Page 1
Chapter II
58
CERIUM(IV) AMMONIUM NITRATE
Cerium(IV) ammonium nitrate (CAN) is one of the most important reagent,
which is used as one-electron oxidant and Lewis acid catalyst among the Lanthanide(IV)
complexes in organic synthesis.1 The reason for its general acceptance as a one-electron
oxidant and Lewis acid is due to its large reduction potential valve of +1.61 V vs NHE
(Normal Hydrogen Electrode) and low affinity for oxygen as compared to other Lewis
acids.2 CAN has been found chemically superior in many respects to widely employed
manganese triacetate for the generation of radicals.3 CAN has the additional advantages
of having a low toxicity besides being inexpensive, reasonably soluble in many organic
media, air stable, easily handled and allowing for a considerable degree of experimental
simplicity. A large number of research papers and several reviews have been published
for CAN mediated reactions.4-13
Cerium has a property, unique among the lanthanides, which explain its ability
to participate in one-electron transfer reactions, its ability to exist in two stable
adjacent oxidation states +3 and +4. Cerium in its ground state has an electronic
configuration of [Xe]4f26s2, where Xe represents the xenon configuration. The
electronic configuration of the Ce+3 ion is [Xe]4f1, while that of Ce+4 ion is [Xe]4f0.
The enhanced stability of the vacant f shell in Ce+4 accounts for the ability of cerium
to exist in the +4 oxidation state. The large reduction potential value of 1.61V (vs
NHE) makes Ce(IV) a very efficient oxidizing reagents compared to other metal ions.
Cerium(IV) ammonium nitrate is used as a catalyst for several types of
transformations. They are classified into the following categories:
1. Reactions involving the formation of carbon-carbon bond.
2. Reactions involving the formation of carbon-heteroatom bond.
3. Miscellaneous transformations.
Page 2
Chapter II
59
1. Reactions Involving the Formation of Carbon-Carbon Bond
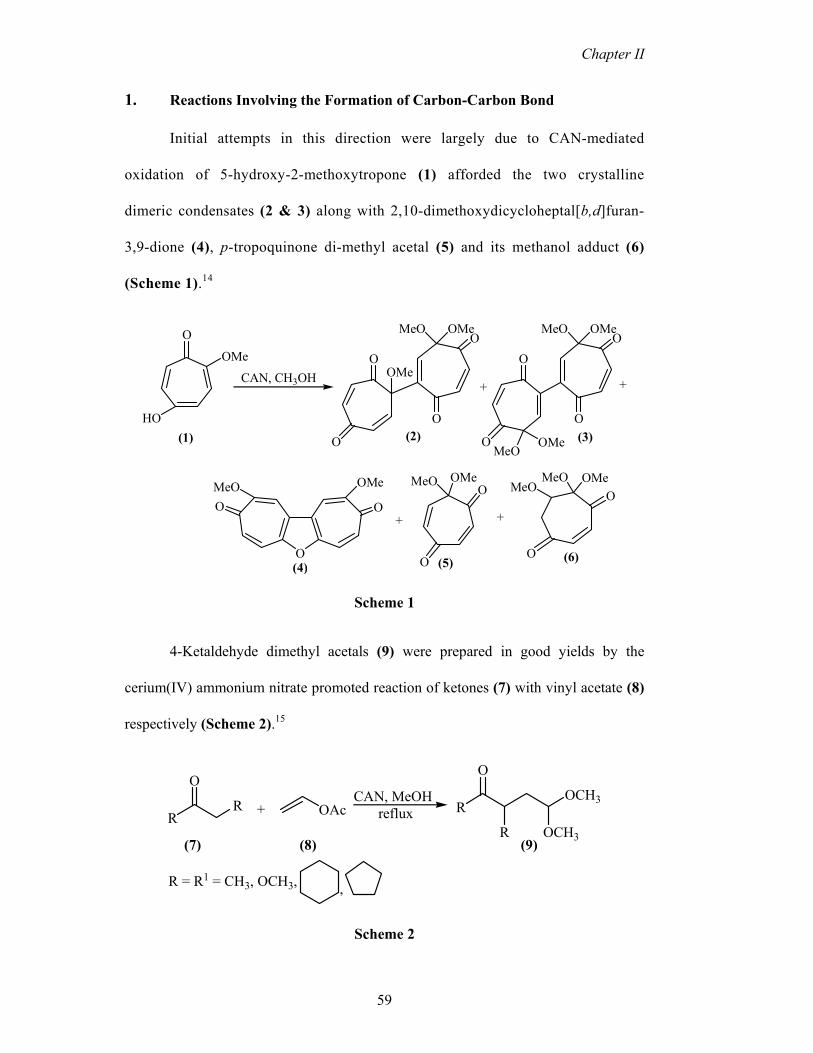
Initial attempts in this direction were largely due to CAN-mediated
oxidation of 5-hydroxy-2-methoxytropone (1) afforded the two crystalline
dimeric condensates (2 & 3) along with 2,10-dimethoxydicycloheptal[b,d]furan-
3,9-dione (4), p-tropoquinone di-methyl acetal (5) and its methanol adduct (6)
(Scheme 1).14
OMe
HO
O
(1)
CAN, CH3OH
O
OMeO
O
OMe
(2)
O
MeOO
OMeMeO
(4)
+
O
O
MeO OMe
(3)
+
+
O OMeMeO
O
O
OOMeMeO
+
O
O
O
OOMeMeO
OMe
(5) (6)
Scheme 1
4-Ketaldehyde dimethyl acetals (9) were prepared in good yields by the
cerium(IV) ammonium nitrate promoted reaction of ketones (7) with vinyl acetate (8)
respectively (Scheme 2).15
RR
O
(7) (8) (9)
+ OAcCAN, MeOH
reflux R
R OCH3
OCH3
O
R = R1 = CH3, OCH3, ,
Scheme 2
Page 3
Chapter II
60
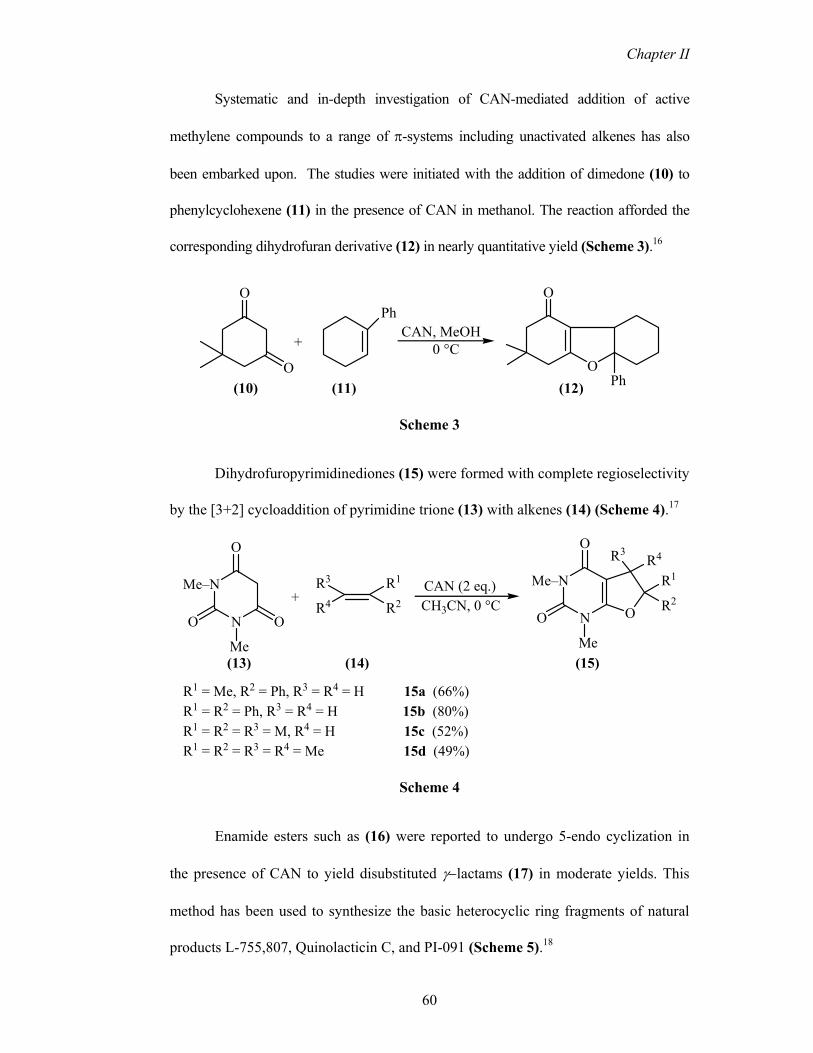
Systematic and in-depth investigation of CAN-mediated addition of active
methylene compounds to a range of π-systems including unactivated alkenes has also
been embarked upon. The studies were initiated with the addition of dimedone (10) to
phenylcyclohexene (11) in the presence of CAN in methanol. The reaction afforded the
corresponding dihydrofuran derivative (12) in nearly quantitative yield (Scheme 3).16
O
O
+
PhCAN, MeOH
0 °CO
Ph
O
(10) (11) (12)
Scheme 3
Dihydrofuropyrimidinediones (15) were formed with complete regioselectivity
by the [3+2] cycloaddition of pyrimidine trione (13) with alkenes (14) (Scheme 4).17
Me–N
NO
Me
O
+
O
R3
R4
R1
R2 CH3CN, 0 °CCAN (2 eq.) Me–N
NO
Me
O
O R2
R1R4R3
R1 = Me, R2 = Ph, R3 = R4 = H 15a (66%)R1 = R2 = Ph, R3 = R4 = H 15b (80%)R1 = R2 = R3 = M, R4 = H 15c (52%)R1 = R2 = R3 = R4 = Me 15d (49%)
(13) (14) (15)
Scheme 4
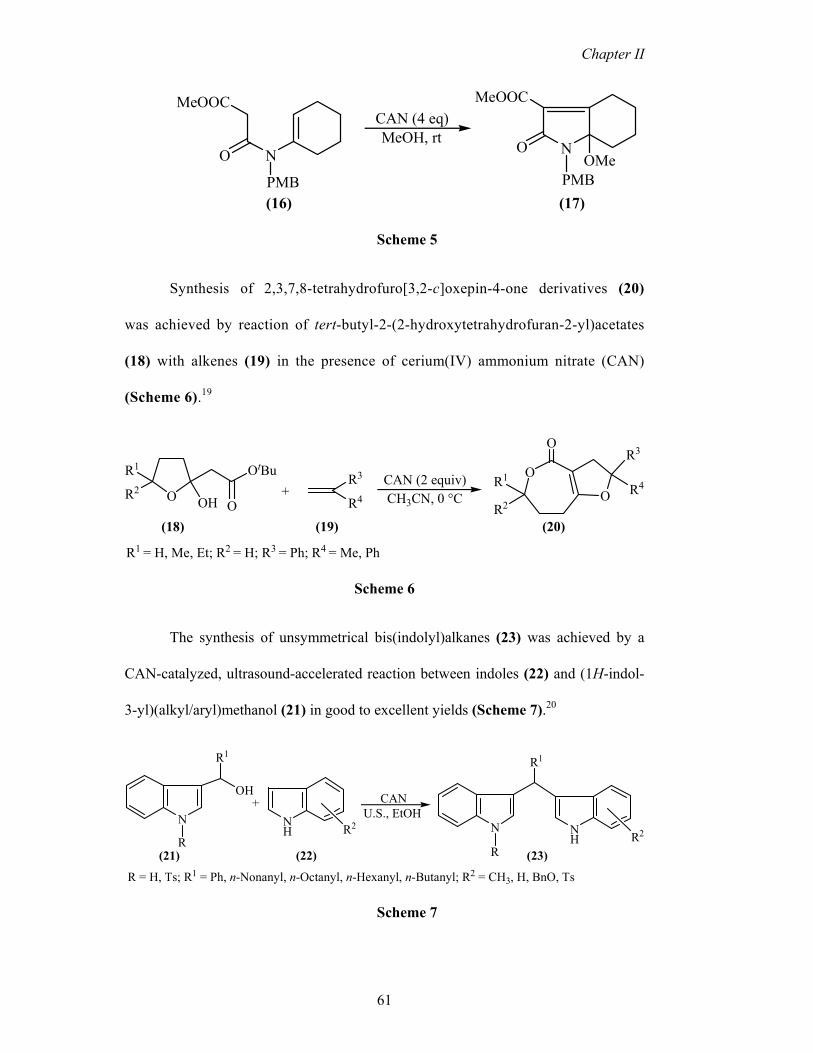
Enamide esters such as (16) were reported to undergo 5-endo cyclization in
the presence of CAN to yield disubstituted γ−lactams (17) in moderate yields. This
method has been used to synthesize the basic heterocyclic ring fragments of natural
products L-755,807, Quinolacticin C, and PI-091 (Scheme 5).18
Page 4
Chapter II
61
O
MeOOC
N
PMB
MeOH, rtCAN (4 eq)
(16) (17)
NO
MeOOC
PMBOMe
Scheme 5
Synthesis of 2,3,7,8-tetrahydrofuro[3,2-c]oxepin-4-one derivatives (20)
was achieved by reaction of tert-butyl-2-(2-hydroxytetrahydrofuran-2-yl)acetates
(18) with alkenes (19) in the presence of cerium(IV) ammonium nitrate (CAN)
(Scheme 6).19
O OH
OtBu
O
R1
R2 +R3
R4
CAN (2 equiv)CH3CN, 0 °C
R1 = H, Me, Et; R2 = H; R3 = Ph; R4 = Me, Ph
O
O
R3
R4R1
R2
O
(18) (19) (20)
Scheme 6
The synthesis of unsymmetrical bis(indolyl)alkanes (23) was achieved by a
CAN-catalyzed, ultrasound-accelerated reaction between indoles (22) and (1H-indol-
3-yl)(alkyl/aryl)methanol (21) in good to excellent yields (Scheme 7).20
NH
N
R
OH
R1
CANU.S., EtOH
N
R
R1
NH
+
(21) (22) (23)
R = H, Ts; R1 = Ph, n-Nonanyl, n-Octanyl, n-Hexanyl, n-Butanyl; R2 = CH3, H, BnO, Ts
R2R2
Scheme 7
Page 5
Chapter II
62
Ethylation of a variety of imines (24) at the methine moiety was accomplished
by CAN-promoted coupling of imines with triethyl aluminium to give the
corresponding products (25) (Scheme 8).21
Ar1 NAr2
CAN (1 equiv.), Et3Al (3 equiv.)C6H6, 25 oC, H2O
Ar1
NH
Ar2
(24) (25)
Ar1 = Ar2 = Ph; Ar1 = 4-MeC6H4, Ar2 = Ph; Ar1 = Ph, Ar2 = 4-MeOC6H4Ar1 = 4-ClC6H4, Ar2 = Ph; Ar1 = Ph, Ar2 = 4-ClC6H4; Ar1 = 4-BrC6H4, Ar2 = PhAr1 = 3-HOC6H4, Ar2 = Ph; Ar1 = 2-C5H4N, Ar2 = Ph; Ar1 = Ph, Ar2 = 2-C10H7Ar1 = 4-NCC6H4, Ar2 = Ph
Scheme 8
Perumal et al. has demonstrated that CAN catalyzed 1,4-addition of pyrimidin-
2(1H)-ones (26) with several nucleophiles (27) in methanol to furnish the highly
functionalized 3,4-dihydropyrimidin-2(1H)-ones (28) in very good yields (Scheme 9).22
N
NHR
O
O
Ar
H
Nu HCe(NH2)2(NO3)6 (5 mol%)
MeOH, rt, 1-3 hN
NHR
O
O
Ar
Nu
+
(26) (27) (28)
R = CH3CO, EtOOC; Ar = C6H5, o-O2NC6H4, o-ClC6H4
NH
NH
NCH3CH3
NH
Ph N Ph
NH
Br
,
,, ,Nu = ,
Scheme 9
α-dehydro-β-amino esters (31) were synthesized from acetates of Baylis-
Hillman adducts (29) with amines (30) in the presence of a catalytic amount of CAN
in tetrahydrofuran (THF) at 60-65 °C. (Scheme 10).23
Page 6
Chapter II
63
ArCOOMe
OAc
R NH2Ar
NHR
COOMe+ CAN (10 mol%)
THF, 60-65 oC, 3 h
(29) (30) (31)
Ar = Ph, 4-MeC6H4, 4-ClC6H4R = Bn, Ph, 4-MeC6H4, 4-MeOC6H4, 3-Cl-4-MeC6H3, 3-O2NC6H4, n-C8H17, n-C16H33, β-Naphthyl, cyclo-C6H11
Scheme 10
The synthesis of 4,4'-(arylmethylene)bis(1H-pyrazol-5-ols) (34) was
accomplished by tandem Knoevenagel–Michael reaction of two equivalents of 5-
methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one (32) with various aromatic aldehydes
(33) catalyzed by ceric ammonium nitrate (CAN) in water at room temperature
(Scheme 11).24 All these compounds were evaluated for in vitro antiviral activity
against peste des petits ruminant virus (PPRV).
NN
H3C
O
R H
OCAN (5 mol%)
H2O, rt
NN
H3C
OH
N
N
OH
H3CR
(32) (33) (34)R = C6H5, 3-H3CC6H4, 4-H3CC6H4, 4-CH3OC6H4, 4-FC6H4, 4-O2NC6H4, 3,4-(OCH3)2C6H3, 3-CH3O-4-HOC6H3, 2-Furfuryl, 2-Pyridyl
+
Scheme 11
2. Reactions Involving the Formation of Carbon-Heteroatom Bond
Carbon-heteroatom bond formation assumes much significance, especially
from the vantage point of heterocyclic compound formation. Recently, there has been
a flurry of activity towards developing novel methods for attaining this goal. The
Page 7
Chapter II
64
utility of CAN in carbon-heteroatom bond formation, particularly C-O, C-S, C-N, C-
Se, C-Br and C-I bonds, is noteworthy in this connection. Mostly these reactions
involve the oxidative addition of heteroatom-centered radicals, formed by the
oxidation of anions by CAN, to alkenes and alkynes.
In his seminal work, which marked the beginning of the use of CAN for the
construction of carbon-heteroatom bonds, Huston has demonstrated that CAN
promotes a facile addition of azide radicals to olefins such as stilbene and
acenaphthene (35) to afford trans-α-azido-β-nitroalkanes (36) (Scheme 12).25
NaN3, CAN
N3
O2NO
CH3CN, rt
(35) (36)
H
H
Scheme 12
Nitration of naphthalene derivatives (37) was carried out with silica gel
supported CAN to give corresponding nitrated products (38) (Scheme 13).26
CAN on silica gel
OR
X2(37) (38)
ORX1
R = H, X1 = H, X2 = NO2; R = CH3, X1 = H, X2 = NO2R = H, X1 = NO2, X2 = H; R = C2H5, X1 = H, X2 = NO2
Scheme 13
Reaction of α,β−unsaturated ketones and esters (39) with iodine and CAN in
methanol, ethanol, or isopropyl alcohol, under reflux conditions afforded the
corresponding β-alkoxy-α-iodoketones and esters (40) in good yields (Scheme 14).27
Page 8
Chapter II
65
OEt
OCAN, I2, MeOH
OEt
OOMe
I(39) (40)
Scheme 14
Reaction of substituted styrenes (41) with ammonium thiocyanate (NH4CN)
(42) was mediated by CAN as catalyst to give the corresponding dithiocyanates (43)
in acetonitrile at room temperature (Scheme 15).28
R rt, 15 minCAN, CH3CN
R
SCN
SCN
(41) (42) (43)
+ NH4SCN
R = CH3, OCH3, COCOCH3, R-C6H4 = ,
Scheme 15
3-(selenocyanato)-indoles (46) were synthesized by the reaction of indoles
(44) with potassium selenocyanate (KSeCN) (45) in methanol at room temperature
(Scheme 16).29
N
R1
R2
(44) (46)
KSeCN (45)N
R1
R2
SeCN
R1 = H, R2 = CH3; R1 = CH3, R2 = H; R1 = H, R2 = C6H5
CAN/MeOH/0 oC
Scheme 16
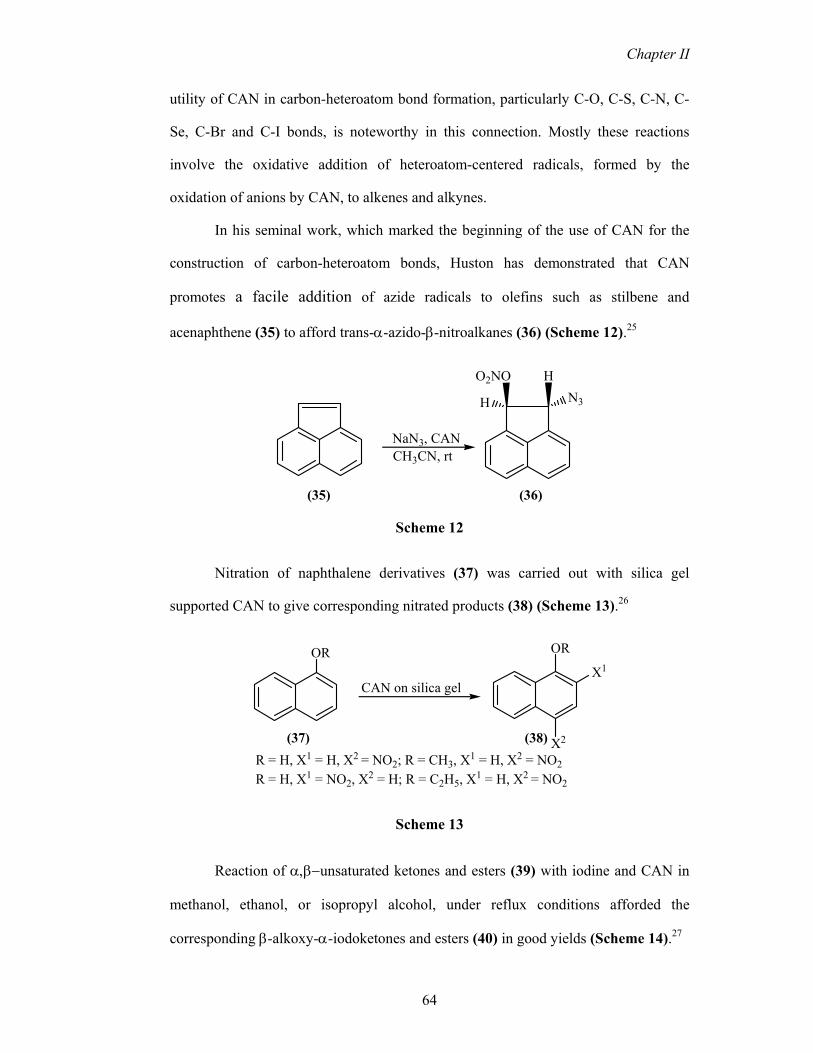
The reaction of styrene (47) with p-toluenesulfinate (48) was catalyzed by ceric
ammonium nitrate (CAN) to give the corresponding α-sulfinato-β-nitrato product (49)
and β-keto-sulfone (50) in anhydrous acetonitrile at room temperature (Scheme 17).30
Page 9
Chapter II
66
O2S
ONO2
Me
O2S
Me
O
SO2Na
Me
CAN, CH3CNrt, 40 min
+
(49)
(50)
(47) (48)
+
Scheme 17
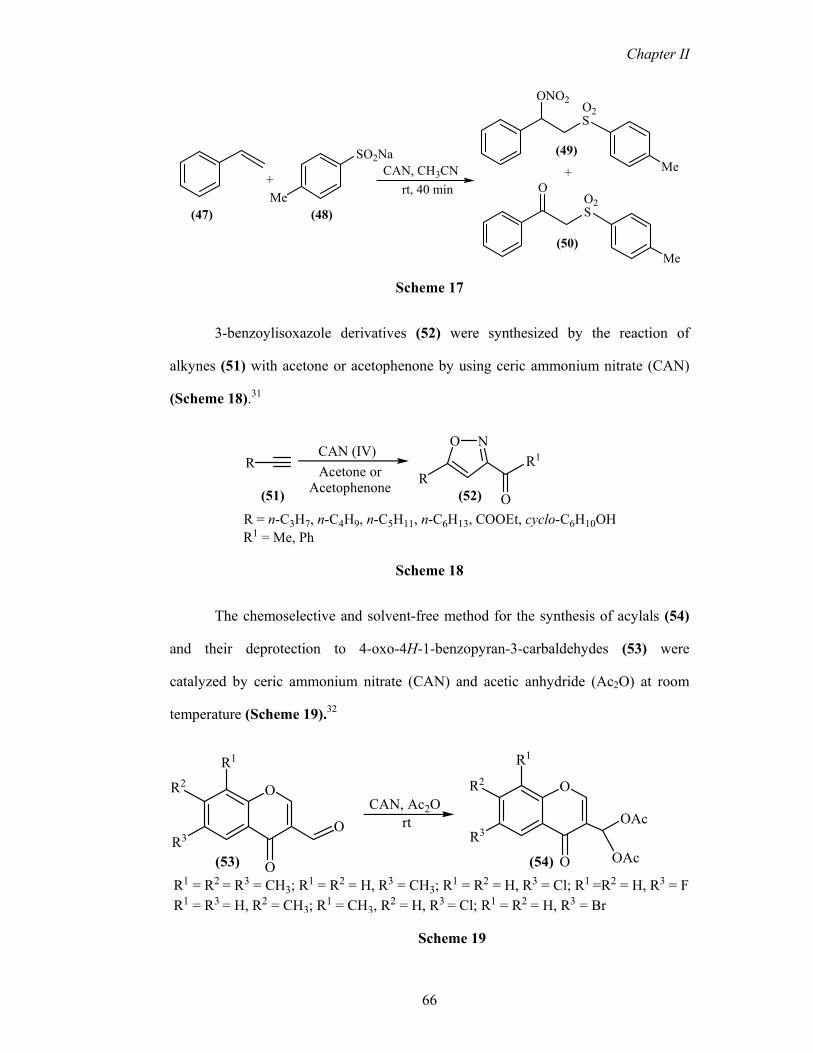
3-benzoylisoxazole derivatives (52) were synthesized by the reaction of
alkynes (51) with acetone or acetophenone by using ceric ammonium nitrate (CAN)
(Scheme 18).31
RCAN (IV)Acetone or
Acetophenone
NO
RR1
O(51) (52)
R1 = Me, PhR = n-C3H7, n-C4H9, n-C5H11, n-C6H13, COOEt, cyclo-C6H10OH
Scheme 18
The chemoselective and solvent-free method for the synthesis of acylals (54)
and their deprotection to 4-oxo-4H-1-benzopyran-3-carbaldehydes (53) were
catalyzed by ceric ammonium nitrate (CAN) and acetic anhydride (Ac2O) at room
temperature (Scheme 19).32
OR2
R3
R1
O
O
O
OAc
OAc
R2
R1
R3
O
CAN, Ac2O
(53) (54)
rt
R1 = R2 = R3 = CH3; R1 = R2 = H, R3 = CH3; R1 = R2 = H, R3 = Cl; R1 =R2 = H, R3 = FR1 = R3 = H, R2 = CH3; R1 = CH3, R2 = H, R3 = Cl; R1 = R2 = H, R3 = Br
Scheme 19
Page 10
Chapter II
67
Ceric ammonium nitrate (CAN) in PEG was used as an efficient and
recyclable solvent system for one-pot three-component Mannich reaction of
acetophenone (55) with aromatic aldehydes (56) and aromatic amines (57) to give the
corresponding β-amino carbonyl compounds (58) at 45 °C (Scheme 20).33
CH3
CHO
R1 R2
NH2HNO
R2
R1
O
+ + CAN (5 mol%)PEG, 45 oC
R1 = H, 4-CH3, 4-OCH3, 4-NO2, 4-Br; R2 = H, 4-CH3, 3,4-(CH3)2, 4-Cl, 4-NO2, 2-NO2
(55) (56) (57) (58)
Scheme 20
2,4,5-Triarylimidazoles (61) were obtained in excellent yields by the one-
pot three-component condensation of benzil/benzoin (59), aldehydes (33) and
ammonium acetate (60) in the presence of catalytic amount of cerium (IV)
ammonium nitrate (CAN) in aqueous media under ultrasound at room temperature
(Scheme 21).34
O
O NH
N
RNH4OAc (60), CAN (5 mol%)Ultrasonication, rt
+
(59) (33) (61)R = C6H5, 2-ClC6H4, 4-ClC6H4, 4-MeC6H4, 4-MeOC6H4, 3,4-(MeO)2C6H3, 4-O2NC6H4, 4-(Me)2NC6H4, 4-HOC6H4, 4-FC6H4, 2-Furyl, 2-Thienyl
RCHO
Scheme 21
Page 11
Chapter II
68
The one-pot synthesis of N-substituted decahydroacridine-1,8-diones (63) was
reported by the reaction of 1,3-dicarbonyl compounds (62) with aromatic aldehydes
(56) and aromatic amines (57) in the presence of cerium(IV) ammonium nitrate
(CAN) as the catalyst in polyethylene glycol (PEG) at 50 °C (Scheme 22).35
R1
R2
O
N
R1
R2
R1
R2
R4
R3
CHO NH2
R3 R4
CAN (5 mol%), PEG 40050 oC, 4 h
+ +
(62) (56) (57) (63)
R1 = R2 = R3 = R4 = H; R1 = R2 = R4 = H, R3 = 4-OCH3, 4-Cl, 3-NO2; R1 = R2 = CH3, R3 = R4 = H R1 = R2 = R3 = H, R4 = 4-Br, C6H5CH2; R1 = R2 = CH3, R4 = H, R3 = 4-OCH3, 4-Cl, 3-NO2
2
O O
Scheme 22
3. Miscellaneous transformations
The serendipitous discovery of some novel and interesting processes turned
out to be one of the exciting aspects of the pursuit of Ce(IV) chemistry. Such
reactions also provide additional insight into the mechanistic details of several CAN-
mediated transformations.
Dethioacetalisation procedure for the transformation of dithioacetals such as
(64) into the parent carbonyl compound (65) by employing CAN in aqueous
acetonitrile at room temperature was developed (Scheme 23).36
SS
CAN, aq. CH3CNrt, 3 min
O
(64) (65)
Scheme 23
Page 12
Chapter II
69
Treatment of acetoacetanilides (66) with CAN in methanol, in
anticipation of an intramolecular reaction to derive the oxindole, nevertheless
afforded the corresponding oxamates (67) in good yield. Substantial
enhancement of the overall yield occurred in an atmosphere of oxygen
(Scheme 24).37
NH
O
O
CAN, MeOH, O2rt, 15 min
NH
COOMe
O
(66) (67)
R3
R2
R1 R1
R2
R3
R1 = R2 = R3 = H; R1 =R2 = H, R3 = CH3; R1 = R2 = H, R3 = OCH3; R1 = R2 = H, R3 = ClR2 = R3 = H, R1 = CH3; R1 = OCH3, R2 = R3 = H; R1 = COOCH3, R2 = R3 = H R1 = R3 = H, R2 = Br
Scheme 24
Hwu and co-workers have reported a highly regioselective, silicon directed
C-C bond cleavage of β-(trimethylsilyl)cycloalkanones (68) to afford the
β-alkenyl carboxylic acids (69) in the presence of CAN and acetonitrile
(Scheme 25).38
R1
R2n
O
CAN, CH3CN, H2O60 °C, 30 min, 2 h
OH
n
O
n = 1, R1 = H, R2 = SiMe3; n = 2, R1 = H, R2 = SiMe3 n = 2, R1 = CH2SiMe3, R2 = H; n = 3, R1 = CH2SiMe3, R2 = H
(68) (69)
Scheme 25
Page 13
Chapter II
70
The reaction of monoterpenes such as (+)-α-pinene (70) with CAN in
acetonitrile afforded the bisamide (71) in good yields (Scheme 26).39
CAN (2.3 equiv)CH3CN, rt, 3 h
HN CH3
NH
CH3
OO
(70)
(71)
Scheme 26
The cerium(IV) ammonium nitrate (CAN) promoted oxidation of 4,5-
diphenyloxazoles (72) in to the corresponding imides (73) in good yields was reported
(Scheme 27).40 This reaction will allow the use of robust oxazole ring system as a
surrogate amide in organic synthesis.
CAN (3.8 eq)CH3CN-H2O Ph N R
H
O O
+
(72) (73) (74)
R = Et, i-Pr, i-Bu, Cy, Bn, t-Bu,
ClMe
,O ,
N
OPh
Ph R PhCOOH
Scheme 27
Three component domino reaction between α,β-unsaturated aldehydes (75),
aromatic amines (76) and ethyl acetoacetate (77) were carried out using CAN as
catalyst in methanol as solvent to give the desired product 1,4-dihydropyridines (78)
at room temperature (Scheme 28).41
Page 14
Chapter II
71
O
+
R4
NH2
+
CH3
Z
O
O
EtOH, rt, 1 hN CH3
Z
O
R4
CAN (5 mol%)R1
R3
R2
R5
R3
R2
R5
R1
(75) (76) (77)
(78)R1 = R2 = R3 = R4 = R5 = H, Z = OC2H5; R3 = CH3, R1 = R2 = R4 = R5 = H, Z = OC2H5R3 = F, R1 =R2 = R4 = R5 = H, Z = OC2H5; R3 = Cl, R1 =R2 = R4 = R5 = H, Z = OC2H5R2 = CH3, R3 = R1 = R4 = R5 = H, Z = OC2H5; R2 =R3 = CH3, R1 = R4 = R5 = H, Z = OC2H5R2 =Cl, R1 = R3 = R4 = R5 = H, Y = OC2H5; R1 = R2 = R3 = R4 = R5 = H, Y = S-C(CH3)3R1 = R2 = R3 = R4 = R5 = H, Y = O-C(CH3)3
Scheme 28
The oxidation of selected metal anions (M+X-) (80) by 2 equivalent of CAN in
the presence of substituted cyclopropyl alcohols (79) provided a novel approach to β-
functionalized ketones (81) (Scheme 29).42
R OHM+X-(80), solvent,
rt, 30 minR X
O
M = K+, Na+, NH4+; X = Br–, I–, N3
–, SCN–
R = Ph, p-CH3OC6H4, Cyclohexyl, CH3
(79) (81)
2 CAN
Scheme 29
CAN catalyzed sequential, one pot reaction between alkylamines (30),
chalcones (82) and β-ketoesters (83) afforded cis-4,6-disubstituted-2-
alkylaminocyclohexene-1-carboxylic esters (84) with complete diastereoselectivity
(Scheme 30).43
Page 15
Chapter II
72
R1–NH2 + O
Ph
Ar
+
O
O
OR2
CH3
EtOH, rtCAN (5%)
HO Ar
Ph
OR2O
NH
R1
R1 = n-Bu, n-C6H13, n-C7H15, (±)-2-Me-Bu, (S)-2-Me-Bu, (±)-sec-Bu, (R)-sec-BuR2 = Et, tBu; Ar = Ph, 4-ClC6H4
(30) (82) (83) (84)
Scheme 30
A variety of highly substituted 3,4-dihydroquinoxalin-2-amine derivatives (88)
were efficiently synthesized by the reaction of o-phenylenediamines (85), ketones
(86) and isocyanides (87) using cerium(IV) ammonium nitrate (CAN) as catalyst in
ethanol at room temperature (Scheme 31).44
NH2
NH2
R1
R2 R3
OR4 N C+ +
N
HN R2
R3
NH
R4R1CAN (5 mol%)
EtOH, rt
(85) (86) (87) (88)R1 = H, 4-NO2, 4-C6H5CO, 5-CH3, 2-NO2; R2 = R3 = CH3R4 = t-Butyl, Cyclohexyl, 2,6-Dimethylphenyl, Benzyl, 1,1,3,3-Tetramethylbutyl
Scheme 31
Ceric ammonium nitrate (CAN) was found to catalyze the one-pot synthesis of
2,2,4-trimethyl-1,2-dihydroquinoline derivatives (90) from substituted anilines (57)
and acetone (89) via a modified Skraup reaction at 50-55 °C (Scheme 32).45
HN
H3C CH3
ONH2
R
CH3
CH3
CH3RCAN, 50-55 C
20-24 h°
(57) (89) (90)R = 4-NO2, 4-MeO, 4-F, 4-Me, 4-CF3, 4-Cl, 4-Br
+
Scheme 32
Page 16
Chapter II
73
Ceric ammonium nitrate (CAN) was found to be an effective catalyst for the
reaction of 1,3-dicarbonyl compounds (62) with aromatic aldehydes (33) and
ammonium acetate (91) by using polyethylene glycol as reaction medium to give the
corresponding decahydroacridine-1,8-diones (92) at 25 °C (Scheme 33).46
R1 H NH
R1
R
R
RR
RR
O
O
O
OO
NH4OAcCAN (5 mol%)
PEG, 25 oC++
(62) (33) (91) (92)R = H, CH3 R1 = C6H5, 2-MeOC6H4, 4-MeOC6H4, 4-ClC6H4, 4-HOC6H4, 2-HOC6H4, 3-O2NC6H4, 2-Thienyl
Scheme 33
An environment friendly method for the synthesis of 2-
oxo/thioxooctahydroquinazolin-5-ones (95) and 2-oxo/thioxo-7,7-
dimethyloctahydroquinazolin-5-ones (96) was reported by the reaction of 5,5-
dimethyl-1,3-cyclohexanedione (10) or 1,3-cyclohexanedione (93) with aldehydes
(33) and urea or thiourea (94) using ceric ammonium nitrate (CAN) as catalyst and
polyethylene glycol (PEG) as solvent at 50 °C (Scheme 34).47
R H H2N NH2
XH3C
H3C
O
O
O
O
O NH
NH
NH
NH
X
X
O
O R
R
H3CH3C
R H H2N NH2
XOCAN (5 mol%)PEG 400, 50 C°
CAN (5 mol%)PEG 400, 50 C°
+ +
++
(93) (33) (94)
(10) (33) (94) (96)
X = O, S; R = C6H5, 4-CH3OC6H4, CH2CH3, C3H8,O
O
(95)
Scheme 34
Page 17
Chapter II
74
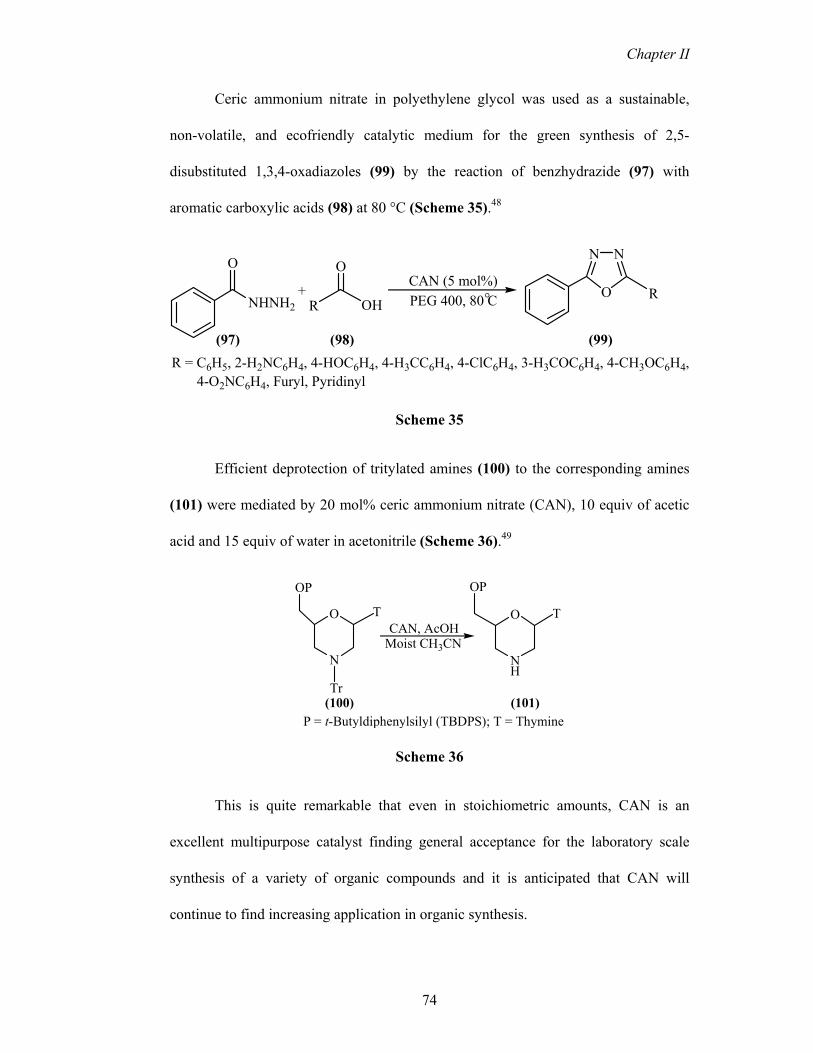
Ceric ammonium nitrate in polyethylene glycol was used as a sustainable,
non-volatile, and ecofriendly catalytic medium for the green synthesis of 2,5-
disubstituted 1,3,4-oxadiazoles (99) by the reaction of benzhydrazide (97) with
aromatic carboxylic acids (98) at 80 °C (Scheme 35).48
NHNH2 R OH
OO
+CAN (5 mol%)
NN
O RPEG 400, 80 C°
(97) (98) (99)R = C6H5, 2-H2NC6H4, 4-HOC6H4, 4-H3CC6H4, 4-ClC6H4, 3-H3COC6H4, 4-CH3OC6H4, 4-O2NC6H4, Furyl, Pyridinyl
Scheme 35
Efficient deprotection of tritylated amines (100) to the corresponding amines
(101) were mediated by 20 mol% ceric ammonium nitrate (CAN), 10 equiv of acetic
acid and 15 equiv of water in acetonitrile (Scheme 36).49
N
O
NH
O
OP
T T
OP
Tr
CAN, AcOHMoist CH3CN
(100) (101)P = t-Butyldiphenylsilyl (TBDPS); T = Thymine
Scheme 36
This is quite remarkable that even in stoichiometric amounts, CAN is an
excellent multipurpose catalyst finding general acceptance for the laboratory scale
synthesis of a variety of organic compounds and it is anticipated that CAN will
continue to find increasing application in organic synthesis.
Page 18
Chapter II
75
DITHIOCARBAMATES
The discovery of the dithiocarbamates early in the history of organosulfur
compounds50 led to important uses in the rubber industry.51 Their profound effects
on biological systems due to their metal-combining capacity52 and their ability to
interact with sulfhydryl-containing compounds have practical applications in the
fields of medicinal chemistry53 and agriculture.54 Of the many dithiocarbamates
synthesized and studied, mainly by the agricultural chemical industry, a few have
been widely used as fungicides. The biological activity of the dithiocarbamates is
increased when they are in the form of heavy-metal salts as versatile classes of
ligands with the ability to stabilize transition metals in a wide range of their
oxidation states. They are also efficient ligands in surface science and
nanomaterial chemistry.55 Furthermore, they are useful building blocks for the
synthesis of biologically active heterocyclic compounds and solid support grafting
materials.56
S-[4,5-dichloro-6(1H)-pyridazinon-1-ylmethyl]-N,N-dialkyldithiocarbamates
(102) were prepared and tested for their fungicidal and acaricidal activities against
Sclerotiniafructicola and Tetranychusurticae.57
NN
H2C S C
S
NR1
R2
Cl
Cl
O
R1 = R2 = Me, Et
(102)
5-(N,N-dimethyldithiocarbamoylmethyl)picolinic acids or amides (103) was
reported as anti-hypersensitive agents.58
Page 19
Chapter II
76
N
CH2
S C
S
NMe2
R
O
R = OH, MeO, NH2, NHMe, NMe2
(103)
Sodium salts of N,N-diethyldithiocarbamate (104) were injected into the renal
artery of dogs did not affect tubular resorption but inhibited glomerular filtration and
evaluated as antidiuretic agents.59
N
C
C2H5 C2H5
SHS
Na
(104)
A series of N-((2-methyl-4(3H)-quinazolinon-6-yl)methyl)dithiocarbamates
(105) were synthesized and evaluated for their cytotoxic activity against five human
cancer cell lines.60
HN
N
O
H3C
NH
SR
S
(105)
R = CH3, CH3CH2, CH3CH2CH2, (CH3)2CH, CH3(CH2)2CH2, CH3(CH2)3CH2, CH3(CH2)4CH2, CyclohexylCH2, Ph2C(CN)CH2CH2, PhCH2, 4-H3CC6H4CH2, 4-FC6H4CH2, 4-BrC6H4CH2, 2,4-F2C6H3CH2, F5C6CH2, 4-O2NC6H4CH2, 4-CNC6H4CH2, 4-HO2CC6H4CH2, Ph2CH, 4-EtO2CC6H4CH2, H2C=CHCH2-, HC=CCH2
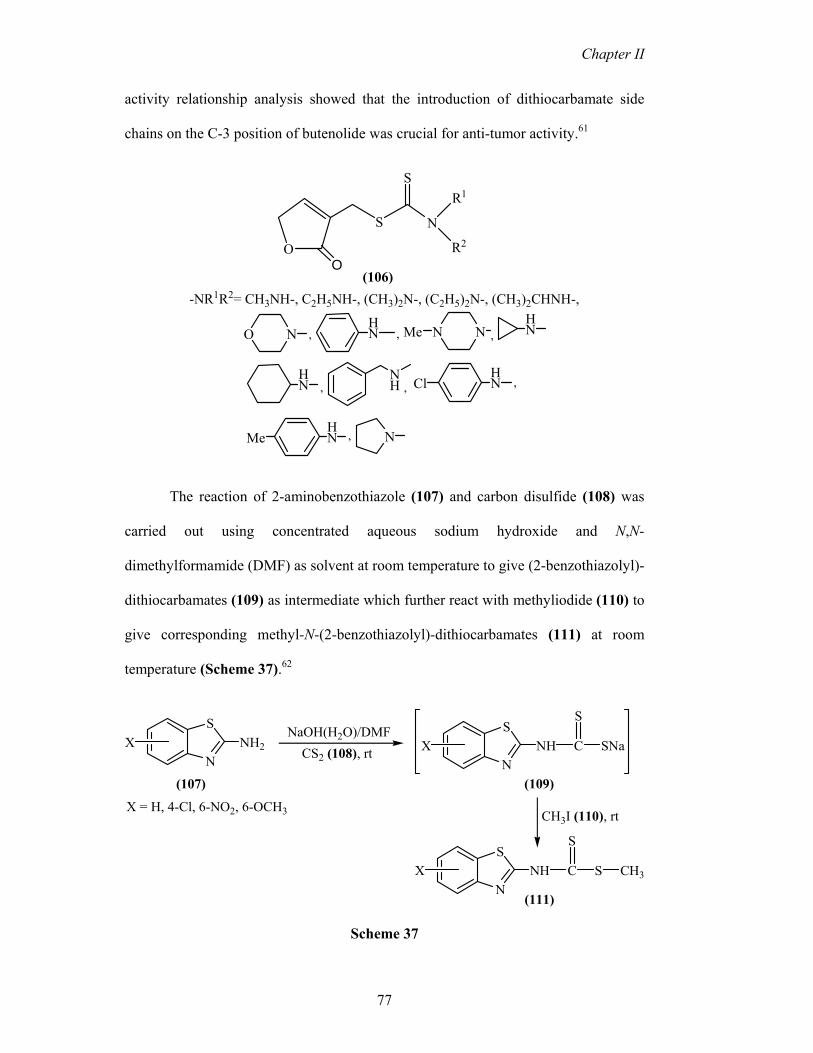
Synthesis and in vitro antitumor activity of new butenolide-containing
dithiocarbamates (106) were evaluated. These compounds exhibited broad spectrum
anti-cancer activity against five human cancer cell lines with IC50 < 30 lM. Structure-
Page 20
Chapter II
77
activity relationship analysis showed that the introduction of dithiocarbamate side
chains on the C-3 position of butenolide was crucial for anti-tumor activity.61
OO
S N
R1
R2
S
(106)-NR1R2= CH3NH-, C2H5NH-, (CH3)2N-, (C2H5)2N-, (CH3)2CHNH-,
N
N NMeHN
HN
HN
Me
ClNH
O N
HN
HN, , ,
,,
,
,
The reaction of 2-aminobenzothiazole (107) and carbon disulfide (108) was
carried out using concentrated aqueous sodium hydroxide and N,N-
dimethylformamide (DMF) as solvent at room temperature to give (2-benzothiazolyl)-
dithiocarbamates (109) as intermediate which further react with methyliodide (110) to
give corresponding methyl-N-(2-benzothiazolyl)-dithiocarbamates (111) at room
temperature (Scheme 37).62
N
SNH2X
NaOH(H2O)/DMFCS2 (108), rt
N
SNHX C SNa
S
(107) (109)
N
SNHX C S
S
CH3
CH3I (110), rt
(111)
X = H, 4-Cl, 6-NO2, 6-OCH3
Scheme 37
Page 21
Chapter II
78
Bis(dithiocarbamate) derivatives of glycerol (114) were obtained by reaction
of dithiocarbamic acid salt (113) with 1,3-dichloro-1,3-dideoxyglycerol (112) in
acetone under reflux condition, which was evaluated as antifungal activity against
Alternaria brassicae, Pseudocercosporella herpotrichoides, Septoria nodorum and
Phytophtora cinnamomi (Scheme 38).63
Cl
Cl
OHR1R2NCSSLi (113) (3 eq.)
Acetone, reflux
N S
S
NR1R2S
S
OH
R1
R2
(112) (114)
R1 = R2 = Ethyl; NR1R2 = Morpho-4-yl; NR1R2 = 1-Piperidyl
Scheme 38
Dithiocarbamate derivatives (117) were prepared by a simple one-pot
procedure from primary or secondary amines (115) carbon disulfide (108) and a
variety of alkyl halides (116) in the presence of anhydrous potassium phosphate under
mild condition in good yields (Scheme 39).64
RNH
R1CS2 R2 X++
K3PO4/CH3COCH310 oC~rt
RNCS2R2
R1
(115) (108) (116) (117)
RR1NH = 1-Methylpiperazine; R2X = C2H5Br, i-C3H7Br, n-C4H9Br, HOCH2CH2Cl, NCCH2Cl, H2C=CHCH2Cl, BrCH2CO2C4H9RR1NH = C6H5CH2NH2; R2X = C2H5Br, C6H5CH2Cl, HOCH2CH2ClRR1NH = 1-Benzoylpiperazine; R2X = 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide, 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide
Scheme 39
Page 22
Chapter II
79
A novel synthesis of functional dithiocarbamates (120) was reported by the
reaction of bis(thiocarbonyl) disulfides (118) with azo-compounds (119) in ethyl
acetate under reflux condition (Scheme 40).65
Z C S S C
S
Z
S+ N N RR
Ethyl acetatereflux, 18 hr Z C S R
S
(118) (119) (120)
Z = C6H5, R = C(CH3)(CN)(CH2CH2COOH), C(CH3)2CN; Z = CH3CH2O, R = C(CH3)2CN
Scheme 40
S-alkyl N,N-dimethyldithiocarbamates (123) were synthesized by the reaction
of N,N-dimethylthiocarbamoyl chloride (121) with the corresponding thiolates (122)
(Scheme 41).66
(H3C)2N Cl
S+ R E M
(H3C)2N ER
S
E = S; M = Li or Na; R = C6H5, C6H5CH2, 4-MeC6H4, 3-MeC6H4, 2-ClC6H4
(121) (122) (123)
Scheme 41
Reactions of 1-(1-hydroxyalkyl)benzotriazoles (125) with alkyl N-
alkyldithiocarbamates (124) gave intermediate N-[1-(benzotriazol-1-yl)alkyl]
dithiocarbamates (126), which further gave N-(1-sulfanylalkyl) dithiocarbamates
(128) or N-[1-(dialkylphosphono)alkyl] dithiocarbamates (130) on treatment with
thiols (127) or trialkyl phosphates (129) respectively, in the presence of zinc bromide
(Scheme 42).67
Page 23
Chapter II
80
RNH
SR3
SOH
R1 Bt
TsOHToluenefeflux
+R
SR3
Bt R1
S
P(OR2)3 (129)ZnBr2
R2SH(127)
RN S
R3
R1R2S
S
R3
SN
R1P
OR2O
R2O
SR
(124) (125) (126)
(128)
(130)(126): R = Me, R1 = H, R3 = Et; R = Me, R1 = Pr, R3 = Et; R = Et, R1 = H, R3 = Me; R = Et, R1 = Pr, R3 = Me; R = Bu, R1 = Pr, R3 = Et; R = Bu, R1= Pr, R3 = Et(128): R = Me, R1 = Pr, R2 = Ph, R3 = Et; R = Et, R1 = Pr, R2 = Ph, R3 = Me; R = Et, R1 = Pr, R2 = Et, R3 = Me; R = Bu, R1 = Pr, R2 = Ph, R3 = Et; R = Bu, R1 = Pr, R2 = R3 = Et(130): R = R2 = R3 = Et, R1 = Pr; R = Me, R1 = Pr, R2 = i-Pr, R3 = Et; R = Et, R1 = Pr, R2 = Et, R3 = Me; R = Et, R1 = Pr, R2 = i-Pr; R3 = Me
ZnBr2
Scheme 42
A simple protocol was reported for the one-pot synthesis of various
structurally divergent dithiocarbamates (132 & 133) in one-pot reaction of amines
(115), carbon disulfide (108) and styrene oxide (131) without using any solvent and
catalyst at room temperature was described (Scheme 43).68
RR1NHO
Ph+CS2 (108), rt
1-4 h R1RN S
OH
PhR1RN S
OH
PhS S
+
(115) (131) (132) (133)
RR1NH = NH NHNH2
NH2NH2
NH, , , , ,
Scheme 43
An efficient, Amberlite IRA 400 (basic resin) mediated, one-pot synthesis of
dithiocarbamates (135) were accomplished in high yields by the reaction of amines
(115), carbon disulfide (108) and α,β-unsaturated compounds (134) via Michael
addition. (Scheme 44).69
Page 24
Chapter II
81
R1
NHR2
CS2
R3
EWG++ Amberlite IRA 400, Dry DMSO
rt, 2-4 h
R1
N SEWG
R3R2
S
(115) (108) (134) (135)
R1 = C2H5, CH2Ph, t-C4H9, PhCH(CH3); R2 = C2H5, H; R1R2NH = Pyrrolidine, Piperidine, Morpholine; R3 = H, CH3 EWG = COCH3, COOCH3, CN, CONH2
Scheme 44
The Ullmann-type coupling reaction of sodium dithiocarbamates (137) with aryl
iodides (136) was catalyzed by CuI/N,N-dimethylglycine proceeds smoothly in DMF at
110 °C to give corresponding dithiocarbamates (138) in good yields (Scheme 45).70
I
R
R1
N S
S
R2Na+
CuI/N,N-dimethylglycineDMF, 110 oC
S
NR1 R2
S
R
(136) (137) (138)
R = H, Cl, Br, Me, MeO; R1 = R2 = CH3; R1 = H, R2 = C6H5; R1R2N = N
Scheme 45
The synthesis of thia-Michael adducts (141) was developed by the reaction of
various organic halides (primary, secondary, tertiary, allylic and benzylic) (116),
structurally diverse electron-deficient alkenes (ketones, esters and acrylonitrile) (140)
and thiourea (139) in the presence of sodium carbonate in wet polyethylene glycol
(PEG 200) at 30-35 °C (Scheme 46).71
R X H2N NH2
S
EWG++ H2O, Na2CO3
PEG 200, 30-35 oC RSEWG
(116) (139) (140) (141)
EWG = OC2H5, OC4H9-n R-X = n-C10H21I, n-C8H17Br, CH3I,
O2N
Br
BrCl
ClClBr
Br
Br, , , ,
,,,
Scheme 46
Page 25
Chapter II
82
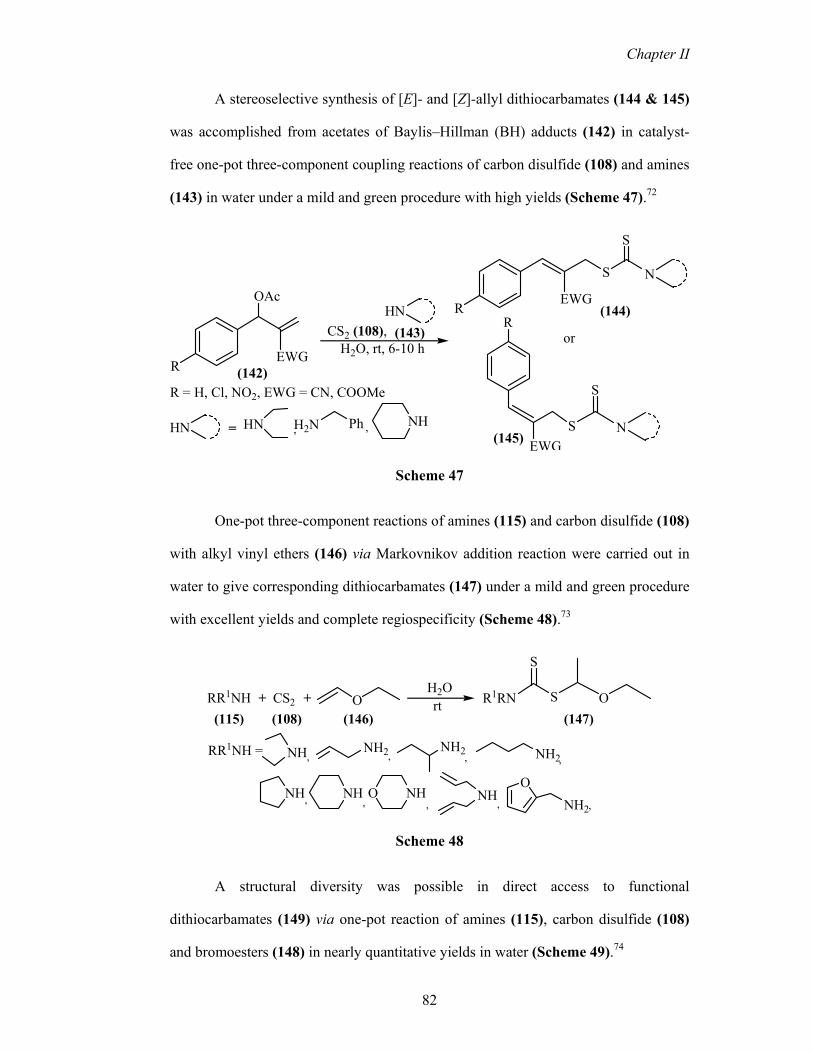
A stereoselective synthesis of [E]- and [Z]-allyl dithiocarbamates (144 & 145)
was accomplished from acetates of Baylis–Hillman (BH) adducts (142) in catalyst-
free one-pot three-component coupling reactions of carbon disulfide (108) and amines
(143) in water under a mild and green procedure with high yields (Scheme 47).72
EWGR
OAc
CS2 (108),HN
H2O, rt, 6-10 h(143)
EWGR
S N
S
(144)
or
EWGS N
S
R
(145)
R = H, Cl, NO2, EWG = CN, COOMe
HN = NHHN H2N Ph, ,
(142)
Scheme 47
One-pot three-component reactions of amines (115) and carbon disulfide (108)
with alkyl vinyl ethers (146) via Markovnikov addition reaction were carried out in
water to give corresponding dithiocarbamates (147) under a mild and green procedure
with excellent yields and complete regiospecificity (Scheme 48).73
RR1NH CS2 OH2Ort+ + R1RN S O
S
(115) (108) (146) (147)
RR1NH =
NH NH
NH NH2NH2
ONH2
NH2
O NH NH
, , , ,
, ,, , ,
Scheme 48
A structural diversity was possible in direct access to functional
dithiocarbamates (149) via one-pot reaction of amines (115), carbon disulfide (108)
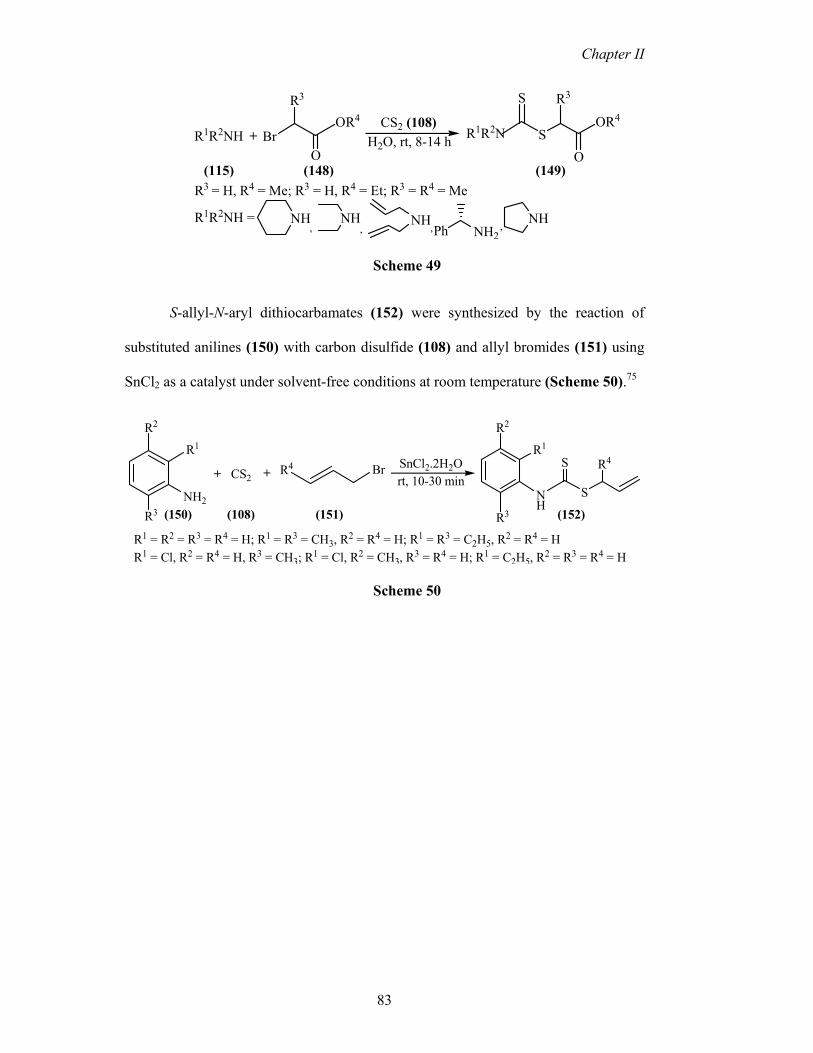
and bromoesters (148) in nearly quantitative yields in water (Scheme 49).74
Page 26
Chapter II
83
R1R2NH
R3
BrOR4
O+
CS2 (108)H2O, rt, 8-14 h R1R2N S
OR4
S
O
R3
(115) (148) (149)
R1R2NH = NH
R3 = H, R4 = Me; R3 = H, R4 = Et; R3 = R4 = Me
NHNH NHPh NH2
, , , ,
Scheme 49
S-allyl-N-aryl dithiocarbamates (152) were synthesized by the reaction of
substituted anilines (150) with carbon disulfide (108) and allyl bromides (151) using
SnCl2 as a catalyst under solvent-free conditions at room temperature (Scheme 50).75
R2
R1
NH2
R3
CS2R4 Br
R2
R1
NH
S
R4S
R3
+ +SnCl2.2H2Ort, 10-30 min
(150) (108) (151) (152)
R1 = R2 = R3 = R4 = H; R1 = R3 = CH3, R2 = R4 = H; R1 = R3 = C2H5, R2 = R4 = HR1 = Cl, R2 = R4 = H, R3 = CH3; R1 = Cl, R2 = CH3, R3 = R4 = H; R1 = C2H5, R2 = R3 = R4 = H
Scheme 50
Page 27
Chapter II
84
OBJECT OF THE PRESENT WORK
The environmentally benign synthesis of organic compounds without using
hazardous reaction conditions has become several steps closer in recent years. Strict
environmental legislations have forced chemists all over the world to develop
alternatives synthesis of biologically and synthetically important compounds. In view
of the potential biological activity of sulfur-nitrogen containing compounds and
substantial reduction in reaction period under greener techniques, it was of interest to
us to prepare the dithiocarbamate compounds as possible drugs effective against the
tropical diseases.
Dithiocarbamates (DTCs) have received considerable attention due to their
interesting chemistry and biological activity. They have widely been used as
pharmaceuticals76 and agrochemicals77 intermediates for the protection of amino
groups in peptide synthesis78, linkers in solid phase synthesis52a and recently in the
synthesis of ionic liquids.79 In rubber industry, dithiocarbamates have been used as
vulcanization accelerators and antioxidants.80 Because of the strong metal binding
capacity, dithiocarbamates can act as inhibitors of enzymes and have profound effect
on biological system and are widely used in medicinal chemistry as well as in cancer
treatment.81 Dithiocarbamates are also used as ligands for soft metal complexation
and are usually prepared by the addition of an amine with carbondisulfide and halides
or α,β-unsaturated compounds.82 Despite their biological activities, no recent progress
on their synthesis has been made. The present study aimed to devise a novel and
environment friendly method for the synthesis of some dithiocarbamate derivatives
and evaluation of their biological activity as potential antimicrobial and anticancer
agents. Only limited preparative methods have been developed for the synthesis of
dithiocarbamates.80b,83 However, these synthetic approaches suffer from the
Page 28
Chapter II
85
drawbacks such as low availability of starting materials, harsh reaction conditions,
high temperatures, unsatisfactory yields and use of expensive and hazardous catalysts
with side product formation that may be harmful to be environment or sophisticated
techniques.
The discovery of new green and more efficient synthetic protocols for the
preparation of industrial and biologically active organo-sulfur compounds via C-S
bond formation have attracted a great deal of attention.84 In this context, the use of
green catalytic system, with the replacement of expensive, toxic and flammable
organic solvents is highly required for the development of environmentally benign
methods.85
The preliminary studies have revelated that PEG could be used as green and
recyclable reaction medium for selective reactions.33,86 A number of reviews have also
explained PEG chemistry and its application in biotechnology and medicine.87 To
address the concerns raised by volatile organic medium, polyethylene glycol as an
efficient reaction medium for CAN catalyzed C-S bond formation. Ceric ammonium
nitrate (CAN) act as a water-compatible Lewis acid in aqueous medium.88
Additionally, ceric ammonium nitrate is one of the most inexpensive, ecofriendly and
greener reagents for several reactions33,35,46-48 involving C-C, C-S, C-N, C-Se and C-
Cl bond formation.12,89
In line with our interest for the application of CAN in PEG and water herein,
we have tried to describe a novel, highly efficient and ecofriendly method for the
synthesis of dithiocarbamates using CAN as catalyst in PEG 400-H2O aqueous system
(Scheme 51).
Page 29
Chapter II
86
RESULTS AND DISCUSSION
Chemistry
In order to investigate the optimum reaction condition, we initiated our study
using diethylamine (1.2 mmol) (153), carbon disulphide (2.5 mmol) (154) and
iodobenzene (1 mmol) (155) as a model reaction. The whole reaction mixture was
stirred at an ambient temperature in ethanol for 15 h, only 45% yield of desired
product was obtained, whereas the same reaction was carried out using PEG 400 as a
green solvent, to give 52% yield of product under similar reaction conditions.
Surprisingly, a significant improvement was observed and the yield of product was
dramatically increased up to 85% when water was added in to the reaction mixture
within 5 h at 50 oC (Table 1, Entry 5).
We have also studied the influence of different solvents (Table 1) on the
reaction rate as well as the yield of products, the best result was obtained with PEG
400-H2O system (Table 1, Entry 5). Minimization of chemical waste, of which 80%
is estimated to be solvents, is a constant challenge as environmental concerns are
increasingly brought into focus. With this purpose, PEG-H2O may be a seemly
choice. However, we used PEG as a reaction medium because it is non-toxic and
thermally stable.
Continuously, to improve the yield of product and also to examine the
catalytic activity of CAN, the same reaction was carried out with similar amount of
reactants in PEG 400-H2O (1:1) system at 50 °C and the yield of product was
increased to 92%, when only 2 mol% of CAN was added in the reaction mixture
(Table 2). Encouraged by this observation, we further analyzed the best reaction
condition by using different amount of CAN. An increase in the quantity of catalyst
from 2 mol% to 5 mol% not only decreased the reaction time from 4 h to 2 h but also
Page 30
Chapter II
87
increased the product yield from 92 to 95%. This revealed that catalyst concentration
plays a major role during optimization of the product yield. Although the use of 10
mol% of CAN permitted the reaction time to be decreased to 1 h, but the yield
unexpectedly decreased to 70%. A possible explanation for decrease the product
yields is that the starting material or product may have been destroyed during the
reaction when excess amount (10 mol%) of CAN was used in the reaction. It shows
that 5 mol% concentration of CAN is the suitable choice for an optimum yield of
dithiocarbamates (Table 2, Entry 3).
After successful optimization of reaction conditions, we screened a variety of
amines and aryl/alkyl halides including iodide and bromide to afford the desired
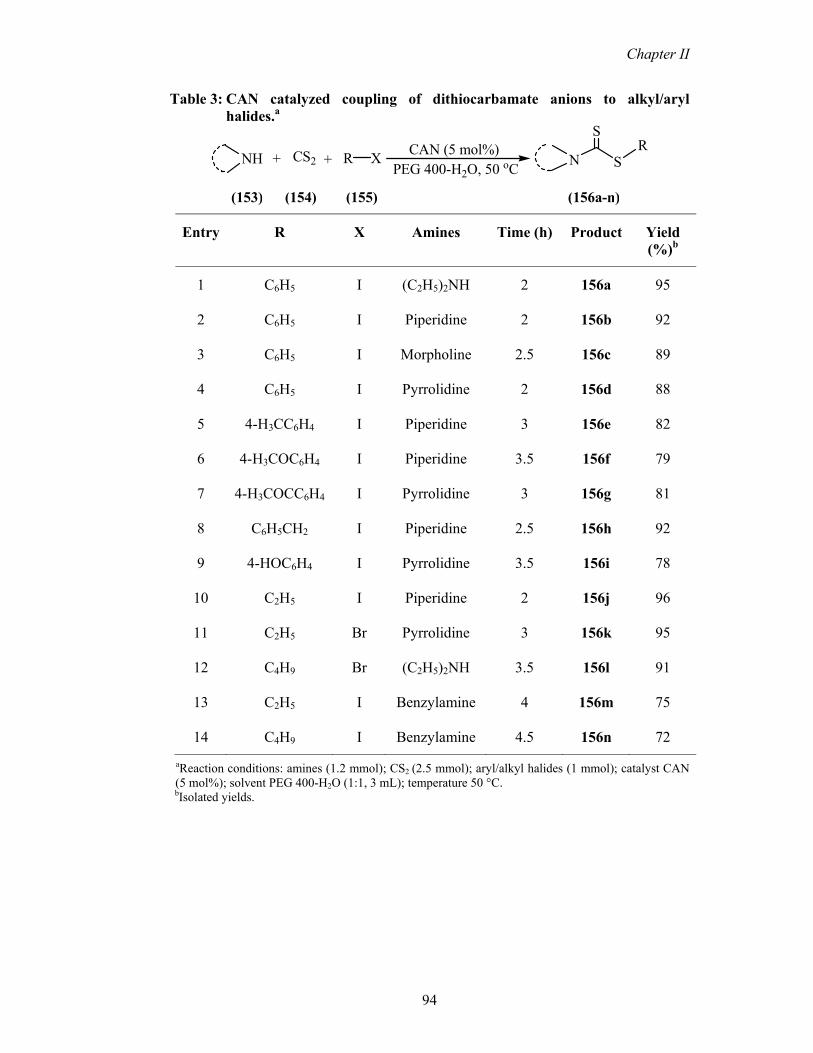
products in good to excellent yields (Table 3). In case of amines, we used various
primary and secondary amines. Generally, secondary amines such as diethyl amine,
piperidine, morpholine and pyrrolidine gave higher yields of products in short
reaction times (Table 3, 156a-l) compared with the primary amines (Table 3, 156m
& n).
To exploit this in situ generated dithiocarbamates anion for other useful
reactions, we used different Michael acceptor (α,β-unsaturated alkenes) and amines
for the synthesis of dithiocarbamates via Michael type addition. The reaction works
well with different electron deficient acceptors such as chalcone and methyl acrylate
to produce the desired Michael products in good to excellent yields. However, when
acrylamide was used as Michael acceptor, low yield of products were obtained. In
addition, we found that the use of 1-Phenylpiperazine also gave the desired Michael
adduct in higher yield (Table 4, Entry 158d).
PEG could be recycled with minimum loss and decomposition after
completion of the reaction. The product was isolated from the reaction mixture by
Page 31
Chapter II
88
extracting with diethyl ether and remaining PEG phase may be used. The solvent
phase was recycled up to 3 runs but approximately 5% weight loss of PEG was
observed from cycle to cycle (Table 5).
Pharmacology
Anticancer Activity
The cytotoxic effects of compounds 156a-n and 158a-l on U87 human glioma
cells were studied. The efficacy of the test compound was compared with control on
the basis of % cytotoxicity. The % cytotoxicity for the compounds at five different
concentrations was calculated as:
% cytotoxicity = (100-test optical density (OD)/control OD) x 100
It was evident from the study that all the dithiocarbamate derivatives had cytotoxic
effect on U87 human glioma cells.
The IC50 values for all the compounds were calculated and tabulated. The IC50
values for compounds 156b, 156d and 158d were found to be similar and lowest amongst
all others. Amongst all the dithiocarbamate derivatives, 156c, 156m, 156n, 158i and 158j
were found to be the most effective and 156a, the least effective (Table 6).
Antibacterial Activity
Screening of all the synthesized compounds for their antibacterial activity was
performed by employing Broth Microdilution MIC method.90 Using sterile microtitre
plates, 0.2 ml of Mueller Hinton Broth was added to each of the 96 wells. By
doubling the dilution of each compound were made in the wells, a plate contained 0.5-
100 μg/ml dilution of 21 different compounds and that of ampicillin. In each plate one
well was kept as positive control (broth + inoculum) and another as negative control
(broth only). The inoculum was adjusted to a turbidity equivalent to McFarland 0.5
turbidity standard. The inoculum was suitably diluted so as to get a final concentration
Page 32
Chapter II
89
of approximately 5 x 105 cfu/ml of bacteria in each well. Each well was inoculated
with 0.01 ml of prepared inoculum using a multichannel micropipette and the plates
were incubated overnight at 37 oC. The MICs of these compounds and ampicillin
were determined by using the standard protocol of NCCLS Broth Microdilution MIC
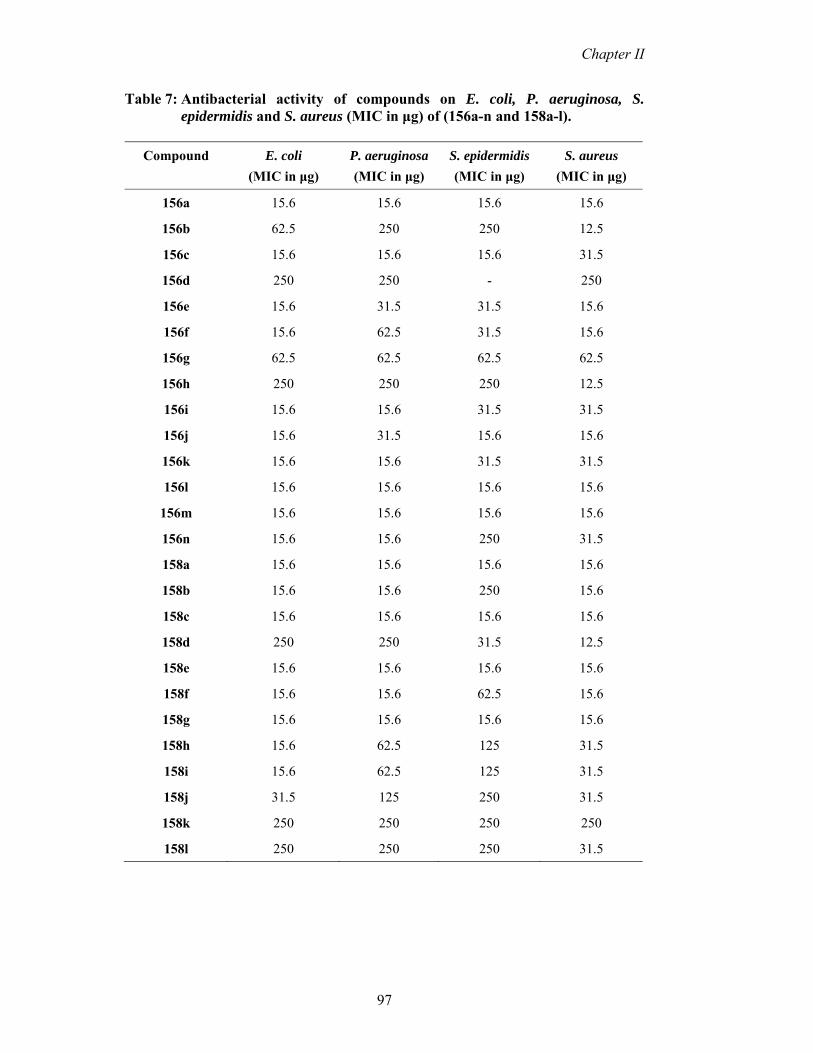
method (Table 7).
Page 33
Chapter II
90
NH + CS2CAN (5 mol%)ice-bath, 5 min N S
S
N S
S
R
N S
SY
R XR= Alkyl, ArylX= I, Br
PEG 400-H2O, 50 oC
YR1 = H, C6H5 Y= COOMe, CONH2, COPh
PEG 400-H2O, 50 oC
(153) (154)
(155)
(156a-n)
(157)
(158a-l)
R1
R1
Scheme 51: CAN catalyzed synthesis of dithiocarbamates.
Page 34
Chapter II
91
EXPERIMENTAL
Chemistry
General procedure for the synthesis of dithiocarbamates
Carbon disulphide (2.5 mmol) was added drop by drop to the stirred mixture
of amines (1.2 mmol) in PEG 400-H2O system (3 mL) at 0-5 °C. This mixture was
stirred for 5 min and ceric ammonium nitrate (CAN) (5 mol%) and aryl/alkyl
halides/α,β-unsaturated alkenes (1 mmol) was added. The reaction mixture was stirred
at 50 °C for an appropriate time. The progress of reaction was monitored by TLC. On
completion of reaction, the reaction mixture was cooled in dry ice-acetone bath to
precipitate the PEG 400 and extracted with ether (3 x 10 ml) (PEG being insoluble in
ether). The upper organic layer was washed with water, brine and dried over
anhydrous sodium sulfate (Na2SO4). The solvent was removed under reduced pressure
to afford the crude products, which was further purified by silica gel column
chromatography using hexane:ethylacetate (80:20) as an eluent to yield the desired
products (156a-n & 158a-l). The structure of all the products was unambiguously
established on the basis of their spectral analysis (IR, 1H NMR, 13C NMR and mass
spectral data).
Pharmacology
(Anticancer Activity)
Cell Culture
U87 human glioma cells and Human Embryonic Kidney 293 (HEK-293) cells
were obtained from the Department of Biocybernatics, Institute of Nuclear Medicine
and Allied Sciences, Defense Research and Development Organization, Delhi, India.
U87 human glioma cells were cultured in low glucose (1 g/l) DMEM (Dulbecco’s
modified Eagle’s medium, Himedia, India) supplemented with 10% fetal bovine
Page 35
Chapter II
92
serum and a mixture of penicillin (100 U/ml) and streptomycin (50 μg/ml) of medium,
under a humidified 5% CO2 atmosphere at 37 °C. Cells were cultured to
approximately 50% confluence at 37 °C with 5% CO2 overnight to insure complete
attachment of cells to the culture matrix. The next day, cells were treated with or
without compounds.
In vitro Cytotoxicity Assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay91
was performed in order to examine the cytotoxic effects of the compounds on U87
cells. 5 x 103 cells per well were seeded in 96 flat bottom well plates. The appropriate
concentrations (μg/ml for U87 cells) of the compounds were then added to the cells.
The cells were continuously treated for 24 h. The cytotoxicity was measured by
adding 10μl of 5 mg/ml of MTT (Sigma-Aldrich Inc., U.S.A.) to each well and
incubated for another 2 h in CO2 incubator. The purple formazan crystals were
dissolved by adding 100 μl of dimethyl sulfoxide (DMSO) to each well. The
absorbance was read at 570 nm in a spectrophotometer (Biotek Synergy HT, U.S.A.).
The cell death was calculated as follows:
% cytotoxicity = (100-test OD/control OD) x 100
The test result is expressed as the concentration of a test compound which
inhibits the cell growth by 50% (IC50).
Page 36
Chapter II
93
Table 1: Screening of solvent.a
Entry Solvent Time (h) Yield (%)b
1 EtOH 15 45
2 H2O 15 15
3 EtOH-H2O (1:1) 10 60
4 PEG 400 15 52
5 PEG 400-H2O (1:1) 5 85
6 PEG 200-H2O (1:1) 5 82
aReaction conditions: diethylamine (1.2 mmol); CS2 (2.5 mmol); iodobenzene (1 mmol); solvent (3 mL); temperature 50 °C. bIsolated yields.
Table 2: Catalytic activity evaluation of CAN for the synthesis of dithiocarbamates.a
Entry CAN (mol%) Time (h) Yield (%)b
1 0 5 85
2 2 4 92
3 5 2 95
4 10 1 70
aReaction conditions: diethylamine (1.2 mmol); CS2 (2.5 mmol); iodobenzene (1 mmol); catalyst CAN (x mol%); solvent PEG 400-H2O (1:1, 3 mL); temperature 50 °C. bIsolated yields.
Page 37
Chapter II
94
Table 3: CAN catalyzed coupling of dithiocarbamate anions to alkyl/aryl halides.a
R X+ CS2 +NH CAN (5 mol%)PEG 400-H2O, 50 oC
RSN
S
(153) (154) (155) (156a-n)
Entry R X Amines Time (h) Product Yield (%)b
1 C6H5 I (C2H5)2NH 2 156a 95
2 C6H5 I Piperidine 2 156b 92
3 C6H5 I Morpholine 2.5 156c 89
4 C6H5 I Pyrrolidine 2 156d 88
5 4-H3CC6H4 I Piperidine 3 156e 82
6 4-H3COC6H4 I Piperidine 3.5 156f 79
7 4-H3COCC6H4 I Pyrrolidine 3 156g 81
8 C6H5CH2 I Piperidine 2.5 156h 92
9 4-HOC6H4 I Pyrrolidine 3.5 156i 78
10 C2H5 I Piperidine 2 156j 96
11 C2H5 Br Pyrrolidine 3 156k 95
12 C4H9 Br (C2H5)2NH 3.5 156l 91
13 C2H5 I Benzylamine 4 156m 75
14 C4H9 I Benzylamine 4.5 156n 72
aReaction conditions: amines (1.2 mmol); CS2 (2.5 mmol); aryl/alkyl halides (1 mmol); catalyst CAN (5 mol%); solvent PEG 400-H2O (1:1, 3 mL); temperature 50 °C.
bIsolated yields.
Page 38
Chapter II
95
Table 4: CAN catalyzed nucleophilic addition of dithiocarbamate anions to Michael acceptor.a
+ CS2 +NH CAN (5 mol%)PEG 400-H2O, 50 oC SN
S
R1 Y
R1
Y
R1 = H, Ph; Y = COOMe, COPh, CONH2
(153) (154) (157) (158a-l)
Entry Michael acceptor Amines Time
(h) Product Yield
(%)b
16 OMe
O (C2H5)2NH 2 158a 92
17 Piperidine 2.5 158b 81
18 Pyrrolidine 2.5 158c 94
19 1-Phenylpiperazine 3.5 158d 82
20 NH2
O (C2H5)2NH 3 158e 88
21 Piperidine 3 158f 86
22 Pyrrolidine 3.5 158g 81
23
Ph Ph
O
(C2H5)2NH 2.5 158h 95
24 Piperidine 2.5 158i 93
25 Pyrrolidine 3 158j 92
26 Benzylamine 4 158k 81
27 n-Butylamine 4.5 158l 83
aReaction conditions: amines (1.2 mmol); CS2 (2.5 mmol); α,β-unsaturated alkenes (1 mmol); catalyst CAN (5 mol%); solvent PEG 400-H2O (1:1, 3 mL); temperature 50 °C. bIsolated yields.
Page 39
Chapter II
96
Table 5: Recyclability of PEG 400.a
No of Cyclesa Fresh Run 1 Run 2 Run 3
Yield (%)b 95 95 94 92
Time (h) 2 2 2 2 aReaction conditions: diethylamine (1.2 mmol); CS2 (2.5 mmol); iodobenzene (1 mmol); catalyst CAN (5 mol%); solvent PEG 400-H2O (1:1, 3 mL); temperature 50 °C. bIsolated yields.
Table 6: Percentage viability at different concentrations and IC50 in μg/ml of compounds (156a-n and 158a-l).
Compd Viability (%) IC50
(µg/ml) 2500 1250 625 312 156 78 39 19 9.8 4.9 2.5
156a 35.333 48.8 49.688 55.822 67.844 81.444 89.35 91.77 100 100 100 625
156b 30.177 42.244 47.822 51.133 63.083 79.111 83.87 97.011 99.11 100 100 312
156c 3.921 3.137 2.549 18.627 19.974 21.568 23.779 37.549 43.581 51.773 78.894 4.9
156d 13.555 13.377 14.177 53.711 65.022 73.511 87.755 99.487 100.6 100 100 312
156e 27.523 32.19 36.19 43.904 47 54.238 91.619 96.761 100 100 100 78
156f 6.686 11.686 17.469 17.988 18.9 20.11 20.542 22.65 93.614 98.795 100 14
156g 4.215 7.352 7.993 8.0 8.0 14.117 44.803 47.45 66.887 100 100 19
156h 1.911 2.556 12.998 24.057 35.113 41.335 55.507 97.246 100 100 100 39
156i 4.215 5.392 6.443 8.443 10.444 10.98 14.705 32.843 53.137 62.549 77.566 98
156j 13.714 17.476 22.904 23.047 74.809 110.666 109.952 91.285 87.476 78.142 80.142 184
156k 2.221 12.667 13.875 17.112 19.443 21.776 25.115 27.407 81.666 91.296 99.259 14.9
156l 36.38 39.285 43 47.619 49.991 51.047 79.523 89.333 99.047 101.809 100 78
156m 15.889 18.666 22.134 28.778 31.011 33.889 39 43.931 47.111 47.988 53.109 2.5
156n 18.11 22.134 23.401 33.89 37 41.556 43.277 49 54.457 88.734 90.06 9.8
158a 35.048 35.998 37.66 40.853 46.999 51.99 69.77 70.331 83.412 87.9620 100 78
158b 30.161 33.838 40.731 47.012 54.301 89.838 96.973 104.30 100 100 100 156
158c 28.104 31.455 39.99 47.882 53.696 86.303 98.704 100 100 100 100 156
158d 36.101 36.909 47.011 53.672 93.686 93.99 100 100 100 100 100 312
158e 32.204 32.904 39.111 39.903 47.873 55.107 93.446 100 100 100 100 78
158f 30.053 37.389 37.998 41.904 56.881 91.047 100 100 100 100 100 156
158g 40.094 41.838 41.989 47.99 49 50.383 88.223 99.44 100 100 100 78
158h 3.887 12.776 13.1324 13.983 15.332 18.223 21.993 24.074 72.407 88.665 100 14.9
158i 8.382 7.205 10.443 19.331 23.556 31.227 37.112 43.778 49.71 72.898 85.797 9.8
158j 8.676 10.823 13.546 19.665 21.11 27.112 31.998 37.391 45.112 51.739 98.443 4.9
158k 33.677 37 41.33 41.99 47.401 49 49.91 52.8 84.463 97.377 100 19
158l 21.871 22.951 32.444 39.55 41 43.367 49 49.834 53.996 74.457 76.385 19
Page 40
Chapter II
97
Table 7: Antibacterial activity of compounds on E. coli, P. aeruginosa, S. epidermidis and S. aureus (MIC in μg) of (156a-n and 158a-l).
Compound
E. coli
(MIC in μg) P. aeruginosa (MIC in μg)
S. epidermidis (MIC in μg)
S. aureus (MIC in μg)
156a 15.6 15.6 15.6 15.6
156b 62.5 250 250 12.5
156c 15.6 15.6 15.6 31.5
156d 250 250 - 250
156e 15.6 31.5 31.5 15.6
156f 15.6 62.5 31.5 15.6
156g 62.5 62.5 62.5 62.5
156h 250 250 250 12.5
156i 15.6 15.6 31.5 31.5
156j 15.6 31.5 15.6 15.6
156k 15.6 15.6 31.5 31.5
156l 15.6 15.6 15.6 15.6
156m 15.6 15.6 15.6 15.6
156n 15.6 15.6 250 31.5
158a 15.6 15.6 15.6 15.6
158b 15.6 15.6 250 15.6
158c 15.6 15.6 15.6 15.6
158d 250 250 31.5 12.5
158e 15.6 15.6 15.6 15.6
158f 15.6 15.6 62.5 15.6
158g 15.6 15.6 15.6 15.6
158h 15.6 62.5 125 31.5
158i 15.6 62.5 125 31.5
158j 31.5 125 250 31.5
158k 250 250 250 250
158l 250 250 250 31.5
Page 41
Chapter II
98
Spectroscopic data of synthesized dithiocarbamates (156a-n and 158a-l)
Diethyl-1-dithiocarbamic acid-n-phenyl ester (156a): IR (KBr/cm-1): νmax = 2978,
1501, 1421, 1271, 1065, 732, 686; 1H NMR (CDCl3, 400 MHz): δ = 7.30-7.69 (m,
5H), 3.99 (q, J = 6.92 Hz, 2H), 3.72 (q, J = 6.92 Hz, 2H), 1.24-1.47 (m, 6H); 13C
NMR (CDCl3, 100 MHz): δ = 196.4, 137.3, 130.2, 130.1, 129.4, 129.3, 127.4, 52.3,
52.5, 12.6, 12.5; m/z 226.0679 (M+1, C11H15NS2 requires 225.0646).
Piperidine-1-carbodithioic acid-n-phenyl ester (156b): IR (KBr/cm-1): νmax =
2918, 1459, 1252, 740, 650; 1H NMR (CDCl3, 400 MHz): δ = 7.05-7.65 (m, 5H), 4.16
(br, 2H), 3.80 (br, 2H), 1.67 (br, 6H); 13C NMR (CDCl3, 100 MHz): δ = 198.8, 135.2,
129.6, 129.5, 129.2, 129.1, 125.6, 50.7, 50.6, 24.5, 24.4, 24.1; m/z 238.0679 (M+1,
C12H15NS2 requires 237.0646).
Morpholine-1-carbodithioic acid-n-phenyl ester (156c): IR (KBr/cm-1): νmax =
2918, 1466, 1267, 1216, 1113, 756, 667; 1H NMR (CDCl3, 400 MHz): δ = 7.49-6.70
(m, 5H), 4.30 (br, 4H), 3.79-3.85 (m, 4H); 13C NMR (CDCl3, 100 MHz): δ = 198.2,
136.3, 131.2, 131.1, 130.6, 130.4, 125.2, 66.5, 66.3, 51.7, 51.6; m/z 240.0472 (M+1,
C11H13NOS2 requires 239.0439).
Pyrrolidine-1-carbodithioic acid-n-phenyl ester (156d): IR (Film/cm-1): νmax =
2920, 1441, 1217, 756, 667; 1H NMR (CDCl3, 400 MHz): δ = 7.07-7.39 (m, 5H), 3.56
(t, J = 6.8 Hz, 2H), 3.43 (t, J = 6.7 Hz, 2H), 1.95-2.02 (m, 2H), 1.84-1.91 (m, 2H); 13C
NMR (CDCl3, 100 MHz): δ = 197.6, 134.7, 129.5, 129.4, 128.6, 128.2, 125.2, 53.3,
53.1, 25.2, 25.1; m/z 224.0523 (M+1, C11H13NS2 requires 223.0489).
Piperidine-1-carbodithioic acid-4-methyl-phenyl ester (156e): IR (KBr/cm-1):
νmax = 2945, 1480, 1279, 1134, 948, 851, 755, 666; 1H NMR (CDCl3, 400 MHz): δ =
7.23-7.48 (m, 4H), 4.21 (br, 2H), 3.94 (br, 2H), 2.38 (s, 3H), 1.74 (br, 6H); 13C NMR
Page 42
Chapter II
99
(CDCl3, 100 MHz): δ = 198.4, 135.2, 131.6, 131.4, 131.3, 129.2, 129.1, 50.5, 50.4,
25.7, 24.3, 24.2, 22.1; m/z 252.0836 (M+1, C13H17NS2 requires 251.0802).
Piperidine-1-carbodithioic acid-4-methoxy-phenyl ester (156f): IR (KBr/cm-1):
νmax = 2960, 1486, 1458, 1286, 913, 819, 744, 670; 1H NMR (CDCl3, 400 MHz): δ =
7.55 (d, J = 8.8 Hz, 2H), 6.68 (d, J = 8.7 Hz, 2H), 4.22 (br, 2H), 3.76 (br, 2H), 1.75
(br, 6H); 13C NMR (CDCl3, 100 MHz): δ = 196.8, 159.3, 138.3, 138.1, 126.3, 116.3,
116.2, 55.2, 52.5, 52.3, 24.8, 24.5, 24.0; m/z 268.0785 (M+1, C13H17NOS2 requires
267.0752).
Pyrrolidine-1-carbodithioic acid-4-acetyl-phenyl ester (156g): IR (KBr/cm-1):
νmax = 2953, 2865, 1685, 1438, 1264, 1153, 1009, 955, 820, 748, 607; 1H NMR
(CDCl3, 400 MHz): δ = 7.80 (d, J = 8.7 Hz, 2H), 7.59 (d, J = 8.1 Hz, 2H), 3.93 (br,
2H), 3.75 (br, 2H), 2.56 (s, 3H), 2.14-2.18 (m, 2H), 1.97-2.01 (m, 2H); 13C NMR
(CDCl3, 100 MHz): δ = 197.5, 195.2, 135.8, 131.9, 129.8, 129.7, 128.3, 128.2, 54.2,
52.6, 52.5, 24.6, 24.2; m/z 266.0629 (M+1, C13H15NOS2 requires 265. 0595).
Piperidine-1-carbodithioic acid-benzyl ester (156h): IR (KBr/cm-1): νmax = 2919,
1426, 1292, 1180, 934, 809, 709; 1H NMR (CDCl3, 400 MHz): δ = 7.47-7.60 (m, 5H),
4.55 (s, 2H), 4.16 (br, 2H), 3.65 (br, 2H), 1.76 (br, 6H); 13C NMR (CDCl3, 100 MHz):
δ = 197.5, 136.4, 130.1, 129.2, 128.4, 127.4, 52.1, 51.5, 43.2, 29.6, 24.7, 24.1; m/z
252.0836 (M+1, C13H17NS2 requires 251.0802).
Pyrrolidine-1-carbodithioic acid-4-hydroxy-phenyl ester (156i): IR (KBr/cm-1):
νmax = 3441, 2953, 1436, 1250, 1154, 1038, 957, 856, 753, 683; 1H NMR (CDCl3,
400 MHz): δ = 7.63 (d, J = 8.6 Hz, 2H), 6.72 (d, J = 8.0 Hz, 2H), 5.25 (s, 1H), 3.94
(br, 2H), 3.61 (br, 2H), 2.09-2.14 (m, 2H), 1.96-2.02 (m, 2H); 13C NMR (CDCl3, 100
MHz): δ = 189.5, 158.6, 136.7, 133.3, 127.2, 123.7, 122.6, 56.9, 50.9, 26.5, 24.1; m/z
240.0473 (M+1, C11H13NOS2 requires 239.0440).
Page 43
Chapter II
100
Piperidine-1-carbodithioic acid-n-ethyl ester (156j): IR (Film/cm-1): νmax = 2917,
1431, 1243, 1108, 1006, 908, 851, 731; 1H NMR (CDCl3, 400 MHz): δ = 4.21 (br,
2H), 3.85 (br, 2H), 3.14 (q, J = 7.0 Hz, 2H), 1.74 (br, 6H), 1.10 (t, J = 2.1 Hz, 3H);
13C NMR (CDCl3, 100 MHz): δ = 198.4, 52.8, 51.6, 31.4, 26.5, 25.2, 24.5, 14.8; m/z
190.0679 (M+1, C8H15NS2 requires 189.0646).
Pyrrolidine-1-carbodithioic acid-n-ethyl ester (156k): IR (Film/cm-1): νmax =
2918, 1435, 1251, 1155, 1002, 910, 731, 646; 1H NMR (CDCl3, 400 MHz): δ = 3.93
(br, 2H), 3.61 (t, J = 6.8 Hz, 2H), 3.27 (q, J = 7.3 Hz, 2H), 2.10-2.15 (m, 2H), 1.95-
2.06 (m, 2H), 1.32 (t, J = 9.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ = 192.6, 50.9,
50.4, 30.6, 26.5, 25.9, 13.9; m/z 177.0447 (M+1, C7H13NS2 requires 175.0489).
Diethyl-1-dithiocarbamic acid-n-butyl ester (156l): IR (Film/cm-1): νmax = 2932,
1418, 1270, 1143, 1007, 914, 732, 646; 1H NMR (CDCl3, 400 MHz): δ = 4.04 (br,
2H), 3.75 (br, 2H), 3.28 (t, J = 7.3 Hz, 2H), 1.64-1.72 (m, 2H), 1.42-1.48 (m, 2H),
1.27-1.30 (m, 6H), 0.92 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ = 195.9,
51.9, 49.1, 36.8, 30.5, 22.0, 13.6, 11.5, 11.3; m/z 206.0992 (M+1, C9H19NS2 requires
205.0959).
Benzylamine-1-carbodithioic acid-n-ethyl ester (156m): IR (Film/cm-1): νmax =
3263, 2927, 2093, 1638, 1506, 1454, 1383, 1329, 1253, 1073, 1002, 912, 740, 699,
648; 1H NMR (CDCl3, 400 MHz): δ = 8.82 (br, 1H, NH), 7.33-7.36 (m, 5H), 4.93 (d,
J = 5.1 Hz, 2H, PhCH2N), 3.26 (q, J = 6.8 Hz, 2H), 1.22 (t, J = 6.8 Hz, 3H); 13C NMR
(CDCl3, 100 MHz): δ = 198.1, 137.4, 128.9, 128.8, 128.2, 127.9, 126.7, 33.5, 25.6,
14.3; m/z 212.0523 (M+1, C10H13NS2 requires 211.0489).
Benzylamine-1-carbodithioic acid-n-butyl ester (156n): IR (Film/cm-1): νmax =
3246, 2959, 2093, 1601, 1508, 1455, 1347, 1278, 1127, 1028, 909, 733, 698, 648; 1H
NMR (CDCl3, 400 MHz): δ = 7.90 (br, 1H, NH), 7.30-7.39 (m, 5H), 4.70 (d, J = 6.2
Page 44
Chapter II
101
Hz, 2H, PhCH2N), 3.19 (t, J = 6.9 Hz, 2H), 1.66-1.80 (m, 2H), 1.35-1.48 (m, 2H),
0.92 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ = 197.5, 134.0, 128.9, 128.3,
128.0, 126.8, 126.1, 48.6, 35.2, 31.8, 21.6, 13.3; m/z 240.0836 (M+1, C12H17NS2
requires 239.0802).
3-Diethylthiocarbamoylsulfanyl-propionic acid methyl ester (158a): IR (Film/cm-1):
νmax = 2935, 1735, 1421, 1355, 1202, 1147, 1008, 909, 732, 648; 1H NMR (CDCl3,
400 MHz): δ = 4.0 (br, 2H, CH2N), 3.67 (br, 2H, CH2N), 3.65 (s, 3H), 3.53 (t, J = 6.9
Hz, 2H), 2.78 (t, J = 6.9 Hz, 2H), 1.22-1.58 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ
= 194.6, 172.5, 51.9, 51.7, 49.3, 33.8, 31.5, 12.3, 11.4; m/z 236.0736 (M+1,
C9H17NO2S2 requires 235.0702).
3-(Piperidine-1-carbothiosulfanyl)-propionic acid methyl ester (158b): IR
(Film/cm-1): νmax = 2919, 1654, 1439, 1217, 1020, 953, 709; 1H NMR (CDCl3, 400
MHz): δ = 4.25 (br, 2H), 3.84 (br, 2H), 3.67 (s, 3H), 3.55 (t, J = 7.1 Hz, 2H), 2.79 (t, J
= 7.1 Hz, 2H), 1.67 (br, 6H); 13C NMR (CDCl3, 100 MHz): δ = 195.2, 172.4, 52.6,
51.8, 51.3, 34.6, 31.8, 25.8, 25.6, 24.3; m/z 248.0734 (M+1, C10H17NO2S2 requires
247.0701).
3-(Pyrrolidine-1-carbothiosulfanyl)-propionic acid methyl ester (158c): IR
(Film/cm-1): νmax = 2917, 1735, 1437, 1221, 1156, 1007, 909, 732, 647; 1H NMR
(CDCl3, 400 MHz): δ = 3.98 (t, J = 6.8 Hz, 2H, CH2N), 3.70 (s, 3H), 3.63 (t, J = 7.2
Hz, 2H, CH2N), 3.57 (t, J = 6.9 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H), 1.96-2.16 (m, 4H);
13C NMR (CDCl3, 100 MHz): δ = 191.9, 172.4, 54.9, 51.7, 50.9, 33.8, 30.9, 26.5,
24.1; m/z 234.0578 (M+1, C9H15NO2S2 requires 233.0544).
3-(4-Phenylpiperazine-1-carbothiosulfanyl)-propionic acid methyl ester (158d):
IR (Film/cm-1): νmax = 3017, 2917, 2850, 1734, 1599, 1492, 1420, 1215, 1145, 1015,
925, 755, 667; 1H NMR (CDCl3, 400 MHz): δ = 7.24-7.29 (m, 2H), 6.84-6.93 (m,
Page 45
Chapter II
102
3H), 4.54 (br, 2H), 4.45 (br, 2H), 3.38 (s, 3H), 3.18 (t, J = 6.5 Hz, 2H), 3.08-3.10 (m,
4H), 2.85 (t, J = 6.5 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ = 193.4, 172.5, 151.1,
129.3, 129.1, 120.3, 116.2, 52.4, 51.3, 49.1, 45.2, 31.8, 29.6; m/z 325.1245 (M+1,
C15H20N2O2S2 requires 324.0976).
3-Diethylcarbamodithioic acid-3-amino-3-oxopropyl ester (158e): IR (KBr/cm-1):
νmax = 3411, 2920, 1667, 1271, 1202, 1147, 1006, 908, 733, 649; 1H NMR (CDCl3,
400 MHz): δ = 5.70 and 5.34 (together, br, 2H, NH2), 4.06 (br, 2H, CH2N), 3.72 (q, J
= 7.3 Hz, 2H, CH2N), 3.56 (t, J = 6.5 Hz, 2H), 2.68 (t, J = 6.9 Hz, 2H), 1.25 (t, J = 7.3
Hz, 3H), 0.85 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ = 196.2, 172.1,
53.8, 52.3, 34.1, 33.5, 13.6, 12.8; m/z 221.0738 (M+1, C10H16N2OS2 requires
220.0704).
3-(Piperidine-1-carbodithioic acid)-3-amino-3-oxopropyl ester (158f): IR
(KBr/cm-1): νmax = 3340, 2917, 1645, 1208, 1154, 1042, 913, 744; 1H NMR (CDCl3,
400 MHz): δ = 7.23 (br, 2H, NH2), 4.21 (br, 2H, CH2N), 3.75 (br, 2H, CH2N), 3.58 (t,
J = 7.1 Hz, 2H), 2.68 (t, J = 7.2 Hz, 2H), 0.85-1.73 (m, 6H); 13C NMR (CDCl3, 100
MHz): δ = 194.3, 173.5, 52.6, 52.2, 34.7, 33.2, 24.3, 24.1, 21.3; m/z 233.0738 (M+1,
C9H16N2OS2 requires 232.0704).
3-(Pyrrolidine-1-carbodithioic acid)-3-amino-3-oxopropyl ester (158g): IR
(Film/cm-1): νmax = 3431, 2979, 1647, 1442, 1216, 1154, 1038, 955, 757, 667; 1H
NMR (CDCl3, 400 MHz): δ = 4.13 and 4.11 (together, br, 2H, NH2), 3.93 (t, J = 6.8
Hz, 2H, CH2N), 3.63 (t, J = 6.9 Hz, 2H, CH2N), 3.54 (t, J = 6.6 Hz, 2H), 2.13 (t, J =
6.6 Hz, 2H), 1.98-2.03 (m, 4H); 13C NMR (CDCl3, 100 MHz): δ = 206.5, 189.1, 56.9,
50.9, 32.1, 30.9, 26.5, 24.2; m/z 219.0581 (M+1, C8H14N2OS2 requires 218.0548).
Diethyl-1-carbamodithioic acid-3-oxo-1,3-diphenylpropyl ester (158h): IR (Film/cm-
1): νmax = 3017, 2981, 1663, 1578, 1450, 1216, 1178, 1018, 916, 754, 689; 1H NMR
Page 46
Chapter II
103
(CDCl3, 400 MHz): δ = 8.00-8.03 (m, 2H), 7.39-7.65 (m, 8H), 4.89 (dd, J = 4.4, 4.4 Hz,
1H), 3.45-4.03 (m, 6H), 1.25-1.47 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ = 192.3,
190.2, 144.6, 137.9, 134.6, 132.6, 130.4, 129.6, 129.1, 128.8, 128.4, 128.3, 128.2, 52.1,
47.2, 29.5, 13.2, 11.2; m/z 358.1255 (M+1, C20H23NOS2 requires 357.1221).
Piperidine-1-carbodithioic acid-3-oxo-1,3-diphenylpropyl ester (158i): IR
(Film/cm-1): νmax = 3060, 2940, 1664, 1606, 1576, 1449, 1216, 1134, 1016, 748, 689;
1H NMR (CDCl3, 400 MHz): δ = 8.00-8.02 (m, 4H), 7.39-7.64 (m, 8H), 4.87 (dd, J =
4.4 Hz, 4.4 Hz, 1H), 4.21 (br, 2H, CH2N), 3.72 (br, 2H, CH2N), 3.57-3.63 (m, 2H),
1.62-2.31 (m, 6H); 13C NMR (CDCl3, 100 MHz): δ = 202.4, 199.2, 138.1, 134.8,
132.7, 130.5, 128.9, 128.7, 128.5, 128.4, 128.3, 128.0, 127.8, 127.6, 52.8, 45.0, 39.1,
25.3, 24.8, 24.1; m/z 370.1255 (M+1, C21H23NOS2 requires 369.1221).
Pyrrolidine-1-carbodithioic acid-3-oxo-1,3-diphenylpropyl ester (158j): IR (KBr/cm-
1): νmax = 3062, 2923, 1664, 1577, 1440, 1215, 1178, 1017, 909, 753, 689; 1H NMR
(CDCl3, 400 MHz): δ = 7.26 -8.03 (m, 10H), 5.75 (dd, J = 4.5, 4.5 Hz, 1H), 3.59-4.15 (m,
6H), 2.01-2.17 (m, 4H); 13C NMR (CDCl3, 100 MHz): δ = 197.2, 190.3, 137.6, 134.6,
130.4, 128.8, 128.6, 128.4 128.3, 128.2, 128.1, 126.5, 126.3, 126.1, 52.9, 52.8, 48.5, 38.7,
26.3, 24.1; m/z 356.1098 (M+1, C20H21NOS2 requires 355.1065).
Benzyl-1-carbamodithioic acid-3-oxo-1,3-diphenylpropyl ester (158k): IR
(Film/cm-1): νmax = 3018, 2918, 2850, 1638, 1664, 1605, 1450, 1336, 1215, 1177,
1016, 747; 1H NMR (CDCl3, 400 MHz): δ = 8.02 (d, J = 7.3 Hz, 2H), 7.82-7.91 (m,
3H), 7.22-7.65 (m, 10H), 4.86 (dd, J = 5.8, 5.8 Hz, 1H), 6.20 (br, 1H, NH), 3.74-3.86
(m, 2H), 4.70 (s, 2H); 13C NMR (CDCl3, 100 MHz): δ = 196.2, 196.1, 144.8, 138.0,
134.7, 133.7, 132.7, 130.5, 128.9, 128.8, 128.5, 128.4, 128.2, 128.0, 127.7, 127.5,
127.3, 126.7, 126.4, 50.5, 48.5, 42.4; m/z 392.1099 (M+1, C23H21NOS2 requires
391.1067).
Page 47
Chapter II
104
Butyl-1-carbamodithioic acid-3-oxo-1,3-diphenylpropyl ester (158l): IR (Film/cm-
1): νmax = 3065, 2918, 2850, 1663, 1605, 1577, 1450, 1336, 1216, 1178, 1017, 908;
1H NMR (CDCl3, 400 MHz): δ = 8.0 (d, J = 7.3 Hz, 2H), 7.39-7.64 (m, 8H), 4.86 (dd,
J = 6.6, 6.6 Hz, 1H), 4.62 (br, 1H, NH), 3.73-3.83 (m, 2H), 3.59 (t, J = 6.8 Hz, 2H),
1.51-1.58 (m, 2H), 1.30-1.38 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 100
MHz): δ = 202.0, 198.9, 138.1, 134.8, 132.7, 128.9, 128.6, 128.4, 127.7, 127.5, 50.4,
48.2, 38.7, 29.5, 15.1; m/z 357.7302 (M+1, C20H23NOS2 requires 357.1221).
Page 48
Chapter II
105
REFERENCES
1. T. L. Ho, Synthesis, 1973, 347.
2. T. Imamoto, M. Nishiura, Y. Yamanoi, H. Tsuruta and K. Yamaguchi, Chem.
Lett., 1996, 25, 875.
3. V. Nair, J. Mathew and K. V. Radhakrishnan, J. Chem. Soc., Perkin Trans. 1,
1996, 1487.
4. T. L. Ho, Organic Synthesis by Oxidation with Metal Compounds; W. J. Mijs,
C. R. H. I de Jonge, Eds; Plenum Press: New York, 1986, 569.
5. H. B. Kagan and J. L. Namy, Tetrahedron, 1986, 42, 6573.
6. G. A. Molander, Chem. Rev., 1992, 92, 29.
7. T. Imamoto, Lanthanides in organic synthesis, Academic Press: London,
1994, chapter 4.
8. V. Nair, J. Mathew and J. Prabhakaran, Chem. Soc. Rev., 1997, 127.
9. J. R. Hwu and K. -Y. King, Curr. Sci., 2001, 81, 1043.
10. V. Nair, S. B. Panicker, L. G. Nair, T. G. George and A. Augustine, Synlett,
2003, 156.
11. V. Nair, L. Balagopal, R. Rajan and J. Mathew, Acc. Chem. Res., 2004, 37, 21.
12. V. Nair and A. Deepthi, Chem. Rev., 2007, 107, 1862.
13. V. Sridharan and J. C. Menendez, Chem. Rev., 2010, 110, 3805.
14. A. Mori, Y. Isayama and H. Takeshita, Bull. Chem. Soc. Jpn., 1986, 59, 511.
15. E. Baciocchi, G. Civitarese and R. Ruzziconi, Tetrahedron Lett., 1987, 28,
5357.
16. A. Citterio, L. Pesce, R. Sebastiano and R. Santi, Synthesis, 1990, 142.
17. K. Kobayashi, H. Tanaka, K. Tanaka, K. Yoneda, O. Morikawa and H.
Konishi, Synth. Commun., 2000, 30, 4277.
Page 49
Chapter II
106
18. A. J. Clark, C. P. Dell, J. M. McDonagh, J. Geden and P. Mawdsley, Org.
Lett., 2003, 5, 2063.
19. K. Kobayashi, H. Umakoshi, K. Hayashi, O. Morikawa and H. Konishi, Chem.
Lett., 2004, 33, 1588.
20. X. -F. Zeng, S. -J. Ji and S. -Y. Wang, Tetrahedron, 2005, 61, 10235.
21. D. Tsvelikhovsky, H. Schumann and J. Blum, Synthesis, 2006, 1819.
22. P. Shanmugam, P. Boobalan and P. T. Perumala, Tetrahedron, 2007, 63,
12215.
23. M. Paira, S. K. Mandal and S. C. Roy, Tetrahedron Lett., 2008, 49, 2432.
24. K. Sujatha, G. Shanthi, N. P. Selvam, S. Manoharan, P. T. Perumal and M.
Rajendran, Bioorg. Med. Chem Lett., 2009, 19, 4501.
25. J. L. Huston, J. Am. Chem. Soc., 1971, 93, 5256.
26. H. M. Chawla and R. S. Mittal, Synthesis, 1985, 70.
27. C. A. Horiuchi, K. Ochiai and H. Fukunishi, Chem. Lett., 1994, 23, 185.
28. V. Nair and L. G. Nair, Tetrahedron Lett., 1998, 39, 4585.
29. V. Nair, A. Augustine and T. G. George, Eur. J. Org. Chem., 2002, 2363.
30. V. Nair, A. Augustine, S. B. Panicker, T. D. Suja and S. Mathai, Res. Chem.
Intermed., 2003, 29, 213.
31. K. -I. Itoh and C. A. Horiuchi, Tetrahedron, 2004, 60, 1671.
32. S. S. Shindalkar, B. R. Madje, R. V. Hangarge and M. S. Shingare, Indian J.
Chem., 2005, 44B, 2409.
33. M. Kidwai, D. Bhatnagar, N. K. Mishra and V. Bansal, Catal. Commun.,
2008, 9, 2547.
34. K. F. Shelke, S. B. Sapkal and M. S. Shingare, Chin. Chem. Lett., 2009, 20,
283.
Page 50
Chapter II
107
35. M. Kidwai and D. Bhatnagar, Tetrahedron Lett., 2010, 51, 2700.
36. T. -L. Ho, H. C. Ho and C. M. Wong, J. Chem. Soc., Chem. Commun., 1972,
791.
37. V. Nair and V. Sheeba, J. Org. Chem., 1999, 64, 6898.
38. J. R. Hwu, S. -S. Shiao and S. -C. Tsay, J. Am. Chem. Soc., 2000, 122, 5899.
39. V. Nair, R. Rajan, L. Balagopal, S. Thomas and K. Narasimlu, Tetrahedron
Lett., 2002, 43, 8971.
40. D. A. Evans, P. Nagorny and R. Xu, Org. Lett., 2006, 8, 5669.
41. V. Sridharan, P. T. Perumal, C. Avendano and J. C. Menendez, Tetrahedron,
2007, 63, 4407.
42. J. Jiao, L. X. Nguyen, D. R. Patterson and R. A. Flowers, Org. Lett., 2007, 9,
1323.
43. V. Sridharan and J. C. Menendez, Org. Lett., 2008, 10, 4303.
44. J. Li, Y. Liu, C. Li and X. Jia, Tetrahedron Lett., 2009, 50, 6502.
45. S. Durgadasa, V. K. Chatare, K. Mukkanti and S. Pal, Lett. Org. Chem., 2010,
7, 306.
46. M. Kidwai and D. Bhatnagar, Chem. Papers, 2010, 64, 825.
47. M. Kidwai, D. Bhatnagar, R. Kumar and P. M. Luthra, Chem. Pharm. Bull.,
2010, 58, 1320.
48. M. Kidwai, D. Bhatnagar and N. K. Mishra, Green Chem. Lett. Rev., 2010, 3, 55.
49. S. Pattanayak and S. Sinha, Tetrahedron Lett., 2011, 52, 34.
50. M. Delepine, Compt. Rend., 1907, 144, 1125.
51. (a) Y. S. Chen, I. Schuphan and J. E. Casida, J. Agric. Food. Chem., 1979, 27,
709; (b) T. Mizuno, I. Nishiguchi, T. Okushi and T. Hirashima, Tetrahedron
Lett., 1991, 32, 6867.
Page 51
Chapter II
108
52. (a) P. J. Nieuwenhuizen, A.W. Ehlers, J. G. Haasnoot, S. R. Janse, J. Reedijk
and E. J. Baerends, J. Am. Chem. Soc., 1999, 121, 163; (b) G. D. Thorn and R.
A. Ludwig, R. A. The Dithiocarbamates and Related Compounds; Elsevier:
Amsterdam, New York, 1962.
53. H. G. Guy, J. Econ. Entomol., 1936, 29, 467.
54. (a) W. Walter and K. D. Bode, Angew. Chem., Int. Ed. Eng., 1967, 6, 281; (b)
G. H. Elgemeie and S. H. Sayed, Synthesis, 2001, 1747; (c) C. Ronconi, C.
Marzano, P. Zanello, M. Corsini, G. Miolo, C. Macca, A. Trevisan and D.
Fregona, J. Med. Chem., 2006, 49, 1648.
55. (a) A. Ivachtchenko, S. Kovalenko, O. V. Tkachenko and O. Parkhomenko, J.
Comb. Chem., 2004, 6, 573; (b) Y. Zhao, W. Perez-segarra, Q. Shi and A.
Wei, J. Am. Chem. Soc., 2005, 127, 7328.
56. (a) T. S. Griffin, T. S. Woods and D. L. Klayman, In Advances in Heterocyclic
Chemistry; A. R. Katritzky, A. J. Boulton, (Eds.); Academic Press: New York,
1975; 18, 99; (b) S. Goubert-Renaudin, R. Schneider and A. Walcarius,
Tetrahedron Lett., 2007, 48, 2113.
57. K. Vaclav, Chem. Abstr., 1974, 80, 117181.
58. I. Matsumoto, J. Yoshizawa and H. Hidaka, JP51105073, 76, 1976; Chem.
Abstr., 1977, 86, 89610v.
59. M. V. Korablev and M. A. Evets, Farmakol. Toksikol., 1977,40, 603 (in
Russian); Chem. Abstr., 87, 1977, 87, 177859j.
60. S. -L. Cao, Y. Wang, L. Zhu, J. Liao, Y. -W. Guo, L. -L. Chen, H. -Q. Liu and
X. Xu, Eur. J. Med. Chem., 2010, 45, 3850.
61. X. -J. Wang, H. -W. Xu, L. -L. Guo, J. -X. Zheng, B. Xu, X. Guo, C. -X.
Zheng and H. M. Liu, Bioorg. Med. Chem. Lett., 2011, 21, 3074.
Page 52
Chapter II
109
62. F. Merchan, J. Garin and E. Melendez, Synthesis, 1982, 590.
63. C. Len, A. -S. Boulogne-Merlot, D. Postel, G. Ronco, and P. Villa, J. Agric.
Food Chem., 1996, 44, 2856.
64. R. -T. Li, P. -Y. Ding, M. Han and M. -S. Cai, Synth. Commun., 1998, 28, 295.
65. S. H. Thang, (Bill) Y. K. Chong, R. T. A. Mayadunne, G. Moad and E.
Rizzardo, Tetrahdron Lett., 1999, 40, 2435.
66. M. Koketsu, T. Otsuka and H. Ishihara, Phosphorus Sulfur and Silicon, 2004,
179, 443.
67. A. R. Katritzky, S. Singh, P. P. Mohapatra, N. Clemens and K. Kirichenko,
Arkivoc, 2005, (ix), 63.
68. N. Azizi, B. Pourhasan, F. Aryanasab and M. R. Saidi, Synlett, 2007, 1239.
69. D. Chaturvedi, N. Mishra and V. Mishra, J. Sulfur Chem., 2007, 28, 39.
70. Y. Liu and W. Bao, Tetrahedron Lett., 2007, 48, 4785.
71. H. Firouzabadi, N. Iranpoor and M. Abbasi, Tetrahedron, 2009, 65, 5293.
72. L. D. S Yadav, R. Patel, Vishnu and P. Srivastava, Tetrahedron Lett., 2009,
50, 1335.
73. A. Z. Halimehjani, K. Marjani and A. Ashouri, Green Chem., 2010, 12, 1306.
74. N. Azizia, F. Aryanasab, L. Tourkian and M. R. Saidi, Synth. Commun., 2011,
41, 94.
75. O. M. Singh and N. S. Devi, Synh. Commun., 2011, 41, 516.
76. A. R. Katritzky, S. Singh, P. P. Mohapatra, N. Clemens and K. Kirichenko,
Arkivoc, 2005, (ix), 63.
77. P. G. M. Wuts, T. P. Greene and T. W. Greene’s Protecting Groups in
Organic Synthesis, 4rd ed.; Wiley Interscience; New York, 2006, Chap. 4.
78. D. Zhang, J. Chen, Y. Liang and H. Zhou, Synth. Commun., 2005, 35, 521.
79. L. Ronconi, C. Marzano, P. Zanello, M. Corsini, G. Miolo, C. Macca, A.
Trevisan and D. Fregona, J. Med. Chem., 2006, 46, 1648.
Page 53
Chapter II
110
80. (a) B. C. Ranu, A. Saha and S. Banerjee, Eur. J. Org. Chem., 2008, 519; (b) A.
Ziyaei-Halimjani and M. R. Saidi, J. Sulfur Chem., 2005, 26, 149.
81. (a) A. Thuillier and P. Metzner, Sulfur Reagents in Organic Synthesis;
Academic: New York, 1994; (b) E. Fujita and Y. Nagao, Bioorg. Chem., 1977,
6, 287.
82. N. Azizi, F. Ebrahimi, E. Aakbari, F. Aryanasab and M. R. Saidi, Synlett,
2007, 2797.
83. A. Ziyaei-Halimajani and M. R. Saidi, Can. J. Chem., 2006, 84, 1515.
84. Y. Quin, G. -Y. Ma, Y. Yang, K. Cheng, Q. -Z. Zheng, W. J. Mao, L. Shi, J.
Zhao and H. L. Zhu, Bio. Org. Med. Chem., 2010, 18, 4310.
85. (a) M. C. Pirrung and K. D. Sarma, J. Am. Chem. Soc., 2004, 126, 444; (b) H.
Firouzabadi, N. Iranpoor and A. Garzan, Adv. Synth. Catal., 2005, 347, 1925;
(c) H. Firouzabadi, N. Iranpoor and A. Khoshnood, J. Mol. Catal. A: Chem.,
2007, 274, 109.
86. M. Kidwai, A. Jahan and D. Bhatnagar, J. Chem. Sci., 2010, 122, 607.
87. (a) J. Chen, S. K. Spear, J. G. Huddleston, R. D. Rogers, Green. Chem., 2005, 7,
64; (b) G. Molineux, Cancer Treat. Rev., 2002, 28, 13; (c) F. M. Veronese, P.
Caliceti, O. Schiavon and M. Sergi, Adv. Drug Delivery Rev., 2002, 54, 587.
88. (a) S. V. More, M. N. V. Sastry and C. -F. Yao, Green Chem., 2006, 8, 91; (b)
A. Dhakshinamoorthy and K. Pitchumani, Catal. Commun., 2009, 10, 872.
89. L. Wang, H. Jing, X. Bu, T. Chang, L. Jin and Y. Liang, Catal. Commun.,
2007, 8, 80.
90. (a). M. Kidwai, S. Saxena, M. K. R. Khan and S. S. Thukral, Eur. J. Med.
Chem., 2005, 40, 816; (b). M. Kidwai, R. Poddar, S. Bhardwaj, S. Singh and
P. M. Luthra, Eur. J. Med. Chem., 2010, 45, 5031.
91. E. Liu, J. Wu, W. Cao, J. Zhang, W. Liu, X. Jiang and X. Zhang, J.
Neurooncol., 2007, 85, 263.














































![SERBIATRIB ‘13tribolab.mas.bg.ac.rs/proceedings/2013/169-176.pdf · cerium [12] chromium [13] or other elements, ... In the case of metal oxide layers, this is an oxidant agent,](https://static.documents.pub/doc/80x56/60643817275b9976cf6d2766/serbiatrib-a-cerium-12-chromium-13-or-other-elements-in-the-case-of-metal.jpg)










