Chapter4 ROLE OF GLYCOSYLATION IN THE ACTIVITY OF HUMAN PANCREATIC RIBONUCLEASE uman pancreatic ribonuclease (HPR), is a glycosylated protein and has three Asn-X-Thr/Ser carbohydrate ! ..-:_attachment sites (Asn 34, Asn 76 and Asn 88) that present a different glycosylation pattern depending on the organ or fluid analyzed. Pancreatic type RNase from urine has each of the three sites glycosylated with complex type oligosaccharide chains (Beintema, et a!., 1988), whereas HPR purified from pancreas has carbohydrates attached to Asn 34 and is only partially glycosylated at the other two positions. A significant degree of microheterogeneity in the carbohydrate moiety is observed among the glycosylated ribonucleases (Ribo, et al., 1994). Differences are also observed in RNases from pancreatic juice (Thomas, eta!., 1984), seminal plasma (De Prisco, et al., 1984) and kidney (Mizuta, et al., 1990). The heterologous expression of HPR using recombinant methods has been reported by different groups using prokaryotic or eukaryotic systems. Futami, et al.,
Transcript
Chapter4
ROLE OF GLYCOSYLATION IN THE ACTIVITY OF HUMAN
PANCREATIC RIBONUCLEASE
uman pancreatic ribonuclease (HPR), is a glycosylated
protein and has three Asn-X-Thr/Ser carbohydrate !
~ ..-:_attachment sites (Asn 34, Asn 76 and Asn 88) that present a
different glycosylation pattern depending on the organ or fluid analyzed. Pancreatic
type RNase from urine has each of the three sites glycosylated with complex type
oligosaccharide chains (Beintema, et a!., 1988), whereas HPR purified from pancreas
has carbohydrates attached to Asn 34 and is only partially glycosylated at the other
two positions. A significant degree of microheterogeneity in the carbohydrate moiety
is observed among the glycosylated ribonucleases (Ribo, et al., 1994). Differences are
also observed in RNases from pancreatic juice (Thomas, eta!., 1984), seminal plasma
(De Prisco, et al., 1984) and kidney (Mizuta, et al., 1990).
The heterologous expression of HPR using recombinant methods has been
reported by different groups using prokaryotic or eukaryotic systems. Futami, et al.,
Expression of glycosylated HP R
(1995) described the expression of HPR in E. coli and its purification with a yield of
1.2 mg/L culture and a purity of 95 .5%. Boix, eta!., (1996) reported the expression of
HPR and its N-terminal variants together with onconase with recoveries of 10-50 mg
recombinant protein per liter of culture. A comparison of the expression of
recombinant HPR between Saccharomyces cerevisiae and E. coli was reported by
Ribo, et al., (1996). They obtained 5-10 fold more protein with the latter than the
former expression system. The E. coli system allowed the production of about lmg of
protein per liter of culture. Finally, our laboratory has reported a yield for HPR of 30-
50 mg/L culture using an E. coli expression system (Bal and Batra, 1997). The
expression of HPR in an eukaryotic expression system was done by Russo, et al.,
(1993). They reported the stable expression of an HPR variant lacking the last C
terminal amino acid (Thr), in chinese hamster ovary (CHO) cells. After its purification
they obtained a yield of 3-5 mg of a mixture of glycosylated and non-glycosylated
HPR per litre of culture. It is quite apparent that the yields of the recombinant proteins
expressed in the prokaryotic expression system are much higher than that obtained
from eukaryotic expression systems. However, the proteins expressed in the
prokaryotes are non-glycosylated.
The aim of this project was to study the effect of carbohydrate moieties on the
ribonucleolytic activity of HPR. In order to obtain the glycosylated form of HPR, the
baculovirus expression system was chosen. Baculoviruses are a diverse group of
viruses found mostly in insects. They are not known to have any non-arthropod hosts.
The baculo portion of the name refers to the rod shaped capsids of the virus particles.
Baculovirus capsids are usually 40-50 nm in diameter and 200-400 nm in length
(Harrap, 1972). The length of the capsids cari extend to accommodate larger DNA
genomes such as those of recombinant viruses carrying large inserts (Fraser, 1986).
Within the capsid, the DNA is condensed into a nucleoprotein structure known as the
core (Tweeten, et al., 1980). The capsid plus the core are collectively referred to as the
nucleocapsid. The DNA genome of a baculovirus is double stranded, covalently
closed and circular (Summers and Anderson, 1972). The two baculoviruses commonly
used for expression vector work are Autographa californica nuclear polyhedrosis
virus (AcMNPV) and the silkworm virus, Bombyx mori nuclear polyhedrosis virus
(BmNPV). The DNA ofboth the viruses are approximately 130 Kbp long.
The biology of the infection process in insect larvae underlies the utility of
91
Expression of glycosylated HP R
baculoviruses as expression vectors (Miller, 1981). During normal infection the virus
produces nuclear inclusion bodies which consists of virus particles embedded in a
protein matrix, the major component of which is a virus encoded protein called
polyhedrin. Large amounts of virus and polyhedrin are produced. Transcription of the
polyhedrin gene is driven by an extremely active promoter, which is therefore ideally
suited for driving expression of foreign genes. This is all the more attractive because
the polyhedrin gene product is not essential for viral replication. However, even in cell
· culture, the polyhedrin gene (polh) is expressed at very high levels during a separate
and final phase of infection. Thus, the basic design of the first baculovirus gene
expression vectors was replacement of polh with the heterologous gene under
polyhedrin promoter control (Miller, et al., 1983; Smith, et al., 1983). One advantage
of substituting the heterologous gene in place of polh is that such recombinant viruses
replicate normally as wild type baculovirus in cell culture and can be distinguished
visually from wild type virus by their occlusion negative (occ-) phenotype. Such occ
recombinant viruses are not efficient at infecting larvae by the natural oral route of
infection and do not persist in the environment. These features have some benefits
from environmental and recombinant DNA safety perspectives (Miller, 1981 ).
Using a eukaryotic system for expressing a gene can be particularly important
in obtaining biologically active proteins. The insect baculovirus expression system
provides a eukaryotic environment that is generally conducive to proper folding,
disulfide bond formation, oligomerisation, and/or other post translational
modifications required for the biological activity of some eukaryotic proteins. The
post-translational modifications that have been reported to occur in baculovirus
infected insect cells include signal cleavage, proteolytic cleavage, N-glycosylation, 0-
glycosylation, acylation, amidation, phosphorylation, prenylation, and
carboxymethylation. The sites of such modifications are usually at identical positions
on the proteins produced in mammalian cells. In general, the insect baculovirus
expression system mimics a vertebrate cell system quite remarkably with regard to
protein post-translational modifications.
The most distinguishing feature of the baculovirus expression vectors is the
potential for a very strong expression of a heterologous gene. The highest expression
levels reported using the baculovirus expression vectors is 25-50% of the total cellular
protein. Such levels are equivalent to polh expression and correspond to
92
Expression of glycosylated HP R
approximately 1 gram of protein product per liter culture. All heterologous proteins
however are not produced at the same level as polyhedrin and levels approaching 25%
of the total cellular protein have been achieved in few cases. Most of these cases
involved expression of structural genes of other virus families, the products of which
are quite stable. On comparing different eukaryotic expression systems, the
baculovirus system has usually outperformed the other expression systems in overall
protein production. Yields of a heterologous gene product must be determined
empirically as it is difficult to provide any guidelines for how a gene will behave in
the expression system unless similar genes have been expressed previously. In this
study, an attempt has been made to express the glycosylated form of HPR using the
baculovirus expression system, in order to elucidate the effect of sugar moieties on the
structure and function of the human ribonuclease.
Experimental Procedures
Construction of the recombinant baculovirus
The AcMNPV genome is a double stranded circular DNA of 128 kb. The size
of the viral DNA makes it difficult to manipulate directly, so the recombinant
baculovirus expression vectors was constructed in two steps. The target DNA
encoding HPR was first cloned into the modified viral polyhedrin locus of the
baculoviral transfer vector BacPak8 (Figure 4.1A). In order to do so, the BamHI and
EcoRI sites in the MCS of the BacPak8 vector were used. A BamHI site was
introduced at the 5' end of HPR gene by PCR using the forward primer JKB7,
5'GAAGGAGATATAGGATCCATGAAGGAATCCCGGGCCAAG3', the reverse
primer ERP, 5'TTATGCTAGTTATTGCTCAG3', and pHPR plasmid as template.
The amplified HPR fragment, having recognition sites for BamHI at the 5' end and
EcoRI at the 3' end, was digested with BamHI and EcoRI, purified by gel
electrophoresis, and cloned into BacPak8 vector restricted with the same enzymes.
The putative clones were confirmed by restriction analysis and DNA sequencing
(Sanger, 1977). The DNA was purified by using the Qiagen column kit (Qiagen Inc.,
USA).
In the second step, 2 x 106 exponentially growing Spodoptera frugiperda (Sf9)
insect cells were seeded in a volume of 2 ml of complete Grace's insect medium in
93
Figure 4.1
A. pBacPak8 transfer vector used for introducing HPR DNA in the viral genome
B. pBacPak6 viral DNA
BacPak6 137 kb
pBacPak8 5.5kb
Bsu36 I
PPolyhedrin
Stu I BstBI
Xbal Bg/11
Kpnl
Sac I £coR!
C. pBacPak-HPR
BacPak HPR
BamHI
£coR!
The target HPR gene was inserted in the BamH I and EcoRI sites in the MCS placed between the polyhedrin promoter and polyadenylation signals in BacPak8 transfer vector (A). The BacPak6 vector(B) is a specially engineered virus, which facilitates the construction and selection of recombinant expression vectors. pBacPak-HPR (C) represents the recombinant viral vector containing HPR DNA.
Expression of glycosylated HP R
two 35 mm tissue culture dishes and incubated at 27°C for 1 hour. The medium was
replaced with 2 ml of Grace's insect cell medium (without serum) and further
incubated for 30 minutes at room temperature. The modified transfer vector BacPak8
containing the HPR gene and Bsu36I-digested linearized BacPak6 viral DNA (Figure
4.1B) were then cotransfected into Sf9 cells using Bacfectin. The BacPak6 is a
specially engineered virus, which facilitates the construction and selection of
recombinant expression vectors.
The transfection mixture was prepared as follows:
Sterile water
BacPak8 transfer vector (20 ng/j.tl)
BacPak6 viral DNA(Bsu36I digest)
Bacfectin
Total
66 j.tl
25 j.tl
', 5 j.tl
4 j.tl
100 j.tl
A control transfection was also done in which BacPak6 viral DNA was not
added. The cells were incubated with the Bacfectin-DNA mixture at 27°C for 5 hours
after which complete medium was added to the dish and further incubation was done
at 27°C for 72 hours. The recombinant HPR baculovirus with the HPR DNA inserted
in its genome is named as pBacPak-HPR (Figure 4.1C). After 72 hours, the medium
containing pBacPak-HPR viruses produced by the transfected cells was transferred to
a sterile container and stored at 4°C. In order to obtain more pBacPak-HPR virus, 1.5
ml of complete insect cell medium was added to each dish, incubated at 27°C for 48
hours and medium was harvested as mentioned above.
Plaque Purification of recombinant pBacPak-HPR virus
Spodoptera frugiperda (Sf9) insect cells were seeded in 35 mm tissue culture
dishes at a density of 2x 106 exponentially growing cells in a volume of 2 ml and
incubated at 27°C for 1 hour. Several dilutions, ranging from 10-1 to 10-7 of the
pBacPak-HPR stocks were prepared in the Grace's insect cell medium, added to the
cell monolayers and incubated at room temperature for 90 minutes. After incubation,
the cells were overlayed with 2 ml of agarose, which was prepared by mixing equal
94
Expression of glycosylated HP R
volume of 3% LGT agarose and complete Grace's insect cell medium at 45°C. The
agarose was allowed to cool after which 1ml of insect cell medium was added on top.
The cells were further incubated at 27°C for 72 hours. After incubation, the liquid
medium was removed from top of the agarose overlay and petridish was dried by
placing it in an inverted position on a paper towel. After drying, 2 ml of neutral red
staining solution (1.2 ml of neutral red in 20 ml plaque assay buffer) was added in the·
petridishes and incubated for 2 hours at room temperature. Thereafter the stain was
drained off and the petridishes were kept inverted overnight for the plaques to become
well formed. The recombinant plaques were scored on the basis of differential
refraction (due to occ- phenotype) by placing it on top of an illuminated box. The
plaques were observed under the light microscope. The occ- plaques appear clear with
some cell debris, while the occ + plaques show polyhedrin that becomes apparent due
to their bright and shiny appearance. The plaques were picked by gentle suction using
a pipet. The agarose plugs picked were placed in 1ml of Grace's insect cell medium
and vortexed well to release the budded virus particles from the plug. These isolates .
were further screened by DNA hybridization analysis.
Identification of recombinant pBacPak-HPR virus by DNA hybridization
The DNA hybridization identifies virus isolates that contain the heterologous
gene integrated into their genomes. A 96 well plate was seeded with 1.5 x 106 Sf9
cells/ well. The cells were infected in duplicate with the putative recombinant
pBacPak-HPR viruses identified by visual screening in the plaque assay and incubated
at 27°C for 72 hours. This time period allowed sufficient viral replication to give rise
to a strong hybridization signal without allowing the infection to proceed to the point
where all the cells lyse and the DNA is lost in the tissue culture fluid. The pBacPak
HPR viral supernatants were removed and stored. The infected cells were lysed and
the DNA was denatured by adding 200 Ill of 0.5 M NaOH and 20 Ill of 10 M
ammonium acetate. The celllysates containing the viral and chromosomal DNA were
applied to the nylon membrane using a dot blot apparatus. The membrane was then
rinsed in 2X SSC for several minutes, air dried and baked under vacuum at 80°C for 2
hours.
The radioactive probe for detecting the HPR DNA in the genome of the
recombinant pBacPak-HPR virus was prepared by random primer labeling, using the
95
Expression of glycosylated HP R
Rediprime DNA labeling system (Amersham Pharmacia Biotech, U.K). This kit
includes reaction tubes consisting of a pre-mix of the nucleotide stocks, random
primers, and Klenow fragment of E.coli DNA polymerase I. The HPR DNA obtained
by restriction digestion of the pHPR plasmid with Nhel and EcoRI was used for
preparing the DNA probe. [a-32P]deTP (NEN) was used to label the probe. The
labeling reaction was set up by dissolving 25 ng of HPR DNA in 45 J.!l Tris-EDTA
buffer, pH 7.5, and further denaturing it by boiling for 5 minutes. The denatured DNA
was immediately cooled on ice for five minutes and centrifuged briefly. The DNA was
added to the pre-mix reaction tube. Further, 5 J.!l of 32P-deTP was added to the
reaction mix and incubated at 37°e for 10 minutes. The labeling reaction was stopped
by adding 5 J.!l of 0.20 M EDT A.
The nylon membrane with the transferred viral DNA was prehybridised for 1
hour at 42°e in a heat sealable plastic bag, to reduce the spurious background signals.
The probe was denatured and added to the bag containing the filter and hybridization
solution and further incubated at 42°e overnight. After incubation, the filter was
washed twice at 55°e with 2X sse, 0.1% SDS for 15-20 minutes and then with O.lX
sse, 0.1% SDS. The filter was dried and exposed to X ray film.
Amplification of the recombinant pBacPak-HPR virus
In order to generate large stocks of the potential recombinant pBacPak:-HPR
virus, viral infections were set up with a large number of cells in tissue culture flasks.
Sf9 cells were grown in 25 cm2 flasks to a confluency of 70% and the monolayer was
infected with 0.5 ml inoculum of the recombinant pBacPak-HPR virus purified by the
plaque assay and identified by DNA hybridization. The cells were incubated at room
temperature for one hour with gentle rocking. 5 ml of Grace's insect cell complete
medium was added to the cells and further incubation was done at 27°e for 5 days.
After incubation, the culture medium was collected and centrifuged at 1 OOOx g for 5
minutes at 4°e to remove cell debris. The supernatant containing the pBacPak-HPR
virus was stored at 4°e.
Characterization of recombinant HPR expression
The expression of recombinant HPR in the infected Sf9 cells was analyzed by
SDS-PAGE and Western blotting. Sf9 cells were seeded in 24 well plate and infected
96
Expression of glycosylated HP R
with 100 J..Ll inoculum of the recombinant pBacPak-HPR as mentioned earlier. The
medium was aspirated and cells were harvested 24, 48 and 72 hours post-infection.
The harvested cells were lysed in 50 J..Ll of IX SDS gel-loading buffer. Uninfected Sf9
cells were used as a control and were processed similarly. Equal volume of each
sample was loaded on a 12.5% polyacrylamide gel and the proteins were visualized by
Coomassie blue staining. The expression of HPR protein was further analyzed by
Western blotting using anti-HPR antibody.
The HPR protein was localised in the virally infected Sf9 cells to check
whether the protein was being expressed in the soluble or insoluble form. The cells
· were harvested after infection and resuspended in 4 ml PBS. The cell suspension was
sonicated and centrifuged at 1 OO,OOOxg for 30 minutes. The supernatant was removed
and the pellet obtained was resuspended in 4 ml of 20 mM Tris-HCl buffer, pH 7.5,
containing O.lmM PMSF, 2% deoxycholate and 5 mM P-mercaptoethanol. The
suspension was further centrifuged at 1 OO,OOOxg for 30 minutes. The supernatant was
removed and the pellet was stored at -70 °C. At each stage, appropriate samples were
taken of both pellet and supernatant for SDS-PAGE and Western blot analysis.
In another experiment, the pellet was resuspended in 4 ml of 20 mM Tris-HCl
mercaptoethanol). The suspension was centrifuged at 1 OO,OOOxg for 30 minutes. The
supernatant was removed and the pellet was solubilised in 2 ml of 6 M guanidine
hydrochloride (GnHCl) using a homogeniser. The pellet in GnHCl was further
incubated at room temperature for 2 hours with vigorous shaking and centrifuged at
1 OO,OOOx g for 30 minutes. The supernatant was removed and the pellet was stored at
-70 °C. The GnHCl supernatant was precipitated with TCA and resuspended in IX
SDS-gel loading buffer. At each stage, appropriate samples were taken of both pellet
and supernatant for SDS-PAGE and Western blot analysis.
Propagation of Sf9 cells in suspension culture
The Sf9 cells were grown in suspension in specially designed spinner flasks,
containing a teflon coated magnetic stir bar suspended from a glass. or stainless steel
rod that is driven from below by a magnetic stirrer. The suspension culture was set up
by adding 200 ml of Grace's insect cell complete medium to the spinner flask. The
medium was inoculated with Sf9 cells from two 70% confluent 75 cm2 flasks such
97
Expression of glycosylated HP R
that the starting density was 3-4 x 105 cells/mi. It 'was further incubated at 27°C with
constant stirring at 60-80 RPM for 72 hours. Sf9 cells were pelleted and resuspended
in culture medium at a cell density of 106 cells/mi. The cells were infected with the
recombinant pBacPak-HPR at an MOl of 20 pfulcell and incubated at room
temperature for 1 hour with gentle shaking. The infected cells were returned to the
spinner flask and the volume was made up to 200 ml with Grace's insect cell
complete medium. The flask was incubated at 27°C with stirring for 72 hours. The
cells were harvested by centrifugation. The supernatant was filtered and stored as
working stock for the recombinant virus whereas the pelleted cells were further
processed for isolation of the recombinant HPR protein.
Isolation of the recombinant HPR protein
The cell pellet obtained on harvesting the infected Sf9 insect cells in the spinner
cultures was resuspended in PBS. The cell suspension was sonicated and centrifuged at
1 OO,OOOxg for 30 minutes. The supernatant was removed and the pellet obtained was
· dissolved in 6 M guanidine hydrochloride and incubated for 2 hours at room
temperature followed by centrifugation at 50,000 gat 4°C, for 30 min. The supernatant
containing the denatured protein was collected and the protein concentration was
adjusted to 10 mg/ml with 6 M guanidine hydrochloride. The denatured protein was
reduced by adding 65 mM dithioerythritol and incubated at room temperature for 2 hrs.
Renaturation was done by diluting the denatured and reduced protein 100-fold in the
refolding buffer. After incubating at l0°C for 36 hrs, the renatured material was
dialyzed against 20 mM Tris-HCl buffer (pH 7.4) containing 100 mM urea. The
renatured material was loaded on a 6 ml S-Sepharose column, and eluted using 70 ml
NaCl gradient 0-1 M in 20 mM Tris-HCl buffer (pH 7.4) using an FPLC system.
Relevant fractions were pooled, concentrated and purified to homogeneity by gel
filtration chromatography on a TSK 3000 column (LKB) in 50 mM PBS (pH 7.4).
Detection of carbohydrate moieties present in the baculovirus expressed HPR
The estimation of total carbohydrate content in the baculovirus expressed HPR
protein was performed by the phenol-sulfuric acid method. 200J.!l of phenol, 5%
wt/vol in water, was added to the sample protein or glucose standard. Concentrated
sulfuric acid, lml, was then added rapidly and directly to the solution surface without
98
Expression of glycosylated HP R
touching sides of the tube. The solution was left undisturbed for 10 minutes at room
temperature after which it was vortexed vigorously. The sample was incubated at
room temperature for additional 30 minutes. The absorbance was measured at 490 nm.
The glycosylated protein phCG was used as a positive control in the assay, whereas
the E. coli expressed HPR protein was taken as negative control.
Results
Construction of the recombinant pBacPak-HPR baculovirus
The baculovirus genome is large (1 00-200 kbp) and usually contains one or
more recognition sites for known restriction endonucleases. When AcMNPV was
being developed as an expression vector, there were no restriction endonucleases that
lacked several recognition sites in this genome, so allelic replacement was adopted to
insert foreign genes in to the genome (Smith, et al., 1984; Pennock, et al., 1984;
Maeda, et al., 1985). Allelic replacement still remains the preferred method for
homologous gene insertion. Following the allelic replacement strategy, we inserted
DNA encoding HPR into a transfer plasmid BacPak8, so that it is downstream of the
required viral promoter and flanked on both sides by viral sequences that will target
the gene and promoter to the desired region in the viral genome. The BacPak8 transfer
vector contains a plasmid origin of replication and an antibiotic resistance gene for
propagation in E. coli (Figure 4.1A), but it is unable to replicate in insect cells. The
viral segment cloned into the transfer vector includes the entire polyhedrin locus with
flanking sequences, but the polyhedrin coding sequence has been deleted and replaced
with a polylinker containing multiple cloning site, MCS (Figure 4.1A). The target
HPR DNA was inserted in the MCS placed between the polyhedrin promoter and
polyadenylation signals.
The modified BacPak8 plasmid and the parental linearized BacPak6 viral
DNA were co-transfected into the Sf9 insect cells. The enzymes in the cells recombine
the DNAs. This primarily involves the homologous recombination and results in the
formation of the recombinant pBacPak-HPR baculovirus with the HPR DNA inserted
in the baculovirus genome (Figure 4.1C). The restriction ofBakPak6 viral DNA with
Bsu36I removes a fragment that includes a part of the essential gene, ORF 1629
99
Expression of glycosylated HP R
(Figure 4.1B). The double recombination events restore the integrity of the essential
gene. The unrecombined large Bsu361 fragment of BacPak6 is unable to produce
viable viruses. This selection results in viral plaques containing recombinant
pBacPak-HPR viruses that have acquired the HPR DNA. The viral stocks, obtained
from the cotransfection experiment, contain the required recombinant virus mixed
with a large excess of non-recombinant and single crossover recombinant viruses. The
pBacPak-HPR virus was purified from cotransfection stocks by plaque purification.
The recombinant pBacPak-HPR virus· are identified based on the plaque phenotype,
occlusion body formation. This screening method is based on the fact that viruses that
lack a polyhedrin gene (polh) are incapable of forming occlusion bodies and,
therefore, form plaques that are visually distinguishable from wt ( occ +) virus plaques.
After repeated plaque assays, we picked sixteen plaques that on the basis of the
phenotype of occlusion body formation appeared to contain recombinant virus.
The presence of the HPR DNA in the genome of the recombinant pBacPak
HPR was confirmed by DNA hybridization. Out of the 16 recombinant viruses picked
on the basis of the occ- phenotype in the plaque assay, 11 were found to possess the
HPR DNA in their viral genome (Figure 4.2). Very high intensity signals were
observed for the viral DNA in lanes 15 and 16 (Figure 4.2), as these recombinant
viruses had undergone one round of amplification compared to the other positive
recombinant viruses shown in lanes 1, 2, 6, 7, 8, 10, 11, 12 and 13 (Figure 4.2). No
signal was observed in case of the uninfected Sf9 cells (Figure 4.2). Similarly, a very
weak signal was observed for the wild type baculovirus infected Sf9 cells (Figure 4.2).
The recombinant pBacPak-HPR viruses were amplified and stored as stocks at 4°C.
Expression and localization of HPR protein in virally infected Sf9 cells
The expression of the HPR was studied by infecting the Sf9 insect cells with
recombinant pBacPak-HPR at various time intervals. The total cell proteins were
isolated from the infected Sf9 cells 24, 48 and 72 hours post-infection and analyzed by
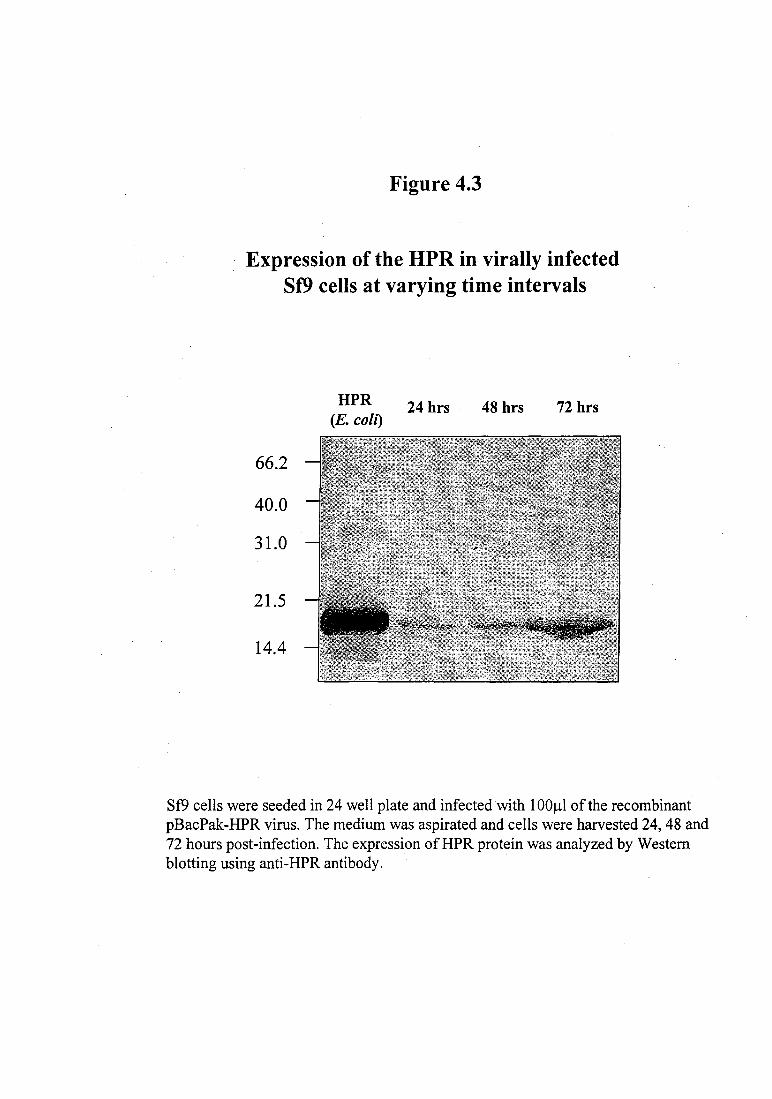
SDS-PAGE and subsequent Western blotting. As seen in Figure 4.3, the expression of
the HPR protein was found to gradually increase with time and was maximum at 72
hours post infection.
To localize the expressed recombinant protein in the virally infected Sf9 cells,
the total cell homogenate was fractionated into membrane and cytosolic fractions. The
100
1
Figure 4.2
Screening of putative recombinant baculovirus by DNA hybridization
2 3 4
Control (Sf9 cells)
5 6 7 8 9 10 11 12
"" duplicates
/
Control (wild type AcMnPV)
Sf9 cells were infected in duplicate ( 1-13) with the putative recombinant viruses, pBacPak-HPR, identified by visual screening in the plaque assay and incubated at 27°C for 72 hours. The infections set up in lanes 14-16 were done with recombinant viruses obtained after one round of amplification. The celllysates were applied to the nylon membrane using a dot blot apparatus and probed with labeled HPR DNA. The membrane was dried and exposed to X ray film.
Figure 4.3
Expression of the HPR in virally infected Sf9 cells at varying time intervals
66.2
40.0
31.0
21.5
14.4
HPR (E. coil)
24 hrs 48 hrs 72 hrs
Sf9 cells were seeded in 24 well plate and infected with 1 00~1 of the recombinant pBacPak-HPR virus. The medium was aspirated and cells were harvested 24, 48 and 72 hours post-infection. The expression ofHPR protein was analyzed by Western blotting using anti-HPR antibody.
Expression of glycosylated HP R
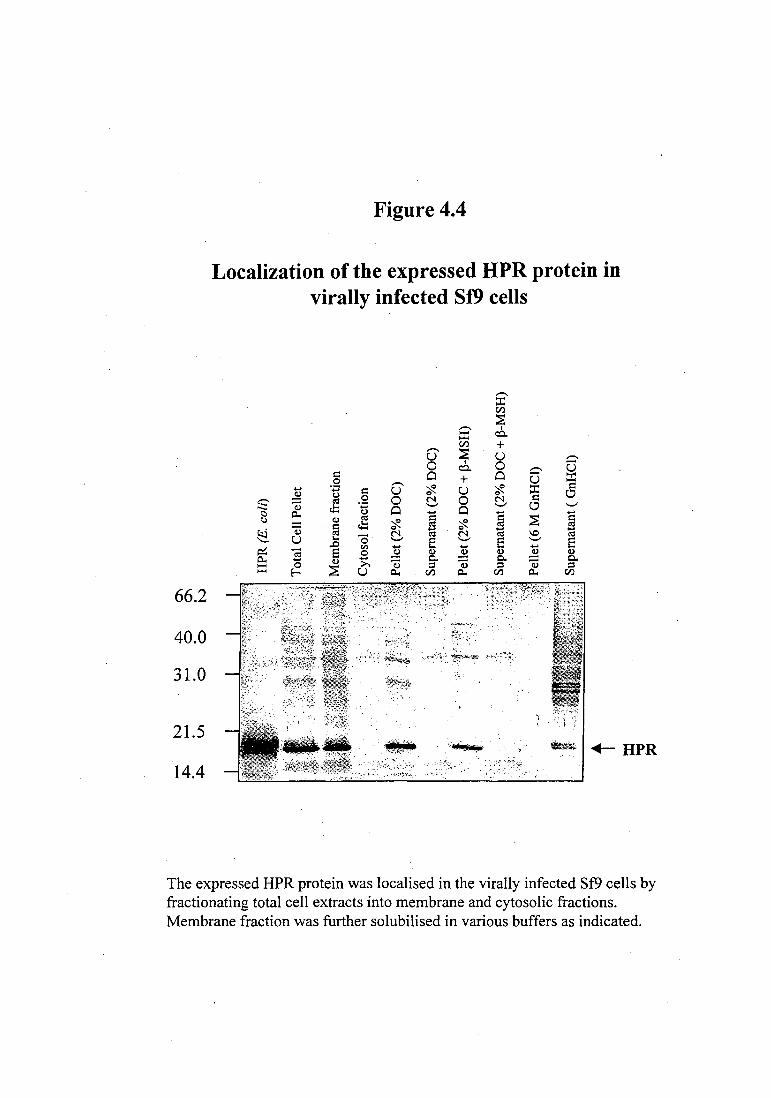
protein was only found to be associated with the membrane fraction (Figure 4.4). To
solubilize the HPR protein, different agents such as 2% deoxycholate (DOC) with or
without P-mercaptoethanol CP-MSH) and 6 M GnHCl were used. As seen in Figure
4.4, the recombinant protein was not soluble in 2% DOC with or without P-MSH and
the protein remained in the pellet (Figure 4.4). However, the HPR protein was soluble
in 6 M GnHCI.
Preparation of HPR protein
To purify HPR protein in a significant amount, Sf9 insect cells were grown in
suspension spinner cultures and infected with HPR recombinant baculovirus at a MOl
of 20 pfulcell. The cells were harvested 72 hours post infection.
The total cell pellet from five spinner cultures (200ml each) was resuspended
in PBS with a tissue homogeniser. The cell suspension was centrifuged and the pellet
was solubilised in 6 M GnHCI. The solubilised protein was refolded by diluting the
protein 1:100 in L-Arginine rich buffer. Oxidized glutathione and dithioerythreitol
were added to help create a redox condition, to facilitate the disulfide bond formation.
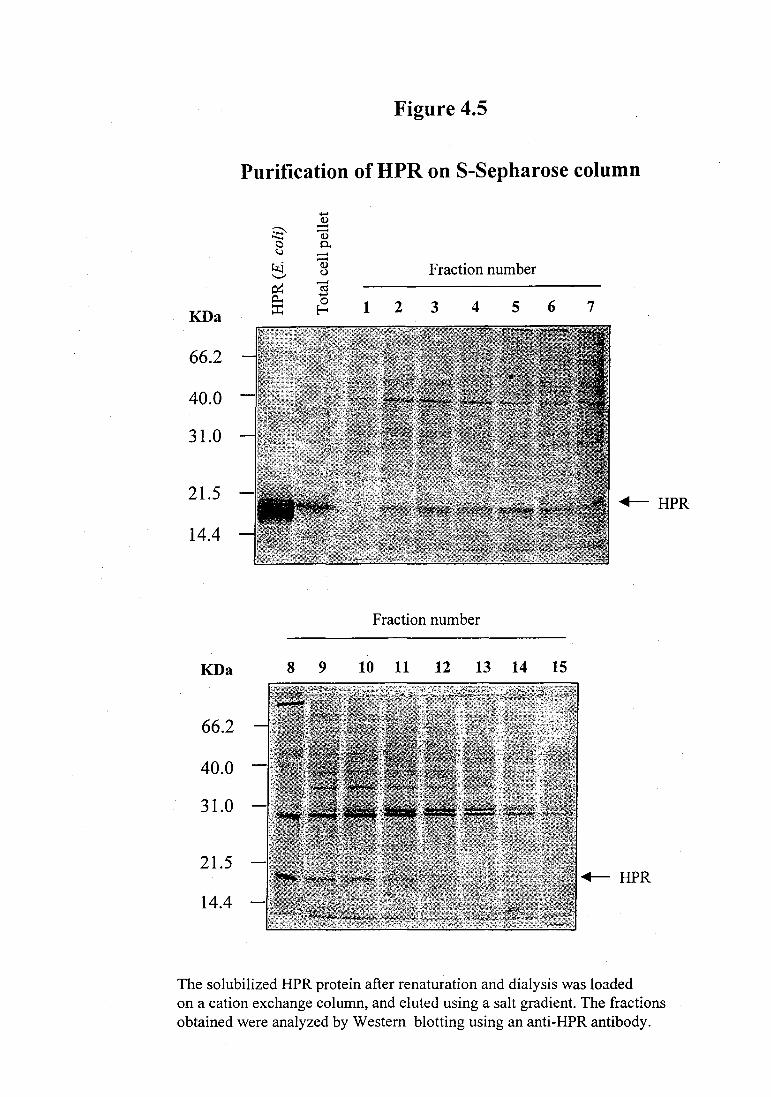
The refolded protein after dialysis was purified by cation exchange chromatography
using S-Sepharose column. The fractions (2-1 0) eluted from the S-Sepharose cation
exchange column contained HPR protein, however the protein was not completely
pure and contained other contaminating proteins as shown in the Western blot in
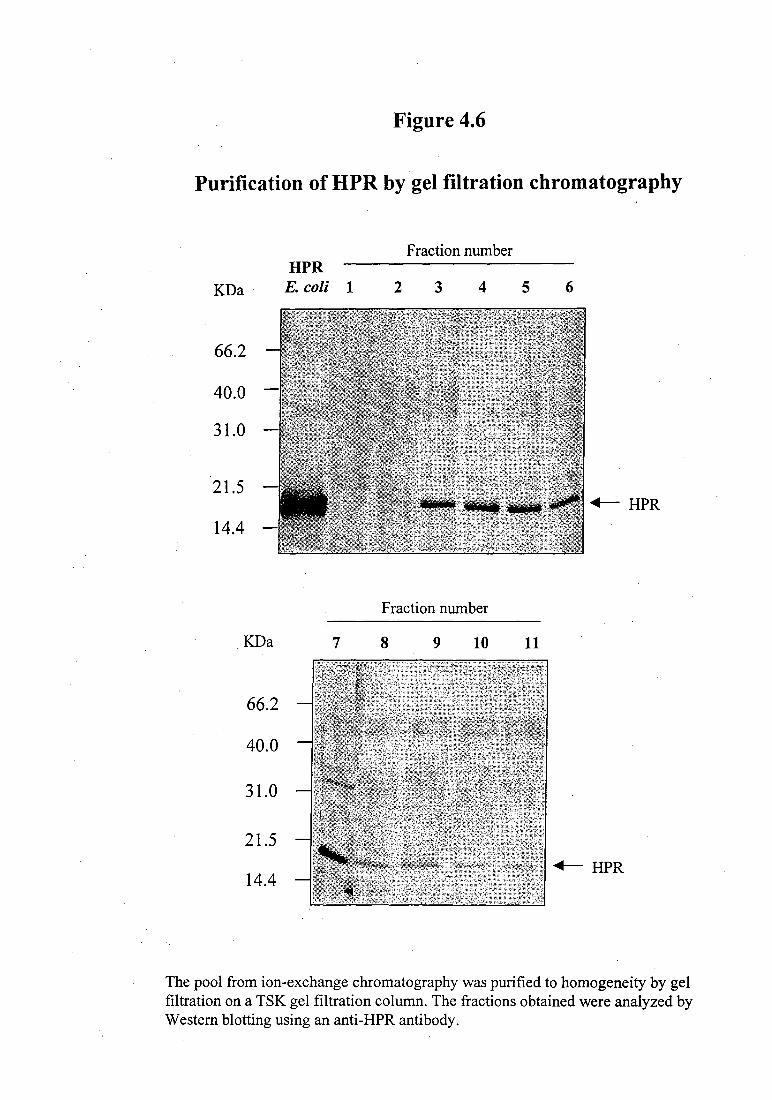
Figure 4.5. The protein pool from S-Sepharose was further purified by gel filtration
chromatography. As can be seen in Figure 4.6, fractions 3-9 from the gel filtration
column contained HPR protein in a pure and homogenous form. The final yield of the
pure protein was low, and 470 f.lg of pure HPR protein was obtained from lliter of
spinner culture.
Structural characterization of the purified HPR protein
I. Secondary structure estimation of HPR using circular dichroism
The structural conformation of the HPR protein expressed in baculovirus
expression system was investigated by CD-spectral analysis. The CD-spectrum of the
baculovirus expressed protein in the far-UV region was compared with that of the
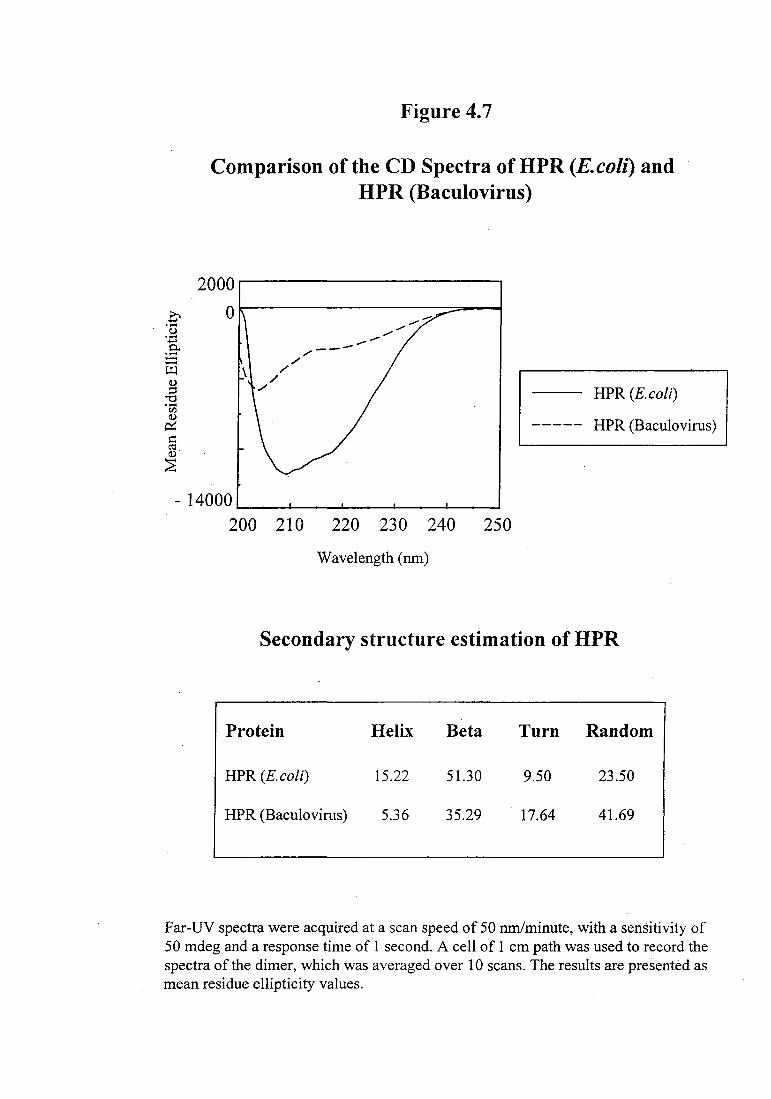
native protein expressed in E. coli. As shown in Figure 4.7, the HPR protein from
101
66.2
40.0
31.0
21.5
14.4
Figure 4.4
Localization of the expressed HPR protein in virally infected Sf9 cells
,-.._
::r:: r/l
:E ,-.._ I
::r:: co.. r/l + ,-.._ ~ u u Q
0 I 0 c:::l.. Q u ~ 0 0 ::r:: .9 ,-.._ + u
~ ..... - ~ u '<f. u '<f. ::r:: 0 ~ (.) Q ~ .9 0 N 0 N ~ - Q) rJ:: - 0
The expressed HPR protein was localised in the virally infected Sf9 cells by fractionating total cell extracts into membrane and cytosolic fractions. Membrane fraction was further solubilised in various buffers as indicated.
Figure 4.5
Purification of HPR on S-Sepharose column
-~
~ -...._ (!)
0 0.. ~ --kj (!) Fraction number
'-" (.)
KDa
0::: -· ro 0... -0 1 2 3 4 5 6 7 ::c: E-<
66.2
40.0
31.0
21.5 +- HPR
14.4
Fraction number
KDa 8 9 10 11 12 13 14 15
66.2
40.0
31.0
21.5 +- HPR
14.4
The solubilized HPR protein after renaturation and dialysis was loaded on a cation exchange column, and eluted using a salt gradient. The fractions obtained were analyzed by Western blotting using an anti-HPR antibody.
Figure 4.6
Purification of HPR by gel filtration chromatography
K.Da
66.2
40.0
31.0
21.5
14.4
.KDa
66.2
40.0
31.0
21.5
14.4
HPR E. coli 1
7
Fraction number
2 3 4 5 6
..._ HPR
Fraction number
8 9 10 11
..._ HPR
The pool from ion-exchange chromatography was purified to homogeneity by gel filtration on a TSK gel filtration column. The fractions obtained were analyzed by Western blotting using an anti-HPR antibody.
Figure 4.7
Comparison of the CD Spectra ofHPR (E.colz) and HPR (Baculovirus)
2000~------------------~
0 0 ·-<.)
·~ :..::: ~ \
.§ ·-en
~ s:: ell (L)
~
HPR (E. coli)
HPR (Baculovirus)
-14000~--~--~--~--~--~ 200 210 220 230 240 250
Wavelength (nm)
Secondary structure estimation of HPR
Protein Helix Beta Turn Random
HPR (E.coli) 15.22 51.30 9.50 23.50
HPR (Baculovirus) 5.36 35.29 . 17.64 41.69
Far-UV spectra were acquired at a scan speed of 50 run/minute, with a sensitivity of 50 mdeg and a response time of 1 second. A cell of 1 em path was used to record the spectra of the dimer, which was averaged over 10 scans. The results are presented as mean residue ellipticity values.
Expression of glycosylated HP R
E. coli appears to be compactly folded with an a+~ conformation, however, the
conformation of HPR protein expressed in baculovirus was found to be significantly
different. The secondary structure estimates, calculated using Yang's reference
parameters showed a dramatic decrease in the a-helical content of the HPR protein
expressed in baculovirus expression system (Figure 4. 7).
II. Detection of carbohydrate content in HPR
The total sugar present in the HPR protein expressed in the baculovirus
expression system was estimated using phenol-sulfuric acid method. Using glucose as
standard, the absorbance of varying concentrations of the test sample (5-50 j...Lg) and
controls was compared with that of the standard. The sugar content was 0.21 mg/mg
protein based on the glucose standard, which translates to 21% sugar in the
baculovirus expressed HPR. This is similar to the sugar content of 20% observed in
case of glycosylated ~hCG protein used as positive control. The E. coli expressed
HPR protein, used as negative control, showed no significant levels of sugar content.
Functional characterization of the purified HPR protein
The ribonuclease activity of the glycosylated HPR was assayed on four
different RNA substrates, poly(C), yeast tRNA, cCMP and poly(A):poly(U). The
glycosylated HPR was less active compared to the bacterially expressed enzyme on all
substrates. On the single stranded, pyrimidine homopolymer substrate, poly(C), the
HPR (baculovirus) displayed 18% activity compared to HPR (E. coli) (Table 4.1). On
yeast tRNA, the baculovirus HPR protein displayed 24% activity compared to HPR
(E. coli) (Table 4.1). The hydrolytic activity of HPR (baculovirus) was found to be
40% of that of HPR (E. coli) (Table 4.1 ). The ribonucleolytic activity of the HPR
(baculovirus) on the double stranded RNA substrate poly(A):poly(U) was only 12 %
activity of the HPR (E. coli) (Table 4.1 ).
Discussion
Human pancreatic ribonuclease, isolated from the human pancreas is a
glycosylated protein. Heterogeneity has been observed in the glycosylation pattern of
HPR. All human secretory RNases are products of the same gene, however they differ
in their post-translational processing. Several glycosylated forms of the human
102
Table 4.1
Catalytic activity of HPR expressed in baculovirus system
RNA substrate ~Absorbance/min./mg protein
HPR (E. colz) HPR (Baculovirus)
poly( C) 219166 (100) 40667 (18)
yeast tRNA 17750 (100) 4250 (24)
cCMP 0.41 (100) 0.17(41)
poly(A):poly(U) 413 (100) 48 (12)
Each substrate was incubated with different concentrations of baculovirus expressed HPR protein for 1 hour at 37°C. The undigested large molecular weight RNA was precipitated with perchloric acid and uranyl acetate on ice and removed by centrifugation. The acid soluble product was quantitated by measuring the absorbance at 260 nm.
Expression of glycosylated HP R
ribonuclease with apparent molecular masses ranging from 14-40 kDa have been
isolated. The significance of glycosylation for the activity of the ribonuclease is not
well understood. Native HPR has been found to be a weaker enzyme compared to
RNase A. Since isolation of HPR from its native source has been a difficult issue,
detailed comparison of activity of recombinant HPR with that of native is not
available. It has been demonstrated that the non-glycosylated form of HPR, expressed
in E. coli, exhibits a very significant ribonucleolytic activity (Bal and Batra, 1997).
HPR exhibits a significant ribonuclease activity on double stranded RNA and it has
been speculated that the carbohydrate moieties may have a role in this activity on
double-stranded RNA (Libonati and Sorrentino, 1992). Extensively glycosylated
ribonucleases, like the enzymes from pig and horse pancreas, show a much higher
activity on double-stranded RNAs than similarly charged, carbohydrate-free RNases
under standard assay conditions of relatively high salt concentrations (Carsana, et al.,
1981). The glycosylated pig and horse pancreas RNases also show a larger
destabilizing effect on double-stranded-RNA, than that displayed by bovine RNase A
under these conditions (Carsana, et al., 1981). A partial enzymic removal of the
heterosaccharide side chains from pig and horse RNases reduces their degradative
activity on double-stranded RNA. These results are tentatively correlated with a
modification of the microenvironment of the enzyme protein caused by its extensive
glycosylation. The effect of sugar residues on the dsRNA cleavage activity of a
glycosylated ribonuclease is strictly dependent on the extent of glycosylation
(Carsana, et al., 1981 ). It was also demonstrated that the dsRNA cleavage activity of
the glycosylated form of human seminal RNase was greater than that of its non
glycosylated form (Sorrentino, et al., 1985). We used the baculovirus vector
expression system to produce the glycosylated form of HPR. The insect cell
baculovirus expression system is a popular means of expressing recombinant proteins.
The insect cell baculovirus system is also a promising tool for the expression of
heterologous glycoproteins because insect cells are capable of both N- and 0-linked
glycosylation. Proteins that are N-glycosylated in vertebrate cells are generally also
glycosylated in insect cells. The insect cell enzymes have the capacity to attach atleast
a Man9GlcNAc2 to the same sites recognized by vertebrate enzymes. Usually, the
Man9GlcNAc2 moiety is trimmed to shorter oligosaccharide structures, such as
Man3GlcNAc2, in both vertebrate and insect cells. In vertebrates, these shorter core
103
Expression of glycosylated HP R
structures serve as the complex oligosaccharide synthesis involving further GlcNAc,
Gal, or sialic acid additions. In insect cells, this additional, complex oligosaccharide
synthesis does not appear to occur in many cases.
The expressed HPR protein could not be secreted and was localized in the
membrane fraction in an insoluble form. The insoluble protein was solubilised and
renatured in vitro. However, the yield of the glycosylated HPR protein was low. The
recombinant protein was found to be glycosylated and the sugar content was found to
be 21% on weight basis. This is similar to the carbohydrate content of HPR isolated
from human pancreas (Beintema, et al., 1984). The expressed glycosylated HPR
protein was significantly less active than the protein expressed in E. coli. This
difference in enzymatic activity appears to be the consequence of conformational
change in the glycosylated form of HPR, compared to the bacterially expressed
protein.
There have been similar reports of proteins such as calpain and thyroid
peroxidase, expressed in an insoluble form in the recombinant baculovirus infected
insect cells (Branca, et al., 1999; Gardas, et a!., 1999). Large amounts of human
muscle-specific calpain p94 protein has been reported to be produced in insect cells
psing recombinant baculovirus expression system, with most of the protein recovered
in an insoluble and catalytically inactive form (Branca, et a!., 1999). Similarly, the
high yield purification of full length recombinant human thyroid peroxidase (TPO)
was reported from baculovirus infected insect cells (Gardas, eta!., 1999). TPO is an
integral membrane protein that catalyses the biosynthesis of human thyroid hormone
from thyroglobulin. The recombinant TPO protein was also resistant to solubilization
in detergents and similar to baculovirus expressed HPR was found to be devoid of
enzymatic activity. The lack of enzymatic activity of TPO has been attributed to
structural changes in the protein backbone surrounding the haem. The human
eosinophil ribonucleases, ECP and EDN, have been expressed as active proteins using
the baculovirus expression system (Domachowske, et a!., 1998). Both these human
RNases were secreted in the culture medium.
104
Expression of glycosylated HP R
The current study will be further extended to obtain a fully functional
glycosylated form of HPR, which should be useful in understanding the role of sugar






















![[60892.1] 2012 10 15 CC AR 11 - Pasadena, Californiaww2.cityofpasadena.net/councilagendas/2012 agendas/Oct_15_12/AR 10.pdfCalifornia Environmental Quality Act (CEQA Guidelines §15308:](https://static.documents.pub/doc/80x56/5fddababec7c100f700e9dc4/608921-2012-10-15-cc-ar-11-pasadena-agendasoct1512ar-10pdf-california.jpg)













