A wide array of chronic inflammatory conditions predisposes susceptible cells to neoplastic transformation. In general, the longer the inflammation persists, the higher the risk of cancer. A mutated cell is a sine qua non for carcinogenesis. Inflammatory processes may induce DNA mutations in cells via oxidative/nitrosative stress. This condition occurs when the generation of free radicals and active intermediates in a system exceeds the system's ability to neutralize and eliminate them. Inflammatory cells and cancer cells themselves produce free radicals and soluble mediators such as metabolites of arachidonic acid, cytokines and chemokines, which act by further producing reactive species. These, in turn, strongly recruit inflammatory cells in a vicious circle. Reactive intermediates of oxygen and nitrogen may directly oxidize DNA, or may interfere with mechanisms of DNA repair. These reactive substances may also rapidly react with proteins, carbohydrates and lipids, and the derivative products may induce a high perturbation in the intracellular and intercellular homeostasis, until DNA mutation. The main substances that link inflammation to cancer via oxidative/nitrosative stress are prostaglandins and cytokines. The effectors are represented by an imbalance between pro-oxidant and antioxidant enzyme activities (lipoxygenase, cyclooxygenase and phospholipid hydroperoxide glutathione-peroxidase), hydroperoxides and lipoperoxides, aldehydes and peroxinitrite. This review focalizes some of these intricate events by discussing the relationships occurring among oxidative/nitrosative/metabolic stress, inflammation and cancer.
Introduction
General concepts on carcinogenesis
Carcinogenesis is a long and multistep process that includes initiation (selection of a mutated cell), promotion (selective expansion of the initiated cell) and progression as a consequence of an imbalance between cell proliferation and cell death. More genetic and epigenetic events are required to drive from initiated cells to malignant tumors, each conferring one or another type of growth advantage, and leads to the progressive conversion of normal human cells into cancer cells. The establishment of a mutation is a sine qua non of cancer. Most notable among proneoplastic mutations are those that result in increased expression of oncogenes (e.g., myc, ras, abl, bcl-2) or decreased activity of tumor-suppressor genes1 (e.g., p53, Rb), conferring a selective growth or survival advantage to the cell. Phenotypic changes representative of preneoplastic mutations include a decreased need for metabolites and growth factors, abnormal
signal transduction, inappropriate expression of receptors for available growth factors (epidermal growth factor receptor, HER2/neu), dysregulation of cell-cycle checkpoints and resistance to apoptosis.1 At this point, any agent that causes increased cell proliferation increases the risk of neoplastic transformation. The damage to the DNA must survive the many DNA-repair processes and must be readable by DNA polymerase, which creates and locks in the mutation. If, by chance, this damage to the DNA results in a selective growth or survival advantage to the cell, it may become a precancerous lesion. On the other side, while normal cells have the ability to turn on and off transiently the genes that help them survive toxic signals (e.g., induction of P450 enzymes for metabolism of toxic chemicals and drugs), under circumstances of prolonged stress, such as during chronic inflammation, a mutation may lock in the growth-advantaged phenotype. Hence, prolonged exposure to stress can result in selection of preneoplastic cells. Some examples of adaptive changes in a cell are increased expression of antioxidant enzymes, matrix metalloproteinases and growth factor receptors; increased anaerobic respiration; and de novo synthesis of angiogenic factors. Studies in experimental tumor models suggest that the inhibition of angiogenesis can impede tumor growth and metastasis and can cause preformed tumors to necrotize and regress.
The role of inflammation on carcinogenesis
There is a large series of reviews that extensively demonstrate the direct relationship between chronic inflammation and cancer.2, 3, 4, 5, 6, 7, 8, 9, 10
In fact, a wide array of chronic inflammatory conditions predispose susceptible cells to neoplastic transformation.11 In general, the longer the inflammation persists, the higher the risk of cancer. Inflammation is a step-by step process that includes injury, repair and resolution. All inflammatory cells (neutrophils, monocytes, macrophages, eosinophils, dendritic cells, mast cells and lymphocytes) are recruited after a damage or an infection, and may contribute to the onset and progression of cancer. Key molecular players that link inflammation to genetic alterations are prostaglandins, cytokines, nuclear factor NFkB, chemokines and angiogenic factors. The main chemical effectors are free radical species derived from oxygen (ROI) and nitrogen (RNI). Free radicals may act as direct or indirect damaging agents through their reaction with other chemical or structural components in cells. ROI and RNI also recruit other inflammatory cells with secondary amplification of the damage. In this review, we describe the role of free radicals and of oxidative stress consequent to a chronic inflammation on the induction and progression of cancer.
Free radicals as a link between inflammation and cancer
General concepts on free radicals and oxidative stress
The main sources of reactive species in all cells are mitochondria, cytochrome P450 and peroxisome. Under physiological conditions, there is a constant endogenous production of reactive intermediates of ROI and RNI that interact as “signaling”
molecules for metabolism, cell cycle and intercellular transduction pathways.12, 13 The balance between beneficial and/or harmful effects of intermediate species is the crucial event in living organisms. In fact, the redox homeostasis is, in vivo, the main protective process from cell death. To control the balance between production and removal of ROI and RNI, there are a series of protective molecules and systems globally defined as “antioxidant defences.”.These include enzymes such as superoxide dismutase, catalase, glutathione peroxidase and glutathione-S-transferase, proteins that sequester transition metals, glutathione (GSH), cysteine, thioredoxin, vitamins, etc.14 Oxidative stress occurs when the generation of free radicals and active intermediates in a system exceeds the system's ability to neutralize and eliminate them.15, 16, 17 At the moment, the concept of oxidative stress originally confined to ROI—such as hydroxyl and superoxide radicals, and hydrogen peroxide and singlet oxygen—has been extended onto reactive nitrogen species (RNI) as nitric oxide (NO), peroxynitrite and, recently, S-nitrosothiols.13 Therefore, the current concept of “oxidative stress” should also include the pathways related to the “nitrosative stress” and, for their implication in cellular and extracellular metabolic events, to the “metabolic stress.” In these conditions, ROI and RNI act as “toxic” substances that may react with proteins, carbohydrates and lipids, with consequent alteration both in the intracellular and intercellular homeostasis, leading to possible cell death and regeneration (see Fig. 1).
Figure 1. Reactions of ROI and RNI with proteins, carbohydrates and lipids, with consequent alteration both in the intracellular and intercellular homeostasis until possible cell death and regeneration.
Sources of free radicals during inflammation
Inflammatory cells secrete a large number of cytokines and chemokines that promote the outgrowth of neoplastic cells, in addition to the autocrine growth factor production by the tumor cells themselves. ROI and RNI are produced under the stimulus of
proinflammatory cytokines in phagocytic and nonphagocytic cells through the activation of protein-kinases signaling. For example, TNF-α enhances the formation of ROI by neutrophils and other cells, while interleukin-1-β (IL-1-β), TNF-α and interferon (IFN)-γ stimulate the expression of inducible nitric oxide synthase in inflammatory and epithelial cells. In experimental animals, pristane-induced plasma cell tumors require IL-6 for their growth, and it is provided by macrophages in the chronically inflamed tissue. Production of IL-6 in this system is stimulated by PGE2 derived from cyclooxygenase-2 (COX-2), which is elevated in inflammatory macrophages; this process is inhibited by the administration of indomethacin, a potent antiinflammatory drug.18, 19 Knockout of the TNF-α gene in mice significantly inhibits the development of skin tumors in response to DMBA and phorbol esters.20 IL-8, an inflammatory chemokine derived from monocytes, macrophages and endothelial cells, is implicated in the progression and metastasis of tumors in the colon, bladder, lung and stomach.21
Chronic inflammation is closely associated with angiogenesis.22 Macrophages, platelets, fibroblasts and tumor cells themselves are a major source of angiogenic factors such as basic fibroblast growth factor, vascular endothelial growth factor, prostaglandins-1 and -2 other than inflammatory cytokines, chemokines and NO.21 There is a significant interaction and synergy among these mediators of inflammation until cancer. As an example, it has been demonstrated that prostaglandins, derived from the oxidative metabolism of arachidonic acid in inflammatory cells, contribute to the development of cancer.23, 24 Prostaglandins induce the expression of certain inflammatory cytokines, which can, in turn, enhance the production of ROI and RNI. COX-2 is the main enzyme that responds to inflammation through the synthesis of prostaglandins in monocytes and macrophages; however, COX-2 is also expressed in noninflammatory cells such as fibroblasts, epithelial and endothelial cells. In vitro, the expression of COX-2 is induced by bacterial cell products and inflammatory cytokines.25, 26 COX-2 is known to be involved in cancer biology, and it is upregulated in many cancer types including carcinomas of the colon, breast, lung, pancreas, esophagus, and head and neck.27, 28 Studies from COX-2 transgenic mice and COX-2 knockout mice confirm that COX-2 plays a role in colonic cancer development, both through angiogenesis29, 30 and through the activation of different oncogenes, including v-src, v-Ha-ras, HER-2/neu and Wnt.31 Notably, prostaglandin synthesis can also be stimulated by peroxynitrite,32 a toxic radical derived from NO.
Free radicals may react with phospholipids of membranes generating hydroperoxides, lipoperoxides and toxic aldehydes such as malondialdehyde (MDA), which in turn may alter membrane permeability and microcirculation. These last events may induce a further adhesion of granulocytes to endothelium that may activate the xantine–xantine–oxidase pathway with an enhanced production of hydroperoxide in a vicious circle. ROI, RNI and their derivative products may also activate nuclear factors such as, for example, NFkB, leading to the production of other amount of proinflammatory cytokines, which in turn enhance inflammation and, therefore, the generation of other reactive species.33 The imbalance in redox status and the enhanced production of intermediate reactive species progressively consumes the antioxidant defences, leading
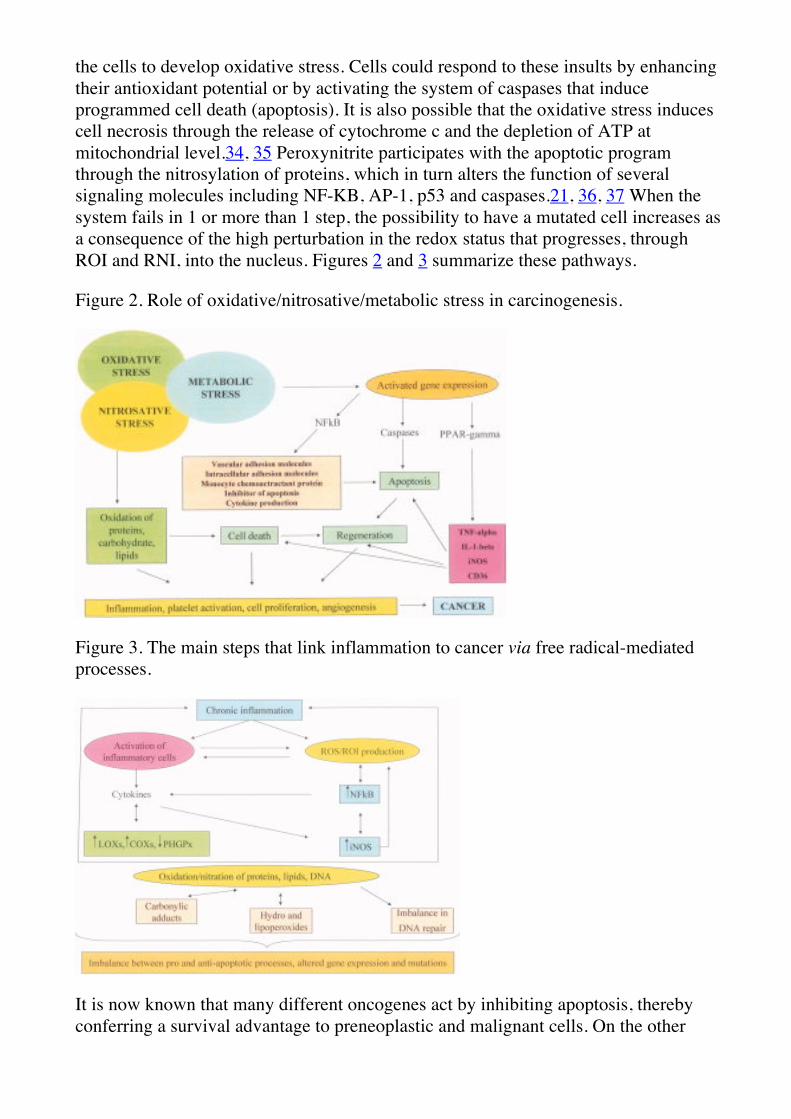
the cells to develop oxidative stress. Cells could respond to these insults by enhancing their antioxidant potential or by activating the system of caspases that induce programmed cell death (apoptosis). It is also possible that the oxidative stress induces cell necrosis through the release of cytochrome c and the depletion of ATP at mitochondrial level.34, 35 Peroxynitrite participates with the apoptotic program through the nitrosylation of proteins, which in turn alters the function of several signaling molecules including NF-KB, AP-1, p53 and caspases.21, 36, 37 When the system fails in 1 or more than 1 step, the possibility to have a mutated cell increases as a consequence of the high perturbation in the redox status that progresses, through ROI and RNI, into the nucleus. Figures 2 and 3 summarize these pathways.
Figure 2. Role of oxidative/nitrosative/metabolic stress in carcinogenesis.
Figure 3. The main steps that link inflammation to cancer via free radical-mediated processes.
It is now known that many different oncogenes act by inhibiting apoptosis, thereby conferring a survival advantage to preneoplastic and malignant cells. On the other
hand, several chemotherapeutic drugs exert their action of cell death promotion by increasing ROI production.38, 39
The start of carcinogenesis mediated by ROI and RNI following chronic inflammation may be direct (oxidation, nitration, halogenation of nuclear DNA, RNA and lipids), or may be mediated by the products of ROI-RNI and proteins, lipids and carbohydrates that are capable of forming DNA adducts. ROI can also increase the expression of transcriptional factors including c-fos and c-jun oncogenes involved in neoplastic transformation.
Direct effect of free radicals on DNA damage and carcinogenesis
To date, more than 100 oxidized DNA products have been identified. ROI-induced DNA damage involves single- or double-stranded DNA breaks; purine, pyrimidine or deoxyribose modifications; DNA intrastrand adducts; and DNA protein crosslinks.40 DNA damage can result in either arrest or induction of transcription, induction of signal transduction pathways, replication errors and genomic instability, all processes associated with carcinogenesis.41, 42 NO reacts with superoxide (O2
−) to form peroxynitrite (ONOO−), a highly reactive species that induce nitrative and oxidative DNA damage. In fact, ONOO− mediates the formation of 8-oxo-7,8-dihydro-2′-deoxyguanosine43 and 8-nitroguanine,44, 45 which has also been suggested as a potential biomarker of inflammation-related carcinogenesis.42, 46 Recent evidences indicate that 8-nitroguanosine is a highly redox-active molecule47, 48 and that 8-nitroguanine is a mutagenic substance, which preferentially leads to G>T transversions.44, 49 Indeed, G>T transversions have been observed in vivo in the ras gene50 and the p53 tumor suppressor gene, in lung and liver cancer.51, 52 These findings indicate that the DNA damage mediated by ROI and RNI may participate to carcinogenesis, both via activation of proto-oncogenes and inactivation of tumor suppressor genes. In the carcinogenesis also, the oxidative damage of mitochondria is strongly involved.42 In fact, hydrogen peroxide and other reactive oxygen species activate nuclear genes that regulate the biogenesis, transcription and replication of the mitochondrial genome. Although the rate of tumor cells that possess mutated mitochondrial DNA and the cancer processes to which mitochondrial DNA alterations participate have not been satisfactorily established, a significant amount of information support the involvement of the mitochondria in carcinogenesis.42 Fragments of mitochondrial DNA have been found to be inserted into nuclear DNA, suggesting a possible mechanism of oncogene activation. Mitochondrial DNA (mtDNA), which is bound to the inner mitochondria membrane, is particularly liable to oxidative damage because of the lack of histone proteins, the presence of incomplete repairing mechanisms53, 54, 55 and its proximity to the mitochondrial electron transport chain. Damage to mtDNA causes mitochondrial respiratory chain dysfunction, which in turn increases the production of hydroxyl radicals which are the major source of oxidative damage for DNA.56, 57, 58 MtDNA also encodes essential proteins involved in the phosphorylation processes,59 and its damage is associated with a cascade of reactions that amplify the oxidative stress.60, 61, 62, 63 Mutations and altered expression in mitochondrialgenes encoding for complexes I, III, IV and V,
and in the hypervariable regions of mitochondrial DNA, have been identified in various human cancers.64, 65
The role of ROI- and RNI-derived products on carcinogenesis
Free radicals immediately react with all components of cells by forming stable products. Proteins are more susceptible to oxidation by free radicals. In the global cellular economy, the oxidation of SH groups of cysteine reduces the activity of various enzymes as well as the synthesis of GSH, which is the main intracellular free radical scavenger.
The oxidation of lipids induces the formation of aldehydes and lipid peroxides. These molecules, at very low and nontoxic concentrations, act as signaling transductors of ROI-mediated metabolic reactions, and therefore, they modulate several cell functions including gene expression and cell proliferation.66 In high concentrations, lipid-derived products are considered the more damaging species because they easily react with proteins, DNA and phospholipids, generating a variety of intra- and intermolecular toxic covalent adducts that lead to the propagation and amplification of oxidative stress.67 This has been demonstrated in human cancerogenesis.68, 69, 70 Lipid hydroperoxides are formed in vivo through the action of ROI on polyunsaturated fatty acids, both directly and as specific products of lipoxygenase (LOX) and COX activities.67 Cell membranes are the main target for lipid peroxidation. Lipid peroxides cause cell damage by its decomposition in breakdown toxic products that are bifunctional aldehydes,71 which are relatively stable and able to diffuse in and outside the cell. The most abundant ROI–lipid-derived product is—in conditions of oxidative stress—the 4-hydroxynonenal (HNE). At low levels it promotes cell proliferation, while at a higher concentration it induces oxidative alterations of DNA72 and apoptosis.73, 74, 75 Therefore, 4-HNE is directly involved in cell cycle control,76 and in human cancer, it causes mutation in the p53 gene expression.68 4-HNE forms etheno adducts with DNA and upregulates COX-2 expression.77 It has previously been stressed that COX2 expression induces an enhanced production of ROI. The breakdown of membrane lipid hydroperoxides also increases the levels of MDA,78 which can react with DNA bases G, A, and C by forming adducts M1G, M1A and M1C, respectively.79 M1G adducts have been found in human tissues at levels as high as 1.2 adducts per 106 bases (which corresponds to ∼6,000 adducts per cell). M1G has been detected in human breast tissue by32phospho-postlabeling as well as in rodent tissues.80 Moreover, M1G has been demonstrated to be mutagenic in Escherichia coli, through the transversions to T and transitions to A.81, 82 There are also other exocyclic DNA adducts that arise from lipid peroxidation. For example, etheno-dA, etheno-dC and etheno-dG have been detected by both32phospho-postlabeling and GC–MS.83 It has been demonstrated that etheno-dA and etheno-dC are strongly genotoxic but weakly mutagenic when introduced on single-stranded vectors in E. coli. In addition, hydroxypropanodeoxyguanosines are present in human DNA in presence of cancer.41 These adducts are most probably derived from the reaction of DNA with acrolein and crotonaldehyde generated by the lipid peroxidation process.
Two enzymes, with opposed activities, control the peroxidation of cell membranes. These are LOXs and the phospholipid hydroperoxide glutathione-peroxidase (PH-GPx). The regulatory relation of these enzymes is of crucial importance to maintain a normal redox status in cells. An imbalance in this regulation leads to the generation of cytotoxic molecules that are involved in carcinogenesis.71 LOXs catalyze the specific dioxygenation of polyenoic fatty acids, forming reactive fatty acid hydroperoxides. PH-GPx is a member of the GPx enzymes family, a selenium-dependent protein family that contains selenocysteine at its active site, and it has the ability to reduce organic and inorganic hydroperoxides by utilizing GSH as a reducing agent.84
Among the three 12-LOX forms that have been identified, both the leukocyte and platelet types have been found in different cancer tissues, including melanoma, prostate and epidermal cancers.85 Inhibition of LOXs induces apoptosis and blocks the proliferation in carcinosarcoma cells.86 The expression of 12-LOX also correlates with tumor cell metastasis. Clinically, the degree of 12-LOX expression in human prostate cancer correlates with the tumor grade and stage, and its expression level is higher in metastasic prostate cancers than in nonmetastatic tumors.87 Some studies have suggested that 15-LOX enhances colonic tumorogenesis by potentiating the cell proliferation and the mitogenic response to EGF.88, 89 12/15 LOX, that is the oxidative counterpart of PH-GPx, is transcriptionally regulated by ILs. In a variety of human cells, the expression of the human 12/15-LOX is upregulated when cells are exposed to IL-4. Other cytokines, such as IFN-γ, IL-6 and IL-10 did not induce the enzyme.90, 91 Experiments with human epidermoid carcinoma A431 cells that were transfected with the cDNA sequence of cytoplasmic PH-GPx revealed that PH-GPx downregulates the 12-LOX activity.92 In animals, platelets 12-LOX, 15-LOX and COX-2 are more sensitive to PH-GPx inhibition than do 5-LOX and COX-1, which are more resistant.93 This inhibition is probably caused by a requirement of fatty acid hydroperoxides for the activation of LOX and COX that are regulated by its own products.94, 95 These results support the hypothesis that cytoplasmic PH-GPx plays a pivotal role in the regulation of arachidonate metabolism. On the other hand, inhibition of PH-GPx activity with the treatment of cells with an antisense oligonucleotide of PH-GPx mRNA increases the enzymatic activity of 12-LOX in A431cells.96 This interplay can be further complicated by stimulation with ILs. When the human lung carcinoma cell line A549 is exposed to IL-4 and 13, an upregulation of the 12/15-LOX and a downregulation of the PH-GPx are observed.90, 91 Mitochondrial PH-GPx protects from cell death induced by phospholipid hydroperoxidation triggered by photodynamic therapy.97 Likewise, 2 arsenic compounds, arsenic trioxide and arsenic disulfide, employed against acute promyelocytic leukemia, induce apoptosis in leukemia cells98 by inhibiting PH-GPx expression.99
In cancer tissues, a great part of the increased lipid peroxidation could be triggered by increased arachidonic acid metabolism, as a result of elevated COXs. On the other hand, MDA can be produced by isomerization of prostaglandins PGH2. Therefore, a high interplay exists among PHGPx, 12/15LOX and COX-2 in cancerogenesis mechanisms. Figure 4 summarizes the role of free radicals on carcinogenesis.
Figure 4. The role of free radicals on carcinogenesis.
Conclusive remarks
ROI and RNI participate in various redox-regulatory mechanisms of cells in order to maintain a normal and constant “redox homeostasis.” When the system fails and the antioxidant control mechanisms are exhausted or overrun, the cellular redox potential shifts toward an oxidative and nitrosative stress, which leads to DNA mutations and genomic instability. A chronic inflammation is the main system capable of inducing an oxidative/nitrosative stress in a self-perpetuating system. The most widely studied and best established of these links are colon carcinoma associated with inflammatory bowel diseases (chronic ulcerative colitis and Crohn's disease), esophageal adenocarcinoma associated with reflux esophagitis (Barrett's esophagus), hepatitis predisposing to liver cancer, schistosomiasis causing an increased risk of bladder and colon carcinomas and chronic Helicobacter infection leading to cancer of the stomach. Inflammatory cytokines also regulate the progression and metastasis of tumors (colon, bladder, lung and stomach). Examples of tumor cell cytokine dependence in human disease are the growth dependence of AIDS- and EBV-associated B-cell lymphomas, B-cell leukemias and multiple myeloma on the inflammatory cytokines IL-6 and IL-15, and the dependence of malignant mesothelioma on platelet-derived growth factor.100, 101
In animals, indomethacin blocks carcinogenesis by reducing the production of inflammatory cytokines. In humans, epidemiological studies documented a lower cancer risk for people regularly taking nonsteroidal antiinflammatory drugs.102 For example, the use of COX-2 inhibitors, like celecoxib, are now being tested as antitumor agents in clinical trials.103 In fact, this drug reduces the number of colorectal polyps in patients with familial adenomatous polyposis.
On the basis of these considerations, future research should be aimed to precociously discover the preneoplastic tranformation of a chronically inflamed tissue. Lipid peroxides and 8-dehoxiguanosine and guanine appear at the moment as promising tools for quantifying the risk of DNA mutations. The evaluation of the levels of these supposed markers should also be useful to monitor therapies aimed to block the carcinogenic potential of the inflammation-related oxidative and nitrosative cascade.
There are studies on these aspects in humans following a diet rich in antioxidants. Depletion of antioxidants, and in particular of GSH, may sensitize tumor cells to some chemotherapy agents, and many of the GSH-depleting agents have a dose-related toxicity. In this context, the development of nontoxic GSH-depleting agent may be strongly relevant in overcoming multidrug resistance. Finally, future researches are needed to better determine the most rational and effective combination of redox-active and antiinflammatory agents to other routine drugs in the treatment of cancer.
References
• 1
Hanahan D,Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70.
• 2
Bartsch H,Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch Surg 2006; 391: 499–510.
• 3
Aggarwal BB,Shishodia S,Sandur SK,Pandey MK,Sethi G. Inflammation and cancer: how hot is the link? Biochem Pharmacol 2006; 72: 1605–21.
• 4
Lu H,Ouyang W,Huang C. Inflammation, a key event in cancer development. Mol Cancer Res 2006; 4: 221–33.
• 5
Moss SF,Blaser MJ. Mechanisms of disease: inflammation and the origins of cancer. Nat Clin Pract Oncol 2005; 2: 90–7.
• 6
Hofseth LJ,Ying L. Identifying and defusing weapons of mass inflammation in carcinogenesis. Biochim Biophys Acta 2006; 1765: 74–84.
• 7
Hussain SP,Hofseth LJ,Harris CC. Radical causes of cancer. Nat Rev Cancer 2003; 3: 276–85.
• 8
Ohshima H,Tatemichi M,Sawa T. Chemical basis of inflammation-induced carcinogenesis. Arch Biochem Biophys 2003; 417: 3–11.
• 9
Coussens LM,Werb Z. Inflammation and cancer. Nature 2002; 420: 860–7.
• 10
Fitzpatrick FA. Inflammation, carcinogenesis and cancer. Int Immunopharmacol 2001; 1: 1651–67.
• 11
Slaga TJ,Lichti U,Hennings H,Elgjo K,Yuspa SH. Effects of tumor promoters and steroidal anti-inflammatory agents on skin of newborn mice in vivo and in vitro. J Natl Cancer Inst 1978; 60: 425–31.
• 12
Nathan C. Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signalling. J Clin Invest 2003; 111: 769–78.
Valko M,Leibfritz D,Moncol J,Cronin MTD,Mazur M,Telser J. Free radicals and antioxidant in normal physiological function and human disease. Int J Biochem Cell Biol 2007; 39: 44–84.
• 15
Sies H. Oxidative stress: Introductory remarks. In: SiesH, ed. Oxidative stress. San Diego: Academic Press, 1985: 1–8.
• 16
Sies H. Biochemistry of oxidative stress. Angew Chem Int Ed Engl 1986; 25: 1058–71.
Direct Link:
• 17
Sies H,Cadenas E. Oxidative stress: damage to intact cells and organs. Philos Trans R Soc Lond B Biol Sci 1985; 311: 617–31.
• 18
Hinson RM,Williams JA,Shacter E. Elevated interleukin 6 is induced by prostaglandin E2 in a murine model of inflammation: possible role of cyclooxygenase-2. Proc Natl Acad Sci USA 1996; 93: 4885–90.
• 19
Shacter E,Arzadon GK,Williams J. Elevation of IL-6 in response to a chronic inflammatory stimulus in mice: inhibition by indomethacin. Blood 1992; 80: 194–202.
• 20
Moore RJ,Owens DM,Stamp G,Arnott C,Burke F,East N,Holdsworth H,Turner L,Rollins B,Pasparakis M,Kollias G,Balkwill F. Mice deficient in tumor necrosis factor-α are resistant to skin carcinogenesis. Nat Med 1999; 5: 828–31.
• 21
Zouki C,Jozsef L,Ouellet S,Paquette Y,Filep JG. Peroxynitrite mediates cytokine-induced IL-8 gene expression and production by human leukocytes. J Leukoc Biol 2001; 69: 815–24.
• 22
Jackson JR,Seed MP,Kircher CH,Willoughby DA,Winkler JD. The codependence of angiogenesis and chronic inflammation. FASEB J 1997; 11: 457–65.
• 23
Baron JA,Sandler RS. Nonsteroidal anti-inflammatory drugs and cancerprevention. Annu Rev Med 2000; 51: 511–23.
Howe LR,Dannenberg AJ. A role for cyclooxygenase-2 inhibitors in the prevention and treatment of cancer. Semin Oncol 2002; 29: 111–19.
• 26
Dannenberg AJ,Altorki NK,Boyle JO,Dang C,Howe LR,Weksler BB,Subbaramaiah K. Cyclooxygenase 2: a pharmacological target for the prevention of cancer. Lancet Oncol 2001; 2: 544–51.
• 27
Lin DT,Subbaramaiah K,Shah JP,Dannenberg AJ,Boyle JO. Cyclooxygenase-2: a novel molecular target for the prevention and treatment of head and neck cancer. Head Neck 2002; 24: 792–9.
• 28
Gupta RA,DuBois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 2001; 1: 11.
• 29
Williams CS,Mann M,DuBois RN. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999; 18: 7908–16.
• 30
Gately S. The contributions of cyclooxygenase-2 to tumor angiogenesis. Cancer Metastasis Rev 2000; 19: 19–27.
• 31
Howe LR,Subbaramaiah K,Brown AM,Dannenberg AJ. Cyclooxygenase-2: a target for the prevention and treatment of breast cancer. Endocr Relat Cancer 2001; 8: 97–114.
• 32
Goodwin DC,Landino LM,Marnett LJ. Effects of nitric oxide and nitricoxide-derived species on prostaglandin endoperoxide synthase and prostaglandin biosynthesis. FASEB J 1999; 13: 1121–36.
• 33
Marnett LJ,Riggins JN,West JD. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J Clin Invest 2003; 111: 583–93.
• 34
Herrera B,Alvarez AM,Sanchez A,Fernandez M,Roncero C,Benito M,Fabregat I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. FASEB J 2001; 15: 741–51.
• 35
Herrera B,Fernandez M,Alvarez AM,Roncero C,Benito M,Gil J,Fabregat I. Activation of caspases occurs downstream from radical oxygen species production, Bcl-xL down-regulation, and early cytochrome C release in apoptosis induced by transforming growth-factor β in rat fetal hepatocytes. Hepatology 2001; 34: 548–56.
• 36
Sata N,Klonowski-Stumpe H,Han B,Haussinger D,Niederau C. Cytotoxicity of peroxynitrite in rat pancreatic acinar AR4-2J cells. Pancreas 1997; 15: 278–84.
• 37
Zhuang S,Simon G. Peroxynitrite-induced apoptosis involves activation of multiple caspases in HL-60 cells. Am J Physiol Cell Physiol 2000; 279: C341–C351.
• 38
Johnstone RW,Ruefli AA,Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002; 108: 153–64.
• 39
Jabs T. Reactive oxygen intermediates as mediators of programmed cell death in plants and animals. Biochem Pharmacol 1999; 57: 231–45.
• 40
Valko M,Morris H,Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem 2005; 12: 1161–208.
• 41
Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis 2000; 21: 361–70.
• 42
Valko M,Rhodes CJ,Moncol J,Izakovic M,Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 2006; 160: 1–40.
• 43
Inoue S,Kawanishi S. Oxidative DNA damage induced by simultaneous generation of nitric oxide and superoxide. FEBS Lett 1995; 371: 86–88.
• 44
Yermilov V,Rubio J,Becchi M,Friesen MD,Pignatelli B,Ohshima H. Formation of 8-nitroguanine by the reaction of guanine with peroxynitrite in vitro. Carcinogenesis 1995; 16: 2045–50.
• 45
Akaike T,Okamoto S,Sawa T,Yoshitake J,Tamura F,Ichimori K,Miyazaki K,Sasamoto K,Maeda H. 8-Nitroguanosine formation in viral pneumonia and its implication for pathogenesis. Proc Natl Acad Sci USA 2003; 100: 685–90.
• 46
Valko M,Leibfritz D,Moncol J,Cronin MT,Mazur M,Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007; 39: 44–84.
• 47
Sawa T,Akaike T,Ichimori K,Akuta T,Kaneko K,Nakayama H,Stuehr DJ,Maeda H. Superoxide generation mediated by 8-nitroguanosine, a highly redox-active nucleic acid derivative. Biochem Biophys Res Commun 2003; 311: 300–6.
• 48
Zaki MH,Akuta T,Akaike T. Nitric oxide-induced nitrative stress involved in microbial pathogenesis. J Pharmacol Sci 2005; 98: 117–29.
• 49
Suzuki N,Yasui M,Geacintov NE,Shafirovich V,Shibutani S. Miscoding events during DNA synthesis past the nitration-damaged base 8-nitroguanine. Biochemistry 2005; 44: 9238–45.
• 50
Bos JL. The ras gene family and human carcinogenesis. Mutat Res 1988; 195: 255–71.
• 51
Takahashi T,Nau MM,Chiba I,Birrer MJ,Rosenberg RK,Vinocour M,Levitt M,Pass H,Gazdar AF,Minna JD. p53: a frequent target for genetic abnormalities in lung cancer. Science 1989; 246: 491–4.
• 52
Hsu IC,Metcalf RA,Sun T,Welsh JA,Wang NJ,Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991; 350: 427–8.
• 53
Albring M,Griffith J,Attardi G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc Natl Acad Sci USA 1977; 74: 1348–52.
• 54
Clayton DA. Transcription of the mammalian mitochondrial genome. Annu Rev Biochem 1984; 53: 573–94.
• 55
Yakes FM,Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA 1997; 94: 514–19.
Berlett BS,Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 1997; 272: 20313–16.
• 58
Esterbauer H,Schaur RJ,Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991; 11: 81–128.
• 59
Kowaltowski AJ,Vercesi AE. Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med 1999; 26: 463–71.
• 60
Fliss MS,Usadel H,Caballero OL,Wu L,Buta MR,Eleff SM,Jen J,Sidransky D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 2000; 287: 2017–19.
• 61
Richard SM,Bailliet G,Paez GL,Bianchi MS,Peltomaki P,Bianchi NO. Nuclear and mitochondrial genome instability in human breast cancer. Cancer Res 2000; 60: 4231–7.
• 62
Polyak K,Li Y,Zhu H,Lengauer C,Willson JK,Markowitz SD,Trush MA,Kinzler KW,Vogelstein B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet 1998; 20: 291–3.
• 63
Habano W,Nakamura S,Sugai T. Microsatellite instability in the mitochondrial DNA of colorectal carcinomas: evidence for mismatch repair systems in mitochondrial genome. Oncogene 1998; 17: 1931–7.
• 64
Tamura G,Nishizuka S,Maesawa C,Suzuki Y,Iwaya T,Sakata K,Endoh Y,Motoyama T. Mutations in mitochondrial control region DNA in gastric tumors of Japanese patients. Eur J Cancer 1999; 35: 316–19.
• 65
Horton TM,Petros JA,Heddi A,Shoffner J,Kaufman AE,Graham SD,Gramlich T,Wallace DC. Novel mitochondrial DNA deletion found in renal cell carcinoma. Genes Chromosomes Cancer 1996; 15: 95–101.
• 66
Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res 2003; 42: 318–43.
Hu W,Feng Z,Eveleigh J,Iver G,Pan J,Amin S,Chung FL,Tang MS. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002; 23: 1781–9.
• 69
Gago-Dominguez M,Castelao JE,Yuan JM,Ross RK,Yu MC. Lipid peroxidation: a novel and unifying concept of the etiology of renal cell carcinoma (United States). Cancer Causes Control 2002; 13: 287–93.
• 70
Sander CS,Hamm F,Elsner P,Thiele JJ. Oxidative stress in malignant melanoma and non-melanoma skin cancer. Br J Dermatol 2003; 148: 913–22.
• 71
Kuhn H,Borchert A. Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes. Free Radic Biol Med 2002; 33: 154–72.
• 72
Nair J,Barbin A,Velic I,Bartsch H. Etheno DNA-base adducts from endogenous reactive species. Mutat Res 1999; 424: 59–69.
• 73
Ruef J,Rao GN,Li F,Bode C,Patterson C,Bhatnagar A,Runge MS. Induction of rat aortic smooth muscle cell growth by the lipid peroxidation product 4-hydroxy-2-nonenal. Circulation 1998; 97: 1071–8.
• 74
Cheng JZ,Singhal SS,Saini M,Singhal J,Piper JT,VanKuijk FJ,Zimniak P,Awasthi YC,Awasthi S. Effects of mGST A4 transfection on 4-hydroxynonenal-mediated apoptosis and differentiation of K562 human erythroleukemia cells. Arch Biochem Biophys 1999; 372: 29–36.
• 75
Dianzani MU,Barrera G,Parola M. 4-Hydroxy-2,3-nonenal as a signal for cell function and differentiation. Acta Biochim Pol 1999; 46: 61–75.
• 76
Fazio VM,Rinaldi M,Ciafre S,Barrera G,Farace MG. Control of neoplastic cell proliferation and differentiation by restoration of 4-hydroxynonenal physiological concentrations. Mol Aspects Med 1993; 14: 217–28.
• 77
Kumagai T,Kawamoto Y,Nakamura Y,Hatayama I,Satoh K,Osawa T,Uchida K. 4-hydroxy-2-nonenal, the end product of lipid peroxidation, is a specific inducer of cyclooxygenase-2 gene expression. Biochem Biophys Res Commun 2000; 273: 437–41.
• 78
Pryor WA,Stanley JP. Letter: a suggested mechanism for the production of malonaldehyde during the autoxidation of polyunsaturated fatty acids. Nonenzymatic production of prostaglandin endoperoxides during autoxidation. J Org Chem 1975; 40: 3615–17.
• 79
Marnett LJ. Lipid peroxidation-DNA damage by malondialdehyde. Mutat Res 1999; 424: 83–95.
• 80
Wang MY,Dhingra K,Hittelman WN,Liehr JG,deAndrade M,Li DH. Lipid peroxidation-induced putative malondialdehyde–DNA adducts in human breast tissues. Cancer Epidemiol Biomark Prev 1996; 5: 705–10.
• 81
Fink SP,Reddy GR,Marnett LJ. Mutagenicity in Escherichia coli of the major DNA adduct derived from the endogenous mutagen malondialdehyde. Proc Natl Acad Sci USA 1997; 94: 8652–57.
• 82
Mao H,Schnetz-Boutaud NC,Weisenseel JP,Marnett LJ,Stone MP. Duplex DNA catalyzes the chemical rearrangement of a malondialdehyde deoxyguanosine adduct. Proc Natl Acad Sci USA 1999; 96: 6615–20.
• 83
Fedtke N,Boucheron JA,Walker VE,Swenberg JA. Vinyl chloride-induced DNA adducts, Part 2: Formation and persistence of 7-2�-oxoethylguanine and N- 2,3-ethenoguanine in rat-tissue DNA. Carcinogenesis 1990; 11: 1287–92.
Steele VE,Holmes CA,Hawk ET,Kopelovich L,Lubet RA,Crowell JA,Sigman CC,Kelloff GJ. Lipoxygenase inhibitors as potential cancer chemopreventives. Cancer Epidemiol Biomarkers Prev 1999; 8: 467–83.
• 86
Tang DG,Honn KV. Apoptosis of W256 carcinosarcoma cells of the monocytoid origin induced by NDGA involves lipid peroxidation and depletion of GSH: role of 12-lipoxygenase in regulating tumor cell survival. J Cell Physiol 1997; 172: 155–70.
• 87
Nie D,Che M,Grignon D,Tang K,Honn KV. Role of eicosanoids in prostate cancer progression. Cancer Metastasis Rev 2001; 20: 195–206.
• 88
Eling TE,Glasgow WC. Cellular proliferation and lipid metabolism: importance of lipoxygenases in modulating epidermal growth factor-dependent mitogenesis. Cancer Metastasis Rev 1994; 13: 397–410.
• 89
Eling TE,Everhart AL,Angerman-Stewart J,Hui R,Glasgow WC. Modulation of epidermal growth factor signal transduction by linoleic acid metabolites. Adv Exp Med Biol 1997; 407: 319–22.
• 90
Schnurr K,Borchert A,Kuhn H. Inverse regulation of lipid-peroxidizing and hydroperoxyl lipid-reducing enzymes by interleukins 4 and 13. FASEB J 1999; 13: 143–54.
• 91
Schnurr K,Brinckmann R,Kuhn H. Cytokine induced regulation of 15-lipoxygenase and phospholipid hydroperoxide glutathione peroxidase in mammalian cells. Adv Exp Med Biol 1999; 469: 75–81.
• 92
Chen CJ,Huang HS,Chang WC. Inhibition of arachidonate metabolism in human epidermoid carcinoma a431 cells overexpressing phospholipid hydroperoxide glutathione peroxidase. J Biomed Sci 2002; 9: 453–9.
• 93
Huang HS,Chen CJ,Suzuki H,Yamamoto S,Chang WC. Inhibitory effect of phospholipid hydroperoxide glutathione peroxidase on the activity of lipoxygenases and cyclooxygenases. Prostaglandins Other Lipid Mediat 1999; 58: 65–75.
• 94
Shitashige M,Morita I,Murota S. Different substrate utilization between prostaglandin endoperoxide H synthase-1 and -2 in NIH3T3 fibroblasts. Biochim Biophys Acta 1998; 1389: 57–66.
• 95
Samuelsson B,Dahlen SE,Lindgren JA,Rouzer CA,Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 1987; 237: 1171–6.
• 96
Chen CJ,Huang HS,Lin SB,Chang WC. Regulation of cyclooxygenase and 12-lipoxygenase catalysis by phospholipids hydroperoxide glutathione peroxidase in A431 cells. Prostaglandins Leukot Essent Fatty Acids 2000; 62: 261–8.
• 97
Wang HP,Qian SY,Schafer FQ,Domann FE,Oberley LW,Buettner GR. Phospholipid hydroperoxide glutathione peroxidase protects against singlet oxygen-induced cell damage of photodynamic therapy. Free Radic Biol Med 2001; 30: 825–35.
• 98
Chen GQ,Zhu J,Shi XG,Ni JH,Zhong HJ,Si GY,Jin XL,Tang W,Li XS,Xong SM,Shen ZX,Sun GL, et al. In vitro studies on cellular and molecular mechanisms of
arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR α/PML proteins. Blood 1996; 88: 1052–61.
• 99
Huang HS,Chang WC,Chen CJ. Involvement of reactive oxygen species in arsenite-induced downregulation of phospholipids hydroperoxide glutathione peroxidase in human epidermoid carcinoma A431 cells. Free Radic Biol Med 2002; 33: 864–73.
• 100
Aoki Y,Yarchoan R,Braun J,Iwamoto A,Tosato G. Viral and cellular cytokines in AIDS-related malignant lymphomatous effusions. Blood 2000; 96: 1599–601.
• 101
Trentin L,Cerutti A,Zambello R,Sancretta R,Tassinari C,Facco M,Adami F,Rodeghiero F,Agostini C,Semenzato G. Interleukin-15 promotes the growth of leukemic cells of patients with B-cell chronic lymphoproliferative disorders. Blood 1996; 87: 3327–35.
• 102
Fosslien E. Molecular pathology of cyclooxygenase-2 in neoplasia. Ann Clin Lab Sci 2000; 30: 3–21.
• 103
Subbaramaiah K,Dannenberg AJ. Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol Sci 2003; 24: 96–102.
































![Skin Inflammation, [Acute, Suppurative, Chronic, Chronic ... · Skin – Inflammation, [Acute, Suppurative, Chronic, Chronic Active, Granulomatous] presence of mononuclear cells (lymphocytes,](https://static.documents.pub/doc/80x56/5f0eb0c97e708231d44075f1/skin-inflammation-acute-suppurative-chronic-chronic-skin-a-inflammation.jpg)





