Page 1
1
Chapter I: Introduction
The quality of a drug plays an important role in ensuring the safety and efficacy of the
drugs. Quality assurance and control of pharmaceutical and chemical formulations is essential
for ensuring the availability of safe and effective drug formulations to consumers. Hence
Analysis of pure drug substances and their pharmaceutical dosage forms occupies a pivotal
role in assessing the suitability to use in patients. The quality of the analytical data depends on
the quality of the methods employed in generation of the data. Hence, development of rugged
and robust analytical methods is very important for statutory certification of drugs and their
formulations with the regulatory authorities.
The quality and safety of a drug is generally assured by monitoring and controlling the
assay and impurities effectively. While assay determines the potency of the drug and
impurities will determine the safety aspect of the drug. Thus, the analytical activities
concerning impurities in drugs are among the most important issues in modern
pharmaceutical analysis. Assay of pharmaceutical products plays an important role in efficacy
of the drug in patients. The impurity profile of pharmaceuticals is of increasing importance as
drug safety receives more and more attention from the public and from the media.
The wide variety of challenges is encountered while developing the methods for
different drugs depending on its nature and properties. Depending on mechanism of action,
drugs are formulated in to different pharmaceutical dosage forms like Tablets, hard gelatin
capsules, soft gelatin capsules, injections. Depending on target site of absorption of the drug,
drugs are formulated into different dosage forms with different delivery mechanisms like
immediate release, delayed release and extended release. Depending on delivery mechanism
needed, different kinds of excipients are used in formulations to achieve target release profile
of the drug in human body. Depending on the formulation matrix chosen, the complexity of
extracting the drug and its impurities from formulations will vary. This along with the
importance of achieving the selectivity, speed, cost, simplicity, sensitivity, reproducibility and
accuracy of results gives an opportunity for researchers to come out with solution to address
the challenges in getting the new methods of analysis to be adopted by the pharmaceutical
industry and chemical laboratories.
Page 2
2
The drugs may be classified according to their chemical structure or therapeutic
action, as chemotherapeutic agents and pharmacodynamic agents. Their action and
classification is the subject of any postgraduate course in pharmacy or medicinal chemistry
and forms the content of many textbooks [1-5] and hence needs no reproduction.
Drugs play a vital role in the progress of human civilisation by curing diseases. The
word drug is derived from the French word drogue, which means a dry herb. In general, a
drug may be defined as a substance used in the prevention, diagnosis, treatment or cure of
diseases in man or other animals. According to World Health Organisation (WHO), a drug
may be defined as any substance or product which is used or intended to be used for
modifying or exploring physiological systems or pathological states for the benefit (physical,
mental as well as economical) of the recipient
An ideal drug when administered to the ailing individual or host, should satisfy the
following requirements: Its action should be localised at the site where it is desired to act,
Should act on a system with efficiency and safety, Should not have any toxicity, Should have
minimum side effects, Should not injure host tissues or physiological processes, Should not
develop tolerance by the tissues even administered for long duration. Such drugs are rare and
hence the search for ideal drug continues.
Importance of Analytical methods for testing potency and impurities in drugs:
Safety and efficacy of pharmaceuticals are two fundamental issues of importance in
drug therapy. The safety of a drug is determined by its pharmacological-toxicological profile
as well as the adverse effects caused by the impurities in bulk and dosage forms. The
impurities in drugs often possess unwanted pharmacological or toxicological effects by which
any benefit from their administration may be outweighed [6]. Therefore, it is quite obvious
that the products intended for human consumption must be characterized as completely as
possible. The quality and safety of a drug is generally assured by monitoring and controlling
the impurities effectively. Thus, the analytical activities concerning impurities in drugs are
among the most important issues in modern pharmaceutical analysis. This has become quite
clear by the recent research articles on this topic [7-10].
Page 3
3
Control is more important today than ever. Until the beginning of the 20th century,
drug products were produced and sold having no imposed control. Quality was generally not
verified. Many products were patent medicines of dubious value. Some were harmful and
addictive. In 1937, ethylene glycol was used as a vehicle for an elixir of sulfanilamide, which
caused more than 100 deaths [11]. Thereupon the Food, Drug and Cosmetic act was revised
requiring advance proof of safety and various other controls for new drugs. The impurities to
be considered for new drugs are listed in regulatory documents of the Food and Drug
Administration (FDA) [12], International Conference on the Harmonization of the Technical
Requirements for Registration of Pharmaceuticals for Human Use (ICH) [13] and United
States Pharmacopoeia (USP). Nevertheless, there are many drugs in existence, which have not
been studied in such detail. The USP and National Formulary (NF) are the recognized
standards for potency and purity of new drugs. These compendia have become official upon
adoption of the first food and drug act. They formulate legal standards of quality, purity and
strength of new drugs. The good manufacturing practices provide minimum quality standards
for production of pharmaceuticals as well as their ingredients [14]. The ICH, which took place
in Yokohama, Japan in 1995 has released new guidelines on impurities in new drug products
[15].
These guidelines have a number of advantages, both for the industry and the regula-
tors. The most critical aspect of the elaboration of the guidelines was the definition of the
levels of impurities for identification and qualification. Qualification is the process of
acquiring and evaluating data for establishing the biological safety of an individual impurity
or a given impurity profile at the levels specified. The level of any impurity present in a new
drug substance that has been adequately tested in safety and clinical studies is considered
qualified. A rationale for selecting impurity limits based on safety considerations has to be
provided. Analytical procedures should be able to separate all the impurities from each other
and the method should be optimized to separate and quantify them in the dosage forms. Such
methods are to be validated demonstrating the accuracy, precision, specificity, limit of
detection, quantification, linearity range and interferences.
The validation of analytical procedures, i.e., the proof of its suitability for the intended
purpose, is an important part of the registration application for a new drug [16,17]. Additional
peak tailing, peak resolution and analyte recoveries are important in case of chromatographic
Page 4
4
methods. The ICH has harmonized the validation requirements in two guidelines [18,19]. The
first one summarizes and defines the validation characteristics needed for various types of test
procedures; the second one extends the previous text to include the experimental data required
and some statistical interpretation. These guidelines serve as a basis worldwide both for
regulatory authorities and industry and bring the importance of a proper validation to the
attention of all those involved in the process of submission of drug master files. The analytical
research and development units in the pharmaceutical industry are responsible for preparation
and validation of test methods. Every country has legislation [20] on bulk drugs and their
pharmaceutical formulations that sets standards and obligatory quality indices for them. These
regulations are presented in separate articles- general and specific- relating to individual
drugs, and are published in the form of book called Pharmacopoeia (e.g. Indian, IP[21],
United States, USP[22], European, EP[23], United Kingdom, BP[24], Martindale Extra
Pharmacopoeia[25], Merck Index[26], etc.).
The monitoring of in-process impurities was an obscure and unidentified field about
20 years ago. Now it has become a major factor in modern pharmaceutical industry. This is
mainly because of the pressure for product quality, and the demand for higher standards of
process reliability. Toxicological issues have also brought about a greater sensitivity to the
significance of impurities at trace levels [27]. New attention has been given to the various
classes of toxicants present as impurities in pharmaceutical products. In view of these changes
it has become necessary to pay more attention to the origins and pathways of a host of
impurities within the process. Frequently, impurities are formed as isomers of the desired
reaction products and a critical impurity can often enter with the feed. Analytical
identification of the problematic compounds is the first step towards the solution of the
problem. Different analytical techniques like preparative HPLC, liquid-liquid extraction,
Flash chromatography, Mass spectrometry, High Resolution mass spectrometry, NMR
spectroscopy for characterization of impurities. Analytical methods used for Monitoring of
the process reactions often lend valuable insight into the types of impurities that may be
present. The purity of the final product may often be aided by controlling the purity of the
materials used in its synthesis. Wherever possible, the levels of impurities originating for the
starting materials should be limited through appropriate in-process controls in order to avoid
the need for their monitoring in the drug substance. The use of chromatographic techniques
Page 5
5
for monitoring the starting materials, intermediates, and the process reactions is an excellent
means for controlling the purity of the final drug and thereby protecting the patient who
ultimately receives it. The best way to characterize the quality of a bulk drug is to determine
its purity. There are two possible approaches to reach this goal. The determination of the
active ingredient content with a highly accurate and precise specific method or the
determination of its impurities. In the early years of drug analysis, when chromatographic
techniques were not yet available the characterization of the purity of drugs was based on the
determination of the active ingredient content by non-specific titrimetric and photometric
methods supported by the determination of physical constants and some limit tests for known
impurities based mainly on colour reactions. The deficiencies of this approach are well
known. In many cases even highly contaminated drug materials could meet the requirements
set in the early editions of different pharmacopoeias. As a consequence of the enormous
development of the analytical technology in the last two decades entirely new
possibilities have been created for the determination of the purity of drug materials [28-
30]. In principle, it is now possible to replace all non-specific assay methods with highly
specific and precise HPLC/UPLC methods thus greatly improving the value of the
determination of the active ingredient content of bulk materials. Nearly all organic
impurities are determined by chromatographic or related methods of which Liquid
chromatography (LC) has been the most important for over the last two decades. A
thorough literature search has revealed that different methods of estimation of drugs and
its impurities based on HPLC / UPLC, capillary electrophoresis (CE), Gas-liquid
chromatography (GLC), SFC, Thin-layer chromatography (TLC) etc. were published. LC
has been the main technique used for analysis of impurities in drugs. Most used the
reversed-phase mode with UV absorbance detection whenever appropriate, because this
provided the best available reliability, analysis time, repeatability and sensitivity. In fact,
this technique has set the standard against which others are compared. Recent advances
like UPLC, U-HPLC coupled with new advances in stationary phases like columns which
are having 1.7 µm and 1.3 µm particles has revolutionized the separation science.
Today a majority of the drugs used are of synthetic origin. These are produced in bulk
and used for their therapeutic effects in pharmaceutical formulations. These biologically
active chemical substances are generally formulated into convenient dosage forms such as
Page 6
6
tablets, capsules, dry syrups, liquid orals, creams or ointments, parenterals (injections in dry
or liquid form), lotions, dusting powders, aerosols, metered dose inhalers (MDI) and dry
powder inhalers (DPI) etc. These formulations deliver the drug substances in a stable, non-
toxic and acceptable form, ensuring its bioavailability and therapeutic activity.
In tablets one or more among the diluents such as lactose monohydrate,
microcrystalline cellulose, hydroxy propyl cellulose, sodium starch glycolate, magnesium
stearate, crospovidone, calcium phosphate, mannitol, sorbitol, sucrose, aerosil, acacia, gelatin,
alginic acid, tragacanth, sodium stearyl fumarate, talc, waxes, methyl paraben, propyl
paraben, meglumine, sodium benzoate, permitted flavors and colors are added. In capsules
one or more among the excipients, certified dyes, gelatin, plasticizers, starch, lactose, talc,
preservatives are added. In dry syrups and liquid orals, sucrose, sorbitol, preservatives,
certified colors and flavors are added. In creams and ointments, waxes, carbopol, petroleum
jelly, surfactants, preservatives, permitted colors and perfumes are added. In parenterals,
water, vegetable oils, mineral oils, simulated oils, propylene glycol, dioxalamines, dimethyl
acetamide are used as vehicles. Any one or more among stabilizers, anti-oxidants, buffering
agents like citrate, acetate, phosphate, co-solvents, wetting, suspending and emulsifying
agents like tween-80, sorbital oleate and preservatives are added. In lotions, dusting powders
and aerosols, talc, silica derivatives, alcohol, preservatives are added.
In view of wide variety of excipients used in formulating drugs for administration to
patients, drug substances can undergo transformation by interacting with one or more
components of the formulation. For example, drugs which contains primary amine as function
group in their structure, can undergo Millard reaction with reducing sugars like lactose. Drug
substances can also react with trace impurities like formic acid, acetic acid, formaldehyde
present in cellulose derivatives and peroxides present in excipients like povidone, cross
povidone. Formulated drugs can degrade due to acidic or basic environments created due to
the formulation matrix. Drugs can degrade due to exposure to temperature, humidity and light
during manufacturing, transportation and storage during its shelf life. Due to this it is essential
to know the degradation pathways of the drugs in acidic, basic, neutral, oxidation conditions
and their susceptibility to temperature and humidity to formulate them in a manner in which
they are stabilized and retains its quality throughout their shelf life. As most drugs contains
functional groups which can participate in reactions in some way or the other, it is essential
Page 7
7
that the analytical methods developed for estimation of the purity and impurities are capable
enough to separate all the desired and undesired components and devoid of any interferences
from the formulation matrix. When analytical methods are able to precisely and accurately
quantify without missing any impurities, without underestimation or over estimation, and
detect all possible impurities and degradants those can form during stability studies with
adequate sensitivity and exactly reflect the quality of drug substances and drug products
(formulated products of drugs), those methods are called stability indicating methods.
In view of the foregoing discussion assaying and stability testing in pharmaceutical
analysis occupies an important role to meet the requirement of statutory certification of drugs
and their formulations by the industry .The complexity of problems encountered in
pharmaceutical analysis coupled with the importance of achieving high selectivity, speed,
cost, simplicity, sensitivity, precision and accuracy, new methods of analysis are being
quickly absorbed by the pharmaceutical industry and chemical laboratories depending upon
the facilities available.
Among the several instrumental techniques [HPLC/UPLC GC, CE (Capillary
electrophoresis), Fluorimetry, NMR, mass spectroscopy, spectrophotometry covering IR,
NIR, Raman, UV and visible regions] available for the assay of drugs, usually visible
spectrophotometric technique is simple and less expensive. The selectivity and sensitivity of
the visible spectrophotometric method depends only on the nature of chemical reactions
involved in color development and not on the sophistication of the equipment.
Spectrophotometric analytical procedures are not generally stability indicating. Most widely
used methods are based on HPLC / UPLC, GC. Capillary electrophoresis and Super critical
fluid chromatography are slowly gaining ground in recent years.
A stability-indicating assay method should accurately measure the active ingredients,
without interference from degradation products, process impurities, excipients, or other
potential impurities. If an industry uses a non-stability indicating analytical procedure for
release testing, then an analytical procedure capable of qualitatively and quantitatively
monitoring the impurities, including degradation products, should complement it. Analytical
procedures for stability studies of assay should be stability indicating. As a result of stability
testing a re-test period for the active substance or a shelf life for the pharmaceutical product
can be established, and storage conditions can be recommended.
Page 8
8
The ICH (International conference on Harmonization) guideline QIA on Stability
Testing of New Drug Substances and Products [31] emphasizes that the testing of those
features which are susceptible to change during storage and are likely to influence quality,
safety and/or efficacy must be done by validated stability indicating testing methods. It is also
mentioned that forced decomposition studies (stress testing) at temperatures in 10 °C
increments above the accelerated temperatures, extremes of pH, under oxidative and
photolytic conditions should be carried out on the drug substance and drug product so as to
establish the inherent stability characteristics and degradation pathways to support the
suitability of the proposed analytical procedures.
Formulations containing various drugs and combinations of drugs for potentiating or
complementing one another in therapy are available in market. Pharmaceutical equivalents
containing identical amounts of the same active ingredient(s) in the same dosage form and
targeted to give in the same route of administration are called as generics drugs. For a generic
drug to be approved it must be shown to be pharmaceutically equivalent and bioequivalent to
the Reference Listed Drug (RLD). They must also meet all relevant standards of strength,
quality, purity, and identity. In some cases, no analytical method is reported and quite often
the reported procedures need improvements or changes keeping in view the findings and
advances. Among the several instrumental techniques [HPLC, GC, NMR, mass spectroscopy,
UV-Vis Spectrophotometry] available for the assay and impurities of drugs, HPLC/UPLC is a
versatile tool for the qualitative and quantitative analysis of drugs and pharmaceuticals and
has become indispensable. HPLC/UPLC technique has been regarded as the best among
various instrumental techniques in spite of its cost and maintenance problems.
Keeping in view the above discussion, author has examined the present state of
development of such analytical methods for some of the widely used drugs. Hence, the author
has proposed to develop stability indicating methods for assay and impurities for five most
widely used drug products made up of drugs, namely, dexlansoprazole, repaglinide,
candesartan cilexetil + hydrochlorothiazide, docetaxel and paricalcitol. The above drugs are
selected for research to whom there is wide scope for the development of new analytical
methods for their assay and impurities determination by HPLC/UPLC by exploiting their
characteristics, physical and chemical properties.
Page 9
9
Dexlansoprazole Capsules:
Dexlansoprazole belongs to the class of proton pump inhibitors. The drug is unstable
to heat, oxidation and acid hydrolysis conditions. During formulation development stability
studies, lot of unknown degradants are observed. Literature does not contain good stability
indicating methods which addresses the separation of all degradation products of either
lansoprazole or dexlansoprazole. When the current known methods are employed, the late
eluting degradants are found to be not separating well. Author has seen this as an opportunity
to work on identification of unknown degradants as well as a stability indicating methods for
assay and impurities.
Repaglinide tablets:
Repaglinide is used for treatment of Type-2 diabetes. When the official pharmacopeal
methods are used, they are found to be deficient in separation of some unknown degradants.
Also, literature did not give a rich information on the possible solutions. This molecule
offered an opportunity to improve the existing methodology and also scope to identify few
unknown degradation products hitherto unreported.
Candesartan cilexetil + Hydrochlorothiazide Tablets:
Candesartan cilexetil is used for treatment of hypertension. Hydrochlorothiazide is
used as diuretic as a combination therapy. Although several methods are reported for analysis
of individual drugs, there was no method which can address the impurities of combination
product. Instead of following two individual methods, author has seen an opportunity to
develop a single method for estimating impurities for this combination product.
Docetaxel Injection:
Docetaxel injection is used for the treatment of cancer. When the published
methodologies are adopted, they were found to be deficient in terms of adequacy and
accuracy. Hence author has seen an opportunity to improve and publish for the benefit of
scientific community.
Page 10
10
Paricalcitol Capsules:
Paricalcitol is a drug used for treatment of secondary hyperparathyroidism. While the
Official pharmacopeal method is available for aqueous based injection formulation, there are
no published methods found in literature for oil based capsules formulation. The
pharmacopoeal method, when adopted for capsules formulation, was found to be not suitable
due to large amount of interferences from placebo matrix. Also, pharmacopeia does not
provide any structural information about degradants. Paricalcitol is administered in very low
doses like 1 µg, 2µg and 4µg capsules. Hence, it is felt that it will be challenging to develop a
suitable sensitive method for low dose oil based capsule formulation and also give an attempt
to understand the nature of degradation products of paricalcitol.
Page 11
11
Section ii: Brief account on drugs selected and the therapy:
Dexlansoprazole, a proton pump inhibitor:
Dexlansoprazole belongs to the class of proton pump inhibitors (PPIs). Proton-pump
inhibitors are a group of drugs whose main action is a pronounced and long-lasting reduction
of gastric acid production. They are the most potent inhibitors of acid secretion available
today. The group followed and has largely superseded another group of pharmaceuticals with
similar effects, but different mode-of-action, called H2-receptor antagonists. These drugs are
among the most widely-selling drugs in the world [32]. The vast majority of these drugs are
benzimidazole derivatives. However, promising new research indicates that imidazopyridine
derivatives may be a more effective means of treatment[33]. The widely used drugs in this
category are omeprazole, esomeprazole, rabeprazole, pantoprazole, lansoprazole and
dexlansoprazole.
These drugs are utilized in the treatment of many conditions such as:
1. Dyspepsia
2. Peptic ulcer disease (PUD)
3. Gastro esophageal reflux disease (GERD)
4. Laryngopharyngeal reflux
5. Barrett's esophagus
6. prevention of stress gastritis
7. Gastrinomas and other conditions that cause hypersecretion of acid
8. Zollinger-Ellison syndrome.
Proton pump inhibitors act by irreversibly blocking the hydrogen/potassium adenosine
triphosphatase enzyme system (the H+/K
+ ATPase, or more commonly gastric proton pump)
of the gastric parietal cells. The proton pump is the terminal stage in gastric acid secretion,
being directly responsible for secreting H+ ions into the gastric lumen, making it an ideal
target for inhibiting acid secretion. Targeting the terminal step in acid production, as well as
Page 12
12
the irreversible nature of the inhibition, results in a class of drugs that are significantly more
effective than H2 antagonists and reduce gastric acid secretion by up to 99%. ("Irreversibility"
refers to the effect on a single copy of the enzyme; the effect on the overall human digestive
system is reversible, as the enzymes are naturally destroyed and replaced with new copies.)
The lack of the acid in the stomach will aid in the healing of duodenal ulcers, and reduces the
pain from indigestion and heartburn, which can be exacerbated by stomach acid. However,
lack of stomach acid is also called hypochlorhydria, the lack of sufficient hydrochloric acid,
or HCl. Hydrochloric acid is required for the digestion of proteins and for the absorption of
nutrients, particularly of vitamin B12 and of calcium.
The proton pump inhibitors are given in an inactive form. The inactive form is
neutrally charged (lipophilic) and readily crosses cell membranes into intracellular
compartments (like the parietal cell canaliculus) that have acidic environments. In an acid
environment, the inactive drug is protonated and rearranges into its active form. As described
above, the active form will covalently and irreversibly bind to the gastric proton pump,
deactivating it. Dexlansoprazole is the first PPI introduced into the market which is
formulated as long acting. It is based on a dual release technology, with the first quick release
producing a plasma peak concentration about one hour after application, and the second
retarded release producing another peak about four hours later [34].
Page 13
13
Repaglinide, an insulin Secretagogue:
Repaglinide is the first drug of the meglitinide class. Meglitinides, aka "Glinides"[35],
are a class of drugs used treat diabetes type 2. They bind to an ATP-dependent K+ (KATP)
channel on the cell membrane of pancreatic beta cells in a similar manner to sulfonylureas but
at a separate binding site. This inhibits a tonic, hyperpolarizing outflux of potassium, which
causes the electric potential over the membrane to become more positive. This depolarization
opens voltage-gated Ca2+
channels. The rise in intracellular calcium leads to increased fusion
of insulin granulae with the cell membrane, and therefore increased secretion of (pro)insulin.
Repaglinide (Prandin), gained US FDA approval in 1997. It was branded Prandin
because of its quick onset and short duration of action concentrates its effect around meal time
(the prandium was the Roman meal which is comparable to the modern lunch).Another type
of drug in this class is nateglinide (Starlix). These drugs should be taken 0-30 minutes prior to
eating.
Diabetes mellitus (DM) is a set of related diseases in which the body cannot regulate
the amount of sugar (specifically, glucose) in the blood. The blood delivers glucose to provide
the body with energy to perform all of a person's daily activities. The liver converts the food a
person eats into glucose. The glucose is then released into the bloodstream. In a healthy
person, the blood glucose level is regulated by several hormones, primarliy insulin. Insulin is
produced by the pancreas, a small organ between the stomach and liver. The pancreas also
makes other important enzymes released directly into the gut that helps digest food. Insulin
allows glucose to move out of the blood into cells throughout the body where it is used for
fuel. People with diabetes either do not produce enough insulin (type 1 diabetes) or cannot use
insulin properly (type 2 diabetes), or both (which occurs with several forms of diabetes)[36].
Type 2 diabetes is usually controlled with diet, weight loss, exercise, and oral medications.
Like Sulfonylureas, for example, glyburide (Diabeta; Glynase; Micronase), glipizide
(Glucotrol), glimepiride (Amaryl), tolbutamide (Orinase), and tolazamide (Tolinase),
repaglinide stimulates cells in the pancreas to produce insulin. Glyburide may be more potent
than repaglinide at increasing insulin release in persons with low or high blood glucose levels,
whereas repaglinide may be more potent in persons with moderate blood glucose levels.
Page 14
14
Repaglinide is unusual in that it has a rapid onset of action and a short duration of action.
When taken just prior to meals, it promotes the release of insulin that normally occurs with
meals and is responsible for preventing blood glucose levels from becoming high. It has been
shown to lower hemoglobin A1c levels by 1.6% to 1.9%. (Hemoglobin A1c is a blood test
which measures the effectiveness of a drug in controlling high blood glucose levels. The
lower the hemoglobin A1c, the better the control.) Today there are various drugs available in
market for treatment of Diabetes. The below table gives various class of compounds classified
according to their mechanism of action for the treatment of diabetes.
Table 1.1: Oral anti-diabetic drugs and Insulin analogs [37].
Insulin
Sensitiz
ers
Biguanides Metformin# , Buformin
‡ , Phenformin
‡
TZDs/"glitazones" (PPAR) Pioglitazone, Rivoglitazone
†,
Rosiglitazone, Troglitazone‡
Dual PPAR agonist Aleglitazar
†, Muraglitazar
§, Tesaglitazar
§,
Ragaglitazar§
Secreta
gogues
K+ ATP -Sulfonylureas
1st generation: Acetohexamide,
Carbutamide, Chlorpropamide,
Metahexamide, Tolbutamide, Tolazamide
2nd generation: Glibenclamide
(Glyburide)# , Glibornuride, Glipizide,
Gliquidone, Glisoxepide, Glyclopyramide,
Glimepiride, Gliclazide
K+ ATP -
Meglitinides/"glinides"
Repaglinide, Nateglinide, Mitiglinide
GLP-1 agonists Exenatide, Liraglutide, Taspoglutide
†,
Albiglutide†, Lixisenatide
DPP-4 inhibitors Alogliptin
†, Gemigliptin, Linagliptin,
Saxagliptin, Sitagliptin, Vildagliptin
Analogs/other insulins fast-acting (Insulin lispro · Insulin aspart ·
Insulin glulisine) · short-acting (Regular
insulin) ·
long-acting (Insulin glargine · Insulin
detemir · NPH insulin) ·
ultra-long-acting (Insulin degludec†) ·
inhalable Exubera‡
Others
Alpha-glucosidase inhibitors Acarbose, Miglitol, Voglibose
Amylin analog Pramlintide
SGLT2 inhibitors Canagliflozin†, Dapagliflozin
†,
Remogliflozin§, Sergliflozin
§
o #WHO-Essential Medicine ,
‡Withdrawn from market , Clinical trials: (
†Phase III ·
§Never to
phase III)
Page 15
15
Candesartan + Hydrochlorothiazide, an anti-hypertensive and Diuretic
Candesartan cilexetil is a drug used for treating high blood pressure (hypertension).
It is in a class of drugs called angiotensin receptor blockers (ARBs). Angiotensin, formed in
the blood by the action of angiotensin converting enzyme (ACE), is a powerful chemical that
attaches to angiotensin receptors found in many tissues but primarily on smooth muscle cells
surrounding blood vessels [38]. Angiotensin II is a very potent chemical that causes muscles
surrounding blood vessels to contract, thereby narrowing blood vessels. This narrowing
increases the pressure within the vessels and can cause high blood pressure (hypertension).
Angiotensin II receptor blockers (ARBs) are medications that block the action of angiotensin
II by preventing angiotensin II from binding to angiotensin II receptors on blood vessels. As a
result, blood vessels enlarge (dilate) and blood pressure is reduced. Reduced blood pressure
makes it easier for the heart to pump blood and can improve heart failure. In addition, the
progression of kidney disease due to high blood pressure or diabetes is slowed. ARBs have
effects that are similar to angiotensin converting enzyme (ACE) inhibitors, but ACE inhibitors
act by preventing the formation of angiotensin II rather than by blocking the binding of
angiotensin II to muscles on blood vessels [39].
ARBs are used for controlling high blood pressure, treating heart failure, and
preventing kidney failure in people with diabetes or high blood pressure. They may also
prevent diabetes and reduce the risk of stroke in patients with high blood pressure and an
enlarged heart. ARBs may also prevent the recurrence of atrial fibrillation. Since these
medications have effects that are similar to those of ACE inhibitors, they often are used when
ACE inhibitors are not tolerated by patients (for example, due to excessive coughing) [39].
Hydrochlorothiazide, is a first-line diuretic drug of the thiazide class that acts by
inhibiting the kidneys' ability to retain water. This reduces the volume of the blood,
decreasing blood return to the heart and thus cardiac output and, by other mechanisms, is
believed to lower peripheral vascular resistance [40]. Thiazides are also used in the treatment
of osteoporosis. Thiazides decrease mineral bone loss by promoting calcium retention in the
kidney, and by directly stimulating osteoblast differentiation and bone mineral formation[41].
Page 16
16
Hydrochlorothiazide is frequently used for the treatment of hypertension, congestive
heart failure, symptomatic edema, diabetes insipidus, renal tubular acidosis, and the
prevention of kidney stones [42]. It is also sometimes used for hypercalciuria, Dent's disease
and Ménière's disease. Hydrochlorothiazide reduces blood volume by acting on the kidneys to
reduce sodium (Na) reabsorption in the distal convoluted tubule. The major site of action in
the nephron appears on an electroneutral Na+-Cl- co-transporter by competing for the
chloride site on the transporter. By impairing Na transport in the distal convoluted tubule,
hydrochlorothiazide induces a natriuresis and concomitant water loss. Thiazides increase the
reabsorption of calcium in this segment in a manner unrelated to sodium transport.[43]. The
candesartan + hydrochlorothiazide fixed dose combination is indicated for the treatment of
hypertension. This fixed dose combination is not indicated for initial therapy [55-56]. There
are several drugs today available in the market and in research for the treatment of
hypertension. The below table gives various class of compounds classified according to their
mechanism of action for the treatment.
Table 1.2: Antihypertensives: agents acting on the renin-angiotensin system.[44]
A II RBs/
("-sartan")
Azilsartan, Candesartan, Candesartan (+HCT), Eprosartan, Irbesartan,
Losartan (+HCT), Olmesartan, Tasosartan§, Telmisartan (+HCT),
Valsartan, Valsartan (+HCT).
ACE inhibitors
("-pril")
Sulfhydryl-containing: Captopril , Zofenopril
Dicarboxylate-containing: Enalapril#, Ramipril, Quinapril, Perindopril,
Lisinopril (+HCT), Benazepril Phosphonate-containing: Fosinopril
Other/ungrouped: Alacepril, Cilazapril, Delapril, Imidapril, Moexipril,
Rentiapril, Spirapril, Temocapril, Trandolapril
Renin inhibitors/
("-kiren")
Aliskiren (+amlodipine), Remikiren§
#WHO-Essential Medicine , ‡Withdrawn from market , Clinical trials: (†Phase III · §Never to phase III)
Page 17
17
Docetaxel, a mitotic inhibitor:
A mitotic inhibitor is a drug that inhibits mitosis, or cell division. These drugs
disrupt microtubules, which are structures that pull the cell apart when it divides. Mitotic
inhibitors are used in cancer treatment, because cancer cells are able to grow and eventually
spread through the body (metastasize) through continuous mitotic division and so are more
sensitive to inhibition of mitosis than normal cells. Mitotic inhibitors are derived from natural
substances such as plant alkaloids, and prevent cells from undergoing mitosis by disrupting
microtubule polymerization, thus preventing cancerous growth. Microtubules are long,
ropelike proteins that extend through the cell and move cellular components around. One of
the important functions of microtubules is to move and separate chromosomes and other
components of the cell for cell division (mitosis). Mitotic inhibitors interfere with the
assembly and disassembly of tubulin into microtubule polymers. This interrupts cell division,
usually during the mitosis (M) phase of the cell cycle when two sets of fully formed
chromosomes are supposed to separate into daughter cells.[45,46].
Docetaxel exhibits cytotoxic activity on breast, colorectal, lung, ovarian, gastric, renal
and prostate cancer cells [48, 54]. Docetaxel does not block disassembly of interphase
microtubules and so does not prevent entry into the mitotic cycle, but does block mitosis by
inhibiting mitotic spindle assembly. Resistance to paclitaxel or anthracycline doxorubicin
does not necessarily indicate resistance to docetaxel. Microtubules formed in the presence of
docetaxel are of a larger size than those formed in the presence of paclitaxel, which may result
in improved cytotoxic efficacy . Abundant formation of microtubules and the prevention to
replicate caused by the presence of docetaxel leads to apoptosis of tumour cells and is the
basis of docetaxel use as a cancer treatment[48,49].
The cytotoxic activity of docetaxel is exerted by promoting and stabilising
microtubule assembly, while preventing physiological microtubule
depolymerisation/disassembly in the absence of GTP[50]. This leads to a significant decrease
in free tubulin, needed for microtubule formation and results in inhibition of mitotic cell
division between metaphase and anaphase, preventing further cancer cell progeny. Because
microtubules do not disassemble in the presence of docetaxel, they accumulate inside the cell
and cause initiation of apoptosis. Apoptosis is also encouraged by the blocking of apoptosis-
Page 18
18
blocking bcl-2 oncoprotein. Both in vitro and in vivo analysis show the anti-neoplastic
activity of docetaxel to be effective against a wide range of known cancer cells, cooperate
with other anti-neoplastic agents activity, and have greater cytotoxicity than paclitaxel,
possibly due to its more rapid intracellular uptake. The main mode of therapeutic action of
docetaxel is the suppression of microtubule dynamic assembly and disassembly, rather than
microtubule bundling leading to apoptosis, or the blocking of bcl-2 [48, 51].
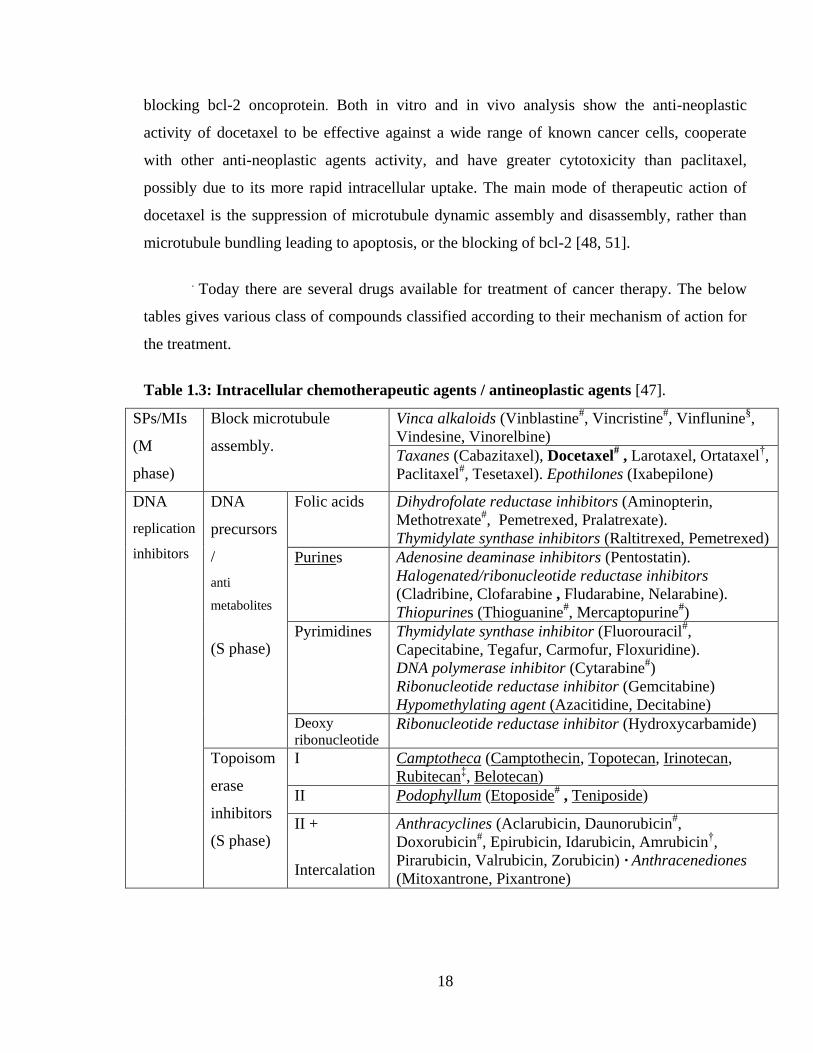
. Today there are several drugs available for treatment of cancer therapy. The below
tables gives various class of compounds classified according to their mechanism of action for
the treatment.
Table 1.3: Intracellular chemotherapeutic agents / antineoplastic agents [47].
SPs/MIs
(M
phase)
Block microtubule
assembly.
Vinca alkaloids (Vinblastine#, Vincristine
#, Vinflunine
§,
Vindesine, Vinorelbine)
Taxanes (Cabazitaxel), Docetaxel# , Larotaxel, Ortataxel
†,
Paclitaxel#, Tesetaxel). Epothilones (Ixabepilone)
DNA
replication
inhibitors
DNA
precursors
/
anti
metabolites
(S phase)
Folic acids Dihydrofolate reductase inhibitors (Aminopterin,
Methotrexate#, Pemetrexed, Pralatrexate).
Thymidylate synthase inhibitors (Raltitrexed, Pemetrexed)
Purines Adenosine deaminase inhibitors (Pentostatin).
Halogenated/ribonucleotide reductase inhibitors
(Cladribine, Clofarabine , Fludarabine, Nelarabine).
Thiopurines (Thioguanine#, Mercaptopurine
#)
Pyrimidines Thymidylate synthase inhibitor (Fluorouracil#,
Capecitabine, Tegafur, Carmofur, Floxuridine).
DNA polymerase inhibitor (Cytarabine#)
Ribonucleotide reductase inhibitor (Gemcitabine)
Hypomethylating agent (Azacitidine, Decitabine)
Deoxy
ribonucleotide Ribonucleotide reductase inhibitor (Hydroxycarbamide)
Topoisom
erase
inhibitors
(S phase)
I Camptotheca (Camptothecin, Topotecan, Irinotecan,
Rubitecan‡, Belotecan)
II Podophyllum (Etoposide# , Teniposide)
II +
Intercalation
Anthracyclines (Aclarubicin, Daunorubicin#,
Doxorubicin#, Epirubicin, Idarubicin, Amrubicin
†,
Pirarubicin, Valrubicin, Zorubicin) · Anthracenediones
(Mitoxantrone, Pixantrone)
Page 19
19
Table 1.3: Intracellular chemotherapeutic agents / antineoplastic agents ( conti.,)
DNA replication
inhibitors
Crosslinking of
DNA (CCNS)
Alkylating Nitrogen mustards: Mechlorethamine,
Cyclophosphamide# (Ifosfamide,
Trofosfamide), Chlorambucil#
(Melphalan, Prednimustine),
Bendamustine, Uramustine,
Estramustine.
Nitrosoureas: Carmustine, Lomustine
(Semustine), Fotemustine, Nimustine,
Ranimustine, Streptozocin.
Alkyl sulfonates: Busulfan
(Mannosulfan, Treosulfan)
Aziridines: Carboquone, ThioTEPA,
Triaziquone, Triethylenemelamine
Platinum-
based
Carboplatin, Cisplatin, Nedaplatin,
Oxaliplatin, Satraplatin
Nonclassical Hydrazines (Procarbazine#) ·
Triazenes (Dacarbazine#,
Temozolomide), Altretamine,
Mitobronitol , Pipobroman
Intercalation Streptomyces (Actinomycin#,
Bleomycin#, Mitomycin, Plicamycin)
Photosensitizers/
PDT
Aminolevulinic acid / Methyl aminolevulinate, Efaproxiral,
Porphyrin derivatives (Porfimer sodium, Talaporfin, Temoporfin,
Verteporfin)
Others Enzyme
inhibitors
FI (Tipifarnib), CDK inhibitors (Alvocidib†,
Seliciclib†), PrI (Bortezomib), PhI (Anagrelide),
IMPDI (Tiazofurine§), LI (Masoprocol), PARP
inhibitor (Olaparib§), HDAC (Panobinostat, Vorinostat,
Romidepsin)
Receptor
antagonists
ERA (Atrasentan) , Retinoid X receptor (Bexarotene),
Sex steroid (Testolactone)
Others
/ungrouped
Amsacrine, Trabectedin, Retinoids (Alitretinoin,
Tretinoin), Arsenic trioxide, Asparagine depleters
(Asparaginase#/Pegaspargase), Demecolcine,
Elesclomol§, Elsamitrucin, Etoglucid, Lonidamine,
Lucanthone , Mitoguazone, Mitotane, Oblimersen†,
Omacetaxine mepesuccinate§, Eribulin
#WHO-Essential Medicine ,
‡Withdrawn from market , Clinical trials: (
†Phase III ·
§Never to
phase III)
Page 20
20
Paricalcitol, an agonist for the vitamin D receptor :
Paricalcitol, is a drug used for the prevention and treatment of secondary
hyperparathyroidism associated with chronic renal failure[55] There are three kinds of
hyperparathyroidism, namely, primary hyperparathyroidism, secondary hyperparathyroidism
and tertiary hyperparathyroidism.
Primary hyperparathyroidism causes hypercalcemia (elevated blood calcium levels)
through the excessive secretion of parathyroid hormone (PTH), usually by an adenoma
(benign tumors) of the parathyroid glands. Signs and symptoms of primary
hyperparathyroidism are those of hypercalcemia. They are classically summarized by the
mnemonic "stones, bones, abdominal groans and psychiatric moans"[56].
Secondary hyperparathyroidism refers to the excessive secretion of parathyroid
hormone (PTH) by the parathyroid glands in response to hypocalcemia (low blood calcium
levels) and associated hypertrophy of the glands. Chronic renal failure is the most common
cause of secondary hyperparathyroidism. Failing kidneys do not convert enough vitamin D to
its active form, and they do not adequately excrete phosphate. When this happens, insoluble
calcium phosphate forms in the body and removes calcium from the circulation. Both
processes lead to hypocalcemia and hence secondary hyperparathyroidism. Secondary
hyperparathyroidism can also result from malabsorption (chronic pancreatitis, small bowel
disease, malabsorption-dependent bariatric surgery) in that the fat soluble vitamin D can not
get reabsorbed. This leads to hypocalcemia and a subsequent increase in parathyroid hormone
secretion in an attempt to increase the serum calcium levels. If left untreated, the disease will
progress to tertiary hyperparathyroidism, where correction of the underlying cause will not
stop excess PTH secretion, i.e. parathyroid gland hypertrophy becomes irreversible.
Paricalcitol acts as an agonist for the vitamin D receptor and thus lowers the blood
parathyroid hormone level [57].
Tertiary hyperparathyroidism is a state of excessive secretion of parathyroid
hormone (PTH) after a long period of secondary hyperparathyroidism and resulting
hypercalcemia[58]. Cinacalcet has greatly reduces the number of patients who ultimately
Page 21
21
require surgery for secondary hyperparathyroidism [59]; however, approximately 5% of
patients do not respond to medical therapy. When secondary hyperparathyroidism is corrected
and the parathyroid glands still can remain hyperfunctioning, it becomes tertiary
hyperparathyroidism. The treatment of choice is surgical removal of three and one half
parathyroid glands.
Page 22
22
Section-iii: Liquid chromatography
Different LC techniques:
The following are the most widely used chromatographic techniques for developing the
stability indicating assay and impurities methods for pharmaceutical formulations.
i). High performance liquid chromatography (HPLC),
ii). Ultra performance liquid chromatography (UPLC).
The following are the most widely used detectors using the above chromatographic
techniques.
i). UV-Vis detector, ii). Diode array detector, iii). Fluorescence detector, iv). Refractive index
detector, v). ELSD detector, vi). Mass detector.
Modes of chromatography :
Modes of chromatography are defined essentially according to the nature of the
interactions between the solute and the stationary phase, which may arise from hydrogen
bonding, vander walls forces, electrostatic forces or hydrophobic forces or basing on the size
of the particles. Most widely used modes of liquid chromatography are as follows:
i). Reversed phase chromatography, ii). Normal phase chromatography, iii). Ion-exchange
chromatography, iv). Size exclusion chromatography.
The essential components of the instruments used for the above techniques, the principles
involved and their application areas are well known and widely published and hence does not
need a reproduction.
Different stationary phases used in LC:
The following are the most widely used LC columns with stationary phases for separation and
quantification of wide variety of drugs and its impurities.
Page 23
23
i). Pure silica and hybrid silica columns.
ii). Silica based columns with different bonding phases like C4, C6, C8, C18, C20 and
bonding phases having functional groups like cyano, phenyl, naphthyl and amino.
iii). Silica based columns with polar embedded phases within chains of C8, C18, NH2.
iii). Hybrid silica based columns like C4, C6, C8, C18, C20 and bonding phases having
functional groups like cyano, phenyl, naphthyl and Amino.
iv). Strong cation exchange (SCX) and strong anion exchange (SAX) columns.
v). Size Exclusion chromtography (SEC) or gel permeation chromatography (GPC) columns.
vi). Silica based monolith columns.
vii). Fused core silica columns with bonding phases like C8, C18, CN, phenyl.
viii). Metal oxide columns like zirconia based and alumina based.
ix). Chiral columns.
The LC columns with the above stationary phases are available from a large number of
suppliers. The most popular brands of LC columns are Inertsil, Hypersil, X-terra, X-bridge,
Sun-fire, Atlantis, Aquity-BEH, Zorbax, Lichrosphere, Purosphere, Sperisorb, Luna,
Kromasil, ACE, YMC, Symmetry, Chiralcel and Chiralpak. These LC columns are supplied
in different dimensions, viz., lengths of 10 mm, 50 mm, 100mm, 150mm, 250mm, 300mm,
500mm and internal diameters of 2.1mm, 3.0mm, 4.0mm, 4.6mm. LC columns with
stationary phases having different particle sizes like 5.0 µm, 4.0 µm 3.5 µm, 3.0 µm, 2.5 µm,
2.0 µm, 1.9 µm, 1.8 µm, 1.7 µm and 1.3 µm are available.
The following properties of the LC column stationary phases play an important role in giving
different selectivity for separations.
i). Particle size, ii). Particle shape, iii). Pore size / Pore volume, iv). Specific surface area,
v). End capping vi). % carbon loading.
Different manufacturers supplies same columns with different properties and hence today
analytical scientists have a wide variety of choice of columns to achieve desired separations.
The properties of above mentioned different stationary phases and their applications are well
known and widely published and hence does not need a reproduction.
Page 24
24
Solvents used in HPLC / UPLC:
Chromatographers have a choice among hundreds of solvents for use as mobile-phase
components, sample solvents or in sample pretreatment. A particular selection is usually
affected by solvent characteristics that relate to detection, separation, flow resistance (column
pressure drop or mobile phase viscosity) and miscibility. The solvent properties such as UV
cutoff, refractive index, viscosity, polarity, miscibility and elutropic values described in
several text books [60] will be useful to chromatographers when it comes to selecting one or
more solvents. Most widely used solvents in reverse phase chromatography are acetonitrile,
methanol, ethanol and THF and widely used solvents in normal phase chromatography are
Hexane, methylene chloride, chloroform, ethyl acetate, isopropyl alcohol, ethanol, methanol.
Applications of LC in pharmaceutical research :
(a) Separation: This can be accomplished using HPLC/UPLC by utilizing the fact that,
certain compounds have different migration rates given a particular column and mobile
phase. The extent or degree of separation is determined by the choice of stationary phase and
mobile phase along with parameters like flow, temperature and gradient programme.
(b) Identification: For this purpose a clean peak of known sample has to be observed from
the chromatogram. Selection of column mobile phase and flow rate matter to certain level in
this process. Identification is generally by comparing with reference compound based on
retention time and also based on UV-Vis spectra in some cases. Identification can be assured
by combining two or more detection methods, where necessary.
(c) Quantification: Analyte concentrations are estimated by measuring the responses ( peak
areas) known reference standards followed by unknown samples. Quantification of known
and unknown components are done by various methods like - area normalization method,
internal standard method, external standard method and diluted standard method along with
relative response factors.
(d) Isolation : It refers to the process of isolation and purification of compounds using
analytical scale or preparative scale HPLC. Volatile buffers and solvents are preferred choice
as mobile phases as it reduces the effort on purification. Solute purity and throughput is the
key challenge in isolation and purification processes.
Page 25
25
Section-iv: Method development and Validation – General methodology:
Literature collection:
Collect and review the literature for method related information already reported on
the molecule. Collect information if available on solubility profile (solubility of Drug in
different solvents and at different pH conditions), analytical profile (Physico-chemical
properties, Eg: pKa, melting point, degradation pathways, etc) and stability profile (sensitivity
of the drug towards light, heat, moisture etc).
Chemical structures :
Collect the structures of the molecule and the impurities likely to be present, starting
material, byproduct, and intermediates in the reaction and degradation products. Compare the
structures of impurities, starting material, byproduct, intermediates and degradation products
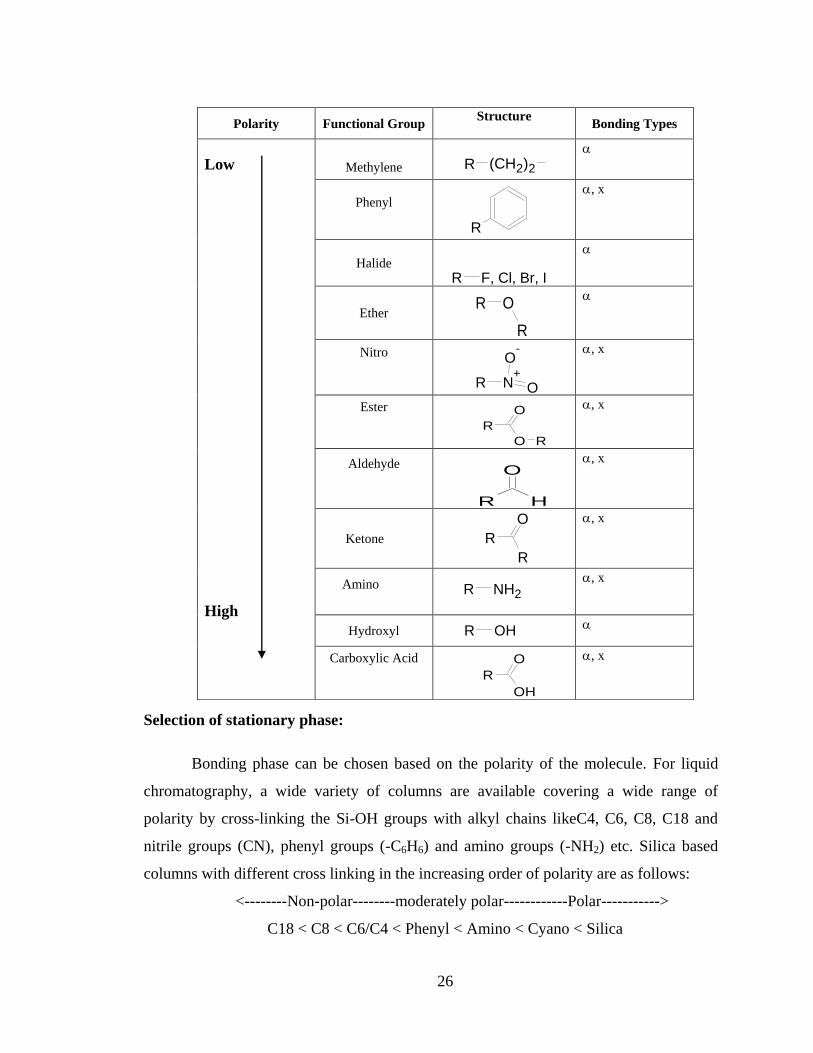
with the structure of drug substances and arrive at the polarity whether they are less polar or
more polar than the compound of interest. The below table provides the polarity index of
various functional groups.
Page 26
26
Selection of stationary phase:
Bonding phase can be chosen based on the polarity of the molecule. For liquid
chromatography, a wide variety of columns are available covering a wide range of
polarity by cross-linking the Si-OH groups with alkyl chains likeC4, C6, C8, C18 and
nitrile groups (CN), phenyl groups (-C6H6) and amino groups (-NH2) etc. Silica based
columns with different cross linking in the increasing order of polarity are as follows:
<--------Non-polar--------moderately polar------------Polar----------->
C18 < C8 < C6/C4 < Phenyl < Amino < Cyano < Silica
Polarity Functional Group Structure
Bonding Types
Low
High
Methylene
Phenyl
, x
Halide
Ether
Nitro
, x
Ester
, x
Aldehyde
, x
Ketone
, x
Amino
, x
Hydroxyl
Carboxylic Acid
, x
R (CH2)2
R
R F, Cl, Br, I
R O
R
N+
O
O-
R
O
O R
R
O
R H
O
R
R
R NH2
R OH
O
OH
R
Page 27
27
150mm x 4.6 mm, 5 µm column column is always a good starting point.
Detector selection:
PDA (photodiode Array) detector is useful for initial method development based
on the chromophores present in the compounds to be separated. Select the initial wavelength
analyzing the UV spectra of the compounds using UV-VIS spectrophotometer. If the
compounds are not having chromophores, other detectors like RI, ELSD/CCAD are useful.
Mobile phase selection:
Buffer pH to be used in mobile phase is based on the pKa of the analyte, which is
based on the structure of the molecule. Assess whether the compound is basic, acidic or
neutral. If compound is acidic, use acidic mobile phase. For a basic compound, use low pH or
basic mobile phase. For a neutral compound, neutral mobile phase is suitable. Assessment of
pKa values based on functional groups; select a pH, which is ± 1 from the pKa values. Buffer
strength of about 10 to 25 mM is advisable for initial experiments. Acetonitrile is the best
organic modifier (Because of favorable UV transmittance and Low viscosity). Methanol is the
second best organic solvent and Third organic solvent is THF. These three solvents are widely
used to control selectivity and separations.
Order of Polarity: Methanol>Acetonitrile >Ethanol>THF>Propanol
Order of Solvent Strength: Propanol>THF> Ethanol >Acetonitrile>Methanol.
THF has some disadvantages, higher UV absorbance, reactivity with oxygen and slower
column equilibration. But sometimes it gives very unique selectivity for closely eluting peaks.
Therefore these solvents are used for solvent type selectivity. Intermediate selectivity (if
needed for a particular sample) can be obtained by blending appropriate amounts of each of
these solvents.
When a peak tailing is observed, increasing the salt concentration can reduce peak
tailing for both bases and acids. For ionizable compounds, an increase in ionic strength can
suppress solute and silica ionization, as well as secondary interactions between them. Increase
beyond 50 mM is not recommended, due to possible solubility problems of the salt in the
Reverse phase Bonded REVERSE PHASE
Bonded
Page 28
28
organic portion of the mobile phase. Triethylamine (TEA) or diethyl amine (DEA) can be
added to the mobile phase to control peak tailing for bases. TEA /DEA acts as a competing
base and minimizes solute-silanol interactions. This is usually a final step to try because
TEA/DEA will reduce retention, modify the column, and complicate the mobile phase.
Whenever TEA or modifier is used to reduce the tailing, use columns with very low level of
silanols. For reducing the peak tailing for acids, 1% acetic acid can be added to the mobile
phase to minimize solute-silanol interactions as it acts as a competing acid. The relatively
high concentration of acetic acid resulted in the noisy base line. Using 0.1% TFA as the
aqueous modifier results in a more transparent mobile phase and still reduces the tailing.
Ion pair reagents to be used when necessary, when a closely related compounds
separation is required. If a mixture of ionic and non-ionic analytes to be separated, start
by optimizing the method for non-ionic compounds. Then select the appropriate ion-pair
reagent to provide the necessary selectivity. Whenever ion pair reagent is used, preferably
HPLC column should be dedicated for that specific analysis only or a cleaning Procedure
for the method with ion pair needs to be ensured every time. Alkyl sulphonates are good
first choice for basic compounds. Quaternary amines are useful for acidic compounds.
Use ion-pair reagents in the concentration range of 0.0005M to 0.02M. Sodium per
chlorate also can be used as an ion-pair reagent to get the required selectivity for acidic
components.
Selection of Elution mode:
If isocratic elution is possible, this means conditions for 0.5<k<20; If the k range
exceeds these limits, gradient elution is necessary. When an initial chromatogram
suggests a wide retention range (0.5>k>20) for an ionic sample, the use of ion pair
reagent often permits isocratic separation with 0.5<k<20. Similar changes in retention
change can also be achieved by a change in pH, to ionize late eluting compounds (for
reduced retention) or reduce the ionization of early eluting sample components (for late
elution). Gradient elution methods are widely being used due to availability of advanced
liquid chromatographic instruments and need for achieve robust separations. It is a good
idea to start method development with the following two default gradient programmes :
Page 29
29
Programme 1 Programme 2
Time Buffer
Organic
phase Time Buffer
Organic
phase
0.01 50 50 0.01 95 5
60 5 95 60 5 95
70 5 95 70 5 95
71 50 50 71 50 50
75 50 50 75 50 50
Understand elution pattern of all the peaks. Based on the elution with these gradient
programmes, further optimization can be done.
Flow rate and column temperature:
Initial flow rate of 1.0 ml-1
min or 1.5 ml-1
min and column temperature as ambient
(25 – 30°C) is preferable.
Diluent selection:
Select a diluent in which impurities, starting material, byproduct, intermediates and
degradation products and the analyte are soluble. It is advisable to check first suitability of
mobile phase as diluent. All the analytes should be completely soluble and solution should be
stable reasonably atleast until its injection into LC. For Finished dosage forms, diluents shall
be selected in such a way that the analyte(s) should be extracted > 95% for impurities and
>98% for Assay. Diluent should be compatible with the mobile phase to obtain the good peak
shape.
Selection of test concentration and injection volume :
Select a test concentration and injection volume to get an appropriate response for
the analytes for an assay method. For impurities method, test concentration and injection
volume shall be based on the sensitivity required for the method. For impurities method,
LOQ of all impurities and analyte shall be less than or equal to the reporting threshold.
Page 30
30
Degradation studies:
Degradation studies or stress testing is conducted in order to investigate the likely
degradation products, which in turn helps to establish the degradation pathways and the
intrinsic stability of the drug molecule and also to provide foundation for developing a
suitable stability indicating method. Stress testing the drug molecule under particular stress
condition generate samples containing degradation products. Use these samples to develop
suitable analytical methods. The degradation products generated in the stressed samples are
termed as “potential” degradation products that may or may not be formed under relevant
storage conditions. Stress drug product, and placebo separately to understand the peaks due
to placebo components, if any. Four major forced degradation studies are (i) Thermolytic
Degradation, (ii). Hydrolytic degradation, (iii). Oxidative degradation, (iv). Photolytic
degradation. Preferably, have the degradation in the range of 1 % to 20%, in order to reflect
the true degradation pathways. Beyond 20%, sometimes secondary degradations will occur,
which may not occur in actual conditions of use. After the satisfactory separations are
achieved, evaluate the peak purity of the analyte peak using PDA detector. Conduct Mass
balance study by assaying the stressed samples apart from quantifying the impurities. Sum of
Assay and impurities obtained in stressed samples is presented as mass balance. A mass
balance of >95% is always considered satisfactory. If the mass balance is less than the
required criteria, investigation to be done inorder to correct or to justify.
The following are the reasons for not achieving the mass balance.
(1). Degradation products are not eluted from the HPLC column,
(2). Degradation products are not detected by the detector used.
(3). Degradation products lost from the sample matrix, due to insolubility, volatility.
(4). Parent compound lost from the sample matrix, due to insolubility, volatility.
(5). Degradation products are co-eluted with the parent compound.
(6). Degradation products are not integrated due to poor chromatography.
(7). Inaccurate quantification due to differences in response factors.
Page 31
31
Finalisation of chromatographic conditions :
Based on the interferences, and based on the separations required the experiments are to be
planned to achieve the following separation goals. A base to base separation between all the
impurities. A base to base separation between impurities and placebo peaks (if any). A base-
to-base separation between all the impurities and Principal analyte peak. A base-to-base
separation between placebo peak(s), if any and Principal analyte peak. Good peak shapes for
all the impurities. Ensure that the separations are affected by small changes in the set
conditions.
Relative response factors :
Establish the relative response factors of all known impurities by linearity method
against the target analyte. Relative response factor is dividing the slope of impurity by slope
of active compound.
Selection of system suitability or Performance calculations:
Calculating one or more of the following values is necessary to access overall system
and method performance. (i). Relative retention times, (ii). Theoretical plates, (iii). Capacity
factor, (iv). Resolution, (v). tailing factor and (vi). % RSD of peak area of replicate standard
injections.
Validation of a stability indicating LC method:
The objective of the method of validation process is to provide evidence that the
method does what it is intended to. All the variables of the method should be considered,
including sample preparation, chromatographic separation, detection and data evaluation. For
chromatographic methods used in analytical applications method validations are to be carried
out in accordance with ICH guideline Q2 (R1) “Validation of Analytical Procedures: Text
(Q2A) and Methodology (Q2B)”. Typical validation characteristics which should be
considered are : accuracy, precision (repeatability, intermediate Precision), specificity,
detection Limit, quantitation limit, linearity, range and robustness. This list should be
considered typical for the analytical procedures cited but occasional exceptions should be
Page 32
32
dealt with on a case-by-case basis. The following is the table given in ICHQ2(R1) which
describe the parameters to be validated for assay and impurities methods [61].
Type of Analytical
Procedure
IDENTIFICATION TESTING FOR IMPURITIES ASSAY
Characteristics Quantitation Limit
Dissolution
( measurement
only)
Content / potency
Accuracy - + - +
Precision
Repeatability - + - +
Intermediate
precision - + (1) - + (1)
Specificity (2) + + + +
Detection limit - - (3) + -
Quantitation limit - + - -
Linearity - + - +
Range - + - +
- Signifies that this characteristic is not normally evaluated.
+ Signifies that this characteristic is normally evaluated. (1) In cases where reproducibility has been performed, intermediate precision is not
needed.
(2) Lack of specificity of one analytical procedure could be compensated by other
supporting analytical procedure(s).
(3) May be needed in some cases.
Definitions of all validation characteristics and the methodology is given in the ICH
guidance and hence does not need a reproduction. Specific procedures followed for each of
the molecules selected is given in the individual chapters. Apart from the ICH prescribed
characteristics or parameters, and stability of mobile phases are routinely studied for
operational convenience.
Page 33
33
References
1. Atherden,L.M., Edt, Bentley and Drivers, Text Book of Pharmaceutical Chemistry,
8th
Edn., Oxford University Press, 1996.
2. Melentyeva, G., Antonova, L., Pharmaceutical Chemistry, Mir Publishers, Moscow,
1988.
3. Wolff, M.E., Edt., Burger’s Medicinal Chemistry, Part IV, 4th
Edn., Wiley Interscience,
New York, 1981.
4. Deorge, R.F., Edt., Wilson and Gisvolds’s Text book of Organic and Medicinal and
Pharmaceutical Chemistry, 8th
Edn., Lippincott Company, 1982.
5. Pandeya, S.N., A Text Book of Medicinal chemistry, Vol.I and II, 2 nd
Edn., 2003.
6. Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W., Gilman, A.G., Edt,
Goodman and Gillman's The Pharmacological Basis of Therapeutics , 9th
Edn.,
McGraw Hill, New York, 1996.
7. Gorog, S., Edt., Determination of Impurities in Drugs, Elsevier Sciences, Amsterdam,
1999.
8. Ahuja, S., Impurities Evaluation of Pharmaceuticals, Marcel Dekker, New York, 1998.
9. Husain, S., Rao, R.N., Monitoring of process impurities in drugs. In: Deyl, Z., Miksik,
I., Tagliaro, F., Tesarova, E., Edt., Advanced Chromatographic and Electromigration
Methods in Biosciences, Elsevier Sciences, Amsterdam, 1998: 834.
10. Saranjit Singh, Tarun Handa, Mallikarjun Narayanam, Archana Sahu, Mahendra
Junwal, Ravi P. Shah1., Journal of Pharmaceutical and Biomedical Analysis 69
(2012), 148– 173.
11. Zanowiak, P., In: Kirk-Othmer, Edt, Encyclopedia of Chemical Technology 18, 4th
Edn.,
Wiley, Chichester, 1996: 480.
12. ANDAs: Impurities in drug products, Guidance for Industry, US Food and Drug
Administration, Center for Drug Evaluation and Research, Department of Health and
Human Services, 2008.
13. ICH. Q3A: Impurities in New Drug Substances (June 2006) and Q3B: Impurities in
New Drug Products, Nov 1999 to June 2006.
14. Maggon, K.K., Mechkovsi, A., Drug News Perspect., 1992; 5:261.
Page 34
34
15. Erni, F., Helboe, P., Pharma times, 1996; 10: 9.
16. Guidelines for Submitting Samples and Analytical Data for Methods Validation,
US Food and Drug Administration, Center for Drugs and Biologics, Department of
Health and Human Services, 1987.
17. CDER Guideline on Validation of Chromatographic Methods, Reviewer Guidance
of Chromatographic Methods, US Food and Drug Administration, Center for Drugs
and Biologics, Department of Health and Human Services, 1994.
18. ICH, Q2A: Validation of Analytical Methods, Definitions and Terminology, 1994.
19. ICH, Q2B: Analytical Validation-Methodology, 1996 & 2005.
20. The Drugs and Cosmetics Act and Rules, Government of India Publications,1984.
21. Indian Pharmacopoeia, Vol I, II, 1996 & Addendum 2005 and 2010 and Vet,
Government of India, Ministry of Health and Family Welfare, Controller of
Publications, Delhi, 2000.
22. United States Pharmacopoeia USP 35 & NF 30 and Supplements, USP
convention Inc., Rockville, 2012.
23. European Pharmacopoeia, 7th
Edn., 2010 and Supplements 2011, Council of Europe,
Strasbourg, 2011.
24. British Pharmacopoeia, Vol. I & II and Vet 2012, HMSO, London, 2012.
25. Sweetman, S.C., Martindale, The Complete Drug Reference (Extra
Pharmacopoeia) 33rd
Edn., The Pharmaceutical Press, London, 2002.
26. The Merck Index, 14th
Edn., Merck & Co Inc, New York, 2006.
27. Saletan, D., In: Creative Troubleshooting in the Chemical Process Industries,
Blackie Academic Professional, New York, 1996: 284.
28. Gorog, S., Brlik, J., Csehi, A., Halmos, Zs., Herenyi, B., Horvath, P., et al, Anal.
Method. Instrum., 1995; 2:154.
29. Cunnif, P., Edt., Official Methods of Analysis of AOAC International, 16th
Edn.,
Vol. 1. AOAC International, Arlington, 1995.
30. Gilpin, R.K., Pharmaceutical and drugs. In: Meyers, R.A., Edt., Encyclopedia of
Analytical Chemistry 8, Wiley, Chichester ,2000.
31. ICH Q1A(R2) : Stability Testing of New Drug Substances and Products, February 2003.
Page 35
35
32. Proton pump Inhibitors: http://en.wikipedia.org/wiki.
33. Sachs, G., Shin, J.M., Howden, C.W., Ailment. Pharmacol. Ther.,2006; 23 (2): 2–8
34. Metz, D.C., Vakily, M., Dixit, T., Mulford, D., Aliment Pharmacol Ther.,2009; 29 (9):
928–37.
35. Blicklé, J.F., Diabetes Metab., 2006; 32 (2): 113–20
36. Diabetes : http://www.emedicinehealth.com/diabetes/article_em.htm
37. Repaglinide : http://en.wikipedia.org/wiki/Repaglinide.
38. Candesartan :http://www.medicinenet.com/candesartan/article.htm
39. Angiotensin II receptors blockers :
http://www.medicinenet.com/angiotensin_ii_receptor_blockers/article.htm
40. Duarte, J.D., Cooper-DeHoff, R.M., Expert Rev. Cardiovasc. Ther.,2010; 8 (6): 793–802.
41. Dvorak, M.M., De Joussineau, C., Carter, D.H.,et al., J. Am. Soc. Nephrol.,2007; 18 (9):
2509–16.
42. Hydrochlorothiazide". The American Society of Health-System Pharmacists.
http://www.drugs.com/monograph/hydrochlorothiazide.html.
43. Hydrochlorothiazide :http://en.wikipedia.org/wiki/Hydrochlorothiazide
44. Candesartan : http://en.wikipedia.org/wiki/Candesartan
45. Mitotic Inhibitors : http://en.wikipedia.org/wiki/Mitotic_inhibitor#cite_note-1
46. Treatment Options: Mitotic Inhibitors". Drug Digest. Archived from the original on
2007-02-16.
47. Docetaxel : http://en.wikipedia.org/wiki/Docetaxel.
48. Lyseng-Williamson, K.A., Fenton, C., Drugs.,2005; 65 (17): 2513–31
49. Eisenhauer, E.A., Vermorken, J.B., Drugs.,1998; 55 (1): 5–30.
50. Docetaxel:Clinical Pharmacology, http://www.rxlist.com/cgi/generic3/docetaxel_cp.htm
51. Yvon, A.M., Wadsworth, P., Jordan, M.A., Mol. Biol. Cell., 1999; 10 (4): 947–59,
52. A. Michael.,K.Syrigos.,H.Pandha., Prostate Cancer and Prostatic Diseases.,2009; 12 (1):
13–16, 2009.
53.Melian, E.B., Jarvis, B., Drugs., 2002; 62(5):787-816.
54. Mengden.,T., Uen, S., Bramlage, P., Vasc Health Risk Manag., 2009; 5:1043-58.
55. Paricalcitol : http://en.wikipedia.org/wiki/Paricalcitol
Page 36
36
56. Primary hyperparathyroidism :
http://en.wikipedia.org/wiki/Primary_hyperparathyroidism
57. Secondary hyperparathyroidism :
http://en.wikipedia.org/wiki/Secondary_hyperparathyroidism
58. Tertiary hyperparathyroidism : http://en.wikipedia.org/wiki/Tertiary_hyperparathyroidism
59. Torres, P.U., J. Ren. Nutr .,2006;16 (3): 253–8.
60. Sethi, P.D., Quantitative Analysis of Drugs in Pharmaceutical Formulations,
Unique Publishers, 1985.
61. ICH guideline Q2 (R1) “Validation of Analytical Procedures: Text and
Methodology, May 1997