Journal of Nutritional Biochemistry 24 (2013) 824–831
Cellular signaling of amino acids towards mTORC1 activation in impaired humanleucine catabolism☆,☆☆
Sonja C. Schrievera, Manuel J. Deutscha, Jerzy Adamskib, Adelbert A. Roschera, Regina Ensenauera,⁎
aResearch Center, Dr. von Hauner Children's Hospital, Ludwig-Maximilians-Universität München, 80337 Munich, GermanybHelmholtz Zentrum München, Institute of Experimental Genetics, Genome Analysis Center, 85764 Neuherberg, Germany
Received 28 August 2011; received in revised form 19 March 2012; accepted 30 April 2012
The mammalian target of rapamycin complex 1 (mTORC1)controls protein synthesis and cell growth in response to nutrientavailability, insulin and growth factors [1]. Amino acids, in particular,the branched-chain amino acid L-leucine, exert powerful modulatoryeffects on the mTORC1 signaling pathway [2]. This major nutrient-sensing mechanism is frequently found to be dysregulated insituations of catabolic stress such as in advanced tumor stages,inflammation or sepsis or in metabolic conditions like insulinresistance [3–5].
☆ Grants: This work was supported by the German Federal Ministry ofEducation and Research (BMBF) Grant 0315088 (S.C.S., M.J.D., R.E.) and inpart by a grant from the BMBF to the German Center Diabetes Research (DZDe.V.) (J.A.).
☆☆ Disclosure summary: The authors confirm that there are no conflictsof interest.
Insulin and growth factors activate mTORC1 via the phosphatidyl-inositide-3-kinase/Akt (protein kinase B) pathway and the tuberoussclerosis complex TSC1/2, leading to phosphorylation of two keytranslational regulators: p70 ribosomal protein S6 kinase 1 (S6K1)and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) [6]. Ithas been shown that amino acids directly stimulate mTORC1-dependent phosphorylation of S6K1 and 4E-BP1, whereas aminoacid deprivation results in rapid dephosphorylation [7].
The exact cellular mechanism whereby amino acids activatemTORC1 signaling still remains undefined [6]. Downstream effectsof leucine-induced mTORC1 activation are well studied, whereas theupstream mechanisms are less precisely defined. Studies designed toaddress the mechanism of leucine-induced mTORC1 activation havefocused on structural characteristics of the leucine molecule for aputative recognition site [8,9] or on the intracellular metabolism ofleucine at the second regulatory step of its catabolic pathway [10,11].Leucine metabolites generated by the degradation of leucine in themitochondria have also been discussed to facilitate mTORC1 activa-tion [12]. However, these studies were not able to differentiate ifleucine itself or a metabolically transformed product of leucinecatabolism activates mTORC1 signaling.
Fig. 1. Leucine catabolism pathway. Enzymatic steps of leucine catabolism areindicated by numbers, and metabolically transformed products of leucinedegradation are shown for each step. The second irreversible step of leucinecatabolism is tightly regulated by branched-chain keto acid dehydrogenase kinaseand phosphatase. 1: BCAT2=branched-chain amino acid transaminase 2, mito-chondrial; 2: BCKDH=branched-chain 2-keto acid dehydrogenase complex, 3:IVD=isovaleryl-CoA dehydrogenase; 4: MCC=3-methylcrotonoyl-CoA carboxylase,5: AUH=3-methylglutaconyl-CoA hydratase (AU RNA binding protein/enoyl-CoAhydratase); 6: HMGCL=3-hydroxymethyl-3-methylglutaryl-CoA lyase.
825S.C. Schriever et al. / Journal of Nutritional Biochemistry 24 (2013) 824–831
Beugnet et al. proposed that the intracellular amino acid levels maybe involved in upstream regulation of mTOR signaling [13]. In Xenopus,direct intracytoplasmic injection of L-leucine stimulated S6K1 phos-phorylation, whereas extracellular leucine did not [14]. Recently, it wasshown that L-glutamine uptake by solute carrier family 1 (SLC1A5)transporter is rate limiting for leucine-dependentmTOR activation [15].Intracellular glutamine acts as an efflux substrate for the bidirectionaltransporter SLC7A5/SLC3A2 (LAT1) that regulates the simultaneousefflux of glutamine out of cells and transport of leucine into cells. Howan increase in intracellular leucine concentration is sensed towardsdownstream signaling effectors is not fully clarified. Some conceptspropose that amino acid transporters also act as sensing receptors forintracellular amino acid concentrations, so-called transceptors [16],whereas others propagate intracellular amino acid sensors [17]. Severalintracellular proteins such as class III phosphoinositide 3-kinase(human vacuolar protein sorting 34), Rag-GTPases and MAP kinasekinase kinase kinase 3 (MAP4K3) have been proposed as sensors ofintracellular amino acid availability that act upstream of mTORC1 inHeLa and HEK293 cells [18–20]. Studies in primary human cells havehitherto not been conducted.
Catabolism of leucine is a highly regulated, six-step degradativepathway that yields acetyl-CoA and acetoacetate which can beutilized for adenosine triphosphate (ATP) generation, fatty acidsynthesis and ketogenesis. Regulation of leucine catabolism isnecessary both to conserve leucine for protein synthesis and toprovide anaplerotic substrate to the citrate cycle [21,22]. In commonhuman pathologies such as hyperinsulinemia [23] or hyperthyroidism[24], leucine catabolism is suppressed, leading to preservation of thisessential amino acid. However, associated regulatory mechanismshave remained undetermined.
Specific monogenic enzyme defects are known for the second tosixth enzymatic step of leucine catabolism in humans, whereas adeficiency of the first reversible step in leucine catabolism (mito-chondrial branched-chain amino acid transferase, BCAT2) has notbeen reported in humans as yet [25]. In this study, we took advantageof human fibroblast cell lines derived from patients with thesedeficiencies in order to address some of the unresolved mechanisticquestions. This unique set of human cell lines also enabled us toinvestigate whether mTORC1 signaling is subject to disturbedregulation in defective leucine degradation.
2. Materials and methods
2.1. Materials
Cell culture media were purchased from PAA Laboratories GmbH (Pasching,Austria). L-[1-14 C] leucine was obtained from American Radioactive Chemicals (St.Louis, MO, USA). Other chemicals were obtained fromMerck (Darmstadt, Germany) orSigma (Steinheim, Germany) unless otherwise noted.
2.2. Cell culture and treatment
Primary human fibroblast cell lines were derived from skin of five healthy children (agerange: 3 months to 7 years) undergoing inguinal hernia repair that served as control celllines and of children (age range: 1 month to 4 years) with enzymatically and/or geneticallyconfirmed inborn errors of leucine catabolism of the second to sixth enzymatic step [26–29],after informed consent was obtained: branched-chain 2-keto acid dehydrogenase complex(BCKDH), isovaleryl-CoA dehydrogenase, 3-methylcrotonyl-CoA carboxylase, 3-methylglu-taconyl-CoA hydratase and 3-hydroxymethyl-3-methylglutaryl-CoA lyase deficiencies(Fig. 1). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (1 g/Lglucose) supplemented with 10% fetal calf serum (FCS) and 2mM stable glutamine at 37 °Cin 5% CO2. All studies were performed on subcultures between the 10th and 25th passage(controls cells: 10th–23rd; defective cells: 10th–25th).Within this range, human fibroblastsretained a normal morphology, and cell viability was confirmed at different passagenumbers using the colorimetric MTS reduction assay according to the manufacturer'sinstructions (Promega, Mannheim, Germany).
For Western blot analysis, cells seeded in 12-well plates and grown to 80%confluency were cultured in MEM supplemented with 0.25% bovine serum albumin(BSA) for 72 h prior to the start of the experimental treatments. Prior to all leucinestimulation experiments, cells were starved for amino acids and serum in Hank's buffer
with 0.1% BSA for 1 h and subsequently in Hank's buffer without BSA for 20 min.Following starvation, cells were stimulated with L-leucine for 20 min. In someexperiments, 100 nM rapamycin was added to Hank's buffer with and without BSAin order to inhibit mTORC1 signaling function. For glutamine preload experiments,Hank's buffer with and without BSA was supplemented with 2 mM L-glutamine.
2.3. Transfection reactions
Magnetofection was applied for siRNA delivery using a protocol modified fromEnsenauer et al. [30]. Transfection mixtures consisted of 1 μl PolyMAG (Chemicell,Berlin, Germany) and 0.3 μg (25 nM) of siGENOME SMARTpool siRNA (BCAT2;MAP4K3) or siGENOME Non-Targeting siRNA #2 (Dharmacon, Lafayette, CO, USA) inserum-free DMEM. After magnetofection, cultivation was continued for 72 h whencells were analyzed for mRNA expression or used for leucine stimulation experiments.
2.4. Western blot and immunofluorescence analysis
Following starvation conditions and leucine stimulation, cells were harvested andlysed in ice-cold lysis buffer (50 mM Tris–HCl, 150 mM sodium chloride, pH 8.0, 1%Tween-20, 25 mM sodium fluoride, 50 mM glycerol-phosphate, 1% Calyculin, 1 mMEDTA, and 1 μg/ml protease inhibitor cocktail). The protein concentration of clearsupernatants was determined using the bicinchoninic acid method (Thermo FisherScientific, Rockford, IL, USA). Equal amounts of protein (8 μg protein/lane) wereresolved by a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis andtransferred electrophoretically to a polyvinylidene fluoride membrane (GE Healthcare,München, Germany). After blocking with 5% BSA, the membranes were probed usingantibodies specific for S6K1, phosphorylated S6K1 (Thr389), 4E-BP1, phosphorylated4E-BP1 (Ser65), Akt, phosphorylated Akt (Thr308) (Cell Signaling Technology,Danvers, MA, USA) and α-tubulin (Acris, Herford, Germany) and were immunode-tected by CDP-Star (Roche, Mannheim, Germany) using appropriate alkalinephosphatase-conjugated secondary antibodies (Promega). Blots were quantifiedusing a DIANA III densitometer (Raytest, Straubenhardt, Germany). The immunoflu-orescence procedure is described in the Supplemental data.
2.5. Amino acid analysis
Analysis was performed using a protocol adapted from Melancon et al. [31]. Cellsseeded in six-well plates were harvested in bidistilled water and disrupted by freeze-thawing. The cell protein was precipitated with 10% sulfosalicylic acid. The aqueous cellextract was cleared by centrifugation through spin columns and then diluted 1:1 withassay buffer containing 1% trifluoroacetic acid (Biochrom, Cambridge, UK). Free cellular
826 S.C. Schriever et al. / Journal of Nutritional Biochemistry 24 (2013) 824–831
amino acid concentrations were measured by photometric estimation with ninhydrinafter chromatographic separation [32] using an amino acid analyzer (Biochrom 30Amino Acid Analyzer, Biochrom).
2.6. L-[1-14 C] leucine oxidation assay
Leucine decarboxylation was measured using a modified protocol of Tanaka et al.[33]. Cells seeded in T-25 culture flasks (Sarstedt, Nümbrecht, Germany) wereincubated with MEM containing 10% FCS for 48 h. Amino acid starvation wasperformed by incubation with Hank's–HEPES buffer for 1 h. Buffer was then replacedwith Hank's–HEPES buffer containing 125 μM leucine plus 0.33 μCi/μmol [1-14 C]leucine for 2 h. Cells inactivated by 10% trichloroacetic acid and then treated asdescribed above were used as background control. The reaction was stopped byaddition of 5 M hydrogen chloride. The amount of 14CO2 produced until 30 minpostincubation was trapped and quantitated by scintillation counting (LS6000IC,Beckman Coulter, Krefeld, Germany).
2.7. RNA isolation and analysis
Total RNA was isolated from fibroblasts using the NucleoSpin RNA II Kit(Macherey-Nagel, Düren, Germany), and 1 μg RNA was reverse transcribed to cDNAusing the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). RelativemRNA abundance was measured by quantitative real-time polymerase chain reaction(PCR) using 2× Power SYBR Green PCR MasterMix (Applied Biosystems, Foster City,CA, USA). The reactions were run in the ABI StepOnePlus (Applied Biosystems). Therelative mRNA abundance was normalized to the endogenous control hypoxanthinephosphoribosyltransferase 1 and calculated relative to the mRNA expression understandard culture conditions or to the nonspecific siRNA control.
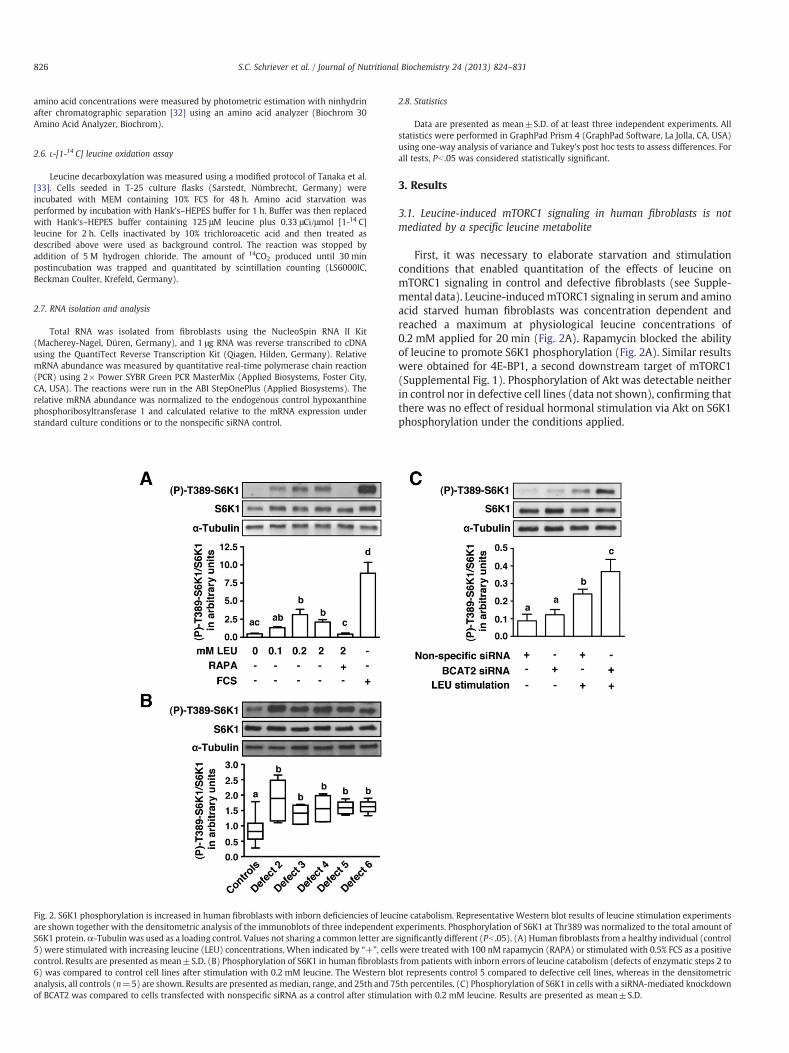
Fig. 2. S6K1 phosphorylation is increased in human fibroblasts with inborn deficiencies of leucare shown together with the densitometric analysis of the immunoblots of three independentS6K1 protein.α-Tubulin was used as a loading control. Values not sharing a common letter are5) were stimulated with increasing leucine (LEU) concentrations. When indicated by “+”, cellscontrol. Results are presented as mean±S.D. (B) Phosphorylation of S6K1 in human fibroblasts6) was compared to control cell lines after stimulation with 0.2 mM leucine. The Western blanalysis, all controls (n=5) are shown. Results are presented asmedian, range, and 25th and 7of BCAT2 was compared to cells transfected with nonspecific siRNA as a control after stimula
2.8. Statistics
Data are presented as mean±S.D. of at least three independent experiments. Allstatistics were performed in GraphPad Prism 4 (GraphPad Software, La Jolla, CA, USA)using one-way analysis of variance and Tukey's post hoc tests to assess differences. Forall tests, Pb .05 was considered statistically significant.
3. Results
3.1. Leucine-induced mTORC1 signaling in human fibroblasts is notmediated by a specific leucine metabolite
First, it was necessary to elaborate starvation and stimulationconditions that enabled quantitation of the effects of leucine onmTORC1 signaling in control and defective fibroblasts (see Supple-mental data). Leucine-inducedmTORC1 signaling in serum and aminoacid starved human fibroblasts was concentration dependent andreached a maximum at physiological leucine concentrations of0.2 mM applied for 20 min (Fig. 2A). Rapamycin blocked the abilityof leucine to promote S6K1 phosphorylation (Fig. 2A). Similar resultswere obtained for 4E-BP1, a second downstream target of mTORC1(Supplemental Fig. 1). Phosphorylation of Akt was detectable neitherin control nor in defective cell lines (data not shown), confirming thatthere was no effect of residual hormonal stimulation via Akt on S6K1phosphorylation under the conditions applied.
ine catabolism. Representative Western blot results of leucine stimulation experimentsexperiments. Phosphorylation of S6K1 at Thr389 was normalized to the total amount ofsignificantly different (Pb .05). (A) Human fibroblasts from a healthy individual (controlwere treated with 100 nM rapamycin (RAPA) or stimulated with 0.5% FCS as a positivefrom patients with inborn errors of leucine catabolism (defects of enzymatic steps 2 to
ot represents control 5 compared to defective cell lines, whereas in the densitometric5th percentiles. (C) Phosphorylation of S6K1 in cells with a siRNA-mediated knockdowntion with 0.2 mM leucine. Results are presented as mean±S.D.
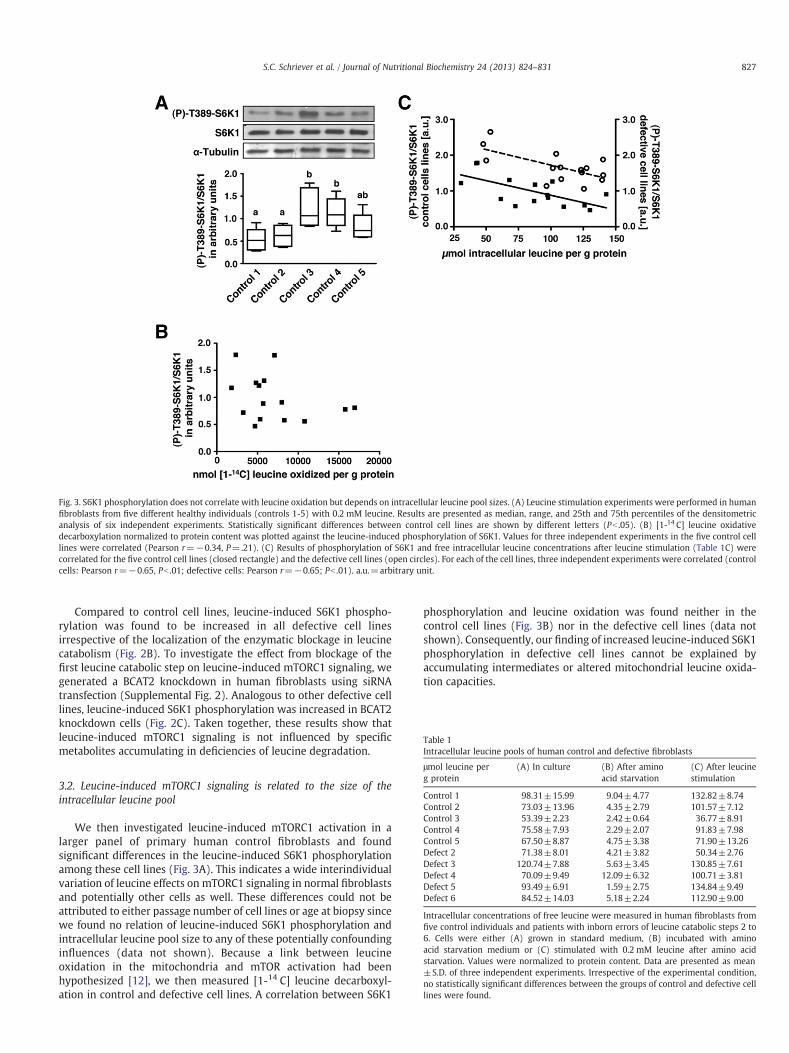
Fig. 3. S6K1 phosphorylation does not correlate with leucine oxidation but depends on intracellular leucine pool sizes. (A) Leucine stimulation experiments were performed in humanfibroblasts from five different healthy individuals (controls 1-5) with 0.2 mM leucine. Results are presented as median, range, and 25th and 75th percentiles of the densitometricanalysis of six independent experiments. Statistically significant differences between control cell lines are shown by different letters (Pb .05). (B) [1-14 C] leucine oxidativedecarboxylation normalized to protein content was plotted against the leucine-induced phosphorylation of S6K1. Values for three independent experiments in the five control celllines were correlated (Pearson r=−0.34, P=.21). (C) Results of phosphorylation of S6K1 and free intracellular leucine concentrations after leucine stimulation (Table 1C) werecorrelated for the five control cell lines (closed rectangle) and the defective cell lines (open circles). For each of the cell lines, three independent experiments were correlated (controlcells: Pearson r=−0.65, Pb .01; defective cells: Pearson r=−0.65; Pb .01). a.u.=arbitrary unit.
Table 1Intracellular leucine pools of human control and defective fibroblasts
Intracellular concentrations of free leucine were measured in human fibroblasts fromfive control individuals and patients with inborn errors of leucine catabolic steps 2 to6. Cells were either (A) grown in standard medium, (B) incubated with aminoacid starvation medium or (C) stimulated with 0.2 mM leucine after amino acidstarvation. Values were normalized to protein content. Data are presented as mean±S.D. of three independent experiments. Irrespective of the experimental condition,no statistically significant differences between the groups of control and defective celllines were found.
827S.C. Schriever et al. / Journal of Nutritional Biochemistry 24 (2013) 824–831
Compared to control cell lines, leucine-induced S6K1 phospho-rylation was found to be increased in all defective cell linesirrespective of the localization of the enzymatic blockage in leucinecatabolism (Fig. 2B). To investigate the effect from blockage of thefirst leucine catabolic step on leucine-induced mTORC1 signaling, wegenerated a BCAT2 knockdown in human fibroblasts using siRNAtransfection (Supplemental Fig. 2). Analogous to other defective celllines, leucine-induced S6K1 phosphorylation was increased in BCAT2knockdown cells (Fig. 2C). Taken together, these results show thatleucine-induced mTORC1 signaling is not influenced by specificmetabolites accumulating in deficiencies of leucine degradation.
3.2. Leucine-induced mTORC1 signaling is related to the size of theintracellular leucine pool
We then investigated leucine-induced mTORC1 activation in alarger panel of primary human control fibroblasts and foundsignificant differences in the leucine-induced S6K1 phosphorylationamong these cell lines (Fig. 3A). This indicates a wide interindividualvariation of leucine effects on mTORC1 signaling in normal fibroblastsand potentially other cells as well. These differences could not beattributed to either passage number of cell lines or age at biopsy sincewe found no relation of leucine-induced S6K1 phosphorylation andintracellular leucine pool size to any of these potentially confoundinginfluences (data not shown). Because a link between leucineoxidation in the mitochondria and mTOR activation had beenhypothesized [12], we then measured [1-14 C] leucine decarboxyl-ation in control and defective cell lines. A correlation between S6K1
phosphorylation and leucine oxidation was found neither in thecontrol cell lines (Fig. 3B) nor in the defective cell lines (data notshown). Consequently, our finding of increased leucine-induced S6K1phosphorylation in defective cell lines cannot be explained byaccumulating intermediates or altered mitochondrial leucine oxida-tion capacities.
828 S.C. Schriever et al. / Journal of Nutritional Biochemistry 24 (2013) 824–831
Next, we evaluated whether the human cell lines show differencesin intracellular amino acid levels that may explain the variations inleucine-induced mTORC1 signaling. Under standard culture condi-tions, the fibroblast strains differed greatly in their leucine pool sizeirrespective of an enzymatic defect in leucine catabolism (Table 1A).After amino acid starvation, all cell lines were confirmed to bedepleted of intracellular free leucine (Table 1B) and glutamine (mean±S.D., controls: 5.31±2.43 μmol/g protein, defective cells: 4.44±2.87 μmol/g protein). Following stimulation with leucine, therewere differences between the cell lines in their ability to replete theintracellular leucine pool; the majority of cell lines had higherintracellular leucine concentrations after leucine stimulation thanunder standard culture conditions (Table 1C). When we related theleucine pool size after leucine repletion to concomitant phosphor-ylation of S6K1 in the different control cell lines, an inversecorrelation was observed (Fig. 3C). Similar to control cells, theintracellular leucine concentrations after leucine stimulation werealso inversely correlated with S6K1 phosphorylation in the defectivecell lines, although at a higher signaling level (Fig. 3C). Therefore,these results suggest that the size of the intracellular leucine pool iscrucial for activation of mTORC1 signaling.
3.3. Leucine/glutamine antiporter expression is related to mTORC1activation capacity
Because cellular leucine uptake involves the leucine/glutamineantiporter SLC7A5/SLC3A2 [15], we assessed the mRNA expression of
Fig. 4. The leucine/glutamine antiporter SLC7A5/SLC3A2 has an influence on leucine uptake andtransporter subunit of the leucine/glutamine antiporter, after amino acid starvation were plot(Fig. 3A). Values for three independent experiments in the five control cell lines were correlatepreloaded with glutamine (GLN) with or without subsequent stimulation with 0.2 mM leucineto leucine stimulation to generate a gradient of intracellular to extracellular glutamine. Freepresented as mean±S.D. of three independent experiments. Statistically significant differencephosphorylation of S6K1 was analyzed by Western blotting in cells that were treated as dindependent experiments. Values not sharing a common letter are significantly different (Pbconditions described above were plotted against the leucine-induced phosphorylation of S6K1Pb .0001).
its catalytic transporter subunit SLC7A5. In all control and defectivefibroblasts, a significant up-regulation of SLC7A5 mRNA expressionwas noted after amino acid starvation (Supplemental Fig. 3A). Theexpression levels remained unchanged during subsequent short-termleucine stimulation (data not shown) and showed a significantpositive correlation with corresponding values on S6K1 phosphory-lation (Fig. 4A), suggesting that the cellular capacity of leucine poolrepletion influences mTORC1 activation.
SLC7A5/SLC3A2 has been reported to use glutamine as effluxsubstrate [15]; thus, experimental manipulation of glutamine con-centrations is expected to have a strong impact on intracellular leucineconcentrations. When we altered leucine/glutamine antiporter activ-ity by preloading control cells with glutamine, a significant increase inintracellular leucine concentrations was observed (Fig. 4B). However,this effectwas only evidentwhen extracellular glutaminewaswashedout prior to stimulation with leucine (Fig. 4B, lane 5), resulting in agradient with higher intracellular than extracellular glutamineconcentrations (data not shown). Leucine-induced S6K1 phosphory-lation was also potentiated by the concentration gradient ofintracellular to extracellular glutamine (Fig. 4C, lane 5). Under thevarious experimental conditions of modifying glutamine levels, thesubsequently observed intracellular leucine concentrations showed aclear relationship to the phosphorylation of S6K1 (Fig. 4D). Usingimmunofluorescence, we confirmed at the subcellular level thatphosphorylation of both mTOR and its target S6K1 was dependenton intracellular leucine concentrations (see Supplemental data,Supplemental Fig. 3B). Together, these results show that modification
S6K1 phosphorylation. (A) Results of relative mRNA expression of SLC7A5, the catalyticted against the leucine-induced phosphorylation of S6K1 in human control fibroblastsd (Pearson r=0.52, Pb .05). (B) Where indicated by “+”, control cells (control 3) were(LEU). In lane 5, extracellular glutamine was removed by washing the cells twice priorintracellular leucine concentrations were normalized to protein content. Results ares between the treatments are shown by different letters (Pb .05). (C) Leucine-inducedescribed above. The graph shows the mean±S.D. of densitometric analyses of three.05). (D) The free intracellular leucine concentrations measured under experimental. For each condition, three independent experiments were correlated (Pearson r=0.88,
829S.C. Schriever et al. / Journal of Nutritional Biochemistry 24 (2013) 824–831
of the leucine/glutamine antiporter expression and thus regulation ofintracellular leucine availability can have a strong influence onmTORC1 activation capacity.
3.4. MAP4K3 expression is required for leucine-induced mTORC1activation in human primary fibroblasts
We then questioned whether MAP4K3, a putative amino acidsensor, can act as a sensor of intracellular leucine concentrations inhuman fibroblasts. RNA interference experiments using humancontrol cells resulted in strongly reduced levels of MAP4K3 transcript(Supplemental Fig. 4A). Subsequent to amino acid starvation, leucine-induced S6K1 phosphorylation was significantly reduced in MAP4K3knockdown cells (Fig. 5A), showing that MAP4K3 is capable ofmodifying leucine-mediatedmTORC1 activation in human fibroblasts.
We went on to compare MAP4K3 mRNA expression in control anddefective cell lines after amino acid starvation relative to standardculture conditions (Fig. 5B). Upon starvation, MAP4K3 mRNA levelswere significantly reduced in control cell lines. In contrast, MAP4K3mRNA expression remained unchanged and displayed no regulatory
Fig. 5. Transcriptional regulation of MAP4K3, a sensor of intracellular leucineavailability, is disturbed in defective human leucine catabolism. (A) Phosphorylationof S6K1 in cells with a siRNA-mediated knockdown of MAP4K3 was compared to cellstransfected with nonspecific siRNA after stimulation with 0.2 mM leucine (LEU). Thegraph shows the mean±S.D. of densitometric analyses of three independentexperiments. Statistically significant differences between the treatments are shownby different letters (Pb .05). (B) Relative MAP4K3 mRNA expression after amino acidstarvation compared to standard culture conditions (indicated by the dotted line) wascalculated for each control and defective cell line. Data are presented as mean±S.D. ofthree independent experiments. Statistically significant reductions of MAP4K3 mRNAlevels after starvation relative to standard culture conditions are indicated by anasterisk (Pb .05). Statistically significant differences in MAP4K3 mRNA expressionbetween cell lines are shown by different letters (Pb .05). Short-term stimulation withleucine after amino acid starvation did not result in further changes of MAP4K3 mRNAexpression (data not shown).
response upon starvation in all enzymatically defective fibroblaststrains. Remarkably, this feature was evident irrespective of theparticular site of deficiency within the leucine catabolic pathway.These findings demonstrate that MAP4K3 is prone to regulation uponreduced amino acid supply. In the defective cell lines, however, whichalso showed increased mTORC1 signaling, this type of transcriptionalregulation was not found.
4. Discussion
Regulation of leucine catabolism and leucine-associated mTORC1activation plays a significant role in a range of human pathologiessuch as in patients with severe burns, inflammation, duringpostoperative catabolism and in certain muscle-wasting condi-tions [5,34]. Equally, in the pathologies associated with nutrientoverload, such as obesity and type 2 diabetes, leucine-mediatedmTORC1 nutrient signaling appears to contribute to the metabolicdysregulations observed [3]. The upstream mechanisms that sensethe level of amino acids and signals to mTORC1 activation are notyet clearly defined. Several lines of indirect evidence suggest thatleucine catabolic pathways that give rise to carboxylic acidproducts of leucine, ATP and/or other metabolic signals might beinvolved [10–12].
To investigate these hypotheses, cultured human fibroblastsderived from patients withmonogenetic defects in leucine catabolismwere used. Depending on the respective enzyme defect (Fig. 1), thesecells accumulate specific metabolites of leucine catabolism andgenerate less acetyl-CoA as the final product of leucine degradation[25]. However, despite these enzyme deficiencies, intracellular freeleucine concentrations are maintained at normal levels (Table 1). Thisis in accordance with an earlier report of normal intracellular leucineconcentrations in BCKDH-deficient cells [35].
We found increased S6K1 phosphorylation in all deficient cell linesas compared to controls and, remarkably, irrespective of thelocalization of the enzymatic defect in leucine catabolism. Evenafter BCAT2 knockdown, which disrupted the conversion of leucine toα-ketoisocaproic acid (KIC), cells displayed increased leucine-induced S6K1 phosphorylation. This implies that the conversion ofleucine to KIC is not essential for mTORC1 activation, although earlierreports have suggested that KIC is more efficacious than leucine atactivating mTORC1 signaling [11]. Our data are consistent with recentin vivo data showing elevated mTORC1 signaling in gastrocnemiusmuscle from BCATm−/− mice [36]. The more distal defects in leucinecatabolism equally showed enhanced mTORC1 signaling whichindicates that accumulation of specific metabolites of leucinedegradation is highly unlikely to be involved in mTORC1 activation.The overall oxidative capacity of leucine decarboxylation to generateATP has also been implicated to be involved in mTORC1 activation[12], but was not found to be a factor influencing leucine-inducedS6K1 phosphorylation in a broad panel of control cell lines. Among theleucine catabolic defects, fibroblasts deficient in BCKDH, the regula-tory step of the pathway (Fig. 1), are insufficient to activate leucinecatabolism and indeed displayed no 14CO2 production from [1-14 C]leucine decarboxylation (data not shown). Nevertheless, they had themost intense signal of all defective cell lines for phosphorylated S6K1after leucine stimulation as compared to controls. Therefore, neither asingle specific metabolite of leucine nor total leucine oxidativecapacity appears to be a crucial determinant of leucine-inducedmTORC1 activation in human fibroblasts.
Despite an interindividual variance in leucine pool sizes (Table 1),leucine-induced S6K1 phosphorylation subsequent to amino acidstarvation showed significant correlation with free intracellularleucine levels in both control and defective cell lines. Cells with aper se small intracellular leucine pool showed a more pronouncedS6K1 phosphorylation. Earlier findings of Christie et al. showed that
830 S.C. Schriever et al. / Journal of Nutritional Biochemistry 24 (2013) 824–831
small changes above the basal levels of intracellular leucineconcentrations are sufficient to increase S6K1 phosphorylation[14]. Our data suggest that cell lines with a small leucine poolmight reach such threshold level of intracellular leucine concentra-tions sufficient to activate mTORC1 signaling faster than cell lineswith a larger leucine pool. However, irrespective of confoundingvariance in leucine pool sizes, the maximum level of S6K1phosphorylation obtained at a defined leucine concentration wasfound to be consistently higher in all fibroblast strains with defects inleucine catabolism.
Then, we addressed the question of which determinants of leucinepool size could potentially be modified in the defective cell lines.Several membrane amino acid transporters are reported to have astrong impact on the size and composition of intracellular amino acidpools and can act as regulators for amino-acid-dependent mTORC1signaling [16]. Among these systems, the SLC7A5/SLC3A2 antiporterhas recently been shown to facilitate the uptake of extracellular leucinein exchange with intracellular glutamine [15]. As this cell surfacetransporter is ubiquitously expressed in mammalian tissues [37], wemeasured the expression of the catalytic transporter subunit in ourhuman control fibroblasts and found the individual capacity to up-regulate SLC7A5 expression after amino acid starvation being correlat-edwith leucine-induced S6K1 phosphorylation. The dependency of thismechanism on glutaminewas confirmed bymanipulating the transportrate of the antiporter system by a glutamine preload that resulted inpotentiation of leucine uptake and mTORC1 activation. These findingsdemonstrate that the leucine/glutamine antiporter system, previouslydescribed in immortalized HeLa and MCF7 cells, is also operative inprimary humanfibroblasts. They also suggest that regulation of leucine/glutamine antiporter expression can exert strong influence on theintracellular leucine availability and thereby on mTORC1 activation.
However, these data could not provide an explanation for thefinding of enhanced mTORC1 activation in cell lines with defectiveleucine catabolism. Leucine/glutamine antiporter expression was notfound to be different between control and defective cell lines,although further confirmation would require studies on transporteractivities that were beyond the scope of this study. Moreover, all ourdata on leucine-induced mTORC1 signaling were obtained subse-quent to short-time amino acid starvation. Under these conditions, nodifferences in intracellular glutamine concentrations between controland defective cells were observed, and thus, these cannot account forthemore pronouncedmTORC1 activation in response to leucine in thedefective cell lines.
Among the various protein complexes implicated as intracellularamino acid sensors [18–20], we considered MAP4K3 as a promisingcandidate for mediating leucine effects. It has recently been shownthat MAP4K3 and Rag-GTPases can interact and might work togetherin mediating amino acid signaling to mTORC1 [38]. In our primaryhuman control fibroblasts, knockdown of MAP4K3 resulted in asignificantly weaker, but not completely blunted, leucine-inducedmTORC1 signaling. This suggests that MAP4K3 is indeed important forfull activation of mTORC1 in response to leucine in human fibroblasts,although its activity may not be absolutely required to maintainresidual mTORC1 activation. Our study has also revealed thatMAP4K3mRNA expression is prone to down-regulation upon reduced aminoacid supply. However, in all of the defective cell lines, which alsoshowed elevated mTORC1 signaling, this type of transcriptionalregulation appeared to be abrogated or operating at a less sensitivethreshold. These data point to adaptive mechanisms that secure andmaintain mRNA expression of this amino acid sensor during catabolicstress due to starvation.
Intermediary metabolism of patients afflicted with leucinecatabolic defects is severely dysregulated and highly vulnerableagainst catabolic stress. In order to avoid endogenous proteolysis andthus accumulation of leucine and leucine-degradative metabolites
before the pathway blockage, promoting anabolism is a cornerstoneof clinical treatment of these disorders [25]. Because nutrientsignaling via leucine is sensed more actively to mTORC1 in enzyme-deficient fibroblasts as compared to controls, this could translate toeffects of mTOR kinase in promoting translational initiation, ribosomebiogenesis and protein synthesis — all processes that contribute to amore anabolic metabolism. Enhanced mTOR activation and resistanceof MAP4K3 transcriptional down-regulation against amino acidrestriction in these cells might be seen as physiologically usefuladaptive mechanisms facilitating some functional compensation forthe metabolic disturbances due to incomplete leucine degradation.Such a regulatory circuit might serve to protect these cells againstdetrimental consequences of reduced nutrient utilization caused bytheir enzyme deficiencies. Besides in monogenic deficiencies ofleucine catabolism, this biological response may also play a roleduring metabolic inactivation of this highly regulated pathway, suchas in hyperinsulinemia or hyperthyroidism [23,24]. Our findingsestablish MAP4K3 and downstream mTOR-S6K1 signaling as novelregulators in impaired human leucine catabolism.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jnutbio.2012.04.018.
Acknowledgment
The authors acknowledge the excellent technical assistance ofBarbara Fink, Susanne Wullinger and Petra Eckel. We are grateful toJerry Vockley for providing the BCKDH-deficient cell line and JörnOliver Sass for the AUH-deficient cell line. We also thank MartinKlingenspor for discussion.
[2] Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. Amino acidregulation of TOR complex 1. Am J Physiol Endocrinol Metab 2009;296:E592–602.
[3] Wullschleger S, Loewith R, Hall MN. TOR signaling in growth andmetabolism. Cell2006;124:471–84.
[4] Tremblay F, Brule S, Hee Um S, Li Y, Masuda K, RodenM, et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance.Proc Natl Acad Sci U S A 2007;104:14056–61.
[5] Frost RA, Lang CH. mTOR signaling in skeletal muscle during sepsis andinflammation: where does it all go wrong? Physiology (Bethesda) 2011;26:83–96.
[6] Wang X, Proud CG. mTORC1 signaling: what we still don't know. J Mol Cell Biol2011;3:206–20.
[7] Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acidsufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a commoneffector mechanism. J Biol Chem 1998;273:14484–94.
[8] Iiboshi Y, Papst PJ, Kawasome H, Hosoi H, Abraham RT, Houghton PJ, et al. Aminoacid-dependent control of p70(s6k). Involvement of tRNA aminoacylation in theregulation. J Biol Chem 1999;274:1092–9.
[9] Lynch CJ, Fox HL, Vary TC, Jefferson LS, Kimball SR. Regulation of amino acid-sensitive TOR signaling by leucine analogues in adipocytes. J Cell Biochem2000;77:234–51.
[10] Lynch CJ, Halle B, Fujii H, Vary TC, Wallin R, Damuni Z, et al. Potential role ofleucine metabolism in the leucine-signaling pathway involving mTOR. Am JPhysiol Endocrinol Metab 2003;285:E854–63.
[11] Xu G, Kwon G, Cruz WS, Marshall CA, McDaniel ML. Metabolic regulation byleucine of translation initiation through the mTOR-signaling pathway bypancreatic beta-cells. Diabetes 2001;50:353–60.
[12] McDaniel ML, Marshall CA, Pappan KL, Kwon G. Metabolic and autocrineregulation of the mammalian target of rapamycin by pancreatic beta-cells.Diabetes 2002;51:2877–85.
[13] Beugnet A, Tee AR, Taylor PM, Proud CG. Regulation of targets of mTOR(mammalian target of rapamycin) signalling by intracellular amino acidavailability. Biochem J 2003;372:555–66.
[14] Christie GR, Hajduch E, Hundal HS, Proud CG, Taylor PM. Intracellular sensing ofamino acids in Xenopus laevis oocytes stimulates p70 S6 kinase in a target ofrapamycin-dependent manner. J Biol Chem 2002;277:9952–7.
[15] Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al.Bidirectional transport of amino acids regulates mTOR and autophagy. Cell2009;136:521–34.
831S.C. Schriever et al. / Journal of Nutritional Biochemistry 24 (2013) 824–831
[16] Hundal HS, Taylor PM. Amino acid transceptors: gate keepers of nutrientexchange and regulators of nutrient signaling. Am J Physiol Endocrinol Metab2009;296:E603–13.
[17] Kim E. Mechanisms of amino acid sensing in mTOR signaling pathway. Nutr ResPract 2009;3:64–71.
[18] Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, et al. Amino acidsmediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol3OH-kinase. Proc Natl Acad Sci U S A 2005;102:14238–43.
[19] Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator–Ragcomplex targets mTORC1 to the lysosomal surface and is necessary for itsactivation by amino acids. Cell 2010;141:290–303.
[20] Findlay GM, Yan L, Procter J, Mieulet V, Lamb RF. AMAP4 kinase related to Ste20 isa nutrient-sensitive regulator of mTOR signalling. Biochem J 2007;403:13–20.
[21] Tischler ME, Desautels M, Goldberg AL. Does leucine, leucyl-tRNA, or somemetabolite of leucine regulate protein synthesis and degradation in skeletal andcardiac muscle? J Biol Chem 1982;257:1613–21.
[22] Li F, Yin Y, Tan B, Kong X, Wu G. Leucine nutrition in animals and humans: mTORsignaling and beyond. Amino Acids 2011;41:1185–93.
[23] Doisaki M, Katano Y, Nakano I, Hirooka Y, Itoh A, Ishigami M, et al. Regulation ofhepatic branched-chain alpha-keto acid dehydrogenase kinase in a rat model fortype 2 diabetes mellitus at different stages of the disease. Biochem Biophys ResCommun 2010;393:303–7.
[24] Kobayashi R, Shimomura Y, Otsuka M, Popov KM, Harris RA. Experimentalhyperthyroidism causes inactivation of the branched-chain alpha-ketoaciddehydrogenase complex in rat liver. Arch Biochem Biophys 2000;375:55–61.
[25] Sweetman L, Williams JC. Branched chain organic acidurias. In: Scriver CR,Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases ofinherited disease. 8th ed. Quebec: McGraw-Hill, Inc.; 2001. p. 2125–64.
[26] Ensenauer R, Fingerhut R, Maier EM, Polanetz R, Olgemöller B, RöschingerW, et al.Newborn screening for isovaleric acidemia using tandemmass spectrometry: datafrom 1.6 million newborns. Clin Chem 2011;57:623–6.
[27] Stadler SC, Polanetz R, Maier EM, Heidenreich SC, Niederer B, Mayerhofer PU, et al.Newborn screening for 3-methylcrotonyl-CoA carboxylase deficiency: populationheterogeneity of MCCA andMCCBmutations and impact on risk assessment. HumMutat 2006;27:748–59.
[28] Shellmer DA, DeVito Dabbs A, Dew MA, Noll RB, Feldman H, Strauss KA, et al.Cognitive and adaptive functioning after liver transplantation for maple syrupurine disease: a case series. Pediatr Transplant 2011;15:58–64.
[29] Ensenauer R, Müller CB, Schwab KO, Gibson KM, Brandis M, Lehnert W. 3-Methylglutaconyl-CoA hydratase deficiency: a new patient with speech retarda-tion as the leading sign. J Inherit Metab Dis 2000;23:341–4.
[30] Ensenauer R, Hartl D, Vockley J, Roscher A, Fuchs U. Efficient and gentle siRNAdelivery by magnetofection. Biotech Histochem 2011;86:226–31.
[31] Melancon SB, Tayco J, Nadler HL. The free amino acid pool of cultivated humanskin fibroblasts. Proc Soc Exp Biol Med 1972;141:391–5.
[32] Moore S, SteinWH.Amodified ninhydrin reagent for the photometric determination ofamino acids and related compounds. J Biol Chem 1954;211:907–13.
[33] Tanaka K, Mandell R, Shih VE. Metabolism of [1-(14)C] and [2-(14)C] leucine incultured skin fibroblasts from patients with isovaleric acidemia. Characterizationof metabolic defects. J Clin Invest 1976;58:164–72.
[34] De Bandt JP, Cynober L. Therapeutic use of branched-chain amino acids in burn,trauma, and sepsis. J Nutr 2006;136:308S–13S.
[35] Shih VE, Mandell R, Levy HL, Littlefield JW. Free amino acids in extracts of culturedskin fibroblasts from patients with various amino acid metabolic disorders. ClinGenet 1975;7:421–5.
[36] She P, Reid TM, Bronson SK, Vary TC, Hajnal A, Lynch CJ, et al. Disruption of BCATmin mice leads to increased energy expenditure associated with the activation of afutile protein turnover cycle. Cell Metab 2007;6:181–94.
[37] Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners incrime? Semin Cancer Biol 2005;15:254–66.
[38] Bryk B, Hahn K, Cohen SM, Teleman AA. MAP4K3 regulates body size andmetabolism in Drosophila. Dev Biol 2010;344:150–7.