25
CHAPTER-1 INTRODUCTION AND LITERATURE SURVEY

CHAPTER-1
INTRODUCTION AND LITERATURE SURVEY

Chapter-1
2
Introduction and Literature Survey
1.0 Introduction: In this discussed on biological active compounds and literature on
synthesis of important active intermediates. Dithiazole1 compounds (1-4) have
excellent inhibiting action of matrix metalloprotease (MMPs) activity, and it is
useful for pharmaceutical, cosmetic and skin external compositions. Inhibition of
MMPs activity is important in protecting the basement membrane and various
extracellular matrixes, and improving and preventing skin aging such as wrinkles and
slacks.
Phenyl pyridine2 compounds (5 to 7) has an excellent controlling activity against
plant diseases (fungicidal).
Chapter-1
3
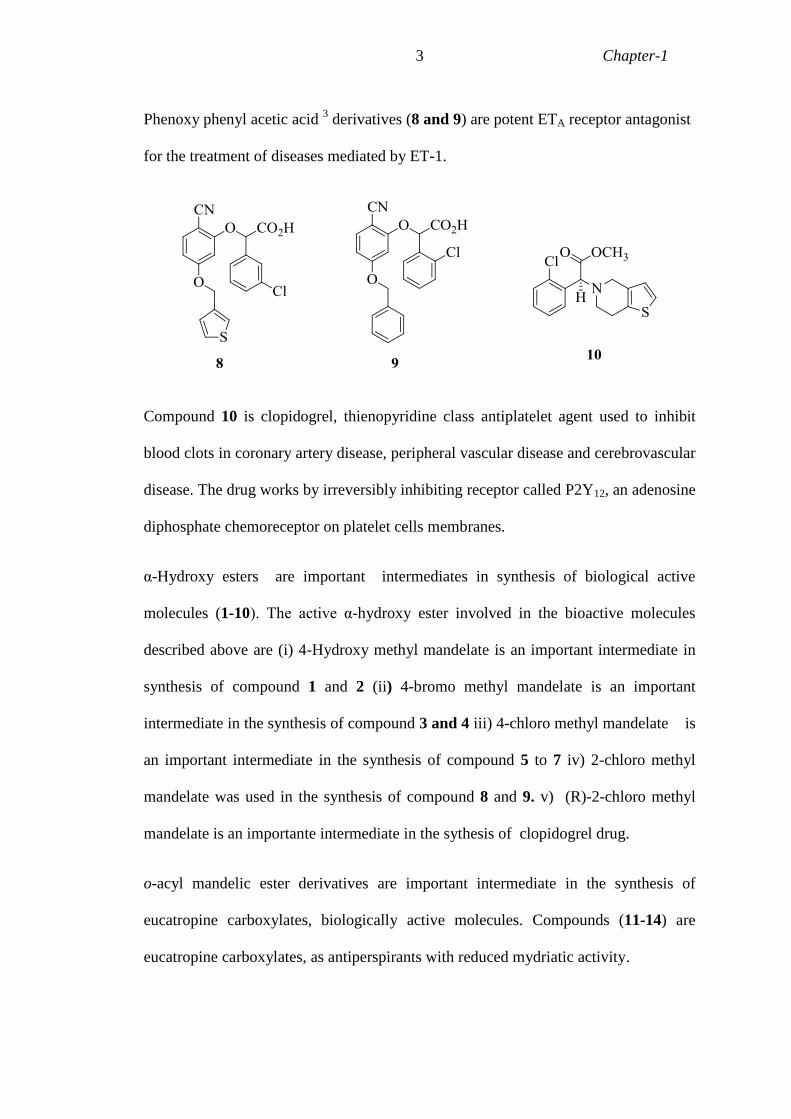
Phenoxy phenyl acetic acid 3 derivatives (8 and 9) are potent ETA receptor antagonist
for the treatment of diseases mediated by ET-1.
Compound 10 is clopidogrel, thienopyridine class antiplatelet agent used to inhibit
blood clots in coronary artery disease, peripheral vascular disease and cerebrovascular
disease. The drug works by irreversibly inhibiting receptor called P2Y12, an adenosine
diphosphate chemoreceptor on platelet cells membranes.
α-Hydroxy esters are important intermediates in synthesis of biological active
molecules (1-10). The active α-hydroxy ester involved in the bioactive molecules
described above are (i) 4-Hydroxy methyl mandelate is an important intermediate in
synthesis of compound 1 and 2 (ii) 4-bromo methyl mandelate is an important
intermediate in the synthesis of compound 3 and 4 iii) 4-chloro methyl mandelate is
an important intermediate in the synthesis of compound 5 to 7 iv) 2-chloro methyl
mandelate was used in the synthesis of compound 8 and 9. v) (R)-2-chloro methyl
mandelate is an importante intermediate in the sythesis of clopidogrel drug.
o-acyl mandelic ester derivatives are important intermediate in the synthesis of
eucatropine carboxylates, biologically active molecules. Compounds (11-14) are
eucatropine carboxylates, as antiperspirants with reduced mydriatic activity.

Chapter-1
4
Both α-hydroxy esters and o-acyl derivatives of mandelic esters are important
synthetic intermediates in bioactive molecules. This thesis emphasize on synthesis of
novel α-hydroxy esters , o-acyl derivatives of mandelic esters and analogues of
mandelic acid with variety of 4-substitued benzoic acids mediated by 1,3-bis(2,4,6-
trimethylphenyl)imidazolium chloride and triethyl amine via generating N-
heterocyclic carbene insitu .
1.1 Literature on α-Hydroxy esters and o-acyl derivatives of mandelic esters:
α-Hydroxy esters are important synthetic intermediates, that can be obtained through
transition metal catalyzed hydrogenation,4
bioreduction with baker’s yeast reductase
or Escherichia coli cells,5 and chemical reduction by β-
chlorodiisopinocampheylborane,6 titanium(II) porphyrincomplexes
7 or sodium
dithionite.8, reduction of α-keto esters with alkyl phosphines, such as
trimethylphosphine or diphenylmethylphosphine,9 reduction of α-keto esters with
aldehyde catalyzed by N-heterocyclic carbenes.10

Chapter-1
5
Only one literature data is available for o-acyl derivatives of mandelic acid esters,
prepared by hydroacylation of α-keto esters with aldehydes catalyzed by N-
heterocyclic carbenes.10
1.1.1 Limitations on synthesis of α-hydroxy ester
Transition metals are expensive and metal recycling is not possible.
Bio reduction using baker’s yeast can be done in gram level. For large scale
require bioreactors which are too expensive.
Titanium (II) phorphyrino complexes are expensive and metal catalyst poisons
the product.
Phosphine complexes resulted less yield and more byproducts were formed.
1.1.2 Synthesis of α-hydroxy esters and o-acyl mandelic esters mediated by NHC
In 2006 Karl A Scheidt10
and co-workers reported hydroacylation of α-keto esters
with aldehydes catalyzed by N-heterocyclic carbenes. In this process the carbene
facilitates selective catalytic oxidation of a C-H bond with concominant reduction of a
ketone. The combination of α-keto ester with benzaldehyde in presence of 15 mol%
triazolium salt and DBU in methanol produces methyl mandelate in excellent yield
96%. In presence of aprotic solvent, such as dichloromethane, and 10 mol%
triazolium salt affords the hydroacylation product in 78% yield.

Chapter-1
6
The proposed reaction path way. A common premise in this process is that tetrahedral
intermediate 6 resulting from addition of NHC to an aldehyde and the collapse of this
intermediate to afford acyl heteroazolium species 7 with concominant formation of a
hydride equivalent. The resulting alcohol 4 produced after a reduction step can
undergo an acylation event with the acyl iminium species formed insitu 7 thus
promoting catalyst turnover and yielding an overall hydroacylation process catalyzed
by an organic molecule.
Mechanism for Hydroacylation of activated ketones(o-acyl mandelic esters
from α-keto esters) catalyzed by NHC.
Limitations on Synthesis of α-hydroxy esters and o-acyl mandelic esters
mediated by NHC (triazolium precatalyst)
1) Triazolium catalyst was costlier and loading quantity was more. 2) Usage of
chlorinated solvents. 3) Lengthy reaction time. 4) Process is capable for electron rich
aldehydes.

Chapter-1
7
1.2 Introduction N-heterocyclic carbenes
In this section presents an overview of literature survey on the synthesis and
applications of NHCs in organic chemistry. The present survey captures the
information on many NHCs as synthetic tool for our research work. N-Heterocyclic
carbene catalyst as recently emerged as an important field in organic chemistry. Two
new strategies have been developed to advance the use of N-heterocyclic carbenes
(NHCs) as Lewis-base organic catalyst. The first approach utilizes NHCs to catalyze
the generation of homoenolates and the conjugated Breslow intermediate is added to
different electrophiles to provide various organic compounds. The second method
employs NHCs to catalyze the hydroacylation of activated ketones by using an
aldehyde as a hydride equivalent and as acylating agent.
1.3 Review on N-heterocyclic carbenes:
In 1958, Breslow recognized the role of N-heterocyclic carbenes as nucleophilic
catalysts in enzymatic reactions. His seminal work showed that the vitamin B1
enzyme cofactor thiamine 111
, a naturally occurring thiazolium salt, plays a crucial
role in biochemical transformations. As thiamine diphosphate, it catalyzes the
decarboxylation of pyruvic acid to active acetaldehyde as well as the benzoin
condensation of aromatic aldehydes. The active species involved in this reaction was
found to be the thiazolylidene 2. In early 1960s, Wanzlick and co-workers realized
that the stability of carbenes could be dramatically enhanced by the presence of
amino substituents, and they attempted to prepare a carbene center at C-2 of the
imidazole ring. However, only the dimeric electron-rich olefin was isolated.

Chapter-1
8
Figure1.1: Thiazolylidene carbene
Later on, Wanzlick’s group demonstrated that potassium tert-butoxide can
deprotonate imidazolium salts to afford imidazol-2-ylidenes, which can be trapped
with phenyl isothiocyanate and mercury salts. However, Wanzlick’s group never
reported the isolation of the free carbene.
1.3.1 Synthesis of Imidazole carbene
Arduengo et al12
, 13
. In 1991 isolated a stable crystalline N-heterocyclic carbene by
the deprotonation of 1,3-di(1-adamantyl)imidazolium chloride with sodium or
potassium hydride in the presence of a catalytic amount of either potassium tert-
butoxide or dimethyl sulfoxide (Scheme1) . The structure was unequivocally
established by single-crystal X-ray analysis, and the carbene was found to be
thermally stable, which stimulated extensive research in this field. Later Arduengo et
al. demonstrated that stable carbenes can be prepared by the deprotonation of
imidazolium salts bearing less bulky substituents in the 1- and 3-positions.

Chapter-1
9
1.3.2 Synthesis of Chloro Imidazolium carbene
In 1997 Arduengo et al14
. prepared first air stable carbene (Scheme2) from 1,3-
dimesityl-4,5-dihydro-1H-imidazol-3-ium chloride 5 with sodium hydride in presence
of catalytic amount of dimethyl sulfoxide resulted 6. The carbene 7 generated from 6
upon treatment with carbon tetrachloride. Carbene 7 exposed to air did not show any
decomposition after two days. The stability of 7 was explained on the basis of the
electronegative effect of the chlorine atoms, which reduces the reactivity and makes
the carbene air stable. (as shown in scheme 2).
1.3.3 Synthesis of Bromo Imidazolium carbene
Cole15
and co-workers have reported the synthesis of bromo analogues of 8 from 6 by
treatment with carbon tetrabromide in tetrahydrofuran solvent at 25 ºC. The carbene 8
(Scheme 3) was characterized by X-ray analysis and found stable in air.

Chapter-1
10
1.3.4 Synthesis of Bis carbenes and Tris carbenes
Hermann16, 17
and co-workers have developed an efficient route for synthesis of bis
and tris carbenes, NaH in liquid ammonia was used to convert the azolium salts to
their corresponding carbenes in a homogeneous phase. An illustration of this strategy
is the synthesis of the stable bis carbene 10 (Scheme 4) and the tris carbene 12
(Scheme 5).
1.3.5 Synthesis of Triazole carbenes
Enders18
and co-workers reported the first synthesis of the crystalline triazole
carbene 15 by the thermal decomposition of the 5-methoxy triazole 14 (Scheme 6).

Chapter-1
11
1.3.6 Synthesis of Pendant Alkene N-heterocyclic carbene
Furstner19
and co-workers reported the synthesis of alkyl-substituted N-heterocyclic
carbenes consisting of pendant alkenes and C-H acidic sites; structure of these
compounds (17&18) has been established by single crystal X-ray analysis
(Scheme 7).
1.3.7 Synthesis of Six membered N-heterocyclic carbenes
Bertrand20
reported a novel procedure using bis(trimethylsilyl)mercury for generating
diaminocarbenes, and it has been applied to the synthesis of the stable six membered
N-heterocyclic carbenes (Scheme 8) . The generality of this reaction remains to be
established.
The electronic properties of NHCs are a key determinant of the unique reactivity of
these catalysts. Lewis bases are normally considered as single electron-pair donors.
However, the singlet carbenes of NHCs are distinct Lewis bases that have both

Chapter-1
12
basicity and acidity characteristics. These attributes allow for the generation of a
second nucleophile in the flask. Nucleophilic addition of the carbene to an aldehyde
results in the formation of a new nucleophile. The “doubly” nucleophilic aspect is
unique to the carbenes. The combination of these characteristics allows NHCs to react
as powerful nucleophiles, which has driven the development of a distinct class of
catalytic processes during the last decade.
1.4 Applications of N-heterocyclic carbenes
1.4.1 Acylation of aryl fluorides catalyzed by imidazol-2- ylidene
Utility of NHCs catalysts in nucleophilic aromatic substitution reactions was
demonstrated by Miya-shita et al.21
on the acylation of aryl fluorides catalyzed by
imidaol-2-ylidene (Scheme 9).
1.4.2 NHC-catalyzed Ring expansion of Oxacycloalkane-2- carboxaldehydes
Imidazolium carbenes catalyze the ring-expansion lactonization of Oxacycloalkane-2-
carboxaldehydes. To oxacycloalkane-2-carboxaldehyde in anhydrous CH2Cl2 was
added 1,3-bis-(2,6-diisopropylphenyl)imidazolinium chloride followed by DBU under
nitrogen at room temperature (Scheme 10)22
.

Chapter-1
13
Figure 1.2: Mechanism for NHC-Catalyzed Ring expansion of Oxalcycloalkane-2-
carboxaldehydes
Mechanism of proposed ring-expansion reaction catalyzed by NHC would combine
with an oxacycloalkane-2-carboxaldehyde to form a “Breslow intermediate” 27
poised for ring opening. Following the Tautomerization of ring-opened intermediate
28 to the activated carboxylate 29, intramolecular attack of the alkoxide moiety
would provide the desired lactone 26 and regenerate the NHC catalyst.
1.4.3 Asymmetric Intramolecular Benzoin Reaction
The asymmetric intramolecular benzoin reaction by using aldehyde-ketone 30, show a
good enantiomeric excess, 67%ee for the resulting acyloin 32 as well as high yields
(68%). First experiment with substrate 33 resulted the five-membered cyclic acyloin
34 resulted in an excellent yield and a good enantiomeric excess (74%). When the
reaction was carried out at 58ºC, the enantiomeric excess could be slightly increased
to 75% (Scheme 12).23

Chapter-1
14
1.4.4 Intermolecular N-Heterocyclic carbene catalyzed Hydroacylation of
Arynes
Reaction of 4-bromobenzaldehyde 35 with the aryne generated in situ from 2-
trimethylsilylaryl triflate 36 and 2.0 equivalents each of KF and [18] crown-6.
Reacting these substrates in the presence of the carbene generated from 37 by
deprotonation using K2CO3 resulted in the formation of 4-bromobenzophenone 38 in
60% yield (Scheme 14)24
.

Chapter-1
15
1.4.5 Redox Amidations of α- Functionalized Aldehydes with Amines
The redox amidation of a formyl cyclopropane 39 with benzyl amine 40, catalyzed by
a combination of triazolium precatalyst 41 and DBU was resulted amide 42. In the
absence of a suitable promoter, less than 5% of the desired amide was isolated, and
NMR analysis revealed that the bulk of the aldehyde starting material was converted
to the corresponding imine 44 (Scheme 15)25
. Reaction pathways for imidazole-
promoted redox amidation of α-functionalized aldehydes (Scheme 16).
1.4.6 β’-amino enones via carbonyl umpolung reaction of enals with aziridines
β’-amino-α,β unsaturated ketones 48 involves carbonyl umpolung of enals 45 in
which carbonyl carbon attacks nucleophilically on electrophilic terminal aziridines
46 regioselectively (Scheme 17)26
.

Chapter-1
16
Scheme18 describes the mechanism for the formation of ketones 48 by initial
addition of NHC 53 to α,β-unsaturated aldehyde 45 followed by proton shift in 49 to
generate conjugated acyl anion equivalent 50. On the basis of the literature reports
and the work of Stetter and co-workers on enals, we reasoned that an appropriately
substituted NHC could shepherd the umpolung reactivity of enals to the carbonyl
group presumably by sterically blocking the β-position without posing any significant
steric hindrance at the acyl carbon. The NHC 47 mediated reaction of enals 45 with
aziridines 46 proceeded well via acyl anion to afford β’-amino ketones.
Figure 1.3: Mechanism for formation β’-amino enones from enals and aziridines
catalyzed by NHC

Chapter-1
17
1.4.7 NHC-catalyzed Oxidative esterification:
Stephen J. Connon27
first reported oxidative esterification of aldehydes with
equimolar amounts of primary and secondary alcohols at room temperature catalyzed
by N-heterocyclic carbene derived from thiazolium salt (Scheme 19).
Initial Investigation began with the oxidative esterification of benzaldehyde with
methanol as solvent catalyzed by the thiazolium salt in presence of catalytic loadings
of base and one equivalent of acridine as oxidant at ambient temperature. Initial
experiments afforded ester 55 in moderate yield. More efficient oxidative
esterification could be carried out in THF using one equivalent of alcohol without
requiring elevated reaction temperatures. Mechanism for nucleophilic carbene
catalyzed oxidative esterification presented in scheme 20. Deprotonation of
thiazolium salt 56 gives rise to carbene 57, the addition of which to benzaldehyde 54
gives the Breslow intermediate 58 which in the presence of suitable oxidant can be
diverted from benzoin condensation pathway through the formation of 2-benzoyl
thiazolium ion 60 which is capable of transferring its acyl group to an alcohol
nucleophile to regenerate the catalyst.

Chapter-1
18
1.4.8 one-pot-four-component synthesis of pyrrole derivatives
Muller and co-workers28
reported four-component synthesis of pyrrole derivatives.
Chalcones 63 formed from electron-poor aryl halides and aryl propargyl alcohols in a
coupling/isomerizaion sequence, can be transformed in a one-pot sequence first to the
corresponding1, 4 diketones 65 and then to pyrrole derivatives 66.
1.4.9 Hydroacylation of activated ketones catalyzed by N-heterocyclic carbenes
One established approach to selectively oxidize C-H bonds utilizes transition metals
capable of undergoing oxidative addition pathways. Rhodium (I) –catalyzed

Chapter-1
19
intramolecular hydroacylations of alkenes reported from Bosnich, Larock, and Jun
etc. Interestingly, the development of analogous catalytic hydroacylation processes
involving aldehydes and carbonyl π systems has not received the same attention. Karl
A Scheidt29
and co-workers reported hydroacylation of α-keto esters 68 with
aldehydes 67 catalyzed by N-heterocyclic carbenes. In this process the carbene
facilitates selective catalytic oxidation of a C-H bond with concominant reduction of a
ketone. The combination of α-keto ester 68a with benzaldehyde 67a in presence of 15
mol% triazolium salt 69 and DBU in methanol produces methyl mandalate 70a in
excellent yield 96%. In presence of aprotic solvent, such as dichloromethane, and 10
mol% 69 affords the hydroacylation product 71a produced 78% yield, which is the
product expected from Scheme 22.
The proposed reaction path way (Scheme 23). A common premise in this process is
that tetrahedral intermediate 72 resulting from addition of NHC to an aldehyde and
the collapse of this intermediate to afford acyl heteroazolium species 73 with
concominant formation of a hydride equivalent. The resulting alcohol 70 produced
after a reduction step can undergo an acylation event with the acyl iminium species
formed insitu 73 thus promoting catalyst turnover and yielding an overall
hydroacylation process catalyzed by an organic molecule.

Chapter-1
20
Figure 1.4: Mechanism for Hydroacylation of activated ketones catalyzed by NHC.
1.4.10 Highly Diastereo- and Enantioselective Additions of Homoenolates to
Nitrones Catalyzed by N-Heterocyclic carbenes
Karl A.Scheidt30
and co-workers reported diastereo and enantioselective addition of
homoenolates to nitrones. The reaction of cinnamaldehyde 74a and diphenyl nitrone
75a screened with different triazolium salts (Scheme 24). While thiazolium and
imidazolium salts did not produce desired products, the use of achiral triazolium salt
76 , generated 78a with high levels of stereoselectivity (8:1 dr, 87% ee at 0°C) but
only moderate yield. Lowering the temperature (-25 °C) provided increased
selectivity (20:1 dr, 93% ee) and yield of 78a with 20mol% of 76 necessary for
consumption of 74a.

Chapter-1
21
Mechanism involves the addition of the homoenolate equivalent (79, formed in situ
from the combination of the NHC and unsaturated aldehyde 74) to the nitrone 75.
After this stereochemical-determining step, catalyst turnover is promoted by an
intramolecular acylation after the tautomerization of enol 80 to acyl azolium 81.
(Scheme 26).
Figure1.5: Mechanism for formation of nitrones catalyzed by NHC.

Chapter-1
22
1.4.11 Conversion of α,β-Unsaturated Aldehydes into Saturated Esters: An
Umpolung Reaction Catalyzed by nucleophilic carbenes
Audrey Chan and Karl A. Scheidt31
reported the conversion of α, β-unsaturated
aldehydes into homoenolates catalyzed by nucleophilic carbenes generated from
precatalyst 83. The use of optimal alcohol would presumably serve a double purpose
as the electrophile source and subsequent nucleophile. To probe this possibility,
cinnamaldehyde 82 and various alcohols (ethanol, phenol and benzyl alcohols) were
heated in toluene in the presence of catalytic quantity of 83 and DBU. With ethanol
isolated desired ester 84 (57% yield).
Mechanism involves the addition of nucleophilic carbene to the carbonyl compound
should generate tetrahedral intermediate 85, and subsequent hydrogen migration
would generate the reactive dienamine 86. The normal nucleophilic character of the
carbonyl carbon is then extended to the β-position and, in the presence of electrophile
should produce enol 87. After tautomerization of 87 to activated ester 88, the attack of
a suitable nucleophile completes the catalytic cycle.

Chapter-1
23
Figure 1.6: Mechanism for conversion of Unsaturated Aldehydes into Saturated
Esters catalyzed by NHC carbenes
1.4.12 Sonogashira coupling catalyzed by Palladium-Carbene Complex for aryl
bromide and alkyne32
Palladium(II) complex bearing an N-heterocyclic carbene and triphenylphosphine
failed to promote Sonogashira cross-coupling, Instead providing diyne and enyne
products, formed through alkyne self-coupling. Because of the possibility of
byproduct generation and other functional group tolerances, instead of applying the
standard reaction conditions with amine as solvent, investigated several other organic
solvents in the palladium-carbene complex 90 catalyzed Sonogashira reaction of 4-
bromo-acetophenone with phenylacetylene. The reaction was carried out at elevated
temperature with Et3N as the base. The use of DMF as solvent gave excellent yields.
The palladium complex 90 was prepared from Pd(OAc)2 and two equiv of carbamoyl

Chapter-1
24
imidazolium salt in refluxing THF. Orange-yellow crystals of 90 were obtained after
purification via flash chromatography. Sonogashira coupling of bromo acetophenone
89 with phenyl acetylene using 1mol% of NHC 90, PPh3 (1 mol%) , CuI (2 mol%)
and Cs2CO3(1.2 eq) in DMF solvent at 80°C for 24h resulted compound 91 in 99%
yield.
1.4.13 Sonogashira coupling using Bulky Palladium –Phenanthryl Imidazolium
carbene catalysis33
NHC ligands, due to their enhanced stability and reactivity, have been successfully
applied to palladium and other transition metal-catalyzed reactions. A Dramatic effect
is now demonstrated with the novel phenanthrenyl NHC ligands, produces
Sonogashira coupling product in high yields at moderate temperature and short
reaction times. Aryl halide (1.0 eq), terminal alkyne (1.4 eq), PdCl2(PPh3)2 (3 mol %),
NHC 92 ( 3 mol %), and 18-crown-6 (0.150 mmol) were treated with tBuOK
(0.150mmol) in anhydrous THF (5 mL). The resulting suspension was stirred at room
temperature for the aryl iodides or at reflux for the bromides. The reaction mixture

Chapter-1
25
was washed (water, 5 mL) and extracted with ethyl acetate followed by silica gel
chromatography (EtOAc/hexanes, 0-10%).
1.1.5 Objective of our work: Our objective is to synthesize novel α-hydroxy esters
and o-acyl mandelic esters using (i) readily available and cheaper N-heterocyclic
carbenes (by screening various types of NHCs) (ii) less quantity of catalyst loading
(iii) usage of non-chlorinated solvents (iv) process is capable for both electron rich
and deficient aldehydes. (v) Construction of small organic molecules by multiple
bond formation mediated by NHC.
















