rules are summarized in Table 1.
D
hn
D
hn
Conrotatory
Disrotatory
Disrotatory
Conrotatory
4n
4n
4n+2
4n+2
# of e- Total Rotation Direction Conditions
WOODWARD-HOFFMAN RULES
Table 1. The Woodward-Hoffman Rules.
7
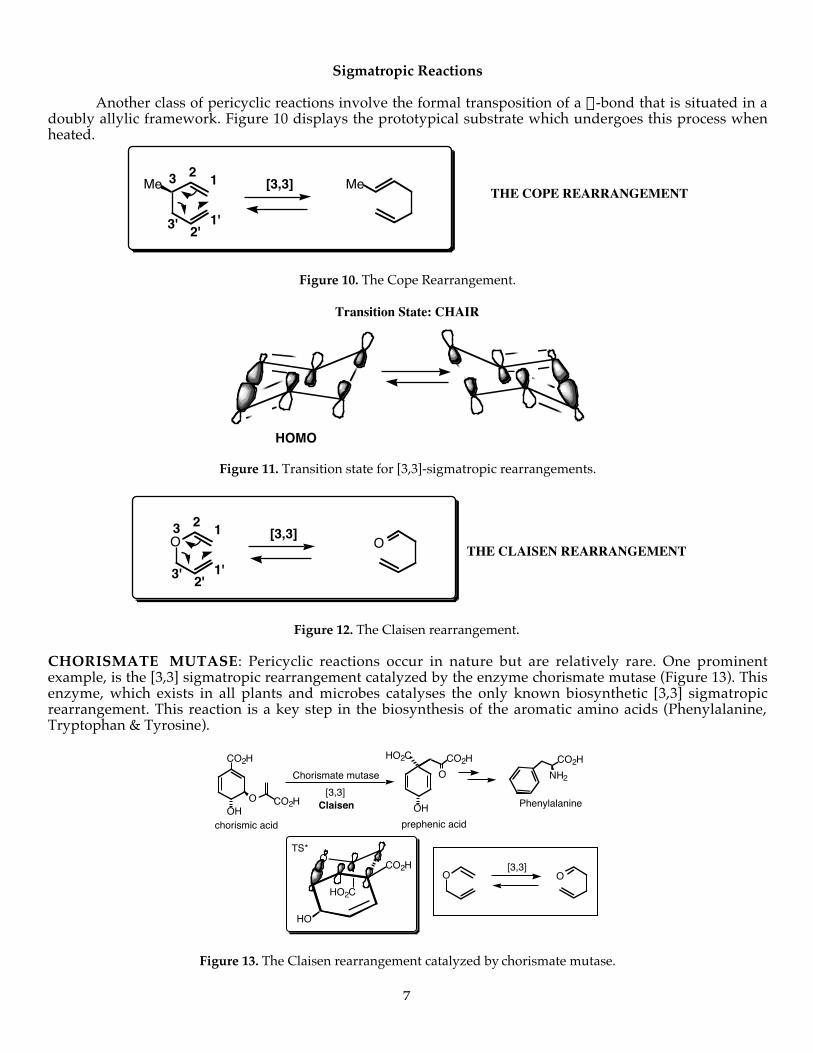
Sigmatropic Reactions
Another class of pericyclic reactions involve the formal transposition of a s-bond that is situated in adoubly allylic framework. Figure 10 displays the prototypical substrate which undergoes this process whenheated.
Me Me THE COPE REARRANGEMENT[3,3]
3' 2'1'
3 2 1
Figure 10. The Cope Rearrangement.
HOMO
Transition State: CHAIR
Figure 11. Transition state for [3,3]-sigmatropic rearrangements.
O O THE CLAISEN REARRANGEMENT[3,3]
3' 2'1'
3 2 1
Figure 12. The Claisen rearrangement.
CHORISMATE MUTASE: Pericyclic reactions occur in nature but are relatively rare. One prominentexample, is the [3,3] sigmatropic rearrangement catalyzed by the enzyme chorismate mutase (Figure 13). Thisenzyme, which exists in all plants and microbes catalyses the only known biosynthetic [3,3] sigmatropicrearrangement. This reaction is a key step in the biosynthesis of the aromatic amino acids (Phenylalanine,Tryptophan & Tyrosine).
O O[3,3]
OHO CO2H
CO2H HO2C CO2HO
OH
NH2
CO2H
O
HO
CO2H
Claisen Phenylalanine
Chorismate mutase[3,3]
HO2C
chorismic acid prephenic acid
TS*
Figure 13. The Claisen rearrangement catalyzed by chorismate mutase.
8
Examples of the Cope Rearrangement:
H3CThis is a degenerative Coperearrangement. (If the reactantwas optically active, the Copewould racemize this compound)
Cope[3,3]
H
H
H
H
H
H
CH3
Cope[3,3]
Figure 14
Degenerative Cope Rearrangements: In situations where the starting material and product are identical (DGo= 0), rapid equilibration between identical structures is often observed. For example, in bullvalene, all CHgroups are equivalent by 1H nmr at 120 oC.
Figure 15. The degenerative Cope rearrangement of bullvalene.
The Diels-Alder Cycloaddition Reaction
By far, the most important use of conjugated dienes are in condensation reactions with otherunsaturated compounds forming six-membered rings. This reaction, discovered in 1928 by Otto Diels andKurt Alder, established that conjugated dienes react with certain unsaturated compounds (called“dienophiles”) with only the application of thermal energy (heat); no other reagents, acids, bases, reducing oroxidizing agents were required. The general reaction is shown below and proceeds through a concerted six-membered ring transition state. This reaction forms two C-C s-bonds simultaneously and concomitantly formsa single new C=C p-bond.
D
Figure 16. The Diels-Alder Cycloaddition Reaction.
The transition state for the Diels-Alder reaction is highly ordered with a “boat-like” geometry. Amolecular orbital picture, shown below (Figure 17), illustrates the interactions. The reaction is a concerted,orbital symmetry-controlled process meaning, that there is a smooth and continuous transformation of theinteracting orbitals in the starting materials to those in the product. There are no discreet chargedintermediates and no exogenous reagents are required (other than heat).
D
=
"DIENOPHILE"
"DIENE"
Figure 17. Interacting molecular orbitals of the Diels-Alder Cycloaddition.
9
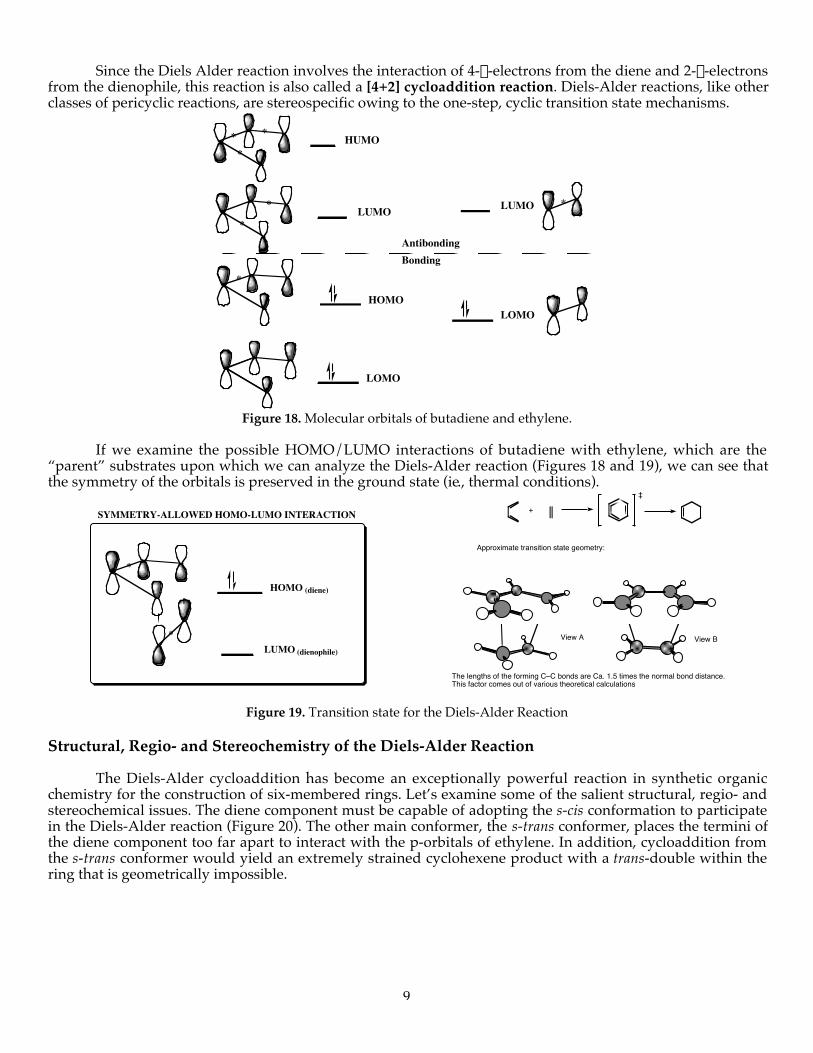
Since the Diels Alder reaction involves the interaction of 4-p-electrons from the diene and 2-p-electronsfrom the dienophile, this reaction is also called a [4+2] cycloaddition reaction. Diels-Alder reactions, like otherclasses of pericyclic reactions, are stereospecific owing to the one-step, cyclic transition state mechanisms.
AntibondingBonding
LUMO *
LOMO
***
HUMO
LUMO*
*
*
HOMO
LOMO
Figure 18. Molecular orbitals of butadiene and ethylene.
If we examine the possible HOMO/LUMO interactions of butadiene with ethylene, which are the“parent” substrates upon which we can analyze the Diels-Alder reaction (Figures 18 and 19), we can see thatthe symmetry of the orbitals is preserved in the ground state (ie., thermal conditions).
LUMO (dienophile)
*
*
HOMO (diene)
SYMMETRY-ALLOWED HOMO-LUMO INTERACTION
The lengths of the forming C–C bonds are Ca. 1.5 times the normal bond distance. This factor comes out of various theoretical calculations
View BView A
Approximate transition state geometry:
+
‡
Figure 19. Transition state for the Diels-Alder Reaction
Structural, Regio- and Stereochemistry of the Diels-Alder Reaction
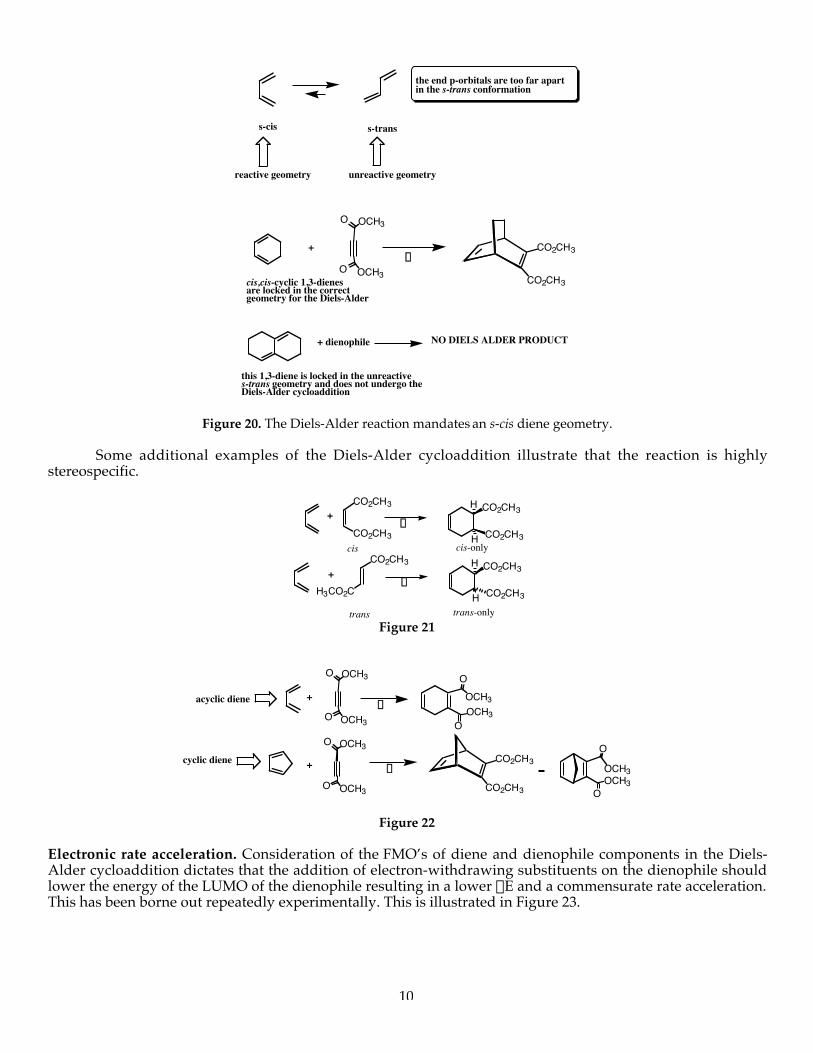
The Diels-Alder cycloaddition has become an exceptionally powerful reaction in synthetic organicchemistry for the construction of six-membered rings. Let’s examine some of the salient structural, regio- andstereochemical issues. The diene component must be capable of adopting the s-cis conformation to participatein the Diels-Alder reaction (Figure 20). The other main conformer, the s-trans conformer, places the termini ofthe diene component too far apart to interact with the p-orbitals of ethylene. In addition, cycloaddition fromthe s-trans conformer would yield an extremely strained cyclohexene product with a trans-double within thering that is geometrically impossible.
10
unreactive geometryreactive geometry
the end p-orbitals are too far apartin the s-trans conformation
s-transs-cis
O OCH3
O OCH3
CO2CH3
CO2CH3
NO DIELS ALDER PRODUCT+ dienophile
this 1,3-diene is locked in the unreactive s-trans geometry and does not undergo theDiels-Alder cycloaddition
D+
cis,cis-cyclic 1,3-dienesare locked in the correctgeometry for the Diels-Alder
Figure 20. The Diels-Alder reaction mandates an s-cis diene geometry.
Some additional examples of the Diels-Alder cycloaddition illustrate that the reaction is highlystereospecific.
CO2CH3
CO2CH3 CO2CH3
CO2CH3
H
H
H3CO2C
CO2CH3 CO2CH3
CO2CH3
H
H
+D
+D
cis cis-only
trans trans-onlyFigure 21
O OCH3
O OCH3
OOCH3OCH3
O
OCH3O
OCH3OCO2CH3
CO2CH3
O
OCH3OCH3
O
D+cyclic diene
acyclic diene D+
Figure 22
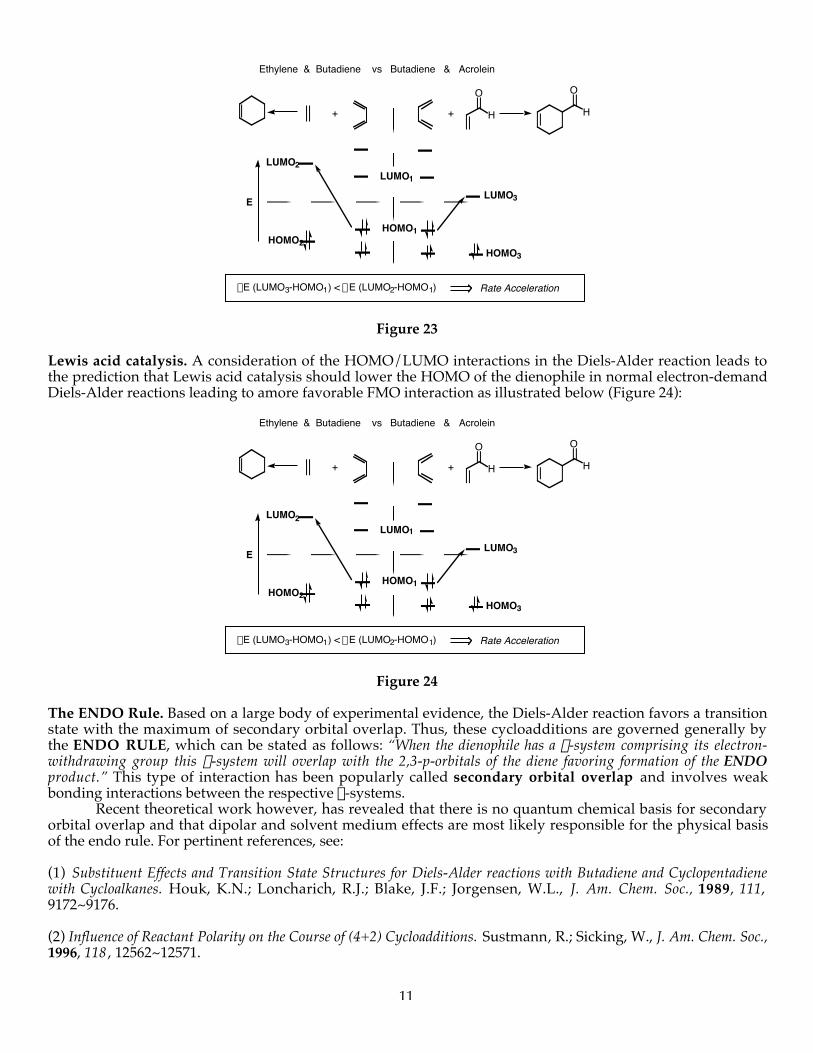
Electronic rate acceleration. Consideration of the FMO’s of diene and dienophile components in the Diels-Alder cycloaddition dictates that the addition of electron-withdrawing substituents on the dienophile shouldlower the energy of the LUMO of the dienophile resulting in a lower DE and a commensurate rate acceleration.This has been borne out repeatedly experimentally. This is illustrated in Figure 23.
11
Ethylene & Butadiene vs Butadiene & Acrolein
Rate AccelerationDE (LUMO3-HOMO1) < DE (LUMO2-HOMO1)
+
LUMO1
HOMO1
+
E
HOMO3
LUMO3
HOMO2
LUMO2
H
O O
H
Figure 23
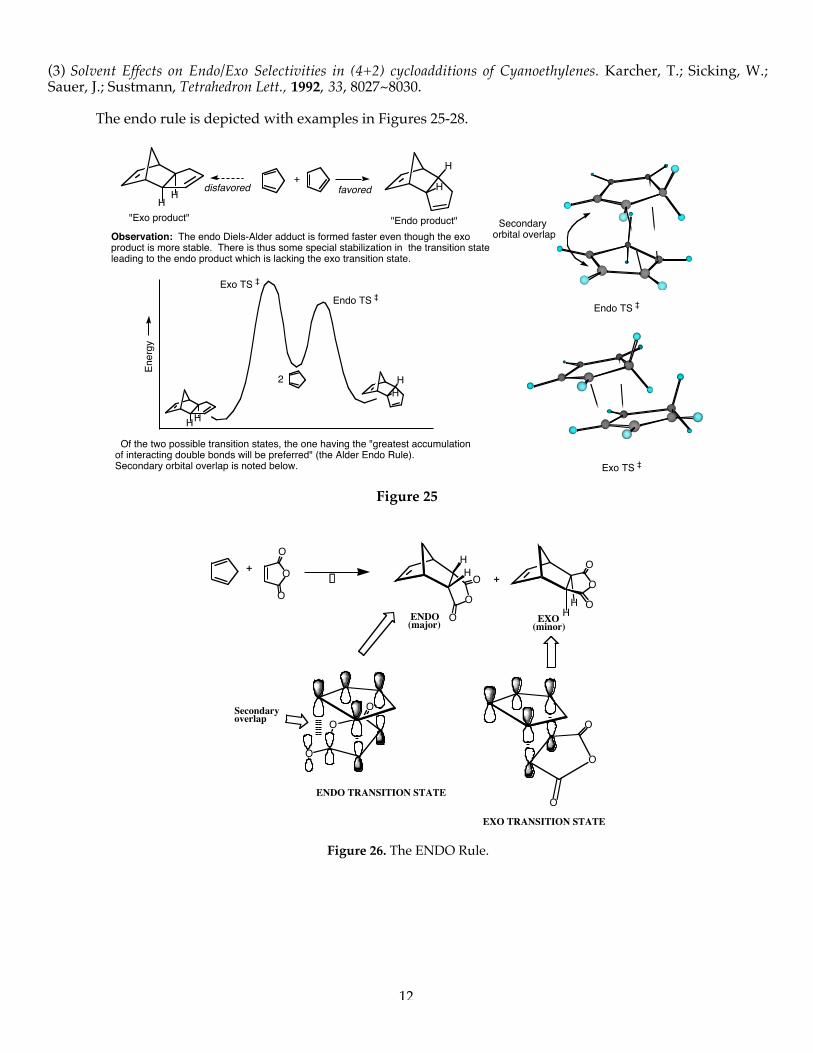
Lewis acid catalysis. A consideration of the HOMO/LUMO interactions in the Diels-Alder reaction leads tothe prediction that Lewis acid catalysis should lower the HOMO of the dienophile in normal electron-demandDiels-Alder reactions leading to amore favorable FMO interaction as illustrated below (Figure 24):
Ethylene & Butadiene vs Butadiene & Acrolein
Rate AccelerationDE (LUMO3-HOMO1) < DE (LUMO2-HOMO1)
+
LUMO1
HOMO1
+
E
HOMO3
LUMO3
HOMO2
LUMO2
H
O O
H
Figure 24
The ENDO Rule. Based on a large body of experimental evidence, the Diels-Alder reaction favors a transitionstate with the maximum of secondary orbital overlap. Thus, these cycloadditions are governed generally bythe ENDO RULE, which can be stated as follows: “When the dienophile has a p-system comprising its electron-withdrawing group this p-system will overlap with the 2,3-p-orbitals of the diene favoring formation of the ENDOproduct.” This type of interaction has been popularly called secondary orbital overlap and involves weakbonding interactions between the respective p-systems.
Recent theoretical work however, has revealed that there is no quantum chemical basis for secondaryorbital overlap and that dipolar and solvent medium effects are most likely responsible for the physical basisof the endo rule. For pertinent references, see:
(1) Substituent Effects and Transition State Structures for Diels-Alder reactions with Butadiene and Cyclopentadienewith Cycloalkanes. Houk, K.N.; Loncharich, R.J.; Blake, J.F.; Jorgensen, W.L., J. Am. Chem. Soc., 1989, 111,9172~9176.
(2) Influence of Reactant Polarity on the Course of (4+2) Cycloadditions. Sustmann, R.; Sicking, W., J. Am. Chem. Soc.,1996, 118 , 12562~12571.
12
(3) Solvent Effects on Endo/Exo Selectivities in (4+2) cycloadditions of Cyanoethylenes. Karcher, T.; Sicking, W.;Sauer, J.; Sustmann, Tetrahedron Lett., 1992, 33, 8027~8030.
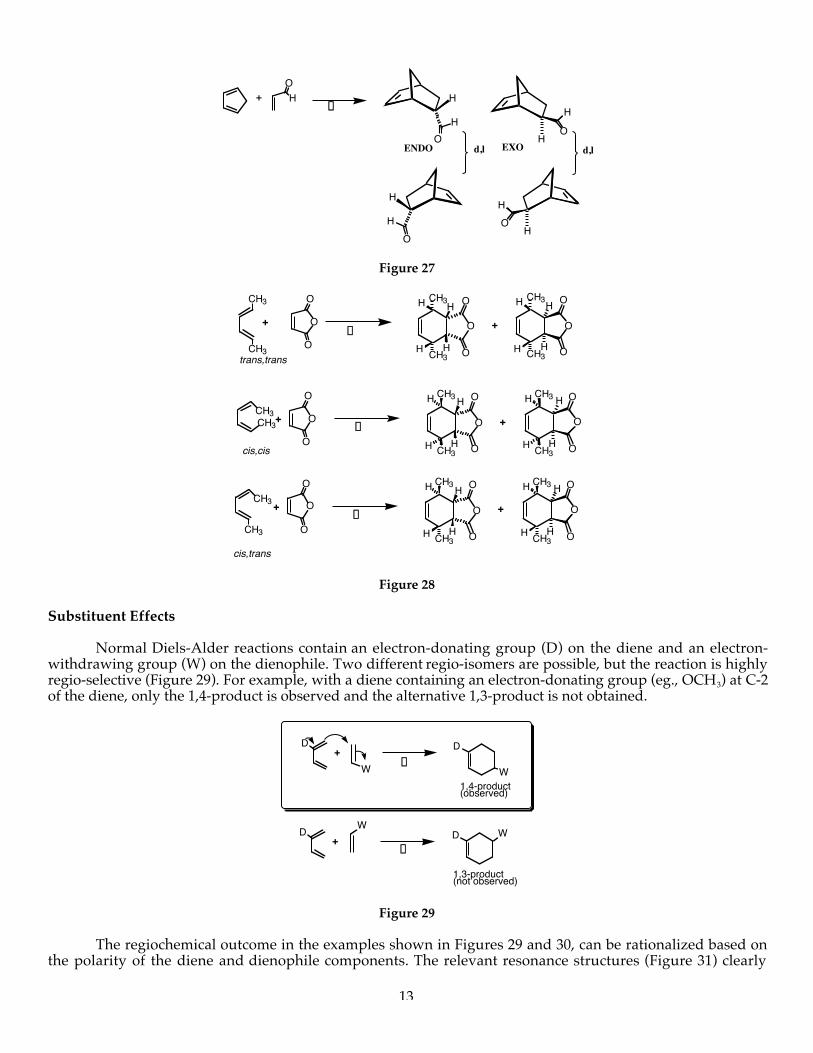
The endo rule is depicted with examples in Figures 25-28.
Ener
gy
disfavored favored+
"Endo product""Exo product"
Observation: The endo Diels-Alder adduct is formed faster even though the exo product is more stable. There is thus some special stabilization in the transition state leading to the endo product which is lacking the exo transition state.
2
Exo TS ‡
Endo TS ‡
Of the two possible transition states, the one having the "greatest accumulation of interacting double bonds will be preferred" (the Alder Endo Rule). Secondary orbital overlap is noted below.
H
HH
H
HH
HH
Secondary orbital overlap
Endo TS ‡
Exo TS ‡
Figure 25
O
O
O O
H
O
OHO
OH
HO
O
O
O
O
O
O
+D
ENDO(major) EXO
(minor)
+
Secondary overlap
ENDO TRANSITION STATE
EXO TRANSITION STATE
Figure 26. The ENDO Rule.
13
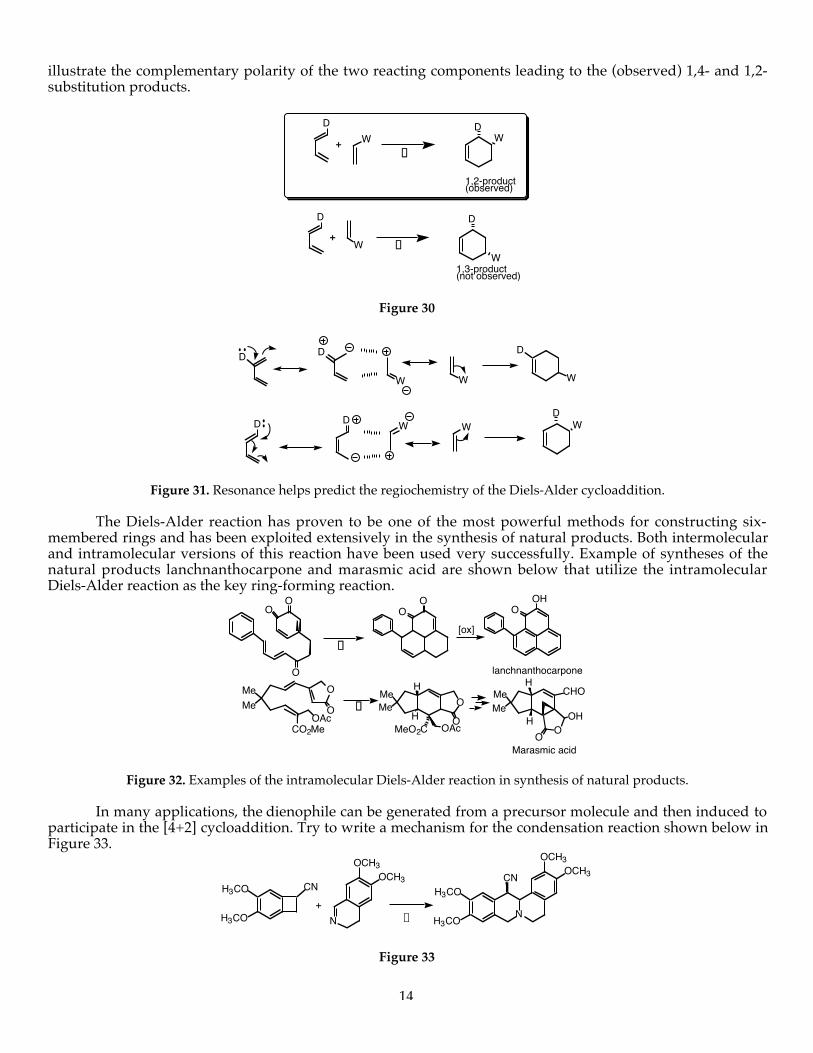
OH H
OH
H
HO
HO
H
OH
H
+D
ENDO EXO d,ld,l
Figure 27
O
O
O H
H
O
O
O
CH3
CH3
CH3
CH3H
H H
H CH3
CH3
O
O
O
H
H
H
CH3H
O
O
O
CH3
CH3H
HH
H CH3
CH3
O
O
O
H
HO
O
O
O
O
O H
H
O
O
O
CH3
CH3H
H H
H CH3
CH3
O
O
O
HCH3
HCH3
CH3
+D +
+D+
+D +
cis,cis
trans,trans
cis,trans
Figure 28
Substituent Effects
Normal Diels-Alder reactions contain an electron-donating group (D) on the diene and an electron-withdrawing group (W) on the dienophile. Two different regio-isomers are possible, but the reaction is highlyregio-selective (Figure 29). For example, with a diene containing an electron-donating group (eg., OCH3) at C-2of the diene, only the 1,4-product is observed and the alternative 1,3-product is not obtained.
D
W
D
W
WDD W
+D
1,4-product(observed)
1,3-product(not observed)
D+
Figure 29
The regiochemical outcome in the examples shown in Figures 29 and 30, can be rationalized based onthe polarity of the diene and dienophile components. The relevant resonance structures (Figure 31) clearly
14
illustrate the complementary polarity of the two reacting components leading to the (observed) 1,4- and 1,2-substitution products.
DW W
D
W
D
D
W
+D
1,2-product(observed)
1,3-product(not observed)
D+
Figure 30
D D WW
WW
D D
WD
D
W
Figure 31. Resonance helps predict the regiochemistry of the Diels-Alder cycloaddition.
The Diels-Alder reaction has proven to be one of the most powerful methods for constructing six-membered rings and has been exploited extensively in the synthesis of natural products. Both intermolecularand intramolecular versions of this reaction have been used very successfully. Example of syntheses of thenatural products lanchnanthocarpone and marasmic acid are shown below that utilize the intramolecularDiels-Alder reaction as the key ring-forming reaction.
OO
O
OO O
OH
lanchnanthocarpone
[ox]D
MeMe
CO2MeOAc
O
OO
MeMe
OMeO2C OAc
H
H H
H
O O
MeMe CHO
OH
Marasmic acid
D
Figure 32. Examples of the intramolecular Diels-Alder reaction in synthesis of natural products.
In many applications, the dienophile can be generated from a precursor molecule and then induced toparticipate in the [4+2] cycloaddition. Try to write a mechanism for the condensation reaction shown below inFigure 33.
H3CO
H3CO
CN
N
OCH3OCH3
+H3CO
H3CO N
OCH3OCH3CN
D
Figure 33
15
[2+2] Photocycloadditions: Cyclobutane Synthesis
The Diels-Alder reaction was an example of a [4n+2] p-electron cycloaddition reaction (where n=1; witha total of 6-p-electrons interacting) that was symmetry allowed thermally. Other types of p-systems with 4n+2p-electrons also undergo symmetry-allowed cycloaddition reactions. Experimentally, it has been known for along time that, ethylene derivatives do not undergo cycloadditions thermally but do undergo cycloadditionsphotochemically. This is known as a photochemical [2+2] cycloaddition reaction and provides a very usefulmethod to make cyclobutane derivatives (Figure 34).
CH2CH2
CH2CH2
+ hn
Figure 34. The photo [2+2] cycloaddition.
We can rationalize why the [2+2] cycloaddition reaction is forbidden thermally by applying orbitalsymmetry rules. The p-molecular orbitals for ethylene in the ground state are shown in Figure 35. Using theanalysis we used above for the [4+2] cycloaddition reaction, the highest occupied molecular orbital (HOMO) ofone ethylene will interact with the lowest unoccupied molecular orbital (LUMO) of another ethylenemolecule. In order to maintain a bonding character of all the orbitals throughout the reaction (ie., conserveorbital symmetry), there should be no nodes between interacting orbitals. As you can see from the figurebelow, the HOMO-LUMO interaction of ground state ethylene is symmetry forbidden due to the appearanceof a node between two of the interacting p-orbitals.
GroundState
AntibondingBonding
HOMO
LUMO*
Figure 35. p-Molecular orbitals of ethylene.
We can the symmetry-forbidden interaction in the ground state-ground state interactions in Figure 36.
LUMO (GS)
HOMO (GS)
Antibondinginteraction(node)
Symmetry FORBIDDEN
Figure 36
We can promote an electron from the lowest energy p-molecular orbital of ethylene to the next highest-energy orbital photochemically generating the excited state of ethylene. As shown in Figure 37, the HOMO ofethylene in the excited state corresponds to the LUMO of ethylene in the ground state.
16
GroundState
ExcitedState
AntibondingBonding
HOMO
LUMO
hn
HOMO
LOMO
* *
Figure 37
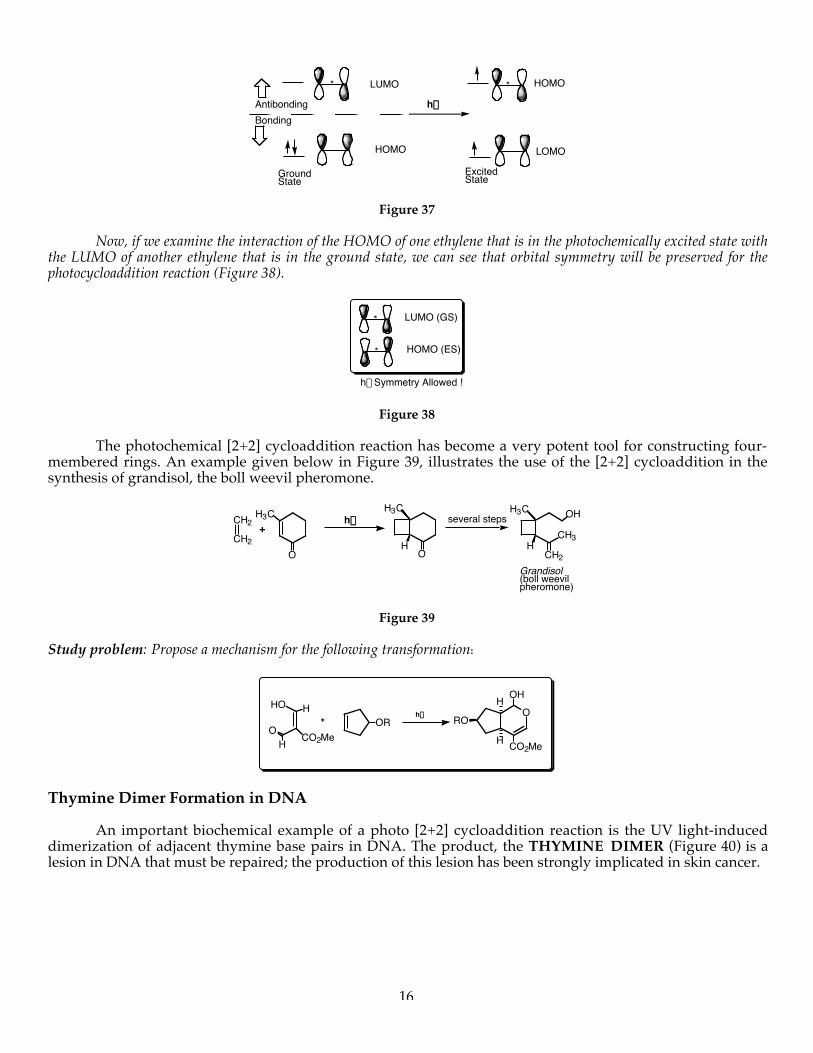
Now, if we examine the interaction of the HOMO of one ethylene that is in the photochemically excited state withthe LUMO of another ethylene that is in the ground state, we can see that orbital symmetry will be preserved for thephotocycloaddition reaction (Figure 38).
LUMO (GS)
HOMO (ES)
hn Symmetry Allowed !
*
*
Figure 38
The photochemical [2+2] cycloaddition reaction has become a very potent tool for constructing four-membered rings. An example given below in Figure 39, illustrates the use of the [2+2] cycloaddition in thesynthesis of grandisol, the boll weevil pheromone.
H3C
O
CH2
CH2O
H3C
H HCH3
OHH3C
CH2
+hn several steps
Grandisol(boll weevil pheromone)
Figure 39
Study problem: Propose a mechanism for the following transformation:
ORHO
CO2MeOH
HRO
O
CO2Me
OHH
H
+ hn
Thymine Dimer Formation in DNA
An important biochemical example of a photo [2+2] cycloaddition reaction is the UV light-induceddimerization of adjacent thymine base pairs in DNA. The product, the THYMINE DIMER (Figure 40) is alesion in DNA that must be repaired; the production of this lesion has been strongly implicated in skin cancer.
17
NRib
N-HO
H
OH3C O
H
O
N
N-H
RibH3C
N
Rib
NRib
N-HO
HO
H3C
H3CO
H
O N-HThe Thymine Dimer
hn
Figure 40. Photo [2+2] formation of the thymine dimer results in damage to DNA.
UV irradiation has been known for a long time as a potent means of damaging cellular DNA and othervital compounds. The mechanism for UV-induced DNA damage is fairly well understood. DNA absorbs lightin the ultraviolet portion of the electromagnetic spectrum. Thymine bases situated directly adjacent on a strandof DNA are p-stacked in the double helix (see Figure 41) and align the p-systems of the heterocyclic thymidinerings proximally. Upon absorption of ultraviolet irradiation, the thymine p-system is excited to a higher energyexcited state and undergoes a subsequent symmetry-allowed [2+2] photocycloaddition reaction resulting inthe cyclobutane photoadduct (shown). The thymine dimer can cause lesions in DNA (strand breaks orreplication mismatches) unless repaired. There are two main mechanisms for repairing such lesions in DNA.An EXINUCLEASE enzyme can detect the presence of the thymine dimer and cut out a twelve base residueincluding the thymine dimer. The gapped DNA is then refilled with DNA POLYMERASE and ligated backtogether with DNA LIGASE. Another repair mechanism involves DNA PHOTOLYASE. This enzyme effectsthe photosensitized symmetry-allowed [2+2] cycloreversion of the thymine dimer back to the original T-Tsequence. Extensive UV light irradiation can, however, damage the genes responsible for directing thesynthesis of these repair enzymes. UV-induced DNA damage (ie., thymine dimer formation) is one of theprimary mechanisms responsible for skin cancer.
N-HN
O
O
NN
NN
H-N
O
H3C H
O
OP O-O
O
O
OP O-O
O
O
OP
OPO O-
-OO
O
O
OPO
N-HN
O
NN
NN
H-N
O
H3C H
N-HN
O
O
N
O-O
N
NN
H-N
H
H
O
H
O
OP
O-O
O
O
OP O-O
ON-H
N
ON
N
NN
H-N
O
H3C H
O
OP
OP
OO-
-OO
O
O
OPO O-O
H3C
hnPhotochemical [2+2]cycloaddition
Figure 41. Photochemical formation of the thymine dimer in duplex DNA.
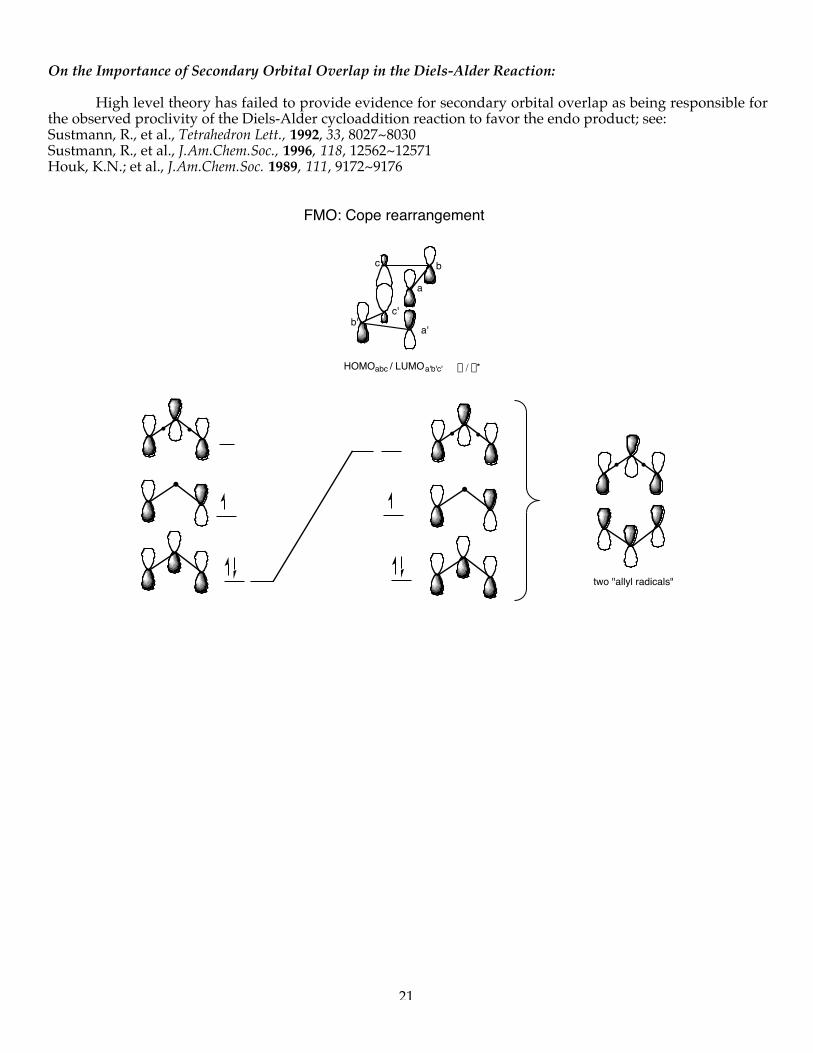
On the Importance of Secondary Orbital Overlap in the Diels-Alder Reaction:
High level theory has failed to provide evidence for secondary orbital overlap as being responsible forthe observed proclivity of the Diels-Alder cycloaddition reaction to favor the endo product; see:Sustmann, R., et al., Tetrahedron Lett., 1992, 33, 8027~8030Sustmann, R., et al., J.Am.Chem.Soc., 1996, 118, 12562~12571Houk, K.N.; et al., J.Am.Chem.Soc. 1989, 111, 9172~9176