55
Chapter-1 Introduction
Chapter-1
Introduction

Chapter 1
PhD Thesis 2
1.1. Introduction
1.1.1. Carbon monoxide
Carbon monoxide (CO) is a colorless, odorless and tasteless gas which is
highly toxic to humans and animals in higher quantities [1-2]. It consists of one
carbon atom and one oxygen atom, connected by a triple bond which consists of two
covalent bonds as well as one dative covalent bond. It is the simplest Oxo carbon in
nature, and is an anhydride of formic acid [3].
Carbon monoxide is the twelfth most abundant gas in the atmosphere with
about 1.2 × 10-5% in the lower atmosphere. Worldwide, the largest source of carbon
monoxide is natural in origin, due to photochemical reactions in the troposphere
which generate about 5 x 1012 kilograms per year [4]. Other natural sources of CO
include volcanoes, forest fires, and other forms of combustion. Carbon monoxide is
produced from the partial oxidation of carbon-containing compounds, it forms when
there is not enough oxygen to produce carbon dioxide (CO2), such as when operating
a stove or an internal combustion engine in an enclosed space. In the presence of
oxygen, carbon monoxide burns with a blue flame, producing carbon dioxide.
Other than natural sources CO was also produced by daily human life. The
typical concentrations of carbon monoxide in natural and artificial environments are
given in Table 1.1.
Table 1.1. Concentration of CO in different human environment
Concentration (ppm)
Source
0.1 Atmosphere level [5] 0.5-5 Average level in homes [6] 5-15 Near properly tuned gas stoves [7] 100-200 Exhaust from automobiles in the central area of
Mexico City [8] 5,000 Home wood fire [7] 7,000 Undiluted warm car exhaust without a catalytic
converter[7]
1.1.2. Biological and physiological properties of CO
Carbon monoxide is produced naturally by the human and animal body as a
signaling molecule. Thus, carbon monoxide may have a physiological role in the body,
such as a neurotransmitter or a blood vessel relaxant [9]. Because of the role of carbon

Chapter 1
PhD Thesis 3
monoxide in the body, abnormalities in its metabolism have been linked to a variety
of diseases, including neurodegenerations, hypertension, heart failure and
inflammation. But CO has adverse effects if it is inhaled. CO forms a very strong
bond with the iron atom of hemoglobin in the blood, and once bonded cannot be
dislodged (unlike oxygen and CO2 which detach easily and reversibly). As more and
more CO is inhaled, more red blood cells get 'used up' leaving fewer and fewer
available to carry the vital oxygen to the muscles, tissues and brain of the animal. If
not treated immediately with O2 and possibly a blood transfusion, the animal would
die of asphyxiation.
1.1.3. Industrial production of CO
A major industrial source of CO is producer gas, a mixture containing mostly
carbon monoxide and nitrogen, formed by combustion of carbon in air at high
temperature when there is an excess of carbon. In an oven, air is passed through a bed
of coke. The initially produced CO2 equilibrates with the remaining hot carbon to give
CO. The final product consists of three gases, carbon monoxide, carbon dioxide, and
nitrogen in the ratio of 6:1:18. Above 800 °C, CO is the predominant product.
O2 + 2 C → 2 CO (∆H = −221 kJ/mol) (1.1)
Another source is water gas, a mixture of hydrogen and carbon monoxide
produced via the endothermic reaction of steam and carbon. The products in this case
are hydrogen (50%), carbon monoxide (40%), carbon dioxide (5%) and other gases
(5%) [10].
H2O + C → H2 + CO (∆H = +131 kJ/mol) (1.2)
Other similar synthesis gases (H2+CO) can be obtained by partial oxidation of
natural gas and other hydrocarbons. Carbon monoxide is also a byproduct of the
reduction of metal oxide ores with carbon, shown in a simplified form as follows.
MO + C → M + CO (1.3)
1.1.4. Applications of CO
CO is very important in industry, since it is a precursor to a number of
important organic chemicals (Fig.1.1). Its application includes the water-gas shift
reaction for the production of high purity hydrogen, Monsanto process for the
synthesis of acetic acid, Tennessee-Eastman acetic anhydride process, Fischer–

Chapter 1
PhD Thesis 4
Tropsch synthesis of hydrocarbons, Mond process for purification of nickel,
hydroformylation and hydroaminomethylation.
CO+H2
CH3OH
CH4
C1-Chemicals
Acetic acid
Acetic anhydride
Vinylacetate
Ethylene glycol
Homologation
Methylformate
CO2
Fine Chemicals Hydrogen or CO
Carbonylation
Hydroformylation
Fischer-Tropsch products
Methylformate
Chemicals
Gasoline Fine chemicals Olefins, Aromatics
Hydromainomethylation
Fig. 1.1. C1 building blocks
1.1.4.1. Water-gas shift reaction
The water-gas shift reaction [11] is the reaction in which carbon monoxide
reacts with water vapor to form carbon dioxide and hydrogen.
CO(g) + H2O(v) → CO2(g) + H2(g) (1.4)
The water-gas shift reaction is an important industrial reaction. It is often used
in conjunction with steam reforming of methane or other hydrocarbons, which is
important for the production of high purity hydrogen for use in ammonia synthesis.
The process is often used in two stages, stage one a high temperature shift (HTS) at
350 °C and stage two a low temperature shift (LTS) at 190–210 °C. Standard
industrial catalysts for this process are iron oxide promoted with chromium oxide for
the HTS step and copper on a mixed support composed of zinc oxide and aluminum
oxide for the LTS shift step.

Chapter 1
PhD Thesis 5
1.1.4.2. Monsanto acetic acid process
The Monsanto process is an important method for the production of acetic acid
by catalytic carbonylation of methanol. This process operates at a pressure of 30–60
atm and a temperature of 150–200 °C gave selectivity greater than 99%. The
Monsanto process has largely been superseded by the Cativa process, a similar
iridium-based process industrialized by BP Chemicals Ltd. which is more economical
and environmentally friendly [12].
1.1.4.3. Tennessee-Eastman acetic anhydride process
Acetic anhydride is produced by carbonylation of methyl acetate in a process
that was inspired by the Monsanto acetic acid synthesis [13].
CH3CO2CH3 + CO → (CH3CO)2O (1.5)
In this process lithium iodide converts methyl acetate to lithium acetate and
methyl iodide, which in turn affords through carbonylation of acetyl iodide. Acetyl
iodide reacts with acetate salts or acetic acid to give the product. Rhodium iodides and
lithium salts are employed as catalysts. The reaction is conducted under anhydrous
conditions in contrast to the Monsanto acetic acid synthesis because acetic anhydride
is not stable in aqueous medium.
1.1.4.4. Fischer–Tropsch synthesis of hydrocarbons
The Fischer–Tropsch process (or Fischer–Tropsch synthesis) is a set of
chemical reactions that convert a mixture of carbon monoxide and hydrogen into
liquid hydrocarbons [14].
(2n+1) H2 + n CO → CnH(2n+2) + n H2O (1.6)
This key process of gas to liquids technology produces a petroleum substitute,
typically from coal, natural gas, or biomass for use as synthetic lubrication oil and as
synthetic fuel. The F-T process has received intermittent attention as a source of low-
sulfur diesel fuel and to address the supply and/or cost of petroleum-derived
hydrocarbons. A variety of synthesis gas compositions can be used. For cobalt-based
catalysts the optimal H2:CO ratio is around 1.8-2.1. Iron-based catalysts stimulate the
water-gas-shift reaction and thus can tolerate significantly lower ratios. This reactivity
can be important for synthesis gas derived from coal or biomass, which tend to have
relatively low H2:CO ratios (<1).

Chapter 1
PhD Thesis 6
1.1.4.5. Mond process
The Mond Process, sometimes known as the Carbonyl process is a technique
created by Ludwig Mond in 1890 to extract and purify nickel [15]. The process was
used commercially before the end of the 19th century. It is done by converting nickel
oxides (nickel combined with oxygen) into pure nickel. This process makes use of the
fact that carbon monoxide complexes with nickel readily and reversibly to give nickel
carbonyl. No other element forms a carbonyl compound under the mild conditions
used in the process.
This process has three steps:
1. Nickel oxide is reacted with syngas at 200 °C to remove oxygen, leaving impure
nickel. Impurities include iron and cobalt.
NiO (s) + H2 (g) → Ni (s) + H2O (g) (1.7)
2. The impure nickel is reacted with excess carbon monoxide at 50-60 °C to form
nickel carbonyl.
Ni (s) + 4 CO (g) → Ni(CO)4 (g) (1.8)
3. The mixture of excess carbon monoxide and nickel carbonyl is heated to 220-
250 °C. On heating, tetracarbonyl nickel decomposes to give nickel:
Ni(CO)4 (g) → Ni (s) + 4 CO (g) (1.9)
1.1.4.6. Hydroformylation and hydroaminomethylation
The reaction between olefinic double bond and the mixture of hydrogen and
carbon monoxide (synthesis gas) leading to the formation of linear and branched
aldehydes as primary products is known as hydroformylation reaction or Oxo reaction
(Scheme 1.1). Adkins has introduced the term hydroformylation, since there is an
attack of hydrogen and formyl group (–CHO) at the unsaturated center of the carbon
chain of the alkene molecule [16]. The primary products of hydroformylation
reactions are aldehydes only. However, the formation of other Oxo products is also
known subsequent to the formation of aldehydes [17].

Chapter 1
PhD Thesis 7
Scheme 1.1. Hydroformylation reaction
Hydroaminomethylation (Scheme 1.2) is the single pot synthesis of amines
from an olefin, amine (alkyl amines, morpholine, pyrollidine etc) and syngas (H2, CO)
[18]. It is an elegant, atom economic, efficient process for the synthesis of amines. It
is the tandem reaction of hydroformylation in which the olefin reacts with CO and H2
to form aldehydes which will then react with amine to give an imine or enamine. This
will in turn get hydrogenated to the final amine. Hydroaminomethylation is closer to
applications as an intermediate for the synthesis of bulk, nylon, drugs and fine
chemicals.
Scheme 1.2. Hydroaminomethylation of olefin (R1 = R2 = R3 = H, alkyl or aryl group)
1.2. Hydroformylation: Synthesis of aldehydes
Otto Roelen in 1938 while investigating the reaction mechanism of the
Fischer–Tropsch (F–T) synthesis by addition of syngas to ethene could observe a
small amount of propanal formation which led to the discovery of hydroformylation
[19–21]. A small amount of propanal (and diethyl ketone) was formed when a mixture
of ethylene and synthesis gas was passed over a fixed bed of the catalyst containing
cobalt at 150 °C and 100 atm. He also observed that the selectivity of propanal
increased significantly when the reaction was carried out at 51 atm and 150 °C. First
real catalyst for the hydroformylation reaction was the conventional F–T catalyst.
Later it was recognized that cobalt afforded higher conversion and selectivity of

Chapter 1
PhD Thesis 8
propanal when used as a catalyst in homogeneous conditions. Hydroformylation is
one of the most important homogeneously catalyzed reactions performed in industries
along with carbonylation of methanol to acetic acid and oxidation of p–xylene to
dimethyl terephthalate (DMT). Consequently, hydroformylation of alkenes is the most
used and well–understood homogeneous catalytic reactions and is also a subject of
exhaustive review [22–38].
The hydroformylation reactions are done in homogeneous conditions and the
catalysts typically, consist of complex of transition metal atom (M), especially
platinum group metals [39, 40]. These transition metal complexes interact with CO
[41-45] and hydrogen to form metal carbonyl hydride species, which is an active
hydroformylation catalyst. Typically, complexes containing carbonyl ligands are
known as unmodified catalysts. On the other hand, introduction of tailor made ligand
to the transition metals results into modified catalysts.
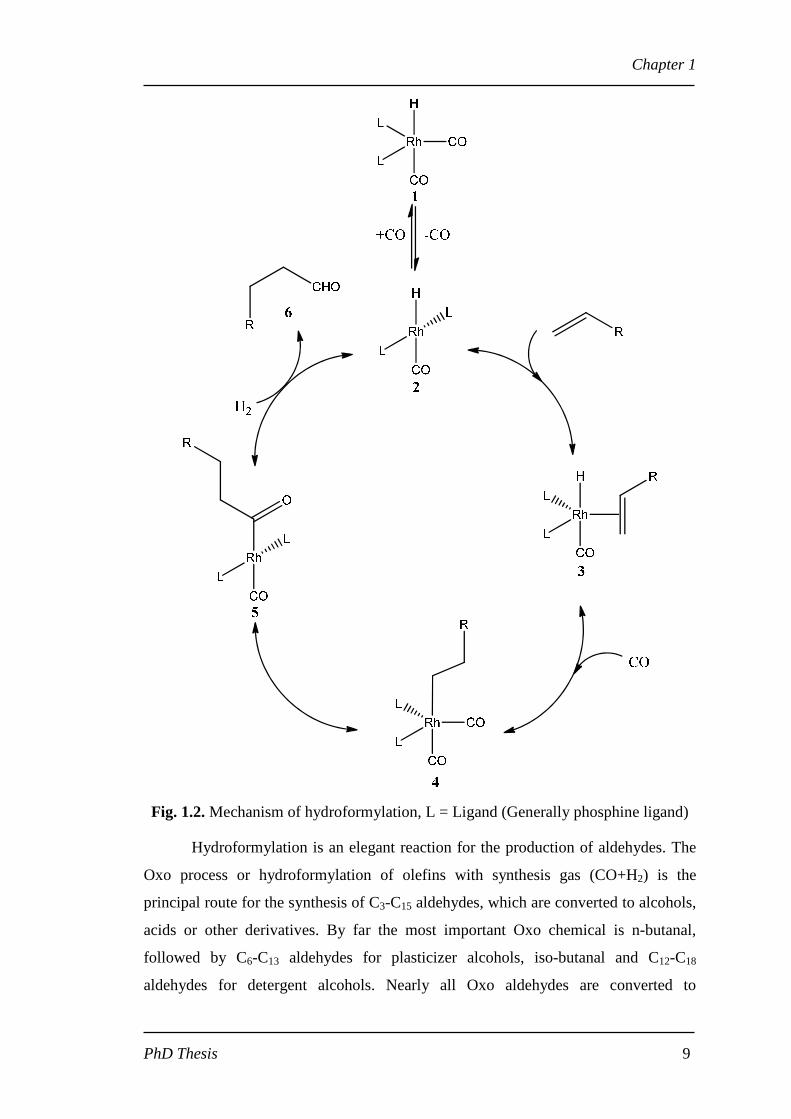
The mechanism for the cobalt-catalyzed hydroformylation was proposed by
Heck and Breslow in 1960s, which is accepted as the general mechanism for Co- and
Rh-catalyzed hydroformylation at present as given in Fig. 1.2 [46]. The first step of
the mechanism is the dissociation of one carbon monoxide ligand from complex (1),
which is the catalyst precursor, leading to the formation of the hydride species
containing an empty coordination site (2). The alkene is then coordinated to complex
(2) and led to the formation of complex (3), and a migratory insertion of the alkene
into the rhodium hydride bond results in the formation of the alkyl species (4).
Subsequently, a CO inserts into the rhodium-alkyl bond resulting in the acyl species
(5) and hydrogenation, via an oxidative addition of hydrogen followed by a reductive
elimination, gives the product aldehyde (6) and regenerates the unsaturated Rh-
complex (2). Furthermore, β-hydrogen elimination of the alkyl species (4) may lead to
isomerization and the formation of less reactive internal alkenes.
Chapter 1
PhD Thesis 9
Fig. 1.2. Mechanism of hydroformylation, L = Ligand (Generally phosphine ligand)
Hydroformylation is an elegant reaction for the production of aldehydes. The
Oxo process or hydroformylation of olefins with synthesis gas (CO+H2) is the
principal route for the synthesis of C3-C15 aldehydes, which are converted to alcohols,
acids or other derivatives. By far the most important Oxo chemical is n-butanal,
followed by C6-C13 aldehydes for plasticizer alcohols, iso-butanal and C12-C18
aldehydes for detergent alcohols. Nearly all Oxo aldehydes are converted to
Chapter 1
PhD Thesis 10
derivatives in plants adjacent to the hydroformylation unit; very small volumes of
Oxo aldehydes are transported.
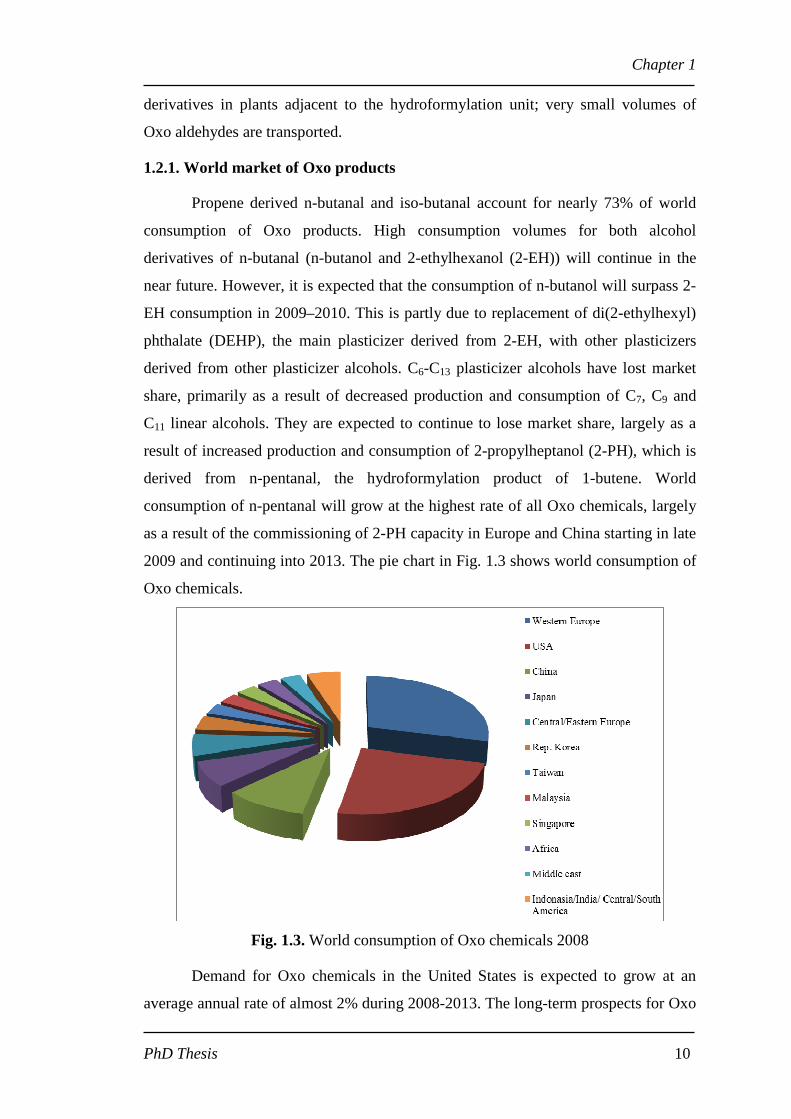
1.2.1. World market of Oxo products
Propene derived n-butanal and iso-butanal account for nearly 73% of world
consumption of Oxo products. High consumption volumes for both alcohol
derivatives of n-butanal (n-butanol and 2-ethylhexanol (2-EH)) will continue in the
near future. However, it is expected that the consumption of n-butanol will surpass 2-
EH consumption in 2009–2010. This is partly due to replacement of di(2-ethylhexyl)
phthalate (DEHP), the main plasticizer derived from 2-EH, with other plasticizers
derived from other plasticizer alcohols. C6-C13 plasticizer alcohols have lost market
share, primarily as a result of decreased production and consumption of C7, C9 and
C11 linear alcohols. They are expected to continue to lose market share, largely as a
result of increased production and consumption of 2-propylheptanol (2-PH), which is
derived from n-pentanal, the hydroformylation product of 1-butene. World
consumption of n-pentanal will grow at the highest rate of all Oxo chemicals, largely
as a result of the commissioning of 2-PH capacity in Europe and China starting in late
2009 and continuing into 2013. The pie chart in Fig. 1.3 shows world consumption of
Oxo chemicals.
Fig. 1.3. World consumption of Oxo chemicals 2008
Demand for Oxo chemicals in the United States is expected to grow at an
average annual rate of almost 2% during 2008-2013. The long-term prospects for Oxo

Chapter 1
PhD Thesis 11
chemicals in Western Europe improved considerably during 2005–2008, as
associations and capacity reductions resulted in improved efficiencies and capacity
utilization. The commissioning of plants for 2-PH and additional iso-nonyl alcohol
(INA) capacity helped to reduce the former dependence on 2-EH. Western European
consumption of Oxo chemicals is predicted to grow at an average annual rate of 2.0%
during 2008–2013. Japanese consumption is forecast to experience 0.9% average
annual growth during 2008–2013. Other Asian consumption, excluding Japan, is
expected to grow at 5.0% annually during the same period; China, India and Taiwan
are the main growth markets in this region. Middle Eastern consumption of Oxo
chemicals is forecast to grow significantly at an average annual rate of 4.8% during
2008–2013, albeit from a small base, largely as a result of increased n-butanol
demand for n-butyl acrylate by late 2010 [47].
1.2.2. Hydroformylation process: Stages of developments
Table 1.2. Developments in hydroformylation processes in different stages
1st Stage 2nd Stage 3rd Stage
Temperature (°C) 120-180 85-130 110-130
Pressure (bar) 200-350 15-60 40-60
Metal concentration
0.4-0.7
(cobalt, wt% of feed)
200-270
(rhodium, ppm)
200-270
(rhodium, ppm)
Ligand Absent or triphenyl
phosphine
Triphenyl
phosphine
Water soluble
ligand
Light products
(wt%)
3-13 - -
Aldehydes (wt%) 70-75 82-95 91-95
Alcohols (wt%) 6-10 5-8 5-9
Heavy ends (wt%) 4-17 - -
As far as hydroformylation catalysts are concerned, three developmental
stages for these catalysts can be visualized (Table 1.2). The first stage of
hydroformylation is exclusively for cobalt based catalytic systems. Second stage is for
the development of ligands, specially triphenyl phosphine, and the shift from cobalt to

Chapter 1
PhD Thesis 12
rhodium as the central metal. Third stage is the development of biphasic catalyst
based on rhodium by modification of ligand [33]. The various ligands used in the
different stages of development are given in Fig. 1.4
Fig. 1.4. Ligands developed at different stages of development in hydroformylation
1.2.2.1. Cobalt based processes (1st stage)
The first stage of development (Table 1.2) is the reaction with cobalt carbonyl
hydrides (HCo(CO)4) as a catalyst, with the pressure range between 200-350 bar to
avoid decomposition of the catalyst and deposition of metallic cobalt. The
temperature was adjusted according to the pressure and the concentration of the
catalyst between 120 and 180 °C to get an acceptable rate of reaction. Cobalt
processes are mostly used in the production of medium to long chain olefins, because
rhodium catalysts dominate the hydroformylation of propene. The presently applied
cobalt processes have reached a high standard of performance. Most of these
processes are pretty similar, the main difference between the cobalt processes are the
separation of product and catalyst. Three of these processes are that of BASF, Exxon
and Shell.
BASF process
The BASF hydroformylation process of propene or higher olefins occurs
under high pressure [48]. The catalyst is in the form of HCo(CO)4. This catalyst will
be separated from the liquid product by addition of oxygen and formic or acetic acid,
leading to an aqueous solution which contains the cobalt predominantly as formate or
acetate. The organic products are withdrawn in a phase separator and the cobalt
solution is concentrated afterwards and sent to the carbonyl generator. The cobalt
losses are compensated. In the reactor stirring is important to accomplish thorough
mixing of the olefins and the aqueous catalyst solution. The best selectivity to linear
aldehydes is obtained at low temperatures. The disadvantage is the process difficulty
and economic aspects of the separation and recyclability of the catalyst.

Chapter 1
PhD Thesis 13
Exxon process
The Exxon process is designed to convert olefins in the range of C6-C12.
Recovery of the catalyst is different than that of BASF process. In the Exxon process
the ‘Kuhlmann’ catalyst cycle technology is applied [49]. This involves two main
steps: the recovery of sodium carbonylate and its regenerative conversion into cobalt
carbonyl hydride. The HCo(CO)4 catalyst reacts with syngas in the reactor under
normal hydroformylation conditions. After the reaction the product mixture is treated
with aqueous alkali, to convert HCo(CO)4 to water-soluble NaCo(CO)4, which is
extracted as aqueous solution from the organic product phase. Then the catalyst is
regenerated by addition of H2SO4. The advantage of the Exxon process is that the
catalyst does not undergo decomposition and enters the reactor in its most active form.
A big disadvantage is that catalyst separation and recovery must be carried out under
CO pressure to preserve the catalyst.
Shell process
In the Shell process reactants in range of C7-C14 are converted using a
phosphine modified cobalt catalyst [50]. In this process the product mixture is
distillated, the organic products leave the distillation column at the top and the
catalyst is recovered at the bottom. Before re-entering the reactor the catalyst recycle
is upgraded with catalyst and phosphine ligand. The benefits of this process are the
high n/iso ratio, the low pressure and the direct formation of alcohols. The drawback
is the low activity of the ligand modified catalyst, which requires a large reactor
volume and difficulties associated with separation of the catalyst.
1.2.2.2. Rhodium based processes (2nd Stage)
Rhodium catalyst processes are used since the 1970s and dominate for
hydroformylation of propene. Rhodium catalysts are more expensive than cobalt
catalysts and have higher activity, but have lower activity in case of branched olefins.
The catalyst used is rhodium with triphenyl phosphine ligand. UCC, BASF and
Mitsubishi use rhodium based hydroformylation process for commercial synthesis.
UCC, BASF and Mitsubishi process
The Union Carbide Corporation (UCC) commercially applies the
hydroformylation of propene in a liquid-recycle process [51]. These plants are called
LPO (Low Pressure Oxo) plants [52]. The reaction takes place in a stainless steel
reactor where the gas and propene are introduced via a feed line and a gas-recycle.

Chapter 1
PhD Thesis 14
The catalyst is dissolved in high-boiling aldehyde condensation products. The product
mixture consists of dissolved gas, aldehydes, rhodium-phosphine complex, free
phosphine ligand and the higher-boiling aldehyde condensation products. The product
mixture passed into a separator and a flash evaporator, where the major part of inert
and unconverted reactants is taken overhead. The flashed-off gases are returned to the
reactor. The liquid stream is heated and is fed to two distillation columns in series.
The gaseous aldehydes are sent over the top and condensed and separated from the
syngas, which is recycled. At the bottom the catalyst solution is separated and
recycled in the reactor. If the feed of the process has a sufficient purity the catalyst
may last more than a year. The BASF and Mitsubishi rhodium based processes are
quite parallel to the UCC process [53, 54].
1.2.2.3. Disadvantages of cobalt and rhodium based homogeneous
hydroformylation and development of biphasic catalysts (3rd Stage)
Both cobalt and rhodium based industrial processes suffer the easy and
effective recycling of the catalyst. The separation of the catalyst and products from
the homogeneous reaction mixture, severe conditions, and low activities of catalysts
were main limitations for cobalt based processes. Development of rhodium-phosphine
catalyst gave higher activities and selectivity with lower temperature and pressure
conditions. But these systems also lack the efficient separation, regeneration and
recyclability of the catalyst.
These disadvantages led to the development of biphasic catalysts. Here the
catalyst will be in one phase (aqueous phase) and reactant and product in the other
phase (organic phase). The reaction occurs at the interface of the two phases as shown
in Fig. 1.5. Development of water soluble ligand triphenylphosphine trisulfonate
sodium salt (TPPTS) was the chief achievement in this stage. The replacement of
triphenyl phosphine with TPPTS led to the development of water soluble rhodium
complex. This complex is used in the Ruhrchemie/Rhône-Poulenc (RCH/RP) process
for hydroformylation of propene.

Chapter 1
PhD Thesis 15
Fig. 1.5. Biphasic hydroformylation of olefin using water soluble
HRhCO(TPPTS)3
Ruhrchemie/Rhône-Poulenc process
The RCH/RP (Ruhrchemie/Rhône-Poulenc) process is an example of a two
phase system. The reactor contains the aqueous catalyst and is fed with propene and
syngas, TPPTS is used as a ligand for the rhodium catalyst complex [55]. After the
reaction the crude aldehyde product is degassed and separated into the aqueous
catalyst solution and the organic aldehyde phase. The heat of the aqueous phase is
then used to produce steam in a heat exchanger. After separation the organic phase is
passed through a stripping column, where the unreacted olefins are separated and sent
back to the reactor. The product mixture is then distilled into n- and iso-butanal. The
produced steam from the reactor is used in the reboiler of the distillation unit, which is
a big advantage. This is the cleanest hydroformylation industrial process because of
the straightforward separation of the organic products from the catalyst.
However the system is limited by the solubility of organic substrates in
aqueous phase. The rate of higher olefins hydroformylation drops dramatically
because of their low solubility in water. Therefore, there is a pressing need to develop
a heterogeneous catalyst to overcome the drawbacks of homogeneous catalyst
(separation and catalyst reuse) and biphasic catalysis (solubility of the higher olefins).
Also the development of heterogeneous catalyst can help in developing flow reactors
for hydroformylation which can improve the process in industries. This lead the

Chapter 1
PhD Thesis 16
researchers to conduct research based on heterogeneous catalyst for hydroformylation
reactions.
1.3. Heterogeneous hydroformylation
Heterogeneous hydroformylation is one of the emerging research areas of last
three decades to overcome the drawbacks of recyclability associated with
homogeneous hydroformylation. In this regard the research has been progressed by
heterogenization of homogeneous catalysts into a solid support like zeolites, activated
carbon, alumina, polymers, supported dendrimers and mesoporous silica materials
(silica, MCM-41, SBA-15 etc.).
1.3.1. Heterogenization of homogeneous catalyst on zeolite materials
Zeolites are materials consisting of Si-Al framework and having uniform
channel size, high surface area, shape selectivity, thermal and chemical stability with
varied acidic and basic properties. The main advantage in the use of zeolites as a
support is the enhanced selectivity due to the well-defined pores structure.
Rhodium exchanged zeolite-X and -Y are found to be active for
hydroformylation of propene at atmospheric pressures [56]. In an another study the
rhodium phosphine complexes was in situ encapsulated in zeolite NaY and was active
for propene hydroformylation at 150 °C and 1 atm [57]. The catalysts were not stable,
but showed an enhancement in linear vs. branched products with an increased
production of alcohols compared to rhodium zeolites without phosphines.
Hydroformylation of 1-hexene by various zeolite-supported rhodium species were
reported [58]. The hydroformylation of 1-hexene was carried out at 50 and 125°C and
300 psig CO:H2 (1:1). The activity of the immobilized catalysts is affected by the type
and amount of phosphine. Chaudhari et al. reported a heterogeneous system of
catalyst made up by tethering of HRh(CO(PPh3)3 in zeolite Y using phosphotungstic
acid as tethering agent [59]. The catalyst showed excellent stability, selectivity to
aldehydes and improved activity for hydroformylation of various olefins. The catalyst
was also recyclable without loss in its activity and selectivity. In an another study by
Chaudhari et al., HRh(CO(PPh3)3 was impregnated on zeolite Y (Scheme 1.3) and
also encapsulated in zeolite Y [60]. The impregnated catalyst was leached out in the
first cycle itself where as the encapsulated catalyst was found to be efficiently active

Chapter 1
PhD Thesis 17
and recyclable. The zeolite based supports had demerits of lower pore diameter which
restricted the encapsulation of bulky complexes and also the hydroformylation of
higher olefins.
Scheme 1.3. Tethering of HRh(CO)(PPh3)3 complex to zeolite Y by phosphotungstic
acid for hydroformylation [60]
1.3.2. Heterogenization of homogeneous catalyst on activated carbon
Activated carbon support is having a very high surface area and has a
possibility of enhancement in surface chemistry. Rh supported on activated carbon of
different origins (peat, wood and coconut shell based) is reported [61]. The catalyst is
tested for ethene hydroformylation and found that the catalyst based on coconut shell
based activated carbon showed better activity and selectivity to propanal. Without any
pretreatment with H2 or CO the catalyst showed better activity. In another study,
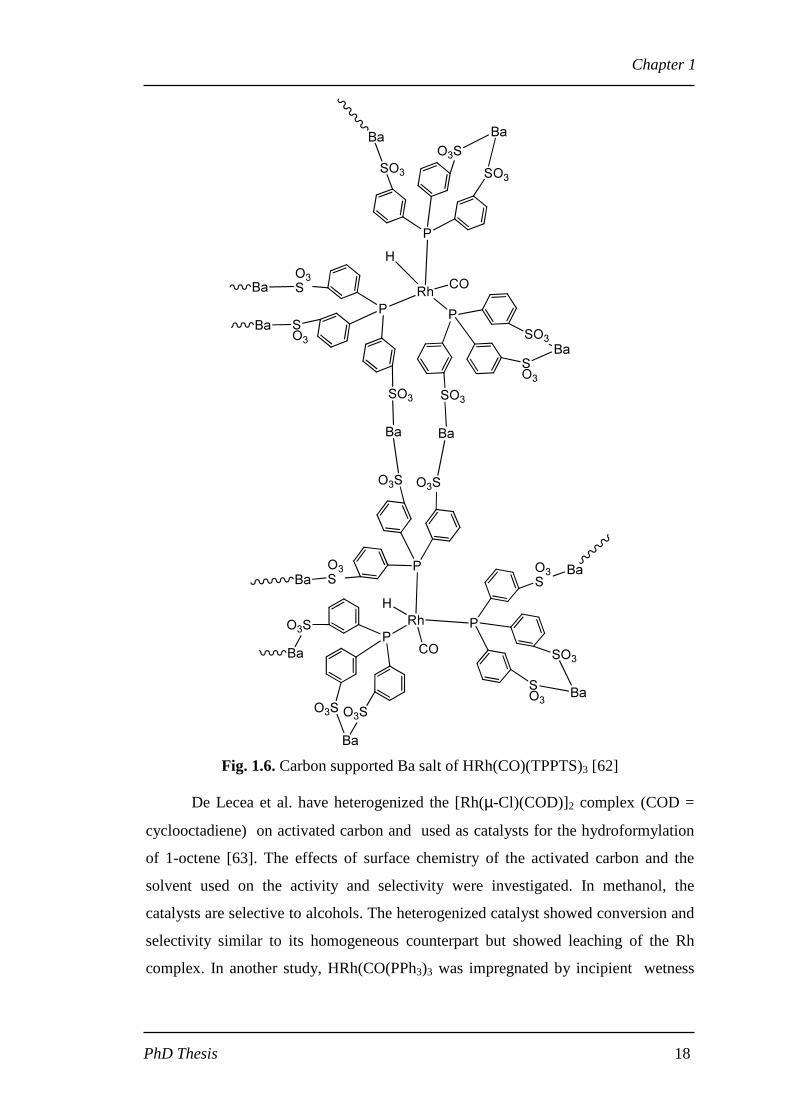
carbon supported Ba salt of HRh(CO)(TPPTS)3 (Fig. 1.6) was used as a highly active
and stable hydroformylation catalyst [62]. The catalyst is prepared by interaction of a
carbon supported barium nitrate with an aqueous solution of HRh(CO)(TPPTS)3 and
NaTPPTS [1:6], [TPPTS = triphenylphosphine trisulfonate] to form a precipitated
Ba2+ salt of HRh(CO)(TPPTS)3 and free TPPTS-Ba3/2 where Ba2+ is adsorbed on the
carbon surface by physical adsorption. Conversion of 95% of 1-decene
hydroformylation with 89.5% aldehyde selectivity was obtained but with a lower n/iso
ratio of 0.51.
Chapter 1
PhD Thesis 18
Rh
P
CO
SO3
SO3
Ba
P
SO3
Ba
O3S
SO3
Ba
P
SO3
O3
SBa
SO3 SO3
Ba Ba
Ba
Rh
PCO
H
O3SO3S
Ba
P
O3
SBa
SO3
SO3
Ba
P
O3S O3S
O3
S
O3S
Ba
Ba
H
Fig. 1.6. Carbon supported Ba salt of HRh(CO)(TPPTS)3 [62]
De Lecea et al. have heterogenized the [Rh(µ-Cl)(COD)]2 complex (COD =
cyclooctadiene) on activated carbon and used as catalysts for the hydroformylation
of 1-octene [63]. The effects of surface chemistry of the activated carbon and the
solvent used on the activity and selectivity were investigated. In methanol, the
catalysts are selective to alcohols. The heterogenized catalyst showed conversion and
selectivity similar to its homogeneous counterpart but showed leaching of the Rh
complex. In another study, HRh(CO(PPh3)3 was impregnated by incipient wetness

Chapter 1
PhD Thesis 19
technique on to carbon nanotube [64]. The catalyst was highly active and selective
for propene hydroformylation with molar n/iso ratio of 12-13.
1.3.3. Heterogenization of homogeneous catalyst on alumina
Heterogenization of rhodium on alumina by ion-exchange and homogeneous
precipitation method is reported [65]. The supported Rh(III) is then reduced to Rh(0)
using molecular H2 or NaBH4. The catalyst was active and selective for styrene
hydroformylation but the leaching of Rh metal was prominent under
hydroformylation conditions. Nano-porous alumina synthesized through sol-gel
method was used as a support for rhodium catalyst impregnation [66]. The catalyst
showed high activity on hydroformylation of ethene. Kawi et al. synthesized a series
of alumina-supported Wilkinson’s catalyst for hydroformylation of styrene [67]. The
support was functionalized by surface amine ligands onto which PAMAM
(polyamidoamine) dendrimers were grafted followed by anchoring of Wilkinson’s
catalyst. Dendritic nanoalumina supported catalysts showed higher activity and
selectivity than that of dendritic α-alumina, β-alumina and SBA-15. The zeroth
generation dendritic nanoalumina showed higher activity while the first generation
dendritic nanoalumina showed higher regioselectivity. The higher surface area of the
support was the reason for zeroth generation catalysts for its higher activity and the
crowdedness around the metal center was the reason for lower activity of higher
generation catalysts.
1.3.4. Heterogenization of homogeneous catalyst on resins and polymers
The research for heterogenization of homogeneous complex on the resins and
other polymeric supports, in which metal complex is immobilized on resin/polymer
surface through their surface functional groups, is one of the current interest for
hydroformylation reaction. Uozumi et al., reported the synthesis and application of
rhodium phosphine complexes supported on amphiphilic resin beads of polystyrene–
poly(ethylene glycol) graft co–polymer (1% divinyl benzene (DVB) cross–linked) for
the hydroformylation of various alkenes in water [68]. The heterogeneous catalyst
showed excellent yields up to 99% for aldehydes. Hydroformylation of 1-hexene in
supercritical CO2 (scCO2) and other organic solvents was reported by Fujita et al.,
using polymer supported rhodium catalyst prepared from polystyrene bound PPh3 and
dicarbonyl acetylacetonato rhodium. The supported catalyst was reused for several

Chapter 1
PhD Thesis 20
cycles by means of simple filtration [69]. Chaudhari et al. studied the kinetics of
hydroformylation of 1-hexene using rhodium–TPPTS bounded on the surface of ion
exchange resin, amberlite IRA–93 as a catalyst [70]. Another method for synthesis of
immobilized homogeneous catalyst by addition of a bi–functional ligand followed by
a metal complex (rhodium, cobalt and platinum/tin) onto ion exchange resin
(sulfonated styrene–divinylbenzene) for hydroformylation of 1-hexene was reported
[71]. Artner et al., have reported metal doped thermosetting epoxy resins, like
triglycidyl derivative of 4–aminophenol, synthesized by polymerization reaction using
molybdenum, palladium, or rhodium complexes as initiators. These metal doped
epoxy resins were observed as highly efficient catalysts for hydroformylation reaction.
These catalysts were recovered by filtration and recycled without much loss in its
activity [72].
1.3.5. Heterogenization of homogeneous catalyst on supported dendrimers
Heterogenization of rhodium metal complexes on supported dendrimers is one
of the research interests in recent times. Dendrimers are macromolecules with
emerging applications in the area of material and biological sciences [73–75]. In
current decades, dendrimers are found to be novel materials for use in catalysis
because of their highly branched and bulky structures which can provide multiple
sites for coordination with metal complexes [76–78]. The dendrimers are generally
soluble in organic solvents, but can be separated by nano filtration from the reaction
mixture in liquid phase due to their large size. The coordination of dendrimers with
transition metal complexes may bring forth enhanced catalytic performance due to
positive dendritic effect [77]. Dendrimers being large (2 to 4 nm) tree–like molecules
with a stubborn globular shape makes them more suitable for ultrafiltration than
soluble polymers. The metal–binding groups are usually on the exteriors of
dendrimers but can also be covered within shape–selective pockets. Advantageously,
dendrimers may exhibit bidentate binding (through two donor atoms on the same
dendrimer arm) to the metal. The chelate (ring–forming) effect will then ensure the
minimum leaching of metal complex.

Chapter 1
PhD Thesis 21
Fig. 1.7. Phosphonated PAMAM-SiO2 dendrimer rhodium complexes of different
generation [79]
Alper et al. have studied supported dendrimers used to anchor the rhodium
metal complexes for hydroformylation reactions. PAMAM dendrimers were grown
over silica in successive generations and is used for the preparation of phosphonated
PAMAM-SiO2 dendrimer rhodium complexes (Fig. 1.7) for hydroformylation of
olefins [79]. The catalyst was highly active and regioselective for hydroformylation of
aryl olefins and vinyl acetate. However, the third- and fourth generation catalysts of
these type displayed low activity compared to the lower generations. This was
believed to be due to incomplete phosphonation reactions at higher generation
dendrimers arising from steric crowding and ultimately resulting in the threshold of
dendrimer growth being reached. It was supposed that extending the chain length of
each generation would relieve the steric crowding and allow for increased catalyst
loading at higher generations. This led this group to extend their work in this regard.
To extend the chain length, the ethylenediamine linker was substituted by 1,4-
diaminobutane, 1,6-diaminohexane and 1,12-diaminododecane [80]. The lengthening
of the monomeric unit of the dendrimer decreased the congestion at the surface (Fig
1.8). The hydroformylation of styrene and vinyl acetate proceeds in up to quantitative
yields, and in fine regioselectivity using these catalysts. These silica-supported
catalysts were recovered by microporous filtration using a 0.45 µm membrane filter
and are reusable for at least three cycles.

Chapter 1
PhD Thesis 22
O
O
O
Si N
O
NH
O
NH
N
N
NH
OHN
O
N
NH
O
N
NH
O
N
N
HN
O
NH
O
N
Ph2P
PPh2
Rh
Ph2P
PPh2
Rh
Ph2P
PPh2
Rh
NPh2P
Ph2P
Rh
O
HN
N
NH
ON
PHPh2
PHPh2
Rh
NH
O
N
NH
O
N
Ph2P
Ph2P
Rh
HN
O
N
Ph2P
PPh2
Rh
NH
O
N
Ph2P
PPh2
Rh
Fig. 1.8. Lengthening of the monomeric unit of the dendrimer in third generation [80]
PAMAM dendrimers were also successfully grown over periodic mesoporous
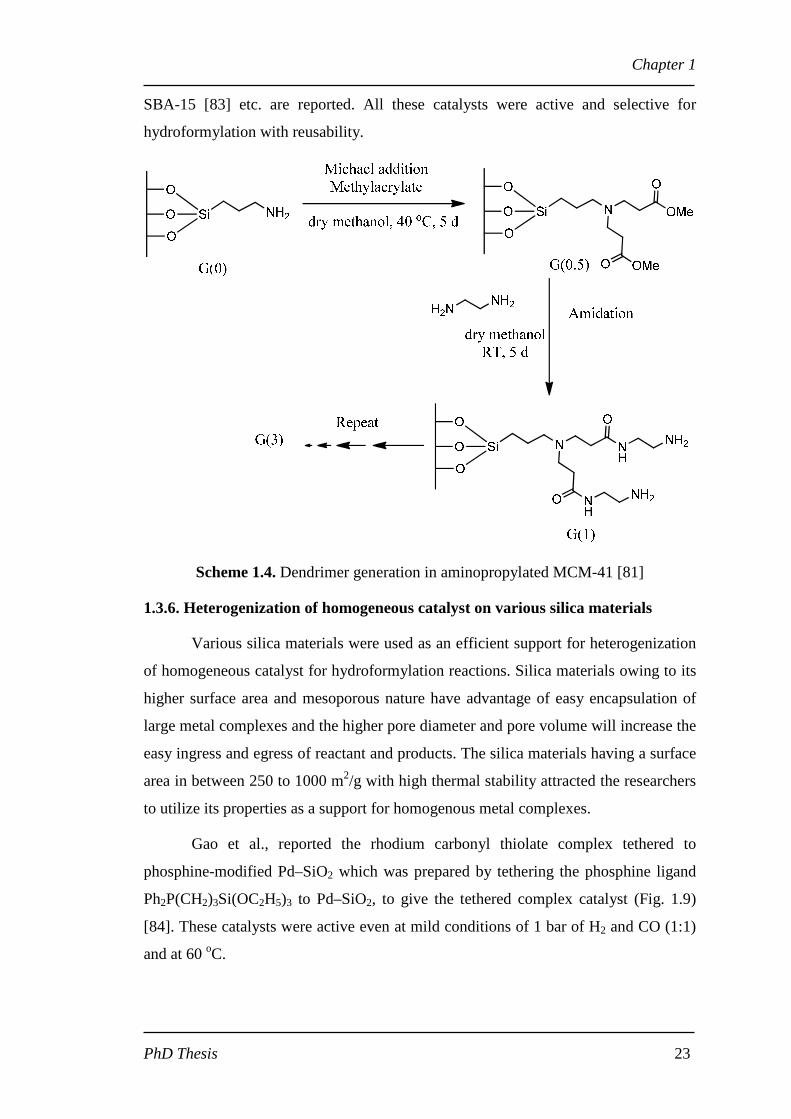
silica like MCM-41 [81]. The dendrimer generation was done on aminopropylated
MCM-41 (Scheme 1.4). This dendrimer supported on MCM-41 was then
phosphinomethylated and then formed complexation with rhodium. The catalyst was
highly active with turn over frequency (TOF) of 1800 h-1 for hydroformylation of 1-
octene. Similar kind of PAMAM dendrimers supported on davasil silica [82] and
Chapter 1
PhD Thesis 23
SBA-15 [83] etc. are reported. All these catalysts were active and selective for
hydroformylation with reusability.
Scheme 1.4. Dendrimer generation in aminopropylated MCM-41 [81]
1.3.6. Heterogenization of homogeneous catalyst on various silica materials
Various silica materials were used as an efficient support for heterogenization
of homogeneous catalyst for hydroformylation reactions. Silica materials owing to its
higher surface area and mesoporous nature have advantage of easy encapsulation of
large metal complexes and the higher pore diameter and pore volume will increase the
easy ingress and egress of reactant and products. The silica materials having a surface
area in between 250 to 1000 m2/g with high thermal stability attracted the researchers
to utilize its properties as a support for homogenous metal complexes.
Gao et al., reported the rhodium carbonyl thiolate complex tethered to
phosphine-modified Pd–SiO2 which was prepared by tethering the phosphine ligand
Ph2P(CH2)3Si(OC2H5)3 to Pd–SiO2, to give the tethered complex catalyst (Fig. 1.9)
[84]. These catalysts were active even at mild conditions of 1 bar of H2 and CO (1:1)
and at 60 oC.

Chapter 1
PhD Thesis 24
Fig. 1.9. Rhodium carbonyl thiolate complex tethered Pd–SiO2, M =Pd [84]
In another study, the hybrid PPh3–Rh/SiO2 catalyst for hydroformylation of
olefins was prepared by doping PPh3 onto the Rh/SiO2. The chemical bond of the
hybrid catalyst was formed between the PPh3 ligand and Rh metal particles on the
surface of SiO2 support [85]. The catalyst was highly active and recyclable for
hydroformylation. The results reveal that hydroformylation of olefins to aldehydes
dominantly take place on the surface of the hybrid catalyst.
P
RhHP
PRhHP
PRhHP
PRhHP
P
RhHP
PRhHP
SiO2
SiO2
P
PH
HP
PH
PH
PH
PH
PHPH
HP
P
P
P
SiO2
Rh particle
RhPh3P PPh3
H
PPh3OC
Fig. 1.10. Model of the PPh3-Rh/SiO2 catalyst [86]
Yan et al., had reported the PPh3 modified Rh/SiO2 catalyst as an active
catalyst for heterogeneous hydroformylation of propene in a fixed-bed reactor [86].
The conducted in situ FTIR studies confirmed that PPh3 molecules could be

Chapter 1
PhD Thesis 25
chemically adsorbed on the heterogeneous Rh/SiO2, and they promote the in situ
formation of HRh(CO)(PPh3)3 species and HRh(CO)2(PPh3)2 species, which are the
active catalytic species that improve the activity and selectivity of propene
hydroformylation (Fig. 1.10). The problem of metal leaching is greatly reduced by
directly fastening Rh nano-particles to the support, and Rh species are tightly bound
by the Rh–O bonds and the very strong metal–metal bonds.
Heterogeneous chiral catalysts were prepared by modifying silica-supported
rhodium (Rh/SiO2) with chiral phosphorus ligands [87]. The chirally modified
Rh/SiO2 catalysts exhibited high activity, regioselectivity, and enantioselectivity for
the asymmetric hydroformylation of styrene and vinyl acetate. Up to 72% ee and
100% selectivity of branched aldehyde for the hydroformylation of vinyl acetate were
obtained for (R)-BINAP–Rh/SiO2 catalysts. Marchetti et al., had reported SiO2
tethered rhodium complexes obtained from Rh(CO)2(acac) and 3-(mercapto)propyl-
and 3-(1-thioureido)propyl- functionalized silica gel [88]. These catalysts were used
for hydroformylation of functionalized olefins of biological interests like styrene, 1,1-
diphenylethene, 2-tosyloxystyrene, 2-benzyloxystyrene and vinyl acetate. The catalyst
showed good activity, with conversion, chemo- and regioselectivity comparable with
homogeneous catalysts. But a small extent of leaching was observed.
Supported aqueous phase catalysts (SAPC) synthesized over SiO2 was
prepared by Zhu et al. for hydroformylation of higher olefins [89]. The Rh was
supported over high surface area SiO2 and TPPTS was introduced as water soluble
ligand. The reaction was conducted in an aqueous biphasic media and the catalyst was
found to reduce the resistance of mass transfer in water/organic biphasic media for the
hydroformylation of higher olefins. The chemical coordination bonds between the
highly dispersed Rh particles and the TPPTS ligands are responsible for its high
catalytic activity for the hydroformylation of olefins.
Apart from the above SiO2 materials the research is more focused on
mesoporous silica like MCM-41, HMS, SBA-15 type materials. The highly porous
nature with high surface area and pore diameters in the range of 2 to 20 nm made
these materials as a suitable support for heterogenization. Mobile Oil Corporation
discovered the new class of mesoporous silica material called Mobile Composite
Materials like MCM-41 and MCM-48 which have been extensively studied as a
support for heterogeneous hydroformylation [90–92].

Chapter 1
PhD Thesis 26
Rh4(CO)12-derived rhodium carbonyls have been successfully anchored to
MCM-41(PPh2), MCM-41(NH2) and MCM-41(SH), which are formed, respectively,
by functionalization of silicate MCM-41 with Cl(CH2)3Si(OMe)3 plus KPPh2,
H2N(CH2)3Si(OEt)3 and HS(CH2)3Si(OMe)3, to produce MCM-41-tethered
unidentified phosphine- and amine-containing rhodium carbonyl clusters [91].
Rh4(CO)12 is mostly converted to Rh6(CO)16 on unfunctionalized MCM-41. All the
Rh4(CO)12-derived catalysts exhibited very high selectivity (>98%) for the formation
of cyclohexane carboxaldehyde in cyclohexene hydroformylation. The
unfunctionalized catalyst was leached out whereas the functionalized ones were stable
and recyclable.
Huang et al. also extended their studies and reported a Rh-complex tethered on
aminated MCM-41 as a heterogeneous catalyst for efficient hydroformylation of 1-
hexene [93], 1-octene and styrene [94]. A nitrogen-containing organosilane coupling
reagent like aminopropyl triethoxy silane, was used for the functionalization of the
MCM-41 support to obtain aminated MCM-41. The tethering of the rhodium
complexes substantially does not result in the change in the mesoporous structural
ordering of MCM-41, although the resultant materials exhibit reduced pore sizes, total
pore volumes and BET surface areas. Two sets of catalysts, tethered PPh3-free Rh-
complex and tethered PPh3-containing Rh-complexes were synthesized. For 1-hexene
and 1-octene hydroformylation, the tethered PPh3-free rhodium complex is of no
advantages in activity, selectivity and n/i aldehyde ratio over the corresponding
untethered one. But, the catalyst tethered with PPh3-containing rhodium complex had
enormous advantage in activity and selectivity over the untethered PPh3-containing
rhodium complex except for n/i aldehyde ratio.
In another study, two triphenyl phosphine analogues, (4-tert-butylphenyl)
diphenyl phosphine and bis-(4-tert-butylphenyl) phenylphosphine ligands, have been
synthesized for the preparation of Rh–P complexes with a formula of Rh(CO)Cl(L)2
(L-ligand). The synthesized complexes were immobilized in amino-functionalized
MCM-41 and MCM-48 [95]. The heterogenized catalysts showed catalytic activity
and n-heptanal selectivity comparable with that of the corresponding homogeneous
complexes for 1-hexene hydroformylation. There interactions had been occurred
between the surface amino-groups and the rhodium complex during the
immobilization, resulting in highly dispersed active Rh-moieties and a significant
modification in the catalytic stability.

Chapter 1
PhD Thesis 27
Ali and co-workers studied the heteropolyacid supported Rh(I) and Rh(III)
complexes supported on MCM-41 for hydroformylation. Heteropolyacids
(H3PW12O40, 25H2O) impregnated with Rh(I) and (III) complexes were prepared and
used as supported catalysts in the hydroformylation of alkyl alkenes [96]. The results
showed that the catalysts with heteropolyacids had better activity than that of the
catalysts without heteropolyacid. In another study the same catalyst is used for
hydroformylation-acetalization domino reactions of aryl alkenes [97]. Rh(I) supported
MCM-41 combined with the heteropolyacid H3PW12O40 showed high catalytic
activity towards the formation of acetals. Whereas the Rh(III) showed better activity
for acetals in the absence of any additives.
Supported ionic liquid-phase catalyst (SILPC) on MCM-41 was investigated
with Rh-TPPTS complex dissolved in various ionic liquids, 1-butyl-3-methyl-
imidazolium tetrafluoroborate (BMI·BF4), 1-butyl-3-methyl-imidazolium
hexafluorophosphate (BMI·PF6) and 1,1,3,3-tetramethylguanidinium lactate (TMGL)
[98]. The supported ionic liquid-phase catalysts synthesized were very active for the
hydroformylation of higher olefins. The catalytic performance of SILPC was almost
independent of the type of ionic liquid used.
Other than MCM-41 or MCM-48 type materials, SBA-15, HMS etc. also were
used as a support for hydroformylation reactions. Rh tethered on Ti modified
hexagonal mesoporous silica functionalized with 2,2’ bipyridine (Rh/Ti-HMS/bipy)
was investigated for heterogeneous hydroformylation of olefins [99]. In this catalyst
system Ti-HMS was calcined in air at 650 °C for 4 h and was functionalized by 2,2’
bipyridine (bipy). The catalyst was active for hydroformylation of alkenes. Yan et al.
have developed SBA-15 as a support for homogenous Rh-complexes [100]. Rh, Rh-
PPh3 and HRh(CO)(PPh3)3 were used for the catalyst synthesis. The catalyst Rh-PPh3
supported on SBA-15 of medium pore size of 6.1 nm gave higher n/iso ratio and
comparable activity. It was confirmed that at the nanometer sized pores the
HRh(CO)2(PPh3)2 active species were formed in situ and increases the activity and
regioselectivity.

Chapter 1
PhD Thesis 28
Si
PH3
Rh
OC
Cl
Si
HP
3
Higher olefins with different lengths,
C=C positions, structures
+CO +H2
Hydroformylation
Fig. 1.11. Rh-PrPPh2-SBA-15 catalysed hydroformylation [101]
In another piece of study, RhCl3 was immobilized to diphenylphosphinopropyl
(–PrPPh2)-modified mesoporous silica SBA-15 through a multi-step-assembly
process (Fig. 1.11) [101]. The catalysts were tested for hydroformylation activity of
several higher olefins of different lengths (C6, C8, and C10), different double bond
positions (terminal or internal) or different structures (linear or branched). Shorter
linear olefin substrates were more easily activated with higher catalyst specific
activity, while the catalyst showed recycling stability in hydroformylation of longer
and branched olefin substrates.
Other heterogenized catalysts are also reported by using hydrotalcite like
materials as a support for heterogenization. Jasra et al., have used these type of
heterogenized catalysts for the synthesis of C8 aldols and alcohols from propene via
hydroformylation, aldol condensation and hydrogenation in a single pot [102–106].
This study has been extended for the synthesis of 2-methylpentanol from ethene by
this multistep reaction performed in single pot using similar catalytic systems [107].
The same catalyst was used for the hydroformylation of various alkenes by changing
the parameters without formation of aldol and/or hydrogenated products [108].

Chapter 1
PhD Thesis 29
1.4. Hydroaminomethylation: Synthesis of Amines
Amines are important class of bulk and fine chemicals in chemical and
pharmaceutical industries with a production of million-ton scale per year [109].
Amines are organic compounds and functional groups that contain a basic nitrogen
atom with a lone pair. Amines are derivatives of ammonia, wherein one or more
hydrogen atoms have been replaced by a substituent such as an alkyl or aryl group
[110].
Amines can be classified as primary, secondary or tertiary, meaning one, two
and three alkyl groups bonded to the nitrogen respectively as shown in Fig. 1.12.
Important amines include amino acids, biogenic amines, methylamine,
trimethylamine, and aniline. Amines play prominent roles in biochemical systems;
they are widely distributed in nature in the form of amino acids, alkaloids, and
vitamins. Many complex amines have pronounced physiological activity, for example,
epinephrine (adrenalin), thiamin or vitamin B1, and Novocaine. The odor of decaying
fish is due to simple amines produced by bacterial action. Amines are used to
manufacture many medicinal chemicals, such as sulfa drugs and anesthetics. The
important synthetic fiber nylon is an amine derivative [111].
NH2
H3C C NH2
CH3
CH3
NH
CH3
NH
N
CH3
CH3
N
Primary amine
Secondary amine
Tertiary amine
Fig. 1.12. Primary, secondary and tertiary amines

Chapter 1
PhD Thesis 30
1.4.1. Synthesis of amines
1.4.1.1. Industrial synthesis of amines
Commercial preparation of aliphatic amines can be accomplished by direct
alkylation of ammonia or by catalytic alkylation of amines with alcohols (Reductive
amination) at elevated temperatures [112].
ROH + NH3 → RNH2 + H2O (1.10)
RX + 2 R'NH2 → RR'NH + [RR'NH2]X (1.11)
Alkylation of ammonia is sometimes employed in industrial processes; the
resulting mixture of amines is separated by distillation. The ultimate starting materials
for the industrial preparation of allylamine are propene, chlorine, and ammonia.
Alkylation of ammonia can lead to a complex mixture of products, it is used to
prepare primary amines only when the starting alkyl halide is not particularly
expensive and the desired amine can be easily separated from the other components of
the reaction mixture.
Reductive amination processes involve reaction of alcohol with ammonia over
a supported metal catalyst, generally nickel or cobalt on silica or alumina [113, 114]
at temperatures ranging from 150 to 210 °C and pressures of 18 to 200 bar. The
reaction is conducted in the presence of hydrogen to maintain catalyst activity [115].
Some industries use aldehydes or ketones, such as acetaldehyde or acetone, as
feedstocks for reasons of availability and cost relative to the alcohol. When these
unsaturated feedstocks are used, the hydrogen requirement exceeds stoichiometric
amounts based on the alkyl feed. Continuous, fixed bed vapor phase or liquid
phase/trickle bed reactors may be used for alcohol or aldehyde/ketone amination. The
heat of reaction is significantly higher for amination of aldehydes/ketones relative to
alcohols, and some means of controlling the reactor temperature generally is required.
In addition to fixed bed reactors, liquid phase stirred tank reactors also are used
commercially, particularly for amination of carbonyl feedstocks. The reactions are
usually run under conditions selected to achieve high conversion of the alkyl
feedstock, and the product selectivities are controlled by equilibrium as the amination
catalysts also catalyze disproportionation.

Chapter 1
PhD Thesis 31
1.4.1.2. Other routes to amine synthesis
Gabriel synthesis
Gabriel synthesis is the reaction that transforms primary alkyl halides into
primary amines using potassium phthalimide [116–118]. The utility of the method is
based on the fact that the alkylation of ammonia is an unselective and inefficient route
to amines in the laboratory. The conjugate base of ammonia, sodium amide (NaNH2),
is more basic than it is nucleophilic. In fact, sodium amide is used to deliberately
obtain the dehydrohalogenation product. In this method, the sodium or potassium salt
of phthalimide is N-alkylated with a primary alkyl halide to give the corresponding N-
alkylphthalimide. The reaction fails with most secondary alkyl halides. Acid
hydrolysis of N-alkylphthalimide liberates primary amine salt [119].
Staudinger reaction
Staudinger reaction is the combination of an azide with a phosphine or phosphite
which produces an iminophosphorane intermediate. The hydrolysis of the aza-ylide
produces a phosphine oxide and an amine, this reaction is a mild method of reducing
an azide to an amine (Scheme. 1.5). Triphenylphosphine is commonly used as the
reducing agent, yielding triphenylphosphine oxide as the side product in addition to
the amine [120, 121].
Scheme. 1.5. Staudinger reaction for the synthesis of pinwheel compound
Schmidt reaction
The Schmidt reaction involves alkyl migration over the carbon-nitrogen
chemical bond in an azide with expulsion of nitrogen [122]. The reagent used is
hydrazoic acid and the reaction product depends on the type of reactant. Carboxylic
acids form amines through an isocyanate intermediate and ketones form amides. A
catalyst is required which can be a protic acid usually sulfuric acid or a Lewis acid. It

Chapter 1
PhD Thesis 32
is a tool regularly used in organic chemistry for the synthesis of new organic
compounds eg. 2-quinuclidone.
Hofmann rearrangement
The Hofmann rearrangement is the rearrangement of a primary amide in
presence of bromine and NaOH to a primary amine with one fewer carbon atom than
the starting primary amide [123, 124]. The reaction of bromine with sodium
hydroxide in situ forms sodium hypobromite, which converts the primary amide into
an intermediate isocyanate. The intermediate isocyanate is hydrolyzed to a primary
amine giving off carbon dioxide.
Nitrile reduction
In nitrile reduction a nitrile is reduced to either an amine or an aldehyde with a
suitable chemical reagent (Scheme 1.6). Reagents for the conversion to amines are
lithium aluminium hydride, Raney nickel/hydrogen/or diborane. This organic reaction
is one of several nitrogen-hydrogen bond forming reactions [125].
Diisopropylaminoborane [BH2N(iPr)2] in the presence of a catalytic amount of
lithium borohydride (LiBH4) reduces a large variety of aliphatic and aromatic nitriles
in excellent yields. BH2N(iPr)2 can also reduce nitriles in the presence of
unconjugated alkenes and alkynes [126].
Scheme.1.6. Nitrile reduction to form amine
Hydroamination
The hydroamination reaction is the addition of an N-H bond across the C=C or
C≡C bonds of an alkene or alkyne (Scheme 1.7) [127, 128].
Scheme 1.7. Hydroamination of alkene and alkyne

Chapter 1
PhD Thesis 33
This is highly an atom economic method for preparing substituted amines that
are attractive targets for organic synthesis and the pharmaceutical industry. The
hydroamination reaction is approximately thermodynamically neutral, there is a high
activation barrier due to the repulsion of the electron-rich substrate and the amine
nucleophile. The reaction also has high negative entropy, making it unfavorable at
high temperatures. As a result, catalysts are necessary for this reaction to proceed
[129]. Despite of substantial effort put in this area, the development of a general and
efficient catalytic process for this reaction remains subtle. Progress has been reported
on the hydroamination of alkynes and alkenes using lanthanides and late transition
metals [130-133].
1.4.2. Hydroaminomethylation-Advancement in last decade
The hydroaminomethylation (HAM), discovered in 1949 in the laboratories of
BASF by Walter Reppe, using [Fe(CO)5] [134, 135] is a promising reaction to fulfill
the requirement of waste reduction since water is the only side product. HAM is a
one-pot cascade reaction, starting with the hydroformylation of an alkene, consecutive
condensation of the intermediate aldehyde with the substrate amine, and subsequent
hydrogenation of the formed enamine or imine to the desired amine product. In this
reaction, primary and secondary amines, as well as ammonia can be used as the amine
substrate. The HAM with ammonia is particularly challenging in terms of
chemoselectivity, since the desired primary amine is more nucleophilic than ammonia,
leading to a higher reactivity towards the intermediate aldehyde, which in turn results
in the formation of a secondary amine.
HAM consists of a domino reaction, hydroformylation and a reductive
amination. This implies that side products of both reactions might be observed in the
HAM reaction (Scheme 1.8). In the HAM of alkenes, both the linear and the branched
amines can be formed. The regioselectivity of the product amines are important in
which one of the isomer is preferred over the other to have easy separation and
commercial viability of the reaction. This regioselectivity is already determined in the
first reaction step, the hydroformylation where the n-aldehyde or iso-aldehyde are
formed. Chemoselectivity, which comprises the selectivity to the amine product, is
mainly determined in the reductive amination step. Very important in this respect is
the hydrogenation of the C=N double bond of the enamine or imine. If the reduction
with H2 is not effective the reaction ends up with imine or enamine.

Chapter 1
PhD Thesis 34
(R1 = R2 = R3= H, alkyl or aryl groups)
Scheme. 1.8. Possible reactions (desired and side products) during
hydroaminomethylation
Although the hydroaminomethylation (HAM) has been discovered already in
the late 1940s, the majority of the reports concerning this reaction stems from the last
10-12 years. The researches on hydroaminomethylation are still in its early stage
having promising potential for its well exploration. In available literatures [136-138]
most of the works are focused on homogeneous hydroaminomethylations using
various ligands keeping Rh as the metal center. Some reports [139, 140] are also
available with biphasic systems. Rather than detail catalytic studies the research is
more focused on synthesis of various amine containing organic compounds using
hydroaminomethylation. The contributions of Elibracht [141-143] and Beller [144,
145] to hydroaminomethylation are outstanding.
1.4.2.1. Contributions of P. Elibracht and co-workers to hydroaminomethylation
Peter Elibracht and co-workers have done an extraordinary research in the
field of homogeneous hydroaminomethylation for the synthesis of various valuable
organic molecules. In the time of starting of their noteworthy research in
hydroaminomethylation symmetrically and unsymmetrically substituted secondary
and tertiary amines are selectively synthesized in high yields by a one-pot

Chapter 1
PhD Thesis 35
hydroaminomethylation of ammonia or primary amines with styrene and/or cyclic
olefins, carbon monoxide and hydrogen in the presence of [Rh(cod)Cl]2, catalyst [146,
147].
Scheme 1.9. Bisalkylation of styrene with ammonia [146]
They could get isolated yields above 70% for methyl styrene with amines like
butylamine, aniline and benzylamine. Bisalkylation of styrene with ammonia was also
possible with the catalyst (Scheme 1.9). In another study using [Rh(cod)Cl]2 and
Rh(acac)(CO)2 Elibracht et al., could synthesize azomacrocycles via ring closing
hydroaminomethylation (Scheme 1.10 ) [148]. The substrates were diolefins and
diamines. The macrocycles of up to 36 ring size were synthesized.
Scheme 1.10. Ring closing hydroaminomethylation: Synthesis of macroheterocycles
The catalyst Rh(acac)(CO)2 was used for the synthesis of α−
and β−aminofunctionalized amines and silanes via hydroaminomethylation of
enamines and silanes [149]. The hydroaminomethylation of vinylsilanes with
morpholine yielded more than 80% of the product. The reaction with benzylamine
and isopropylamine gave low yields because of the cleavage of vinylsilane bond
leading to the generation of (diphenyl)-methylsilanol. This vinylsilane bond cleavage
is depending on the substituents in the silyl moiety. If trialkyl substituents instead of
aryl functionalities are introduced, the hydroaminomethylation sequence can be
performed in medium yields without vinylsilane cleavage. Hydroaminomethylation of
Chapter 1
PhD Thesis 36
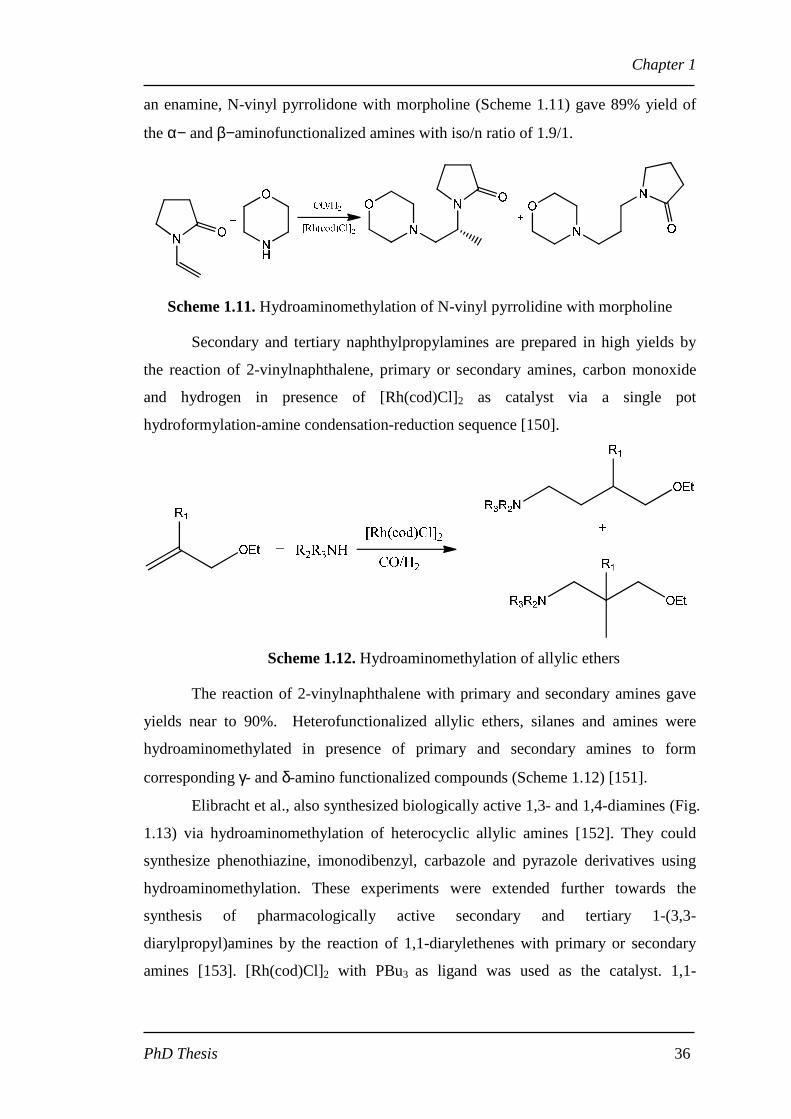
an enamine, N-vinyl pyrrolidone with morpholine (Scheme 1.11) gave 89% yield of
the α− and β−aminofunctionalized amines with iso/n ratio of 1.9/1.
Scheme 1.11. Hydroaminomethylation of N-vinyl pyrrolidine with morpholine
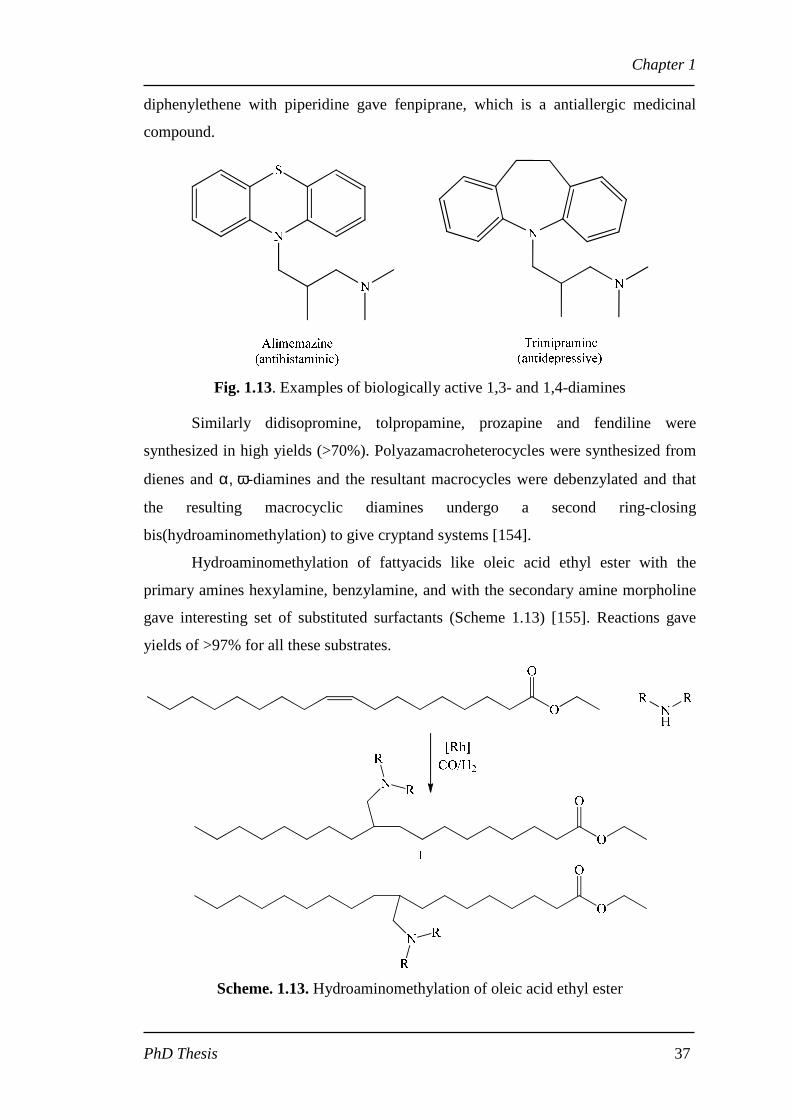
Secondary and tertiary naphthylpropylamines are prepared in high yields by
the reaction of 2-vinylnaphthalene, primary or secondary amines, carbon monoxide
and hydrogen in presence of [Rh(cod)Cl]2 as catalyst via a single pot
hydroformylation-amine condensation-reduction sequence [150].
Scheme 1.12. Hydroaminomethylation of allylic ethers
The reaction of 2-vinylnaphthalene with primary and secondary amines gave
yields near to 90%. Heterofunctionalized allylic ethers, silanes and amines were
hydroaminomethylated in presence of primary and secondary amines to form
corresponding γ- and δ-amino functionalized compounds (Scheme 1.12) [151].
Elibracht et al., also synthesized biologically active 1,3- and 1,4-diamines (Fig.
1.13) via hydroaminomethylation of heterocyclic allylic amines [152]. They could
synthesize phenothiazine, imonodibenzyl, carbazole and pyrazole derivatives using
hydroaminomethylation. These experiments were extended further towards the
synthesis of pharmacologically active secondary and tertiary 1-(3,3-
diarylpropyl)amines by the reaction of 1,1-diarylethenes with primary or secondary
amines [153]. [Rh(cod)Cl]2 with PBu3 as ligand was used as the catalyst. 1,1-
Chapter 1
PhD Thesis 37
diphenylethene with piperidine gave fenpiprane, which is a antiallergic medicinal
compound.
Fig. 1.13. Examples of biologically active 1,3- and 1,4-diamines
Similarly didisopromine, tolpropamine, prozapine and fendiline were
synthesized in high yields (>70%). Polyazamacroheterocycles were synthesized from
dienes and α, ω-diamines and the resultant macrocycles were debenzylated and that
the resulting macrocyclic diamines undergo a second ring-closing
bis(hydroaminomethylation) to give cryptand systems [154].
Hydroaminomethylation of fattyacids like oleic acid ethyl ester with the
primary amines hexylamine, benzylamine, and with the secondary amine morpholine
gave interesting set of substituted surfactants (Scheme 1.13) [155]. Reactions gave
yields of >97% for all these substrates.
Scheme. 1.13. Hydroaminomethylation of oleic acid ethyl ester

Chapter 1
PhD Thesis 38
Rhodium(I) catalyzed regioselective hydroformylation of diolefins and
subsequent reductive amination of the dialdehydes in the presence of α,ω-diamines is
applied to azamacroheterocyclic ring synthesis [156]. Starting from aromatic diallyl
ethers of hydroquinone, biphenol and binaphthol 20–28 membered macroheterocycles
were obtained up to 78% yield. Synthesis of hydroquinone-, biphenol-, and
binaphthol-azamacro heterocycles via regioselective hydroaminomethylation were
conducted using Rh(acac)(CO)2 and then was studied with ligands BIPHEPHOS and
XANTPHOS (Fig. 1.14) for the required regioselectivity [157]. The experiment with
BIPHEPHOS was unsuccessful because of the instability of ligand under harsh
conditions of hydroaminomethylation. Therefore XANTPHOS ligand was used which
is stable under hydroaminomethylation conditions with good regioselectivity.
OO
O O
P P
O
O
O
O
O
P P
BIPHEPHOS XANTPHOS
Fig. 1.14. Structure of BIPHEPHOS and XANTPHOS ligands
The pharmaceutically important 3,3-diarylpropylamine was synthesized using
hydroaminomethylation [158]. This product finds application in the treatment of
urinary incontinence and other spasmogenic conditions. Many other significant
contributions of Eilbracht and co-workers are reported in literature [159–162].
1.4.2.2. Contributions of M. Beller and co-workers to hydroaminomethylation
Matthias Beller et al. have significant contributions for
hydroaminomethylation reactions. They have used different ligands and improved
protocols for the hydroaminomethylation [163]. The first efficient
hydroaminomethylation with ammonia using dual metal catalysts and two-phase
catalysis to primary amines were successfully introduced [164]. The catalyst used was
Rh/Ir dual metal with TPPTS ligand. The important objective of the research was a

Chapter 1
PhD Thesis 39
more rapid hydrogenation of the imine to amine and thus fewer side reactions of
intermediates by the use of dual metal catalysts (Rh/Ir), and a simple catalyst
separation through phase separation and control of selectivity with respect to the
formation of primary, secondary, and tertiary amines [165] by the use of the principle
of biphasic catalysis. The use of BINAS (sulphonated NAPHOS) ligand [166] with
the dual metal could give higher yields than TPPTS with n/iso ratio of 99:1. In
another study, hydroformylation was conducted with various ligands like triphenyl
phosphine (TPP), IPHOS, XANTPHOS, dppe (1,2-bisdiphenyl phosphinoethane),
dppb (1,2-bisdiphenyl phosphine butane) with Rh(cod)2BF4 (cationic) as catalyst
precursor [167]. Hydroaminomethylation of 1-pentene with piperidine was conducted
and XANTPHOS was found as a better ligand with high yields and selectivity. This
study on aliphatic olefins gave for the first time the corresponding linear amines in
general with regioselectivities >98:2.
Fig. 1.15. Pharmaceutical pheniramine drugs synthesized by hydroaminomethylation

Chapter 1
PhD Thesis 40
A rhodium carbene complex Rh(cod)(Imes)Cl (Imes=1,3-dimesitylimidazol-2-
ylidene, cod=cyclooctadiene) was prepared from [Rh(cod)Cl]2 and used as a
hydroaminomethylation catalyst [168]. Similarly five different rhodium carbene
complexes were synthesized and used for the synthesis of 3,3-diarylpropylamines.
Various pharmacologically active 3,3-diarylpropylamines were synthesized using
these catalysts as shown in Fig. 1.15. The hydroaminomethylation of 1-pentene with
piperidine (Scheme 1.14) in different solvents toluene, methanol and THF showed
that THF is having, higher hydrogenation activity than that of toluene and higher
chemoselectivity than that of methanol for the rhodium carbene complex.
Scheme. 1.14. Hydroaminomethylation with Rh-carbene catalyst
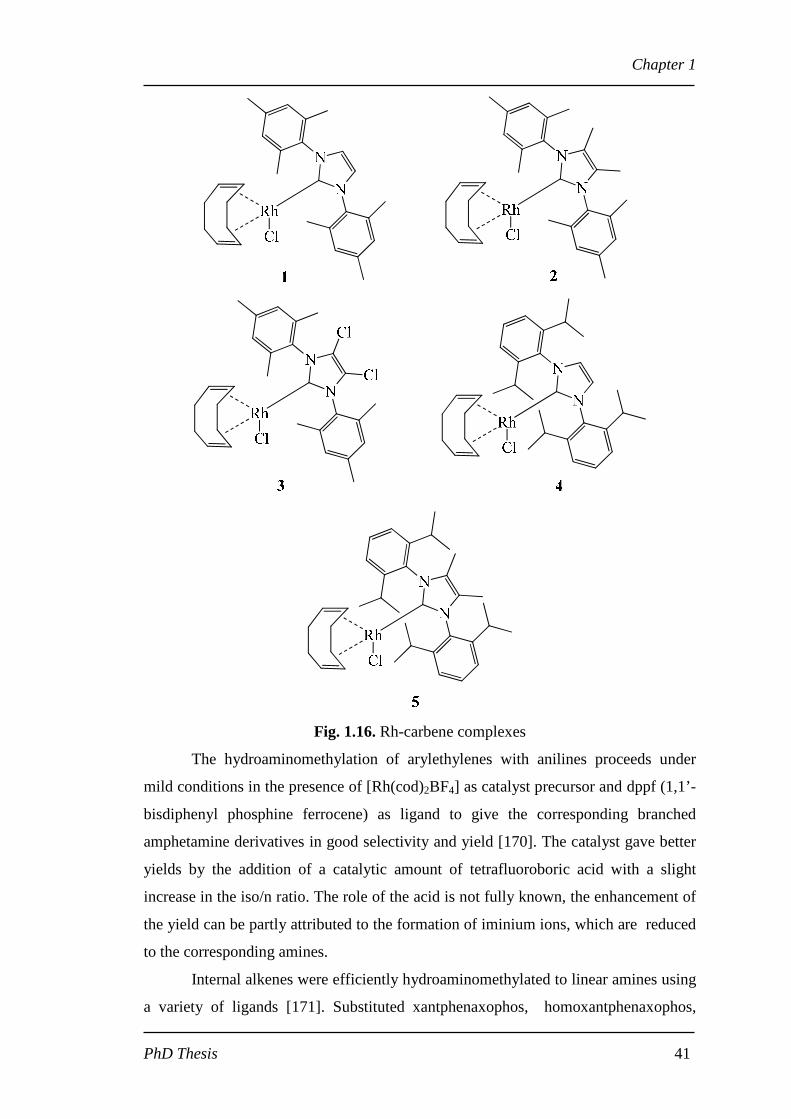
The catalysts were synthesized from Rh(cod)Cl2 precursor with five different
substituted 1,3-dimesitylimidazol-2-ylidene (Fig. 1.16) [169]. The synthesis of
pheniramines were yielded with maximum TOF (turn over frequency) of 288 h-1 with
rhodium carbene catalyst, 4 . The conversion levels were >90% with high n/iso ratio
of 99:1.
Chapter 1
PhD Thesis 41
Fig. 1.16. Rh-carbene complexes
The hydroaminomethylation of arylethylenes with anilines proceeds under
mild conditions in the presence of [Rh(cod)2BF4] as catalyst precursor and dppf (1,1’-
bisdiphenyl phosphine ferrocene) as ligand to give the corresponding branched
amphetamine derivatives in good selectivity and yield [170]. The catalyst gave better
yields by the addition of a catalytic amount of tetrafluoroboric acid with a slight
increase in the iso/n ratio. The role of the acid is not fully known, the enhancement of
the yield can be partly attributed to the formation of iminium ions, which are reduced
to the corresponding amines.
Internal alkenes were efficiently hydroaminomethylated to linear amines using
a variety of ligands [171]. Substituted xantphenaxophos, homoxantphenaxophos,

Chapter 1
PhD Thesis 42
thixantphenaxophos, nixantphenaxophos etc., were used as ligands for this reaction.
Pent-2-ene was used as internal alkene with piperidine to give linear amines with
>65% selectivity in a toluene/methanol mixed solvent. [Rh(cod)2]BF4 was used as the
catalyst precursor. A conversion of 100% with linear amine selectivity of 95% for 2-
pentene and piperidine was obtained using xantphenaxophos ligand.
Similarly other reports using supercritical ammonia [172], effect of lewis acid
on rhodium complexes with phosphine ligands bearing donor sites [173] were
investigated by Beller et al.
1.4.2.3. Important contributions to hydroaminomethylation by other groups
The contributions of Alper et al. to hydroaminomethylations are noteworthy.
A novel route to 1,2,3,4-tetrahydroquinolines via rhodium(I) catalyzed
hydroaminomethylation of 2-isopropenylanilines was an important achievement
[174]. Tetrahydroquinolines play an important role in the fields of natural products
and medicinal chemistry. They are of synthetic interest for the preparation of
pharmaceuticals and agrochemicals, as well as in material sciences. Ionic diamine
rhodium(I) complexes were used as the catalyst (Fig. 1.17). The catalysis could give
tetrahydroquinolines in high yields.
Fig. 1.17. Structure of ionic diamine rhodium(I) complex
The same catalyst (Fig. 1.17) was used for the synthesis of 2,3,4,5-Tetrahydro-
1H-2-benzazepines [175]. Notable features of the method were, the air stable catalyst
not requiring added phosphine, the ease of access of substrates and the high product
yields.
Hydroaminomethylation of long chain alkenes with dimethylamine catalyzed
by a water-soluble rhodium–phosphine complex, RhCl(CO)(TPPTS)2 in an aqueous–
organic two-phase system in the presence of the cationic surfactant
cetyltrimethylammonium bromide (CTAB) was investigated by Luo and co-workers
Chapter 1
PhD Thesis 43
[176]. The addition of the cationic surfactant CTAB accelerated the reaction due to
the micelle effect. Lou et al. also extended their studies on biphasic
hydroaminomethylation in ionic liquids (1-n-alkyl-3-methylimidazolium tosylates)
with Rh-BISBIS (sulfonated 2,2’-bis(diphenylphosphinomethyl)-1,1’ -biphenyl)
complex as catalyst [177, 178]. High activity and selectivity for amines were achieved
using this biphasic hydroaminomethylation. The ionic liquid containing catalyst was
easily separated and recycled several times with only a slight decrease in activity.
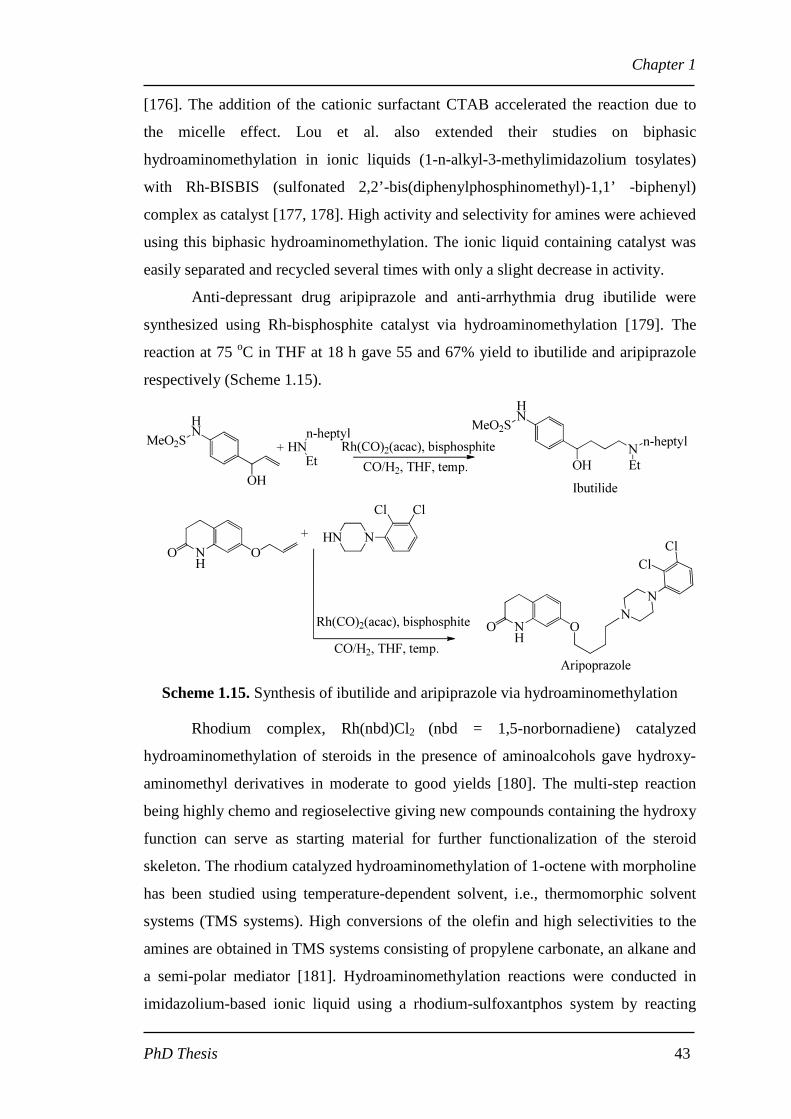
Anti-depressant drug aripiprazole and anti-arrhythmia drug ibutilide were
synthesized using Rh-bisphosphite catalyst via hydroaminomethylation [179]. The
reaction at 75 oC in THF at 18 h gave 55 and 67% yield to ibutilide and aripiprazole
respectively (Scheme 1.15).
HN
MeO2S
OH
+ HNn-heptyl
EtRh(CO)2(acac), bisphosphite
CO/H2, THF, temp.
HN
MeO2S
OH
Nn-heptyl
Et
Ibutilide
NH
O O
+ HN N
Cl Cl
Rh(CO)2(acac), bisphosphite
CO/H2, THF, temp.
NH
O ON
N
Cl
Cl
Aripoprazole
Scheme 1.15. Synthesis of ibutilide and aripiprazole via hydroaminomethylation
Rhodium complex, Rh(nbd)Cl2 (nbd = 1,5-norbornadiene) catalyzed
hydroaminomethylation of steroids in the presence of aminoalcohols gave hydroxy-
aminomethyl derivatives in moderate to good yields [180]. The multi-step reaction
being highly chemo and regioselective giving new compounds containing the hydroxy
function can serve as starting material for further functionalization of the steroid
skeleton. The rhodium catalyzed hydroaminomethylation of 1-octene with morpholine
has been studied using temperature-dependent solvent, i.e., thermomorphic solvent
systems (TMS systems). High conversions of the olefin and high selectivities to the
amines are obtained in TMS systems consisting of propylene carbonate, an alkane and
a semi-polar mediator [181]. Hydroaminomethylation reactions were conducted in
imidazolium-based ionic liquid using a rhodium-sulfoxantphos system by reacting

Chapter 1
PhD Thesis 44
piperidine with different alkenes and have given yields >95% of the product amine
with turnover frequencies of up to 16,000 h-1, along with high regioselectivity for the
linear amines with linear/branch ratios up to 78 [182]. Hydroaminomethylation using
various olefins and amines substrates and metal complexes were studied and
investigated in detail [183–187].
Hence in view of the above, hydroformylation which is extensively
investigated and hydroaminomethylation which is scantly investigated, both in
homogeneous conditions are the topic of interest to carry out in heterogeneous
conditions. Therefore the scope in this regards are very much. Attempt in the present
work is towards the development of suitable heterogeneous catalysts using a suitable
solid support for the synthesis of aldehydes (hydroformylation) and amines
(hydroaminomethylation). The heterogenization of existing homogenous catalyst was
done into the pores of a porous support which could act as nanophase reactors for
hydroformylation and hydroaminomethylation reactions.
1.5. Aim and objectives of the present work
The aim of the present work in this thesis is to synthesize rhodium based
heterogeneous catalyst for utilization in two important reactions, (1)
hydroformylation for the synthesis of aldehydes and (2) hydroaminomethylation for
the synthesis of amines. This heterogenization is done using a solid inorganic support.
Heterogenized catalyst has the potential of easy separation from the product mixture
and recyclability.
Hexagonal mesoporous silica (HMS) is an ordered mesoporous material which
is not well explored for heterogenization for hydroformylation reactions. Synthesis of
HMS is very elegant and done at room temperature. Here the objective is to
heterogenize HRh(CO)(PPh3)3 and Rh(Cl)(TPPTS) complexes in to the pores of HMS.
The complexes are in situ encapsulated thus avoiding the calcination and
functionalization of the support. The complex is trapped in the reverse micelles of the
surfactant or structure directing agent, C10 to C18 amines, which is in the pores of
HMS.
The catalyst is synthesized by in situ method as shown in Fig. 1.18. The
synthesized catalyst has shown the potential to act as nanophase reactor.

Chapter 1
PhD Thesis 45
Fig. 1.18. Synthesis of HRh(CO)(PPh3)3 encapsulated HMS
The synthesized heterogeneous catalyst using HRh(CO)(PPh3)3 is used for the
hydroformylation of propene. Propene hydroformylation is industrially high
demanding. The reaction is investigated for parametric variations and reaction
kinetics. A study on chemical engineering parameters is useful for the understanding
of the extent of mass transfer resistance. The same catalyst is then investigated for the
effect of different chain length of surfactant amine and thus the pore structure of HMS
on hydroformylation. Three different substrates, 1-hexene, styrene and cylcloheptene,
are hydroformylated. A detailed kinetic studies and kinetic modeling is investigated
for 1-hexene hydroformylation.
Fig. 1.19. Synthesis of RhCl(TPPTS)3 in HMS
Similarly RhCl(TPPTS)3 is in situ encapsulated in HMS as shown in Fig. 1.19.
The water soluble complex may have the better solubility thus increasing the better
encapsulation capacity inside the pores of HMS. Hydroformylation of functionalized
olefin, vinyl esters, are important in the production of industrially important

Chapter 1
PhD Thesis 46
intermediates. RhCl(TPPTS)3 complex is encapsulated in the hexagonal mesoporous
silica for this purpose. A study on parametric variations is conducted with vinyl
acetate as a representative vinyl ester.
Hydroaminomethylation is atom economic synthesis of amines which is less
studied in heterogeneous catalysis. Calcined HMS functionalized with aminopropyl
trimethoxy silane (APTMS) was used as a support for heterogenization of
HRh(CO)(PPh3)3 complex. The methodology of functionalization and encapsulation
of complex is given in Scheme. 1.16. The catalyst is investigated in detail for
hydroaminomethylation with various alkene and amine substrates.
Scheme 1.16. Functionalization of HMS and encapsulation of HRh(CO)(PPh3)3
complex
The investigations are further carried out with Rh exchanged Na-ETS-4 and
Na-ETS-10 (titanosilicates). The acidic nature of the ETS support can help in the
formation of enamine intermediates that facilitates the hydroaminomethylation. The
activity of both the catalysts is evaluated for various alkenes and amines for
hydroaminomethylation.

Chapter 1
PhD Thesis 47
1.6. References
[1] R. J. Jaeger, Bull. N.Y. Acad. Med., 57 (1981) 860.
[2] Carbon monoxide poisoning OSHA Fact sheet, U.S. Department of Labor,
Occupational Safety and Health Administration, (2002).
[3] W. Moffitt, The Electronic Structures of Carbon Monoxide and Carbon
Dioxide, Proceedings of the Royal Society of London. Series A, Mathematical
and Physical Sciences, 196 (1949) 524.
[4] B. Weinstock, H. Niki, Science, 176 (1972) 290.
[5] Committee on Medical and Biological Effects of Environmental Pollutants:
Carbon Monoxide, Washington, D.C., National Academy of Sciences, (1977).
pp. 29.
[6] W. Green, An Introduction to Indoor Air Quality: Carbon Monoxide (CO),
United States Environmental Protection Agency,
http://www.epa.gov/iaq/co.html.
[7] T. Gosink, What Do Carbon Monoxide Levels Mean?, Alaska Science Forum,
Geophysical Institute, University of Alaska Fairbanks, (1983).
http://www.gi.alaska.edu/ScienceForum/ASF5/588.html
[8] S. F. Singer, The Changing Global Environment, D. Reidel Dardrecht, pp. 90.
[9] L. Wu, R. Wang, Pharmacol. Rev. 57 (2005) 585.
[10] http://science.jrank.org/pages/1211/Carbon-Monoxide-Sources.html.
[11] N. Schumacher, S. Dahl, A. Gokhale, S. Kandoi, L. Grabow, J. Dumesic, M.
Mavrikakis, J. Catal. 229 (2005) 265.
[12] J. H. Jones, Platinum Metals Rev. 44 (2000) 94.
[13] J. R. Zoeller, V. H. Agreda, S. L. Cook, N. L. Lafferty, S. W. Polichnowski,
D. M. Pond, Catal. Today, 13 (1992) 73.
[14] A. Y. Khodakov, W. Chu, P. Fongarland, Chem. Rev., 107 (2007) 1692.
[15] L. Mond, C. Langer, F. Quincke, J. Chem. Soc., (1890) 749.
[16] H. Adkins, G. Krsek, J. Am. Chem. Soc., 70 (1948) 383.
[17] C. L. Aldridge, H. B. Jonassen, J. Am. Chem. Soc., 85 (1963) 886.
[18] M. Ahmed, A. M. Seayad, R. Jackstell, M. Beller, J. Am. Chem. Soc., 125
(2003) 10311.
[19] O. Roelen, DE Patent 849548, (1938/1952) to Chemische
Verwertungsgesellschaft Oberhausen.

Chapter 1
PhD Thesis 48
[20] O. Roelen, US Patent 2327006, (1943) to Chemische Verwertungsgesellschaft
Oberhausen.
[21] O. Roelen, Chem. Exp. Didakt., 3 (1977) 119.
[22] A.J. Chalk, J.F. Harrod, Adv. Organomet. Chem., 11 (1968) 119.
[23] B. Cornils, W. A. Herrmann, Applied homogeneous catalysis with
organometallic complexes, VCH, Weinheim, Germany, (1996).
[24] B. Cornils, W. A. Herrmann, M. Rasch, Angew. Chem. Int. Ed., 33 (1994)
2144.
[25] P. Pino, F. Piacenti, M. Bianchi, in: Organic synthesis via metal carbonyls,
(Eds. I. Wender, P. Piano), Wiley, New York, (1977) Chapter 2
[26] R. L. Pruett, Adv. Organomet. Chem. 17 (1979) 1.
[27] S. K. Sharma, V. K. Srivastava, R. V. Jasra, in: Catalysis in hydrocarbon
processing and fertilizer industry, Eds. M. A. Siddiqui, R. P. Verma, Lovraj
Kumar Memorial Trust, New Delhi.
[28] B. Cornils, J. Mol. Catal. A. Gen., 143 (1999) 1.
[29] H. W. Bohnen, B. Cornils, Adv. Catal., 47 (2002) 1.
[30] B. Cornils, W.A. Herrmann, J. Catal., 216 (2003) 23.
[31] L. Marko, in: Aspects of homogeneous catalysis, Eds. R. Ugo, D. Riedel,
Dardrecht, (1974) Chapter 1.
[32] V. K. Srivastava, D. U. Parmar, R. V. Jasra, Chem. Weekly, July 8 (2003) 173.
[33] V. K. Srivastava, D. U. Parmar, R. V. Jasra, Chem. Weekly, July 15 (2003)
181.
[34] Chemical Economic Handbook, Oxo chemicals report, SRI International,
(2003).
[35] S. Liu, J. Xiaom, J. Mol. Catal. A: Chem., 270 (2007) 1.
[36] F. Ungvary, Coord. Chem. Rev., 251 (2007) 2087.
[37] F. Ungvary, Coord. Chem. Rev., 251 (2007) 2072.
[38] A. M. Trzeciak, J. J. Ziolkowski, Coord. Chem. Rev., 190–192 (1999) 883.
[39] V. K. Srivastava, S. D. Bhatt, R. S. Shukla, H. C. Bajaj, R. V. Jasra., React.
Kinet. Catal. Lett., 85 (2005) 3.
[40] V. K. Srivastava, R. S. Shukla, H. C. Bajaj, R. V. Jasra., J. Mol. Catal. A.
Chemical., 202 (2003) 65.
[41] V. K. Srivastava, R. S. Shukla, S. D. Bhatt, R. V. Jasra., React. Kinet. Catal.
Lett., 81 (2004) 99.

Chapter 1
PhD Thesis 49
[42] R. S.Shukla, S. D.Bhatt, R. B.Thorat, R. V.Jasra., App. Catal. A. Gen., 294
(2005) 111.
[43] V. K. Srivastava, R. S. Shukla, H. C. Bajaj, R. V. Jasra, Appl. Catal. A: Gen.,
282 (2005) 31.
[44] V. K. Srivastava, S. K. Sharma, R. S. Shukla, N. Subrahmanyam, R. V. Jasra
Ind. Eng. Chem. Res., 44 (2005) 1764.
[45] R. S. Shukla, V. K. Srivastava, M. D. Khokhar, R. V. Jasra, Intern. J. Chem.
Kinet., 40 (2008) 359.
[46] R. F. Heck, D. S. Breslow, J. Am. Chem. Soc., 83 (1961) 4023.
[47] Chemical Economic Handbook, Oxo chemicals report, SRI International,
(2009).
[48] F. H. –J. Blankertz, M. A. V. Grenacher, O. F. Sauer, W. H. Schwahn, L. W.
Schönmann, W. M. Röper, US patent 6331656, (2001) to BASF.
[49] P. W. N. M. van Leeuwen, Homogeneous catalysis: understanding the art,
Kluwer Academic Publishers, The Netherlands, (2004) pp. 130.
[50] P. W. N. M. van Leeuwen, Homogeneous catalysis: understanding the art,
Kluwer Academic Publishers, The Netherlands, (2004) pp. 131.
[51] C. Erkey, D. R. Palo, S. Haji, Fuel Chem. Div. Preprints 47 (2002) 144.
[52] K. Weissermel, H. –J. Arpe, Industrial organic chemistry, fourth ed. Wiley-
VCH Verlag GmbH & Co. KGaA, Weinheim (2003) pp. 134.
[53] P. W. N. M. van Leeuwen, Z. Freixa, in: Activation of small molecules:
organometallic and bioinorganic perspectives, Eds: W. B. Tolman, Wiley-
VCH Verlag GmbH & Co. KGaA, Weinheim, (2006) pp. 322
[54] P. Arnoldy, in: Rhodium Catalyzed Hydroformylation, Eds: P. W. N. M. van
Leeuwen, C. Claver, Kluwer Academic Publishers, The Netherlands, (2002)
pp. 223.
[55] K. Weissermel, H. –J. Arpe, Industrial organic chemistry, fourth ed. Wiley-
VCH Verlag GmbH & Co. KGaA, Weinheim (2003) pp. 135.
[56] E. J. Rode, M. E. Davis, B. E. Hanson, J. Catal., 96 (1985) 563.
[57] E. Rode, M. E. Davis, B. E. Hanson, J. Chem. Soc. Chem. Commun., (1985)
1477.
[58] M. E. Davis, J. Schnitzer, J. A. Rossin, D. Taylor, B. E. Hanson, J. Mol.
Catal., 39 (1987) 243.
[59] K. Mukhopadhyay, R. V. Chaudhari, J. Catal., 213 (2003) 73.

Chapter 1
PhD Thesis 50
[60] K. Mukhopadhyay, A. B. Mandale, R. V. Chaudhari, Chem. Mater., 15 (2003)
1766.
[61] T. A. Kainulainen, M. K. Niemel, A. O. I. Krause, J. Mol. Catal. A. Chem.,
140 (1999) 173.
[62] N. S. Pagar, R. M. Deshpande, R. V. Chaudhari, Catal. Lett., 110 (2006) 129.
[63] J. A. D´ıaz-Auñón, M. C. Román-Mart´ınez, C. S. –M. de Lecea, J. Mol.
Catal. A. Chem. 170 (2001) 81.
[64] Y. Zhang, H. –B. Zhang, G. –D. Lin, P. Chen, Y. –Z. Yuan, K. R. Tsai, Appl.
Catal. A. Gen., 187 (1999) 213.
[65] S. Alini, A. Bottino, G. Capannelli, A. Comite, S. Paganelli, Appl. Catal. A.
Gen., 292 (2005) 105.
[66] Y. J Kim, J. B. Joo, H. C. Kim, J. Yi, J. Nanosci. Nanotechnol., 10 (2010) 269.
[67] P. Li, W. Thitsartarn, S. Kawi, Ind. Eng. Chem. Res., 48 (2009) 1824.
[68] Y. Uozumi, M. Nakazono, Adv. Synth. Catal., 344 (2002) 274.
[69] S. I. Fujita, S. Akihara, S. Fujisawa, M. Arai, J. Mol. Catal. A. Chem., 268
(2007) 244.
[70] M. M. Diwakar, R. M. Deshpande, R. V. Chaudhari, J. Mol. Catal. A. Chem.,
232 (2005) 179.
[71] S. C. Tang, T. E. Paxson, L. Kim, J. Mol. Catal., 9 (1980) 313.
[72] J. Artner, H. Bautz, F. Fan, W. Habicht, O. Walter, M. Doring, U. Arnold, J.
Catal., 255 (2008) 180.
[73] H. T. Chang, J. M. J. Frechet, J. Am. Chem. Soc., 121 (1999) 2313.
[74] J. M. J. Frechet, Science, 263 (1994) 1710.
[75] J. R. McElhanon, D. V. McGarth, J. Am. Chem. Soc., 120 (1998) 1647.
[76] J. W. J. Knapen, A. W. van der Made, J. C. de Wilde, P. W. N. M. van
Leeuwen, P. Wijkens, D. M. Grove, G. van Koten, Nature, 372 (1994) 659.
[77] M. T. Reetz, G. Lohmer, R. Schwickardi, Angew. Chem., Int. Ed., 36 (1997)
1526.
[78] M. T. Reetz, Topics Catal., 4 (1997) 187
[79] S. C. Bourque, F. Maltais, W. –J. Xiao, O. Tardif, H. Alper, P. Arya, L. E.
Manzer, J. Am. Chem. Soc., 121 (1999) 3035.
[80] S. C. Bourque, H. Alper, J. Am. Chem. Soc., 122 (2000) 956.
[81] J. P. K. Reynhardt, Y. Yang, A. Sayari, H. Alper, Chem. Mater., 16 (2004)
4095.

Chapter 1
PhD Thesis 51
[82] J. P. K. Reynhardt, Y. Yang, A. Sayari, H. Alper, Adv. Synth. Catal., 347
(2005) 1379.
[83] P. Li, S. Kawi, Catal. Today, 131 (2008) 1.
[84] H. Gao, R. J. Angelici, J. Mol. Catal. A. Chem., 145 (1999) 83.
[85] H. J. Zhu, Y. J. Ding, L. Yan, J. M. Xiong, Y. Lu, L. W. Lin, Catal. Today,
93–95 (2004) 389.
[86] L. Yan, Y. J. Ding, H. J. Zhu, J. M. Xiong, T. Wang, Z. D. Pan, L. W. Lin, J.
Mol. Catal. A. Chem., 234 (2005) 1.
[87] D. Han, X. Li, H. Zhang, Z. Liu, J. Li, C. Li, J. Catal., 243 (2006) 318.
[88] M. Marchetti, S. Paganelli, E. Viel, J. Mol. Catal. A. Chem., 222 (2004) 143.
[89] H. Zhu, Y. D. F. Yang, L. Yan, J. Xiong, H. Yin, L. Lin, J. Nat. Gas Chem., 13
(2004) 87.
[90] Y. Yang, H. Q. Lin, C. X. Deng, J. R. She, Y. Z. Yuan, Chem. Lett., 34 (2005)
220.
[91] L. Huang, J. C. Wu, S. Kawi, J. Mol. Catal. A. Chem., 206 (2003) 371.
[92] M. Nowotny, T. Maschmeyer, B. F. G. Johnson, P. Lahuerta, J. M. Thomas, J.
E. Davies, Angew. Chem. Int. Ed., 40 (2000) 955.
[93] L. Huang, Y. He, S. Kawi, Appl. Catal. A. Gen., 265 (2004) 247.
[94] L. Huang, Y. He, S. Kawi, J. Mol. Catal. A. Chem., 213 (2004) 241.
[95] Q. Peng, Y. Yang, Y. Yuan, J. Mol. Catal. A. Chem., 219 (2004) 175.
[96] B. E. Ali, J. Tijani, M. Fettouhi, J. Mol. Catal. A. Chem., 250 (2006) 153.
[97] B. E. Ali , J. Tijani, M. Fettouhi, Appl. Catal. A. Gen., 303 (2006) 213.
[98] Y. Yang, C. Deng, Y. Yuan, J. Catal., 232 (2005) 108.
[99] A. N. Ajjou, H. Alper, Mol. Online, 2 (1998) 53.
[100] L. Yan, Y. J. Dinga, L. W. Lina, H. J. Zhu, H. M. Yin, X. M. Li, Y. Lu, J.
Mol. Catal. A. Chem., 300 (2009) 116.
[101] W. Zhou, Y. Li, D. He, Appl. Catal. A. Gen., 377 (2010) 114.
[102] R. V. Jasra, V. K. Srivatava, R. S. Shukla, H. C. Bajaj, S. D. Bhatt., US patent
7294745 B2, (2007) to CSIR.
[103] V. K. Srivastava, S. K. Sharma, R. S. Shukla, R. V. Jasra., Catal. Commun., 7
(2006) 881
[104] S. K. Sharma, V. K. Srivastava, P. A. Parikh, R. S. Shukla, R.V. Jasra., New J.
Chem., 2 (2007) 277.

Chapter 1
PhD Thesis 52
[105] S. K. Sharma, R. S. Shukla, P. A. Parikh, R. V. Jasra, J. Mol. Catal. A. Chem.,
304 (2009) 33.
[106] V. K. Srivastava, S.K. Sharma, R.S. Shukla and R. V. Jasra. Ind. Eng. Chem.
Res., 47 (2008) 3795.
[107] S. K. Sharma, P. A. Parikh, R. V. Jasra, J. Mol. Catal. A. Chem., 301 (2009)
31.
[108] S. K. Sharma, P. A. Parikh, R. V. Jasra, J. Mol. Catal. A. Chem., 316 (2010)
153.
[109] Chemicals market report, December (1998).
[110] J. McMurry, Fundamentals of Organic Chemistry, Seventh ed., Brooks/Cole,
Cengage Learning, USA, pp. 404.
[111] McGraw-Hill Concise Encyclopedia of Science and Technology, Fifth Ed.,
McGraw-Hill Publishers, New York.
[112] Y. Zhang, X. Qi, X. Cui, F. Shi, Y. Deng, Tetrahedron Lett., 52 (2011) 1334.
[113] D. A. Gardner, R. T. Clark, US Patent 4255357, (1981) to Pennwalt
Corporation.
[114] J. V. M. de Pinillos, R. L. Fowlkes, US Patent 4314084, (1982) to Air
Products and Chemicals Inc.
[115] A. Baiker, J. Kijenski. Cat. Rev. Sci. Eng., 27 (1985) 653.
[116] J. C. Sheehan, V. A. Bolhofer, J. Am. Chem. Soc., 72 (1950) 2786.
[117] M. S. Gibson, R.W. Bradshaw, Angew. Chem. Int. Ed., 7 (1968) 919.
[118] O. Mitsunobu, Comp. Org. Syn., 6 (1991) 79.
[119] M. N. J. Khan, Org. Chem., 60 (1995) 4536.
[120] H. Staudinger, J. Meyer, , Helv. Chim. Acta, 2 (1919) 635.
[121] Y. G. Gololobov, Tetrahedron, 37 (1981) 437.
[122] H. Wolff, Org. React., (1946) 3.
[123] E. S. Wallis, J. F. Lane, Org. React., 3 (1949) 267.
[124] T. Shioiri, Comp. Org. Syn., 6 (1991) 800.
[125] B. Klenke, I. H. Gilbert, J. Org. Chem., 66 (2001) 2480.
[126] D. Haddenham, L. Pasumansky, J. DeSoto, S. Eagon, B. Singaram, J. Org.
Chem., 74 (2009) 1964.
[127] K. C. Hultzsch, Org. Biomol. Chem., 3 (2005) 1819.
[128] J. F. Hartwig, Pure Appl. Chem., 76 (2004) 507.
[129] T. E. Müller, M. Beller, Chem. Rev., 98 (1998) 675.

Chapter 1
PhD Thesis 53
[130] S. Hong, T. J. Marks, Acc. Chem. Res., 37 (2004) 673.
[131] P. W. Roesky, T. E. Müller, Angew. Chem. Intern. Ed., 42 (2003) 2708.
[132] F. Pohlki, S. Doye, Chem. Soc. Rev., 32 (2003) 104.
[133] G. A. Molander, E. D. Dowdy, J. Org. Chem., 64 (1999) 6515.
[134] W. Reppe, H. Vetter, Liebigs Ann. Chem., 582 (1953) 133.
[135] W. Reppe, Experientia, 5 (1949) 93.
[136] B. Hamers, E. K-. Morizet, C. Müller, D. Vogt, ChemCatChem, 1 (2009) 103.
[137] J. R. Briggs, G. T. Whiteker, J. Klosin, US patent 0215825 A1, (2005) to The
Dow Chemical Company.
[138] G. T. Whiteker, Top. Catal., 53 (2010) 1025.
[139] A. Behr, M. Becker, S. Reyer, Tetrahedron Lett. 51 (2010) 2438.
[140] Y. Y. Wang, M. M. Luo, Y. Z. Li, H. Chen, X. J. Li, Chinese Chem. Lett., 15
(2004) 774.
[141] F. Koç, M. Wyszogrodzka, P. Eilbracht, R. Haag, J. Org. Chem., 70 (2005)
2021.
[142] C. L. Kranemann, P. Eilbracht, Synthesis, 1 (1998) 71.
[143] H. Greiving, P. Eilbracht, C. Mersch, EP 1265844, 2002 to BAYER AG.
[144] D. Hollmann, S. Bähn, A. Tillack, M. Beller, Angew. Chem. Intern. Ed., 46
(2007) 8291.
[145] M. Beller, J. Seayad, A. Tillack, H. Jiao, Angew. Chem. Intern. Ed., 43 (2004)
3368.
[146] P. Eilbracht, T. Rische, Synthesis, (1997) 1331.
[147] T. Rische, B. Kitsos-Rzychon, P. Eilbracht, Tetrahedron, 54 (1998) 2723.
[148] C. L. Kranemann, B. Costisella, P. Eilbraeht, Tetrahedron Lett. 40 (1999)
7773.
[149] L. Bilrfacker, T. Rische, P. Eilbracht, Tetrahedron, 55 (1999) 7177.
[150] T. Rische, P. Eilbracht, Tetrahedron, 55 (1999) 7841.
[151] T. Rische, L. Bärfacker, P. Eilbracht, Eur. J. Org. Chem., (1999) 653.
[152] T. Rische, K. –S. Miiller, P. Eilbracht, Tetrahedron, 55 (1999) 9801.
[153] T. Rische, P. Eilbracht, Tetrahedron, 55 (1999) 1915.
[154] C. L. Kranemann, P. Eilbracht, Eur. J. Org. Chem., (2000) 2367.
[155] A. Behr, M. Fiene, C. Buß, P. Eilbracht, Eur. J. Lipid Sci. Technol., 102
(2000) 467.
[156] G. Angelovski, P. Eilbracht, Tetrahedron, 59 (2003) 8265.

Chapter 1
PhD Thesis 54
[157] M. Donsbach, P. Eilbracht, C. Buss, A. Schmidt, US patent 6809225, (2004)
to Schwarz Pharma AG.
[158] K. –S. Müller, F. Koç, S. Ricken, P. Eilbracht, Org. Biomol. Chem., 4 (2006)
826.
[159] F. Koç¸ P. Eilbracht, Tetrahedron, 60 (2004) 8465.
[160] A. Schmidt, M. Marchetti, P. Eilbracht, Tetrahedron, 60 (2004) 11487.
[161] V. K. Srivastava, P. Eilbracht, Catal. Commun., 10 (2009) 1791
[162] G. Angelovski, M. D. Keränen, P. Eilbracht, Tetrahedron: Asymm., 16 (2005)
1919.
[163] A. Seayad, M. Ahmed, H. Klein, R. Jackstell, T. Gross, M. Beller, Science,
297 (2002) 1676.
[164] B. Zimmermann, J. Herwig, M. Beller, Angew. Chem. Int. Ed., 38 (1999)
2372.
[165] T. Prinz, W. Keim, B. Driessen-Hölscher, Angew. Chem., 108 (1996) 1835.
[166] H. Bahrmann, K. Bergrath, H. –J. Kleiner, P. Lappe, C. Naumann, D. Peters,
D. Regnat, J. Organometallic Chem., 520 (1996) 97.
[167] M. Ahmed, A. M. Seayad, R. Jackstell, M. Beller, J. Am. Chem. Soc., 125
(2003) 10311.
[168] A. M. Seayad, K. Selvakumar, M. Ahmed, M. Beller, Tetrahedron Lett., 44
(2003) 1679.
[169] M. Ahmed, C. Buch, L. Routaboul, R. Jackstell, H. Klein, A. Spannenberg, M.
Beller, Chem. Eur. J., 13 (2007) 1594.
[170] L. Routaboul, C. Buch, H. Klein, R. Jackstell, M. Beller, Tetrahedron Lett., 46
(2005) 7401.
[171] M. Ahmed, R. P. J. Bronger, R. Jackstell, P. C. J. Kamer, P. W. N. M. van
Leeuwen, M. Beller, Chem. Eur. J., 12 (2006) 8979.
[172] H. Klein, R. Jackstell, M. Kant, A. Martin, M. Beller, Chem. Eng. Technol.,
30 (2007) 721.
[173] Y. Sun, M. Ahmed, R. Jackstell, M. Beller, W. R. Thiel, Organometallics, 23
(2004) 5260.
[174] T. O. Vieira, H. Alper, Chem. Commun., (2007) 2710.
[175] T. O. Vieira, H. Alper, Org. Lett., 10 (2008) 485.
[176] Y. Y. Wang, M. M. Luo, Y. Z. Li, H. Chen, X. J. Li, Appl. Catal. A. Gen. 272
(2004) 151.

Chapter 1
PhD Thesis 55
[177] Y. Y. Wang, M. M. Luo, Q. Lin, H. Chen, X. J. Li, Green Chem., 8 (2006)
545.
[178] Y. Wang, J. Chen, M. Luo, H. Chen, X. Li, Catal. Commun., 7 (2006) 979.
[179] J. R. Briggs, J. Klosin, G. T. Whiteker, Org. Lett., 7 (2005) 4795.
[180] E. Nagy, B. Heil, S. Tőrös, J. Organometallic Chem., 586 (1999) 101.
[181] A. Behr, R. Roll, J. Mol. Catal. A. Chem., 239 (2005) 180.
[182] B. Hamers, P. S. Bäerlein, C. Müller, D. Vogt, Adv. Synth. Catal., 350 (2008)
332.
[183] B. Breit, Tetrahedron Lett., 39 (1998) 5163.
[184] I. D. Kostas, J. Chem. Res. (S), (1999) 630.
[185] C. S. Graebin, V. L. Eifler-Lima, R. G. da Rosa, Catal. Commun., 9 (2008)
1066.
[186] S. K. Potluri, A. R. Ramulu, M. Pardhasaradhi, Synth. Commun., 35 (2005)
971.
[187] E. Petricci, A. Mann, J. Salvadori, M. Taddei, Tetrahedron Lett., 48 (2007)
8501.

















