33
Corrective and Preventive Action Pitfalls and Keys to Success Geoff Habiger, G Habiger Consulting LLC
| Date post: | 09-Aug-2015 |
| Category: |
Documents |
| Upload: | geoff-habiger |
| View: | 60 times |
| Download: | 0 times |

Corrective and Preventive Action
Pitfalls and Keys to Success
Geoff Habiger, G Habiger Consulting, LLC

In FY2014, 21CFR211.192 was the 3rd most citedparagraph in 483 observations. (Source: FDA Inspectional Observation Summaries)
“There is a failure to thoroughly review [any unexplained discrepancy] [the failure of a batch or any of its components to meet any of its specifications] whether or not the batch has been already distributed.”
Why are CAPAs important?
Section 211.192 says (in part):All drug production and control records…shall be reviewed and approved by the quality control unit to determine compliance with all established, approved written procedures before a batch is released or distributed. Any unexplained discrepancy…or the failure of a batch or any of its components to meet any of its specifications shall be thoroughly investigated, whether or not the batch has already been distributed. The investigation shall extend to other batches of the same drug product and other drug products that may have been associated with the specific failure or discrepancy. A written record of the investigation shall be made and shall include the conclusions and followup.

Okay, so we know that CAPAs are important, and we have a CAPA system in place. Isn’t that enough?
Simply put – no. The FDA looks at CAPAs during their inspections and will cite a company for CAPAs that do not meet established procedures.
Why are CAPAs important?
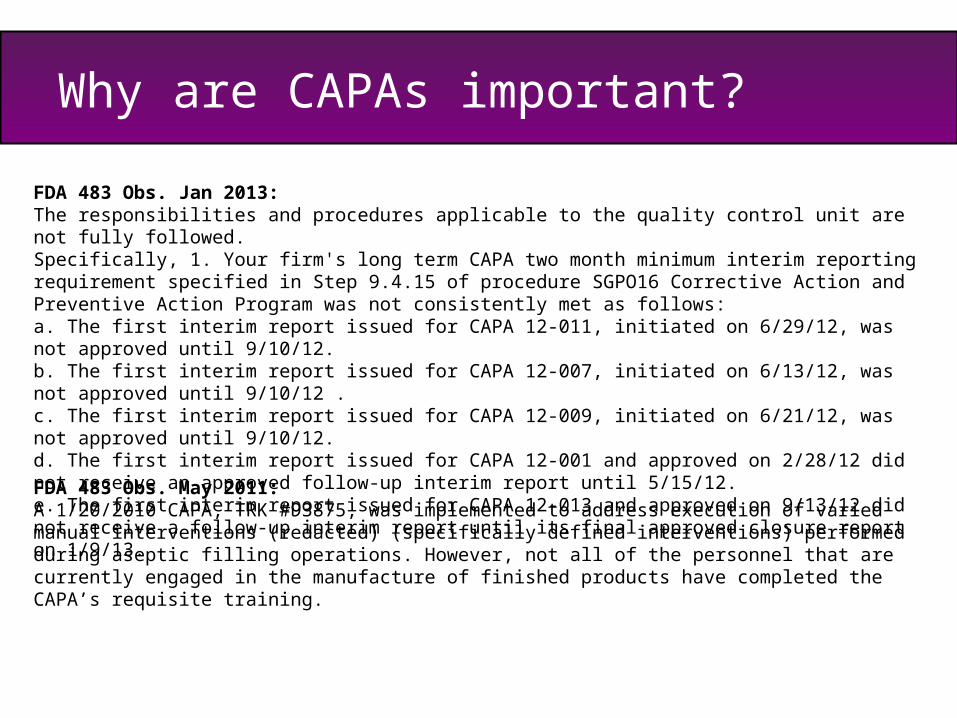
FDA 483 Obs. Jan 2013:The responsibilities and procedures applicable to the quality control unit are not fully followed. Specifically, 1. Your firm's long term CAPA two month minimum interim reporting requirement specified in Step 9.4.15 of procedure SGPO16 Corrective Action and Preventive Action Program was not consistently met as follows: a. The first interim report issued for CAPA 12-011, initiated on 6/29/12, was not approved until 9/10/12. b. The first interim report issued for CAPA 12-007, initiated on 6/13/12, was not approved until 9/10/12 . c. The first interim report issued for CAPA 12-009, initiated on 6/21/12, was not approved until 9/10/12. d. The first interim report issued for CAPA 12-001 and approved on 2/28/12 did not receive an approved follow-up interim report until 5/15/12. e. The first interim report issued for CAPA 12-013 and approved on 9/13/12 did not receive a follow-up interim report until its final approved closure report on 1/9/13.
FDA 483 Obs. May 2011:A 1/20/2010 CAPA, TRK #93875, was implemented to address execution of varied manual interventions (redacted) (specifically defined interventions) performed during aseptic filling operations. However, not all of the personnel that are currently engaged in the manufacture of finished products have completed the CAPA’s requisite training.
Why are CAPAs important?

FDA 483 Obs July 2012:b) According to the firm's Deviation and CAPA Management Procedure, status reports are required every (redacted) calendar days and "any delays shall be promptly documented and justified in the respective section/form" (See Section titled, "Overall Timing Requirements"). The status report #87754 states in the Section titled "Extension Request / Justification of Delay", "In addition, this Status Report covers Status Report missed on May 2012" and gives no explanation as to why the status report was "missed."
We know that the FDA will look at our quality systems, including CAPA system. And your point is?
Why are CAPAs important?

Knowing the FDA will look at your CAPA system is the first step to making that system robust.
How do you make it robust?
By avoiding common Pitfalls and making use of Keys to Success!
Why are CAPAs important?

Try not to be like Calvin and avoid these CAPA pitfalls. We want to fix our problems, not blame somebody else or the process.
© Bill Watterson
Why are CAPAs important?

Opening a CAPA without buy in from the responsible lead or department that the CAPA is necessary.
• Have clear ownership of the CAPA and clarity of roles. (owner, lead, reviewer, approver, facilitator, etc.) – results in accountability and empowerment.
• Have management engagement. QA shouldn’t be the only driver.
• Start the CAPA process with a strategy meeting with key personnel to discuss the event and make plans.
CAPA Initiation

CAPAs shouldn’t be a surprise that they are coming. The responsible lead and stakeholders should know about the CAPA before it is opened. Discuss the need for the CAPA in advance with all stakeholders to avoid opening unnecessary or repeat CAPAs.
CAPA Initiation

• Opening without notifying stakeholders.• Not notifying when project gates/CAPA stages are
met, next part is ready.
• Timely and accurate notification of all stakeholders for all stages of the CAPA.
• Communication and removing silos are important to keep all parties updated on the CAPA and to allow for quick response in the event roadblocks or problems are identified.
CAPA Notification

• Lack of/poor root cause. (Only identifying causal factors or symptoms and not true cause.)
• Opening for immediate corrections. (Jumping to solutions.)• Not identifying a preventive action, then having the issue
reoccur.
• Use root cause analysis (Brainstorming, 5-Why, Fishbone, 6M, Process Mapping, Is/Is Not) consistently to find true root cause.
• Have a well defined problem statement for the event/cause.
Root Cause

Know the differences between a correction (action taken immediately for containment), corrective action (action taken to correct the problem), and preventive action (action taken to prevent recurrence on same or other systems/process, etc.)
Root Cause

If you have many CAPAs for similar events (not just the same event), then you are only putting on Band-Aids and not fixing the problem. You may need to re-evaluate the original root cause.
Root Cause

Define the root cause method used in the investigation so it is clear to a reader how you reached the conclusion. e.g. Chronology of Events, 5-Why Analysis, and 6M Analysis were used to determine the root cause. These tools provide a comprehensive look at the event to be able to determine the root cause. Cross-functional team members included Manufacturing Investigator (name), Manufacturing Supervisor (name), and API Scientist (name).
Root Cause

• Action items are not specific.• Changing/modifying action items without
notifying responsible person. • Working on action items alone – within a silo.
• Well defined action plan with roles, due dates, and anticipated effectiveness checks. Follow S.M.A.R.T process (Specific, Manageable, Attainable, Realistic, Timely).
• Action items – even simple ones – will require input and assistance from multiple people in different departments. It’s critical to work together as a team to meet the CAPA goals.
Action Items

Action items should name the person who will do the work (not just department), exactly what will be done, due date for completion, and proposed effectiveness check. Avoid generalities and vague statements. This allows for ease of implementation and provides accountability.
Action Items

Issue: Scale was set to the wrong units of measure.
CAPA ##### (Parent PR####) was previously initiated on 26Nov2012 to revise procedure (number) Rev. (rev.) (effective date 20Dec2011) “(title)”. The revision will include instruction for performing a daily weight check before initial daily use of the top loader balance and bench top scale in manufacturing. The results of the weight check will be documented in a log book. The revision to the procedure does not impact the validated state, regulatory filing, product quality, or product stability. The revision will improve the process for using scales and balances in the manufacturing area, alerting operators to potential issues with a scale or balance before use.
Responsible Person: Manufacturing Investigator (name): Due Date: 26Mar2014. This due date was determined to be acceptable based on Status Report #### as allowing enough time to complete the necessary tasks of revising the procedure and creating the logbooks.
An effectiveness check was identified in CAPA #####. The effectiveness check will be performed 30 days after the revision of (procedure) becomes effective and will ensure logbooks are present and being used for each balance and scale.
Responsible Person: Manufacturing Investigator (name): Due Date: 26Apr2014
Examples

Issue: Foreign material found during packaging of product batch.
CAPA ###### was opened on 25Nov2013 to remove cardboard boxes from Processing Area XX (PA-XX). Specifically, the box containing drum covers stored in a supply cabinet will be removed and replaced with a plastic storage bin or tote. Other cardboard boxes, not including boxes used to hold gloves or kimwipes, will be removed and replaced with a plastic storage bin or tote. For boxes removed and replaced, place a label on the new container to identify the contents of the material stored in the container. Perform an inspection of other Processing Areas at the facility for similar cardboard boxes and replace them with plastic bins or totes as necessary, being sure to label the container for the contents.
This CAPA will not impact product quality, product stability, regulatory filing, or the validated state. Replacing the cardboard boxes is a preventive measure to improve the housekeeping in the Processing Areas and reduce the risk of extraneous matter from cardboard sources. Responsible Person: Manufacturing Supervisor (name): Due Date: 31Dec2013. This due date has been determined to be appropriate based on the manufacturing schedule.
Examples

Effectiveness Check ##### has been opened to verify the effectiveness of CAPA ######. The effectiveness check will perform a review 6 months after completion of CAPA ######. Perform a (system) query for extraneous matter investigations where the material was identified or linked to a cardboard source. There will be no investigations for cardboard being identified as the source of an extraneous matter investigation in the manufacturing Processing Areas. If an event for extraneous matter related to cardboard is identified conduct an investigation to determine the source and cause.
Examples

• To short or “immediate”• Not long enough based on processes and
procedures.• Unrealistic – overpromising and underdelivering
• Timely completion.• Setting realistic due dates based on sound reasoning
and accounting for variables such as production schedule, procedures, vacations, holidays, etc.
Responsible Person: Manufacturing Supervisor (name): Due Date: 31Dec2013. This due date has been determined to be appropriate based on the manufacturing schedule.
Due Dates

• Measurement not defined or vague• Not timely (performed late, or not allowing enough
time to properly determine)
Creating and implementing continuous monitoring into the system/process to measure effectiveness. e.g. continuous monitoring of equipment over the use of chart recorders, continuous fill weight monitoring versus periodic fill checks, etc.
Effectiveness Checks

• Lack of documentation• Documents not up to date• Documents sloppy and unclear.• CAPA logs are not up to date
• Quantitative and Objective outcomes are fully documented. • Include copies of all relevant documents and records. Should
be identified as attachments following documentation procedures.
• CAPA log is updated in real-time when actions are completed.
Documentation

If you didn’t write it down, it didn’t happen!
Create your own CAPA file and put copies of all documents there. This creates a backup and allows you to have a record of everything you worked on. This can have benefits outside of just CAPA or Investigations, such as for performance evaluations. (What did you work on the last quarter/year?)
Documentation

Treat your CAPA records like any other original records. Are the final records controlled? Are they stored like other records to be protected from fire and/or water? What is the retention policy for completed CAPAs?
Documentation

The FDA and other auditors will make judgements about your entire Quality System based upon the CAPA and Exception Event (investigation) logs used to track these systems. If these logs are a mess, the auditor will assume there are other problems in the system.
Documentation

• Not containing the problem.• Not documenting the containment.• Not properly assessing risk – containing the wrong
thing or not enough containment.
Perform a thorough risk assessment•Determine scope of impact (fence setting)•Clearly documenting and performing the containment for the duration of the CAPA.•Assess risk to all systems – validation, regulatory, product, stability.
Containment

Containment actions might involve using planned deviations or temporary changes to document the containment and rationale. It’s important to document the containment and the justification why the containment is acceptable. (How will this protect the product/customer until the problem is fixed by the CAPA?)
Containment

A proper risk assessment should happen as part of the investigation. A separate risk assessment is not needed for the CAPA, unless the CAPA is stand alone and not tied to an investigation.
Containment
Not aligning a software system (tool) with the CAPA process.
Not all investigations will require a CAPA, and not all CAPAs must come from an investigation!
CAPA Tools

Communication and education with staff on the changes and the reason for them. This is more than just training, but an understanding of what the problem was and why the change was implemented. This creates buy in with the end users of the system/process.
Communication

Make your CAPA reports easy to read. Consider using short sentences, small words, and avoid (or spell out) acronyms – even if you think they are common. Use headings and subheadings to break up the text and convey information quickly. Write reports using an active voice.
“On 15Apr15, John Doe, manufacturing operator, reported that the product method in batch record XXXXX did not match the method stored in the Chromatography System.”
CAPA Reports

Create a CAPA Review Board (CRB)
• Meet periodically (monthly/quarterly) to discuss CAPAs.• A formal process for updates to CAPAs, reviews, progress, issue
identification and mitigation.• Monitors actions, supporting documentation, outputs, and metrics. • An ongoing assessment of corrective actions and implementation of
preventive actions.• Involve Quality Management (minimum) and other management as needed.• Create metrics to monitor CAPA system (cycle time closure, CAPA type
(SOP/BR revision, training, equipment, etc.), department, missed gates/due dates)
Review Board

Questions?

















