Calcium and phosphate compatibility: Revisited again
DaviD W. NeWtoN aND DaviD F. Driscoll
Am J Health-Syst Pharm. 2008; 65:73-80
DaviD W. NeWtoN, B.S.Pharm., Ph.D., FaPha, is Professor and Chairman, Depart-ment of Biopharmaceutical Sciences, Ber-nard J. Dunn School of Pharmacy, Shenan-doah University, Winchester, VA. DaviD F. DriScoll, B.S.Pharm., Ph.D., is Senior Researcher, Department of Medicine, Beth Israel Deaconess Medical Center, and Assis-tant Professor of Medicine, Harvard Medical School, Boston, MA.
Address correspondence to Dr. Newton at the Bernard J. Dunn School of Phar-macy, Shenandoah University, 1460 Univer-sity Drive, Winchester, VA 22601 ([email protected]).
The subject of the compatibility between calcium and phosphates was revisited in an April 1994
FDA safety alert,1,2 6–16 years after the four seminal research articles appeared in 1978,3 1980,4 1982,5 and 1988.6 In the 1980s there were two case reports of nonfatal adverse events involving calcium phosphate precipitation in total parenteral nu-trient (TPN) admixtures.7,8 A review of the main determinants of paren-teral drug and admixture compat-ibility and stability also appeared during that decade.9 Soon after the April 1994 safety alert, several publications on calcium phosphate precipitation in TPN formulations appeared.10-18 Thus, this article is yet another revisit of calcium and phosphate compatibility with i.v. formulations.
This article discusses the chem-istry and practical compatibility or solubility factors relevant to the safe administration of combination therapy with calcium gluconate and potassium or sodium phosphate in-jections. Patient case reports that led to adverse events and pharmaceuti-cal and clinical factors important to calcium phosphate solubility are also presented.
pH and pKa equilibria relevant
to calcium and phosphate compat-ibility. The keys to understanding the chemical reactions and relative risks for calcium phosphate precipitation are as follows:
• The clinically relevant dissocia-tion equilibria for which the pK
a2 of
phosphoric acid is 7.2 (i.e., the pH at which the concentrations or, ther-modynamically, the ionic activities of HPO
42– and H
2PO
4– are equal) (Table
1):
OH– + H2PO
4– ↔ HPO
42– + H
2O; shifts
to right when pH increases (1) H
2O + H
2PO
4– ↔ HPO
42– + H
3O+;
shifts to left when pH decreases (2)
• T h e H e n d e r s o n – H a s s e l b a c hequations9,19:
pH = pKa + log ([A–]/[HA]); percent
ionized, A–, = 100/(1 + antilog [pK
a – pH]) = 100{[A–]/([A–] +
[HA])} (3)pH = pK
a + log ([HPO
42– ]/[H
2PO
4–]);
percent HPO4
2– = 100/(1 + anti-log [pK
a – pH]) = 100{[HPO
42–]/
([HPO4
2–] + [H2PO
4–])} (4)
• Thecompatibilitycurvesforcalciumgluconate versus phosphate concen-trations in clinical mixtures.4,5,17,18
• The influence of other drugs andnutrients.4-8,10-18
The application of knowledge about calcium and phosphate com-patibility in i.v. therapy has been fa-cilitated by four hallmark articles,3-6 several editions of the Handbook on Injectable Drugs17 since 1983, and Trissel’s Calcium and Phosphate Com-patibility in Parenteral Nutrition.18 Despite the availability of these literature sources, calcium and phos-phate compatibility continues to be a clinical enigma.
Physicochemical factors. Calcium and phosphate solubility chemistry. The aqueous chemistry and solubil-ity of the two phosphate anions and their calcium salts that are important to the safety of i.v. therapy are sum-marized in Table 1. The main facts are as follows: The lower the solution pH is below 7.2, which is the critical pK
a2 of phosphoric acid in practice,
the greater is the majority percentage of the desired H
2PO
4– anion (dihy-
drogen or monobasic phosphate). H
2PO
4–, with two dissociable pro-
tons, is an acid relative to HPO4
2–, and HPO
42– (i.e., monohydrogen
or dibasic phosphate) is a base or weaker acid relative to H
2PO
4–.
Ca[H2PO
4]
2 (calcium dihydrogen
phosphate) is 60 times more soluble
than CaHPO4 (calcium monohydro-
COMMENTARy Calcium and phosphates
74 Am J Health-Syst Pharm—Vol 65 Jan 1, 2008
gen phosphate), because CaHPO4
is less dissociated.19,20 Note that, typi-cal of most divalent cation–divalent anion salts, CaHPO
4 is minimally
dissociated into its constituent ions. Consequently, most of the Ca2+ and HPO
42–
ions cannot be solvated by
dipolar water molecules via ion–dipole intermolecular forces, result-ing in 0.3-mg/mL solubility in water.
Ion–dipole forces generally result in greater solubility in water than do other types of solute–water inter-molecular forces.19,20 The contrasting high solubility of the divalent cation–divalent anion, magnesium sulfate, at more than 500 mg/mL, results from dipole–dipole forces between water and the mostly nondissociated MgSO
4 ion pairs, which are dipoles.
The efficient water solubility of some nonionic organic compounds (e.g., sugars) results from accepting and donating multiple intermolecular hydrogen bonds with water (i.e., one hydrogen bond for at least every four carbon atoms).20
The percentages of H2PO
4– and
HPO4
2– decrease and increase, re-spectively, by 1.6% to 5.7% for each 0.1 pH unit increase over the pH range of 6.0–7.6.14 Because 1 meq of HPO
42– corresponds to 2 meq of
H2PO
4–, phosphate concentration
should be expressed in millimoles per liter, not in milliequivalents per liter. In the article by Schuetz and King,3 phosphates were reported in milliequivalents per liter but without specific concentrations of H
2PO
4–
and HPO4
2–. The appendix shows the calculation for milliequivalents of potassium and for millimoles of phos-phates per milliliter in commercial Potassium Phosphates Injection, USP, and for milliequivalents of calcium per milliliter in commercial 10% Cal-cium Gluconate Injection, USP.
Before the transition to the Pharm.D. degree began achieving national momentum in the 1970s, most U.S. pharmacy schools required courses in qualitative and quantita-tive chemical analysis and inorganic pharmaceutical chemistry. Those courses were particularly pertinent to the solubility of calcium salts, as illustrated by the following excerpt from a monograph on CaHPO
4
in a standard pharmacy textbook from 1967: “Because this salt is al-most insoluble in water, its chemi-cal reactions are few and relatively unimportant. It is soluble in diluted hydrochloric acid.”19 That CaHPO
4
is more soluble at increasingly acidic pH represents the leftward shift in equation 2, and the “unimportance”
of CaHPO4
reactions stated in the 1967 source ended in 1968 with the report that launched TPN,21 which made reactions between calcium and phosphates in i.v. formulations a matter of life and death.
Calcium and phosphate solubility for i.v. therapy. It is unlikely that any patient-specific i.v. admixture con-taining calcium and phosphates will exactly duplicate the compatibility results of published studies. Three common variables are (1) practition-er and device volume-measurement accuracy and precision, (2) content and pH ranges from The United States Pharmacopeia and The Na-tional Formulary (USP) for calcium gluconate injection (i.e., 95–105% of labeled content and pH 6.0–8.2) and for potassium and sodium phosphate injections (i.e., 95–105% of labeled content),22 and (3) other drugs and nutrients that may be included in i.v. admixtures (i.e., the variable compo-sition of TPN formulations, which are often patient specific). Even small differences in the USP-allowed percent content ranges of calcium gluconate and potassium or sodium phosphate injections may contribute to the precipitation or nonprecipita-tion of CaHPO
4 in clinical practice.
The main factors that are impor-tant to ensuring total solubility or compatibility of calcium and phos-phates in TPN and other i.v. therapy are as follows1-18:
• The mixture should be agitated toachieve homogeneity after each ingre-dient is added.
• Potassium or sodium phosphateinjection should be added early, and calcium gluconate injection should be added last or nearly last to the most dilute phosphate concentration possible.1,2,17,18
• A 0.2-mm air-eliminating sterile inline filter should be used for non-fat-emulsion-containing i.v. admix-tures, and a 1.2-mm filter should be used for fat-emulsion-containing i.v. admixtures.1-3,10,13,14,17,18
Table 1.Chemistry and Water Solubility of Phosphates and Calcium Phosphates
Ion or Salta Names Solubility (mg/mL)5,10
H2PO
4–-
HPO4
2–
Ca[H2PO
4]
2
CaHPO4
Monobasicb phosphate, dihydrogen
phosphateDibasicd phosphate, monohydrogen
phosphateMonobasic calcium
phosphate,
calcium dihydrogen phosphateDibasic calcium
phosphate, calcium
monohydrogen phosphate
NAc
NA
18
0.3
aThe phosphoric acid aqueous equilibria H3PO
4 ↔ H
2PO
4– + H+ (for which pK
a1 = 2.1) and HPO
42– ↔ PO
43– + H+
(for which pKa3
= 12.319) are clinically negligible.14 bMonobasic refers to neutralization of the –1 charge on H
2PO
4– by one +1 cation (e.g., K+ or Na+, from bases
[alkali] such as potassium hydroxide or sodium hydroxide or carbonate). cNA = not applicable.dDibasic refers to neutralization of the –2 charge on HPO
42– by two +1 cations (e.g., 2 K+ or 2 Na+, or one +2
cation, e.g., Ca2+).
COMMENTARy Calcium and phosphates
75Am J Health-Syst Pharm—Vol 65 Jan 1, 2008
• Calcium chloride injection shouldnever be the calcium source in i.v. therapy that contains phosphate injections, because calcium chloride dissociates more extensively than calcium gluconate, resulting in more Ca2+ available to react with HPO
42–,
thus increasing the likelihood of CaHPO
4 precipitation.4,5
• The intersection of final calculatedcalcium and phosphate concentra-tions in clinical i.v. admixtures must be below the typical solubility curve (Figure 1).4,5,7,18
• A single sum or product of calciumand phosphate concentrations must not be used as the sole criterion for judging compatibility, because prod-ucts of calcium concentration (in mil-liequivalents per liter) and phosphate concentration (in millimoles per liter) vary inconsistently as calcium con-centration decreases and phosphate concentration increases.4,5
• The calculated concentrations ofcalcium and phosphates in TPN for-mulations must include all sources (e.g., amino acids injection) and not just the obvious calcium gluconate and potassium or sodium phosphate injections.
• The lower the final pH, the greaterthe percentage of H
2PO
4– at which
H2PO
4– forms more soluble calcium
dihydrogen phosphate salt with Ca2+. Higher final concentrations of dex-trose and the age-essential amino acid cysteine hydrochloride and lower final i.v. fat-emulsion concentrations favor lower admixture pH.
• Thehigherthefinalaminoacidcon-centration, the less likely CaHPO
4 is
to precipitate. Some amino acids se-quester Ca2+ (i.e., form stable soluble complexes). While most pharmacists are aware that disodium ethylene-diaminetetraacetic acid (EDTA) se-questers divalent ions, including Ca2+,
fewer of them identify EDTA as an amino acid.14
• The rates of crystalline growth andprecipitation of CaHPO
4 in clini-
cal admixtures may be variable and low in supersaturated mixtures. For
example, in one study of a simulated TPN admixture, the measured cal-cium concentration declined expo-nentially from 22 to 7 meq/L over 14 days in 0.2-mm membrane filtrates of the original admixture.14 In another study of a simulated TPN admix-ture, an increase in CaHPO
4 particles
larger than 5 mm was measured over 48 hours by using light obscuration, and the precipitates were confirmed as such by petrography and infrared spectroscopy.23
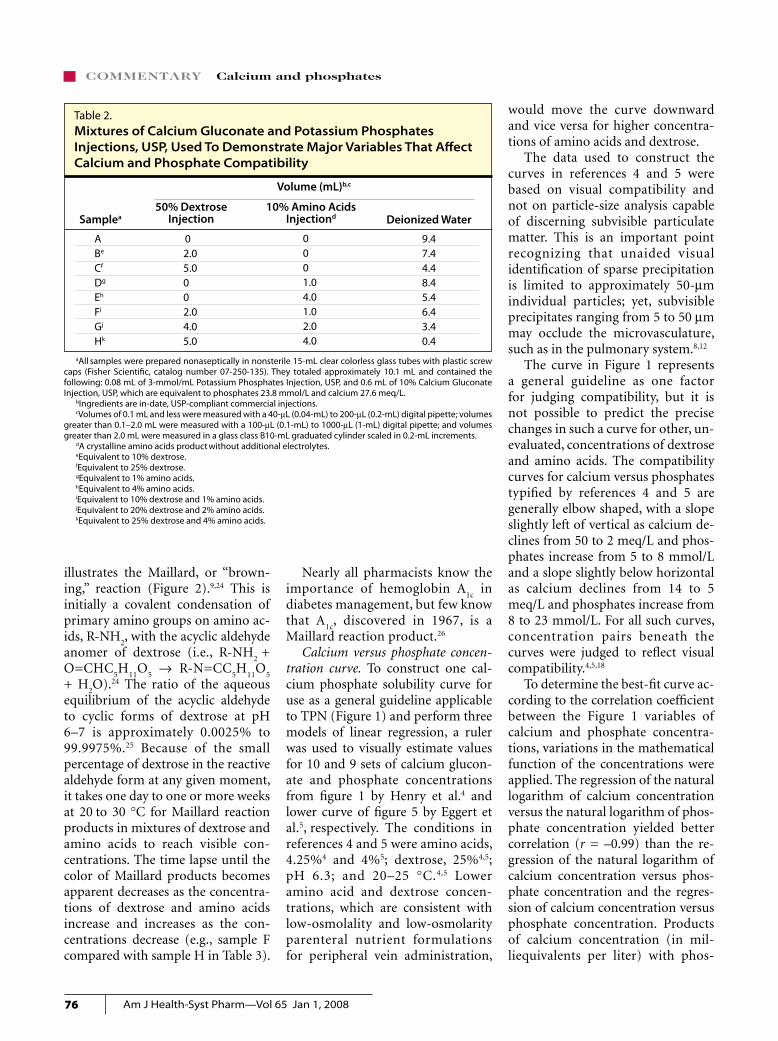
Demonstration samples of calcium gluconate and potassium phosphate injections. Table 2 illustrates the beneficial effects of the acidic pH of dextrose injection and of calcium sequestration by amino acids on the compatibility of i.v. calcium and phosphates. The approximate cal-cium and phosphate concentrations of 28 meq/L and 24 mmol/L, respec-tively, were chosen to intersect well above recommended compatibility curves (Figure 1), so that visible pre-
cipitation would occur quickly and convincingly in samples with little or no content of dextrose and amino acids.18 After thorough mixing, the ingredients were added in this order: potassium phosphates, 50% dextrose injection, sterile water for injection (nonbacteriostatic), amino acids, and calcium gluconate. The sample tubes were stored at 22–24 °C and each day were exposed to ceiling fluorescent illumination for 10 hours and to darkness for 12 hours.
The typical results for the samples listed in Table 2 are presented in Table 3. Adding a few drops of 1.9% (0.05 M) disodium EDTA to sample A or D illustrates calcium sequestra-tion by amino acids when the precip-itated CaHPO
4 dissolves, and adding
a few drops of 1 N hydrochloric acid to sample A or D illustrates the left-shifted equilibrium in equation 2, which favors calcium and phosphate compatibility. The change from col-orless to pale yellow to yellow-amber in samples F, G, and H over 14 days
Figure 1. Composite curve for compatibility of calcium (as gluconate) with phosphates at 20–25 °C and pH 6.3 in 25% dextrose injection and 4–4.25% amino acids injection.4,5 The farther concentrations are below the curve, the greater the probability of nonprecipita-tion is, and the closer to or farther above the curve concentrations are, the greater the probability of precipitation of CaHPO
4 is.
Cal
ciu
m (
meq
/L)
50
45
35
30
20
0 5 10 15 20
40
25
15
10
0
5
25
Phosphates (mmol/L)
COMMENTARy Calcium and phosphates
76 Am J Health-Syst Pharm—Vol 65 Jan 1, 2008
illustrates the Maillard, or “brown-ing,” reaction (Figure 2).9,24 This is initially a covalent condensation of primary amino groups on amino ac-ids, R-NH
2, with the acyclic aldehyde
anomer of dextrose (i.e., R-NH2
+ O=CHC
5H
11O
5 → R-N=CC
5H
11O
5
+ H2O).24 The ratio of the aqueous
equilibrium of the acyclic aldehyde to cyclic forms of dextrose at pH 6–7 is approximately 0.0025% to 99.9975%.25 Because of the small percentage of dextrose in the reactive aldehyde form at any given moment, it takes one day to one or more weeks at 20 to 30 °C for Maillard reaction products in mixtures of dextrose and amino acids to reach visible con-centrations. The time lapse until the color of Maillard products becomes apparent decreases as the concentra-tions of dextrose and amino acids increase and increases as the con-centrations decrease (e.g., sample F compared with sample H in Table 3).
Nearly all pharmacists know the importance of hemoglobin A
1c in
diabetes management, but few know that A
1c, discovered in 1967, is a
Maillard reaction product.26
Calcium versus phosphate concen-tration curve. To construct one cal-cium phosphate solubility curve for use as a general guideline applicable to TPN (Figure 1) and perform three models of linear regression, a ruler was used to visually estimate values for 10 and 9 sets of calcium glucon-ate and phosphate concentrations from figure 1 by Henry et al.4 and lower curve of figure 5 by Eggert et al.5, respectively. The conditions in references 4 and 5 were amino acids, 4.25%4 and 4%5; dextrose, 25%4,5; pH 6.3; and 20–25 °C.4,5 Lower amino acid and dextrose concen-trations, which are consistent with low-osmolality and low-osmolarity parenteral nutrient formulations for peripheral vein administration,
would move the curve downward and vice versa for higher concentra-tions of amino acids and dextrose.
The data used to construct the curves in references 4 and 5 were based on visual compatibility and not on particle-size analysis capable of discerning subvisible particulate matter. This is an important point recognizing that unaided visual identification of sparse precipitation is limited to approximately 50-mm individual particles; yet, subvisible precipitates ranging from 5 to 50 mm may occlude the microvasculature, such as in the pulmonary system.8,12
The curve in Figure 1 represents a general guideline as one factor for judging compatibility, but it is not possible to predict the precise changes in such a curve for other, un-evaluated, concentrations of dextrose and amino acids. The compatibility curves for calcium versus phosphates typified by references 4 and 5 are generally elbow shaped, with a slope slightly left of vertical as calcium de-clines from 50 to 2 meq/L and phos-phates increase from 5 to 8 mmol/L and a slope slightly below horizontal as calcium declines from 14 to 5 meq/L and phosphates increase from 8 to 23 mmol/L. For all such curves, concentration pairs beneath the curves were judged to reflect visual compatibility.4,5,18
To determine the best-fit curve ac-cording to the correlation coefficient between the Figure 1 variables of calcium and phosphate concentra-tions, variations in the mathematical function of the concentrations were applied. The regression of the natural logarithm of calcium concentration versus the natural logarithm of phos-phate concentration yielded better correlation (r = –0.99) than the re-gression of the natural logarithm of calcium concentration versus phos-phate concentration and the regres-sion of calcium concentration versus phosphate concentration. Products of calcium concentration (in mil-liequivalents per liter) with phos-
Table 2.Mixtures of Calcium Gluconate and Potassium Phosphates Injections, USP, Used To Demonstrate Major Variables That Affect Calcium and Phosphate Compatibility
Volume (mL)b,c
50% Dextrose Injection Deionized Water
ABe
Cf
Dg
Eh
Fi
Gj
Hk
9.47.44.48.45.46.43.40.4
Samplea10% Amino Acids
Injectiond
02.05.0002.04.05.0
aAll samples were prepared nonaseptically in nonsterile 15-mL clear colorless glass tubes with plastic screw caps (Fisher Scientific, catalog number 07-250-135). They totaled approximately 10.1 mL and contained the following: 0.08 mL of 3-mmol/mL Potassium Phosphates Injection, USP, and 0.6 mL of 10% Calcium Gluconate Injection, USP, which are equivalent to phosphates 23.8 mmol/L and calcium 27.6 meq/L.
bIngredients are in-date, USP-compliant commercial injections. cVolumes of 0.1 mL and less were measured with a 40-mL (0.04-mL) to 200-mL (0.2-mL) digital pipette; volumes
greater than 0.1–2.0 mL were measured with a 100-mL (0.1-mL) to 1000-mL (1-mL) digital pipette; and volumes greater than 2.0 mL were measured in a glass class B10-mL graduated cylinder scaled in 0.2-mL increments.
dA crystalline amino acids product without additional electrolytes.eEquivalent to 10% dextrose.fEquivalent to 25% dextrose.gEquivalent to 1% amino acids. hEquivalent to 4% amino acids. iEquivalent to 10% dextrose and 1% amino acids.jEquivalent to 20% dextrose and 2% amino acids.kEquivalent to 25% dextrose and 4% amino acids.
0001.04.01.02.04.0
COMMENTARy Calcium and phosphates
77Am J Health-Syst Pharm—Vol 65 Jan 1, 2008
phate concentration (in millimoles per liter) vary inconsistently from 130 to 1704 and from 100 to 1905 as calcium concentration decreases and phosphate concentration increases. This is why a single product should not be used as a sole criterion for judging compatibility.
Case reports. The calcium and phosphate concentrations that re-
sulted in patient harm or death are reviewed below (Figure 3).
Report by Robinson and Wright.7
A right subclavian catheter became occluded after 64 days of continuous TPN therapy. The TPN admixture consisted of 500 mL of 8.5% amino acids injection and 500 mL of 50% dextrose injection in a 1000-mL formula that also contained calcium
gluconate 10 meq/L and phosphate 80 mmol/L (evenly divided between the sodium and potassium salts). This phosphate concentration greatly exceeds the right-hand limit of 25 mmol/L on the phosphate axis in Figure 3. The patient survived, prob-ably because a 0.22-mm inline filter was used.
Report by Knowles et al.8 A patient who had been receiving home TPN therapy for five years developed diffuse granulomatous interstitial pneumonitis due to exposure to pre-cipitated CaHPO
4. The TPN formu-
lation contained 4.25% amino acids injection and 5% dextrose injection; this is a low-osmolality and low-osmolarity formulation that would be expected to be more susceptible to calcium and phosphate precipita-tion than, for example, the dextrose concentrations described by Henry et al.,4 Eggert et al.,5 and Fausel et al.14
Report by Hill et al.12 This report, which prompted the FDA safety alert,1,2 involved four patients who had been receiving a low-osmolality TPN admixture via a peripheral vein during hospitalization at Tripler Army Medical Center in Honolulu and who developed sudden and un-
Figure 2. Calcium gluconate and potassium phosphate injection samples A–H (see Tables 2 and 3) photographed at 14 days.
14 Days3d
001e
0, Y0, Y
0, YA1
0, YA3
Sample 10 minABCDEFGH
3c
001c
0000
30010000
Visual Appearance at Interval Indicateda,b
1 hr 1 Day 5 Days 9 Days3d
001e
000
0, Y
3d
001e
00
0, Y0, YA
1
3d
001e
0Y
0, YA1
0, YA2
a0 = no precipitate or color change, 1 = faint turbidity from CaHPO4 precipitate, 3 = intense turbidity from
CaHPO4 precipitate, Y = pale yellow, YA
1 = pale yellow-amber, YA
2 = darker yellow-amber than YA
1, YA
3 = darker
yellow-amber than YA2.
bSample tubes were gently agitated at each observation time to swirl any possible scant crystalline precipitate from the bottoms. White or black fungi and mold may appear as fluffy masses after several days in dextrose-containing samples, but those are easily distinguished from precipitated CaHPO
4.
cPrecipitation occurred instantly upon the addition of calcium gluconate injection.dClear supernatant over approximately 0.75 in-thick sediment of gelatinous-appearing precipitate.eClear supernatant over approximately 0.75 in-thick sediment of gelatinous-appearing precipitate.
Table 3.Appearance of Samples of Calcium Gluconate and Potassium Phosphates Injections, USP, after Standing at 22–24 °C
COMMENTARy Calcium and phosphates
78 Am J Health-Syst Pharm—Vol 65 Jan 1, 2008
explained respiratory distress, which was fatal in two cases. Postmortem examination of lung tissue identified CaHPO
4 crystals
in the pulmonary
microvasculature. Table 4 compares the institution’s peripheral-vein and central-vein TPN formulations and illustrates most of the important calcium and phosphate compatibility factors. The deaths were attributed to an unfavorable mixing sequence, lack of inline filtration, and a short time from compounding to admin-istration.12 The calcium and phos-phate concentrations did not exceed the solubility limit in the final TPN admixture volume, but CaHPO
4
precipitated when calcium gluconate was added before 70% dextrose injec-tion to only 46% of the final volume of the TPN admixture. There was not adequate time between the comple-tion of compounding and the start of infusion for the precipitated CaHPO
4
to dissolve, nor was the formulation agitated sufficiently.
Report by Shay et al.15 This retro-spective cohort study reviewed all hospitalized patients who received a low-osmolality and low-osmolarity formulation (peripheral-vein par-enteral nutrient [PN] formulation) containing calcium and phosphate over a 16-month period. The defini-tion for possible calcium phosphate precipitation and harm was met if “while receiving [peripheral-vein] PN during the study period, [the patient] developed unexplained chest pain, dyspnea, or cardiopul-monary arrest of noncardiac etiology or had new, unexplained bilateral interstitial infiltrates noted on chest radiograph.” Of the 50 patients who received the therapy, 5 met this defi-nition, and 4 of them died.
Report by author. One of the au-thors (D.W.N.) served as a consultant
in a lawsuit involving a baby’s death (after 2001) caused by precipitation of CaHPO
4 during an i.v. dextrose in-
fusion. The confidential information provided indicated that (1) relevant literature sources4,5,17,18 were either misinterpreted or not reviewed, (2) the curve for calcium concentration versus phosphate concentration was interpreted as a downward-slanting straight line,4,5,17,18 (3) a compat-ibility chart for amounts of calcium gluconate and potassium phosphate injected was based on a final volume of x mL, but the actual volume com-pounded was 0.5x mL, resulting in twice the assumed concentrations of calcium and phosphates, and (4) an inline filter was not used. One phy-sician who attempted to rescue the baby stated “Ten to 15 minutes into resuscitation, the lower 1–2 cm of the baby’s i.v. fluid bag, as well as the i.v. tubing, showed precipitation.”
Figure 3. Calcium and phosphate concentrations that resulted in patient harm or death, superimposed over the compatibility curve shown in Figure 1. Ref. 12a represents values for the total volume of total parenteral nutrient (TPN) formulation to which calcium glu-conate injection was added; Ref. 12 represents values for only 46% of the total TPN admixture volume.
Cal
ciu
m (
meq
/L)
50
45
35
30
20
40
25
15
10
0
5
Phosphates (mmol/L)
Ref. 8
Ref. 12
Ref. 15
Report by author D.W.N.
Ref. 12a
0 5 10 15 20 25
COMMENTARy Calcium and phosphates
79Am J Health-Syst Pharm—Vol 65 Jan 1, 2008
Preventing future harm. All institutions must establish calcium and phosphate mixing guidelines that are supported by peer-reviewed literature and the manufacturers’ product information. The compat-ibility guidelines should be based on actual clinical conditions and be reviewed and approved by the phar-macy and therapeutics committee. Low-osmolality and low-osmolarity formulations, such as PN admixtures administered through a peripheral vein, are notorious for calcium and phosphate incompatibility; thus, they should be avoided when pos-sible. A recent investigation of such compatibility for peripheral-vein PN admixtures (≤3% amino acids and ≤5% dextrose) showed that the upper limit of compatibility was calcium gluconate 5 meq/L and sodium phos-phates 15 mmol/L, or approximately half the parenteral equivalent of the recommended daily allowance of these minerals.23
In the early TPN studies used to construct the curve in Figure 1,4,5 a limited range of macronutrient con-
centrations was employed, and only visual identification of precipitation, which can be highly variable, was performed. Recent studies employing particle detection and size measure-ment by light obscuration provide objective evidence of subvisible microprecipitation,23 which can be clinically dangerous.
Careful interpretation of the cal-cium and phosphate compatibility literature is necessary before applica-tion to clinical practice. For example, Wong et al.27 recently suggested that calcium and phosphate con-centrations in TPN admixtures for neonates could be doubled to meet fetal accretion rates by using a for-mulation containing only monoba-sic potassium phosphate, KH
2PO
4.
This claim was based on the correct premise that the divalent phosphate anion, HPO
42–, is the culprit in cal-
cium phosphate precipitation in TPN formulations. However, it did not emphasize that increasing pH (e.g., pH in TPN formulations that is much higher than pH in the KH
2PO
4
injection product) will cause the
monobasic anion, H2PO
4–, to convert
to the dibasic anion, HPO4
2–, as de-picted in equation 1. In the study by Wong et al., samples were evaluated on three occasions between 0 and 27 hours after admixture preparation. Only 1 of 45 sample measurements exceeded pH 6 (i.e., 6.06) whereas most of the TPN admixtures studied by Henry et al.,4 Eggert et al.,5 and Fausel et al.14 had a pH of 6.3. Wong et al.27 would have identified CaHPO
4
precipitation in more samples if the pH had been higher.
Conclusion. Understanding the chemical and practical compatibility of calcium gluconate and potassium or sodium phosphate injections is critical to ensuring the safe i.v. ad-ministration of these supplements and preventing patient harm.
References1. Lumpkin MM, Burlington DB. FDA safe-
ty alert: hazards of precipitation associ-ated with parenteral nutrition. Rockville, MD: Food and Drug Administration; 1994 Apr 18.
2. Food and Drug Administration. Safety alert: hazards of precipitation associ-ated with parenteral nutrition. Am J Hosp Pharm. 1994; 51:1427-8.
3. Schuetz DH, King JC. Compatibility and stability of electrolytes, vitamins and an-tibiotics in combination with 8% amino acids solution. Am J Hosp Pharm. 1978; 35:33-44.
4. Henry RS, Jurgens RW Jr, Sturgeon R et al. Compatibility of calcium chloride and calcium gluconate with sodium phosphate in a mixed TPN solution. Am J Hosp Pharm. 1980; 37:673-4.
5. Eggert LD, Rusho WJ, Mackay MW et al. Calcium and phosphorus compatibility in parenteral nutrition solutions for neo-nates. Am J Hosp Pharm. 1982; 39:49-53.
6. Lenz GT, Mikrut BA. Calcium and phosphate solubility in neonatal par-enteral nutrient solutions containing TrophAmine. Am J Hosp Pharm. 1988; 45:2367-71.
7. Robinson LA, Wright BT. Central venous catheter occlusion caused by body-heat-mediated calcium phosphate precipita-tion. Am J Hosp Pharm. 1982; 39:120-1.
8. Knowles JB, Cusson G, Smith M et al. Pulmonary deposition of calcium phos-phate crystals as a complication of home total parenteral nutrition. JPEN J Parenter Enteral Nutr. 1989; 13:209-13.
9. Newton DW. Introduction: physico-chemical determinants of incompatibility and instability of drugs for injection and infusion. In: Trissel LA, ed. Handbook on
Peripheral-Vein Formulation12
7%b
33 gc
39 gd
15 mmolf
10 meq/Lh
Table 4.Calcium and Phosphate Compatibility Factors in Central- and Peripheral-Vein Parenteral Nutrient Formulations at Tripler Army Medical Center
Ingredient Central-Vein Formulationa
Dextrose Freamine III with
electrolytesFat from i.v.
emulsion Phosphoruse Calciumg
24%
41 g
28 g14 mmol5 meq/L
aFrom confidential documents provided to author (D.W.N.) as a consultant for a lawsuit in 1997.bA lower final dextrose concentration favors a higher final mixture pH, which favors a higher percentage of
phosphate as HPO4
2–, which favors greater formation of the least-soluble CaHPO4 salt.
cA lesser amino acids concentration reduces Ca2+ sequestration, which increases the free Ca2+ concentration available to react with phosphates.
dA higher fat content favors a higher final mixture pH, from the alkaline pH of fat emulsion, which favors a higher percentage of phosphate as HPO
42–, which favors greater formation of the least-soluble CaHPO
injectable drugs. 3rd ed. Bethesda, MD: American Society of Hospital Pharma-cists; 1983:xiii-xv.
10. Driscoll DF, Newton DW, Bistrian BR. Precipitation of calcium phosphate from parenteral nutrient fluids. Am J Hosp Pharm. 1994; 51:2834-6. Letter.
11. Maswoswe JJ, Okpara AU, Hilliard MA. An old nemesis: calcium and phosphate interaction in TPN admixtures. Hosp Pharm. 1995; 30:579-86.
12. Hill SF, Heldman LS, Goo ED et al. Fatal microvascular pulmonary emboli from precipitation of a total nutrient admix-ture solution. JPEN J Parenter Enteral Nutr. 1996; 20:81-7.
13. Newton DW. Rx: calcium, phosphate, and secundum artem. Nutrition. 1997; 13:390-1.
14. Fausel CA, Newton DW, Driscoll DF et al. Effect of fat emulsion and supersatu-ration on calcium phosphate solubility in parenteral nutrient admixtures. Int J Pharm Compound. 1997; 1:54-9.
15. Shay DK, Fann LM, Jarvis WR. Respira-tory distress and sudden death associated with receipt of a peripheral parenteral nutrition admixture. Infect Control Hosp Epidemiol. 1997; 18:814-7.
16. Reedy JS, Kuhlman JE, Voytovich M. Mi-crovascular pulmonary emboli second-ary to precipitated crystals in a patient receiving total parenteral nutrition: a case report and description of the high-resolution CT findings. Chest. 1999; 115:892-5.
17. Trissel LA. Handbook on injectable drugs. 12th ed. Bethesda, MD: American Society of Health-System Pharmacists; 2007:242-58.
18. Trissel LA. Trissel’s calcium and phos-phate compatibility in parenteral nutri-tion. Houston: Tripharma; 2001.
19. Soine TO, Wilson CO. Roger’s inorganic pharmaceutical chemistry. 8th ed. Phila-delphia: Lea & Febiger; 1967:141-2,170-2,388.
20. Allen LV Jr, Popovich NG, Ansel HC. Ansel’s pharmaceutical dosage forms and delivery systems. 8th ed. Balti-more: Lippincott Williams & Wilkins; 2005:338,341.
21. Dudrick SJ, Wilmore DW, Vars HM et al. Long-term total parenteral nutrition with growth, development, and positive nitro-gen balance. Surgery. 1968; 64:134-42.
22. The United States pharmacopeia, 30th rev., and The national formulary, 25th ed. Rockville, MD: United States Pharmaco-peial Convention; 2006:1600,2993.
23. Driscoll DF, Joy J, Silvestri AP et al. Cal-cium (Ca) and phosphate (P) compatibil-ity in low osmolality parenteral nutrition (PN) mixtures. Clin Nutr. 2005; 24:695.
24. Newton DW, Narducci WA. Extempo-raneous formulations. In: King RE, ed. Dispensing of medication. 9th ed. Chap 12. Easton, PA: Mack; 1984:258-88.
25. Murray RK, Granner DK, Mayes PA et al. Harper’s biochemistry. Stamford, CT: Appleton & Lange; 1996:137-9.
26. Rahbar S. The discovery of glycated he-moglobins. In: Baynes JW, Monnier VM, Ames JM et al., eds. The Maillard reac-tion: chemistry at the interface of nutri-
tion, aging, and disease. Ann N Y Acad Sci. 2005; 1043:9-19.
27. Wong JC, McDougal AR, Tofan M et al. Doubling calcium and phosphate concen-trations in neonatal parenteral nutrition solutions using monobasic potassium phosphate. J Am Coll Nutr. 2006; 25:70-7.
Appendix—Calculation of calcium concentration in Calcium Gluconate Injection, USP, and phosphorus and potassium concentrations in Potassium Phosphates Injection, USPCalcium Gluconate Injection, USP1. Selected information from The United States Pharmacopeia and the National Formulary (USP) 22: Contains 95–105% of labeled strength of calcium gluconate. A small amount of calcium
content from the gluconate salt may be replaced by calcium saccharate or other calcium salts for stabilization.
5. Sum of answers for steps 4a and 4b is 3 mmol/mL.6. Potassium calculation
a. KH2PO
4 contribution
b. K2HPO
4 contribution
7. Sum of answers for steps 6a and 6b is 4.36 or 4.4 meq/mL.
a1 mmol of any compound contains 1 mmol of each of its constituent atoms or ions.
0.224 gmL
mol136.09 g
1000 mmolmol
1.65 mmolmL=
_ ____ ___
0.236 gmL
mol174.18 g
1000 mmolmol
1.35 mmolmL=
_ ____ ___
98 mgmL
g1000 mg
mol448.39 g
2 eqmol
1000 meqeq
0.437 meqmL=
_ ____ _____
_____
__
4.6 mgmL
g1000 mg
mol320.26 g
2 eqmol
1000 meqeq
0.029 meqmL=
_ ____
_________
0.224 gmL
mol136.09 g
1000 meqeq
1.65 meqmL=
_ ____
eqmol_____
__
0.236 gmL
mol174.18 g
1000 meqeq
2.71 meqmL=
_ ____ ___
2 eq mol __
__
· · · ·
· · · ·
· ·
· ·
· · ·
· · ·
AbstractSince the early 1960s, parenteral nutrition
(PN) has been used as a primary source of
nutrition for patients who are unable to use
their gastrointestinal tracts. Over the years,
clinical experience and research have
improved patient care during nutrition
support that includes an interdisciplinary
approach to address and closely monitor
each patient’s individual nutrition needs.
The uniqueness of PN solutions for each
patient not only increases the complexity
of solution compounding but necessitates
that all clinicians, including physicians,
dietitians, pharmacists, pharmacy
technicians, and nurses, work closely
together. The role of pharmacists is
especially important in overseeing issues of
composition, compatibility, stability, sterility,
and safety during PN preparation. Additional
issues involve proper storage conditions
and intravenous (IV) delivery. This article
highlights the chemical properties important
for chemical compatibility and stability of PN
admixtures and the regulations set by the
United State Pharmacopeia (USP) Chapter
797 for personnel and facilities where PN
admixtures are compounded. Safety issues
related to ordering and compounding PN
prescriptions also are addressed and
Compounding Compatibility, Stability, and Safety in Parenteral NutritionNing-Tsu Kuo, PharmD, PhD
examples of fatal mistakes in units of
weight or volume are provided.
IntroductionFor more than 50 years, PN has been part of
the therapeutic treatment for people who
are unable to use their gastrointestinal tracts
adequately to meet their nutrition needs (1).
A successful PN treatment plan requires
collaboration among physicians, dietitians,
nurses, and pharmacists. The dietitian works
with the physician in assessing the patient’s
medical conditions and nutrition needs to
produce a PN formula for the pharmacist,
who compounds the ingredients into a
single bag for IV infusion. A typical PN
admixture is composed of amino acids,
dextrose, fatty acids, electrolytes (sodium,
potassium, calcium, magnesium, phosphate,
acetate, and chloride), trace elements
(chromium, copper, manganese, selenium,
and zinc), multivitamins, water, and often
other additives. Because each ingredient has
unique physical and chemical properties,
mixing all of them together might result in
precipitation or emulsion destabilization (2).
Therefore, the compatibility and stability of
the ingredients must be carefully evaluated
before compounding a PN formulation.
Contributing factors to precipitation or
destabilization include amino acid
concentration, pH of the formulation,
dextrose concentration, concentration of the
electrolytes, order of mixing, temperature
during mixing, storage temperature, and
calcium salt used (3). A PN formulation can be
prepared either manually or by a
computerized automated compounder,
although an automated compounder is
preferred because of its efficiency and
accuracy (4). To ensure absolute sterility in
preparing IV infusion products, the USP
Chapter 797 sets quality standards for
personnel and the facility where
compounding sterile products are made,
stored, and packaged (5). The purpose of this
article is to describe compatibility and
stability issues related to PN compounding
and to address safety issues in compounding
PN at health care facilities.
Compatibility and StabilityCalcium and PhosphateThe most important issue to consider in compounding PN solutions is the possibility of forming a calcium-phosphate precipitate, which could occlude microvasculature and be potentially fatal (6). The risk of developing calcium-phosphate precipitation increases
Calcium salts Calcium gluconate Calcium chloride or calcium acetate
Mixing temperature Room temperature, 25.0°C Other than room temperature
Mixing order Add calcium gluconate as the last ingredient in compounding formula
Final concentration of Final amino acid concentration between 2% and 6% Final amino acid concentration >6% and <2% amino acids and dextrose Final dextrose concentration between 4% to 25% Final dextrose concentration >25% and <4%
Total concentration of Calcium salt (mEq/L) x phosphate salt (mEq/L) Calcium-phosphate product >200 calcium and phosphate salts product ≤200
Presence of other additives Any additives that might increase pH to >7 in 2-in-1 formula and >6.5 in 3-in-1 formulation
Support Line ❙ Volume 33 No. 6 ❙ 13
(Continued on page 16)
with the following factors: 1) high pH, 2) use of calcium chloride instead of calcium gluconate, 3) high temperature of solution, 4) improper mixing order in preparing PN solution, 5) insufficient final concentration of amino acids and dextrose, 6) high concentration of calcium and phosphate salts, and 7) presence of other additives such as lipid emulsion (Table 1) (7).
The amino acid is the primary factor determining the final pH of a PN solution (8). Several commercial amino acid mixtures are available, and each differs in composition, pH, and calcium-phosphate solubility curve. Calcium-phosphate solubility curves were generated by plotting the maximum concentrations of calcium and phosphate using different commercially available amino acid products at room temperature. Some products, such as FreAmine® (B. Braun Medical, Inc, Irvine, CA), contain intrinsic phosphate, which is an important contributor to potential calcium-phosphate precipitation. Calcium gluconate is preferred over calcium chloride. Calcium chloride has a solubility of 74.5 g/L water at 20.0°C, while calcium gluconate has a solubility of 30 g/L water at 20.0°C (9). A desirable calcium salt should have less ionized calcium in a PN solution, and calcium chloride has more than twice the amount of ionized calcium in 1 L of water than calcium gluconate at 20.0°C. Less ionized calcium in a solution reduces the chance of such ions colliding with phosphate molecules to form precipitates. The same logic applies to high temperature. A higher temperature of PN solution increases the dissociation of calcium and phosphate, which combine and precipitate out of solution (10).
The mixing order in preparing a PN solution is also important for inhibiting calcium-phosphate precipitation. Calcium gluconate should be added to a PN admixture after all other ingredients have been mixed (11) to reduce dissociation of calcium gluconate into free ionized calcium and gluconate. At the Cleveland Clinic Home Care (CCHC) Infusion Pharmacy, where more than 100 PN bags are mixed daily, phosphate salt is added as the fifth ingredient and calcium gluconate is added as the last ingredient.
The final concentration of amino acid and dextrose can affect the formation of calcium-phosphate precipitate, largely due to the influence of pH of the PN solution. A lower amino acid concentration means a higher pH that, in turn, means lower solubility of calcium-phosphate. A final concentration of amino acid below 2% is not recommended; the final amino acid concentration should be between 2% and 6% (12). Higher final concentrations of dextrose not only can lower pH but also can increase viscosity, which can have a positive impact of slowing molecule collision between calcium and phosphate (13). Table 2 describes a method of calculating final concentrations of amino acid, dextrose, and lipid in a total nutrient admixture (TNA) solution.
Formation of calcium-phosphate precipitate is directly related to the total concentration of both calcium and phosphate salts. Calcium-phosphate solubility product has been used as a general guideline in PN formulation to predict the possibility of calcium-phosphate precipitate formation (14). A simple formula is used for calculation: calcium salt (mEq/L) x phosphate salt (mEq/L) = calcium-phosphate solubility product. At the CCHC Infusion Pharmacy, a calcium-phosphate solubility product of 200 or less is considered acceptable. Alternatively, a calcium salt (mEq/L) x phosphate salt (mM/L) product of less than 75 is also considered safe in preventing calcium-phosphate precipitation (15). Finally, other additives, especially lipid emulsions, can increase pH and the possibility of calcium-phosphate precipitation (16).
In summary, the most important limiting factor in calcium-phosphate precipitation is the total concentration of calcium salt and phosphate salt. Restricting the calcium-phosphate solubility product to less than 200 is the first step to avoid precipitation.
Destabilization in Total Nutrient AdmixtureAdding lipid emulsion to a PN solution in a single compartment is frequently referred to as 3-in-1 (3:1) or TNA. Commercially available lipid emulsions consist of soybean oil triglycerides or a combination of both soybean and safflower oil. Using egg phospholipid as the emulsifier allows formulating 10%, 20%, and 30% weight by volume oil-in-water emulsions. The resulting chylomicron-like particle exhibits an inner core of triglyceride covered by the hydrophilic portion of phospholipids that carries negative charges on the surface. Electrostatic repulsion maintains the integrity of lipid emulsion (17). Thus, any factor that disrupts such electrostatic repulsion can result in destabilization of the TNA.
TNA destabilization progresses through four stages (18). Stage I is creaming, in which triglyceride particles accumulate at the top of the emulsion. This stage is generally reversible by gentle agitation. Stage II is aggregation, which occurs when triglyceride particles in the creaming stage are too large to be redispersed within the emulsion. Stage III is coalescence. In this stage, triglyceride particles fuse, enlarging the size of particles. In stage IV, which is called cracking, the aqueous and oil phases separate permanently. At stage I, the TNA bag still is safe for infusion following gentle agitation to redisperse the lipid droplets. Once aggregation occurs (stage II), the TNA bag is no longer safe to use. The TNA formula must be reevaluated carefully and adjustments made to avoid any factors that might contribute to the destabilization. Another possible method to avoid TNA destabilization is to infuse the lipid solution in a piggyback bag with a Y-site device along with 2:1 PN solution.
Table 2. Example of Calculating Final Percentage of Amino Acid, Dextrose and Lipid in Total Nutrient Admixture Solution
Total Solution Bag Volume Final Concentration per Bag = 3 L (g/L) Final Percentage per Bag
Amino acid per bag = 90 g 90/3 = 30 g/L 30/1,000 x 100% = 3%
Dextrose per bag = 150 g 150/3 = 50 g/L 50/1,000 x 100% = 5%
Lipid per bag = 100 g 100/3 = 33.3 g/L 33.3/1,000 x 100% = 3.33%
16 ❙ Support Line ❙ December 2011
Possible factors that may contribute to destabilization of TNA include: 1) pH of TNA, 2) final concentration of amino acid and dextrose, 3) final concentration of lipid, 4) compounding sequence, 5) electrolyte concentration, 6) other additives, and 7) storage temperature (19).
The challenge of keeping TNAs stable resides primarily with the final pH of the formula. Because lipid emulsion carries negative charges on the phospholipid, the integrity of the emulsion state can be disturbed simply by lowering the pH to below 5 or through the presence of cations. As the solution becomes more acidic, more hydrogen ions become available to bind to the negative charges on the phospholipid, compromising the electrostatic repulsion state. In a TNA formula, amino acids not only have a major role in determining the final pH, but they provide a buffering capacity through a unique isoelectric point. Each amino acid molecule has a positive charge on one end and a negative charge on the other, called zwitterions. The presence of both a positive charge and a negative charge in the same molecule of the amino acid strengthens the integrity of a lipid emulsion (20). The desired pH of a TNA should be in the range of 5.4 to 6.0 (21). Unlike the TNA solution, the pH for PN (2:1) formulation is largely correlated with the pH of the selected commercial amino acid solutions, which range from 4.5 to 7 (8).
The final concentration of amino acid and dextrose is crucial to keeping the lipid emulsion stable. If the final concentration of amino acid is less than 2% in TNA, the pH is higher than 6.5, and the buffering capacity provided by the amino acid solution is minimal, the risk for lipid destabilization increases. As mentioned earlier, a higher final concentration of dextrose in PN solution increases the viscosity, thereby enhancing lipid stability. Furthermore, the pH for dextrose solution is about 4.0. Therefore, a higher dextrose concentration can compensate for lower amino acid content in a formula that has a low final amino acid concentration but a much higher final dextrose concentration. The recommended final concentration of amino acid is between 20 and 60 g/L at the CCHC Infusion Pharmacy, and the final dextrose concentration is 40 to 250 g/L (Table 3).
The final concentration of the lipid emulsion is also important in TNA stability. If the final concentration is less than 2%, the TNA preparation tends to be unstable due to the dilution effect of the emulsifier. Based on the information provided by Baxter Inc., the CCHC Infusion Pharmacy uses lipid emulsion with a final concentration between 20 and 60 g/L in TNA. As with calcium and phosphate, the order of the compounding sequence can be a factor in TNA stability. Lipid emulsion is added before amino acids and dextrose. The rationale is that the amino acid solution provides a buffering effect for the lipid emulsion, thereby strengthening its electrostatic repulsion state (20). Because the negative charge on phospholipid is the primary force of stability, cations are potentially capable of neutralizing the negative charge on phospholipids. Divalent cations such as calcium and magnesium are of great concern in TNA preparation. The nutrition support team at the Cleveland Clinic sets the upper limit of calcium plus magnesium to 20 mEq/L or less based on the lipid manufacturer’s recommendation. Trivalent compounds such as iron dextran are contraindicated in TNA because they can neutralize the negative charge on phospholipid, but iron dextran is compatible with the PN solution. Table 3 describes acceptable ranges of amino acid, dextrose, lipid, calcium plus magnesium, and calcium-phosphate product in TNAs.
Finally, a storage temperature that is either higher or lower than refrigerator temperature might compromise TNA stability via the breakdown of lipid emulsion (6). TNA bags should not be placed in freezers because ice crystals might disrupt the phospholipid barriers, and increases in solute concentration during slow freezing could cause precipitation. Therefore, TNA should be stored in the refrigerator at 4.0°C
and never frozen. According to USP 797, PN and TNA bags are safe for 7 days from the day they are compounded if they are stored in the refrigerator between 2º and 8°C (6). Both PN and TNA bags should be removed from the refrigerator and warmed at room temperature for 2 to 3 hours before infusion. When lipid is being infused via a Y-site device, it is only safe for 12 hours at room temperature, according to Centers for Disease Control and Prevention (CDC) guidelines (22).
Compatibility With MedicationsIn addition to the standard ingredients mentioned previously, other medications, such as multivitamins, insulin, famotidine, heparin, ranitidine, vitamin K, octreotide, and methylprednisolone, are added to the PN or the TNA bag before infusion. Based on the information provided from the Handbook on Injectable Drugs (23) and King Guide to Parenteral Admixtures (3), many other medications are compatible with PN or TNA infusion when administered via a Y-site device, such as ceftazidime, clindamycin, and diphenhydramine (3). When no literature is available regarding medication compatibility with PN solution, no assumptions should be made.
SafetySafety in compounding PN starts with the writing of a PN prescription. A PN order should be clearly legible, with minimum calculations for converting between units. A very good example of a PN Physician Order Form is included in Safe Practices for Parenteral Nutrition from the American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.) (6). Incorrect conversions between units is the most common overall mistake in hospitals (24). Two recent cases related to PN compounding highlight the safety issue.
Table 3. Total Nutrient Admixture Stability Guide for the Cleveland Clinic Home Infusion Pharmacy Divalent Cations Calcium x (Calcium + Phosphate Amino Acid Dextrose Lipid Emulsion Magnesium) Product
20 to 60 g/L 40 to 250 g/L 20 to 60 g/L <20 mEq/L <200 (2% to 6%) (4% to 25%) (2% to 6%)
Support Line ❙ Volume 33 No. 6 ❙ 17
(Continued on next page)
Case 1: Converting Milligrams of Calcium Gluconate to Milliequivalents. In April 2011, The Institute for Safe Medication Practice (ISMP) published a report of an infant who died from cardiac arrest due to sodium overdose from his PN solution (25). The original order asked for 14.7 mEq of sodium chloride and 982 mg of calcium gluconate. The pharmacy technician misread the order as 982 mEq sodium chloride and 14.7 mEq calcium gluconate. The pharmacist did not catch the mistake and the nurse failed to recognize the mistake. If the units of electrolytes were all unified as milliequivalents (14.7 mEq of sodium chloride and 4.5 mEq of calcium gluconate), the mistake might not have been fatal. The incorrect reading of units of measure with electrolyte would have been 4.5 mEq of sodium chloride and 14.7 mEq of calcium gluconate, which would have lessened the severity of the mistake.
This incident highlights a pressing safety issue associated with PN orders. In the same publication, ISMP called for a standardized PN order as the top recommendation to avoid a mistake such as that reported. A standardized PN form using grams for amino acid and dextrose, milliliters for lipid, and milliequivalents for all electrolytes would be ideal. The A.S.P.E.N. Safe Practices Guideline made a similar recommendation, with the exception of phosphate salts, which they recommended to be ordered as mmol/day (6).
In addition, the initial physician order containing each listed ingredient with its appropriate dosage units must synchronize with the pharmacy computer system, the compounder software, and the final PN label. Any PN order must be reviewed by a pharmacist before handing it to a pharmacy technician for computer entry. A pharmacist also should check the accuracy of the entry before sending the order to the IV room for compounding. A second pharmacist in the IV room should double-check the order before sending the PN solution to the floor for infusion. PN solutions must be compounded by experienced and well-trained personnel. Any experienced technician and pharmacist would have recognized that 982 mEq of sodium
chloride is absolutely too much for any adult patient, let alone an infant.
Case 2. Converting Micrograms of Zinc Chloride to Milligrams. A second case published by ISMP on September 6, 2007, described another fatal mistake involving a 1,000-fold overdose of zinc chloride administered to a 26-week-old infant (26). The initial order asked for zinc chloride 330 µg/100 mL. A pharmacist entering the order into the computer mistakenly chose milligrams (mg) instead of micrograms (µg) as the correct unit. During the compounding, the technician had noticed that 11 vials of zinc chloride solution were requested (correct order should have requested only 0.33 mL) but failed to inform the pharmacist on duty in the IV room. The pharmacist who checked the order in the IV room did not realize that 11 vials of zinc chloride was an unusually large quantity for an order. The floor nurse did not match the initial physician order with the pharmacy label on the zinc chloride bag. The mistake was not identified for 3 hours, and the patient died of cardiac failure due to zinc overdose. This mistake highlights the urgency of setting a standardized ordering form for PN, TNA, and IV fluid orders. Knowing the maximum daily doses for electrolytes or trace element such as zinc chloride in adult and pediatric patients is important for ensuring medication safety.
Another reported mistake in compounding PN is confusion between heparin and insulin, which has resulted in death (27). The United States Food and Drug Administration (FDA) and ISMP have compiled a list of high-alert medications, which includes insulin, hypertonic sodium chloride, potassium chloride, potassium phosphate, and magnesium sulfate. One approach to avoid confusion in reaching for the wrong bottles when compounding PN solutions is to minimize the number of different strength solutions and separate or color-code different strength stock solutions.
ISMP has compiled a list of error-prone abbreviations, symbols, and dosage designations (28). Two abbreviations in the list that are most relevant to PN formulating are U and µg. ISMP recommends substituting
Unit and mcg for U and µg. For dose designation, they recommend not using trailing zeros for whole number doses (e.g., write 1 mg instead of 1.0 mg). They also recommend using a zero before a decimal point when the dose is less than a whole unit (e.g., 0.5 mg rather than .5 mg).
Automated CompounderBecause a typical PN solution contains at least 10 to 15 ingredients, mixing it manually into a single bag has the potential for many mistakes. An automated compounder can remove much of the human error. Accuracy in the automated compounder relies on bar coding computer technology and a mechanical pump that delivers a specific volume based on the number of revolutions. Using bar coding technology, a PN order is scanned and sent electronically to a compounder, which eliminates possible transcription errors that can occur with manual data entry. At least 20 ports are available in a compounder for handling 20 ingredients. Each ingredient is assigned a specific port with a designated bar code. This step allows for temporal separation of incompatible ingredients, thus reducing the chance of calcium and phosphate precipitating. Each bottle of stock solution already has its own National Drug Code (NDC), and scanning the NDC number and matching the correct port assigned through bar code scanning further ensures the accuracy of source solution identification.
The mechanical pump in an automated compounder can deliver a single ingredient within ±3% of requested volume as long as the needed ingredient volume for compounding is greater than 1 mL. The smallest volume an automated compounder can deliver is 0.2 mL. To ensure the accuracy of a volumetric pump further, an independent weight scale is used to check the final volume pumped. This double-fault protection mechanism is inherently designed in an automated compounder, making the PN compounding process accurate and consistent (4).
Even though an automated compounder is superior to manual compounding, errors still are possible if policies for the entire process are not carried out vigilantly by
18 ❙ Support Line ❙ December 2011
the pharmacy. The most significant disadvantage of an automated compounder is its cost. The American Society of Health-System Pharmacists (4) published a guideline that included specific objectives related to cost justification:•Enhancedefficiencyandworker
safety… and patient safety with PN use.•Reductioninlaborassociatedwith
If a health facility has a high PN patient census, minimizing or eliminating the possibility of medication errors and improving efficiency in overall costs for labor and materials can justify the long-term cost of an automated compounder.
Sterility, USP 797One of the biggest concerns for patients who receive PN is infection. The most common infection in patients receiving PN is catheter-related bloodstream infection; rarely is infection due to PN solution contamination. However, ISMP recently published a report on an outbreak of Serratia marcescens bacteremia in 19 patients who all received PN from a compounding pharmacy in Alabama in March 2011 (29). Nine of the affected 19 patients died. The Alabama Department of Public Health (ADPH) and the CDC found traces of S marcescens in the compounding room at the pharmacy. The pharmacy has stopped all production and recalled all compounded preparations. This incident highlights the danger of inadvertently introducing pathogens into PN bags during the compounding process.
Absolute sterility must be maintained in IV rooms used for compounding PN. To achieve the sterility goal, the USP issued General Chapter 797 Pharmaceutical Compounding-Sterile Preparations regulations in 2004 that were revised in 2008. The regulations apply to all persons who compound sterile preparations and all places where such preparations are compounded, stored, and transported (5).
Specifically, every person who works in the
IV room must be properly trained and able to demonstrate good aseptic techniques such as gowning, gloving, and hand washing. Each facility must develop an action plan that includes written procedures to standardize daily tasks such as gowning, gloving, and hand washing. In addition, quality assurance procedures such as sampling protocols, spill cleanup, personnel training, and regular maintenance logs must be well defined and documented.
Each day, before compounding, pharmacy personnel must clean countertops, shelving, vents, anteroom sinks, and storage bin surfaces using 70% isopropyl alcohol. Trash in the IV room must be cleared out twice daily. Sharps containers with needles and syringes and biohazard containers with chemotherapeutic wastes also need to be cleared out twice daily. By the end of the day, floors must be thoroughly cleaned. Every month, IV rooms, anterooms, walls, ceilings, and storage shelving must be cleaned using the procedures for cleaning a surgical suite.
Monitoring sterility for both airborne and surface area contaminants is another important part of USP 797. Using International Organization for Standardization (ISO) as a guide, USP 797 sets different standards for the number of particles per cubic meter allowed in different areas of an IV room. For example, areas where the actual compounding occurs, such as the hood area, is designated as ISO 5 air environment and the allowed particles are no greater than 3,520 particles/M3. Supporting areas could be classified as ISO 7 or 8 air environment, with allowed concentrations of 10,000 particles/M3 and 100,000 particles/M3, respectively. To ensure that air particles circulating in the clean room contain no contaminants from the external environment, USP 797 requires the clean room to be have negative pressure and to have all entering air passed through a high-efficiency particle air (HEPA) filter.
USP 797 recommends the use of contact plates or swabs to take surface samples from all ISO-classified air environments. Two tests are recommended for testing aseptic technique of personnel operating
in the IV room: media-fill test and gloved finger test. Both tests are designed to detect any weakness in aseptic knowledge and technique.
Although seemingly tedious, all of these regulations and standards are absolutely necessary for ensuring patient and worker safety. The challenge for the CDC and APDH is to identify which step or procedure was skipped or missed at the previously cited pharmacy that led to S marcescens contamination in PN bags, resulting in nine deaths.
The FDA is the federal agency that is designated to enforce USP 797 in each health care facility. However, the FDA defers such authority to individual states for regulation and inspection. Some state boards of pharmacy adopt USP 797 fully, but other states only adopt USP 797 partially. The interests of public safety would be best served by universal adoption of USP 797.
SummaryCompounding PN solutions is a complex and complicated process that has inherent safety risks and a very low margin for mistakes. The health care team, including physicians, dietitians, pharmacists, and nurses, must work together to deliver optimum care. Each team member must be vigilant about avoiding harms that might occur during PN order writing, PN formula compounding, or PN solution infusion. Physicians, dietitians, and pharmacists should check every PN formula for compatibility, stability, and safety.
Ning-Tsu Kuo, PharmD, PhD, is a staff pharmacist at Cleveland Clinic Home Infusion Pharmacy, Independence, OH.
References 1. Dudrick SJ. Early developments and clinical
applications of total parenteral nutrition. JPEN J Parenter Enter Nutr. 2003;27:291–299.
2. FDA safety alert: hazards of precipitation associated with parenteral nutrition. Am J Hosp Pharm. 1994;51:1427–1428.
3. King JC, Catania PN, eds. King Guide to Parenteral Admixtures. Napa, CA: King Guide Publications; 2003.
4. American Society of Health-System Pharmacists. ASHP guidelines on the safe use of automated compounding devices for
Support Line ❙ Volume 33 No. 6 ❙ 19
(Continued on next page)
Ainsley Malone, MS, RD, CNSC, as moderator.
We had several break-out assistants,
including Jennifer Wooley, MS, RD, CNSC,
Louise Merriman, MS, RD, CDN, Teresa
Scollard, MBA, RD, LD, and ADA staff
members Marsha Schofield, PhD, RD, and
Mara Bujnowski, MAEd, RD. I especially
appreciated the engaged participants from
this session and their recognition of the
importance in determining a consistent
definition of malnutrition.
For those DNS members who plan to attend
A.S.P.E.N.’s Clinical Nutrition Week, January
21-24, 2012, in Orlando, FL, we hope to see
you at the DNS sessions on Tuesday, January
24, Abdominal Assessment and Interpretation
of Abdominal Radiography for Feeding Tube
Placement and Handgrip Dynamometry and
Interpretation of Radiography for Central Line
Placement. Please stop by our booth in the
exhibit area as well.
Chair’s Column(Continued from page 2)
DNS was also very proud to recognize David
Frankenfield, MS, RD, chief clinical dietitian,
Penn State Milton S. Hershey Medical Center,
Hershey, PA, for winning ADA’s prestigious
Excellence in Practice-Dietetics Research
Award. DNS nominated David for his diligent
and thorough research in predictive energy
requirements, which led to the development
of the Penn State equations. No doubt every
nutrition support dietitian is familiar with his
outstanding work. Another proud moment
DNS shared was the selection of Mark
DeLegge, MD, as an ADA Honorary Member.
Dr. DeLegge is a professor of medicine and
director of the Digestive Disease Center and
the nutrition section as well as the medical
director of nutrition for the Medical
University of South Carolina. As a
gastroenterologist, he has devoted his career
to the care of patients requiring nutrition
support. He has specifically provided support
for our DNS skills workshops, been a great
advocate for our profession, and always
encouraged dietitians to be part of the
medical team. Very hearty congratulations to
both David and Dr. DeLegge.
Our Spotlight Session was a huge success,
with Kathy Barco, RD, CNSC, and Neha
Parekh, MS, RD, CNSC, presenting Navigating
Intestinal Surgery: How to Access and Feed the
Altered GI Tract. They had a packed room and
stayed long afterward to answer questions
from attendees. We were also involved in the
pre-FNCE workshop Documenting Severe
and Non-Severe Malnutrition-A Hands-On
Approach. This session was a joint effort of
ADA’s Coding and Coverage Committee
Malnutrition Workgroup and DNS. Jane
White, PhD, RD, FADA, Bob DeChicco, MS,
RD, CNSC, and I were the speakers, with
the preparation of parenteral nutrition admixtures. Am J Health Syst Pharm. 2000; 57:1343–1348.
5. Lee LD. Compliant compounding: meeting USP 797 pharmacy regulations. Health Facil Mgmt. 2010 February. http://www.hfmmagazine.com/hfmmagazine_app/jsp/articledisplay.jsp?dcrpath=HFMMAGAZINE/Article/data/02FEB2010/1002HFM_FEA_ES&domain=HFMMAGAZINE. Accessed August 2011.
6. Mitallo J, Canada T, Johnson D, et al; Taskforce for the Revision of Safe Practices for Parenteral Nutrition. Safe practices for parenteral nutrition. JPEN J Parenter Enter Nutr. 2004;28:S39–S70.
7. Maswoswe JJ, Okpara AU, Hilliard AM. An old nemesis: calcium and phosphate interaction in TPN admixtures. Hosp Pharm. 1995;30:580.
8. Collins CJ. General pharmacologic issues. In: Matarese LE, Gottschlich MM, eds. Contemporary Nutrition Support Practice: A Clinical Guide. 2nd ed, St. Louis, MO: WB Saunders Co; 2003:315-336.
10. Robinson LA, Wright BT. Central venous catheter occlusion caused by body heat-mediated calcium-phosphate precipitation. Am J Hosp Pharm. 1982;39:120–121.
11. The University of Michigan Hospital and Health Centers. Parenteral nutrition for adolescent and adult patients. In: Parenteral and Enteral Nutrition Manual. 8th ed. Ann Arbor, MI: The University of Michigan Hospital and Health Centers; 2003:22–23.
12. Poole RL, Rupp CA, Kerner JA. Calcium and phosphorus in neonatal parenteral nutrition solutions. JPEN J Parenter Enteral Nutr. 1983; 7:358–360.
13. Driscoll DF. Total nutrition admixtures: theory and practice. Nutr Clin Pract. 1995; 10:114–118.
15. Mierzwa MW. Stability and compatibility in preparing TPN solutions. In: Lebenthal E, ed. Total Parenteral Nutrition: Indications, Utilization, Complications, and Pathophysiological Considerations. New York, NY: Raven Healthcare Publishers; 1986:219–244.
16. Eggert LD, Rusho WJ, MacKay MW, Chan GM. Calcium and phosphorus compatibility in parenteral nutrition solutions for neonates. Am J Hosp Pharm. 1982;39:49–53.
17. Barnett MI. Physical stability of all in one admixtures: factors affecting fat droplets. Nutrition. 1989;5:348–349.
18. Knutsen OH. Stability of intralipid fat emulsion in amino acid solutions. Crit Care Med. 1986; 14:638–641.
19. Bettner FS, Stennett DJ. Effects of pH, temperature, concentration, and time on particle counts in lipid-containing total parenteral nutrition admixtures. JPEN J Parenter Enteral Nutr. 1986;10:375–380.
20. Takamura A, Ishii F, Noro S, et al. Study of intravenous hyperalimentation: effect of selected amino acids on the stability of intravenous fat emulsions. J Pharm Sci. 1984; 73:91–94.
21. Black CD, Popovich NG. A study of intravenous emulsion compatibility effects of dextrose, amino acids, and selected electrolytes. Drug Intell Clin Pharm. 1981; 15:184–193.
22. O’Grady NP, Alexander M, Dellinger EP, et al. Guidelines for the prevention of intravascular catheter-related infections. MMWR Morbid Mortal Wkly Rep. 2002;51(RR10):1–26.
23. Trissel LA. Handbook on Injectable Drugs. 16th ed. Bethesda, MD: American Society for Hospital Pharmacists; 2010.
24. Wong IC, Ghaleb MA, Franklin BD, Barber N. Incidence and nature of dosing errors in pediatric medications: a systematic review. Drug Saf. 2004;27:661–670.
25. Institute for Safe Medicine Practices. Another tragic parenteral nutrition compounding error. ISMP Medication Safety Alert. 2011; 16(8):1–3.
26. Institute for Safe Medicine Practices. Fatal 1000-fold overdoses can occur, particularly to neonates, by transposing mcg and mg. ISMP Medication Safety Alert. 2007;12(18):1–4.
27. Institute for Safe Medicine Practices. Action needed to prevent dangerous heparin insulin confusion. ISMP Medication Safety Alert. 2007;12(9):1–3.
28. Institute for Safe Medicine Practices. List of Error-Prone Abbreviations, Symbols, and Dose Designations. http://www.ismp.org/Tools/errorprone abbreviations.pdf. Accessed August 2011.
29. Institute for Safe Medicine Practices. TPN-related deaths call for FDA guidance and pharmacy board oversight of USP Chapter 797. ISMP Medication Safety Alert. 2011;16(7):1–3.
20 ❙ Support Line ❙ December 2011
http://ncp.sagepub.com/Nutrition in Clinical Practice
http://ncp.sagepub.com/content/24/4/441The online version of this article can be found at:
DOI: 10.1177/0884533609339070
2009 24: 441Nutr Clin PractCaitlin Curtis and Gordon S. Sacks
Compounding Parenteral Nutrition: Reducing the Risks
Published by:
http://www.sagepublications.com
On behalf of:
The American Society for Parenteral & Enteral Nutrition
can be found at:Nutrition in Clinical PracticeAdditional services and information for
Parenteral and Enteral Nutrition10.1177/0884533609339070
http://ncp.sagepub.comhosted at
http://online.sagepub.com
Invited Review
The production of fluids for intravenous use is opti-mally performed in an aseptic environment, and the finished product should be free of microbes,
spores, endotoxins, chemical contamination, and physical matter. Compounding such a product requires trained personnel, an adequate environment, and effective tech-nique.1 However, over the years there have been many reports of contaminated intravenous compounds, includ-ing contaminated parenteral nutrition (PN).2-5 PN is especially at risk of contamination because it is a mixture of multiple additives and is an excellent growth medium for microbes.
The United States Pharmacopeia (USP), a nongovern-mental, nonprofit healthcare organization, is charged with developing national standards for drug purity and safety for all prescription and over-the-counter medicines manufac-tured or sold in the United States. These standards are published in a book, the United States Pharmacopeia: National Formulary (USP-NF), and healthcare practitioners not familiar with its organization need to understand the significance of each chapter number. Book chapters num-bered ≤999 are regarded as medication standards that must be followed and are enforceable by the U.S. Food and Drug
From 1University of Wisconsin Hospital and Clinics, Madison, Wisconsin, and 2School of Pharmacy, Auburn University, Auburn, Alabama.
Adress correspondence to: Caitlin Curtis, PharmD, BCNSP, University of Wisconsin Hospital and Clinics, Department of Pharmacy, 600 Highland Avenue, CSC-1530 F6/133, Madison, WI 53792; e-mail: [email protected].
Administration (FDA). Those book chapters numbered 1000-1999 are considered informational, whereas chapters assigned numbers ≥2000 are related to nutrition supple-ments. Because of a low compliance with voluntary guide-lines, the USP became involved with issuing standards on the pharmaceutical compounding of sterile products. An expert committee of USP drafted Chapter <797>, the first official monograph enforceable by regulatory agencies con-cerning the procedures and requirements for pharmacy-prepared sterile products, and it became the official minimum standard in January 2004. Much of the same information was previously published as recommendations in the nonenforceable Chapter <1206>, which focused on dispensing for home care and guidelines related to sterile product preparation published by the American Society of Health-System Pharmacists (ASHP).6 Although Chapter <797> discusses standards applying to all sterile dosage forms that are compounded, only information pertinent to PN and its implications are reviewed here.
Preparation of PN falls under the category of a com-pounded sterile preparation (CSP) by the definition of USP Chapter <797>.1 The revised version of USP Chapter <797>, Pharmaceutical Compounding: Sterile Preparation, was released in December 2007 and became official on June 1, 2008. The chapter describes new safety standards for facilities that compound sterile products in an attempt to improve production practices and reduce the risk of contamination. All compounding areas have defined levels of cleanliness and limits of particulate matter, which are defined by International Standards (ISO) Classifications. Table 1 provides these classifications. USP Chapter <797>
Compounding parenteral nutrition, either manually or with an automated compounding device, requires aseptic conditions and trained personnel. The revised version of United States Pharmacopeia Chapter <797> is a comprehensive document that describes standards and procedures to minimize the risk of con-tamination of compounded parenteral products. The chapter includes evidence-based instructions for pharmacy design, washing, garbing, cleaning, quality assurance, and personnel training and evaluation designed to improve compounding practices in all
pharmacies that compound parenteral products. Because parenteral nutrition is a compounded product mixed from multiple additives, it is important to maintain these standards, especially when using an automated compounding device. This article is an overview of United States Pharmacopeia Chapter <797>, with special empha-sis on parenteral nutrition.(Nutr Clin Pract. 2009;24:441-446)
Keywords: parenteral nutrition; parenteral nutrition, total; pharmaceutical preparations; drug compounding; drug packaging
Compounding Parenteral Nutrition: Reducing the Risks
Caitlin Curtis, PharmD, BCNSP1; and Gordon S. Sacks, PharmD, BCNSP, FCCP2
Financial disclosure: none declared.
by Karrie Derenski on April 1, 2013ncp.sagepub.comDownloaded from
442 Nutrition in Clinical Practice / Vol. 24, No. 4, August/September 2009
is an official document of the USP-NF and is therefore enforceable by the FDA.6 Because the FDA allows indi-vidual states to regulate the practice of pharmacy, it is usually up to the state board of pharmacy to adopt the standards and inspect pharmacies for compliance.7 However, the FDA does have the power to inspect phar-macies and enforce the standards in the interest of public health. Medication recalls and additional legal sanctions can occur upon failure to comply with USP standards.
Compliance with the USP Chapter <797> standards may require multiple changes in the way that pharmacy personnel operate while preparing CSPs. The standards describe in detail the environments for preparation of sterile products, cleaning of equipment and surfaces, per-sonnel training, washing and garbing procedures, and quality assurance checks. This article summarizes the new standards and also outlines the specific sections which pertain to compounding PN.
Pharmacy Design
USP Chapter <797> outlines a 3-tiered approach for assigning the potential risk of contamination associated with compounding of CSPs (Table 2). PN is classified as a “medium-risk-level” CSP when it is prepared from crystal-line amino acids and commercially available monohydrated dextrose, injectable lipid emulsions, electrolytes, multiple vitamins, trace elements, and sterile water. It carries a higher level of risk because it requires the mixing of mul-tiple small doses of different sterile products into 1 large bag.1 When PN is compounded using powdered amino acids, it is classified as a “high-level” CSP because its preparation involves the use of nonsterile ingredients and carries the highest risk for contamination by microbial, chemical, or physical matter. As a result, high-risk-level CSPs must undergo some type of sterilization process prior to administration. All risk-level compounds should be pre-pared in an environment in which particulate matter and microorganisms are minimized, and the air quality must meet certain standards. Pharmacies must be designed spe-cifically to meet these standards, which require an ante-room and a buffer area to separate the general environment from the area of compounding. The lower the number of classification, the cleaner the air must be. The anteroom is an area where personnel can perform washing and garbing procedures prior to entering the compounding area, and it must meet ISO class 8 requirements. After personnel have garbed and washed, they must enter the buffer area. The buffer area must be separated from the anteroom by demarcation or a physical barrier. If the buffer area is physically separated from the anteroom, it must be an area of positive pressure and meet ISO class 7 requirements. A positive-pressure room is defined as a room that has a higher pressure than adjoining rooms so that the air flows out of the room.1 The buffer area must be kept as clean and as free of particulates as possible, because this is the area
Table 2. ISO Classification of ParticulateMatter in Room Air (Limits Given in
Particles of ≥0.5 mcm/m3)
ISO Class Particle Limit per m3
3 35.24 3525 3,5206 35,2007 352,0008 3,520,000
ISO, International Standards.Adapted from Chapter <797> Pharmaceutical compounding: sterile preparations. In: Revision Bulletin, The United States Pharmacopeia: The United States Pharmacopeial Convention 2008, p 2. Adapted with permission. Copyright 2009 The United States Pharmacopeial Convention. All rights reserved.
Table 1. Risk-Level Classificationand Beyond-Use Dating Guidelines for
Compounded Sterile Preparations
Beyond-Use Dating
Risk Level
Examples
Room (20°-25° C)
Refrigeration (2°-8° C)
Low Reconstitution of a single-dose vial of lyophilized powder with sterile diluents for transfer into another container (eg, pediatric multivitamins)
48 h 14 d
Medium Mixing of multiple manufactured additives for transfer into a large-volume parenteral solution (eg, PN formulations)
30 h 9 d
High Preparation of nonsterile powder for intravenous infusion (eg, extemporaneously compounded L-glutamine for supplementation in a PN formulation)
24 h 3 d
PN, parenteral nutrition.Reprinted from Barber JR, Rollins CJ, Sacks GS. Parenteral nutrition formulations. In: Gottschlich MM, ed. The A.S.P.E.N. Nutrition Support Core Curriculum: A Cased-Based Approach—The Adult Patient. Silver Spring, MD: A.S.P.E.N.; 2007:277-299.
by Karrie Derenski on April 1, 2013ncp.sagepub.comDownloaded from
where products are gathered directly before compounding takes place. All materials in this area, including furniture, equipment and supplies, must be nonpermeable, nonshed-ding, cleanable, and resistant to damage by disinfectants. All materials should be cleaned and disinfected before entering the buffer area. Finally, the cleanest area must be the area where compounding occurs, called the direct compounding area (DCA), which must meet ISO class 5 standards. The DCA airflow must be controlled by 1 of the following devices: laminar airflow work benches, biological safety cabinets, compounding aseptic isolators, or com-pounding aseptic containment isolators.
All areas should be constructed so that the space minimizes dust and dirt collection. Counters, shelves, and carts should be constructed of smooth, nonporous, nonshedding materials such as stainless steel or molded plastic that are able to be cleaned and disinfected on a regular basis. Walls and ceilings must be constructed of nonporous materials, such as heavy-gauge polymer or epoxy-coated gypsum board. Other fixtures, such as hang-ing light fixtures or pipes, should be minimized.
Anterooms are to be free of food, drinks, and materials that have been exposed to patient care areas. In the ante-room, all package compounding supplies, including nee-dles, syringes, tubing sets, and small- and large-volume parenteral products, must be taken out of cartons and wiped with sterile 70% isopropyl alcohol before they are transferred to the buffer area. An apparent demarcation must separate the anteroom and the buffer area, whether it is a line on the floor or a physical separation, such as a door.
Cleaning and Garbing Procedures
To maintain ISO class 5, 7, and 8 standards, personnel must strictly adhere to washing and garbing procedures before entering the areas. First, in the anteroom, before entering the buffer area, personnel should remove all outer garments and any hand, wrist, or ear jewelry. Next, personnel should put on appropriate garb, in the follow-ing order: shoe covers, head covers, and face masks. Personnel should wash hands and forearms with soap and water for 30 seconds and dry with lint-free disposal towels or electronic hand dryer. After the hand-cleansing proce-dure, personnel should put on a nonshedding gown with cuffs at the wrists and closure at the neck. They may enter the buffer area after appropriate garbing and must use waterless alcohol-based surgical hand scrub to clean their hands before donning sterile, powder-free gloves. Gloves should be kept clean throughout the day by wiping or rubbing 70% isopropyl alcohol to all surface areas of the gloves, and this cleansing should take place after the gloves make contact with any nonsterile surface.
After personnel have finished with cleansing and gar-bing procedures, they should begin cleaning the work
area. Personnel should clean and disinfect surfaces in the primary compounding area at the beginning of every shift, before a new batch is started, every 30 minutes during continuous compounding, when there are spills, and when known contamination is present or suspected. Counters and floors must be cleaned daily with nonshed-ding wipers and mops dedicated to each area. For instance, sponges used to clean the buffer area should stay in the buffer area. Cleaning elements should be disposable if possible. If cleaning elements are not disposable, then they should be cleaned by standard procedures on a regu-lar basis. Walls, ceilings, and shelving must be cleaned monthly. All cleaning should be done by trained personnel with approved cleaning and disinfecting agents.
To maintain ISO standards, it is important to avoid extraneous particles and microbes. All supplies brought into the buffer area must be removed from cardboard cartons in an anteroom before being passed into the buffer area in order to minimize airborne particulates. To decrease microbial contamination, all supplies including needles, syringes, tubing sets, vials, and small- and large-volume parenteral products should be cleaned and disin-fected with sterile 70% isopropyl alcohol before being transferred to the buffer area. All equipment that is brought into the buffer area must be cleaned and disin-fected, including carts and storage containers.
Personnel Training and Evaluation
To be compliant with USP Chapter <797>, institutions must show that all personnel are trained in garbing procedures, aseptic work practices, and cleansing/disinfection proce-dures. Institutions must document that personnel com-plete didactic training, pass written competencies, and undergo skill assessment using observational audit tools and media-fill testing. Annual documentation of success-ful completion of written and media-fill testing is required for all personnel who perform low- and medium-risk–level compounding, and semiannual documentation is required for personnel who perform high-risk–level compounding. Appendices in USP Chapter <797> provide excellent tools for assessment of procedures by direct observation.1 To ensure proper hand hygiene, garbing procedures, and cleaning and disinfection procedures, a pharmacy super-visor directly observes the compounding personnel while they perform the procedure. To pass the aseptic tech-nique competency, compounding personnel must pass both a direct observation test and a media-fill test. Media-fill testing assesses the quality of aseptic technique and provides objective evidence of production of a sterile prod-uct. To pass this competency, personnel use commercially available sterile fluid culture media, such as soybean-casein digest medium, to simulate a drug product for compounding. If the medium is not transferred from 1
by Karrie Derenski on April 1, 2013ncp.sagepub.comDownloaded from
444 Nutrition in Clinical Practice / Vol. 24, No. 4, August/September 2009
container to another aseptically, bacteria will grow and will be visibly turbid. For successful completion, the com-pound must be incubated and must not be turbid for 14 days after manipulation.1 Compounding personnel who fail written or observational assessment, or whose aseptic technique results in contamination, must be reinstructed and retested until they pass all portions of assessment.
Quality Assurance
All of the compounding and associated areas must be tested periodically to prove that the areas meet particulate and cleanliness standards. Except for pressure differen-tials, this environmental sampling should occur and be documented as part of the certification of new facilities or equipment as well as for recertification, which should occur every 6 months. Environmental sampling should also occur after any equipment is serviced and after physical changes have been made to the area (eg, new construc-tion, rearrangement). Sampling must occur in response to patient-care–related infections when the sterile product area may be at fault or in response to suspected subopti-mal work practices of compounding personnel.
The environment should be sampled for airflow between areas, nonviable particulates, and viable particu-lates (viable refers to microorganisms). Airflow should be measured between the buffer area and the anteroom, and between the anteroom and the general environment out-side the anteroom. Airflow should be monitored and documented at least every work shift. Nonviable and via-ble particulates are measured less often, as stated previ-ously. Nonviable particulates are measured to make sure that the various levels of cleanliness are being maintained by the devices that maintain ISO class 5 environments. Viable particulates are measured in select sampling sites to ensure the lowest levels of antimicrobial invasion into the compounding area. Sampling sites should include areas of the greatest risk of contamination, such as work areas near passageways or counters located in an area of high air turbulence. The chapter outlines in detail the materials needed for testing, including viable air sampling devices, type of agar or growth medium plates, and incu-bation conditions and time.1
Environmental sampling should be part of the standard operating procedure of the pharmacy. As such, the proce-dure for measuring and documenting environmental sam-pling should include a sampling plan that defines acceptable limits for pressure gradients and nonviable and viable parti-cles. The plan should instruct pharmacy personnel on what steps to take if the samples exceed the acceptable limits.
Quality Assurance for PN Compounding
It has been estimated that 65% of all PN admixtures in the United States are prepared with automated compounding
devices (ACDs), a practice that is permissible under USP Chapter <797> standards.8 Guidelines for ensuring the accuracy and precision of ACDs are addressed in this chapter. Compounding personnel should test the device for accuracy on a daily basis, document the results, and then review the results weekly. Personnel should perform both volumetric and weight testing using sterile water for the fluid test. Other additives may be tested if desired. Additional tests using the hospital laboratory may be used to ensure accurate compounding, such as testing for dex-trose or for potassium concentration in the final com-pounded preparation. Other fundamental elements related to the safe and cost-effective inclusion of this technology into pharmacy operations have been addressed by ASHP guidelines published in 2000.8 As many as 20 single-source containers are attached to an ACD during PN compounding, all of which are pumped into a single large-volume plastic bag. The order in which individual ingredients are added to the final PN admixture is very important because specific components may be incom-patible if added too close together in the sequence (eg, calcium gluconate and sodium/potassium phosphate). Use of the ACD for purposes other than PN compound-ing should be discouraged because this will likely require reprogramming of configurations and increase the risk for error. Compounder tubing changes must occur at appro-priate time intervals in keeping with manufacturer recom-mendations, or mortality from infected solutions can result.9 During the course of a work shift, single-source containers may be replaced several times during the com-pounding process—all of which is performed within an ISO class 5 environment. Strict aseptic practices must be followed by the pharmacist during these changes of addi-tive source containers to reduce the risk of contamination. Minimum performance standards for quality assurance of compounding procedures should also be established. If changes in the accuracy of delivering a single additive (eg, potassium chloride) exceed a predetermined percent error, a plan for implementing corrective actions should be in place. All large-volume PN components (ie, amino acids, dextrose, and lipid emulsions) should be acquired from 1 manufacturer unless published data exist to sub-stantiate the safety, compatibility, and stability of alterna-tive final admixture formulations.
Compounding personnel should inspect the physical appearance of the final PN formulation.10 Although microprecipitates and incompatibilities cannot be seen by the naked eye, large-particle contaminants or precipitates are the most likely to immediately harm the patient, and they can easily be detected before the product is dispensed. For PN formulations without lipid emulsions, the product should be inspected against a dark back-ground for the presence of particulates, such as fibers from alcohol swabs, cores of additive vials, or insoluble precipitates. Compounding personnel should also look for
by Karrie Derenski on April 1, 2013ncp.sagepub.comDownloaded from
any signs of crystallization or for haziness of the product. For PN admixtures with lipid emulsions, compounding personnel should examine the bag for any signs of a cracked emulsion, including separation of components into an oil layer or into fat droplets. Finally, access and use of any ACD equipment for PN preparation should only be available to personnel who have been appropri-ately trained and demonstrated competency in this area.8
Final Checks and Tests
Labeling
The American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.) Safe Practice guidelines outline best prac-tices for labeling PN (Figure 1).10 This format provides the most straightforward way of displaying ingredients in the formulation, as well as other required elements for intra-venous compounds, including route of administration, date and time for administration, beyond-use date, dosing weight, and rate. Compounding personnel should have a standard procedure for verifying the contents of the fin-ished product by comparing the finished labeled PN product to the original order.1 The best way is to have a double-checking procedure in place. If additives are added manually to the bag, original containers (ie, vials) should be grouped with the final product; if the entire vial is not used, a syringe pulled back to the volume of the product measured should accompany the vial and the final product.
Storage and Use of PN Admixtures
A beyond-use date should appear on the PN label. A beyond-use date is different from an expiration date. Whereas expiration dates are based on rigorous tests for the chemical and stability of manufactured sterile products, beyond-use dates are based on the risk level of the CSP with consideration given to the time and temperature at which the preparation will be stored. According to USP Chapter <797> standards, a beyond-use date for a PN admixture prepared for inpatient administration is 30 hours based on storage at room temperature and a medium-risk–level (see Table 1). In the home-care environment, the beyond-use date can be extended to 9 days as long as PN admixtures are stored at 2°-8° C (36°-46° F) until use. The 30-hour time limit still applies once PN infusion is initiated.
Summary
Compounding of PN is not without its risks, but the stan-dards outlined in USP Chapter <797> greatly reduce risks of contamination of microbial or particulate matter. Most pharmacies will need to change their physical
design to provide space for an anteroom and buffer area surrounding direct compounding areas. Washing and garbing are important steps in maintaining a clean envi-ronment, as is regular cleaning with approved agents. Quality assurance tests, including testing of air; aseptic technique; and personnel compliance with standard oper-ating procedures are important to maintain a clean envi-ronment with minimal airborne particulates. Although many of these changes may be difficult and expensive to achieve, they will all lead to safer compounding practices and provide the patient with the best end product.
References
1. Chapter <797> Pharmaceutical compounding—sterile preparations. In: Revision Bulletin, The United States Pharmacopeia. Rockville, MD: The United States Pharmacopeial Convention; 2008:1-61.
2. Campos L, Lobianco L, Seki L, Santos RM, Asensi MD. Outbreak of Enterobacter hormaechei septicaemia in newborns caused by contaminated parenteral nutrition in Brazil. J Hosp Infect. 2007; 66:95-97.
3. Habsah H, Zeehaida M, Van Rostenberghe H, et al. An outbreak of Pantoea spp in a neonatal intensive care unit secondary to con-taminated parenteral nutrition. J Hosp Infect. 2005;61:213-218.
4. Marais E, Stewart R, Duse A, Rosekilly IC, de Jong G, Aithma N. Candida parapsilosis detected in TPN using the BacT/Alert system
Figure 1. Reprinted from Mirtallo J, Canada T, Johnson D, et al; Task Force for the Revision of Safe Practices for Parenteral Nutrition. Safe practices for parenteral nutrition [erratum in JPEN 2006;30:177]. JPEN J Parenter Enteral Nutr. 2004;28:S39-S70.
by Karrie Derenski on April 1, 2013ncp.sagepub.comDownloaded from
446 Nutrition in Clinical Practice / Vol. 24, No. 4, August/September 2009
and characterized by randomly amplified polymorphic DNA. J Hosp Infect. 2004;56:291-296.
5. Selenic D, Dodson D, Jensen B, Arduino MJ, Panlilio A, Archibald LK. Enterobacter cloacae bloodstream infections in pediatric patients traced to a hospital pharmacy. Am J Health Syst Pharm. 2003;60:1440-1446.
6. The ASHP Discussion Guide on USP Chapter <797> for Compounding Sterile Preparations: Summary of Revisions to USP Chapter <797>, developed by the American Society for Health-System Pharmacists in collaboration with Baxter Healthcare Corporation. Bethesda, MD: ASHP, Inc.
7. U.S. Food and Drug Administration. Compliance policy guides manual. Sec. 460.200 pharmacy compounding. Reissued May 29,
2002. http://www.fda.gov/ora/compliance_ref/cpg/cpgdrg/cpg460-200.html. Accessed January 29, 2009.
8. American Society of Health-System Pharmacists. ASHP guidelines on the safe use of automated compounding devices for the prepa-ration of parenteral nutrition admixtures. Am J Health Syst Pharm. 2000;57:1343-1348.
9. Two children die after receiving infected TPN solutions. Pharm J. 1994;252:596.
10. Mirtallo J, Canada T, Johnson D, et al; Task Force for the Revision of Safe Practices for Parenteral Nutrition. Safe practices for parenteral nutrition [published correction appears in JPEN J Parenter Enteral Nutr. 2006;30:177]. JPEN J Parenter Enteral Nutr. 2004;28:S39-S70.
by Karrie Derenski on April 1, 2013ncp.sagepub.comDownloaded from