Page 1
70
CHAPTER 2
EXPERIMENTAL
In this chapter, the main experimental techniques employed will be
briefly introduced. At the beginning of the chapter the complementarities of
these techniques and their relevance to the study of LC systems will be
specifically addressed.
2.1 MATERIALS
Hexane, benzene, dichloromethane, chloroform, ethylacetate,
ethanol, methanol, tetrahydrofuran, acetone, N,N-dimethylformamide,
water and thionylchloride were purified by the reported
procedure (Perrin and Armarego 1988, Furniss et al 1994). Potassium
hydroxide, sodium hydroxide, potassium carbonate, hydrochloric
acid (35%), absolute ethanol were purchased from Merck,
India. 4-Hydroxybenzoic acid, 4-hydroxybenzaldehyde, 4-hydroxyacetanilide,
potassium iodide and triethylamine were purchased Spectrochem, India.
Palladium carbon (10%), potassium dichromate, resorcinol, benzyl chloride,
N,N-dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine
(DMAP) were purchased from Aldrich. All other reagents and chemicals were
used as received.
Page 2
71
2.2 PURIFICATION OF SOLVENTS
2.2.1 Benzene
Benzene (500 mL) was shaken with about 15% of its volume of
concentrated sulphuric acid until free from thiophene, then washed with water
and 10% sodium carbonate solution, again with water and dried in fused
calcium chloride and distilled. The fraction boiling at 80 C was collected and
stored over metallic sodium wire (lit.b.p.80.1 C, Perrin and Armarego 1988).
2.2.2 Dichloromethane
Dichloromethane (100 mL) was shaken with portions of
concentrated sulphuric acid until the acid layer remains colorless and washed
with aqueous 5% sodium carbonate solution then with water. Pre-dried with
calcium chloride and distilled over phosphorus pentoxide. The fraction
boiling at 40 C was collected (lit.b.p.40 C, Perrin and Armarego 1988).
2.2.3 Chloroform
Chloroform (SRL) (500 mL) was shaken several times with half of
its volume of 10% aqueous sodium bicarbonate and followed by distilled
water; the chloroform layer was separated, dried over fused calcium chloride
for 48 h and distilled. The fraction boiling at 62 C was collected and
redistilled with P2O5 to get dry chloroform (lit.b.p.62 C, Furniss et al 1994).
2.2.4 Ethylacetate
Ethyl acetate (Spectrochem, India) (1L) was washed with aqueous
5% sodium carbonate then washed several times with sodium chloride and
dried with potassium carbonate. The fraction boiling at 77 C was collected.
(lit.b.p.77.1 C, Perrin and Armarego 1988).
Page 3
72
2.2.5 Ethanol
Rectified spirit (1000 mL) was refluxed with calcium oxide for 6 h,
set aside overnight and distilled. The fraction distilling at 80 °C was collected
(lit.b.p.80 °C, Furniss et al 1994).
2.2.6 Methanol
Dried methanol was obtained by distilling the commercial
methanol (SRL) (1 L) which was refluxed over anhydrous calcium oxide. The
distilled methanol was treated with magnesium metal and re-distilled. The
fraction boiling at 65 C was collected (lit.b.p.65 C, Furniss et al 1994)
2.2.7 Acetone
Acetone (Merck) (1L) was refluxed with successive quantities of
potassium permanganate until the violet color persisted. It was then dried with
anhydrous potassium carbonate and distilled. The fraction boiling at 57 C
was collected (lit.b.p.57 C, Furniss et al 1994).
2.2.8 N,N'-Dimethylformamide (DMF)
To 500 mL of N,N'-Dimethylformamide (SRL), freshly roasted
copper sulphate (20 g) was added and stirred. This was left for 24 h till green
colored solution was obtained and filtered. The filtrate was then distilled
under reduced pressure and the fraction boiling at 75 C/12mm Hg, was
collected (lit. b.p.75-76 C/12 mm Hg, Furniss et al 1994).
2.2.9 Thionyl Chloride
Commercial thionyl chloride was first fractionated in an all glass
apparatus from quinoline to remove acid impurities (50 g thionyl chloride
Page 4
73
from 10 mL of quinoline), the receiver is protected from the entry of moisture
by using a guard-tube, filled with anhydrous calcium chloride. The distillate
was then refractionated from boiled linseed oil the fraction boiling between
76 to 78 oC collected.
2.2.10 Water
Water (1 L) was distilled with 10 g of potassium permanganate and
sodium hydroxide. The distilled water was collected and then re-distilled to
get double distilled water (b.p.100 °C, Furniss et al 1994).
2.3 SYNTHESIS OF KEY INTERMEDIATES
2.3.1 Synthesis of 4-(10-Undecenoyloxy)biphenyl-4-carboxylic Acid
(1)
Figure 2.1 Synthesis of compound 1
10-Undecenoic acid (0.2117 mol) was dissolved in benzene (150
mL) with one drop of dimethylformamide, then thionyl chloride (75 mL;
0.6351 mol) added drop wise to the reaction mixture. The resultant mixture
was refluxed for 6 h with constant stirring. The benzene and excess thionyl
chloride were removed under vacuum to get acid chloride as colorless liquid
(yield 95%) (Petersen 1953). 10-Undecenoyl chloride (0.1mol) dissolved with
Page 5
74
100 mL dry tetrahydrofuran (THF) and 4-hydroxybiphenyl-4-carboxylic acid
(0.1 mol) followed by dry triethylamine (0.12 mol) were added and stirred at
20 C for 12 h under nitrogen atmosphere. The precipitated triethylamine
hydrochloride was removed and product dissolved in THF and filtered. The
filtrate was evaporated under vacuum to get crude product then recrystallized
in ethanol to yield white crystals (yield 92 %).
2.3.2 Synthesis of 4-(Alkyloxy)benzoic Acids (2a-2g)
Figure 2.2 Synthesis of compound 2a
4-(Alkyloxy)benzoic acids (2a-2g) were synthesized by the
following method and as a representative synthetic procedure for the series,
the synthesis of compound 4-(hexyloxy)benzoic acid (2a) is as follows: . 4-
Hydroxybenzoic acid (6.4 g, 46 mmol) and potassium hydroxide (19 g,
138 mmol) were dissolved in ethanol (30 ml) and stirred for 1h at room
temperature. 1-Bromohexane (7.6 g, 46 mmol) was then added drop wise to
this solution followed by addition of potassium iodide (0.8 g, 0.0046 mmol)
in one portion. The solution refluxed for 24 h with constant stirring. The
reaction mixture was concentrated by distillation of ethanol. Then the mixture
was poured in ice water (500 mL) and neutralized with 10% hydrochloric acid
solution. Resultant precipitate was filtered and recrystallized from absolute
ethanol to get the desired product (yield 65%). A similar procedure was
Page 6
75
adopted for preparation of other alkyl series such as heptyl, octyl, nonyl,
decyl, undecyl, and dodecyl.
2.3.3 Synthesis of 4-(4-n-Alkyloxybenzoyloxy)benzaldehydes (3a-3g)
Figure 2.3 Synthesis of compound 3a
4-(4-n-Alkyloxybenzoyloxy)benzaldehydes (3a-3g) were
synthesized by the following method and as a representative
synthetic procedure for the series, the synthesis of compound
4-(4-n-hexyloxybenzoyloxy)benzaldehyde (3a) is as follows: To a mixture
of 4-n-hexyloxybenzoic acid (5 g, 22 mmol)), 4-hydroxybenzaldehyde (1.8g
22 mmol), dicyclohexylcarbodiimide (DCC) (3g, 25mmol), and 5% w/w of
4-(N,N' dimethylamino)pyridine (DMAP) were dissolved in methylene
chloride (200 mL), and the resulting solution was stirred for 12 h at room
temperature under nitrogen atmosphere. Precipitated by product urea was
filtered from the reaction mixture and filtrate was concentrated by vacuum
evaporation. Crude product was purified by silica gel column chromatography
using chloroform as eluent. The product 4-(4-n-hexyloxybenzoyloxy)
benzaldehyde (5.8 g; 86 %) was obtained as a white powder. A similar
procedure was adopted for preparation of other alkyl series such as heptyl,
octyl, nonyl, decyl, undecyl, and dodecyl containing compounds.
COOHC6H13O CHOHO+
COOC6H13O CHO
DCC, DMAP/DCM
(3a)
Page 7
76
2.3.4 Synthesis of 4-(4-n-Alkyloxybenzoyloxy)benzoic Acids (4a-4g)
Figure 2.4 Synthesis of compound 4a
4-(4-n-Alkyloxybenzoyloxy)benzoic acids (4a-4g) were
synthesized by the following method and as a representative synthetic
procedure for the series, the synthesis of compound 4-(4-n-
hexyloxybenzoyloxy) benzoic acid (4a) is as follows: 4-(4-n-
hexyloxybenzoyloxy)benzaldehyde (5 g, 15 mmol) was dissolved in acetone
(20 mL) and diluted with water to make the final volume of 100 mL.
Addition of Jones reagent [mixture of chromium oxide (26.72 g) with
concentrated sulfuric acid (23 mL)] was continued till red color persisted for
at least 1 min. Resultant mixture was stirred at room temperature for 30 min
to ensure the completion of oxidation. Excess oxidizing reagent was
quenched with 2-propanol. Reaction solution was then diluted with water and
repeated extraction with ether. Combined organic extracts were dried over
anhydrous sodium sulfate, filtered and concentrated in vacuum. Crude
product was purified by column chromatography (4.25 g; 77%). A similar
procedure was adopted for the preparation of other alkyl series such as heptyl,
octyl, nonyl, decyl, undecyl, and dodecyl containing compounds.
Page 8
77
2.3.5 Synthesis of Resorcinolmonobenzylether (5)
Figure 2.5 Synthesis of compound 5
Resorcinol (11 g, 100 mmol) was dissolved in acetone (100 mL).
To this solution, powdered potassium carbonate (13.8 g, 300 mmol) and
potassium iodide (pinch) were added slowly. The mixture was refluxed and
benzyl chloride (11.6 mL, 100 mmol) added drop wise to the refluxing
mixture over a period of 30 min. The reaction was carried at for 48 h and
cooled the reaction to room temperature and potassium carbonate filtered and
washed with acetone, the collected filtrates concentrated by vacuum
distillation. Product thus obtained was purified by column chromatography
using chloroform and hexane (3:7) mixture as eluent to yield brown color
viscous liquid (12 g; 72%).
2.3.6 Synthesis of 4-((3-(Benzyloxy)phenoxy)carbonyl)phenyl-4-
(alkyloxy)benzoates (6a-6g)
Figure 2.6 Synthesis of compound 6a
Page 9
78
4-((3-(Benzyloxy)phenoxy)carbonyl)phenyl-4-(alkyloxy)benzoates
(6a-6g) were synthesized by the following method and as a representative
synthetic procedure for the series, the synthesis of compound 4-((3-
(benzyloxy)phenoxy)carbonyl)phenyl-4-(hexyloxy)benzoate (6a) is as
follows: To a mixture of 4-(4-n-hexyloxybenzoyloxy)benzoic acid (3.42 g,
10 mmol), resorcinol monobenzylether (2 g, 10 mmol) DCC (2.5 g,
12 mmol), and a catalytic amount of DMAP in dry dichloromethane (50 ml)
were stirred for 12 h. The precipitated N, N -9-dicyclohexylurea was filtered,
washed with excess of dichloromethane and the filtrate concentrated in a
rotary evaporator. The residue was purified by silica gel column using
chloroform as eluent. The product obtained on removal of chloroform was
further purified by recrystallization using mixture of chloroform and hexane
(1:3) to get 4.5 g yield (85%). A similar procedure was adopted for
preparation of other alkyl series such as heptyl, octyl, nonyl, decyl, undecyl,
and dodecyl containing compounds.
2.3.7 Synthesis of 4-((3-Hydroxyphenoxy)carbonyl)phenyl-4-
(alkyloxy)benzoates (7a-7g)
Figure 2.7 Synthesis of compound 7a
Page 10
79
4-((3-Hydroxyphenoxy)carbonyl)phenyl-4-(alkyloxy)benzoates
(7a-7g) were synthesized by the following method and as a representative
synthetic procedure for the series, the synthesis of compound 4-((3-
hydroxyphenoxy)carbonyl)phenyl 4-(hexyloxy)benzoate (7a) is as follows:
4-((3-(benzyloxy)phenoxy)carbonyl)phenyl-4-(hexyloxy)benzoate (4.2 g, 8.0
mmol) was dissolved in ethyl acetate (150 ml) containing a suspension of
palladium carbon(Pd/C) catalyst (10% Pd/C). The mixture was stirred for
24 h under hydrogen atmosphere, filtered and concentrated under vacuum.
Crude product thus obtained was purified by silica gel column
chromatography using ethyl acetate-hexane (1:3) as eluent to yield 88%. A
similar procedure was adopted for the preparation of other alkyl series such as
heptyl, octyl, nonyl, decyl, undecyl, and dodecyl containing compounds.
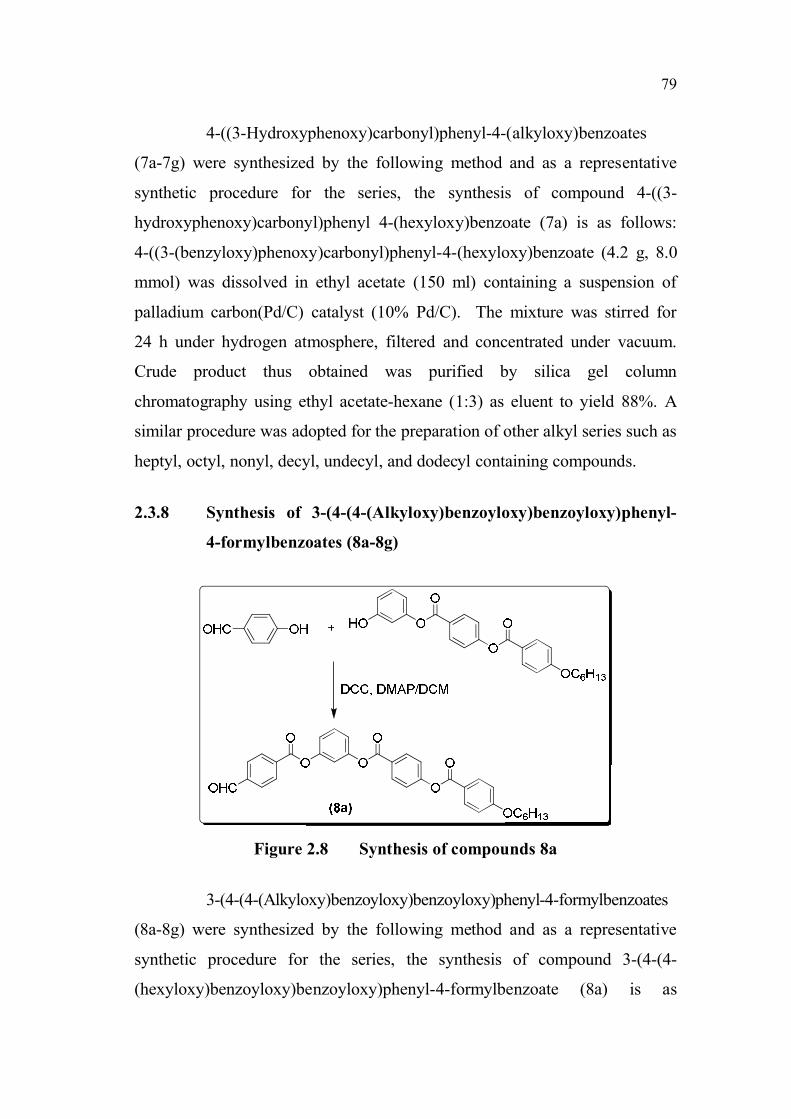
2.3.8 Synthesis of 3-(4-(4-(Alkyloxy)benzoyloxy)benzoyloxy)phenyl-
4-formylbenzoates (8a-8g)
Figure 2.8 Synthesis of compounds 8a
3-(4-(4-(Alkyloxy)benzoyloxy)benzoyloxy)phenyl-4-formylbenzoates
(8a-8g) were synthesized by the following method and as a representative
synthetic procedure for the series, the synthesis of compound 3-(4-(4-
(hexyloxy)benzoyloxy)benzoyloxy)phenyl-4-formylbenzoate (8a) is as
Page 11
80
follows: A mixture of 4-((3-hydroxyphenoxy)carbonyl)phenyl-4-(hexyloxy)
benzoate (4.34 g, 10 mmol), and a 4-formylbenzoic acid (1.5 g, 10 mmol)
and a catalytic amount of 4-(N,N-dimethylamino)pyridine (DMAP) in dry
dichloromethane (25 mL) was stirred for 10 min. To this mixture,
N,N'- dicyclohexylcarbodiimide (DCC) (2.5 g, 12 mmol) was added and
stirred for about 12 h at room temperature. The precipitated
N,N'-dicyclohexyl urea was filtered off and washed with excess of
dichloromethane. The combined organic solution was washed with ice-cold
aqueous 5% sodium hydroxide solution (NaOH) (3 75 mL), 5% hydrochloric
acid (HCl) (3 75 mL) and finally with water (3 75 mL) and dried over
anhydrous sodium sulphate. The residue obtained on removal of solvent was
chromatographed on silica gel using chloroform as eluent to yield 4.9 g
(86.5 %). A similar procedure was adopted for the preparation of other alkyl
series such as heptyl, octyl, nonyl, decyl, undecyl, and dodecyl containing
compounds.
2.3.9 Synthesis of 4-(Alkyloxy)acetanilides (9a-9g)
Figure 2.9 Synthesis of compound 9e
4-(Alkyloxy)acetanilides (9a-9g) were synthesized by the following
method and as a representative synthetic procedure for the series, the
synthesis of compound 4-(hexyloxy)acetanilide (9e) is as follows: A mixture
of 4-hydroxyacetanilide (3 g, 20 mmol), anhydrous potassium carbonate
(K2CO3) (8.4 g, 60 mmol), 1- bromodecane (3.9 g, 19 mmol) and pinch of
potassium iodide (KI) in 200 mL of acetone were stirred at 70 °C for 48 h.
Then the reaction mixture was cooled to room temperature, filtered, washed
Page 12
81
with excess of acetone. The solvent was removed under vacuum to give white
solid. The solid obtained was dissolved in diethyl ether and washed with
water (3 300 mL) to remove the unreacted 4-hydroxyacetanilide. The
organic layer was dried over anhydrous sodium sulphate, solvent removed
under vacuum and recrystallized from methylene chloride to give the 3.3 g of
bright-white crystals of 4-(decyloxy)acetanilide (Yield 60%) (Henderson et al
2001). Similar procedure was adopted for the preparation of hexyloxy,
heptyloxy, octyloxy nonyloxy, undecyloxy and dodecyloxy acetanilides.
2.3.10 Synthesis of 4-(Alkyloxy)anilines (10a-10g)
Figure 2.10 Synthesis of compounds 10e
4-(Alkyloxy)anilines (10a-10g) were synthesized by the following
method and as a representative synthetic procedure for the series, the
synthesis of compound 4-(decyloxy)aniline (10e) is as follows: The
compound 4-decyloxyacetanilide (5.82 g, 20 mmol) was dissolved in ethanol
(150 mL), 20 mL of concentrated HCl in ethanol (25 mL) added drop wise to
the mixture. The reaction mixture was heated to reflux for 12 h, cooled,
poured into ice-water mixture. The white solid thus obtained was extracted
with ether, washed with water (3 100 mL), brine solution (3 100 mL) and
dried over anhydrous sodium sulphate. The solvent was removed under rotary
evaporator to give 3.9 g of white crystal (Yield 81%). Similar procedure was
adopted for the preparation of hexyloxy, heptyloxy, octyloxy nonyloxy,
undecyloxy and dodecyloxy anilines.
Page 13
82
2.4 SYNTHESIS OF TARGET COMPOUNDS
2.4.1 Synthesis of 1,3-Substituted Phenylenebis(4-(10-undecenoyl
oxy)-1,1'-biphenyl-4-carboxylate)s (Ia-Ie)
Figure 2.11 Synthesis of compounds Ia-Ie
A typical procedure for the synthesis of Ia-Ie is as follows:
4'-(10-undecenoyloxy)biphenyl-4-carboxylic acid (3.8 mmol), resorcinol (1.9
mmol) and dicyclohexylcarbodiimide (DCC) (8.7 mmol) were dissolved in
dry dichloromethane (25 mL), to this solution N,N'-(dimethylamino)pyridine
(DMAP; 5.0 mmol) added and then stirred at room temperature for 5 h. The
precipitate thus obtained was removed by filtration and the precipitate
dissolved in dichloromethane and filtered. The filtrate was washed with 5%
HCl (3 50 mL), saturated NaCl (3 50 mL), and followed by water. The
separated organic layer was dried over anhydrous sodium sulphate and
solvent removed under reduced pressure. The product was purified by column
chromatography using chloroform as eluent, and recrystallized from ethanol
O O
O O
O
O O
OR4
R2
DCC/DMAPDCM
8 8
HO
O
O
O
8
HO OH
R4
R2
+
Ia (R2=R4=H)
Ib (R2=H; R4=COOCH3)
Ic (R2=H; R4=Cl)
Id (R2=NO2; R4=H)
Ie (R2=NO2;R4=COOCH3)
Page 14
83
to give white solid (yield 85 %). The above synthetic procedure was adopted
for all other homologoues.
2.4.2 Synthesis of 3-(4-(4-(Alkyloxy)benzoyloxy)benzoyloxy)phenyl
4'-(10-undecenoyloxy)biphenyl-4-carboxylates (IIa-IIg)
Figure 2.12 Synthesis of compounds IIa-IIg
A typical procedure for the synthesis of IIa-IIg is as follows: To a
mixture of 4'-(10-undecenoyloxy)biphenyl-4-carboxylic acid (10 mmol),
4-((3-hydroxyphenoxy)carbonyl)phenyl 4-(hexyloxy)benzoate (10 mmol)
DCC (15 mmol), and a catalytic amount of DMAP in dry dichloromethane
(100 mL) were stirred for 12 h under N2 atmosphere. Precipitated
N, N -9-dicyclohexylurea was filtered out, washed with excess of
dichloromethane and solvent removed in a rotary evaporator. The residue was
purified by silica gel column using chloroform as eluent. The product
obtained on removal of chloroform was further purified by column
chromatography using a mixture of chloroform and hexane (1:3) to get 80%
yield. The similar procedure was adopted for the preparation of other alkyl
Page 15
84
series such as hexyl, heptyl, octyl, nonyl, decyl, undecyl, and dodecyl
containing compounds.
2.4.3 Synthesis of 3-(4-(4-(Alkyloxy)benzoyloxy)benzoyloxy)phenyl
4-((4-(alkyloxy)phenylimino)methyl)benzoates (IIIa-IIIg)
Figure 2.13 Synthesis of compounds IIIa-IIIg
A typical procedure for the synthesis of IIIa-IIIg is as follows:
A solution of 4-decyloxyaniline (1.43 g, 6.4 mmol) and 3-(4-(4-
(hexyloxy)benzoyloxy)benzoyloxy)phenyl-4-formylbenzoate (1.20 g,
3.2 mmol) in chloroform (100 mL) was heated under reflux for 4 h. The
reaction mixture was concentrated and recrystallized from ethanol to get
(1.78 g, 71%) yellow crystals.
Compound IIIa IIIb IIIc IIId IIIe IIIf IIIg
n 6 7 8 9 10 11 12
Page 16
85
2.4.4 Synthesis of 3-(4-(4-(Alkyloxy)benzoyloxy)benzoyloxy)phenyl-
4-((4-(decyloxy)phenylimino)methyl)benzoates (IVa-IVg)
Figure 2.14 Synthesis of compounds IVa-IVg
A typical procedure for the synthesis of compounds IVa-IVg is
similar to the above discussed compounds IIIa-IIIg.
2.5 CHARACTERIZATION OF COMPOUNDS
In our studies, a combination of different experimental techniques
has been used to characterize the structural and phase behavior of liquid
crystalline materials. They include direct space techniques such as
spectroscopy (to ascertain the chemical structure), polarized optical
microscopy (POM) (to identification of mesophase) and X-ray diffraction
study (to conformation of mesophase), electro-optical study (to identify polar
property of the compound). Differential scanning calorimetry (DSC) was
employed to study the thermal stability and thermal transitions occurring in
liquid crystalline systems during heating and cooling ramps respectively.
Compound IVa IVb IVc IVd IVe IVf IVg
n 6 7 8 9 10 11 12
Page 17
86
2.5.1 Fourier Transform-Infrared Spectroscopy
Fourier transform-infrared spectroscopy (FT-IR) is
multidisciplinary analytical tool yields information pertaining to the structural
details of a material. FT-IR involves the absorption of electromagnetic
radiation in the infrared region of the spectrum which results changes in the
vibrational energy of a molecule. It is a valuable and formidable tool in
identifying organic compounds has polar chemical bonds such as OH, NH,
CH, etc., with good charge separation. Since every functional group has
unique vibrational energy, the IR spectra can be seen as their fingerprints.
FT-IR spectrometer (Perkin Elmer PE 1600 FT-IR) was used to substantiate
the formation of products in this study. The spectra recorded for liquid
samples were made into a thin film in between two KBr windows. On the
other hand, solid samples were recorded by making KBr (Merck, IR Grade)
pellets. About 10 mg of the sample was ground with about 70 mg of spectral
grade KBr to form a mixture, which was then made into a pellet using a
hydraulic press. All the spectra were recorded in the range 4000-400 cm-1 at a
resolution of 4 cm-1 with a maximum of 100 scans. A background spectrum
was run before recording the spectra for each sample. The spectral calibration
of the instrument was made using a polystyrene film at regular intervals of
time.
2.5.2 Nuclear Magnetic Resonance Spectroscopy
Nuclear Magnetic Resonance (NMR) is a spectroscopic method is
even more important to the organic chemist than infrared spectroscopy. Many
nuclei may be studied by NMR techniques, but hydrogen and carbon are most
commonly investigated. Whereas infrared spectroscopy reveals the types of
functional groups present in a molecule, NMR gives information about the
number of magnetically distinct atoms of the type being studied.
Page 18
87
High-resolution 1H and 13C-NMR spectra were recorded using
Brucker MSL 300P, 300 and 75.4 MHz NMR spectrometer. Deutriated
chloroform [Aldrich, CDCl3, 99.8% containing 0.03% v/v tetramethylsilane
(TMS)] was used as solvents for recording NMR spectra. The proton NMR
spectra were recorded using broadband inverse probe where the inner coil for
the protons and outer coil for ‘X’ nuclei. Solvent suppression was applied in
some cases where the solvent signal is very strong compared to the sample
signals. 13C spectra were recorded in the dual (13C/1H) probe where the inner
coil for 13C and the outer coil for protons.
2.5.3 Differential Scanning Calorimetry
Differential scanning calorimeter (DSC) has become a method of
choice for quantitative studies of thermal transition in polymers. Differential
scanning calorimetry was performed using the Perkin Elmer DSC-7 model
and Mettler Toledo STAR@ system thermal analysis unit attached to a DSC
module. The experiments were carried out in nitrogen atmosphere at a heating
rate of 10 C/min from ambient to 500 C with a nitrogen flow of 10 mL/min.
Generally, DSC measures the power released or absorbed by
materials during temperature treatments that can include dynamic (i.e.,
heating or cooling ramps) or isothermal segments. The measurement is
performed by comparing the temperature of the sample and that of the
reference materials. The instantaneous heat flux is computed from this
temperature difference using instrumental calibration constants. Standard
samples like pure indium or zinc with known transition enthalpies and
temperatures are used for the calibration.
The measuring cell of a calorimeter includes the sample and
reference materials enclosed in a single furnace. The DSC furnace is made of
silver and separated from the DSC sensor by a ceramic plate. The temperature
Page 19
88
of each of the two containers (pans) is measured by thermocouples connected
in series and located around each of them.
The measure of the enthalpy variation can allow assigning a given
thermal event to a polymorphic crystal to crystal or to a mesophase to
mesophase transition in LC systems. This is based on the fact that the
enthalpy variation associated with crystal melting by far more important than
the one corresponding to the mesophase to mesophase or mesophase to
isotropic phase transitions. The assignment can become difficult when one
deals with ordered mesophases, sometimes called “soft crystals”, which
exhibit transition enthalpies comparable to the one corresponding to crystal
melting.
The DSC is a convenient tool to measure the temperatures and
transition enthalpies to determine the phase diagram of the system and to
study the kinetics of transitions as a function of heating/cooling rates or as a
function of time. Differential scanning calorimeter has become a method of
choice for quantitative studies of thermal transition in polymers.
2.5.4 Polarizing Optical Microscope
Polarizing Optical Microscope (POM) was carried out to find the
texture analysis and also to determine the phase transition with sensitivity of
0.1 C. Polarizing microscope studies were performed with a Euromex
polarizing microscope attached with a Linkem HFS 91 heating stage and a
TP-93 temperature programmer. Samples were placed in between two thin
glass cover slips and melted with heating and cooling at the rate of 2°C/min.
The photographs were taken from Nikon FM10 camera and printed on a
Konica 400 film. All the microphotographs were taken from the second
cooling stage of isotropic transition temperature up to melting temperature.
Page 20
89
2.5.5 X-Ray Diffraction Measurements
X-Ray diffraction measurements were carried out to investigate the
texture of the mesophase. Generally powder samples and in a few cases
oriented samples were used to obtain diffraction patterns of the mesophases of
bent-core compounds. The powder samples held in sealed Lindemann
capillaries (diameter: 0.7 mm; wall thickness: 0.01 mm) were cooled from
isotropic state to the mesophase and irradiated. Oriented patterns were
obtained by slow cooling of a drop of sample on a glass plate from the
isotropic state.
The X-rays were generated by a 4 kW rotating anode generator
(Rigaku Ultrax 18). The beam was monochromated to obtain a radiation of
wavelength 1.54 Å (Cu-K radiation) by using a graphite crystal. A double slit
arrangement was used to collimate the beam, which subsequently interacted
with the sample in a sample holder. The temperature of the sample holder was
controlled by a computer to an accuracy of 0.1 ttern of
the mesophase was collected on a two-dimensional image plate detector. A
schematic representation of the X-ray set- up used is shown in Figure 2.15.
The layer spacing of the mesophase was calculated using Bragg’s Equation
(2.1),
(2.1)
where n = 1 (for first order reflection)
= 1.54 Å (wavelength of Cu-K radiation)
d = measured layer spacing
-1(R/D)
R = Radius of the diffraction pattern
D = Distance between the sample and the detector
Page 21
90
Figure 2.15 Block diagram showing the X-ray diffraction experimental
set-up
The tilt angle of the molecules in the mesophase can be calculated
using the following Equation (2.2).
-1(d/l) (2.2)
where l = calculated molecular length.
2.5.6 Electro-Optical Investigations
To study the polar properties of the banana mesophases and to
measure the polarization value for the mesophase, the standard triangular-
wave method was used (Miyasato et al 1983). A block diagram of the
experimental set-up used is shown in Figure 2.16. The electro-optical
experiments were carried out using sample cells made up of transparent glass
plates, coated with indium tin oxide (ITO). In order to obtain a planar
alignment of the sample, the inner surfaces of these conducting glass plates
were coated with polyimide and unidirectionally rubbed. The thickness of the
cell was controlled by using Mylar spacers and the thickness was measured by
interferrometric technique. In some experiments, the commercially obtained
Page 22
91
(EHC, Japan) cells were also used. The sample was filled in to the cell in the
isotropic phase and cooled slowly to get a good alignment of the sample. The
triangular wave of definite amplitude and frequency was produced by using a
wave from generator (Wavetek model 39) amplified hundred times using an
amplifier (Trek model 601-B). The output from the amplifier was divided into
two channels CH1 and CH2. The waveform Channel CH1 was directly
connected to the oscilloscope, which acted as a reference signal; CH2, the
output signal from the sample connected to the oscilloscope (Agilent 54621A)
via a 10 (or 1) k resistance. The resultant curve obtained on the oscilloscope
screen was a plot of switching current versus time. The polarization value was
calculated by integrating the area under the peaks obtained in the experiment
using the relation.
Figure 2.16 Block diagram of a circuit used for the measurement of
polarization