Cardiovascular, utero- and fetoplacental function in mice during normal
pregnancy and in the absence of endothelial nitric oxide synthase
(eNOS)
Shathiyah Kulandavelu
Doctor of Philosophy
Department of Physiology University of Toronto
2010
Abstract
In pregnancy, the maternal cardiovascular and placental circulation undergoes structural
and functional changes to accommodate the growing fetus, but the mechanisms involved are not
fully understood. Nitric oxide (NO) increases in normal pregnancy and lack of NO has been
implicated in pregnancy related complications, preeclampsia and fetal growth restriction. Thus,
the objective of the thesis was to determine if cardiovascular, uteroplacental and fetoplacental
changes observed in human pregnancy also occur in mice and to assess the obligatory role of
eNOS in mediating these changes.
I showed that like humans, mice exhibit increases in maternal cardiac output, stroke
volume, plasma volume, and uterine arterial blood flow, and a transient decrease in arterial
pressure during pregnancy. Importantly, I showed that endothelial nitric oxide synthase (eNOS)
plays an important role in promoting the progressive increase in maternal cardiac chamber
dimensions and output and the enlargement of the aorta during pregnancy in mice. Another
novel finding was that eNOS plays an important role in remodeling of the uterine and umbilical
vasculatures during pregnancy. The remodeling of the uterine vasculatures, including the uterine
iii
and spiral arteries, were blunted in the eNOS KO mice with ko fetuses (KO(ko)) and this likely
contributed to elevated vascular resistance and reduced perfusion of the uterine circulation
during pregnancy. Impaired spiral artery remodeling may be caused by a deficiency in decidual
uterine natural killer cells. Fetal placental vascularization was also impaired in eNOS KO(ko)
mice, which likely increased vascular resistance and thereby reduced fetoplacental perfusion.
Reduced vascularization may be due to decreased VEGF mRNA and protein expression in
KO(ko) placentas. Decreased perfusion in both the uterine and umbilical circulations most likely
contributed to elevated placental and fetal hypoxia in the eNOS KO(ko) mice. Interestingly,
despite placental hypoxia, eNOS KO(ko) mice do not show the classical signs of preeclampsia
including hypertension and proteinuria nor are maternal plasma sFlt1 levels elevated.
Nevertheless, eNOS KO(ko) pups are growth restricted at term, and this is mainly due to the fetal
genotype. These findings suggest that eNOS plays an essential role during pregnancy in
remodeling of the maternal heart, aorta, and uterine and umbilical vasculatures thereby
augmenting blood flow to the maternal and fetal sides of the placenta and thereby promoting
fetal growth in mice.
iv
Acknowledgements
All that I have accomplished during my PhD years would not have been possible without
the guidance, encouragement and support of many wonderful people. I would like to take this
opportunity to express my appreciation and to acknowledge these individuals, to whom I am
greatly indebted. First and foremost, I would like to express my sincere gratitude to my
supervisor, Dr. S. Lee Adamson – for her keen scientific training, steadfast guidance and
mentorship, and on a personal level, for being incredibly supportive and understanding
throughout my PhD adventures. It has been a pleasure working in your lab as a volunteer,
summer student and as a PhD student for nearly a decade. Thank you for providing me with the
foundation for my scientific training.
I would also like to thank my supervisory committee members Dr. Theodore Brown, Dr.
Steve Lye and the late Dr. Lowell Langille for their scientific guidance, experimental advice,
helpful criticism and honest commitment in supporting my development as a scientist.
Throughout the years, I have had the opportunity to work with some wonderful labmates
who have become my lifelong friends. In particular, I wish to thank Zorana Berberovic, Nora
Jones, Igor Vukobradovic, Carol Akirav, Jennifer Whiteley and Dr. Carole Watson and Dr. Nana
Sunn. Special thanks to Dr. Beth Acton and Dr. Maryam Yeganegi for being my “PhD buddies”
and for providing me with both personal and scientific advice. Thank you all for your
unwavering support, stimulating discussions and most of all your friendship. It has been a
pleasure working with each and every one of you, and I hope that our friendship will last for
many years to come.
Technical support was instrumental to many of my experiments, for which I would like to
thank Dr. Dawei Qu (for his amazing surgical skills, patience and kindness), Dr. Junwu Mu and
Dr. Yuqing Zhou (for being my ultrasound teachers), Kathie Whiteley (for her amazing attention
to detail), and Dr. Qiang Xu (for his immunohistochemistry expertise). I would also like to
recognize all the members of the Adamson lab (both past and present) who made it a pleasure to
go into work each day.
I would like to express my gratitude to all the funding sources for the work contained in
my thesis. Funding for this work was provided by Canadian Institute of Health Research, Heart
v
and Stroke Foundation of Ontario Fellowship, Ontario Graduate Scholarship, Lorne Phenix
Award, University of Toronto Open Scholarship and Genesis Research Foundation from the
Department of Physiology, Al and Hannah Perly Graduate Student Scholarship and Heart &
Stroke/Richard Lewar Centre of Excellence Fellowship. Also, thanks to Cardiovascular Sciences
Collaborative program and Samuel Lunenfeld Research Insitute for providing funding for
numerous travel awards.
Finally, I would like to express my heartfelt thanks and appreciation to my family. To
my amazing parents, thank you for your continued and unwavering support. Without your love,
strength, encouragement and guidance, I would not be where I am today. It is an honor being
your daughter and my achievements are the result of your love and dedication.
vi
Table of Contents
ACKNOWLEDGEMENTS ............................................................................................. IV
TABLE OF CONTENTS ................................................................................................ VI
LIST OF TABLES........................................................................................................... X
LIST OF FIGURES ........................................................................................................ XI
LIST OF ABBREVIATIONS AND ACRONYMS.......................................................... XIII
CHAPTER 1 – LITERATURE REVIEW...........................................................................1
1.1 General Introduction ....................................................................................................................................2
1.2 Cardiovascular and placental changes in human pregnancy ....................................................................3 1.2.1 Maternal cardiovascular changes in human pregnancy ..............................................................................3 1.2.2 Uteroplacental changes during pregnancy..................................................................................................9 1.2.3 Umbilico-placental changes during pregnancy.........................................................................................14
1.3 Nitric oxide and its role in pregnancy........................................................................................................16 1.3.1 Nitric oxide...............................................................................................................................................16 1.3.2 Nitric oxide as it relates to pregnancy ......................................................................................................18 1.3.3 Regulation of eNOS expression and activity............................................................................................21 1.3.4 Regulators of eNOS enzymatic activity ...................................................................................................25 1.3.5 Nitric oxide signaling ...............................................................................................................................31
1.4 Nitric oxide and complications of pregnancy............................................................................................33 1.4.1 Preeclampsia.............................................................................................................................................33 1.4.2 Nitric oxide in preeclampsia.....................................................................................................................37 1.4.3 Intrauterine growth restriction ..................................................................................................................38 1.4.4 Nitric oxide in intrauterine growth restriction ..........................................................................................40
1.5 Mice as a models of human pregnancy......................................................................................................41 1.5.1 Similarities and differences between mice and humans ...........................................................................42 1.5.2 eNOS knockout mice................................................................................................................................45
vii
1.6 Thesis hypothesis and objectives................................................................................................................48
CHAPTER 2 - CARDIOVASCULAR FUNCTION IN MICE DURING NORMAL PREGNANCY AND IN THE ABSENCE OF ENOS .......................................................50
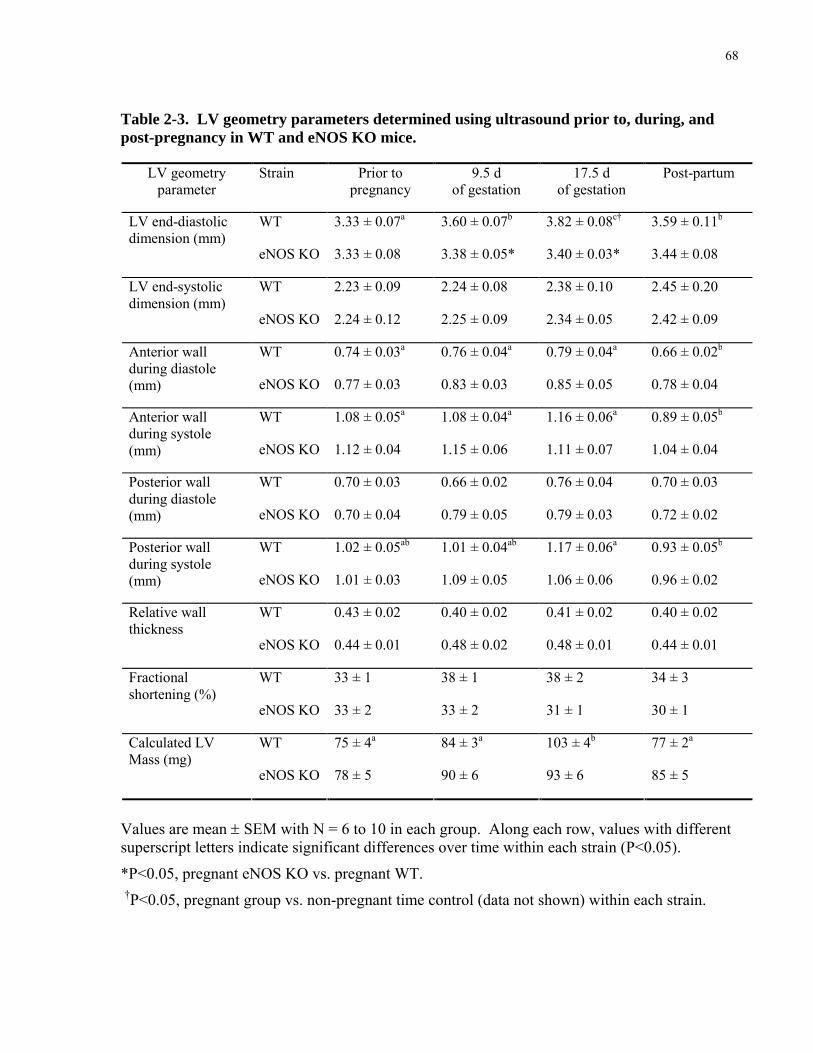
2.2 MATERIAL AND METHODS..................................................................................................................52 2.2.1 Breeding and genotyping..........................................................................................................................52 2.2.2 Hemodynamics.........................................................................................................................................53 2.2.3 Left ventricular geometry .........................................................................................................................56 2.2.4 Arterial blood pressure and heart rate in awake mice...............................................................................56 2.2.5 Hematology of maternal blood .................................................................................................................57 2.2.6 Plasma Volume determination..................................................................................................................57 2.2.7 Statistical Analysis ...................................................................................................................................58
2.3 RESULTS ....................................................................................................................................................58 2.3.1 Cardiovascular changes during pregnancy in WT mice are similar to humans........................................58 2.3.2 eNOS is required for the normal increase in cardiac output during pregnancy ........................................60
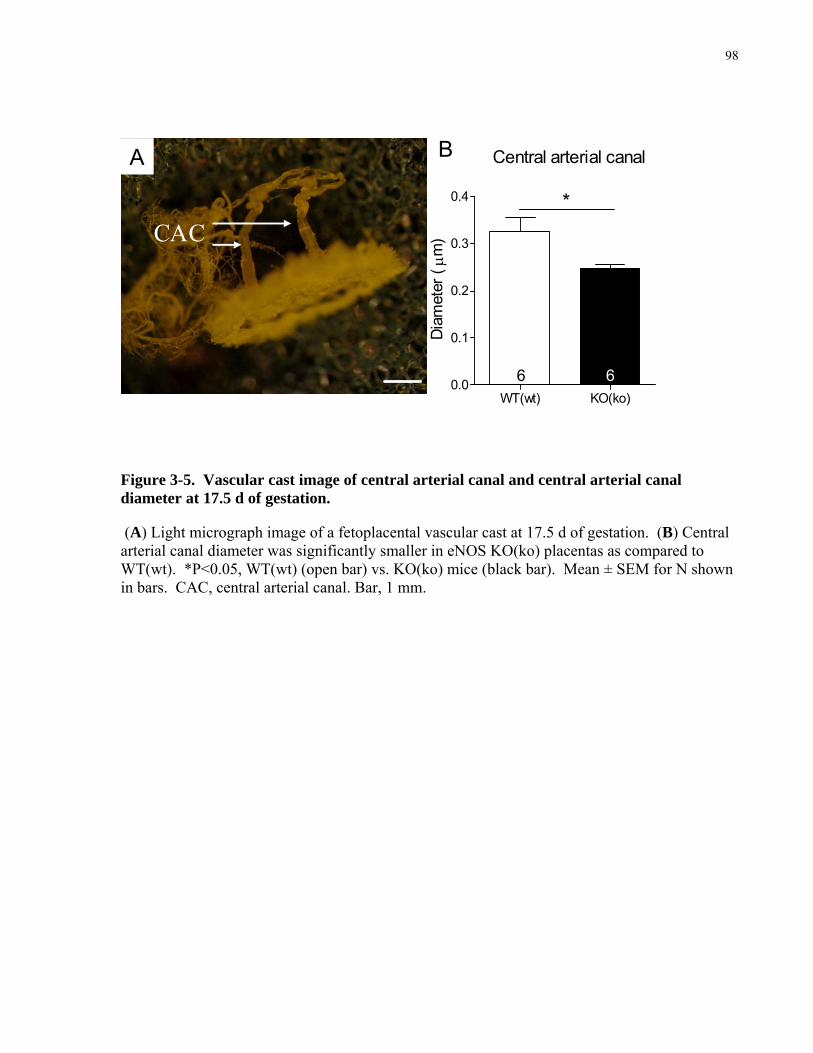
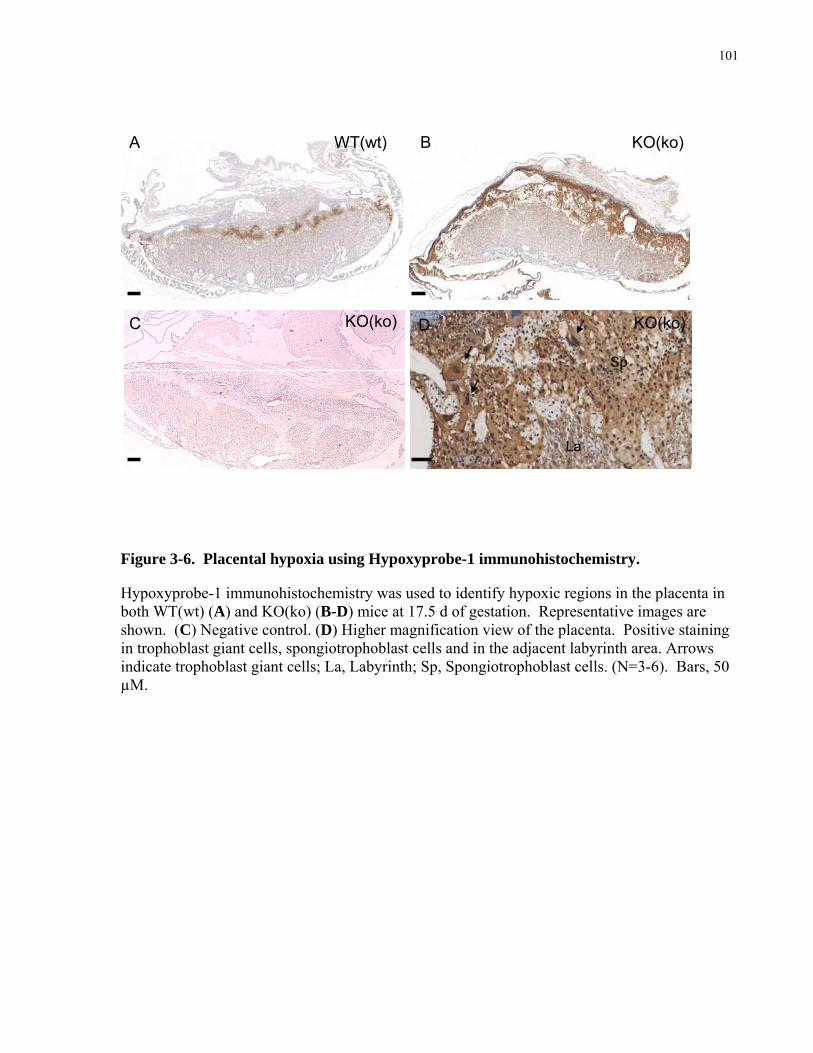
3.2 MATERIAL AND METHODS..................................................................................................................82 3.2.1 Breeding ...................................................................................................................................................82 3.2.2 Uterine Arterial Hemodynamics...............................................................................................................82 3.2.3 Uteroplacental Vascular Casts..................................................................................................................83 3.2.4 Detection of Placental Hypoxia................................................................................................................84 3.2.5 Immunohistochemistry of vascular smooth muscle cells and histochemistry of uNK cells. ....................85 3.2.6 RT-qPCR for sFlt1 mRNA and Flt1 mRNA ............................................................................................86 3.2.7 ELISA of plasma sFlt1 .............................................................................................................................87 3.2.8 Clinical Biochemistry of maternal blood..................................................................................................87 3.2.9 Statistical Analysis ...................................................................................................................................87
3.3 RESULTS ....................................................................................................................................................88 3.3.1 Fetal, placental, and maternal growth in late gestation in eNOS KO(ko) mice........................................88
viii
3.3.2 Reduced uteroplacental blood flow and elevated uteroplacental vascular resistance at mid- and late-
gestation in eNOS KO(ko) mice .............................................................................................................................90 3.3.3 Reduced remodeling of the spiral and central arterial canals in eNOS KO(ko) mice ..............................94 3.3.4 Role of maternal versus fetal genotype on uteroplacental phenotype. .....................................................99 3.3.5 Increased placental hypoxia in eNOS KO(ko) mice.................................................................................99 3.3.6 Reduced placental expression of sFlt1 mRNA levels and no significant changes in maternal sFlt1 levels
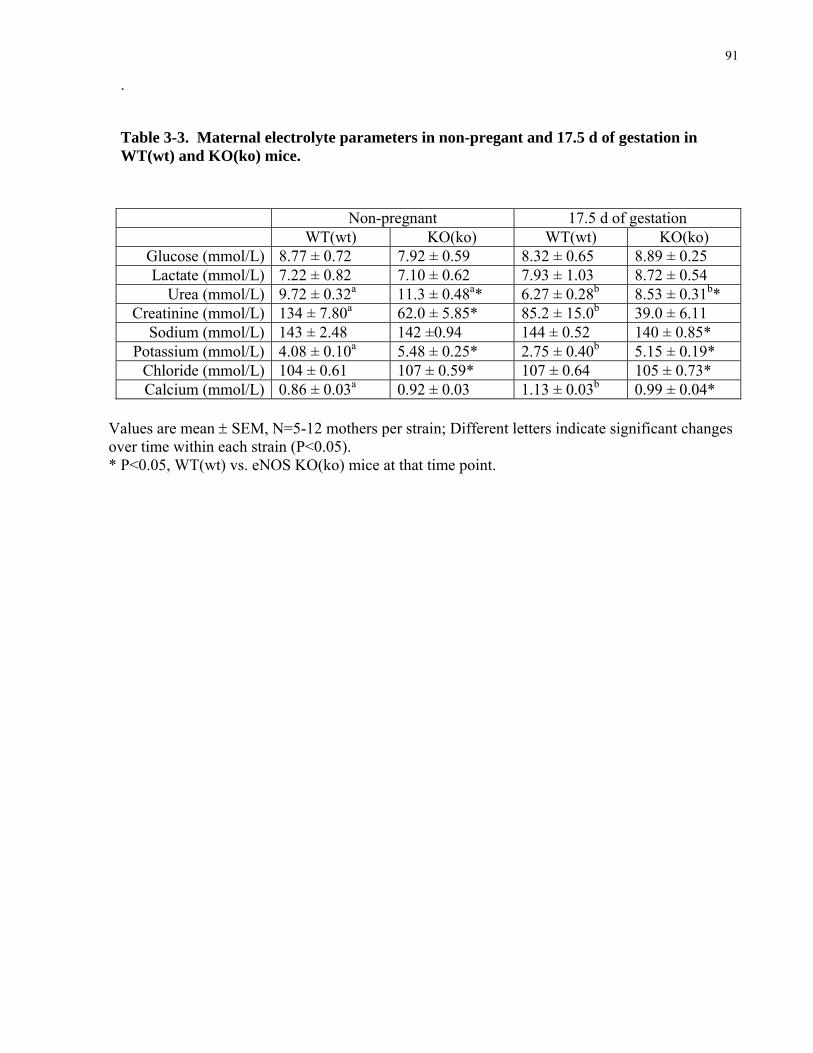
in eNOS KO(ko) mice ..........................................................................................................................................100 3.3.7 Maternal electrolyte balance is altered in pregnant eNOS KO(ko) mice................................................103
CHAPTER 4 – UMBILICO-PLACENTAL STRUCTURAL AND FUNCTIONAL CHANGES IN MICE DURING PREGNANCY IN WILD-TYPE AND IN ENOS KNOCKOUT MICE ......................................................................................................112
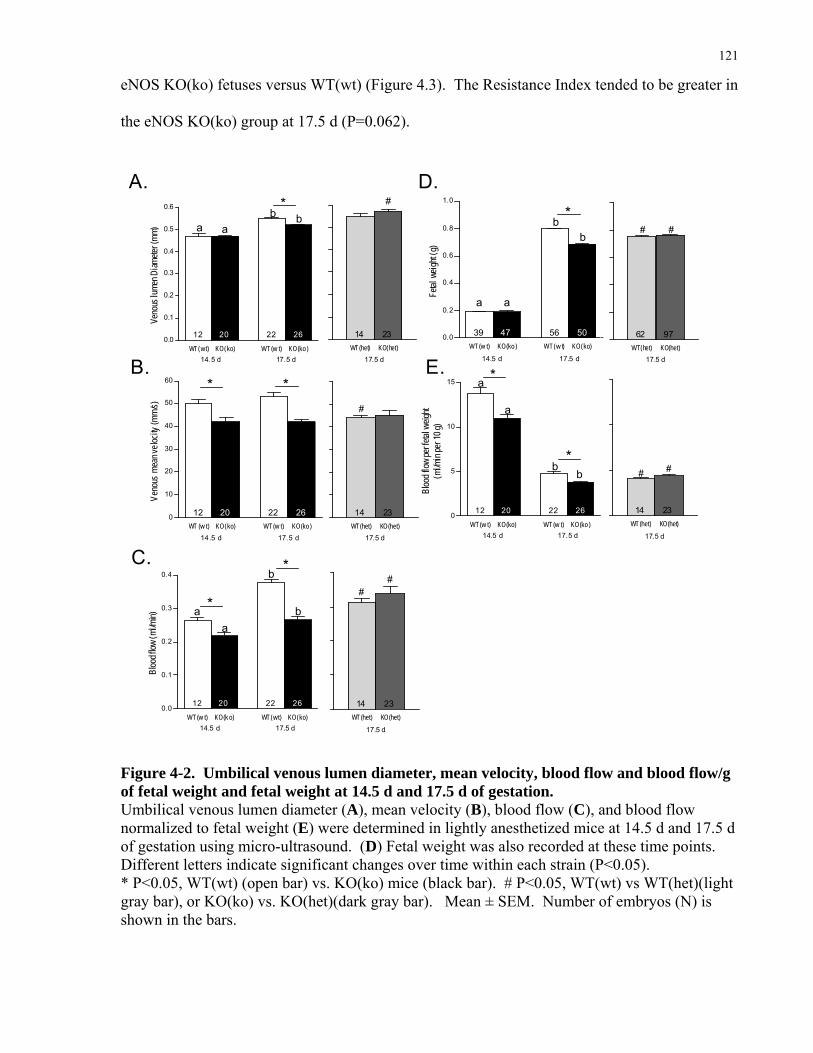
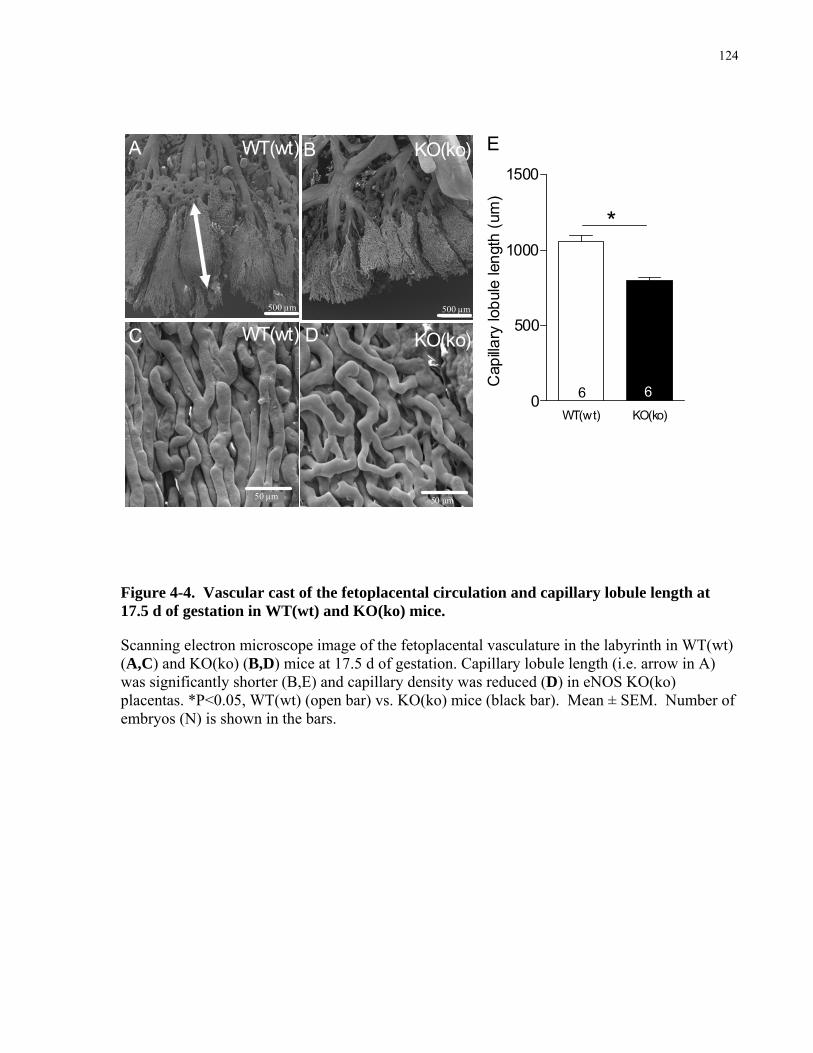
4.2 MATERIAL AND METHODS................................................................................................................114 4.2.1 Breeding .................................................................................................................................................114 4.2.2 Umbilico-placental Hemodynamics .......................................................................................................115 4.2.3 Fetoplacental vascular casts ...................................................................................................................117 4.2.4 Detection of Hypoxia in the embryo ......................................................................................................118 4.2.5 Immunohistochemistry and RT-qPCR for VEGF ..................................................................................118 4.2.6 Hematology of fetal blood......................................................................................................................119 4.2.7 Statistical Analysis .................................................................................................................................119
4.3 RESULTS ..................................................................................................................................................120 4.3.1 Reduced fetoplacental blood flow at mid- and late gestation in eNOS KO(ko) mice. ...........................120 4.3.2 Fetoplacental vascularization and placental expression of VEGF are reduced in eNOS KO(ko) fetuses.
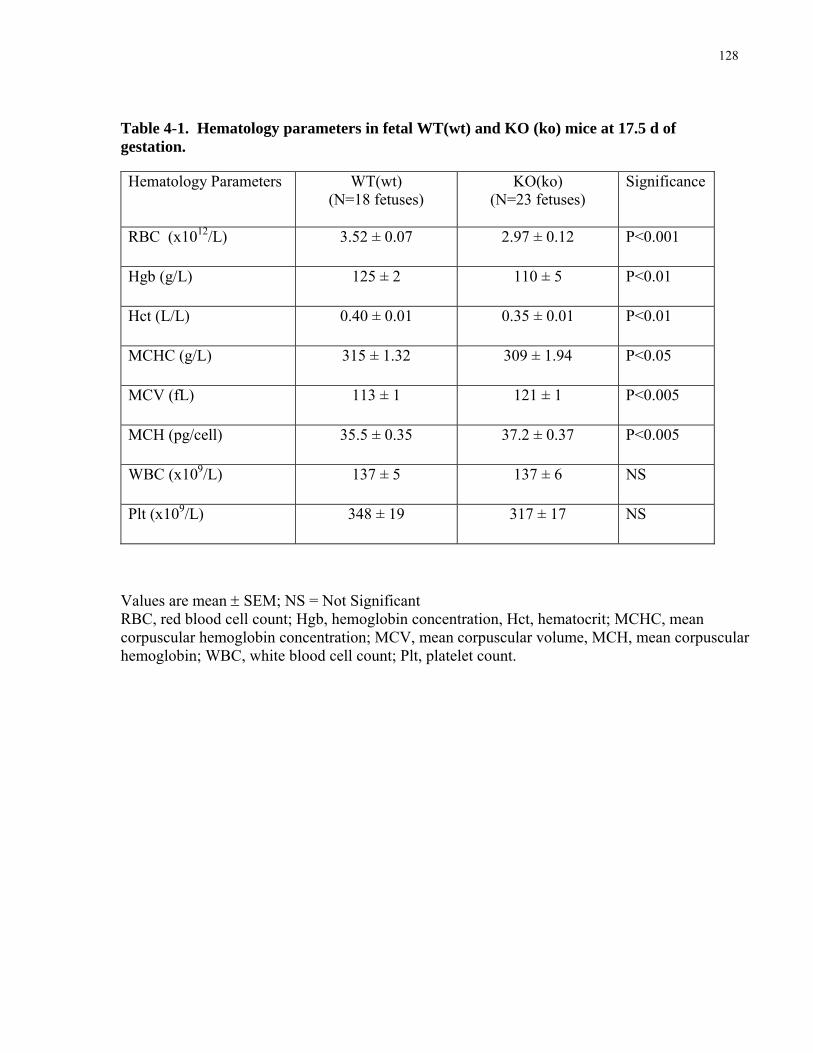
..................................................................................................................................................................123 4.3.3 eNOS KO(ko) pups are hypoxic and anemic and show increased erythrocyte size. ..............................126 4.3.4 Fetal growth is determined by fetal genotype.........................................................................................129
CHAPTER 5 – GENERAL DISCUSSION & FUTURE DIRECTION ............................137
5.1 General Discussion ....................................................................................................................................138
5.2 Future Direction ........................................................................................................................................143
In normal pregnancy, the maternal cardiovascular, uteroplacental, and fetoplacental
systems undergo structural and functional changes to accommodate the increased circulatory
requirements placed on the mother by the growing fetus. A marked, early decrease in peripheral
vascular resistance is thought to be the primary event [1-3] leading to marked increases in
cardiac output, uterine arterial blood flow, and blood volume, and to a decrease in blood
pressure during pregnancy [1-3]. The fall in vascular resistance is aided by structural
reorganization of many vascular beds including the aorta, uterine and placental vasculatures [3-
5]. The mechanisms mediating these changes are poorly understood but important because their
failure likely underlies two of the most common and serious complications of human pregnancy,
preeclampsia and fetal growth restriction.
Pregnancy increases nitric oxide (NO) production in humans and in other species
including rats and sheep [6-8]. Beyond its vasodilatory effect, NO has a number of other
beneficial roles, including promoting remodeling of the vasculature and angiogenesis. These
effects are most likely mediated specifically by the endothelial nitric oxide (eNOS) isoform
because eNOS and NO levels are elevated in the aorta, myocardium, uterine and umbilical
vasculature and in the placenta during pregnancy, whereas nNOS and iNOS levels remain
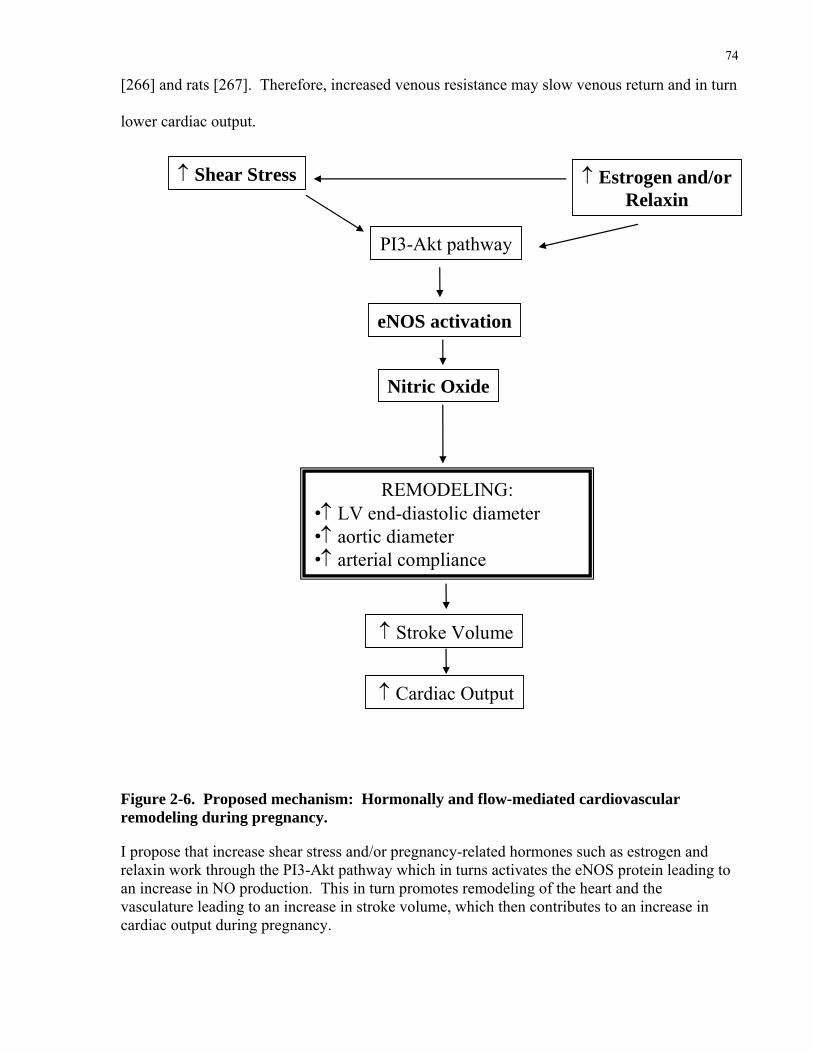
unchanged [7, 9-12]. eNOS activity is elevated by factors such as shear stress, estrogen and
vascular endothelial-derived factor (VEGF) [13-15], all of which increase in these tissues during
pregnancy [16-20]. Furthermore, inhibition of NOS using non-selective NOS inhibitors caused
preeclamptic symptoms including hypertension, decrease in plasma volume and fetal growth
restriction [21, 22]. Therefore, now with the availability of eNOS KO mice, we can study the
obligatory role of eNOS in mediating cardiovascular, uteroplacental, and fetoplacental changes
during normal pregnancy and establish its role in pregnancy-related complications.
3
1.2 Cardiovascular and placental changes in human pregnancy
1.2.1 Maternal cardiovascular changes in human pregnancy
There are striking physiological cardiovascular changes during human pregnancy. The
ultimate goal of the hemodynamic response to pregnancy is to provide adequate uteroplacental
perfusion for fetal development without compromising maternal function. Pregnancy-induced
alterations in cardiovascular function are due to a complex interplay between circulating
humoral factors and functional and structural alterations that occur within the heart and the
vascular tissue.
Cardiovascular function is presumably augmented in pregnancy to meet the increasing
metabolic demands of the conceptus; however, interestingly, most of the cardiovascular changes
begin during the first eight weeks of gestation, and therefore precede any major increase in
metabolic demand [2, 18, 23]. Also, women in their post-ovulatory or luteal phase of the
menstrual cycle demonstrate systemic hemodynamic changes identical to early pregnancy [24].
Thus, the initial changes in cardiac performance do not require the presence of the conceptus
and are likely mediated by hormones derived from maternal tissues such as the ovaries and
decidua [2, 25, 26]. The conceptus likely plays a larger role during late gestation because the
increase in cardiac output is redistributed to the uteroplacental unit to provide nutrients to the
growing fetus [18]. The growing fetus and placenta also secrete hormones such as estrogens
and progesterone, that augment and/or sustain changes in maternal cardiovascular function [18,
26, 27].
A marked, early decrease in peripheral vascular resistance (30%) is thought to be the
primary event [1-3]. However, arterial pressure decreases only slightly (10-15%) because of a
4
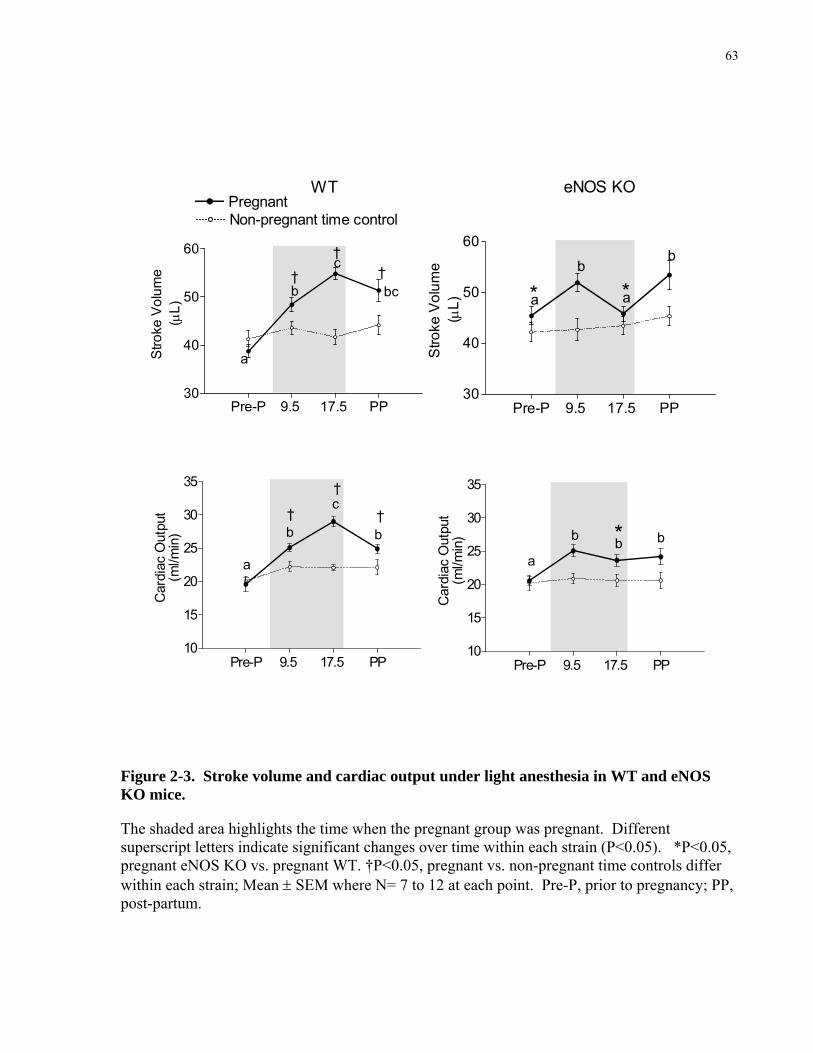
concurrent increase in cardiac output (30-60%) [2, 3, 18]. This increase in cardiac output is due
to increases in both heart rate (20-30%) and stroke volume (30-35%) [2, 3, 18, 28]. Heart rate
increases gradually throughout pregnancy [2, 28]. This rise may be attributed to changes in the
autonomic nervous system: increased sympathetic and decreased parasympathetic activity [2].
In addition to the nervous system, relaxin, a pregnancy related hormone, may also be involved
in regulating heart rate [29, 30]. Stroke volume is increased in normal pregnancy by a
combination of factors, including increased preload, decreased afterload, improved myocardial
function (diastolic & systolic) and structural growth of the heart (Figure 1.1).
Preload & Afterload: The early decrease in peripheral vascular resistance is thought to be
caused by vasodilation which contribute to a fall in afterload [1]. Enlargement of the
cardiovascular system caused by vasodilation induces arterial and venous underfilling that
initiates nonosmotic release of arginine vasopressin, and activation of the renin-angiotensin-
aldosterone system [31]. This in turn leads to sodium and water retention resulting in an
increase in plasma volume (45-55%) [18, 31, 32]. The systemic venous system undergoes
vasodilation which enhance venous capacitance and thereby accommodate this increase in
plasma volume [33, 34]. There is also enhanced erythropoiesis [35] which leads to an increase
in the total volume of circulating red blood cells (15-20%) [18, 32]. These increases in plasma
and red blood cell volumes cause an increase in blood volume (40-60%) and therefore increases
cardiac preload [18]. Both the increase in preload and decrease in afterload contribute to a rise
in stroke volume in pregnancy.
5
Cardiac output Arterial pressure
Heart rate Stroke volume
Peripheral vascular resistance
Sinoatrial Node
Autonomic Nervous system
Geometry Preload Afterload
Anatomy
Ventricular relaxation & compliance
End-diastolic radius
Blood VolumeContractility
Aortic diameter & compliance
Venoustone
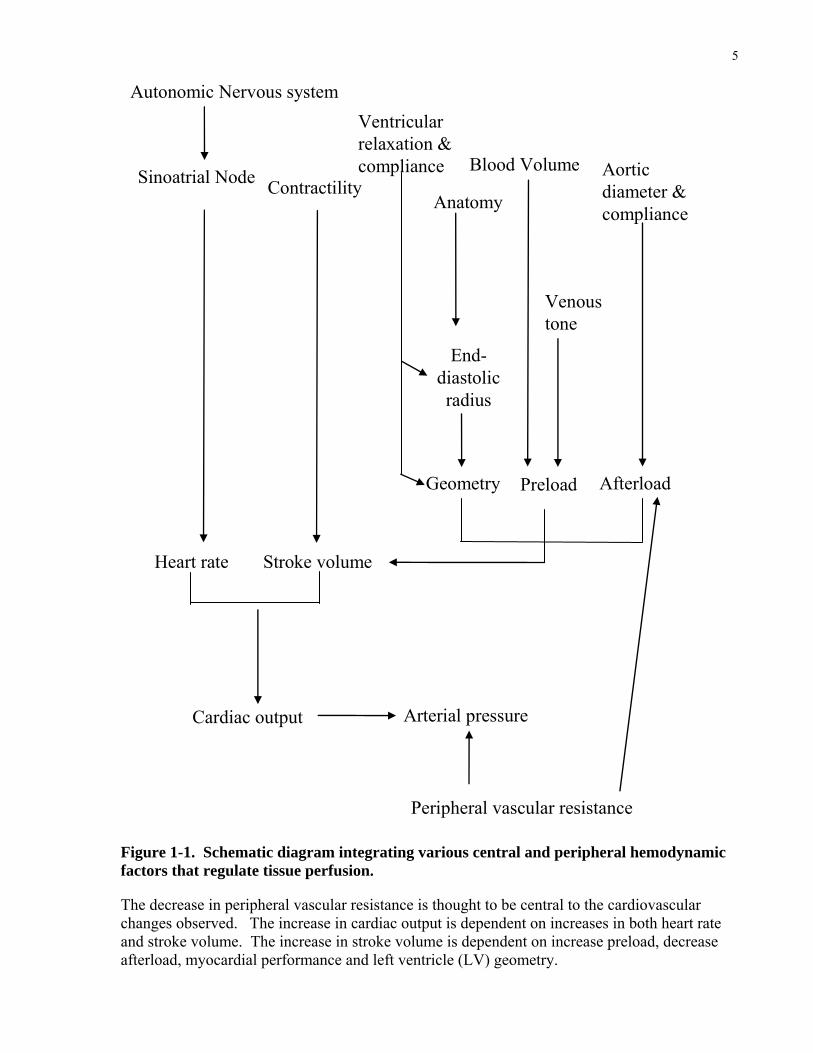
Figure 1-1. Schematic diagram integrating various central and peripheral hemodynamic factors that regulate tissue perfusion.
The decrease in peripheral vascular resistance is thought to be central to the cardiovascular changes observed. The increase in cardiac output is dependent on increases in both heart rate and stroke volume. The increase in stroke volume is dependent on increase preload, decrease afterload, myocardial performance and left ventricle (LV) geometry.
6
Diastolic function: Diastolic filling of the heart depends on a complex sequence of interrelated
events. In early diastole, ventricular filling is due to myocardial relaxation and passive recoil.
In late diastole, filling depends on strength of atrial contraction, and myocardial viscoelastic
properties [36, 37]. These interrelated contributing factors are highly sensitive to changes in
loading conditions, heart rate, contractility, and nonuniformity of myocardial relaxation [36, 37].
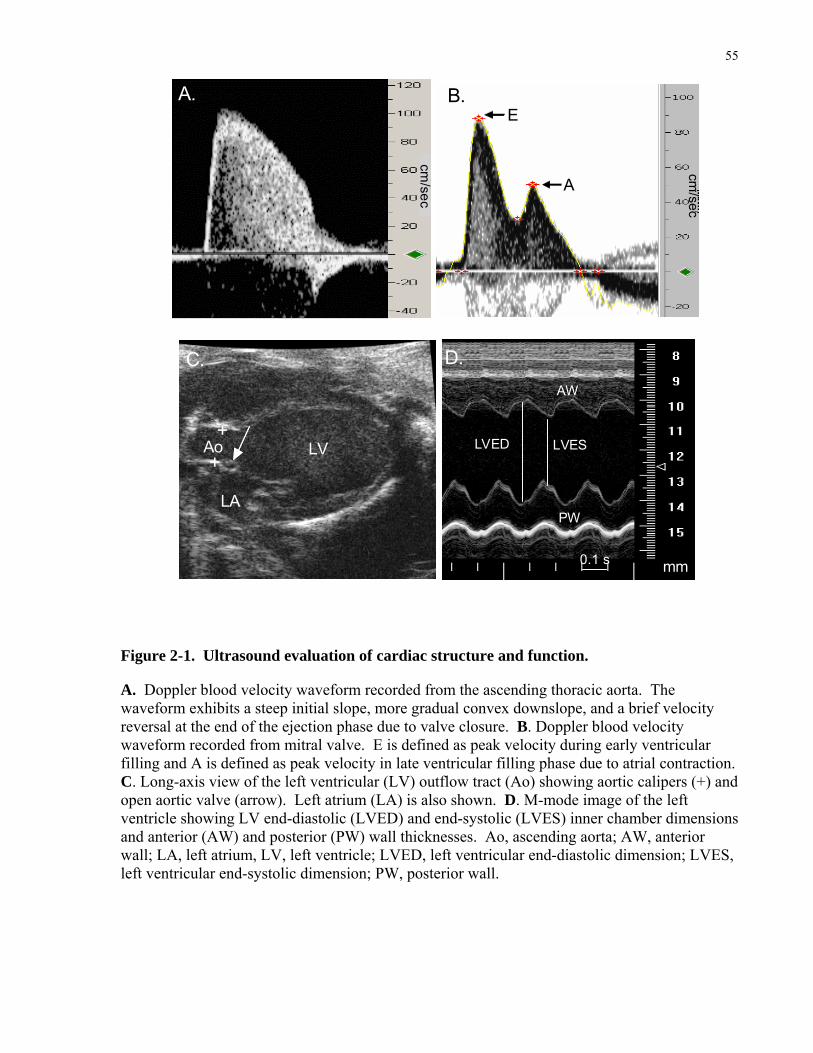
Diastolic function is routinely quantified using peak E and A waves and peak E/A ratio. E wave
is defined as peak velocity during early ventricular filling and A wave is defined as peak
velocity in late ventricular filling phase due to atrial contraction. Therefore, peak E/A ratio is
most often used to quantify ventricular diastolic function. During human pregnancy, there is an
increase in peak E wave velocity during the first trimester and it remains elevated till term,
whereas the peak A wave velocity increases maximally in third trimester [36-38]. Therefore the
E/A ratio is highest during the first trimester [38]. The E wave is high in early gestation because
during this time LV elastic recoil is vigorous and myocardial relaxation is swift so filling is
completed during the early diastole period and only a small amount of filling occurs at atrial
contraction [36, 38]. The A wave is increased late in gestation because there is a greater plasma
volume and hence a greater atrial volume to be moved during atrial contraction [36, 38].
Myocardial contractility is the ability of the ventricle to eject blood against a given load. It is
determined by the number of muscle cells activated (a function of ventricular mass and
conduction) and the force of contraction of individual muscle cells. Increases in myocardial
contractility could contribute to the increase in cardiac output in pregnancy. However, the
evaluation of contractility in pregnancy has produced conflicting results. Some studies have
found that LV myocardial contractility either increased [39], decreased [40, 41] or remained
unchanged [42] during pregnancy. This controversy could be because most ultrasound
7
measures of myocardial performance do not accurately quantify intrinsic contractility due to
their dependence on loading conditions.
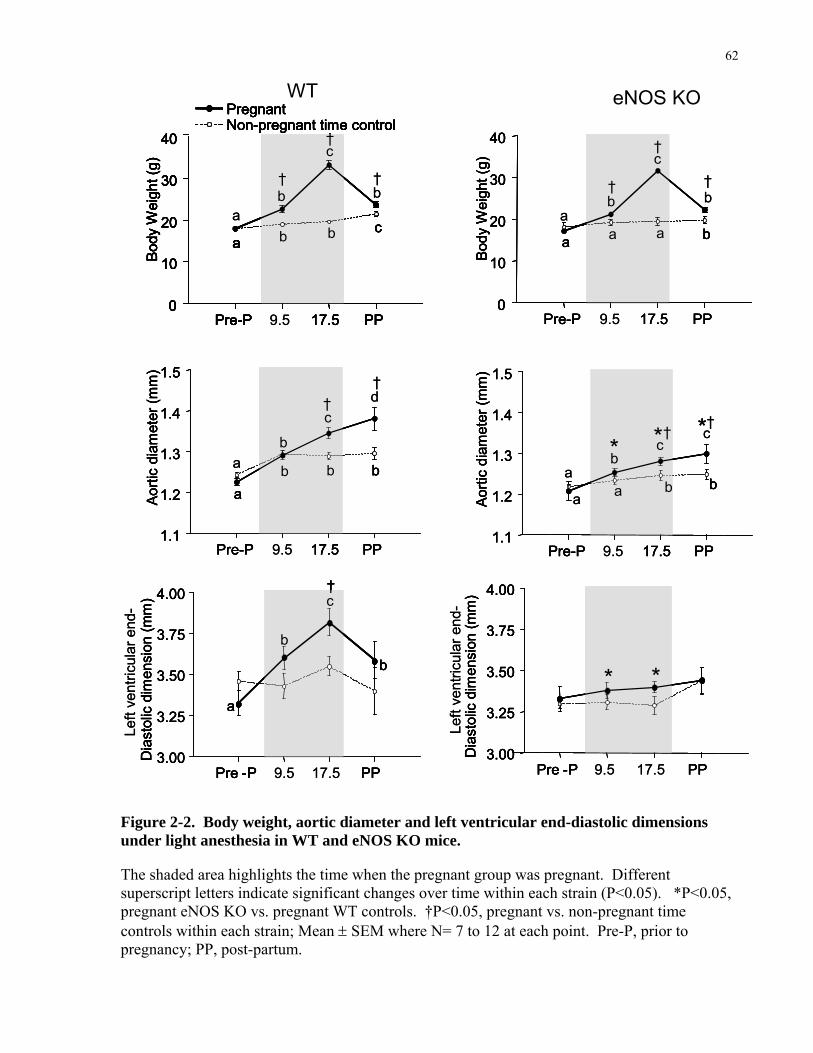
Structural changes of the heart: Left ventricular (LV) mass increases by about 50% during
pregnancy due to 15-25% increases in LV wall thicknesses and 10-20% increases in LV end-
systolic and end-diastolic dimensions [3, 37]. Cardiac hypertrophy of the heart along with the
mechanisms involved are discussed next.
Cardiac Hypertrophy during pregnancy: To accomplish the increase in cardiac output during
normal pregnancy, the maternal heart modifies its shape and its volume [37]. But since the heart
is a terminally differentiated organ [43, 44], its adaptations to increased workload are
accomplished mainly by increasing muscle mass through hypertrophic remodeling (i.e. increase
in cell size rather then cell number). Recently, it has been proposed that a small subpopulation
of cycling cardiomyocyte coming from the differentiation of cardiac stem-like cells could
marginally contribute to cardiac adaptation [44, 45]. However, it is widely accepted that cardiac
hypertrophy rather than regeneration is responsible in large part for the adaptation to increased
demands for cardiac work.
Cardiac hypertrophy is defined as an increase in cardiomyocyte size that can be a
beneficial and adaptive (physiological) or a maladaptive (pathological) phenomenon to
compensate for the hemodynamic stress resulting from pressure or volume overload [46].
Pressure overload, as seen in chronic hypertension and aortic valve stenosis, induces concentric
hypertrophy which is characterized by increases in wall thickness without significant changes in
8
chamber size [47]. Volume overload, as seen in pregnancy, exercise and post-natal
development, induces eccentric hypertrophy characterized by chamber enlargement with a
proportional change in wall thickness [47]. Physiological hypertrophy is reversible and occurs
without morbid effects on cardiac function, whereas pathological hypertrophy can lead to
morbid effects on cardiac function [46, 47]. The mechanisms leading to hypertrophy during
both pathological and physiological states are distinct but, in general, evidence indicates that
hypertrophy results from the interaction of mechanical forces and hormonal factors.
Stimuli for myocardial hypertrophy include stretching of the myocardial fibers, growth
and gut (for increased absorption). Particularly in late gestation, a critical end-organ for
perfusion is the uterus.
1.2.2 Uteroplacental changes during pregnancy
The uteroplacental vascular bed undergoes the most dramatic cardiovascular alternations
during pregnancy. Uterine blood flow increases from <100 mL/min at 10 weeks of gestation to
700-800 mL/min at term [18, 54, 55]. This increase in uteroplacental blood flow is also directly
related to the number of concepti (e.g., triplets > twins > singletons) [18].
Since blood pressure normally decreases during pregnancy, the increase in uterine
arterial blood flow is principally effected by a profound decrease in uterine vascular resistance.
This is accomplished by several different but complimentary mechanisms, including enhanced
vasodilation of uterine and uteroplacental vessels, enlargement of the uterine artery and
downstream vascular tree, and angiogenesis [4, 56-61].
10
It is difficult to measure uterine vascular resistance directly in human pregnancy, so
simple non-invasive uterine arterial Doppler indices have been used to assess successful
pregnancies. From uterine arterial blood velocity waveforms, peak systolic velocity and end-
diastolic velocity are measured, from which the Resistance Index is calculated. Resistance
Index is an indicator of resistance in the downstream vasculature [62]. In humans, a non-
pregnant uterine artery waveform has a prominent diastolic notch which is taken as another
indicator of high downstream vascular resistance [63]. The diastolic notch in the uterine artery
waveform is normally not detected past 26 weeks of pregnancy. Also, end-diastolic blood
velocity increases more rapidly with gestational age than systolic blood velocity, such that
Resistance Index decreases progressively, reaching ~0.5 at term [63, 64]. This suggests a
decrease in vascular resistance in the uteroplacental circulation with gestation.
Role of Blood Flow in Vascular Remodeling: The vascular system is continuously exposed to
changes in hemodynamic forces. The endothelial layer is located between the flowing blood
and the smooth muscle cells and the connective tissue of the tunica media. The endothelium is
critical in sensing changes in flow and signaling these changes to the underlying and
downstream smooth muscle cells. These signals are translated into a wide range of biological
and biochemical reactions that control smooth muscle tone. The types of vascular remodeling
as proposed by Mulvany [65] can be broadly categorized as changes in vessel diameter (inward
or outward) and/or changes in wall mass (increased i.e. hypertrophic; decreased i.e. hypotrophic;
unchanged i.e. eutrophic).
Alterations in blood flow alter shear stress resulting in the release of endothelium-
derived factors that diffuse to the underlying smooth muscle cells [66, 67]. Acute changes in
11
blood flow lead to short-term changes in luminal diameter caused by vasodilation and
vasoconstriction [67]. When this is sustained chronically, this leads to synthesis and activation
of compounds that influence cell growth, apoptosis, migration and reorganization of the
extracellular matrix [66, 68-70]. These changes result in architectural modifications in the
vessel wall. The arterial restructuring is most likely mediated by matrix metalloproteinases
(MMPs), because the expression of MMP-2 and MMP-9 increases after enhanced blood flow
and chronic inhibition of MMPs prevents the expansive remodeling [71]. The vascular response
to both acute and chronic changes in blood flow tends to normalize wall shear stress.
Uterine, arcuate and radial artery remodeling: To accommodate the increase in blood flow
during pregnancy, the uterine artery undergoes circumferential enlargement [4]. The pattern of
circumferential remodeling is outward hypertrophic [4]. The diameters of the uterine artery, and
the arcuate and radial arteries that it supplies, all increase in size during pregnancy [72-74].
Luminal enlargement is mainly accomplished by increases in vascular smooth muscle cell
length (axial hypertrophy). This has been shown in the uterine vasculature of guinea pigs, rats
and sheep [75-77]. Surprisingly, no human data are available. In addition to cellular
hypertrophy, there is also strong evidence for hyperplasia within the vascular wall; increased
rates of smooth muscle cell division occur in pregnancy in uterine arteries and veins in rats and
guinea pigs [75, 78] and increased rates of endothelial cell division have been documented in
rats [75]. Thus, an increase in cell number also contributes to the enlargement of the uterine
artery.
12
Spiral artery remodeling: Downstream of the uterine artery, the spiral arteries undergo
modifications that are an essential feature of normal pregnancy. These physiological
transformations include: 1) elongation; 2) dilation; 3) loss of the muscular and elastic tissue of
the arterial wall; and 4) replacement with a thick layer of fibrinoid material [79-81]. These
changes create a high-flow, low-resistance vessel, and the destruction of the muscle layer leads
to loss of vasomotor control [79-81]. Collectively, these changes are thought to maximize the
delivery of maternal blood to the intervillous space by widening the arterial lumen, and by
reducing the responsiveness of these vessels to vasoconstrictor agents, thereby maintaining
continuous supply.
The mechanisms underlying spiral artery remodeling are incompletely understood. This
is largely because of the difficulty in obtaining human tissue. But it has been postulated that in
human pregnancy, the invading cytotrophoblasts play an essential role in remodeling of the
spiral arteries [81]. The invasive cytotrophoblast cells are derived from the conceptus [56, 57].
They are a differentiated form of the trophoblast cells that are responsible for the formation of
the placenta [57, 82]. The invading cytotrophoblast cells cause apoptosis of the vascular smooth
muscle cells triggered by paracrine signals [83, 84]. Elegant studies by Cartwright and
colleagues [83, 84] have shown that activation of the Fas/fas Ligand (FasL) and tumor necrosis
factor apoptosis inducing ligand (TRAIL) pathways are involved in trophoblast-induced
endothelial and smooth muscle cell apoptosis.
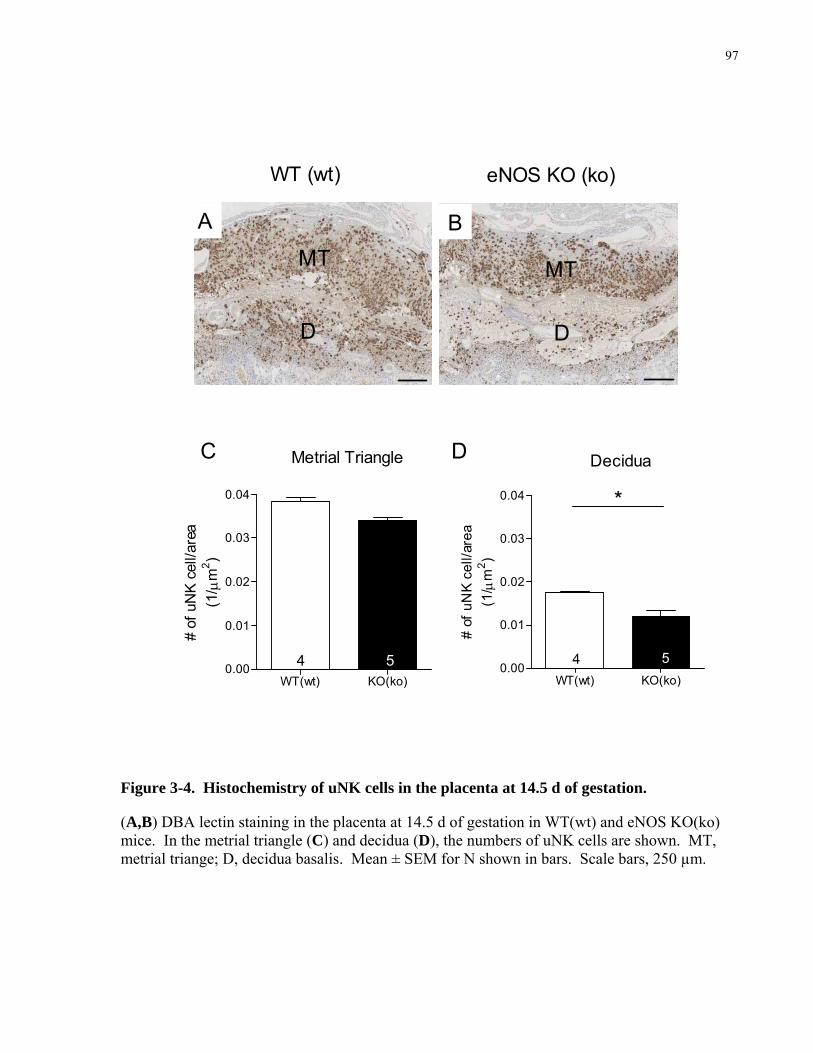
In addition to trophoblast cells, uterine natural killer (uNK) cells have also been
implicated in spiral artery remodeling [85, 86]. During the first trimester of human pregnancy,
uNK cells are a major cell population in the decidua, and account for 70% of the local
lymphocytes [87]. Four major possible roles of uNK cells in spiral artery remodeling have been
proposed.
13
(i) uNK cells control spiral artery remodeling by controlling trophoblast invasion. uNK
cells attract trophoblast by releasing chemokines, interleukin-8 (IL-8) and
interferon-inducible protein-10 (IL-10), which bind to receptors expressed on
invasive trophoblast cells [86].
(ii) uNK produce interferon- γ (IFN-γ), which is thought to participate in spiral artery
remodeling [85, 88]. IFN-γ may initiate this process by antagonizing transforming
growth factor- β (TGF-β) which normally functions to stabilize the blood vessel.
(iii) uNK also express angiopoietin-2 (Ang-2) [89]. Ang-1 and Ang-2 are both ligands
for Tie-2, a tyrosine kinase receptor. Ang-1 mediated phosphorylation of Tie-2
promotes endothelial cell survival and recruitment of pericytes and smooth muscle
cells that help to stabilize the newly formed capillaries. Ang-2 is a competitive
inhibitor of Ang-1, destabilizing the vessels and rendering them more susceptible to
the angiogenic stimulus of vascular growth factor (VEGF) and other growth factors
[5, 61, 85].
(iv) uNK produce proangiogenic factors, including VEGF and placental growth factor
(PlGF) [86, 90]. Both of these factors promote vessel elongation and dilation by
increasing growth. VEGF will be discussed in more detail in section 1.3.4.
In summary, uteroplacental blood flow is elevated during pregnancy due to decreased
vascular resistance. This decrease in vascular resistance is due in part to enlargement of the
uteroplacental vasculatures including uterine and spiral arteries. In addition to the
uteroplacental circulation, the fetoplacental circulation also undergoes tremendous alterations
during pregnancy.
14
1.2.3 Umbilico-placental changes during pregnancy
The umbilical circulation is crucial for fetal development and growth. Umbilical blood
flow increases from 100 mL/min at 22 weeks of gestation to 300 mL/min at 38 weeks [91] due
to increases in mean velocity and lumen diameter [92]. The blood flow increases throughout
pregnancy to meet the increased oxygen and nutrient demand placed by the rapidly growing
fetus.
Umbilical velocity waveform patterns have been used to assess adverse perinatal
outcome. Several indices have been used including (1) Resistance Index (RI): (RI = (peak
systolic velocity (S) – end-diastolic velocity (D))/S where S is the systolic maximum and D is
the diastolic minimum); (2) Systolic/Diastolic (S/D) ratio, and (3) Pulsatility Index (PI): (PI =
(S-D)/M where M is mean velocity over the cardiac cycle) [62, 92]. These indices tend to be
elevated when downstream vascular resistance is elevated [62]. In early human pregnancy,
when the placenta is superficial, and fetoplacental resistance is high, umbilical arterial end-
diastolic velocity is zero. Between 13 and 17 weeks, end-diastolic velocity progressively
increases and is normally present in all fetuses after 18 weeks of gestation [93]. The appearance
of end-diastolic velocity coincides with the end of organogenesis (~10 weeks), and therefore
appears to be caused by changes associated with the maturation phase of the placenta and/or
cardiovascular development [62, 94, 95].
Placental vascularity is increased throughout pregnancy and this contributes to a
decrease in peripheral vascular resistance [5]. Both vasculogenesis and angiogenesis are critical
for normal placental development [5]. Vasculogenesis involves de novo formation of blood
vessel from precursor cells, whereas angiogenesis involves the creation of new vessels from
already existing ones [5, 61, 95, 96]. Vasculogenesis is evident by about 21 days post
15
conception (dpc). During vasculogenesis, hemangiogenic stem cells differentiate to
hemangioblastic stem cells. These cells in turn differentiate into endothelial cells forming new
vascular networks. Shortly after the endothelial tubes are formed, they associate with pericytes
(future vascular smooth muscle cells). These pericytes then proliferate and migrate, coating the
endothelial cell tubes and forming new vessels [5, 61, 95, 96]. Angiogenesis is evident by about
32 dpc in the placenta [5, 61, 95, 96]. Angiogenesis is accomplished by either migration of
endothelial cells from preexisting vessels through the sprouting of endothelial cells (branching
angiogenesis) to form new vessels or by the elongation of the existing vessels (non-branching
angiogenesis) [5]. Branching angiogenesis predominantly occurs from day 32 dpc to 24 weeks
of gestation, whereas non-branching angiogenesis is observed from 24 weeks to term [5, 61, 95,
96]. Several factors have been identified as important regulators for both vasculogenesis and
angiogenesis, including vascular VEGF, PlGF, basic fibroblast growth factor, Ang-1 and Ang-2
[5, 61, 95, 96].
Altogether the described studies demonstrate that pregnancy is associated with extensive
anatomical and functional changes in the cardiovascular and placental systems to accommodate
the increased circulatory demands placed on the mother by the growing fetus. Essential factors
involved in mediating these changes are vasodilation and remodeling of the vasculature. The
endothelium releases a number of vasorelaxing compounds including nitric oxide (NO),
prostaglandins, and endothelium-derived hyperpolarization factor (EDHF) [97, 98]. These
vasodilating signals act on the vascular smooth muscle cell via two intracellular messengers,
cyclic guanosine monophosphate (cGMP) and cyclic adenosine monophophate (cAMP) [99,
100]. Nitric oxide is an important vasodilatory factor present in the vasculature. It has been
implicated as an essential mediator of normal pregnancy-related changes, and reduced NO
16
activity has been implicated in pregnancy-related complications such as preeclampsia and
intrauterine growth restriction. The role of NO in pregnancy-related cardiovascular and
placental changes is the main focus of my thesis.
1.3 Nitric oxide and its role in pregnancy
1.3.1 Nitric oxide
Furchgott and Zawadzki [98] were the first in 1980 to suggest the existence of
endothelium-derived relaxing factor (EDRF). Subsequently, Moncada and colleagues [101]
identified EDRF as NO. NO is a free radical gas that was initially identified as a vasodilator
produced in the endothelium. However, it is now known that NO is generated by a family of
enzymes known as nitric oxide synthases (NOS) that catalyze the conversion of cationic amino
acid L-arginine to L-citrulline and NO.
To date, three NOS isoforms have been identified that share 50-60% amino acid
sequence homology [102-105]. Two isoforms are constitutively expressed, although their
expression may be modulated: neuronal NOS (also known as nNOS, Type I, NOS I, NOS 1)
was the first isoform identified and is predominately expressed in neurons, but also in vascular
smooth muscle cells [102-105]. Endothelial NOS (also known as eNOS, Type III, NOS III,
NOS 3) is predominately expressed in arterial and venous endothelial cells, lymphatic
endothelial cells, endocardial cells, cardiac myocytes and platelets [102-105]. For a complete
list of cell types that express eNOS, the reader is referred to a review by Li et al [14]. The third
isoform is inducible NOS (also known as iNOS, Type II, NOS II, NOS 2), which is expressed
mainly in macrophages, but its activity has also been detected in other cell types including
17
endocardial and endothelial cells, vascular smooth muscle cells, fibroblasts, and neonatal and
adult cardiac myocytes [102-105].
All three NOS isoforms share a carboxy-terminal reductase domain homologous to the
cytochrome P-450 reductases and an amino-terminal oxygenase domain containing a heme
prosthetic group, which are linked roughly in the middle of the protein by a calmodulin-binding
domain [104, 106]. For structure of the eNOS isoform, see Figure 1.2.
Figure 1-2. Domains present in the eNOS isoform.
Electrons are donated by NADPH bound at the reductase domain, which are subsequently shuttled through the calmodulin-binding domain towards the heme-containing eNOS oxygenase domain, which results in the formation of enzyme products citrulline and NO. Post-translational modification sites: Myristoylation (Myr) and palmitoylation (Palm) sites. Arg (L-arginine), BH4 (tetrahydrobiopterin), FAD (flavin adenine dinucleotide), FMN (flavin mononuclotide), NADPH (nicotinamide adenine dinucleotide phosphate), Zn (zinc).
18
Binding of calmodulin appears to act as a molecular switch that enables electron flow
from flavin prosthetic groups in the reductase domain to heme, thereby facilitating the
conversion of O2 and L-arginine to NO and L-citrulline [104, 106, 107]. For eNOS and nNOS,
the physiological concentration of calcium regulates calmodulin binding and the flow of
electrons to heme, whereas for iNOS, calmodulin is tightly bound, even at lower concentrations
of calcium such that this molecular switch is always on [104, 107]. In addition to the binding of
calmodulin, activation of all three NOS isoforms requires tetrahydrobiopterin (BH4). BH4
appears to stabilize the dimeric structure of NOS and enhance the binding of L-arginine [104,
106, 108]. Reduced bioavailability of BH4 results in uncoupling of NOS, leading to superoxide
(O2-) and hydrogen peroxide (H2O2) production [108, 109]. Superoxide (·O2
-) radical is a
powerful oxidant which functions to inhibit mitochondrial electron transport, oxidizes proteins,
initiates lipid peroxidation and nitrates aromatic amino acids [110, 111].
NO has a number of beneficial roles in the vessel wall, including vasodilation [6],
Figure 1-3. Cellular events involved in the regulation of eNOS activity.
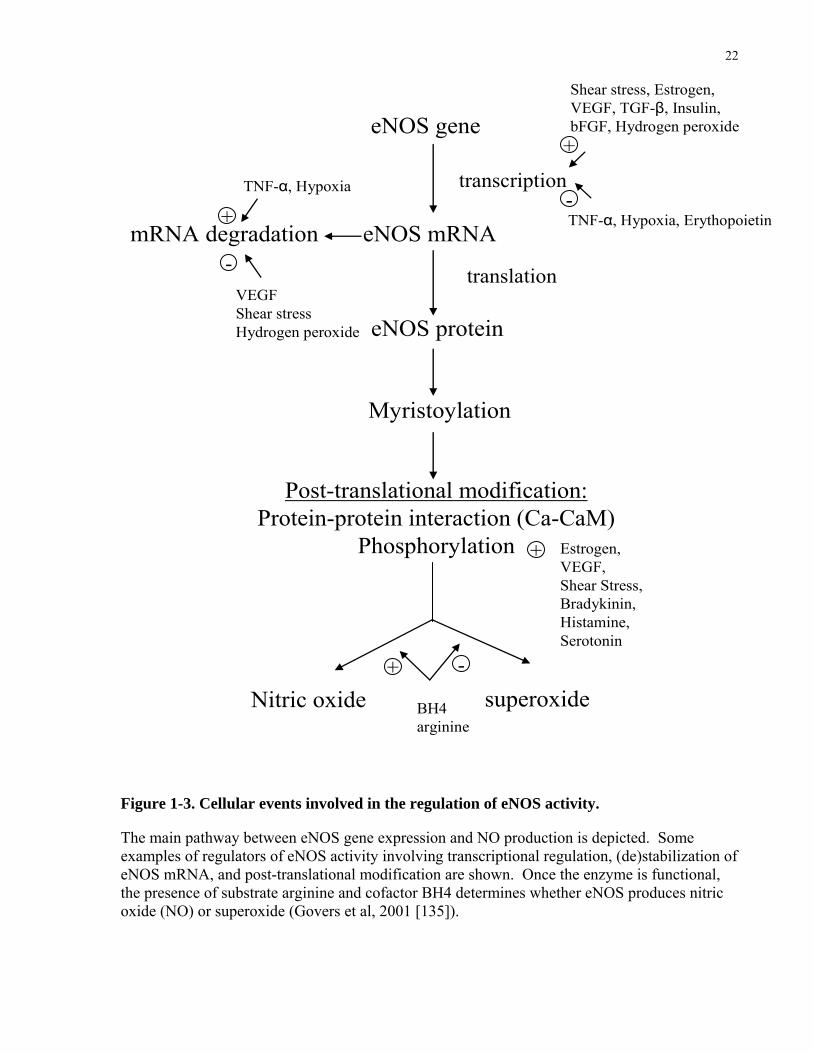
The main pathway between eNOS gene expression and NO production is depicted. Some examples of regulators of eNOS activity involving transcriptional regulation, (de)stabilization of eNOS mRNA, and post-translational modification are shown. Once the enzyme is functional, the presence of substrate arginine and cofactor BH4 determines whether eNOS produces nitric oxide (NO) or superoxide (Govers et al, 2001 [135]).
23
Translational, Co-translational, and Post-translational regulation of eNOS:
Among NOS isoforms, eNOS is unique, as it contains a myristoyl group. Myristoylation
facilates eNOS anchoring to the plasma membrane. The presence of eNOS at the membrane
may serve an important purpose. It may bring eNOS in close proximity to factors which are
required for its proper function, including arginine, calcium and cofactor BH4 [135].
Phosphorylation is a post-translational modification that regulates eNOS activity (Figure
1.4) [135, 136]. eNOS is primarily phosphorylated on serine (S) residues and to a lesser extent
on tyrosine (Y) and threonine (T) residues [135, 136]. Shear stress acting via G proteins can
activate several signal transduction pathways, including PI3K and adenylate cyclase (AC)
pathway, leading to eNOS activation via phosphorylation of serine residues (S617 and S1177
for Akt, and S635 and S1177 for PKA) [135-137]. Additional stimuli such as by VEGF or
estrogens can also alter eNOS phosphorylation. These substances bind to their cognate
receptors and stimulate the PI3K/Akt pathway, thereby augmenting eNOS phosphorylation as
above [15, 138]. They also activate phopholipase C-γ (PLC- γ) which increases cytoplasmic
can activate CaM kinase II, which phosphorylates eNOS on S1177. Increase in DAG levels also
can activate PKC to phosphorylate T497, which may negatively regulate eNOS or influence its
coupling to BH4 [139].
eNOS activity is also regulated by changes in the cytosolic Ca2+ concentration and is
therefore activated by hormones that induce a rise in intracellular calcium levels, such as VEGF,
estrogens, bradykinin, serotonin and histamine [135]. Increases in cytoplasmic concentration of
Ca2+ triggers the binding of Ca2+ to CaM and this complex then interacts with eNOS resulting in
increased eNOS activity [104, 135].
24
Figure 1-4. Protein phosphorylation is a post-translational modification that regulates eNOS activity.
eNOS is primarily phosphorylated on serine (S) and threonine (T) residues. Shear stress, estrogen and VEGF acting via their receptors activate various signal transduction pathways, including phosphoinoside 3-kinase (P13K), adenylate cyclase (AC) and phopholipase C-γ (PLC-γ) which lead to phosphorylation of eNOS protein leading to increased eNOS activity. DAG, diacylglcerol, IP3, inositol triphophate, PKC, protein kinase C, CaM, calmodulin, Akt, protein kinase B, CaMKII, calmodulin-dependent protein kinase, PKA, protein kinase A, ATP, adenosine triphophate, cAMP, cyclic adenosine monophophate (Sessa et al, 2004 [140]).
25
Shear stress, estrogen and VEGF regulate eNOS at the transcriptional, post-
transcriptional and post-translational levels leading to increase NO production. In pregnancy,
these regulators have been shown to play an essential role in mediating vasodilation, remodeling
and angiogenesis in the cardiovascular and placental circulation; therefore, these regulators will
be discussed next.
1.3.4 Regulators of eNOS enzymatic activity
Shear stress:
One of the most potent regulators of eNOS mRNA and eNOS protein expression in
endothelial cells is shear stress [132, 141]. Chronic exposure of endothelial cells to shear stress
increases eNOS expression by both transcriptional induction and stabilization of mRNA [105,
132]. Acutely, shear stress increases eNOS protein activity within seconds. This is regulated by
several different mechanisms involving eNOS-interacting proteins such as Ca2+/CaM, caveolin-
1 and Hsp90; posttranslational regulation (phosphorylation); cofactors and substrates and
In pregnancy, the increases in cardiac output and blood flow to many organs would tend
to elevate shear stress at the endothelial and endocardial surface. Langille [66, 70, 142] and
others [69] have firmly established that increases in shear stress stimulate remodeling in both
large and small arteries in a number of vascular beds. eNOS has been postulated to be an
important mediator.
26
The evidence to support this idea is as follows:
(i) eNOS mRNA and protein levels, and NO production in endothelial cells are increased
by shear stress [141].
(ii) Volume overload by arteriovenous shunt in the rabbit common carotid artery leads to
chronic increases in cardiac output, left ventricular dilation, and arterial enlargement
which were all inhibited by L-NAME [52, 68].
(iii) L-NAME virtually abolishes expansive remodeling in the main uterine artery and the
smaller radial arteries in pregnant rats [122].
(iv) Mice lacking the eNOS gene fail to reduce lumen diameter in response to a reduction in
blood flow in the carotid artery [143].
These studies suggest that eNOS is an important mediator in shear stress mediated responses.
Estrogens:
Estrogens are increased during pregnancy in the maternal circulation in humans and
mice [12, 144]. Estrogens increases eNOS mRNA expression and activity, and increase NO
bioavailability by reducing the rate of NO destruction in the endothelium [10, 14, 145].
Estrogens influence cardiovascular and uteroplacental vasodilation and remodeling by direct and
indirect effects on the vascular wall by working through the NO pathway.
Estrogens acts via estrogen receptor (ER) alpha and beta. Estrogen receptors are
expressed in the heart [146], aorta [147] and endothelium and vascular smooth muscle of the
uterine artery [16, 17] during pregnancy. E2β infusion increased uterine arterial blood flow and
27
cGMP production, and these effects were inhibited with L-NAME [148, 149] indicating that
they were mediated by activation of a NOS pathway. Estrogens may mediate vasodilation
indirectly by acting on endothelial cells to increase eNOS activation and NO production via the
PI3K and PLC-γ pathways [113, 138].
Estrogens may also induce vasodilation directly by acting on vascular smooth muscle
cells. They may target vascular smooth muscle cells through various strategies that include
cGMP and calcium-activated K+ channels (BKca) [113]. Estrogen increases the opening
potential of BKca in the uterine artery myocytes [150]. Potassium channels regulate basal
arterial tone and myotrophic response to various agonists through hyperpolarization of smooth
muscle membranes, which inactivates Ca2+ entry through potential-gated channels and results in
vasorelaxation [151]. Selective blockage of BKca in the uterine artery attenuated E2β-induced
rise in uterine arterial blood flow, which was similar to the effect of L-NAME infusion alone
[151]. Blocking both BKca and NO led to complete inhibition of the E2β-induced rise in uterine
arterial blood flow, suggesting that these two pathways are complementary [151].
VEGF:
VEGF (also referred to as VEGF-A) belongs to a gene family that includes PlGF,
VEGF-B, VEGF-C, and VEGF-D [152]. VEGF-A exerts its effects principally via its two
receptors, VEGFR1 (fms-like tyrosine kinase-1 (Flt1)) and a VEGFR2 (kinase domain region
(KDR/Flk)), respectively [15, 153], whereas VEGF-C and VEGF-D exert their effects
principally via their receptor VEGFR3. VEGF-C and VEGF-D regulate lymphatic angiogenesis
[154], whereas VEGF-A is a potent angiogeneic factor and vasodilator that plays an important
role in vascular remodeling and angiogenesis during pregnancy (Figure 1.5). VEGF-A mRNA
28
levels are elevated in the uterine artery and the placenta during pregnancy in rats and mice [19,
20, 155]. Four different isoforms of VEGF-A are present (VEGF121, VEGF165, VEGF189,
VEGF206), having 121, 165, 189 and 206 amino acids respectively [156].
VEGF mediates endothelium-dependent vasodilation by exerting its effects in part
through the NO pathway. Injection of adenoviral construct encoding VEGF-A into the uterine
artery of pregnant sheep increased uterine arterial blood flow by enhancing vasodilation [157].
Furthermore, dilation of the uterine arcuate arteries in response to VEGF was diminished by L-
NAME in pregnant rats, suggesting that this effect is mediated through NO [155]. In
endothelial cells, VEGF binds to VEGFR1 and VEGFR2 receptors and activates PI3K and PLC-
γ pathways, which lead to Akt dependent phosphorylation of eNOS on serine 1177 [15]. This
activation of eNOS increases NO production [15] (Figure 1.5).
VEGF plays an important role in angiogenesis, mediated in part via the eNOS-NO
pathway. Ziche et al [158] showed that VEGF-induced angiogenesis was blocked by systemic
administration of L-NAME. These studies were extended by Murohara et al [159] who showed
that eNOS KO mice exposed to hind limb ischemia showed markedly lower blood flow in the
ischemic regions and decreased capillary density. In this model, VEGF administration or VEGF
gene therapy failed to restore angiogenesis in eNOS KO mice, supporting the notion that NO is
an essential downstream element regulating VEGF-induced angiogenesis in adult mice [159].
In addition to being a downstream mediator of VEGF, NO also acts upstream to stimulate
VEGF expression. NO has been shown to activate the VEGF promoter in vascular smooth
muscle cells [160, 161] and skeletal muscle [162]. Decreased VEGF mRNA levels in the left
ventricular myocardium [163] and lungs [164] have been reported in non-pregnant eNOS KO
mice, which is consistent with a stimulatory effect of NO on VEGF expression.
29
The precise mechanism where by VEGF-NO mediates angiogenesis is not clear, but it
has been shown that VEGF activates the eNOS enzyme which then leads to increased NO
production. NO then activates cGMP which in turn activates kinase cascades including protein
kinase G (PKG) and MAPK [114]. Activation of these kinases leads to transcription of specific
genes such as fibroblast growth-factor (FGF-2), and to MMP activation and upregulation [114].
This leads to cellular remodeling events associated with angiogenesis such as cell proliferation,
migration and extracellular matrix degradation [114]. NOS inhibitors have been shown to block
VEGF-induced angiogenic processes including endothelial cell proliferation and migration in
vitro and in vivo [165, 166].
In summary, shear stress, estrogens and VEGF increase eNOS activity. Once the
enzyme is functional, it catalyzes the conversion of L-arginine to L-citrulline and NO in the
endothelial cell. This NO then diffuses out to the adjacent smooth muscle cell to mediate
vasodilation.
30
Figure 1-5. VEGF pathway and NO. In endothelial cells, VEGF binds to VEGFR1 and VEGFR2 receptors and activates PI3K and PLCγ pathways, which lead to activation of Akt, phosphorylation of eNOS on serine 1177. Activated eNOS increases NO production which then plays a role in vasodilation and angiogenesis. NO may activate MMPs and growth factors such as FGF2 that mediate angiogenesis. In addition to being a downstream mediator of VEGF, NO is also an upstream promoter of VEGF expression.
31
1.3.5 Nitric oxide signaling
Once NO is produced by the endothelium, it diffuses to the adjacent smooth muscle cells
where it targets soluble guanylate cyclase (sGC) which catalyzes the conversion of guanosine
triphophate (GTP) into secondary messenger, cyclic guanosine 3’5’-monophosphate (cGMP)
[99]. This secondary messenger then activates downstream elements including cGMP-
dependent protein kinases, cGMP-regulated phosphodiesterases and cGMP-gated ion channels
resulting in the relaxation of the vascular smooth muscle cells [99, 167](Figure 1.6).
The cGMP-activated family of serine/threonine protein kinases phosphorylate target
proteins including Ca2+ -ATPase-regulating protein phospholamban, IP3 receptor and other Ca2+
transporters and channels such as Ca2+ -dependent K+ channels leading to a decrease in
intracellular Ca2+ and thus hyperpolarization of the plasma membrane leading to relaxation
[102, 103].
NO has a very short half life (3-5 seconds). This short half-life is due to its rapid
oxidation to nitrite and nitrate by reactions with O2 and superoxide anion ·O2- [168]. NO also
binds to thiol groups forming S-nitroso-compounds [140, 169]. These nitrosylation reactions
are involved in regulating apoptosis and cell proliferation [140].
In addition, NO produced by eNOS in the endothelium may diffuse into the vascular
lumen [170-172](Figure 1.6). The majority of this NO enters the erythrocyte and reacts with
oxyhemoglobin (oxyHb) to form nitrate (NO3-). In the presence of oxygen, NO is also rapidly
oxidized to nitrite (NO2-) (which is a major storage source of NO in the blood and tissues) [170-
172]. In the erythrocyte, nitrite reacts with deoxyhemoglobin (deoxyHb) to form NO and
methemoglobin (metHg) and other NO adducts [170-172]. NO can then diffuse out of the
erythrocyte and exert “endocrine” effects distal from the site of its production.
32
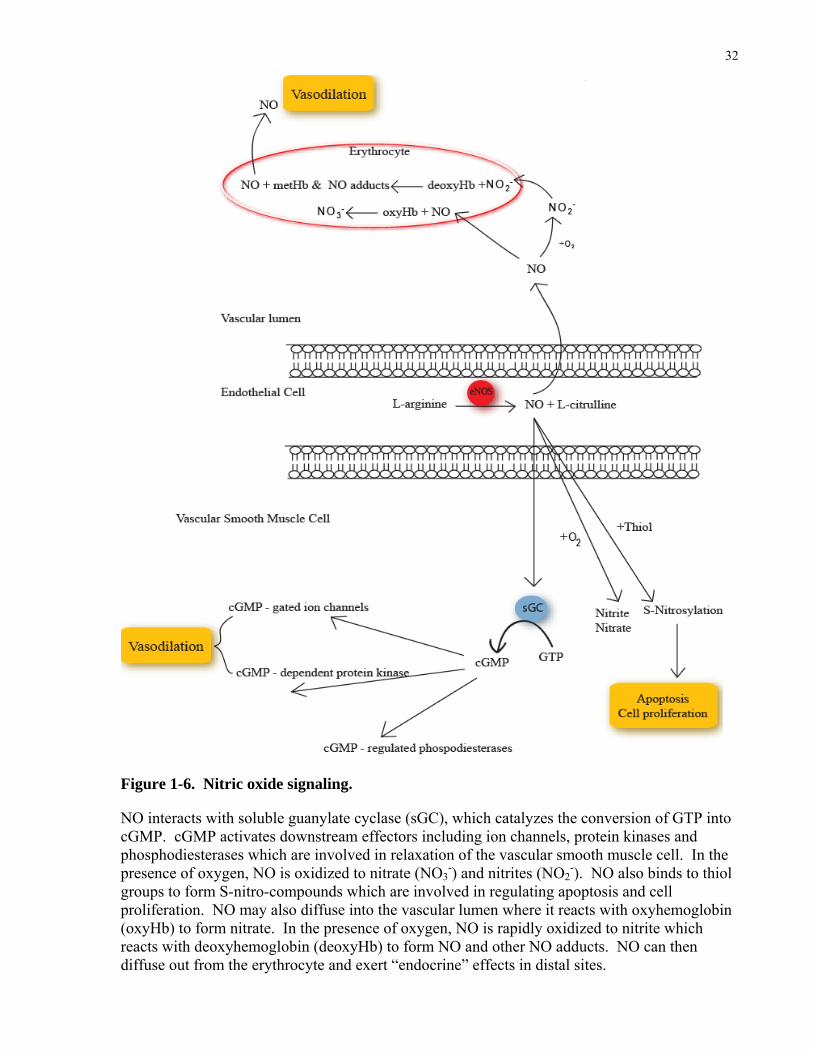
Figure 1-6. Nitric oxide signaling.
NO interacts with soluble guanylate cyclase (sGC), which catalyzes the conversion of GTP into cGMP. cGMP activates downstream effectors including ion channels, protein kinases and phosphodiesterases which are involved in relaxation of the vascular smooth muscle cell. In the presence of oxygen, NO is oxidized to nitrate (NO3
-) and nitrites (NO2-). NO also binds to thiol
groups to form S-nitro-compounds which are involved in regulating apoptosis and cell proliferation. NO may also diffuse into the vascular lumen where it reacts with oxyhemoglobin (oxyHb) to form nitrate. In the presence of oxygen, NO is rapidly oxidized to nitrite which reacts with deoxyhemoglobin (deoxyHb) to form NO and other NO adducts. NO can then diffuse out from the erythrocyte and exert “endocrine” effects in distal sites.
33
1.4 Nitric oxide and complications of pregnancy
As described above, nitric oxide mediates many vital tasks of pregnancy including
placentation, placental vascular remodeling and hemodynamic changes. It has therefore been
the target of investigation as an underlying mediator in several disorders of pregnancy. The
bulk of the work completed to date has focused on the pivotal role of NO in two of the most
serious and common complications of pregnancy, preeclampsia and intrauterine growth
restriction.
1.4.1 Preeclampsia
Preeclampsia is a multisystem disorder of pregnancy associated with elevated maternal
blood pressure, proteinuria, elevated blood flow pulsatility in the uterine artery,
thrombocytopenia, decreased plasma volume and renal glomerular endotheliosis [173, 174]. It
occurs in 5% of human pregnancies and is one of the leading causes of maternal and
fetal/neonatal mortality and morbidity world wide. The only effective treatment to prevent the
disease from progressing to maternal seizures, permanent end-organ damage and death is to end
the pregnancy but this may place the neonate at risk for complications of prematurity. The
mother is also at elevated risk for cardiovascular disease later in life [175].
The pathogenesis of preeclampsia is incompletely understood. However, it is proposed
to occur in two stages [176-178]. In stage 1 of preeclampsia, the root cause is considered to be
reduced placental perfusion. In some, but not all women, this leads to stage 2, which is the
multi-systemic maternal syndrome of preeclampsia. Poor placental perfusion is thought to be
secondary to failed remodeling of the maternal spiral arteries that supply the intervillous space.
34
Recently, Huppertz et al [179] challenged this concept. He proposed that the underlying
placental abnormality associated with preeclampsia occurred prior to the remodeling of the
vessels supplying the placenta. His concept was based on abnormalities in placental proteins
observed in the first trimester (i.e. ≤13 weeks). He proposed that abnormalities in the
differentiation of the trophoblast prior to implantation or the cytotrophoblasts and
synctiotrophoblast after implantation may be involved [179]. These concepts as proposed by
Roberts [176-178] and Huppertz [179] are not mutually exclusive. It is possible that aberrant
trophoblast differentiation in early pregnancy may be the root cause for both abnormal
implantation/placentation in early pregnancy, and abnormal placental bed vascular remodeling
in later pregnancy.
Abnormal remodeling of the vasculature is thought to contribute to reduced placental
perfusion leading to placental hypoxia. Hypoxic placentas are thought to release circulating
factors [180, 181] and/or reactive oxidative species [110, 182] that act on the endothelium to
cause the maternal syndrome of preeclampsia (Figure 1.7). A rat model of reduced placental
perfusion and ischemia created by reducing uterine perfusion pressure (RUPP) caused maternal
signs of preeclampsia, including hypertension, proteinuria, endothelial dysfunction, and reduced
renal plasma flow [183, 184]. These animals also showed increased total peripheral resistance,
decreased cardiac index, and decreased uterine and placental blood flow [183]. The linkage
between reduced placental perfusion and the maternal syndrome of preeclampsia is thought to
be circulating factors which are released from the hypoxic placenta. In the RUPP pregnant rat
reduced uterine artery and spiral artery remodeling↓
elevated uteroplacental vascular resistance↓
reduced rise in uteroplacental blood flow↓
placental hypoxia↓
release of circulating factor such as sFlt1 and endoglin and/or reactive oxidative species
↓damage to maternal endothelium
↓Maternal signs of preeclampsia including hypertension, decrease
in plasma volume, thrombocytopenia
Figure 1-7. Proposed mechanism leading to the pathogenesis of preeclampsia.
It has been proposed that poor placental perfusion secondary to failed remodeling of the uteroplacental vasculatures leads to placental hypoxia. Hypoxic placentas release circulating factors such as sFlt1 and endoglin and/or reactive oxidative species that act on the endothelium to cause the maternal signs of preeclampsia.
36
Angiogenic balance:
sFlt1 results from alternative splicing of Flt1 (VEGF-R1), an endothelial receptor for
VEGF and PIGF. It consists of extracellular ligand-binding domain, but lacks the
transmembrane and intracellular signaling domain, thus it is secreted into the extracellular
circulation [188]. sFlt1 is secreted primarily by the syncytiotrophoblast into the maternal
circulation where it binds to VEGF and PIGF preventing them from interacting with their
endogenous cognate receptors [189]. Recently, it has been shown that the human placenta
expresses a family of sFlt1 splice variants that are identical in their N-terminus but contain
unique C-terminus [190]. These splice variants are upregulated in preeclampsia [190]. The
increase in maternal sFlt1 levels has been shown to precede the onset of the clinical disease
[191], and is correlated with disease severity [192].
sFlt1 administrated to pregnant rats induced preeclampsia-like syndrome including
hypertension, proteinuria and glomerular endotheliosis [193, 194], suggesting sFlt1 may
contribute to endothelial damage in preeclampsia. Furthermore, these hallmarks of
preeclampsia are associated with reduced free VEGF in the maternal plasma [193] and are
reversed by augmenting maternal VEGF levels in this model [194].
In addition to sFlt1, soluble endoglin (sEng) is upregulated in preeclampsia in a pattern
similar to sFlt1 [195]. sEng is a cell surface receptor for TGF-β, and is highly expressed in
endothelial cells and syncytiotrophoblast and impairs the actions of TGF-β [195, 196].
Although increased sEng alone is insufficient to cause preeclampsia, it acts synergistically with
increased sFlt1 to cause preeclampsia-like symptoms in animal models [195].
TGF-β and VEGF stimulate the activity of eNOS via dephosphorylation at Thr497 and
phosphorylation at Ser1177 of the eNOS protein [15, 195]. Therefore, sEng and sFlt1 may
37
oppose physiological NO-dependent vasodilation leading to vasoconstriction, thereby causing
maternal and placental end-organ ischemia, which are hallmarks of preeclampsia.
1.4.2 Nitric oxide in preeclampsia
Given the strength of evidence supporting a crucial role for NO as a vasodilator in the
systemic circulation in pregnancy, many investigators have tested the possibility that abnormal
NO levels may contribute to preeclampsia [197, 198]. Women with Glu298Asp variant in exon
7 of the eNOS gene show increased risk for preeclampsia [197, 198]. This variant results in
selective proteolytic cleavage in the endothelial cell and vascular tissues leading to reduced NO
generation [199, 200]. In addition, acute inhibition of NOS caused a dose-response increase in
blood pressure [201], and long-term NOS inhibition produced preeclampsia-like symptoms in
pregnant rats [21]. Furthermore, expression and/or activity of various molecules that are
involved in the regulation of NOS activity are altered in preeclampsia. For example, G-protein-
coupled receptors, such as corticotrophin-releasing hormone (CRH) receptors type 1 and type 2
are reduced in preeclamptic placentas [202]. This downregulation may dampen the action of
CRH and urocortin on eNOS mRNA expression, NOS activation and cGMP production [128].
In addition, arginase II expression and total L-arginine-transporter activity are elevated in
preeclamptic pregnancies [203, 204]. These changes might reduce L-arginine availability for
NOS in trophoblast cells and in the villous endothelium. Therefore, alterations in the NO
signaling pathway may be involved in the pathogenesis of preeclampsia.
38
1.4.3 Intrauterine growth restriction
Intrauterine growth restriction (IUGR) also occurs in about 5% of human pregnancies,
with or without associated maternal preeclampsia [205, 206]. IUGR is a serious disorder
because it places the fetus at high risk of intrauterine death and perinatal morbidity and
mortality [207, 208]. Currently, premature delivery is the only effective treatment, but this
places the baby at high risk of prematurity-related complications and expensive hospital care
[208]. Furthermore, IUGR predisposes one to disease later in life, including increased risk of
coronary artery disease, hypertension and diabetes [209].
Etiology of IUGR is multi-factoral. It is associated with various maternal, fetal and
placental factors [210]. Maternal factors include hypertensive diseases, autoimmune disorders,
certain medications, severe malnutrition, and maternal lifestyle including smoking and alcohol
and multiple gestation. Placental factors includes anatomical, vascular, chromosomal and
morphological abnormalities [210].
A common cause of human IUGR is abnormal placental development associated with
abnormal umbilical artery hemodynamics and reduced fetoplacental perfusion detected by
Doppler ultrasound [211, 212]. Perinatal mortality and morbidity are markedly increased in the
presence of absence or reversed end diastolic velocity and elevated Pulsatility Index in the
umbilical artery [212]. Abnormal placental vascularization is also a significant contributor to
IUGR. Histology and scanning electron microscopy have revealed long, thin, poorly branched
villi and reduced villous capillary length and surface area [211, 213-215]. Impaired
fetoplacental vascularization is widely thought to elevate fetoplacental vascular resistance
39
causing reduced placental perfusion, thereby reducing oxygen and nutrient transfer to the fetus
which impairs fetal growth (Figure 1.8).
Impaired fetoplacental vascularization↓
elevated fetoplacental vascular resistance↓
reduced rise in fetoplacental blood flow↓
Reduced oxygen and nutrient delivery to fetus↓
Fetal hypoxia↓
Decreased fetal growth
Figure 1-8. Proposed mechanism leading to the pathogenesis of IUGR.
Impaired fetoplacental vascularization increases fetoplacental vascular resistance thereby decreasing fetoplacental perfusion. This reduces the transfer of oxygen and nutrients across the placenta, thereby contributing to fetal hypoxia and limiting fetal growth.
40
1.4.4 Nitric oxide in intrauterine growth restriction
The fetoplacental circulation lacks autonomic innervation [216]; therefore, circulating
and locally released vasoactive molecules like NO are likely critically involved in determining
fetoplacental hemodynamics [217]. NO also plays an important role in vasculogenesis and
angiogenesis [15, 114]; therefore, NO may play an important role in the etiology of IUGR.
Nitric oxide appears to play important roles in fetal growth based on results from animal
and human studies. Long-term inhibition of NO synthase causes IUGR in gravid rats [21], and
eNOS-targeted mutagenesis causes fetal growth restriction in mice [112, 218]. In humans,
reduced eNOS expression in the umbilical vessels [219], and lower eNOS activity in placental
villous tissue [220] have been reported in IUGR pregnancies. Furthermore, endothelial cells
isolated from the human umbilical vein of fetuses with IUGR exhibit reduced synthesis of L-
citrulline from L-arginine, reduced levels of nitrite, and reduced cGMP levels [221] all of which
likely reflect impaired NOS activity in endothelial cells from fetuses with this pathology. These
findings suggest that eNOS-derived NO plays an important role in the etiology of IUGR.
Most of the experiments examining the role of NO in normal pregnancy and in
pregnancy-related complications have used animal models where all three NOS isoforms are
inhibited by non-specific NOS inhibitors such as L-NAME. In addition, human studies cannot
be used to show cause and effect and to isolate the roles of specific factors, such as eNOS.
The availability of mice lacking the eNOS gene offers the opportunity to examine the role
played specifically by this isoform in mediating these pregnancy-related changes.
41
1.5 Mice as a models of human pregnancy
Genetically engineered mice are attractive models to study development and physiology
because of the ability to specifically and independently control genetic and environmental
influences. Unfortunately our knowledge of the physiology of pregnancy in this species is
limited because they are relatively difficult to study due to their small size, and their high heart
rates (~600 bpm). However, the advent of a high-resolution ultrasound imaging technique,
micro-ultrasound, enables us to overcome this barrier of small size.
Prior to availability of micro-ultrasound, a 20-MHz pulsed Doppler system with a
transcutaneous probe had been used to measure Doppler blood velocities in the mitral inflow
and ascending aortic outflow tracts of the left ventricle of mice [222]. This system does not
create an image so the appropriate positioning of the sample volume is uncertain. Also, vessel
diameter cannot be measured precluding the calculation of volume blood flow rate.
Nevertheless, this method proved adequate to detect maternal cardiovascular changes during
pregnancy in the mouse [222]. Clinical ultrasound systems with frequencies of ~15 MHz or less
have also been used to non-invasively assess cardiovascular and fetal development in mice [223,
224]. However, the resolution of images created using clinical ultrasound systems is poor (200-
500 µm) compared to the resolution (~50 µm) of images generated by the much higher
frequency transducers (~40 MHz) of micro-ultrasound systems [225]. Another advantage of
micro-ultrasound is the integration of pulsed Doppler capabilities at these high frequencies (19
to 55 MHz) [225]. This allows for detection and measurement of low blood velocities which is
important when examining very small vessels such as in the embryonic circulation [224]. Our
lab has pioneered the application of micro-ultrasound to monitor and quantify structure and
hemodynamics of the maternal cardiovascular, uteroplacental, and fetoplacental circulations in
mice [224-227].
42
1.5.1 Similarities and differences between mice and humans
Our laboratory was one of the first to illustrate that during pregnancy mice show
cardiovascular changes similar to those of humans. Experiments in an out-bred strain of mice
[222], showed that mice exhibited hypotension in early pregnancy, a blunted pressor response to
angiotensin II, a decrease in hematocrit, and a marked increase in cardiac output in late
pregnancy [222]. Our laboratory also showed that blood flow velocity waveforms in the uterine
and umbilical arteries are very similar in shape and show similar changes during gestation to
those of human pregnancy [227]. The mechanisms involved in mediating these changes during
pregnancy are not well understood. The first goal of my thesis was to further document the
normal cardiovascular and placental changes during pregnancy in mice, and my second goal was
to examine the role played by eNOS in mediating these changes using mice lacking the eNOS
gene (eNOS KO mice).
Although the placentas of no two mammalian species are the same, the placentas of
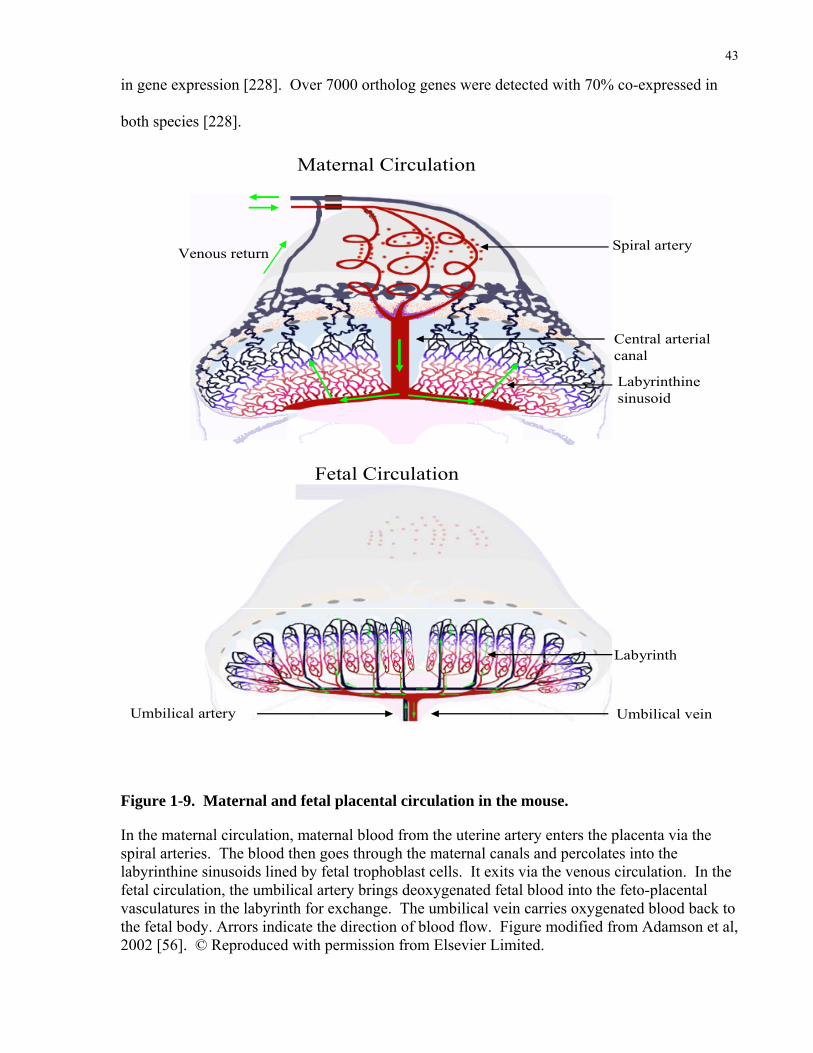
human and mice have strong similarities [56, 57]. In both species, the maternal blood from the
uterine artery enters the placenta through dilated, amuscular spiral arteries (Figure 1.9). The
maternal blood then moves through a dense mesh of channels created and lined by fetal
trophoblast cells in which an equally dense network of fetal capillaries is localized. This region
is the site for exchange between the mother and the fetus and is called the villous tree in humans
and the labyrinth in the mice [57]. In the mouse, one unique feature is that the maternal blood is
confined to a few trophoblast-lined arterial canals that direct blood to the basal side [56] (Figure
1.9). Canal-like structures have not been described in humans. On the fetal side of the placenta
in both species, the umbilical vessels connect the fetal capillaries of the placental exchange
region with the fetal body circulation [56, 57]. Detailed proteomics and transcriptomic
comparison of the placental exchange region of human and mouse showed striking similarities
43
in gene expression [228]. Over 7000 ortholog genes were detected with 70% co-expressed in
both species [228].
Maternal Circulation
Fetal Circulation
Spiral artery
Central arterial canal
Labyrinthine sinusoid
Umbilical veinUmbilical artery
Labyrinth
Venous return
Figure 1-9. Maternal and fetal placental circulation in the mouse.
REFERENCE: Kulandavelu, S., D. Qu, and S.L. Adamson, Cardiovascular function in mice during normal pregnancy and in the absence of endothelial NO synthase. Hypertension, 2006. 47(6): p. 1175-82.
2
51
2.1 INTRODUCTION
This chapter of my thesis is dedicated to examining whether control C57Bl/6J (WT)
mice show similar cardiovascular changes during pregnancy as that of humans, and to assessing
the obligatory role of endothelial nitric oxide synthase (eNOS) in mediating these changes by
studying eNOS knockout (KO) mice.
In the first half of pregnancy, the maternal cardiovascular system preadapts in
anticipation of the physiological demands of pregnancy and the growing perfusion and exchange
requirements of the conceptus, and changes further in the last half of gestation when the most
rapid growth of the conceptus occurs. Failure to make or to sustain these changes may result in
impaired fetal growth and/or preeclampsia, the two most common and serious complications of
human pregnancy [251, 252]. Although the mechanisms are not fully understood, there is
considerable evidence that NO plays an important role in mediating maternal cardiovascular
changes during pregnancy in humans, rats and other species [6, 21, 22, 116]. During pregnancy
in humans, there is a 30% decrease in the circulating levels of asymmetric dimethylarginine
[118], an endogenous inhibitor of nitric oxide synthase (NOS) activity. Furthermore, a non-
selective NOS inhibitor caused a greater decrease in blood flow in the forearm circulation of
pregnant versus non-pregnant women [116], which suggests that an increase in bioactive NO is
present during pregnancy and contributes to the decrease in peripheral vascular resistance during
pregnancy in humans. NO also appears to be important in rats during pregnancy because
plasma and urinary levels of nitrites and nitrates (metabolites of NO), and cGMP (second
messenger of NO) are increased in pregnant rats [6, 8] although whether similar changes occur
in human pregnancy are less certain [6, 121]. Furthermore, treatment of rats in late pregnancy
with non-selective NOS inhibitors blunts or reverses the normal decrease in arterial blood
52
pressure [21, 22]; abolishes the normal increase in plasma volume [22]; and causes fetal
intrauterine growth restriction and preeclamptic-like changes in the mother [21].
While there is considerable experimental evidence supporting a role for NO in mediating
the normal cardiovascular changes during pregnancy, the NOS isoform responsible is less well
established. Most studies investigating a role for NO have used L-arginine analogs that are non-
selective competitive inhibitors of iNOS, nNOS and eNOS. Of the 3 isoforms, eNOS is likely
the most important isoform in that increases in eNOS protein and mRNA levels have been
shown in the myocardium [9], aorta and the mesenteric artery, whereas iNOS and nNOS levels
remain unchanged [10, 11]. In addition, eNOS is an important mediator of cardiovascular
remodeling. For example, activation of eNOS in endothelial cells exposed to high shear stress
promotes arterial vasodilation and eventual structural enlargement [52, 68, 69].
In the current study, I hypothesized that eNOS plays a central role in mediating
cardiovascular adaptations to pregnancy. Therefore, I determined the effect of pregnancy on
cardiac structure and function using ultrasound in lightly anesthetized mice, and on arterial
blood pressure, heart rate, and plasma volume in awake mice in both the eNOS KO and in the
background strain for the KO mice, C57Bl/6J (WT).
2.2 MATERIAL AND METHODS
2.2.1 Breeding and genotyping
All procedures were approved by the Animal Care Committee of Mount Sinai Hospital
and were conducted in accordance with the guidelines of the Canadian Council of Animal Care.
53
Virgin female WT mice and eNOS KO mice were either purchased at 4-6 weeks of age
from Jackson Laboratories (Maine, USA) or raised in-house from the same stock. Between 8-12
wk of age, eNOS KO females were bred with eNOS KO males or with WT males. For the
control strain, WT females were mated with WT males. The presence of a sperm plug was
defined as day 0.5 of pregnancy. Age-appropriate non-pregnant mice of both strains were
studied at equivalent intervals to serve as time-controls (N=7-8). Experimental time-points
included prior to breeding, day 9.5 (mid-gestation, start of umbilico-placental perfusion), day
17.5 (late gestation, two days prior to normal term delivery) and 3 weeks after delivery (at
weaning).
2.2.2 Hemodynamics
eNOS KO females (N=12) were bred with eNOS KO males or with WT males. Male
strain caused no significant differences so the data were pooled. WT females (N=8) were bred
with WT males. Mice were lightly anesthetized with 1-2% isoflurane in oxygen. This
anesthetic minimally affects cardiovascular function in mice [253]. Body temperature was
monitored using a rectal probe and was maintained between 37ºC and 38ºC. Heart rate was
monitored by taping paws to electrodes (Indus Instruments, Houston, TX). With appropriate
body temperature and anesthetic depth, heart rate was kept above 400 min-1. A 20 MHz pulsed
Doppler system (model VF-1; Valpey Fisher, Hopkinton, MA) with a hand-held probe (Matec
Instrument; Northborough, MA) was used to obtain transcutaneous blood velocity waveforms
over three second time intervals from the ascending thoracic aorta (Figure 2.1A) and mitral
orifice (Figure 2.1B) as previously described [222, 254]. Ten consecutive waveforms for each
animal were saved and later analyzed (Doppler Signal Processing Workstation, Indus
Instruments, Houston, TX). Aortic blood velocity and ECG waveforms were analyzed to
54
obtain: 1) heart rate; 2) mean velocity (velocity envelope averaged over the cardiac cycle); 3)
contraction time. Diastolic filling time was calculated by taking the sum of E and A time
durations.
I then measured ascending aortic diameter during systole from an image of the long-axis
of the left ventricular outflow tract obtained using an micro-ultrasound (Model VS40;
VisualSonics, Toronto, ON) with a 19 MHz transducer (resolution ~100 µm) (Figure 2.1C).
The mean value of 10 aortic diameter measurements obtained during systole was used to
calculate vessel cross-sectional area [π (diameter/2)2].
Cardiac output and stroke volume were calculated as the product of aortic luminal cross-
sectional area [π (diameter/2)2] and mean velocity and stroke distance, respectively. In a
separate group of 7 isoflurane-anesthetized, non-pregnant adult WT mice, the coefficient of
variation of aortic diameter measurements obtained using this method daily for 4 consecutive
days was <2%.
55
D.
LVED LVES
AW
PW
0.1 s mm
+LV
LA
C.
LV
LA
Ao+
+
A. A. A.
cm/sec
A.E
A
cm/sec
B.
D.
LVED LVES
AW
PW
0.1 s mm
D.
LVED LVES
AW
PW
0.1 s mm
+LV
LA
C.
LV
LA
Ao+
++
LV
LA
C.
LV
LA
Ao+
+
A. A. A.
cm/sec
A.A. A. A.
cm/sec
A.E
A
cm/sec
B.
Figure 2-1. Ultrasound evaluation of cardiac structure and function.
A. Doppler blood velocity waveform recorded from the ascending thoracic aorta. The waveform exhibits a steep initial slope, more gradual convex downslope, and a brief velocity reversal at the end of the ejection phase due to valve closure. B. Doppler blood velocity waveform recorded from mitral valve. E is defined as peak velocity during early ventricular filling and A is defined as peak velocity in late ventricular filling phase due to atrial contraction. C. Long-axis view of the left ventricular (LV) outflow tract (Ao) showing aortic calipers (+) and open aortic valve (arrow). Left atrium (LA) is also shown. D. M-mode image of the left ventricle showing LV end-diastolic (LVED) and end-systolic (LVES) inner chamber dimensions and anterior (AW) and posterior (PW) wall thicknesses. Ao, ascending aorta; AW, anterior wall; LA, left atrium, LV, left ventricle; LVED, left ventricular end-diastolic dimension; LVES, left ventricular end-systolic dimension; PW, posterior wall.
56
2.2.3 Left ventricular geometry
In a separate cohort of animals, eNOS KO and WT females were bred with males of the
same strain. A newer model of the micro-ultrasound (Model Vevo660, 30 MHz transducer) was
used to measure left ventricular geometry (parasternal long-axis view) in lightly isoflurane-
2.2) caused the increase in stroke volume, as fractional shortening did not change significantly
(Table 2.3). These findings indicate that pronounced maternal cardiovascular changes occur
early in gestation in mice, as in humans [2, 18].
By day 17.5 of gestation, maternal body weight increased by 85% (Figure 2.2). Cardiac
output increased significantly by 48% relative to pre-pregnancy due to a significant 41%
increase in stroke volume, whereas heart rate in anesthetized mice remained unchanged (Figure
2.3, Table 2.1). Heart rate in awake mice studied using the tail-cuff system also did not change
significantly during pregnancy (Figure 2.4). The increase in stroke volume was associated with
increases in LVED dimension by 15%, aortic diameter by 10%, plasma volume by 27%, and a
decrease in hematocrit by 13% (Figure 2.2 and 2.5, all changes significant). Arterial pressure in
awake mice was slightly but significantly reduced throughout pregnancy with a nadir of 15% in
mid-pregnancy (Figure 2.4). At day 17.5, calculated LV mass was 37% higher, whereas
fractional shortening remained unchanged when compared to prior to pregnancy (Table 2.3).
Unlike humans [257], platelet count was 39% higher when compared to prior to pregnancy
(P<0.05) (Table 2.4).
By 3 weeks postpartum, body weight (+32%), aortic diameter (+13%), stroke volume
(+33%), and cardiac output (+27%) remained significantly elevated when compared to prior to
pregnancy and to the time controls (Figures 2.2, 2.3). The magnitude of the cardiovascular
changes in pregnancy and the delayed recovery postpartum are similar to that of humans [2,
222].
60
2.3.2 eNOS is required for the normal increase in cardiac output during
pregnancy
Before pregnancy, eNOS KO mice were similar to WT mice in their body weight,
cardiac output, aortic diameter and LV geometry parameters but they had significantly elevated
arterial pressures and stroke volumes and lower heart rates (Figures 2.2 - 2.4, Tables 2.1, 2.3).
By day 9.5 of gestation, weight gain in eNOS KO mice was similar to WT mice but the
increase in aortic diameter and LVED were significantly reduced (Figure 2.2). Cardiac output
increased by 22% in eNOS KO mice but, compared with WT mice, this was achieved by a
smaller increase in stroke volume (14%) and by an increase in heart rate (9%) measured under
light anesthesia (Figure 2.3, Table 2.1). Heart rate also significantly increased when studied in
awake mice (10%) (Figure 2.4). The increase in calculated stroke volume in eNOS KO mice
was primarily due to the small increase in aortic luminal diameter (4%) whereas the smaller
increase in stroke distance was not statistically significant (Figure 2.2, Table 2.1). These results
indicate that in early pregnancy, the remodeling of the heart is absent, the enlargement of the
aorta is blunted and, unlike controls, an increase in heart rate is an important contributor to the
increase in cardiac output in eNOS KO mice.
By day 17.5 of gestation, the gain in maternal body weight (84%) in the eNOS KO mice
was almost identical to that of WT mice (Figure 2.2). In contrast, the increase in aortic diameter
was significantly blunted and there was still no significant enlargement of LVED in the eNOS
KO mice (Figure 2.2). Also at late gestation, cardiac output in the eNOS KO mice was
significantly lower than WT mice due to a significantly lower stroke volume (Figure 2.3). This
occurred even though fractional shortening was not significantly different and the peak E/A
ratio was significantly improved (due to significantly lower peak A) suggesting that the lower
61
stroke volume was not caused by an impairment in cardiac systolic or diastolic function (Tables
2.1- 2.2). The failure of cardiac output to increase in late gestation in the eNOS KO mice may
account for the significant continued decline in arterial pressure in late gestation in these mice,
which contrasted with the fairly stable decrement in arterial pressure throughout pregnancy in
the WT mice (Figure 2.4). Nevertheless, the 26% increase in plasma volume, the 13% decrease
in hematocrit, and the 37% increase in platelet count observed at 17.5 d of gestation in eNOS
KO mice did not differ significantly from the values observed in WT mice at the same stage of
gestation (Figure 2.5, Table 2.4). In contrast to the substantial (37%) gain in LV mass observed
in WT mice, no significant change relative to pre-pregnancy was observed in eNOS KO mice
(Table 2.3). These findings indicate an essential role for eNOS in maintaining an increase in
cardiac output in late gestation by promoting LV chamber enlargement.
By 3 weeks postpartum, as in WT mice, body weight (+30%), aortic diameter (+7%),
stroke volume (+18%), and cardiac output (+18%) of eNOS KO mice remained significantly
elevated relative to pre-pregnant levels (Figures 2.2, 2.3). The strains differed, however, in that
stroke volume increased from late gestation to post-partum, whereas heart rate decreased back to
its pre-pregnancy level in eNOS KO mice only (Figure 2.3, Table 2.1). The increase in stroke
volume was sufficient to offset the decrease in heart rate so that cardiac output remained stable
postpartum in eNOS KO mice, in contrast to the postpartum decrement in WT mice.
62
Pre-P 9.5 17.5 PP1.1
1.2
1.3
1.4
1.5
a b b ba
b
cd††
Aorti
c di
amet
er (m
m)
Pre-P 9.5 17.5 PP1.1
1.2
1.3
1.4
1.5
a a b ba
bc
c* **
†
Aorti
c di
amet
er (m
m)
Pre-P 9.5 17.5 PP0
10
20
30
40
a a a ba b
c
b†
†
†
Body
Wei
ght (
g)
B6 eNOS -/-
Pre-P 9.5 17.5 PP0
10
20
30
40
a b ca
b
c
b†
†
†
b
Bod
y W
eigh
t (g)
PregnantNon-pregnant time control
Pre-P 17.5 PP1.1
1.2
1.3
1.4
1.5
b b ba
b
c†
Aorti
c di
amet
er (m
m)
Pre-P 17.5 PP1.1
1.2
1.3
1.4
1.5
a b b
bc**†
†
Aorti
c di
amet
er (m
m)
Pre-P 17.5 PP0
10
20
30
40
a a a b
b
c
†
†
Body
Wei
ght (
g)
B6 eNOS -/-
Pre -P 9.5 17.5 PP3.00
3.25
3.50
3.75
4.00
a
b
c
b
†
Left
vent
ricul
ar e
nd-
Dia
stol
ic d
imen
sion
(mm
)
Pre -P PP3.00
3.25
3.50
3.75
4.00
a
b
c
b
†
Dia
stol
ic d
imen
sion
(mm
)
Pre-P 17.5 PP0
10
20
30
40
a b c
b
c
†
†
b
Bod
y W
eigh
t (g)
PregnantNon-pregnant time control
Pre-P 17.5 PP0
10
20
30
40
a b c
b
c
†
†
b
Bod
y W
eigh
t (g)
PregnantNon-pregnant time control
Pre -P 9.5 17.5 PP3.00
3.25
3.50
3.75
4.00
* *
Pre -P PP3.00
3.25
3.50
3.75
4.00
* *
Left
vent
ricul
ar e
nd-
Dia
stol
ic d
imen
sion
(mm
)D
iast
olic
dim
ensi
on (m
m)
Pre-P 9.5 17.5 PP1.1
1.2
1.3
1.4
1.5
a b b ba
b
cd††
Aorti
c di
amet
er (m
m)
Pre-P 9.5 17.5 PP1.1
1.2
1.3
1.4
1.5
a a b ba
bc
c* **
†
Aorti
c di
amet
er (m
m)
Pre-P 9.5 17.5 PP0
10
20
30
40
a a a ba b
c
b†
†
†
Body
Wei
ght (
g)
B6 eNOS -/-
Pre-P 9.5 17.5 PP0
10
20
30
40
a b ca
b
c
b†
†
†
b
Bod
y W
eigh
t (g)
PregnantNon-pregnant time control
Pre-P 17.5 PP1.1
1.2
1.3
1.4
1.5
b b ba
b
c†
Aorti
c di
amet
er (m
m)
Pre-P 17.5 PP1.1
1.2
1.3
1.4
1.5
a b b
bc**†
†
Aorti
c di
amet
er (m
m)
Pre-P 17.5 PP0
10
20
30
40
a a a b
b
c
†
†
Body
Wei
ght (
g)
B6 eNOS -/-
Pre -P 9.5 17.5 PP3.00
3.25
3.50
3.75
4.00
a
b
c
b
†
Left
vent
ricul
ar e
nd-
Dia
stol
ic d
imen
sion
(mm
)
Pre -P PP3.00
3.25
3.50
3.75
4.00
a
b
c
b
†
Dia
stol
ic d
imen
sion
(mm
)
Pre-P 17.5 PP0
10
20
30
40
a b c
b
c
†
†
b
Bod
y W
eigh
t (g)
PregnantNon-pregnant time control
Pre-P 17.5 PP0
10
20
30
40
a b c
b
c
†
†
b
Bod
y W
eigh
t (g)
PregnantNon-pregnant time control
Pre -P 9.5 17.5 PP3.00
3.25
3.50
3.75
4.00
* *
Pre -P PP3.00
3.25
3.50
3.75
4.00
* *
Left
vent
ricul
ar e
nd-
Dia
stol
ic d
imen
sion
(mm
)D
iast
olic
dim
ensi
on (m
m)
WT eNOS KO
Figure 2-2. Body weight, aortic diameter and left ventricular end-diastolic dimensions under light anesthesia in WT and eNOS KO mice.
The shaded area highlights the time when the pregnant group was pregnant. Different superscript letters indicate significant changes over time within each strain (P<0.05). *P<0.05, pregnant eNOS KO vs. pregnant WT controls. †P<0.05, pregnant vs. non-pregnant time controls within each strain; Mean ± SEM where N= 7 to 12 at each point. Pre-P, prior to pregnancy; PP, post-partum.
63
Pre-P 9.5 17.5 PP30
40
50
60
a
b
c
bc†
††
Stro
ke V
olum
e(μ
L)
Pre-P 9.5 17.5 PP30
40
50
60
a
b
a
b
**
Stro
ke V
olum
e(μ
L)
WT eNOS KOPregnantNon-pregnant time control
Pre-P 9.5 17.5 PP10
15
20
25
30
35
a
b
c
b†
†
†
Car
diac
Out
put
(ml/m
in)
Pre-P 9.5 17.5 PP10
15
20
25
30
35
a
bbb *
Car
diac
Out
put
(ml/m
in)
Figure 2-3. Stroke volume and cardiac output under light anesthesia in WT and eNOS KO mice.
The shaded area highlights the time when the pregnant group was pregnant. Different superscript letters indicate significant changes over time within each strain (P<0.05). *P<0.05, pregnant eNOS KO vs. pregnant WT. †P<0.05, pregnant vs. non-pregnant time controls differ within each strain; Mean ± SEM where N= 7 to 12 at each point. Pre-P, prior to pregnancy; PP, post-partum.
64
Pre-P Early Mid Late PP100
120
140
160
a
b b b
ab†
Arte
rial P
ress
ure
(mm
Hg)
Pre-P Early Mid Late PP100
120
140
160*a
acbc
b
ac
††
Arte
rial P
ress
ure
(mm
Hg)
Pre-P Early Mid Late PP400
500
600
700
800
Hea
rt R
ate
(min
-1)
Pre-P Early Mid Late PP400
500
600
700
800
aa
b ba*
†
Hea
rt R
ate
(min
-1)
WT eNOS KOPregnantNon-pregnant time control
Figure 2-4. Arterial pressure and heart rate measured using tail-cuff system in awake WT and eNOS KO mice.
The shaded area highlights the time when the pregnant group was pregnant. Different superscript letters indicate significant changes over time within each strain (P<0.05). *P<0.05, pregnant eNOS KO vs. pregnant WT. †P<0.05, pregnant vs. non-pregnant time controls differ within each strain; Mean ± SEM where N= 7 to 10 at each point. Pre-P, prior to pregnancy; early = days 2.5 and 5.5; mid = day 9.5 and 11.5; late = days 13.5 and 17.5; PP, post-partum.
65
WT eNOS KO0
25
50
75 Pregnant
† †Non-pregnant
Pla
sma
Volu
me
(μL/
g)
A.
WT eNOS KOB.
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a aa
b
a
† † †
Hem
atoc
rit (L
/L)
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a ab
b
a††
Hem
atoc
rit (L
/L)
PregnantNon-pregnant time control
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a aa
b
a
†† †
Hem
atoc
rit (L
/L)
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a ab
b
a†
Hem
atoc
rit (L
/L)
PregnantNon-pregnant time control
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a aa
b
a
† † †
Hem
atoc
rit (L
/L)
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a ab
b
a††
Hem
atoc
rit (L
/L)
PregnantNon-pregnant time control
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a aa
b
a
†† †
Hem
atoc
rit (L
/L)
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a ab
b
a†
Hem
atoc
rit (L
/L)
PregnantNon-pregnant time control
WT eNOS KO0
25
50
75 Pregnant
† †Non-pregnant
Pla
sma
Volu
me
(μL/
g)
WT eNOS KO0
25
50
75 Pregnant
† †Non-pregnant
Pla
sma
Volu
me
(μL/
g)
A.
WT eNOS KOB.
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a aa
b
a
† † †
Hem
atoc
rit (L
/L)
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a ab
b
a††
Hem
atoc
rit (L
/L)
PregnantNon-pregnant time control
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a aa
b
a
†† †
Hem
atoc
rit (L
/L)
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a ab
b
a†
Hem
atoc
rit (L
/L)
PregnantNon-pregnant time control
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a aa
b
a
† † †
Hem
atoc
rit (L
/L)
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a ab
b
a††
Hem
atoc
rit (L
/L)
PregnantNon-pregnant time control
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a aa
b
a
†† †
Hem
atoc
rit (L
/L)
Pre-P 9.5 13.5 17.5 PP0.3
0.4
0.5
0.6
a ab
b
a†
Hem
atoc
rit (L
/L)
PregnantNon-pregnant time control
Figure 2-5. Plasma volume and plasma hematocrit levels at non-pregnant and during pregnancy.
A. Plasma volume in non-pregnant, awake mice (N=6, open bars) and at 17.5 d of gestation (N=6, closed bars). B. Hematocrit (N=7-12) in awake mice where the shaded area highlights the time when the pregnant group was pregnant. Different superscript letters indicate significant changes over time within each strain (P<0.05). †P<0.05, pregnant vs. non-pregnant time controls within each strain. Pregnant eNOS KO mice did not significantly differ from pregnant WT. Mean ± SEM. Pre-P, prior to pregnancy; PP, post-partum.
66
Table 2-1. Aortic Doppler parameters in WT and eNOS KO mice prior to, during, and post-pregnancy.
Values are mean ± SEM with N = 7 to 12 in each group. Along each row, values with different superscript letters indicate significant differences over time within each strain (P<0.05).
TPVR, Total peripheral vascular resistance estimated using anesthetized cardiac output and awake blood pressure.
*P<0.05, pregnant eNOS KO vs. pregnant WT.
†P<0.05, pregnant group vs. non-pregnant time control (data not shown) within each strain.
67
Table 2-2. Mitral Doppler parameters determined using ultrasound prior to, during, and post-pregnancy in WT and eNOS KO mice.
Values are mean ± SEM with N = 7 to 12 in each group. Along each row, values with different superscript letters indicate significant differences over time within each strain (P<0.05).
TVI, time-velocity interval was calculated by taking area under the E and A wave, and diastolic filling time was calculated by taking the sum of E and A time durations.
*P<0.05, pregnant eNOS KO vs. pregnant WT. †P<0.05, pregnant group vs. non-pregnant time control (data not shown) within each strain.
68
Table 2-3. LV geometry parameters determined using ultrasound prior to, during, and post-pregnancy in WT and eNOS KO mice.
LV geometry Strain Prior to 9.5 d 17.5 d Post-partum parameter pregnancy of gestation of gestation
Values are mean ± SEM with N = 6 to 10 in each group. Along each row, values with different superscript letters indicate significant differences over time within each strain (P<0.05).
*P<0.05, pregnant eNOS KO vs. pregnant WT.
†P<0.05, pregnant group vs. non-pregnant time control (data not shown) within each strain.
69
Table 2-4. Maternal hematology parameters prior to, during, and post-pregnancy in WT and eNOS KO mice.
Hematology Strain Prior to 9.5 d 13.5 d 17.5 d Post-partum parameter pregnancy of gestation of gestation of gestation
Values are mean ± SEM with N=7 to 12 in each group. Along each row, values with different superscript letters indicate significant differences over time within each strain (P<0.05). Pregnant eNOS KO mice did not differ significantly from pregnant WT.
†P<0.05, pregnant group vs. non-pregnant time control (data not shown) within each strain.
Hct, Hematocrit; RBC, red blood cell count; Hgb, hemoglobin concentration; Plt, platelet count; WBC, white blood cell count; MCV, mean corpuscular volume, MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration.
70
2.4 DISCUSSION
My study showed that mice model the human cardiovascular changes during pregnancy
including increases in cardiac output, stroke volume, plasma volume, left ventricular and aortic
inner dimensions, and decreases in arterial pressure and hematocrit, and many of these changes
are present early in pregnancy. The primary novel finding of my study was that eNOS was
shown to play an important role in mediating maternal cardiovascular adaptations during
pregnancy in the mouse. The normal increase in cardiac output was blunted at late gestation by
the absence of a functional eNOS gene due to a reduction in stroke volume, which was partially
offset by an increase in heart rate. Lower stroke volume in late gestation was likely caused by
inadequate ventricular remodeling.
Effects of pregnancy on cardiovascular function in eNOS KO mice:
Arterial pressure: Arterial blood pressure was elevated in non-pregnant eNOS KO mice,
as in prior reports [218, 258], presumably due to reduced smooth muscle vasorelaxation
mediated by a reduction in endothelium-derived NO [258] which was not completely offset by
augmented roles of other vasodilators [258] including, endothelium-derived hyperpolarizing
factor (EDHF), prostaglandin and nNOS. Increased vasoconstrictor stimulus may have
contributed because plasma renin levels are elevated in eNOS KO mice [218, 258], which may
lead to an increase in circulating levels of the vasoconstrictor, angiotension II. Interestingly,
despite being chronically hypertensive, the left ventricular wall was not hypertrophied in non-
pregnant eNOS KO mice similar to a prior report [259].
Arterial pressure in eNOS KO mice decreased during pregnancy, to become similar to
that of pregnant WT controls. Interestingly, arterial pressure also decreases during pregnancy in
71
chronically hypertensive women [260] thus eNOS KO mice may provide a useful model for
studying why this occurs. The study was limited in that arterial pressure and cardiac output
were not measured simultaneously and under the same experimental conditions (awake or
anesthetized). However, when values obtained in the same animal on the same gestational day
were used to estimate peripheral vascular resistance, it was found that both strains exhibited
similar decreases in peripheral vascular resistance in early pregnancy (WT -29%, eNOS KO -
26%) whereas in late gestation, the percent decrease was greater in WT (-36%) than in eNOS
KO mice (-24%) (P<0.05 by unpaired t-test) (Table 2.1). Thus results suggest that eNOS-
derived NO is less important in mediating maternal peripheral vasodilation in early than in late
pregnancy at which stage it appears to mediate ~40% of the response. This is in agreement with
prior work showing a role for other vasodilators such as EDHF [261] and prostaglandins [262]
in mediating peripheral vasodilation in pregnancy.
Non-specific NOS inhibitors cause preeclamptic-like symptoms in pregnant rats
including hypertension, thrombocytopenia, and a blunted rise in plasma volume [21, 22]. My
results suggest that these changes may be due to iNOS or nNOS inhibition, or the acute effects
of eNOS inhibition, because they did not occur in eNOS KO mice. My finding that eNOS KO
mice do not exhibit a further significant rise in blood pressure during pregnancy is in agreement
with prior reports [263, 264]. On the other hand, eNOS appears to be important in maintaining
normal fetal growth because embryo weight at term was significantly reduced in eNOS KO
pregnancies (-15%, data not shown) as reported previously in eNOS KO mice [112, 164] and in
pregnant rats treated with NOS inhibitors [21]. Thus, my results suggest that inadequate
maternal cardiovascular changes may contribute to intrauterine growth restriction in eNOS KO
pregnancies.
72
Cardiac output: Although blood pressure in pregnancy did not differ, the maternal
hemodynamic response to pregnancy was abnormal in eNOS KO mice. Cardiac output in eNOS
KO mice was lower than WT controls in late pregnancy due to a significantly lower stroke
volume and this occurred despite a preload increase (i.e. increased plasma volume), afterload
Figure 2-6. Proposed mechanism: Hormonally and flow-mediated cardiovascular remodeling during pregnancy.
I propose that increase shear stress and/or pregnancy-related hormones such as estrogen and relaxin work through the PI3-Akt pathway which in turns activates the eNOS protein leading to an increase in NO production. This in turn promotes remodeling of the heart and the vasculature leading to an increase in stroke volume, which then contributes to an increase in cardiac output during pregnancy.
75
Heart rate: In awake, non-pregnant eNOS KO mice, heart rate was lower than in the control
strain as in previous reports [218, 258] although there was no significant difference in cardiac
output before pregnancy in the two strains. The lower heart rate in eNOS KO hearts is due to
extrinsic factors because heart rates of isolated hearts in vitro do not differ from controls [268].
Lower heart rates may be due to a baroreflex-mediated augmentation of vagal tone caused by
systemic hypertension in eNOS KO mice. Interestingly, other mouse models with chronic
hypertension have normal heart rates [269]. This suggests that eNOS may be required for
baroreceptor resetting in response to chronic changes in arterial pressure. The progressive
increase in heart rate during pregnancy in both awake and anesthetized eNOS KO mice may be
a baroreceptor-mediated response to the progressive decrease in arterial pressure. In contrast,
heart rate was unchanged during pregnancy and postpartum in WT mice. If vascular eNOS
expression is enhanced during pregnancy in mice as in other species [9-11], then results suggest
this increase may blunt baroreceptor sensitivity during pregnancy in normal but not in eNOS
KO mice. Heart rate did not increase during pregnancy in WT B6 mice as in a prior report [53]
whereas we previously observed a significant increase in heart rate during pregnancy in ICR
mice [222] suggesting there are strain-dependent differences in this response.
Limitations: Knockout mouse models provide useful tools for studying the role of
specific gene products in mediating physiologic responses because elimination of the product is
specific and complete. However, in any physiologic system removal of one element can induce
compensatory changes in others. Compensatory changes in other NOS isoforms and in other
vasodilatory pathways have been described in adult eNOS KO mice [258]. In addition, single
genes may serve multiple functions during development and in the adult. In the case of eNOS
KO mice, ventricular septal defects and bicuspid aortic valves are more common in neonates
76
with this genotype [164, 247, 270] and pulmonary hypovascularity is a common embryonic
phenotype that leads to heart failure and death of ~85% of neonates [164, 247]. The eNOS KO
mice used in the current study were the subset that escaped neonatal lethality and therefore were
those that more effectively compensated for the role of eNOS in these developmental pathways.
How this selection process, or the presence of residual developmental effects would impact on
adult cardiovascular function is unknown. Another limitation is that ultrasound measurements
were determined under light isoflurane anesthesia. Isoflurane has fewer systemic hemodynamic
effects in mice than other nonvolatile anesthetics [253]. Cardiac index and cardiac output in
anesthetized non-pregnant control mice in the current study (0.90 ml/min/g, 20 ml/min) were
slightly higher than previous reports in awake mice (0.75 ml/min/g [253], 16 ml/min [271])
suggesting that light isoflurane anesthesia had minimal cardiodepressive effects.
Effects of pregnancy on cardiovascular function in WT mice:
This study also provides novel information on maternal cardiovascular changes during
pregnancy in WT C57Bl/6J mice, a commonly used inbred strain. Half the total increase in
cardiac output during pregnancy in WT mice occurred by 9.5 days of gestation, at a time when
maternal weight gain was modest and embryos were at an early stage of organogenesis. Results
suggest that pronounced peripheral vasodilation had occurred by this stage, because, although
not measured simultaneously, arterial pressure had decreased and cardiac output had increased.
Therefore, as in humans, pronounced maternal cardiovascular changes occur early in gestation
[2, 18] at a stage when the conceptus presents minimal perfusion demands. By late gestation,
calculated LV mass increased 37% and left ventricular end-diastolic dimensions by 15%, similar
to a prior report in C57Bl/6J mice [53]. I further showed that in late gestation, cardiac output
had increased 48%, plasma volume by 27%, aortic diameter by 10%, and hematocrit had
77
decreased by 13%. Similar changes are observed during pregnancy in human, rats and other
species [2, 18, 37]. Thus, results suggest that genetically-altered mice will provide useful new
models for expanding our limited understanding of the mechanisms responsible for
cardiovascular changes during pregnancy.
In conclusion, mice, like humans show similar cardiovascular changes during pregnancy
including an increase in cardiac output due to an increase in stroke volume. The increase in
stroke volume was associated with increases in plasma volume, and in left ventricular and aortic
end-diastolic dimensions. In mice lacking the eNOS gene, cardiac output was blunted in late
gestation due to a decrease in stroke volume back to its pre-pregnant levels, which was offset by
an increase in heart rate. Stroke volume failed to increase in late gestation despite increased
plasma volume and decreased arterial pressure, apparently due to inadequate aortic and
ventricular remodeling. Thus, I speculate that eNOS plays a critical role in maintaining the
increase in stroke volume in late gestation by contributing to flow and/or hormonally induced
cardiac and aortic remodeling.
Perspectives:
My results in eNOS KO mice highlight the inadequacy of using arterial pressure alone to
demonstrate the normalcy of hemodynamic changes during pregnancy in mouse models, and by
extension, in human pregnancy. Most women with chronic hypertension exhibit a decline in
arterial pressure during pregnancy but nevertheless the risk of perinatal death and fetal growth
restriction is twice that of women who are normotensive prior to pregnancy [260]. Whether
increases in cardiac output and stroke volume, and decreases in peripheral vascular resistance
are blunted during pregnancy in such women, as in our chronically-hypertensive eNOS KO
mice, is not known and should be explored. Interestingly, women who have intrauterine
78
growth-restricted fetuses without preeclampsia exhibit significantly reduced increases in cardiac
output, stroke volume, and LV mass and diastolic volume [251]. Thus, it is possible that in
human pregnancy inadequate maternal hemodynamic changes may contribute to fetal growth
restriction as may be the case in the eNOS KO mouse. Finally, although a missense
polymorphism in the eNOS gene has been associated with preeclampsia in some human
populations [197], my results show that eliminating the function of this gene fails to generate a
preeclamptic phenotype in mice, suggesting that other genetic and environmental factors are of
primary importance.
ACKNOWLEDGMENT:
I would like to thank Dr. Dawei Qu for his assistance in the plasma volume experiments.
Chapter 3 - Uteroplacental structural and functional changes in mice during normal pregnancy: the impact of absence of eNOS
BA-1000, Vurlingame, CA, USA) diluted 1:200 was used as the secondary antibody. The
standard ABC method was applied with a Vector ABC Staining Kit. Slides were counterstained
by 3,3 – Diaminobenzidine (DAB).
In a fourth cohort of pregnant eNOS KO(ko) and WT(wt) mice, placentas with the
uterine wall still attached were collected at 14.5 d of gestation for lectin histochemistry to
identify uterine natural killer cells (uNK). Sections were blocked and probed with 50 ug/mL
biotinylated Bandeiraea simplicifolia (BS-I, no. L3759; Sigma) for 1 hour at room temperature.
This was followed by peroxidase quenching using 1% H2O2 in Tris buffer (TBS), and washing
in TBS. Detection of the reaction was done using ABC complex and the slides were
counterstained with DAB. One midline section per placenta per pregnancy was examined
(N=6). The longitutional layer of the myometrium was identified and was used as a marker to
separate the decidua from that of the metrial triangle. Each section was examined using Leica
DM 4500B microscope at 100x magnification and the number of lectin-positive cells per
placental section was determined.
86
3.2.6 RT-qPCR for sFlt1 mRNA and Flt1 mRNA
In a fifth cohort of pregnant eNOS KO(ko) and WT(wt) mice, placentas with any
adherent decidua were collected for RNA isolation at 14.5 d and 17.5 d of gestation (N=3 at
each age for each strain). There were no significant changes with gestational age so results were
combined to test for effect of genotype.
Total RNA was extracted using TRIzol (Gibco BRL, Burlington, ON, Canada) according
to manufacturer’s instructions. RNA samples were purified using RNeasy MinElute Cleanup kit
(Qiagen #74204, Mississauga, ON) and treated with 2.5 mL DNAse 1 (Qiagen #79254). Using
TaqMan Reverse Transcription Reagent (Applied Biosystems/Roche #N808-0234), 1 µg of
RNA in 10 µL water was reverse transcribed using random hexamers at 25°C for 5 minutes,
42°C for 30 minutes, and 95°C for 5 minutes. cDNA was diluted with DEPC-water to 25 ng/µl.
sFlt1 splice variant retain a portion of intron 13, therefore sFlt1 primer was designed to
probe over intron 13, whereas Flt1 primer was designed to span exon 13 and exon 14 [277].
qPCR primer: sFlt1 (Forward: AGA AGA CTC GGG CAC CTA TG, Reverse: GCA GTG CTC
ACC TCT AAC GA), and Flt1 (Forward: TCG TTA GAG ATT CGG AAG CG, Reverse: GGT
CGT AGA GCC ACT GAT GG). β-actin was used as control (Forward: TCG TGC GTG ACA
TCA AAG AGA, Reverse: GAA CCG CTC GTT GCC AAT A).
cDNA was subjected to real time PCR in an optical 96-well plate with the Mastercycler
ep realplex (Eppendorf) using SYBR Green detection chemistry. To each well PCR plate, 12 µl
SYBR Green, 0.25 µl Forward primer, 0.25 µL Reverse Primer, 7 µL water, and 5 µL diluted
cDNA were added. The PCR reaction was run at 95°C for 2 minutes, then 45 cycles at 95°C for
15 s and 60°C for 1 minute. Samples were run in duplicates. The transcript level was
normalized to β-actin, and the data was expressed as fold-change relative to WT(wt) controls.
87
3.2.7 ELISA of plasma sFlt1
In a sixth cohort of eNOS KO(ko) and WT(wt) mice, blood was collected by cardiac
puncture in heparinized-coated capillary tubes from non-pregnant (N=8 per strain) and day 17.5
mice (N=5 per strain). To keep the values within the standard curve range, non-pregnant
samples were diluted 2-fold and 17.5 d pregnant samples were diluted 10-fold. Total sFlt1 in
plasma was measured in duplicate using an ELISA kit (MVR100, R&D Systems).
3.2.8 Clinical Biochemistry of maternal blood
In a seventh cohort of mice, blood (~120 µL) was collected from the saphenous vein of
fed mice prior to pregnancy and on 17.5 d of gestation (N=7-12 per strain) and analyzed using
Nova stat profile M7 for glucose, lactate, urea, creatinine, and electrolytes.
3.2.9 Statistical Analysis
Results are reported as mean ± SEM, where N is number of mothers. Significance was
tested using 2-way ANOVA followed by Holm-Sidak test for multiple comparisons. mRNA
levels, number of uNK cells and central arterial canal diameters were analyzed using a Student’s
t-test. P<0.05 was considered statistically significant.
88
3.3 RESULTS
3.3.1 Fetal, placental, and maternal growth in late gestation in eNOS
KO(ko) mice
Fetal body weight and number of fetuses per litter were significantly reduced at 17.5 d of
gestation in eNOS KO(ko) pregnancies (Table 3.1). Maternal body weight was significantly
lower at 14.5 d and 17.5 d of gestation in eNOS KO(ko) mice (Table 3.1). However, weight
gain over this interval was similar between the two strains. Weights of maternal organs
including the kidney, spleen, heart and the brain decreased similarly from non-pregnant to 17.5
d of gestation when normalized to maternal body weight in eNOS KO(ko) and WT(wt) mice
(Table 3.2). Placental weights were not different between the two strains, and increased
similarly during pregnancy (Table 3.1).
Table 3-1. Placental and maternal body weight in WT and KO mice at 14.5 d and 17.5 d of gestation.
14.5 d of gestation 17.5 d of gestation 17.5 d of gestation WT(wt) KO(ko) WT(wt) KO(ko) WT(het) KO(het)
Maternal body weight (g)
29.9 ± 0.97a
26.3 ± 0.79a*
36.3 ± 0.90b
31.7 ± 1.05b*
33.7 ± 1.48
32.1 ± 1.63
Fetal number
8.0 ± 1.2
6.7 ± 0.5
9.4 ± 0.8
6.6 ± 0.3*
7.8 ± 0.7
7.1 ± 0.4
Fetal weight (g)
0.192 ± 0.003a
0.195 ± 0.003a
0.795 ± 0.008b
0.686 ± 0.09b*
0.755 ± 0.008#
0.761 ± 0.009#
Placental weight (g)
0.174 ± 0.004a
0.166 ± 0.004a
0.213 ± 0.004b
0.206 ± 0.005b
0.214 ± 0.005
0.197 ± 0.005*
Maternal genotype in upper case and conceptus genotype in lower case. Values are mean ± SEM, N=5-13 mothers per strain; 22-97 fetuses; Different letters indicate significant changes over time within each strain (P<0.05). * P<0.05, WT(wt) vs. KO(ko) mice and WT(het) vs. KO(het). # P<0.05, WT(wt) vs. WT(het) or KO(ko) vs. KO(het).
89
Table 3-2. Maternal organ weights in non-pregnant and 17.5 d of gestation in WT(wt) and KO(ko) mice.
Non-pregnant 17.5 d of gestation WT(wt) KO(ko) WT(wt) KO(ko)
Actual (g) 0.117 ± 0.003a 0.115 ± 0.004a 0.142 ± 0.003b 0.133 ± 0.005b Kidney Per maternal body
Values are mean ± SEM, N=5-12 mothers per strain; Different letters indicate significant changes over time within each strain (P<0.05). * P<0.05, WT(wt) vs. eNOS KO(ko) mice at that time point.
92
WT(w t) KO(ko) WT(w t) KO(ko)0.0
0.1
0.2
0.3
14.5 d 17.5 d
a
b
ab
5 57 7
* *
Lume
n diam
eter (
mm)
WT(w t) KO(ko) WT(w t) KO(ko)0
100
200
300
14.5 d 17.5 d
aa
b
b
**
5 57 7
Mean
veloc
ity (m
m/s)
WT(w t) KO(ko) WT(w t) KO(ko)0.0
2.5
5.0
7.5
10.0
14.5 d 17.5 d
a
a
b
b
5 57 7
* *
Blood
flow
per u
nit w
eight
(ml/m
in/kg
)
WT(het) KO(het)0.0
0.1
0.2
0.3 *#
5 7
17.5 d
WT(het) KO(het)0
100
200
300
##
5 7
17.5 d
WT(het) KO(het)0.0
2.5
5.0
7.5
10.0
#
5 7
17.5 d
A.
B.
C.
Figure 3-1. Uterine arterial lumen diameter, mean velocity and blood flow/g at 14.5 d and 17.5 d of gestation.
Uterine arterial diameter (A), mean velocity (B) and blood flow normalized to maternal body weight (C) at 14.5 and 17.5 d of gestation in homozygous (WT(wt) and KO(ko) mice) and heterozygous (WT(het) and KO(het)) mice. Different letters indicate significant changes over time within each strain (P<0.05). * P<0.05, WT(wt) (open bar) vs. KO(ko) mice (black bar) or WT(het) (light gray bar) vs. KO(het) (dark gray bar) mice. # P<0.05, WT(wt) vs WT(het) or KO(ko) vs. KO(het). Mean ± SEM for N shown in bars.
93
WT(w t) KO(ko) WT(w t) KO(ko)0
100
200
300
400
14.5 d 17.5 d
a a
b b
5 57 7Peak
Sys
tolic
Velo
city (
mm/s)
WT(w t) KO(ko) WT(w t) KO(ko)0
50
100
150
200
14.5 d 17.5 d
aa
b
b
*
5 57 7End-
Dias
tolic
Velo
city (
mm/s)
WT(w t) KO(ko) WT(w t) KO(ko)0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
14.5 d 17.5 d
5 57 7
* *
Resis
tanc
e In
dex
A.
B.
C.
Figure 3-2. Uterine artery peak systolic and end-diastolic velocities and Resistance Index at 14.5 d and 17.5 d of gestation.
Peak systolic (A) and end-diastolic (B) velocities were used to calculate Resistance Index (C) at 14.5 and 17.5 d of gestation in WT(wt) and KO(ko) mice. Different letters indicate significant changes over time within each strain (P<0.05). * P<0.05, WT(wt) (open bar) vs KO(ko) (black bar). Mean ± SEM for N shown in bars.
94
3.3.3 Reduced remodeling of the spiral and central arterial canals in
eNOS KO(ko) mice
eNOS deficiency also resulted in blunted remodeling of the uteroplacental circulation
downstream of the uterine arteries, specifically the spiral arteries and central arterial canals. In
normal pregnancy, the spiral arteries that supply blood to the placental exchange region undergo
significant morphological changes, including dilation, elongation and a gradual loss of smooth
muscle cells, such that they become non-vasoactive, high caliber, low resistance vessels [58,
131]. Failure of the spiral arteries to show this transformation is thought to be a common
underlying cause of preeclampsia [279]. Vascular corrosion casts showed reduced spiral artery
coiling in the eNOS KO(ko) mothers relative to controls (Figure 3.3 A,B). This may be due at
least in part to a significant reduction in spiral artery length at 14.5 d and 17.5 d of gestation (by
- 18-30%, Figure 3.3E) whereas the diameter of the spiral arteries were similar between the two
strains at both gestational ages (140 ± 6 µm in KO vs. 169 ± 2 µm in controls at 14.5 d; 176 ± 6
µm in KO vs 170 ± 8 µm in controls at 17.5 d). In addition, immunoreactive desmin, a marker
of vascular smooth muscle, was positive around the spiral arteries in the decidua at 17.5 d in the
eNOS KO(ko) placentas (Figure 3.3 C,D). This suggested that these arteries retained their
muscular wall and therefore may have maintained responsiveness to vasoconstrictor stimuli.
In the mouse, spiral artery remodeling is promoted by granulated uNK lymphocytes
which are abundant in the mouse placenta in early pregnancy [229]. Therefore, I examined the
distribution of uNK cells at 14.5 d of gestation using DBA lectin staining, and found a 30%
reduction in their number in the decidua in eNOS KO(ko) placentas (Figure 3.4 C,D). Thus,
impaired spiral artery remodeling may be secondary to impaired uNK cell recruitment and/or
retainment and/or activity in the decidua in eNOS KO(ko) pregnancies.
95
The maternal spiral arteries are invaded by trophoblast giant cells from the conceptus
[56, 57]. These cells breech the spiral artery wall thereby permitting maternal blood to enter the
trophoblast-derived central arterial canals. These canals direct maternal blood into the
trophoblast-lined sinusoids of the labyrinthine exchange region [56]. The maternal spiral
arteries supplied between 1 and 4 central arterial canals in the placentas in both strains (2.7 ± 0.3
in eNOS, 2.7 ± 0.5 in controls). Despite having similar numbers, the diameters of the arterial
canals were significantly lower in eNOS KO(ko) pregnancies than in controls at 17.5 d of
gestation (Figure 3.5B). These findings suggest that eNOS plays an important role in
remodeling the maternal spiral arteries, and in promoting enlargement of the conceptus-derived
arterial canals during pregnancy in mice.
96
A BWT (wt) eNOS KO (ko)
WT(wt) KO(ko) WT(wt) KO(ko)0
1
2
3
4
5
6
7
8
14.5 d 17.5 d
* *
5 74 6
Spira
l arte
rial le
ngth
(mm)
WT(het) KO(het)0
1
2
3
4
5
6
7
8 *
5 5
17.5 dWT(wt) KO(ko) WT(wt) KO(ko)
0
1
2
3
4
5
6
7
8
14.5 d 17.5 d
* *
5 74 6
Spira
l arte
rial le
ngth
(mm)
WT(het) KO(het)0
1
2
3
4
5
6
7
8 *
5 5
17.5 d
E
D
1 mm 1 mm
C
+ +
Figure 3-3. Vascular cast image of the spiral arteries, spiral artery length, and immunohistochemistry of desmin.
(A,B) Scanning electron micrograph of maternal vascular cast partially filled from the arterial side at 17.5 d of gestation in WT(wt) and KO(ko) mice. (C,D) Desmin staining at 17.5 d of gestation in the placenta. (E) Spiral artery length was determined from vascular cast images. *P<0.05, WT(wt) (open bar) vs. KO(ko) mice (black bar) or WT(het) (light gray bar) vs. KO(het) mice (dark gray bar). +, spiral artery. Mean ± SEM for N shown in bars.
97
A B
D D
MT MT
WT (wt) eNOS KO (ko)
WT(wt) KO(ko)0.00
0.01
0.02
0.03
0.04
4 5
Metrial Triangle
# of
uN
K ce
ll/ar
ea(1
/ μm
2 )
WT(wt) KO(ko)0.00
0.01
0.02
0.03
0.04
4 5
Decidua
*#
of u
NK
cell/
area
(1/ μ
m2 )
C D
Figure 3-4. Histochemistry of uNK cells in the placenta at 14.5 d of gestation.
(A,B) DBA lectin staining in the placenta at 14.5 d of gestation in WT(wt) and eNOS KO(ko) mice. In the metrial triangle (C) and decidua (D), the numbers of uNK cells are shown. MT, metrial triange; D, decidua basalis. Mean ± SEM for N shown in bars. Scale bars, 250 µm.
98
CAC
WT(wt) KO(ko)0.0
0.1
0.2
0.3
0.4
6 6
Central arterial canal
*
Dia
met
er (μm
)
A B
Figure 3-5. Vascular cast image of central arterial canal and central arterial canal diameter at 17.5 d of gestation.
(A) Light micrograph image of a fetoplacental vascular cast at 17.5 d of gestation. (B) Central arterial canal diameter was significantly smaller in eNOS KO(ko) placentas as compared to WT(wt). *P<0.05, WT(wt) (open bar) vs. KO(ko) mice (black bar). Mean ± SEM for N shown in bars. CAC, central arterial canal. Bar, 1 mm.
99
3.3.4 Role of maternal versus fetal genotype on uteroplacental
phenotype.
To understand the extent to which the maternal and fetal genotypes determined the
uteroplacental phenotype, I performed a crossbreeding study so that the mothers were either
eNOS KO or WT, and their fetuses were all heterozygotes. I found that maternal genotype was
a significant factor in determining uterine arterial lumen diameter (Figure 3.1) and spiral artery
length (Figure 3.3), whereas mean blood velocity was independent of maternal genotype (Figure
3.1). Maternal genotype did not significantly influence uterine arterial blood flow/g maternal
weight (Figure 3.1), but there was a strong trend (P=0.06). Fetal genotype was a significant
factor in determining uterine arterial lumen diameter, uterine arterial mean blood velocity and
uterine flow/g because these parameters were significantly decreased in WT(het) as compared to
WT(wt) mice (Figure 3.1). Fetal genotype, however, had no significant influence on spiral
artery length (Figure 3.3). Thus, spiral artery length was a maternal-genotype dependent
phenotype, mean uterine artery blood velocity was a fetal-genotype dependent phenotype, and
both maternal and fetal genotypes contributed to the remaining uteroplacental phenotypes of
eNOS KO pregnancies at 17.5 d of gestation.
3.3.5 Increased placental hypoxia in eNOS KO(ko) mice
Decreased uterine arterial blood flow would be anticipated to decrease oxygen delivery
to the placenta. I therefore hypothesized that the placentas in eNOS KO(ko) pregnancies would
be hypoxic in vivo. To test this, I used the hypoxia marker Hypoxyprobe-1. As shown in Figure
3.6, strong immunoreactivity was detected in the spongiotrophoblast and trophoblast giant cell
100
layers of the junctional zone of the eNOS KO(ko) placentas, whereas faint staining primarily in
the spongiotrophoblast cell layer was detected in controls.
3.3.6 Reduced placental expression of sFlt1 mRNA levels and no
significant changes in maternal sFlt1 levels in eNOS KO(ko) mice
Hypoxia stimulates placental trophoblast cells to increase production of the
antiangiogenic factor, sFlt1 which binds to and neutralizes proangiogenic, VEGF [180, 181,
280]. Indeed, placental hypoxia is thought to cause elevated sFlt1 levels in maternal plasma in
preeclamptic pregnancies [180]. However, despite apparent placental hypoxia in eNOS KO(ko)
pregnancies (Figure 3.6), there was no significant increase in placental sFlt1 mRNA levels
measured by RT-qPCR. Indeed, sFlt1 mRNA level was significantly decreased in eNOS
KO(ko) placentas (Figure 3.7). In maternal plasma, sFlt1 levels increased from non-pregnant to
late pregnancy in both strains, and was not significantly different between the two strains at late
gestation (Figure 3.7).
101
B
Sp
La
A
DC
WT(wt) KO(ko)
KO(ko)KO(ko)
Figure 3-6. Placental hypoxia using Hypoxyprobe-1 immunohistochemistry.
Hypoxyprobe-1 immunohistochemistry was used to identify hypoxic regions in the placenta in both WT(wt) (A) and KO(ko) (B-D) mice at 17.5 d of gestation. Representative images are shown. (C) Negative control. (D) Higher magnification view of the placenta. Positive staining in trophoblast giant cells, spongiotrophoblast cells and in the adjacent labyrinth area. Arrows indicate trophoblast giant cells; La, Labyrinth; Sp, Spongiotrophoblast cells. (N=3-6). Bars, 50 µM.
102
WT(wt) KO(ko)0.0
0.2
0.4
0.6
0.8
1.0
1.2
6 6
Flt1
(fold
cha
nge
rela
tive
to W
T(w
t))
WT(wt) KO(ko)0.0
0.2
0.4
0.6
0.8
1.0
1.2*
6 6
sFlt1
(fold
cha
nge
rela
tive
to W
T(w
t))
A B
Non-pregnant 17.5 d0
10000
20000
30000
40000
8 8 5 5
a a
bb WT(wt)
KO(ko)
sFlt1
(pg/
mL)
C
Figure 3-7. sFlt1 mRNA and Flt1 mRNA levels and plasma sFlt1 levels in WT(wt) and KO(ko) mice.
mRNA levels in the placenta normalized to β-actin are shown for sFlt1 (A), and Flt1 (B) in WT(wt) and KO(ko) mice. (C) ELISA for sFlt1 plasma analysis at non-pregnant and 17.5 d of gestation. Mean ± SEM for N shown in bars; Different letters indicate significant changes over time within each strain (P<0.05). *P<0.05, WT(wt) (open bar) vs. KO(ko) mice (black bar).
103
3.3.7 Maternal electrolyte balance is altered in pregnant eNOS KO(ko)
mice
Alterations in maternal blood biochemistry occur during normal pregnancy [281], and
are abnormal in the pathogenesis of preeclampsia [282]. Therefore, maternal blood was
collected from the saphenous vein and subjected to biochemical analysis. Plasma calcium level
increased by 30% during pregnancy in WT(wt) mothers only (Table 3.3). Therefore, at 17.5 d
of gestation, plasma calcium levels were significantly lower in eNOS KO(ko) as compared to
thereby decreasing fetoplacental perfusion, and decreases the surface area for feto-maternal
exchange. Both effects would decrease the transfer of oxygen and nutrients across the placental
barrier, thereby limiting fetal growth. The human fetoplacental circulation exhibits low vascular
resistance and lacks autonomic innervation [216]; therefore, circulating and locally released
vasoactive molecules are likely critically involved in determining fetoplacental hemodynamics
[217]. On a longer time scale, vasculogenesis and angiogenesis are undoubtedly also important
determinants [5, 95]. NO is a potent vasodilator synthesized by the eNOS isoform in the
fetoplacental arterial endothelium [12, 292]. It is also an important factor promoting
angiogenesis and vasculogenesis [15, 114]. In normal human pregnancy, eNOS expression
increases with gestational age in the fetoplacental circulation [293], whereas eNOS mRNA,
protein, and activity are reduced in IUGR placentas [294-296]. Thus, evidence suggests eNOS
is an important factor determining fetoplacental vascular resistance.
114
In animal models, inhibition of NOS using non-selective competitive inhibitors such as
L-NAME, elicited fetal growth restriction and preeclampsia like symptoms in gravid rats and
sheep [21, 22, 124]. Recently, I (Chapter 3) and others [112, 218] have shown that mice lacking
the eNOS gene also exhibit fetal growth restriction in late gestation. However eNOS KO(ko)
mothers have impaired uteroplacental remodeling and a blunted rise in uteroplacental perfusion
so whether IUGR in eNOS KO(ko) fetuses is secondary to reduced uteroplacental perfusion is
not clear.
In the current study I hypothesized that eNOS plays an important role in promoting
growth and remodeling of the umbilico-placental vasculature during late gestational
development and that this is a fetal effect independent of maternal genotype. To evaluate this
hypothesis, I quantified umbilical blood flow and Resistance Index using micro-ultrasound,
visualized the fetoplacental vasculature using vascular corrosion casts, and evaluated hypoxia in
the fetus and placenta in eNOS KO mice and their background strain, C57Bl/6J (WT).
4.2 MATERIAL AND METHODS
4.2.1 Breeding
Experiments were approved by the Animal care committee of Mount Sinai Hospital
(Toronto, ON, Canada) and were conducted in accordance with guidelines established by
Canadian Council on Animal Care. eNOS KO mice (KO) and C57Bl/6J wildtype (WT) controls
were obtained from Jackson Laboratories (Maine) or raised in-house. Females were bred at 8-
14 weeks of age and were studied in their first pregnancies. The day that a vaginal copulation
plug was detected was designated day 0.5 of pregnancy. KO and WT refer to the adult
genotype, and ko, het, and wt refer to the conceptus genotype. In the first cohort, mice were
115
bred with their cognate strain (N=8 eNOS KO mothers; N=6 WT mothers). The mice from
these experiments are thus referred to as eNOS KO(ko) and WT(wt). They were studied at days
14.5 (end of organogenesis) or 17.5 d of pregnancy (2 days before normal term delivery). In the
second cohort, eNOS KO females (N=12) were bred with WT males to obtain eNOS KO(het)
mice and WT females (N=10) were bred with eNOS KO males to obtain WT(het) mice.
Crossbred pregnancies were studied at 17.5 d of gestation.
4.2.2 Umbilico-placental Hemodynamics
In the first and second cohort of animals, the fetoplacental circulation was examined
using transcutaneous micro-ultrasound (Model 770 with 30-MHz transducer; VisualSonics,
Toronto, Canada) while pregnant mice were lightly anesthetized with ~ 1.5% isoflurane in
oxygen by face mask. Maternal heart rate and rectal temperature were monitored. Rectal
temperature was maintained between 37oC and 38 oC. Doppler waveforms in the umbilical vein
and artery were obtained near the placental end of the umbilical cord (Figure 4.1 B,C). Peak
systolic velocity (PSV) and end-diastolic velocity (EDV), stroke distance (area under the curve)
and R-R interval were measured from three consecutive cardiac cycles and the results were
averaged. Umbilical venous diameter was measured from power Doppler images (Figure 4.1
A). Mean velocity (MV) was calculated by dividing stroke distance by R-R interval. Umbilical
artery Resistance Index (RI = (PSV-EDV)/PSV) was calculated to quantify arterial blood flow
pulsatility. A parabolic blood velocity distribution was assumed so that umbilical venous blood
flow was determined by the formula: F= ½ MV x п x (D/2)2 (where MV = mean velocity
(cm/s); D = diameter (cm); F = Blood flow (ml/min)).
116
P
Em
A B
C
Figure 4-1. Ultrasound evaluation of umbilico-placental vascular structure and hemodynamics.
(A) Power Doppler image of the fetoplacental circulation in an anesthetized pregnant mouse in vivo. The color Power Doppler image of the umbilical vessels was used for caliper measurements of umbilical arterial and venous diameters. Doppler blood velocity waveforms from (B) umbilical artery and (C) umbilical vein are shown. Em = embryo; P = placenta.
117
4.2.3 Fetoplacental vascular casts
In the first cohort of animals at 17.5 d of gestation, vascular corrosion casts of the
fetoplacental vasculature were prepared using published methods [56, 297]. The pregnant
mother was sacrificed by cervical dislocation and the uterus was rapidly removed and immersed
in ice cold PBS. An implantation site was cut from the uterus and the uterine muscle was cut
along the antimesometrial edge to expose the yolk sac. The yolk sac and the amniotic
membrane were then cut near the placenta to expose the embryo and the placental surface. The
exposed embryo and the placenta were then bathed in warm PBS to resume cardiac function and
placental blood flow. Drops of 3% PFA were applied to the umbilical vessels to decrease
vasospasm. A double-lumen tapered glass cannula was inserted into the umbilical artery with
the vein nicked to serve as a vent. Warm 2% xylocaine in 0.9% NaCl and 100 IU heparin/ml
was perfused through the cannula to displace blood from the fetoplacental vasculature. Methyl
methacrylate casing compound (Batson’s no. 17; Polyscience inc., Warrington, PA) was then
perfused into the arterial vasculature via the umbilical artery until it was seen to exit via the
umbilical vein. The umbilical cord was then tied off to maintain pressure during
polymerization. Tissue was then digested using 20% KOH and removed with distilled water
washes. Casts were examined by scanning electron microscopy (FEI XL30, Toronto, ON,
Canada). The lengths of the capillary lobules (mag. 250X) were measured at four orthogonal
locations on the chorionic surface of each cast, and diameters of fetal capillaries (mag. 1200x)
were measured at 20-40 arbitrary locations on each cast. The results from 6 placental casts from
4 pregnancies in WT(wt) mice and 6 placental casts from 3 pregnancies in eNOS KO(ko) mice
were averaged.
118
4.2.4 Detection of Hypoxia in the embryo
In a third cohort of pregnant eNOS KO(ko) and WT(wt) mice, the hypoxia marker,
pimonidazole hydrocholoride (HypoxyprobeTM –1, 60 mg/kg mice, Chemicon) was injected
intraperitoneally at day 17.5 (N=5-6 mothers in each group). Two hours later, the mother was
sacrificed, and placentas and fetuses were collected and processed for immunohistochemistry.
See section 3.2.4 for more detail.
4.2.5 Immunohistochemistry and RT-qPCR for VEGF
In the third cohort of pregnant animals, placentas with the uterine wall still attached were
collected and immersion-fixed overnight at 4oC in 4% PFA for immunohistochemistry.
Placental sections at 17.5 d of gestation were deparaffinized in xylene, rehydrated and
underwent microwave antigen retrieval in 10 mM sodium citrate. Slides were then stained for
VEGF (Rabbit anti-VEGF, Thermo Scientific, RB-222-P1, Fremont, CA, USA; 1:200).
Biotinylated goat anti-rabbit IgG (Vector laboratories, BA-1000, Burlingame, CA, USA)
diluted 1:200 was used as the secondary antibody. The standard ABC method was applied with
a Vector ABC Staining Kit. Slides were counterstained by DAB. One midline section per
placenta per pregnancy was examined (N=6 pregnancies per group).
In a fourth cohort of pregnant eNOS KO(ko) and WT(wt) mice, placentas with any
adherent decidua were collected for RNA isolation at 14.5 d and 17.5 d of gestation (N=3 at
each age for each strain). The method described in section 3.2.6 was followed. There were no
significant changes with gestational age so data were combined to test for effect of genotype.
119
qPCR primer was designed specifically to include all splice varients of VEGF-A
(Forward: GAG CAG AAG TCC CAT GAA CTG, Reverse: TGT CCA CCA GGG TCT CAA
TC). β-actin was used as control (Forward: TCG TGC GTG ACA TCA AAG AGA, Reverse:
GAA CCG CTC GTT GCC AAT A). Samples were run in duplicates. The transcript level was
normalized to β-actin level, and the data were expressed as fold-change relative to WT(wt)
controls.
4.2.6 Hematology of fetal blood
In a fifth cohort of pregnant eNOS KO(ko) and WT(wt) mice (N=3-7 mothers), fetal
blood (~ 10 µL) was collected from the umbilical vessels in EDTA-coated capillary tubes at
17.5 d of gestation and analyzed in a Hematology Analyzer (AcT Diff, Beckman Coulter,
Toronto, Canada) to obtain red blood cell, platelet and white blood cell counts, hematocrit,
hemoglobin, mean corpuscular volume, mean corpuscular hemoglobin, and mean corpuscular
hemoglobin concentration.
4.2.7 Statistical Analysis
Results are reported as mean ± SEM. Significance was tested using a 2-way ANOVA
followed by Holm-Sidak test for multiple comparisons. Hematology parameters, VEGF mRNA
levels and capillary lobule length were analyzed using a Student’s t-test. P<0.05 was considered
statistically significant.
120
4.3 RESULTS
4.3.1 Reduced fetoplacental blood flow at mid- and late gestation in
eNOS KO(ko) mice.
Fetal body weight was not significantly different between the two groups at 14.5 d of
gestation but fetal weight was significantly lower in eNOS KO(ko) fetuses than WT(wt) fetuses
at 17.5 d of gestation (Figure 4.2 D). However, at both ages umbilical venous blood flow was
significantly reduced in eNOS KO(ko) fetuses even when expressed per unit fetal weight
(Figure 4.2 C,E). At 14.5 d, the 20% reduction in umbilical venous blood flow/g in eNOS
KO(ko) fetuses was primarily due to a significant reduction in mean velocity (-10%) whereas
umbilical venous diameter was not significantly altered (Figure 4.2). At 17.5 d of gestation,
umbilical venous blood flow/g remained significantly lower in the eNOS KO(ko) fetuses (21%
lower than WT(wt)) due to a significant reduction in mean velocity as well as blunted growth of
the umbilical vein lumen diameter (Figure 4.2). These findings indicate an essential role for
eNOS in supporting fetoplacental perfusion and fetal growth in late gestation in mice.
In human IUGR, reduced placental blood flow is associated with increased blood flow
pulsatilility in the umbilical artery [298]. I therefore quantified pulsatility in the umbilical artery
using Resistance Index at 14.5 d and 17.5 d of gestation. I observed significant increases in
peak systolic and end-diastolic velocities in WT(wt) fetuses from 14.5 d to 17.5 d of gestation
(Figure 4.3 A,B). There was a much smaller increase in end-diastolic velocity over this interval
in eNOS KO(ko) fetuses with the result that the Resistance Index significantly decreased with
age in the WT(wt) and not the eNOS KO(ko) group (Figure 4.3). At 17.5 d of gestation, both
end-diastolic (-30%) and peak systolic (-13%) blood velocities were significantly decreased in
121
eNOS KO(ko) fetuses versus WT(wt) (Figure 4.3). The Resistance Index tended to be greater in
the eNOS KO(ko) group at 17.5 d (P=0.062).
WT(wt) KO(ko) WT(w t) KO(ko )0.0
0.1
0.2
0.3
0.4
0.5
0.6
14.5 d 17.5 d
a ab b
12 2220 26
*
Veno
us lu
men D
iamete
r (mm
)
WT(het) KO(het)0.0
0.1
0.2
0.3
0.4
0.5
0.6 #
14 23
17.5 d
WT(w t) KO(ko ) WT(w t) KO(ko)0.0
0.2
0.4
0.6
0.8
1.0
14.5 d 17.5 d
a a
bb
39 5647 50
*
Fetal
weig
ht (g
)
WT(het) KO(het)0.0
0.2
0.4
0.6
0.8
1.0
62 97
17.5 d
# #
WT (w t) KO(ko) WT(w t) KO(ko )0
10
20
30
40
50
60
14 .5 d 17.5 d
12 2220 26
*
Veno
us m
ean v
elocit
y (mm
/s)
*
WT(het) KO(het)0
10
20
30
40
50
60
#
14 23
17.5 d
WT(w t) KO(ko) WT(w t) KO(ko )0
5
10
15
14.5 d 17.5 d
a
a
bb
12 2220 26
*
*
Bloo
d flow
per fe
tal w
eight
(ml/m
in pe
r 10 g
)
WT(het) KO(het)0
5
10
15
#
14 23
17.5 d
#
WT(w t) KO(k o) WT(wt) KO(ko)0.0
0.1
0.2
0.3
0.4
14.5 d 17.5 d
aa
b
b
12 2220 26
*
*
Bloo
d flow
(ml/m
in)
WT(het) KO(het)0.0
0.1
0.2
0.3
0.4
14 23
17.5 d
##
A.
B.
C.
D.
E.
Figure 4-2. Umbilical venous lumen diameter, mean velocity, blood flow and blood flow/g of fetal weight and fetal weight at 14.5 d and 17.5 d of gestation. Umbilical venous lumen diameter (A), mean velocity (B), blood flow (C), and blood flow normalized to fetal weight (E) were determined in lightly anesthetized mice at 14.5 d and 17.5 d of gestation using micro-ultrasound. (D) Fetal weight was also recorded at these time points. Different letters indicate significant changes over time within each strain (P<0.05). * P<0.05, WT(wt) (open bar) vs. KO(ko) mice (black bar). # P<0.05, WT(wt) vs WT(het)(light gray bar), or KO(ko) vs. KO(het)(dark gray bar). Mean ± SEM. Number of embryos (N) is shown in the bars.
122
A.
WT(w t) KO(ko) WT(w t) KO(ko)0
50
100
150
14.5 d 17.5 d
ab
*
11 1919 19Peak
Sys
tolic
Velo
city (
mm/s
)
WT(w t) KO(ko) WT(w t) KO(ko)0
2
4
6
8
10
12
14.5 d 17.5 d
a
b
ab
*
11 1919 19End-
Dias
tolic
Velo
city (
mm/s
)
B.
WT(w t) KO(ko) WT(w t) KO(ko)0.900
0.925
0.950
0.975
1.000
14.5 d 17.5 d
a
b
11 1919 19
Resis
tance
Inde
x
C.
Figure 4-3. Umbilical artery peak systolic and end-diastolic blood velocities, and Resistance Index at 14.5 d and 17.5 d of gestation.
Peak systolic (A) and end-diastolic (B) velocities were determined and Resistance Index (C) was calculated in lightly anesthetized mice using micro-ultrasound at 14.5 d and 17.5 d of gestation. Different letters indicate significant changes over time within each strain (P<0.05). *P<0.05, WT(wt) (open bar) vs. KO(ko) mice (black bar). Mean ± SEM. Number of embryos (N) is shown in the bars.
123
4.3.2 Fetoplacental vascularization and placental expression of VEGF are
reduced in eNOS KO(ko) fetuses.
Because the Resistance Index failed to decrease from mid to late gestation in eNOS
KO(ko) fetuses, I suspected that fetoplacental vascularization might be reduced in late gestation
in eNOS KO(ko) placentas. To investigate this possibility, I prepared vascular corrosion casts
of the fetoplacental circulation of eNOS KO(ko) fetuses at 17.5 d and found shorter capillary
lobules at the chorionic surface (-24%) (Figure 4.4 B,E) but similar mean capillary diameters
(11.7 ± 0.5 µm vs. 12.6 ± 0.9 µm in WT(wt)). Capillary density appeared to be decreased in the
KO(ko) placentas (Figure 4.4). These findings suggest that eNOS promotes fetoplacental
vascularization.
To determine whether impaired angiogenesis is associated with a reduction in expression
of the proangiogenic factor VEGF, I next performed RT-qPCR and immunohistochemistry to
detect VEGF mRNA and protein levels in the placenta. I found significantly decreased VEGF
mRNA levels, and an apparent reduction in VEGF immuno-reactivity in eNOS KO(ko)
placentas (Figure 4.5). Therefore, decreased VEGF levels may contribute to impaired
fetoplacental vascularization in the eNOS KO(ko) placentas.
124
A B
C D
WT(wt)
WT(wt)
KO(ko)
KO(ko)
A B
C D
WT(wt)
WT(wt)
KO(ko)
KO(ko)
WT(wt) KO(ko)0
500
1000
1500
*
6 6
Cap
illary
lobu
le le
ngth
(um
)
E
500 µm 500 µm
50 µm 50 µm
Figure 4-4. Vascular cast of the fetoplacental circulation and capillary lobule length at 17.5 d of gestation in WT(wt) and KO(ko) mice.
Scanning electron microscope image of the fetoplacental vasculature in the labyrinth in WT(wt) (A,C) and KO(ko) (B,D) mice at 17.5 d of gestation. Capillary lobule length (i.e. arrow in A) was significantly shorter (B,E) and capillary density was reduced (D) in eNOS KO(ko) placentas. *P<0.05, WT(wt) (open bar) vs. KO(ko) mice (black bar). Mean ± SEM. Number of embryos (N) is shown in the bars.
125
WT(wt) KO(ko)0.0
0.2
0.4
0.6
0.8
1.0
1.2 *
6 6
VEG
F(fo
ld c
hang
e re
lativ
e to
WT(
wt))
A B
C
WT(wt)
KO(ko)
D
S
L
D
S
L
Figure 4-5. VEGF mRNA by RT-qPCR and protein by immunohistochemistry in the placenta at 17.5 d of gestation.
(A) VEGF mRNA levels normalized to β-actin in WT(wt) and KO(ko) placentas. VEGF immunohistochemistry staining in WT(wt) (B) and KO(ko) mice (C) at 17.5 d of gestation. Representative images of placentas from N=6 mothers per genotype are shown. Mean ± SEM. Number of embryos (N) is shown in the bars. *P<0.05, WT(wt) (open bars) vs. KO(ko) mice (black bars). L, labyrinth; S, spongiotrophoblast; D, decidua. Bars, 250 µm.
126
4.3.3 eNOS KO(ko) pups are hypoxic and anemic and show increased
erythrocyte size.
Decreased fetoplacental vascularity and decreased umbilical venous blood flow would
be anticipated to decrease fetal oxygen delivery unless there is a compensatory increase in the
oxygen carrying capacity of the fetal blood. To test this, fetal cord blood was collected and
subjected to hematological analysis. Surprisingly, the erythrocyte count and hematocrit were
significantly lower in eNOS KO(ko) fetuses (Table 4.1) which would tend to exacerbate low
oxygen delivery caused by low umbilical blood flows. I next used the hypoxia marker
Hypoxyprobe-1 to seek direct evidence of fetal tissue hypoxia. I examined the fetal heart, lung,
kidney and liver and found strong immunoreactivity for the hypoxia marker in these tissues in
eNOS KO(ko) fetuses suggesting that fetoplacental oxygen delivery was inadequate to support
normal tissue oxygenation (Figure 4.6). Mean corpuscular volume of erythrocytes was elevated
in eNOS fetuses (Table 4.1) suggesting an increase in the proportion of immature erythrocytes.
This finding is consistent with increased erythropoiesis [35]. An increased proportion of
immature erythrocytes is also observed in human IUGR fetuses [299].
127
WT(wt) KO(ko)
A
B
C
D
heart
lung
kidney
liver
Figure 4-6. Fetal hypoxia using Hypoxyprobe-1 immunohistochemistry.
Hypoxyprobe-1 immunohistochemistry was used to identify hypoxic regions in the fetus. Strong immunoreactivity was detected in the fetal heart (A), lung (B), kidney (C) and liver (D) of WT(wt) as compared to KO(ko) at E17.5d. Representative images obtained from N=4 fetuses from 4 different litters are shown.
128
Table 4-1. Hematology parameters in fetal WT(wt) and KO (ko) mice at 17.5 d of gestation.
Values are mean ± SEM; NS = Not Significant RBC, red blood cell count; Hgb, hemoglobin concentration, Hct, hematocrit; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume, MCH, mean corpuscular hemoglobin; WBC, white blood cell count; Plt, platelet count.
129
4.3.4 Fetal growth is determined by fetal genotype.
eNOS KO(ko) mothers have decreased cardiovascular adaption to pregnancy in late
gestation (Chapter 2), so whether this contributed to IUGR in eNOS KO(ko) fetuses was not
clear. To determine the extent to which maternal genotype determines fetal phenotype, I
performed a crossbreeding study and examined heterozygous placentas and fetuses at 17.5 d of
gestation. In this study, heterozygous fetuses had either homozygous eNOS KO mothers
(KO(het)) or control mothers (WT (het)). Umbilical venous blood flow/g fetal weight, and the
parameters from which it was derived (i.e. mean venous velocity, venous lumen diameter, and
fetal weight) were not significantly affected by maternal genotype at 17.5 days of gestation
(Figure 4.2). Fetal body weight and umbilical venous blood flow/g in heterozygous fetuses
were intermediate and significantly different than both homozygous KO(ko) and WT(wt)
fetuses. Thus, deficits in fetoplacental perfusion and fetal growth are primarily determined by
the fetal genotype. Interestingly, heterozygous placental weights were significantly smaller
when the mother was an eNOS KO(het) (0.197 ± 0.01 g) rather than a WT(het) (0.214 ± 0.01 g)
suggesting an influence of maternal genotype on this parameter.
4.4 DISCUSSION
In this chapter I showed that eNOS promotes vascularization, and contributes to the
increase in umbilical venous blood flow and decrease in umbilical arterial vascular resistance in
the fetoplacental circulation. eNOS KO(ko) mice showed reduced capillary density and
capillary lobule length at late gestation relative to WT(wt) mice. This reduced vascularization
may be due to decreased VEGF mRNA levels and protein expression in the eNOS KO(ko)
placentas. These changes likely contributed to fetal hypoxia and fetal growth restriction at term.
130
I also showed using cross-breeding studies that fetal growth was primarily determined by the
fetal eNOS genotype and that the maternal eNOS genotype was not a significant factor.
Umbilical venous blood flow was significantly reduced at both 14.5 d and 17.5 d of
gestation in eNOS KO(ko) fetuses as compared to WT(wt) fetuses. This decrease in
fetoplacental blood flow may be due to reduced vasodilatory effects of NO on the fetoplacental
circulation, reduced angiogenesis (vascularization) of the placenta, hypoxic vasoconstriction,
and/or a reduced fetal requirement for fetoplacental flow due to fetal growth inhibition caused
by fetal eNOS deficiency.
In the eNOS KO(ko) fetuses, the enlargement of the umbilical venous diameter was
blunted and umbilical venous blood flow was decreased at late gestation. Similar decreases
have been reported in human IUGR pregnancies [300, 301]. Failure to enlarge the umbilical
vein during pregnancy may be due to impaired vasodilation and outward hypertrophic
remodeling caused by the loss of eNOS. The fetoplacental circulation lacks autonomic
innervation [216]; therefore, vascular relaxation of the blood vessels are most likely mediated by
circulating and locally released vasodilators. One of the most potent but labile vasodilators
produced by the fetoplacental endothelium is NO produced by the eNOS isoform. Basal NO
release is important in maintaining low vascular resistance in the fetoplacental circulation in
sheep and humans [124, 302, 303]. In humans, the NO synthesis inhibitor, L-NAME, caused
vasoconstriction in isolated placental arteries, and isolated and perfused placental cotyledons
[302, 303]. In sheep, L-NAME decreased fetoplacental blood flow by increasing vascular
resistance [124]. Therefore, eNOS deficiency could blunt vasodilation in the umbilical and/or
fetoplacental vasculature, thereby leading to increased vascular resistance. High resistance is
supported by the observation that the Resistance Index failed to decrease from mid to late
gestation, and the trend toward an elevation in Resistance Index at late gestation in eNOS
131
KO(ko) fetuses. Therefore, eNOS plays a role in maintaining low vascular resistance and
thereby promoting increased blood flow in the fetoplacental circulation.
Hormones such as estrogen use the NO pathway to elicit vasodilation and thus may be
involved in maintaining low vascular resistance in the fetoplacental circulation. In the uterine
circulation, estrogen mediates vasodilation directly by acting on the vascular smooth muscle
cells via the cGMP pathway and/or mediates vasodilation indirectly by acting on the endothelial
cells to increase eNOS activation and NO production [113]. In the uterine circulation, estradiol-
17β infusion in vivo increased uterine arterial blood flow and these effects were inhibited with
L-NAME [148, 149]. However, umbilical arteries in mice failed to vasodilate in response to
estrogen in vitro [304], although they do respond to the NO donor, sodium nitroprusside (SNP)
[304]. The lack of relaxation response to estrogen may be a function of receptor expression;
estrogen receptor localization in the umbilical vessels in the mouse is unknown.
Shear stress is likely a major stimulus for NO release in the umbilico-placental
circulation as in other vascular beds [6, 217]. In normal pregnancy, increased vascularization of
the placenta would decrease downstream vascular resistance and this would be an effective
stimulus for increasing velocity of flow and shear stress in upstream vessels. Increased shear
stress exerted by blood flow on the endothelium increases eNOS expression, enzyme activity,
and NO production thereby increasing NO-mediated smooth muscle relaxation and vasodilation
in response to increases in blood flow [13, 69]. Thus, loss of eNOS would be predicted to blunt
vasodilation in response to elevated shear stress. However, this does not appear to be the case in
systemic arteries including carotid, coronary and skeletal muscle arteries in non-pregnant eNOS
KO mice [283]. In these vessels, compensatory mechanisms mediated by nNOS, EDHF, and/or
prostaglandins maintained shear-mediated vasodilation in eNOS KO animals [283]. Whether
such compensation occurs in the umbilical and placental vasculature of eNOS KO mice is
132
unknown. Therefore, in the mouse fetoplacental circulation, eNOS may work through pathways
other than shear stress and/or estrogen to mediate vasodilation, such as the VEGF pathway
[157]. VEGF is a well known endothelial growth factor, and endothelium-dependent
vasodilator, and NO is an important mediator of its effects [305, 306].
As well as being a vasodilator, VEGF is also a key mediator of angiogenesis and its
effects are predominantly mediated by eNOS [307]. VEGF is thought to play an important role
during normal placental development in humans [5]. In mice, placental vascularization was
reduced in eNOS KO(ko) fetuses as shown by reduced capillary length and reduced capillary
density at late gestation. In prior work, vascularization of other capillary beds were also shown
to be reduced including the hindlimb [159], and left ventricular myocardium [163] in non-
pregnant eNOS KO mice. VEGF-mediated angiogenesis requires NO because endothelial cell
differentation, migration and formation of capillary networks in response to VEGF are reduced
by NOS inhibition in vitro [158, 165]. VEGF works by binding to VEGFR1 and VEGFR2
receptors and activates the PI3K and phospholipase Cγ1 pathways, which lead to activation of
Akt and subsequently to phosphorylation of eNOS on serine 1177 [15]. This increases NO
production in human umbilical venous endothelial cells [15]. The increase in NO production
appears to be critical for VEGF’s angiogenic effects because in eNOS KO mice, recombinant
VEGF protein or adenovirus-mediated VEGF gene transfer failed to improve the impaired
angiogenesis found in the hindlimb of these mutants [159]. Therefore, eNOS-derived NO is an
important downstream mediator of the angiogenic effects of VEGF. NO production from eNOS
also appears to play a role as an upstream promoter of VEGF expression [160-162].
Administration of a NO donor or transfection with a DNA plasmid encoding eNOS increased
VEGF protein levels in vascular smooth muscle cells in humans and rats [160, 161] and in
skeletal muscle in rats [162]. Therefore, it is possible that reduced NO production in mice
133
caused the decrease in placental VEGF mRNA levels and protein expression in eNOS KO(ko)
placentas. Similarly, others have reported decreased VEGF mRNA levels in the LV
myocardium [163] and lungs [164] of eNOS KO mice. Therefore, decreased VEGF levels
and/or impaired function of VEGF due to lack of eNOS may contribute to decreased placental
vascularization leading to impaired placental perfusion in eNOS KO(ko) fetuses.
Using the hypoxia marker, Hypoxyprobe-1, I showed an increase in hypoxic
immunoreactivity in the junctional zone and adjacent labyrinth in eNOS KO(ko) placentas as
compared to WT(wt) placentas (Chapter 3). Hypoxia would be anticipated to constrict the
fetoplacental vessels, to increase their vasoconstrictor reactivity, and to increase fetoplacental
vascular resistance as observed in human and rat placentas [302, 308]. Hypoxic-
vasoconstriction is thought to function physiologically to divert fetoplacental blood flow away
from regions that are poorly perfused, thereby optimizing exchange between the maternal and
fetal placental circulations. Hypoxia-induced fetoplacental vasoconstriction in the human
placenta was prevented via treatment with L-NAME, suggesting that vasoconstriction was
mediated by an hypoxia-induced decrease in NO production [302]. Therefore, low NO in eNOS
KO(ko) mice may prevent fetal-maternal flow matching in the placenta thereby impairing
exchange and this may contribute to elevated placental hypoxia. Alternatively, the placenta
responds to hypoxia by increasing angiogenesis, as shown in high altitude pregnancies [309].
Whether eNOS levels are elevated in these high altitude pregnancies is unknown. However,
experimentally-induced hypoxia in pregnant mice elevated eNOS protein expression in the
placenta and prevented placental hypoxia suggesting a protective role for eNOS [290]. Thus,
the absence of eNOS in eNOS KO(ko) placentas may prevent the placenta from responding to
hypoxia with eNOS-mediated increases in angiogenesis and/or vasodilation, thereby sustaining
placental hypoxia.
134
eNOS KO(ko) fetuses are hypoxic as shown by increased Hydroxyprobe-1
immunoreactivity in the fetal heart, lung, liver and kidney. Decreased fetal tissue oxygenation
in eNOS KO(ko) fetuses may be due in part to decreased placental vascularity, reduced fetal
tissue vascularity, and/or decreased erythropoiesis. Decreased placental vascularity may
decrease the fetal-placental exchange surface and the umbilical blood flow rate, both of which
would tend to decrease the delivery of oxygen and nutrients to the fetus. eNOS may also play a
direct role in fetal tissue vascularization. eNOS is expressed in endothelial cells of fetal kidney,
liver, aorta and in the endocardium during development [310] and eNOS KO(ko) pups show
impaired myocardial capillary density, and pulmonary hypovascularity [163, 164]. Decreased
capillary density in eNOS KO(ko) fetuses suggest that the fetal tissues are further away from the
capillaries, so diffusion distance is increased, and this may impair tissue oxygenation leading to
fetal hypoxia.
Erythrocyte count and hematocrit levels were decreased in eNOS KO(ko) fetuses
suggesting that the oxygen carrying capacity of the fetal blood was impaired. Blunted
erythropoiesis in eNOS KO(ko) fetuses may be due to reduced production and/or function of
erythropoietin. Erythropoeitin is synthesized in the fetal kidney and liver [170], both of which
are hypoxic in the eNOS KO(ko) fetuses. Hypoxia stimulates erythropoietin production [311]
and this process may be eNOS mediated, and therefore lacking in eNOS KO(ko) mice. NO is
also involved in the formation of hematopoietic stem cells which are involved in erythrocyte
formation [170, 312]. Hematopoietic stem cell production was decreased by L-NAME
treatment whereas it was increased by NO donor, S-nitroso-N-acetyl-DL-penicillamine (SNAP)
in zebrafish [312]. In addition, intrauterine NOS inhibition and embryonic eNOS deficiency in
mice resulted in a reduction in hematopoietic clusters [312]. Furthermore, eNOS is also
expressed in the bone marrow stromal cells [313]. It influences recruitment of stem and
135
progenitor cells because adult eNOS KO mice show defects in progenitor cell mobilization in
response to VEGF [313]. Therefore, failure in erythropoietin production, reduced hematopoietic
stem cell production, and/or defects in progenitor cell mobilization may be responsible for low
erythrocyte count in eNOS KO(ko) fetuses. Interestingly, adult eNOS KO(ko) mice show no
significant differences in erythrocyte count and hematocrit levels as compared to WT(wt) at
both non-pregnant and 17.5 d of gestation (Chapter 2). This could be because eNOS may
function differently in erythropoiesis during development and after birth. Erythrocyte formation
takes place in the bone marrow after birth, whereas the liver is the primary site of erythropoiesis
in the fetus in late gestation [314].
In the eNOS KO(ko) mice, fetal factors such as decreased vascularity in the placenta
and fetal tissues, and decreased erythropoiesis may be mechanisms by which fetal growth is
impaired. Alternatively, impaired fetal growth may be secondary to maternal effects of eNOS
deficiency including blunted uterine arterial perfusion. In the eNOS KO(ko) mothers, the
remodeling of the uteroplacental vasculatures, specifically the uterine arteries, spiral arteries and
central arterial canals were blunted and this was associated with reduced uterine arterial blood
flow and increased uterine arterial Resistance Index. Thus decreased maternal perfusion of the
placenta (Chapter 3) may contribute to reduced fetal growth. To determine the relative roles of
the fetal and/or maternal genotype in controlling fetal growth I performed a crossbreeding study.
Results showed that fetal body weight, and umbilical venous blood flow/g fetal weight (and all
the parameters from which it was derived) did not significantly differ beween heterozygous
fetuses with eNOS KO mothers or WT mothers indicating that the maternal genotype was not a
significant factor. Heterozygous fetal body weights were intermediate between fetal KO and
fetal WT weights showing that defects in fetoplacental perfusion and fetal growth were
primarily determined by the fetal genotype. Interestingly, prior work showed that eNOS
136
KO(het) mice despite having equivalent birth weights exhibited functional and structural
abnormalities in the carotid and mesenteric arteries [315], and elevated diastolic arterial pressure
[316] in adulthood as compared to WT(het) mice. This suggests that the maternal environment
in utero or during lactation does have great impact on the development of the offspring, but that
this influence is not necessarily detectable as a change in body weight at birth.
In conclusion, eNOS plays an essential role in augmenting blood flow in the
fetoplacental circulation. This is likely due to the role of NO in maintaining low vascular
resistance and promoting vascularization in the fetoplacental circulation. eNOS KO(ko)
placentas show reduced vascularization and this could be due to decreased VEGF mRNA and
protein expression. These factors along with decreased erythropoiesis in eNOS KO(ko) fetuses
most likely contributed to the reduced fetal tissue oxygenation and reduced fetal growth at term.
Interestingly, although uterine perfusion was reduced in eNOS KO mothers, this was not a
significant factor affecting fetal growth. Results indicate that fetal growth was primarily
determined by the fetal eNOS genotype rather than that of the mother.
ACKNOWLEDGMENTS:
I would like to thank Dr. Dawei Qu for his assistance in tissue collection, Ms. Kathie
Whiteley for her assistance in the vascular cast experiments, Dr. Qiang Xu for doing the
immunohistochemistry and Dr. Shannon Bainbridge for RT-qPCR of VEGF mRNA levels.
1. Duvekot JJ, Cheriex EC, Pieters FA, Menheere PP, Peeters LH. Early pregnancy changes in hemodynamics and volume homeostasis are consecutive adjustments triggered by a primary fall in systemic vascular tone. Am.J.Obstet.Gynecol. 1993; 169: 1382-1392.
2. McLaughlin MK, Roberts JM, Lindheimer MD, Roberts JM, Cunningham FG. Hemodynamic Changes. In: Chesley's Hypertensive Disorders in Pregnancy, vol. Second Edition. Stamford, Connecticut: Appleton & Lange; 1999: 69-102.
3. Robson SC, Hunter S, Boys RJ, Dunlop W. Serial study of factors influencing changes in cardiac output during human pregnancy. Am.J.Physiol 1989; 256: H1060-H1065.
4. Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology (Bethesda) 2009; 24: 58-71.
5. Charnock-Jones DS, Kaufmann P, Mayhew TM. Aspects of human fetoplacental vasculogenesis and angiogenesis. I. Molecular regulation. Placenta 2004; 25: 103-113.
7. Magness RR, Sullivan JA, Li Y, Phernetton TM, Bird IM. Endothelial vasodilator production by uterine and systemic arteries. VI. Ovarian and pregnancy effects on eNOS and NO(x). Am.J.Physiol Heart Circ.Physiol 2001; 280: H1692-H1698.
8. Conrad KP, Joffe GM, Kruszyna H, Kruszyna R, Rochelle LG, Smith RP, Chavez JE, Mosher MD. Identification of increased nitric oxide biosynthesis during pregnancy in rats. FASEB J. 1993; 7: 566-571.
9. Linke A, Li W, Huang H, Wang Z, Hintze TH. Role of cardiac eNOS expression during pregnancy in the coupling of myocardial oxygen consumption to cardiac work. Am.J.Physiol Heart Circ.Physiol 2002; 283: H1208-H1214.
10. Goetz RM, Morano I, Calovini T, Studer R, Holtz J. Increased expression of endothelial constitutive nitric oxide synthase in rat aorta during pregnancy. Biochem.Biophys.Res.Commun. 1994; 205: 905-910.
11. Xu DL, Martin PY, St John J, Tsai P, Summer SN, Ohara M, Kim JK, Schrier RW. Upregulation of endothelial and neuronal constitutive nitric oxide synthase in pregnant rats. Am.J.Physiol 1996; 271: R1739-R1745.
12. Andersen MR, Simonsen U, Uldbjerg N, Aalkjaer C, Stender S. Smoking cessation early in pregnancy and birth weight, length, head circumference, and endothelial nitric oxide synthase activity in umbilical and chorionic vessels: an observational study of healthy singleton pregnancies. Circulation 2009; 119: 857-864.
14. Li H, Wallerath T, Forstermann U. Physiological mechanisms regulating the expression of endothelial-type NO synthase. Nitric.Oxide. 2002; 7: 132-147.
15. Ahmad S, Hewett PW, Wang P, Al-Ani B, Cudmore M, Fujisawa T, Haigh JJ, le Noble F, Wang L, Mukhopadhyay D, Ahmed A. Direct evidence for endothelial vascular endothelial growth factor receptor-1 function in nitric oxide-mediated angiogenesis. Circ Res 2006; 99: 715-722.
16. Byers MJ, Zangl A, Phernetton TM, Lopez G, Chen DB, Magness RR. Endothelial vasodilator production by ovine uterine and systemic arteries: ovarian steroid and pregnancy control of ERalpha and ERbeta levels. J Physiol 2005; 565: 85-99.
17. Liao WX, Magness RR, Chen DB. Expression of estrogen receptors-alpha and -beta in the pregnant ovine uterine artery endothelial cells in vivo and in vitro. Biol Reprod 2005; 72: 530-537.
18. Magness RR, Bazer FW. Maternal Cardiovascular and Other Physiologic Responses to the Endocrinology of Pregnancy. In: The Endocrinology of Pregnancy. Totowa, NJ: Humana Press Inc.; 1997: 507-539.
19. Hoffmann P, Feige JJ, Alfaidy N. Placental expression of EG-VEGF and its receptors PKR1 (prokineticin receptor-1) and PKR2 throughout mouse gestation. Placenta 2007; 28: 1049-1058.
20. Hewitt DP, Mark PJ, Waddell BJ. Glucocorticoids prevent the normal increase in placental vascular endothelial growth factor expression and placental vascularity during late pregnancy in the rat. Endocrinology 2006; 147: 5568-5574.
21. Molnar M, Suto T, Toth T, Hertelendy F. Prolonged blockade of nitric oxide synthesis in gravid rats produces sustained hypertension, proteinuria, thrombocytopenia, and intrauterine growth retardation. Am.J.Obstet.Gynecol. 1994; 170: 1458-1466.
22. Zhang Y, Kaufman S. Effect of nitric oxide synthase inhibition on cardiovascular and hormonal regulation during pregnancy in the rat. Can.J.Physiol Pharmacol. 2000; 78: 423-427.
23. Slangen BF, Out IC, Verkeste CM, Peeters LL. Hemodynamic changes in early pregnancy in chronically instrumented, conscious rats. Am.J.Physiol 1996; 270: H1779-H1784.
24. Chapman AB, Abraham WT, Zamudio S, Coffin C, Merouani A, Young D, Johnson A, Osorio F, Goldberg C, Moore LG, Dahms T, Schrier RW. Temporal relationships between hormonal and hemodynamic changes in early human pregnancy. Kidney Int. 1998; 54: 2056-2063.
25. Slangen BF, Out IC, Verkeste CM, Smits JF, Peeters LL. Hemodynamic changes in pseudopregnancy in chronically instrumented conscious rats. Am.J.Physiol 1997; 272: H695-H700.
154
26. Das A, Mantena SR, Kannan A, Evans DB, Bagchi MK, Bagchi IC. De novo synthesis of estrogen in pregnant uterus is critical for stromal decidualization and angiogenesis. Proc Natl Acad Sci U S A 2009; 106: 12542-12547.
27. Malassine A, Frendo JL, Evain-Brion D. A comparison of placental development and endocrine functions between the human and mouse model. Hum Reprod Update 2003; 9: 531-539.
28. Hunter S, Robson SC. Adaptation of the maternal heart in pregnancy. Br.Heart J. 1992; 68: 540-543.
29. Ward DG, Thomas GR, Cronin MJ. Relaxin increases rat heart rate by a direct action on the cardiac atrium. Biochem Biophys Res Commun 1992; 186: 999-1005.
30. Conrad KP, Novak J. Emerging role of relaxin in renal and cardiovascular function. Am.J.Physiol Regul.Integr.Comp Physiol 2004; 287: R250-R261.
31. Schrier RW, Briner VA. Peripheral arterial vasodilation hypothesis of sodium and water retention in pregnancy: implications for pathogenesis of preeclampsia-eclampsia. Obstet Gynecol 1991; 77: 632-639.
32. Longo LD. Maternal blood volume and cardiac output during pregnancy: a hypothesis of endocrinologic control. Am.J.Physiol 1983; 245: R720-R729.
33. Wedel Jones C, Mandala M, Barron C, Bernstein I, Osol G. Mechanisms underlying maternal venous adaptation in pregnancy. Reprod Sci 2009; 16: 596-604.
34. Spaanderman ME, Willekes C, Hoeks AP, Ekhart TH, Peeters LL. The effect of pregnancy on the compliance of large arteries and veins in healthy parous control subjects and women with a history of preeclampsia. Am J Obstet Gynecol 2000; 183: 1278-1286.
35. Manasc B, Jepson J. Erythropoietin in plasma and urine during human pregnancy. Can Med Assoc J 1969; 100: 687-691.
36. Mesa A, Jessurun C, Hernandez A, Adam K, Brown D, Vaughn WK, Wilansky S. Left ventricular diastolic function in normal human pregnancy. Circulation 1999; 99: 511-517.
37. Kametas NA, McAuliffe F, Hancock J, Chambers J, Nicolaides KH. Maternal left ventricular mass and diastolic function during pregnancy. Ultrasound Obstet.Gynecol. 2001; 18: 460-466.
38. Mabie WC, DiSessa TG, Crocker LG, Sibai BM, Arheart KL. A longitudinal study of cardiac output in normal human pregnancy. Am.J.Obstet.Gynecol. 1994; 170: 849-856.
39. Gilson GJ, Samaan S, Crawford MH, Qualls CR, Curet LB. Changes in hemodynamics, ventricular remodeling, and ventricular contractility during normal pregnancy: a longitudinal study. Obstet.Gynecol. 1997; 89: 957-962.
155
40. Geva T, Mauer MB, Striker L, Kirshon B, Pivarnik JM. Effects of physiologic load of pregnancy on left ventricular contractility and remodeling. Am.Heart J. 1997; 133: 53-59.
41. Mone SM, Sanders SP, Colan SD. Control mechanisms for physiological hypertrophy of pregnancy. Circulation 1996; 94: 667-672.
42. Lang RM, Pridjian G, Feldman T, Neumann A, Lindheimer M, Borow KM. Left ventricular mechanics in preeclampsia. Am Heart J 1991; 121: 1768-1775.
43. MacLellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annu Rev Physiol 2000; 62: 289-319.
44. Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev 2007; 87: 521-544.
45. Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med 2001; 344: 1750-1757.
49. Schannwell CM, Zimmermann T, Schneppenheim M, Plehn G, Marx R, Strauer BE. Left ventricular hypertrophy and diastolic dysfunction in healthy pregnant women. Cardiology 2002; 97: 73-78.
50. Miyamoto T, Takeishi Y, Takahashi H, Shishido T, Arimoto T, Tomoike H, Kubota I. Activation of distinct signal transduction pathways in hypertrophied hearts by pressure and volume overload. Basic Res.Cardiol. 2004; 99: 328-337.
51. Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol.Cell Biol. 2002; 22: 2799-2809.
52. Miyamoto T, Takeishi Y, Shishido T, Takahashi H, Itoh M, Kubota I, Tomoike H. Role of nitric oxide in the progression of cardiovascular remodeling induced by carotid arterio-venous shunt in rabbits. Jpn.Heart J. 2003; 44: 127-137.
53. Eghbali M, Deva R, Alioua A, Minosyan TY, Ruan H, Wang Y, Toro L, Stefani E. Molecular and functional signature of heart hypertrophy during pregnancy. Circ.Res. 2005; 96: 1208-1216.
156
54. Assali NS, Douglass RA, Jr., Baird WW, Nicholson DB, Suyemoto R. Measurement of uterine blood flow and uterine metabolism. IV. Results in normal pregnancy. Am J Obstet Gynecol 1953; 66: 248-253.
55. Metcalfe J, Romney SL, Ramsey LH, Reid DE, Burwell CS. Estimation of uterine blood flow in normal human pregnancy at term. J Clin Invest 1955; 34: 1632-1638.
56. Adamson SL, Lu Y, Whiteley KJ, Holmyard D, Hemberger M, Pfarrer C, Cross JC. Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev.Biol. 2002; 250: 358-373.
57. Georgiades P, Ferguson-Smith AC, Burton GJ. Comparative developmental anatomy of the murine and human definitive placentae. Placenta 2002; 23: 3-19.
58. Kaufmann P, Black S, Huppertz B. Endovascular trophoblast invasion: implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol Reprod 2003; 69: 1-7.
59. Anderson CM, Lopez F, Zhang HY, Pavlish K, Benoit JN. Reduced uteroplacental perfusion alters uterine arcuate artery function in the pregnant Sprague-Dawley rat. Biol Reprod 2005; 72: 762-766.
60. Carbillon L, Challier JC, Alouini S, Uzan M, Uzan S. Uteroplacental circulation development: Doppler assessment and clinical importance. Placenta 2001; 22: 795-799.
61. Zygmunt M, Herr F, Munstedt K, Lang U, Liang OD. Angiogenesis and vasculogenesis in pregnancy. Eur.J.Obstet.Gynecol.Reprod.Biol. 2003; 110 Suppl 1: S10-S18.
62. Adamson SL. Arterial pressure, vascular input impedance, and resistance as determinants of pulsatile blood flow in the umbilical artery. Eur.J.Obstet.Gynecol.Reprod.Biol. 1999; 84: 119-125.
63. Schulman H, Fleischer A, Farmakides G, Bracero L, Rochelson B, Grunfeld L. Development of uterine artery compliance in pregnancy as detected by Doppler ultrasound. Am J Obstet Gynecol 1986; 155: 1031-1036.
64. Bower S, Vyas S, Campbell S, Nicolaides KH. Color Doppler imaging of the uterine artery in pregnancy: normal ranges of impedance to blood flow, mean velocity and volume of flow. Ultrasound Obstet Gynecol 1992; 2: 261-265.
66. Langille BL, O'Donnell F. Reductions in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science 1986; 231: 405-407.
67. Kaiser L, Sparks HV, Jr. Mediation of flow-dependent arterial dilation by endothelial cells. Circ Shock 1986; 18: 109-114.
157
68. Tronc F, Wassef M, Esposito B, Henrion D, Glagov S, Tedgui A. Role of NO in flow-induced remodeling of the rabbit common carotid artery. Arterioscler.Thromb.Vasc.Biol. 1996; 16: 1256-1262.
69. Lehoux S, Tronc F, Tedgui A. Mechanisms of blood flow-induced vascular enlargement. Biorheology 2002; 39: 319-324.
70. Langille BL, Bendeck MP, Keeley FW. Adaptations of carotid arteries of young and mature rabbits to reduced carotid blood flow. Am.J.Physiol 1989; 256: H931-H939.
71. Tronc F, Mallat Z, Lehoux S, Wassef M, Esposito B, Tedgui A. Role of matrix metalloproteinases in blood flow-induced arterial enlargement: interaction with NO. Arterioscler.Thromb.Vasc.Biol. 2000; 20: E120-E126.
72. Palmer SK, Zamudio S, Coffin C, Parker S, Stamm E, Moore LG. Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet Gynecol 1992; 80: 1000-1006.
73. Osol G, Cipolla M. Pregnancy-induced changes in the three-dimensional mechanical properties of pressurized rat uteroplacental (radial) arteries. Am J Obstet Gynecol 1993; 168: 268-274.
74. St-Louis J, Pare H, Sicotte B, Brochu M. Increased reactivity of rat uterine arcuate artery throughout gestation and postpartum. Am J Physiol 1997; 273: H1148-1153.
75. Cipolla M, Osol G. Hypertrophic and hyperplastic effects of pregnancy on the rat uterine arterial wall. Am J Obstet Gynecol 1994; 171: 805-811.
76. Hees H, Moll W, Wrobel KH, Hees I. Pregnancy-induced structural changes and trophoblastic invasion in the segmental mesometrial arteries of the guinea pig (Cavia porcellus L.). Placenta 1987; 8: 609-626.
77. Annibale DJ, Rosenfeld CR, Stull JT, Kamm KE. Protein content and myosin light chain phosphorylation in uterine arteries during pregnancy. Am J Physiol 1990; 259: C484-489.
78. Keyes LE, Majack R, Dempsey EC, Moore LG. Pregnancy stimulation of DNA synthesis and uterine blood flow in the guinea pig. Pediatr Res 1997; 41: 708-715.
79. Espinoza J, Romero R, Mee Kim Y, Kusanovic JP, Hassan S, Erez O, Gotsch F, Than NG, Papp Z, Jai Kim C. Normal and abnormal transformation of the spiral arteries during pregnancy. J Perinat Med 2006; 34: 447-458.
80. Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta 2006; 27: 939-958.
81. Lyall F, Bulmer JN, Kelly H, Duffie E, Robson SC. Human trophoblast invasion and spiral artery transformation: the role of nitric oxide. Am J Pathol 1999; 154: 1105-1114.
158
82. Lunghi L, Ferretti ME, Medici S, Biondi C, Vesce F. Control of human trophoblast function. Reprod Biol Endocrinol 2007; 5: 6.
83. Keogh RJ, Harris LK, Freeman A, Baker PN, Aplin JD, Whitley GS, Cartwright JE. Fetal-derived trophoblast use the apoptotic cytokine tumor necrosis factor-alpha-related apoptosis-inducing ligand to induce smooth muscle cell death. Circ Res 2007; 100: 834-841.
85. Croy BA, Ashkar AA, Minhas K, Greenwood JD. Can murine uterine natural killer cells give insights into the pathogenesis of preeclampsia? J.Soc.Gynecol.Investig. 2000; 7: 12-20.
86. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, Gazit R, Yutkin V, Benharroch D, Porgador A, Keshet E, Yagel S, Mandelboim O. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med 2006; 12: 1065-1074.
87. Moffett-King A. Natural killer cells and pregnancy. Nat Rev Immunol 2002; 2: 656-663.
88. Ashkar AA, Di Santo JP, Croy BA. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J Exp Med 2000; 192: 259-270.
89. Goldman-Wohl DS, Ariel I, Greenfield C, Lavy Y, Yagel S. Tie-2 and angiopoietin-2 expression at the fetal-maternal interface: a receptor ligand model for vascular remodelling. Mol Hum Reprod 2000; 6: 81-87.
90. Lash GE, Schiessl B, Kirkley M, Innes BA, Cooper A, Searle RF, Robson SC, Bulmer JN. Expression of angiogenic growth factors by uterine natural killer cells during early pregnancy. J Leukoc Biol 2006; 80: 572-580.
91. Gill RW, Trudinger BJ, Garrett WJ, Kossoff G, Warren PS. Fetal umbilical venous flow measured in utero by pulsed Doppler and B-mode ultrasound. I. Normal pregnancies. Am J Obstet Gynecol 1981; 139: 720-725.
92. Goldkrand JW, Moore DH, Lentz SU, Clements SP, Turner AD, Bryant JL. Volumetric flow in the umbilical artery: normative data. J Matern Fetal Med 2000; 9: 224-228.
93. Guzman ER, Schulman H, Karmel B, Higgins P. Umbilical artery Doppler velocimetry in pregnancies of less than 21 weeks' duration. J Ultrasound Med 1990; 9: 655-659.
94. Coan PM, Ferguson-Smith AC, Burton GJ. Developmental dynamics of the definitive mouse placenta assessed by stereology. Biol Reprod 2004; 70: 1806-1813.
159
95. Kaufmann P, Mayhew TM, Charnock-Jones DS. Aspects of human fetoplacental vasculogenesis and angiogenesis. II. Changes during normal pregnancy. Placenta 2004; 25: 114-126.
96. Reynolds LP, Redmer DA. Angiogenesis in the placenta. Biol Reprod 2001; 64: 1033-1040.
97. Henrich WL. The endothelium--a key regulator of vascular tone. Am J Med Sci 1991; 302: 319-328.
98. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980; 288: 373-376.
99. Schmidt HH, Lohmann SM, Walter U. The nitric oxide and cGMP signal transduction system: regulation and mechanism of action. Biochim Biophys Acta 1993; 1178: 153-175.
100. Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev 1994; 46: 205-229.
101. Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987; 327: 524-526.
102. Kelly RA, Balligand JL, Smith TW. Nitric oxide and cardiac function. Circ.Res. 1996; 79: 363-380.
103. Balligand JL, Cannon PJ. Nitric oxide synthases and cardiac muscle. Autocrine and paracrine influences. Arterioscler.Thromb.Vasc.Biol. 1997; 17: 1846-1858.
104. Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem.J. 2001; 357: 593-615.
105. Groves JT, Wang CC. Nitric oxide synthase: models and mechanisms. Curr.Opin.Chem.Biol. 2000; 4: 687-695.
106. Roman LJ, Martasek P, Masters BS. Intrinsic and extrinsic modulation of nitric oxide synthase activity. Chem.Rev. 2002; 102: 1179-1190.
107. Su Z, Blazing MA, Fan D, George SE. The calmodulin-nitric oxide synthase interaction. Critical role of the calmodulin latch domain in enzyme activation. J Biol Chem 1995; 270: 29117-29122.
108. Gross SS, Levi R. Tetrahydrobiopterin synthesis. An absolute requirement for cytokine-induced nitric oxide generation by vascular smooth muscle. J Biol Chem 1992; 267: 25722-25729.
109. Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 2003; 111: 1201-1209.
160
110. Hubel CA. Oxidative stress in the pathogenesis of preeclampsia. Proc.Soc.Exp.Biol.Med. 1999; 222: 222-235.
111. Myatt L, Cui X. Oxidative stress in the placenta. Histochem Cell Biol 2004; 122: 369-382.
112. van der Heijden OW, Essers YP, Fazzi G, Peeters LL, De Mey JG, van Eys GJ. Uterine artery remodeling and reproductive performance are impaired in endothelial nitric oxide synthase-deficient mice. Biol.Reprod. 2005; 72: 1161-1168.
113. Chang K, Zhang L. Steroid Hormones and Uterine Vascular Adaptation to Pregnancy. Reproductive Sciences 2008; 15: 336-348.
114. Ziche M, Morbidelli L. Nitric oxide and angiogenesis. J Neurooncol 2000; 50: 139-148.
115. Kim PK, Zamora R, Petrosko P, Billiar TR. The regulatory role of nitric oxide in apoptosis. Int Immunopharmacol 2001; 1: 1421-1441.
116. Anumba DO, Robson SC, Boys RJ, Ford GA. Nitric oxide activity in the peripheral vasculature during normotensive and preeclamptic pregnancy. Am.J.Physiol 1999; 277: H848-H854.
117. Williams DJ, Vallance PJ, Neild GH, Spencer JA, Imms FJ. Nitric oxide-mediated vasodilation in human pregnancy. Am.J.Physiol 1997; 272: H748-H752.
118. Holden DP, Fickling SA, Whitley GS, Nussey SS. Plasma concentrations of asymmetric dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal pregnancy and preeclampsia. Am.J.Obstet.Gynecol. 1998; 178: 551-556.
119. Dorup I, Skajaa K, Sorensen KE. Normal pregnancy is associated with enhanced endothelium-dependent flow-mediated vasodilation. Am.J.Physiol 1999; 276: H821-H825.
120. Baylis C, Vallance P. Measurement of nitrite and nitrate levels in plasma and urine--what does this measure tell us about the activity of the endogenous nitric oxide system? Curr.Opin.Nephrol.Hypertens. 1998; 7: 59-62.
121. Conrad KP, Kerchner LJ, Mosher MD. Plasma and 24-h NO(x) and cGMP during normal pregnancy and preeclampsia in women on a reduced NO(x) diet. Am.J.Physiol 1999; 277: F48-F57.
122. Osol G, Barron C, Gokina N, Mandala M. Inhibition of Nitric Oxide Synthases Abrogates Pregnancy-Induced Uterine Vascular Expansive Remodeling. J Vasc Res 2009; 46: 478-486.
123. Miller SL, Jenkin G, Walker DW. Effect of nitric oxide synthase inhibition on the uterine vasculature of the late-pregnant ewe. Am J Obstet Gynecol 1999; 180: 1138-1145.
161
124. Chang JK, Roman C, Heymann MA. Effect of endothelium-derived relaxing factor inhibition on the umbilical-placental circulation in fetal lambs in utero. Am J Obstet Gynecol 1992; 166: 727-734.
125. Nathan L, Cuevas J, Chaudhuri G. The role of nitric oxide in the altered vascular reactivity of pregnancy in the rat. Br.J.Pharmacol. 1995; 114: 955-960.
126. Kassab S, Miller MT, Hester R, Novak J, Granger JP. Systemic hemodynamics and regional blood flow during chronic nitric oxide synthesis inhibition in pregnant rats. Hypertension 1998; 31: 315-320.
127. Khalil RA, Crews JK, Novak J, Kassab S, Granger JP. Enhanced vascular reactivity during inhibition of nitric oxide synthesis in pregnant rats. Hypertension 1998; 31: 1065-1069.
128. Vatish M, Randeva HS, Grammatopoulos DK. Hormonal regulation of placental nitric oxide and pathogenesis of pre-eclampsia. Trends Mol Med 2006; 12: 223-233.
129. Sladek SM, Kanbour-Shakir A, Watkins S, Berghorn KA, Hoffman GE, Roberts JM. Granulated metrial gland cells contain nitric oxide synthases during pregnancy in the rat. Placenta 1998; 19: 55-65.
130. Zhou Y, Fisher SJ, Janatpour M, Genbacev O, Dejana E, Wheelock M, Damsky CH. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest 1997; 99: 2139-2151.
131. Nanaev A, Chwalisz K, Frank HG, Kohnen G, Hegele-Hartung C, Kaufmann P. Physiological dilation of uteroplacental arteries in the guinea pig depends on nitric oxide synthase activity of extravillous trophoblast. Cell Tissue Res 1995; 282: 407-421.
132. Balligand JL, Feron O, Dessy C. eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev 2009; 89: 481-534.
133. Searles CD. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am J Physiol Cell Physiol 2006; 291: C803-816.
134. Fish JE, Marsden PA. Endothelial nitric oxide synthase: insight into cell-specific gene regulation in the vascular endothelium. Cell Mol Life Sci 2006; 63: 144-162.
135. Govers R, Rabelink TJ. Cellular regulation of endothelial nitric oxide synthase. Am.J.Physiol Renal Physiol 2001; 280: F193-F206.
136. Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol Cell Physiol 2003; 285: C499-508.
137. Michell BJ, Harris MB, Chen ZP, Ju H, Venema VJ, Blackstone MA, Huang W, Venema RC, Kemp BE. Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide synthase at serine 617 and serine 635. J Biol Chem 2002; 277: 42344-42351.
162
138. Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ.Res. 2000; 87: 677-682.
139. Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res 2001; 88: E68-75.
140. Sessa WC. eNOS at a glance. J Cell Sci 2004; 117: 2427-2429.
143. Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J.Clin.Invest 1998; 101: 731-736.
144. Zhang L, Fishman MC, Huang PL. Estrogen mediates the protective effects of pregnancy and chorionic gonadotropin in a mouse model of vascular injury. Arterioscler.Thromb.Vasc.Biol. 1999; 19: 2059-2065.
145. Florian M, Freiman A, Magder S. Treatment with 17-beta-estradiol reduces superoxide production in aorta of ovariectomized rats. Steroids 2004; 69: 779-787.
146. Jankowski M, Wang D, Mukaddam-Daher S, Gutkowska J. Pregnancy alters nitric oxide synthase and natriuretic peptide systems in the rat left ventricle. J.Endocrinol. 2005; 184: 209-217.
147. Rubanyi GM, Freay AD, Kauser K, Sukovich D, Burton G, Lubahn DB, Couse JF, Curtis SW, Korach KS. Vascular estrogen receptors and endothelium-derived nitric oxide production in the mouse aorta. Gender difference and effect of estrogen receptor gene disruption. J.Clin.Invest 1997; 99: 2429-2437.
148. Rosenfeld CR, Cox BE, Roy T, Magness RR. Nitric oxide contributes to estrogen-induced vasodilation of the ovine uterine circulation. J Clin Invest 1996; 98: 2158-2166.
149. Van Buren GA, Yang DS, Clark KE. Estrogen-induced uterine vasodilatation is antagonized by L-nitroarginine methyl ester, an inhibitor of nitric oxide synthesis. Am J Obstet Gynecol 1992; 167: 828-833.
150. Rosenfeld CR, White RE, Roy T, Cox BE. Calcium-activated potassium channels and nitric oxide coregulate estrogen-induced vasodilation. Am.J.Physiol Heart Circ.Physiol 2000; 279: H319-H328.
163
151. Rosenfeld CR, Cornfield DN, Roy T. Ca(2+)-activated K(+) channels modulate basal and E(2)beta-induced rises in uterine blood flow in ovine pregnancy. Am J Physiol Heart Circ Physiol 2001; 281: H422-431.
152. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9: 669-676.
153. Ferrara N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol 2001; 280: C1358-1366.
154. Karkkainen MJ, Makinen T, Alitalo K. Lymphatic endothelium: a new frontier of metastasis research. Nat Cell Biol 2002; 4: E2-5.
155. Ni Y, May V, Braas K, Osol G. Pregnancy augments uteroplacental vascular endothelial growth factor gene expression and vasodilator effects. Am J Physiol 1997; 273: H938-944.
156. Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, Abraham JA. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem 1991; 266: 11947-11954.
157. David AL, Torondel B, Zachary I, Wigley V, Abi-Nader K, Mehta V, Buckley SM, Cook T, Boyd M, Rodeck CH, Martin J, Peebles DM. Local delivery of VEGF adenovirus to the uterine artery increases vasorelaxation and uterine blood flow in the pregnant sheep. Gene Ther 2008; 15: 1344-1350.
158. Ziche M, Morbidelli L, Choudhuri R, Zhang HT, Donnini S, Granger HJ, Bicknell R. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest 1997; 99: 2625-2634.
159. Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest 1998; 101: 2567-2578.
160. Jozkowicz A, Cooke JP, Guevara I, Huk I, Funovics P, Pachinger O, Weidinger F, Dulak J. Genetic augmentation of nitric oxide synthase increases the vascular generation of VEGF. Cardiovasc Res 2001; 51: 773-783.
161. Dulak J, Jozkowicz A, Dembinska-Kiec A, Guevara I, Zdzienicka A, Zmudzinska-Grochot D, Florek I, Wojtowicz A, Szuba A, Cooke JP. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2000; 20: 659-666.
162. Benoit H, Jordan M, Wagner H, Wagner PD. Effect of NO, vasodilator prostaglandins, and adenosine on skeletal muscle angiogenic growth factor gene expression. J Appl Physiol 1999; 86: 1513-1518.
164. Han RN, Babaei S, Robb M, Lee T, Ridsdale R, Ackerley C, Post M, Stewart DJ. Defective lung vascular development and fatal respiratory distress in endothelial NO synthase-deficient mice: a model of alveolar capillary dysplasia? Circ.Res. 2004; 94: 1115-1123.
165. Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest 1997; 100: 3131-3139.
166. Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, Geppetti P, Ledda F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest 1994; 94: 2036-2044.
167. Goy MF. cGMP: the wayward child of the cyclic nucleotide family. Trends Neurosci 1991; 14: 293-299.
168. Saran M, Michel C, Bors W. Reaction of NO with O2-. implications for the action of endothelium-derived relaxing factor (EDRF). Free Radic Res Commun 1990; 10: 221-226.
169. Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A 1992; 89: 444-448.
170. Cokic VP, Schechter AN. Effects of nitric oxide on red blood cell development and phenotype. Curr Top Dev Biol 2008; 82: 169-215.
171. Dejam A, Hunter CJ, Schechter AN, Gladwin MT. Emerging role of nitrite in human biology. Blood Cells Mol Dis 2004; 32: 423-429.
172. Dejam A, Hunter CJ, Pelletier MM, Hsu LL, Machado RF, Shiva S, Power GG, Kelm M, Gladwin MT, Schechter AN. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood 2005; 106: 734-739.
173. Parra M, Rodrigo R, Barja P, Bosco C, Fernandez V, Munoz H, Soto-Chacon E. Screening test for preeclampsia through assessment of uteroplacental blood flow and biochemical markers of oxidative stress and endothelial dysfunction. Am J Obstet Gynecol 2005; 193: 1486-1491.
174. Roberts JM. Preeclampsia: what we know and what we do not know. Semin.Perinatol. 2000; 24: 24-28.
175. Smith GN, Walker MC, Liu A, Wen SW, Swansburg M, Ramshaw H, White RR, Roddy M, Hladunewich M. A history of preeclampsia identifies women who have underlying cardiovascular risk factors. Am J Obstet Gynecol 2009; 200: 58 e51-58.
177. Roberts JM, Lain KY. Recent Insights into the Pathogenesis of Pre-eclampsia. Placenta 2002; 23: 359-372.
178. Roberts JM, Hubel CA. The two stage model of preeclampsia: variations on the theme. Placenta 2009; 30 Suppl A: S32-37.
179. Huppertz B. Placental origins of preeclampsia: challenging the current hypothesis. Hypertension 2008; 51: 970-975.
180. Nagamatsu T, Fujii T, Kusumi M, Zou L, Yamashita T, Osuga Y, Momoeda M, Kozuma S, Taketani Y. Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: an implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology 2004; 145: 4838-4845.
181. Munaut C, Lorquet S, Pequeux C, Blacher S, Berndt S, Frankenne F, Foidart JM. Hypoxia is responsible for soluble vascular endothelial growth factor receptor-1 (VEGFR-1) but not for soluble endoglin induction in villous trophoblast. Hum Reprod 2008; 23: 1407-1415.
182. Roggensack AM, Zhang Y, Davidge ST. Evidence for peroxynitrite formation in the vasculature of women with preeclampsia. Hypertension 1999; 33: 83-89.
183. Sholook MM, Gilbert JS, Sedeek MH, Huang M, Hester RL, Granger JP. Systemic hemodynamic and regional blood flow changes in response to chronic reductions in uterine perfusion pressure in pregnant rats. Am J Physiol Heart Circ Physiol 2007; 293: H2080-2084.
184. Alexander BT, Kassab SE, Miller MT, Abram SR, Reckelhoff JF, Bennett WA, Granger JP. Reduced uterine perfusion pressure during pregnancy in the rat is associated with increases in arterial pressure and changes in renal nitric oxide. Hypertension 2001; 37: 1191-1195.
185. Gilbert JS, Babcock SA, Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension 2007; 50: 1142-1147.
186. Gilbert JS, Gilbert SA, Arany M, Granger JP. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension 2009; 53: 399-403.
187. Taylor RN, de Groot CJ, Cho YK, Lim KH. Circulating factors as markers and mediators of endothelial cell dysfunction in preeclampsia. Semin Reprod Endocrinol 1998; 16: 17-31.
188. He Y, Smith SK, Day KA, Clark DE, Licence DR, Charnock-Jones DS. Alternative splicing of vascular endothelial growth factor (VEGF)-R1 (FLT-1) pre-mRNA is important for the regulation of VEGF activity. Mol Endocrinol 1999; 13: 537-545.
166
189. Clark DE, Smith SK, He Y, Day KA, Licence DR, Corps AN, Lammoglia R, Charnock-Jones DS. A vascular endothelial growth factor antagonist is produced by the human placenta and released into the maternal circulation. Biol Reprod 1998; 59: 1540-1548.
190. Heydarian M, McCaffrey T, Florea L, Yang Z, Ross MM, Zhou W, Maynard SE. Novel splice variants of sFlt1 are upregulated in preeclampsia. Placenta 2009; 30: 250-255.
191. McKeeman GC, Ardill JE, Caldwell CM, Hunter AJ, McClure N. Soluble vascular endothelial growth factor receptor-1 (sFlt-1) is increased throughout gestation in patients who have preeclampsia develop. Am J Obstet Gynecol 2004; 191: 1240-1246.
192. Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 2004; 350: 672-683.
193. Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 2003; 111: 649-658.
194. Li Z, Zhang Y, Ying Ma J, Kapoun AM, Shao Q, Kerr I, Lam A, O'Young G, Sannajust F, Stathis P, Schreiner G, Karumanchi SA, Protter AA, Pollitt NS. Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension 2007; 50: 686-692.
195. Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D'Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M, Karumanchi SA. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 2006; 12: 642-649.
196. St-Jacques S, Forte M, Lye SJ, Letarte M. Localization of endoglin, a transforming growth factor-beta binding protein, and of CD44 and integrins in placenta during the first trimester of pregnancy. Biol Reprod 1994; 51: 405-413.
197. Serrano NC, Casas JP, Diaz LA, Paez C, Mesa CM, Cifuentes R, Monterrosa A, Bautista A, Hawe E, Hingorani AD, Vallance P, Lopez-Jaramillo P. Endothelial NO synthase genotype and risk of preeclampsia: a multicenter case-control study. Hypertension. 2004; 44: 702-707.
199. Tesauro M, Thompson WC, Rogliani P, Qi L, Chaudhary PP, Moss J. Intracellular processing of endothelial nitric oxide synthase isoforms associated with differences in severity of cardiopulmonary diseases: cleavage of proteins with aspartate vs. glutamate at position 298. Proc Natl Acad Sci U S A 2000; 97: 2832-2835.
167
200. Persu A, Stoenoiu MS, Messiaen T, Davila S, Robino C, El-Khattabi O, Mourad M, Horie S, Feron O, Balligand JL, Wattiez R, Pirson Y, Chauveau D, Lens XM, Devuyst O. Modifier effect of ENOS in autosomal dominant polycystic kidney disease. Hum Mol Genet 2002; 11: 229-241.
201. Molnar M, Hertelendy F. N omega-nitro-L-arginine, an inhibitor of nitric oxide synthesis, increases blood pressure in rats and reverses the pregnancy-induced refractoriness to vasopressor agents. Am.J.Obstet.Gynecol. 1992; 166: 1560-1567.
202. Karteris E, Vatish M, Hillhouse EW, Grammatopoulos DK. Preeclampsia is associated with impaired regulation of the placental nitric oxide-cyclic guanosine monophosphate pathway by corticotropin-releasing hormone (CRH) and CRH-related peptides. J Clin Endocrinol Metab 2005; 90: 3680-3687.
203. Noris M, Todeschini M, Cassis P, Pasta F, Cappellini A, Bonazzola S, Macconi D, Maucci R, Porrati F, Benigni A, Picciolo C, Remuzzi G. L-arginine depletion in preeclampsia orients nitric oxide synthase toward oxidant species. Hypertension 2004; 43: 614-622.
204. Speake PF, Glazier JD, Ayuk PT, Reade M, Sibley CP, D'Souza SW. L-Arginine transport across the basal plasma membrane of the syncytiotrophoblast of the human placenta from normal and preeclamptic pregnancies. J Clin Endocrinol Metab 2003; 88: 4287-4292.
205. Baschat AA, Cosmi E, Bilardo CM, Wolf H, Berg C, Rigano S, Germer U, Moyano D, Turan S, Hartung J, Bhide A, Muller T, Bower S, Nicolaides KH, Thilaganathan B, Gembruch U, Ferrazzi E, Hecher K, Galan HL, Harman CR. Predictors of neonatal outcome in early-onset placental dysfunction. Obstet Gynecol 2007; 109: 253-261.
206. Xiong X, Mayes D, Demianczuk N, Olson DM, Davidge ST, Newburn-Cook C, Saunders LD. Impact of pregnancy-induced hypertension on fetal growth. Am J Obstet Gynecol 1999; 180: 207-213.
207. Wollmann HA. Intrauterine growth restriction: definition and etiology. Horm Res 1998; 49 Suppl 2: 1-6.
208. Mari G, Hanif F. Intrauterine growth restriction: how to manage and when to deliver. Clin Obstet Gynecol 2007; 50: 497-509.
209. Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet 1993; 341: 938-941.
210. Maulik D. Fetal growth restriction: the etiology. Clin Obstet Gynecol 2006; 49: 228-235.
211. Todros T, Sciarrone A, Piccoli E, Guiot C, Kaufmann P, Kingdom J. Umbilical Doppler waveforms and placental villous angiogenesis in pregnancies complicated by fetal growth restriction. Obstet Gynecol 1999; 93: 499-503.
168
212. Karsdorp VH, van Vugt JM, van Geijn HP, Kostense PJ, Arduini D, Montenegro N, Todros T. Clinical significance of absent or reversed end diastolic velocity waveforms in umbilical artery. Lancet 1994; 344: 1664-1668.
213. Sagol S, Sagol O, Ozdemir N. Stereological quantification of placental villus vascularization and its relation to umbilical artery Doppler flow in intrauterine growth restriction. Prenat Diagn 2002; 22: 398-403.
214. Kingdom J, Huppertz B, Seaward G, Kaufmann P. Development of the placental villous tree and its consequences for fetal growth. Eur J Obstet Gynecol Reprod Biol 2000; 92: 35-43.
215. Krebs C, Macara LM, Leiser R, Bowman AW, Greer IA, Kingdom JC. Intrauterine growth restriction with absent end-diastolic flow velocity in the umbilical artery is associated with maldevelopment of the placental terminal villous tree. Am J Obstet Gynecol 1996; 175: 1534-1542.
216. Fox SB, Khong TY. Lack of innervation of human umbilical cord. An immunohistological and histochemical study. Placenta 1990; 11: 59-62.
217. Myatt L. Control of vascular resistance in the human placenta. Placenta 1992; 13: 329-341.
218. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc.Natl.Acad.Sci.U.S.A 1996; 93: 13176-13181.
219. Rutherford RA, McCarthy A, Sullivan MH, Elder MG, Polak JM, Wharton J. Nitric oxide synthase in human placenta and umbilical cord from normal, intrauterine growth-retarded and pre-eclamptic pregnancies. Br J Pharmacol 1995; 116: 3099-3109.
221. Casanello P, Sobrevia L. Intrauterine growth retardation is associated with reduced activity and expression of the cationic amino acid transport systems y+/hCAT-1 and y+/hCAT-2B and lower activity of nitric oxide synthase in human umbilical vein endothelial cells. Circ Res 2002; 91: 127-134.
222. Wong AY, Kulandavelu S, Whiteley KJ, Qu D, Langille BL, Adamson SL. Maternal cardiovascular changes during pregnancy and postpartum in mice. Am.J.Physiol Heart Circ.Physiol 2002; 282: H918-H925.
223. Leatherbury L, Yu Q, Lo CW. Noninvasive phenotypic analysis of cardiovascular structure and function in fetal mice using ultrasound. Birth Defects Res C Embryo Today 2003; 69: 83-91.
169
224. Kulandavelu S, Qu D, Sunn N, Mu J, Rennie MY, Whiteley KJ, Walls JR, Bock NA, Sun JC, Covelli A, Sled JG, Adamson SL. Embryonic and neonatal phenotyping of genetically engineered mice. Ilar J 2006; 47: 103-117.
225. Zhou YQ, Foster FS, Qu DW, Zhang M, Harasiewicz KA, Adamson SL. Applications for multifrequency ultrasound biomicroscopy in mice from implantation to adulthood. Physiol Genomics 2002; 10: 113-126.
226. Kulandavelu S, Qu D, Adamson SL. Cardiovascular function in mice during normal pregnancy and in the absence of endothelial NO synthase. Hypertension 2006; 47: 1175-1182.
227. Mu J, Adamson SL. Developmental changes in hemodynamics of uterine artery, utero- and umbilicoplacental, and vitelline circulations in mouse throughout gestation. Am J Physiol Heart Circ Physiol 2006; 291: H1421-1428.
228. Cox B, Kotlyar M, Evangelou AI, Ignatchenko V, Ignatchenko A, Whiteley K, Jurisica I, Adamson SL, Rossant J, Kislinger T. Comparative systems biology of human and mouse as a tool to guide the modeling of human placental pathology. Mol Syst Biol 2009; 5: 279.
229. Leonard S, Murrant C, Tayade C, van den Heuvel M, Watering R, Croy BA. Mechanisms regulating immune cell contributions to spiral artery modification -- facts and hypotheses -- a review. Placenta 2006; 27 Suppl A: S40-46.
230. Makikallio K, Tekay A, Jouppila P. Yolk sac and umbilicoplacental hemodynamics during early human embryonic development. Ultrasound Obstet Gynecol 1999; 14: 175-179.
231. Carter AM. Animal models of human placentation--a review. Placenta 2007; 28 Suppl A: S41-47.
232. Kanayama N, Takahashi K, Matsuura T, Sugimura M, Kobayashi T, Moniwa N, Tomita M, Nakayama K. Deficiency in p57(Kip2) expression induces preeclampsia-like symptoms in mice. Mol.Hum.Reprod. 2002; 8: 1129-1135.
233. Furuya M, Ishida J, Inaba S, Kasuya Y, Kimura S, Nemori R, Fukamizu A. Impaired placental neovascularization in mice with pregnancy-associated hypertension. Lab Invest 2008; 88: 416-429.
234. Davisson RL, Hoffmann DS, Butz GM, Aldape G, Schlager G, Merrill DC, Sethi S, Weiss RM, Bates JN. Discovery of a spontaneous genetic mouse model of preeclampsia. Hypertension 2002; 39: 337-342.
235. Kanasaki K, Palmsten K, Sugimoto H, Ahmad S, Hamano Y, Xie L, Parry S, Augustin HG, Gattone VH, Folkman J, Strauss JF, Kalluri R. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature 2008; 453: 1117-1121.
170
236. Li Y, Behringer RR. Esx1 is an X-chromosome-imprinted regulator of placental development and fetal growth. Nat Genet 1998; 20: 309-311.
237. Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A, Sibley C, Reik W. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002; 417: 945-948.
238. Watson CS, Bialek P, Anzo M, Khosravi J, Yee SP, Han VK. Elevated circulating insulin-like growth factor binding protein-1 is sufficient to cause fetal growth restriction. Endocrinology 2006; 147: 1175-1186.
239. Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br.J.Pharmacol. 1990; 101: 746-752.
241. Abram SR, Alexander BT, Bennett WA, Granger JP. Role of neuronal nitric oxide synthase in mediating renal hemodynamic changes during pregnancy. Am.J.Physiol Regul.Integr.Comp Physiol 2001; 281: R1390-R1393.
242. Zhao H, Shimokawa H, Uragami-Harasawa L, Igarashi H, Takeshita A. Long-term vascular effects of Nomega-nitro-L-arginine methyl ester are not soley mediated by inhibition of endothelial nitric oxide synthesis in the rat mesenteric artery. J.Cardiovasc.Pharmacol. 1999; 33: 554-566.
244. Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995; 377: 239-242.
245. Gregg AR, Schauer A, Shi O, Liu Z, Lee CG, O'Brien WE. Limb reduction defects in endothelial nitric oxide synthase-deficient mice. Am.J.Physiol 1998; 275: H2319-H2324.
246. MGD. In: Mouse Genome Informatics website in Jackson Laboratory, Bar Harbor, Maine. Available from http://www.informatics.jax.org.; September 2009.
247. Feng Q, Song W, Lu X, Hamilton JA, Lei M, Peng T, Yee SP. Development of heart failure and congenital septal defects in mice lacking endothelial nitric oxide synthase. Circulation 2002; 106: 873-879.
248. Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation 2000; 101: 2345-2348.
249. Kojda G, Laursen JB, Ramasamy S, Kent JD, Kurz S, Burchfield J, Shesely EG, Harrison DG. Protein expression, vascular reactivity and soluble guanylate cyclase
activity in mice lacking the endothelial cell nitric oxide synthase: contributions of NOS isoforms to blood pressure and heart rate control. Cardiovasc.Res. 1999; 42: 206-213.
250. Faraci FM, Sigmund CD, Shesely EG, Maeda N, Heistad DD. Responses of carotid artery in mice deficient in expression of the gene for endothelial NO synthase. Am.J.Physiol 1998; 274: H564-H570.
251. Vasapollo B, Valensise H, Novelli GP, Larciprete G, Di Pierro G, Altomare F, Casalino B, Galante A, Arduini D. Abnormal maternal cardiac function and morphology in pregnancies complicated by intrauterine fetal growth restriction. Ultrasound Obstet.Gynecol. 2002; 20: 452-457.
253. Janssen BJ, De Celle T, Debets JJ, Brouns AE, Callahan MF, Smith TL. Effects of anesthetics on systemic hemodynamics in mice. Am.J.Physiol Heart Circ.Physiol 2004; 287: H1618-H1624.
254. Zhou YQ, Foster FS, Parkes R, Adamson SL. Developmental changes in left and right ventricular diastolic filling patterns in mice. Am.J.Physiol Heart Circ.Physiol 2003; 285: H1563-H1575.
255. Pacileo G, Paladini D, Pisacane C, Palmieri S, Russo MG, Calabro R. Role of changing loading conditions on atrioventricular flow velocity patterns in normal human fetuses. Am J Cardiol 1994; 73: 991-993.
256. Kiatchoosakun S, Restivo J, Kirkpatrick D, Hoit BD. Assessment of left ventricular mass in mice: comparison between two-dimensional and M-mode echocardiography. Echocardiography. 2002; 19: 199-205.
257. Sejeny SA, Eastham RD, Baker SR. Platelet counts during normal pregnancy. J Clin Pathol 1975; 28: 812-813.
258. Ortiz PA, Garvin JL. Cardiovascular and renal control in NOS-deficient mouse models. Am.J.Physiol Regul.Integr.Comp Physiol 2003; 284: R628-R638.
259. Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J.Clin.Invest 2005; 115: 1221-1231.
260. Rey E, Couturier A. The prognosis of pregnancy in women with chronic hypertension. Am.J.Obstet.Gynecol. 1994; 171: 410-416.
261. Keyes L, Rodman DM, Curran-Everett D, Morris K, Moore LG. Effect of K + ATP channel inhibition on total and regional vascular resistance in guinea pig pregnancy. Am.J.Physiol 1998; 275: H680-H688.
172
262. Danielson LA, Conrad KP. Prostaglandins maintain renal vasodilation and hyperfiltration during chronic nitric oxide synthase blockade in conscious pregnant rats. Circ.Res. 1996; 79: 1161-1166.
263. Shesely EG, Gilbert C, Granderson G, Carretero CD, Carretero OA, Beierwaltes WH. Nitric oxide synthase gene knockout mice do not become hypertensive during pregnancy. Am.J.Obstet.Gynecol. 2001; 185: 1198-1203.
264. Hefler LA, Tempfer CB, Moreno RM, O'Brien WE, Gregg AR. Endothelial-derived nitric oxide and angiotensinogen: blood pressure and metabolism during mouse pregnancy. Am.J.Physiol Regul.Integr.Comp Physiol 2001; 280: R174-R182.
265. Huang A, Sun D, Wu Z, Yan C, Carroll MA, Jiang H, Falck JR, Kaley G. Estrogen elicits cytochrome P450--mediated flow-induced dilation of arterioles in NO deficiency: role of PI3K-Akt phosphorylation in genomic regulation. Circ.Res. 2004; 94: 245-252.
266. Magder S, Kabsele K. Evidence for constitutive release of nitric oxide in the venous circuit of pigs. J.Cardiovasc.Pharmacol. 1998; 32: 366-372.
267. Leung SW, Cheng X, Lim SL, Pang CC. Augmented pulmonary vascular and venous constrictions to N(G)-nitro-L-arginine methyl ester in rats with monocrotaline-induced pulmonary hypertension. Pharmacology 2003; 69: 164-170.
268. Hannan RL, Hack BD, Matherne GP, Laubach VE. Deletion of endothelial nitric oxide synthase exacerbates myocardial stunning in an isolated mouse heart model. J.Surg.Res. 2000; 93: 127-132.
269. Melo LG, Veress AT, Ackermann U, Pang SC, Flynn TG, Sonnenberg H. Chronic hypertension in ANP knockout mice: contribution of peripheral resistance. Regul.Pept. 1999; 79: 109-115.
270. Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation 2000; 101: 2345-2348.
271. Barbee RW, Perry BD, Re RN, Murgo JP. Microsphere and dilution techniques for the determination of blood flows and volumes in conscious mice. Am.J.Physiol 1992; 263: R728-R733.
272. Brosens I, Robertson WB, Dixon HG. The physiological response of the vessels of the placental bed to normal pregnancy. J Pathol Bacteriol 1967; 93: 569-579.
273. Nelson SH, Steinsland OS, Wang Y, Yallampalli C, Dong YL, Sanchez JM. Increased nitric oxide synthase activity and expression in the human uterine artery during pregnancy. Circ.Res. 2000; 87: 406-411.
275. Martin D, Conrad KP. Expression of endothelial nitric oxide synthase by extravillous trophoblast cells in the human placenta. Placenta 2000; 21: 23-31.
277. Huckle WR, Roche RI. Post-transcriptional control of expression of sFlt-1, an endogenous inhibitor of vascular endothelial growth factor. J Cell Biochem 2004; 93: 120-132.
278. Murakoshi T, Sekizuka N, Takakuwa K, Yoshizawa H, Tanaka K. Uterine and spiral artery flow velocity waveforms in pregnancy-induced hypertension and/or intrauterine growth retardation. Ultrasound Obstet Gynecol 1996; 7: 122-128.
279. Khong TY, De Wolf F, Robertson WB, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br J Obstet Gynaecol 1986; 93: 1049-1059.
280. Ahmed A, Dunk C, Ahmad S, Khaliq A. Regulation of placental vascular endothelial growth factor (VEGF) and placenta growth factor (PIGF) and soluble Flt-1 by oxygen--a review. Placenta. 2000; 21 Suppl A: S16-S24.
281. van Buul EJ, Steegers EA, Jongsma HW, Eskes TK, Thomas CM, Hein PR. Haematological and biochemical profile of uncomplicated pregnancy in nulliparous women; a longitudinal study. Neth.J.Med. 1995; 46: 73-85.
282. Kisters K, Barenbrock M, Louwen F, Hausberg M, Rahn KH, Kosch M. Membrane, intracellular, and plasma magnesium and calcium concentrations in preeclampsia. Am J Hypertens 2000; 13: 765-769.
283. Godecke A, Schrader J. Adaptive mechanisms of the cardiovascular system in transgenic mice--lessons from eNOS and myoglobin knockout mice. Basic Res.Cardiol. 2000; 95: 492-498.
284. Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors alpha and beta. J Biol Chem 2005; 280: 19704-19710.
285. Quinlan TR, Li D, Laubach VE, Shesely EG, Zhou N, Johns RA. eNOS-deficient mice show reduced pulmonary vascular proliferation and remodeling to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 2000; 279: L641-650.
286. Hassid A, Arabshahi H, Bourcier T, Dhaunsi GS, Matthews C. Nitric oxide selectively amplifies FGF-2-induced mitogenesis in primary rat aortic smooth muscle cells. Am J Physiol 1994; 267: H1040-1048.
287. Keyes LE, Moore LG, Walchak SJ, Dempsey EC. Pregnancy-stimulated growth of vascular smooth muscle cells: importance of protein kinase C-dependent synergy between estrogen and platelet-derived growth factor. J Cell Physiol 1996; 166: 22-32.
174
288. Kelly BA, Bond BC, Poston L. Gestational profile of matrix metalloproteinases in rat uterine artery. Mol.Hum.Reprod. 2003; 9: 351-358.
289. Inoue N, Venema RC, Sayegh HS, Ohara Y, Murphy TJ, Harrison DG. Molecular regulation of the bovine endothelial cell nitric oxide synthase by transforming growth factor-beta 1. Arterioscler Thromb Vasc Biol 1995; 15: 1255-1261.
290. Schaffer L, Vogel J, Breymann C, Gassmann M, Marti HH. Preserved placental oxygenation and development during severe systemic hypoxia. Am J Physiol Regul Integr Comp Physiol 2006; 290: R844-851.
291. Victor VM, Nunez C, D'Ocon P, Taylor CT, Esplugues JV, Moncada S. Regulation of oxygen distribution in tissues by endothelial nitric oxide. Circ Res 2009; 104: 1178-1183.
292. Buttery LD, McCarthy A, Springall DR, Sullivan MH, Elder MG, Michel T, Polak JM. Endothelial nitric oxide synthase in the human placenta: regional distribution and proposed regulatory role at the feto-maternal interface. Placenta 1994; 15: 257-265.
293. Dotsch J, Hogen N, Nyul Z, Hanze J, Knerr I, Kirschbaum M, Rascher W. Increase of endothelial nitric oxide synthase and endothelin-1 mRNA expression in human placenta during gestation. Eur J Obstet Gynecol Reprod Biol 2001; 97: 163-167.
294. Giles W, O'Callaghan S, Read M, Gude N, King R, Brennecke S. Placental nitric oxide synthase activity and abnormal umbilical artery flow velocity waveforms. Obstet Gynecol 1997; 89: 49-52.
295. Schiessl B, Mylonas I, Hantschmann P, Kuhn C, Schulze S, Kunze S, Friese K, Jeschke U. Expression of endothelial NO synthase, inducible NO synthase, and estrogen receptors alpha and beta in placental tissue of normal, preeclamptic, and intrauterine growth-restricted pregnancies. J Histochem Cytochem 2005; 53: 1441-1449.
296. Rutherford RA, McCarthy A, Sullivan MH, Elder MG, Polak JM, Wharton J. Nitric oxide synthase in human placenta and umbilical cord from normal, intrauterine growth-retarded and pre-eclamptic pregnancies. Br.J.Pharmacol. 1995; 116: 3099-3109.
297. Whiteley KJ, Pfarrer CD, Adamson SL. Vascular corrosion casting of the uteroplacental and fetoplacental vasculature in mice. Methods Mol Med 2006; 121: 371-392.
298. Verburg BO, Jaddoe VW, Wladimiroff JW, Hofman A, Witteman JC, Steegers EA. Fetal hemodynamic adaptive changes related to intrauterine growth: the Generation R Study. Circulation 2008; 117: 649-659.
299. Martinelli S, Francisco RP, Bittar RE, Zugaib M. Hematological indices at birth in relation to arterial and venous Doppler in small-for-gestational-age fetuses. Acta Obstet Gynecol Scand 2009; 88: 888-893.
300. Rigano S, Bozzo M, Padoan A, Mustoni P, Bellotti M, Galan HL, Ferrazzi E. Small size-specific umbilical vein diameter in severe growth restricted fetuses that die in utero. Prenat Diagn 2008; 28: 908-913.
175
301. Kiserud T, Kessler J, Ebbing C, Rasmussen S. Ductus venosus shunting in growth-restricted fetuses and the effect of umbilical circulatory compromise. Ultrasound Obstet Gynecol 2006; 28: 143-149.
302. Byrne BM, Howard RB, Morrow RJ, Whiteley KJ, Adamson SL. Role of the L-arginine nitric oxide pathway in hypoxic fetoplacental vasoconstriction. Placenta 1997; 18: 627-634.
303. McCarthy AL, Woolfson RG, Evans BJ, Davies DR, Raju SK, Poston L. Functional characteristics of small placental arteries. Am J Obstet Gynecol 1994; 170: 945-951.
304. Kusinski LC, Baker PN, Sibley CP, Wareing M. In vitro assessment of mouse uterine and fetoplacental vascular function. Reprod Sci 2009; 16: 740-748.
305. Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995; 146: 1029-1039.
306. Brownbill P, McKeeman GC, Brockelsby JC, Crocker IP, Sibley CP. Vasoactive and permeability effects of vascular endothelial growth factor-165 in the term in vitro dually perfused human placental lobule. Endocrinology 2007; 148: 4734-4744.
307. Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A 2001; 98: 2604-2609.
308. Jakoubek V, Bibova J, Herget J, Hampl V. Chronic hypoxia increases fetoplacental vascular resistance and vasoconstrictor reactivity in the rat. Am J Physiol Heart Circ Physiol 2008; 294: H1638-1644.
309. Mayhew TM, Charnock-Jones DS, Kaufmann P. Aspects of human fetoplacental vasculogenesis and angiogenesis. III. Changes in complicated pregnancies. Placenta 2004; 25: 127-139.
310. Teichert AM, Scott JA, Robb GB, Zhou YQ, Zhu SN, Lem M, Keightley A, Steer BM, Schuh AC, Adamson SL, Cybulsky MI, Marsden PA. Endothelial nitric oxide synthase gene expression during murine embryogenesis: commencement of expression in the embryo occurs with the establishment of a unidirectional circulatory system. Circ Res 2008; 103: 24-33.
311. Beleslin-Cokic BB, Cokic VP, Yu X, Weksler BB, Schechter AN, Noguchi CT. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 2004; 104: 2073-2080.
312. North TE, Goessling W, Peeters M, Li P, Ceol C, Lord AM, Weber GJ, Harris J, Cutting CC, Huang P, Dzierzak E, Zon LI. Hematopoietic stem cell development is dependent on blood flow. Cell 2009; 137: 736-748.
176
313. Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med 2003; 9: 1370-1376.
314. Moritz KM, Lim GB, Wintour EM. Developmental regulation of erythropoietin and erythropoiesis. Am J Physiol 1997; 273: R1829-1844.
315. Longo M, Jain V, Vedernikov YP, Bukowski R, Garfield RE, Hankins GD, Anderson GD, Saade GR. Fetal origins of adult vascular dysfunction in mice lacking endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol 2005; 288: R1114-1121.
316. Van Vliet BN, Chafe LL. Maternal endothelial nitric oxide synthase genotype influences offspring blood pressure and activity in mice. Hypertension 2007; 49: 556-562.
317. Joyce JM, Phernetton TM, Magness RR. Effect of uterine blood flow occlusion on shear stress-mediated nitric oxide production and endothelial nitric oxide synthase expression during ovine pregnancy. Biol.Reprod. 2002; 67: 320-326.
319. Sharp BR, Jones SP, Rimmer DM, Lefer DJ. Differential response to myocardial reperfusion injury in eNOS-deficient mice. Am.J.Physiol Heart Circ.Physiol 2002; 282: H2422-H2426.
320. Meng W, Ma J, Ayata C, Hara H, Huang PL, Fishman MC, Moskowitz MA. ACh dilates pial arterioles in endothelial and neuronal NOS knockout mice by NO-dependent mechanisms. Am J Physiol 1996; 271: H1145-1150.
321. Huang A, Sun D, Shesely EG, Levee EM, Koller A, Kaley G. Neuronal NOS-dependent dilation to flow in coronary arteries of male eNOS-KO mice. Am.J.Physiol Heart Circ.Physiol 2002; 282: H429-H436.
322. Godecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Godecke S, Schrader J. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ.Res. 1998; 82: 186-194.
323. Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta JA, Falck JR, Shesely EG, Koller A, Kaley G. EDHF mediates flow-induced dilation in skeletal muscle arterioles of female eNOS-KO mice. Am.J.Physiol Heart Circ.Physiol 2001; 280: H2462-H2469.
324. Waldron GJ, Dong H, Cole WC, Triggle CR. Endothelium-dependent hyperpolarization of vascular smooth muscle: role for a non-nitric oxide synthase product. Zhongguo Yao Li Xue Bao 1996; 17: 3-7.
325. August P, Lindheimer MD, Lindheimer MD, Roberts JM, Cunningham FG. Chronic Hypertension and Pregnancy. In: Chesley's Hypertensive Disorders in Pregnancy, vol. Second. Stamford, Connecticut: Appleton & Lange; 1999: 605-634.
177
326. Liao Y, Day KH, Damon DN, Duling BR. Endothelial cell-specific knockout of connexin 43 causes hypotension and bradycardia in mice. Proc Natl Acad Sci U S A 2001; 98: 9989-9994.
327. Lee P, Morley G, Huang Q, Fischer A, Seiler S, Horner JW, Factor S, Vaidya D, Jalife J, Fishman GI. Conditional lineage ablation to model human diseases. Proc Natl Acad Sci U S A 1998; 95: 11371-11376.
328. Park F. Lentiviral vectors: are they the future of animal transgenesis? Physiol Genomics 2007; 31: 159-173.
329. Okada Y, Ueshin Y, Isotani A, Saito-Fujita T, Nakashima H, Kimura K, Mizoguchi A, Oh-Hora M, Mori Y, Ogata M, Oshima RG, Okabe M, Ikawa M. Complementation of placental defects and embryonic lethality by trophoblast-specific lentiviral gene transfer. Nat Biotechnol 2007; 25: 233-237.
330. Yu J, deMuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci U S A 2005; 102: 10999-11004.
331. Scotland RS, Morales-Ruiz M, Chen Y, Yu J, Rudic RD, Fulton D, Gratton JP, Sessa WC. Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion. Circ Res 2002; 90: 904-910.
332. Ahokas RA, Reynolds SL, Anderson GD, Lipshitz J. Maternal organ distribution of cardiac output in the diet-restricted pregnant rat. J Nutr 1984; 114: 2262-2268.
333. Conrad KP. Renal hemodynamics during pregnancy in chronically catheterized, conscious rats. Kidney Int. 1984; 26: 24-29.