105
CHRONIC LIVER DISEASE BY DR MITHILA DAS MODERATOR : DR PAVAN
| Date post: | 07-Aug-2015 |
| Category: |
Health & Medicine |
| Upload: | mithila-das-mazumder |
| View: | 87 times |
| Download: | 4 times |

CHRONIC LIVER DISEASE
BY DR MITHILA DAS
MODERATOR : DR PAVAN

INTRODUCTION
• Definition• Pathophysiology• Aetiology of chronic liver disease• Symptoms and signs of chronic liver disease• Investigations in chronic liver disease• Treatment • Complications in chronic liver disease

DEFINATION
Cirrhosis is derived from Greek word kirros=orange or tawny and osis=condition
-WHO definition : it is defined as a diffuse process characterized by liver necrosis and fibrosis and conversion of normal liver architechture into structurally abnormal nodules that lack normal lobular organisation.

• Cirrhosis is a form of chronic liver injury that represents an end stage of virtually any progressive liver disease.
• cirrhosis may be superimposed on the primary liver disease and obscure the nature of the original insult.
• As cirrhosis advances, it results in distortion of liver architecture and compression of hepatic vascular and biliary structures. These critical architectural changes lead to irregular delivery of nutrients, oxygen, and metabolites to various areas of the liver and may perpetuate the cirrhotic process even after the original insult has been brought under control or has ceased .


CLASSIFICATION
Morphological classification
Etiological classification
It can be classified on the basis of morphology and etiology

• Micronodular cirrhosis : In micronodular cirrhosis the nodules are regular and small nodules and It is <3mm in diameter . There is diffuse involvement of all the hepatic lobules separated by the thick diffuse septa.
In its early stages extra hepatic Billiary atresia is micronodular.
Morphological classification
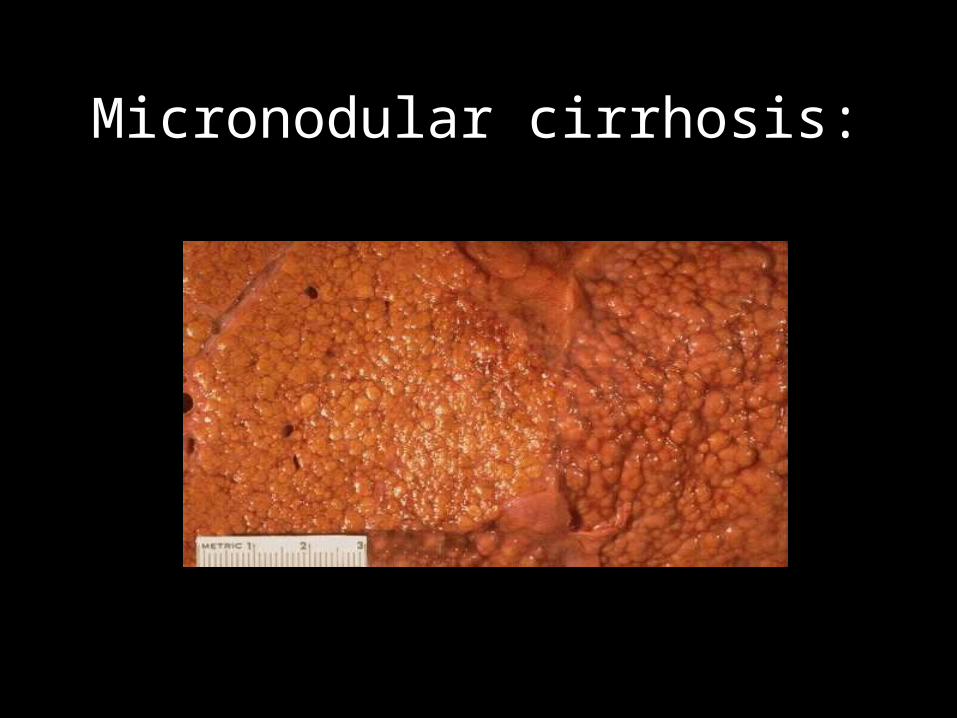
Micronodular cirrhosis:

• Macronodular cirrhosis : in this type the nodules are of various size and are generally larger than 5mm in diameter and the nodules are separated by irregular septa of varying width.
• Macronodular cirrhosis is a feature of advanced Wilson’s disease, α-1 antitrypsin deficiency, chronic active hepatitis

Macronodular Cirrhosis

• Mixed cirrhosis : Some parts of the liver show micronodular appearance and some part show macronodular pattern.

Etiological classification
Laennec Cirrhosis (most common 60-70%)
Post necrotic cirrhosis (10%)
Biliary cirrhosis (5 -10%)
Cardiac cirrhosis
Indian childhood cirrhosis

BILIARY CIRRHOSIScirrhosis is characterized by bile stasis, general
reduction of bile ducts, and increased amounts of connective tissue within and extending from portal tracts .
The lobular structure generally is preserved.
This pattern often is ultimately seen in children with biliary atresia, cystic fibrosis, and progressive familial intrahepatic cholestasis I.

POST NECROTIC CIRRHOSISPostnecrotic cirrhosis, also known as irregular
cirrhosis, is the result of chronic and recurrent liver cell destruction.
It is usually not a result of a single necrotic injury and is characterized by piecemeal necrosis that occurs at the interface between hepatocytes and portal tracts or fibrous septa.
Bridging fibrosis, collapsed hepatic lobules, and regenerative nodules of varying sizes develop, usually producing micronodular cirrhosis .

In children, postnecrotic cirrhosis may occur as a sequela of neonatal hepatitis.
It is associated with chronic active hepatitis, caused by viral hepatitis B or C, or a result of autoimmune or idiopathic inflammation .
Drugs such as methyldopa or isoniazid, which may cause chronic active hepatitis, also can cause irregular or postnecrotic cirrhosis .

CARDIAC CIRRHOSIS Cardiac cirrhosis develops as a result of
centrilobular hemorrhagic necrosis.
Increased right atrial pressure (resulting from congestive heart failure, congenital heart disease, or constrictive pericarditis) results in increased hepatic vein pressure and congestion of blood flow in centrilobular areas.
Necrosis leads to formation of fibrous bridges between
central veins.

Vaso occlusive disorders and the Budd–Chiari syndrome, which results from the congential or acquired obstruction of the hepatic veins, also result in cardiac cirrhosis.

ETIOLOGY
α1-antitrypsin deficiency Cystic fibrosis Fructosemia Galactosemia Gaucher’s disease Glycogen storage disease, type III Glycogen storage disease, type IV
Metabolic causes

Hemochromatosis Indian childhood cirrhosis Histiocytosis X Niemann–Pick disease, Tyrosinemia Wilson’s disease Wolman’s disease
Infectious disease : Viral hepatitides Cytomegalovirus Chronic hepatitis B +/−
delta agent Chronic hepatitis C

Herpes simplex virus Rubella Ascending cholangitis Neonatal sepsis
Inflammatory disease : Autoimmune chronic active
hepatitis Primary sclerosing
cholangitis

Billiary malformations Biliary atresia Arteriohepatic dysplasia (Alagille
syndrome) Intrahepatic biliary hypoplasia Choledochal cyst Congenital hepatic fibrosis Intrahepatic cystic biliary
dilatation (Caroli’s disease)

Vascular lesions : Budd–Chiari syndrome Congestive heart failure Congestive pericarditis Veno-occlusive liver
disease Venacaval web
Toxic disorder : Toxins found in nature
(mushrooms) Organic solvents Hepatotoxic drugs

Nutritional Disorder: Hypervitaminosis A Total parenteral
alimentation Malnutrition
Idiopathic : Cerebrohepatorenal
syndrome (Zellweger’s) Familial intrahepatic
cholestasis (Byler’s disease)
Neonatal hepatitis

Pathophysiology
Cell injury leads to cell death (necrosis), which is followed by scar formation (fibrosis) and, in some cases, nodule formation (regeneration).
Cirrhosis is the result of these three processes :
Cell injury ( extracellular matrix) Fibrosis Regeneration

Extracellular Matrix
The first step in the process that leads to cirrhosis is direct injury to the hepatocyte.This injury may occur as a result of almost any insult, including viral invasion, ischemia, and toxin exposure.
After injury the parenchymal cells regenerate and replace the necrotic cell . This is associated with inflammation and deposition of additional extracellular matrix (ECM), including collagen.

The cells responsible for intrahepatic fibrosis have not all been identified. The hepatic stellate cell is of high importance. Other cells like endothelial cell, hepatic stellate cell, kupffer cells are responsible for fibrosis.
If stimulated by inflammatory cells or by various cytokines, hepatocytes and their supportive cells secrete an altered ECM. The ECM is vital to the survival and proper function of each cell; the ECM provides a stable environment within tissue compartments.

In cirrhosis, the ECM is altered qualitatively and quantitatively.
In the normal liver, connective tissue proteins are seen along the basement membranes surrounding lymphatic and blood vessels, and around bile ducts. There is also scanty collagen in the perisinusoidal space. The hepatocytes and greater part of the portal triads are virtually free of connective tissue.
In cirrhosis, there are increased collagen types III and IV in the perisinusoidal space. The sinusoids are lined with a new basement membrane intimately associated with laminin and collagen type 3

The altered proteins (mostly synthesized by hepatocyte stellate cells) are laid down at the space of Disse, and initially there is no effect on the function of the hepatocyte.
As the collagen network becomes thicker, it becomes a barrier to the intimate interface between blood and hepatocytes and interferes with exchange of substances across the hepatocyte membranes.
Laminin deposition increases along sinusoids and is related to the formation of a sinusoidal basement membrane . This is so called capillarization of sinusoids.

The reduction in the amount of viable, well-vascularized hepatic tissue leads to compensatory growth, or nodule formation. As hepatic nodules form, they increasingly impede blood flow to the lobules by direct compression of the hepatic arterial and venous blood flow.

HEPATIC STELLATECELL
Normal Hepatic SInusoid
Space of Disse
FENESTRATION
SINUICIDAL ENDOTHELIALCELL
Hepatocytes

· Activation of stellate cells· Collagen deposition in space of Disse· Constriction of sinusoids· Defenestration of sinusoids
Alterations in Microvasculature in Cirrhosis

FIBROGENESIS
Although initially considered to be permanent, there is evidence suggesting that fibrosis is reversible, at least in some cases .
Some reversal of liver fibrosis has been seen in some liver diseases, such as chronic hepatitis C and nonalcoholic steatohepatitis (NASH) .

Hepatic stellate cells (HSCs) are responsible for the excess production of ECM components, thus activation of HSC appears to be a key step toward the development of hepatic fibrosis. Inhibition of HSC activation has been of interest in reversing or preventing the development of hepatic fibrosis.
Such treatments include the inhibition of transforming growth factor-β (TGF-β) , platelet-derived growth factor , and herbal extracts such as silymarin.
Antioxidants such as vitamin E and silymarin protect hepatocytes from necrotic injury, inhibit hepatocyte stellate cell activation, and may result in decreased rates of fibrogenesis .

Attempts are being made to treat liver fibrosis. The removal of the causative agent when possible is the first step in treating liver fibrosis and in some cases, may result in some reversal of the fibrotic process.
In general, most treatments have not been widely applied or have not been tested in adults or children and thus are not used in children with progressive liver disease.
Because inflammation is associated with the fibrotic process, anti-inflammatory medications such as corticosteroids have been used in patients with chronic liver disease.
These drugs are especially effective in treating patients with autoimmune hepatitis .

REGENERATION
In response to insult by viral invasion, cirrhosis, ischemia, trauma, or partial hepatectomy, hepatocyte proliferation increases to replace lost cells. Hepatic regeneration is a com- plex process, highly regulated at the cellular level in an autocrine or paracrine manner by cytokines, some of which themselves are regulated by prostaglandins or by other cytokines.

Many hormones are known to influence the process of hepatic regeneration, including insulin, glucagon, growth hormone, adrenocorticotropic hormone (ACTH), vasopressin, cortisol, thyroxin, and estrogen.
All of these can promote hepatic proliferation, but none appears to be the initiator of the process. The recent isolation and identification of specific cytokine hepatic growth factors has shed light on the initiation and control of hepatocyte proliferation as well as fibrogenesis.

Salivary glands, Brunner’s glands
Initiator of hepatic regenerative response. Stimulates hepatocyte DNA synthesis.
Hepatocyte growth factor (HGF)
Unknown Stimulates hepatocyte DNA synthesis
Fibroblast growth factor (FGF)
Endothelial cells Stimulates hepatocyte regeneration at low levels. High levels regulate epidermal growth factor EGF-induced proliferation.
Cytokines Associated with Hepatic Regeneration
CYTOKINE SOURCE ACTIONEpidermal growth factor (EGF)

Interleukin-6 (IL-6) Kupffer cells Promotes hepatocyte proliferation
Transforming growth factor-α (TGF-α)
Macrophage, hepatocyte
Promotes hepatic regeneration; also binds to EGF receptor

Mechanism of fibrosis and cirrhosis of the
liver

CLINICAL FEATURES The clinical presentation of cirrhosis depends on the cause
of the primary liver disease as well as on the pace of progression of hepatocellular failure and fibrosis. Many children and adolescents present with findings incidentally during routine physical examinations. In such patients, the cirrhosis is referred to as latent or compensated.
In others, chronic liver disease may be sudden and dramatic, such as with the onset of hematemesis, encephalopathy, ascites, or infection. If the signs and symptoms of cirrhosis are apparent and progressive, the term decompensated or active cirrhosis applies

The common mode of presentation include : Failure to thrive Anorexia , nausea , vomiting Prolonged or recurrent jaundice Pruritis Pain abdomen ( Colicky type) Abdominal distension Oedema Features of portal hypertension
( hementemesis , malena, epitaxis.) Fever is generally presented in
decompensated liver disease.

Other points in history
• Fever, prodrome (anorexia ,vomiting ,nausea) –which disappears with onset of jaundice – acute viral hepatitis
• jaundice • Young individual with extrapyramidal symptoms,
neuropsychiatric manifestation, anemia• Colicky abdomen pain, jaundice, fever - gall
stones • Drug history ( acetoaminophen toxicity)

Cholestatic disease
• fatigue, malaise• anorexia, nausea• Biliary colic• Deep jaundice• +++ pruritus• +++ abdominal pain and pancreatitis• +++ gray or clay-colored stools


CIRRHOSIS
• Fatigue• Muscle wasting• Hematemesis • Ascites • Easy bruising• Edema

General Poor growth, malnutrition, fever, muscle wasting
Skin and extremities Jaundice, flushing or pallor, palmar erythema, spider angiomata, digital clubbing
Abdomen Distention, caput medusa, ascites, larger tender liver or shrunken liver, large spleen, rectal varices
Central nervous system Asterixis, positive Babinski’s reflex, prolonged relaxation phase of deep tendon refluxes, mental status changes
Physical finding related to chronic Liver disease

Miscellaneous Gynecomastia, testicular atrophy, feminization, delayed puberty



Extra hepatic manifestation ofChronic liver diesese
Gastrointestinal tract : cirrhosis is associated with esophageal and
gastric varices, which form as a consequence of portal hypertension.
Hematemesis secondary to variceal bleeding .
Bleeding from hemorrhoids, which arise from portosystemic shunting through the inferior mesenteric venous collateral system, is much less common.

Chronic gastritis and peptic ulcer disease
Excess gastric acid may be secreted in response to direct gastric stimulation by histidine, the decarboxylated form of histamine. In healthy people, histidine is cleared from the circulation by the liver; with hepatic disease, serum histidine levels are elevated .
Gastroesophageal reflux is seen in cirrhosis and is caused by increased intra-abdominal pressure secondary to ascites and hepatosplenomegaly.
Diarrhea is a common manifestation of liver disease and may be a result of malabsorption, bile acid deficiency, or malnutrition.

pulmonary manifestation :
Development of arteriovenous shunts, which lead to cyanosis and dyspnea. Hypoxemia is seen in up to 30% of children with cirrhosis and can be severe enough to cause cyanosis .
Digital clubbing often accompanies longstanding intrapulmonary shunting
These shunts may result from increased vasodilation caused by vasoactive substances such as ferritin released from the diseased liver

Hematologic changes : cirrhosis include anemia and coagulopathy.
The cause of the anemia of cirrhosis may be multifactorial and may include blood loss due to GI bleed, hemolysis secondary to hypersplenism, iron and folic acid deficiency secondary to malabsorption , anorexia, and dilution of red blood cell volume as a result of sodium and water retention .
The coagulopathy of cirrhosis also is multifactorial . A decrease in the synthesis of liver derived clotting proteins, including prothrombin and factors VII and IX, and increased consumption of clotting factors through increased fibrinolysis and disseminated intravascular coagulation (DIC) occur commonly in late, decompensated cirrhosis.

Cardiovascular manifestations:
cirrhosis include a high cardiac output state related to changes in systemic vascular resistance, pulmonary vascular resistance, and hepatic blood flow (portal hypertension).
The sustained increase in cardiac output results in the flushed appearance of patients with cirrhosis. Systemic hypertension is not common in cirrhosis

Skin manifestations :
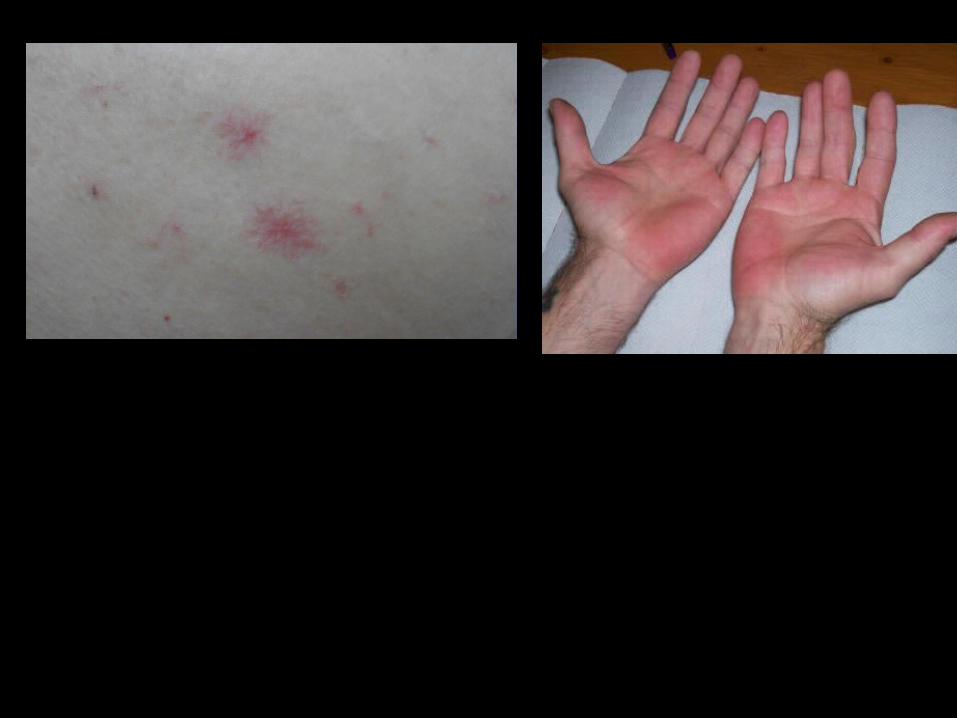
Spider angiomata and palmar erythema.
Spider angiomata are easily recognizable as small, raised, dark lesions with radially distributed convoluted vascular branches. They arise because of vasoactive substances such as estradiol in the circulation of patients with cirrhosis.
The presence of more than five in the body region drained by the superior vena cava is abnormal and is suggestive of cirrhosis.

Palmar erythema, a well-recognized sign of cirrhosis, is not specific to cirrhosis but is seen in other conditions associated with increased cardiac output or altered sex hormone metabolism .
Caput medusae may also be present. This refers to prominent abdominal wall veins seen in patients with portal venous hypertension.
Nail changes characterized by horizontal white bands (Muehrcke’s nails) may be seen in cirrhosis or in other conditions resulting in hypoalbuminemia.
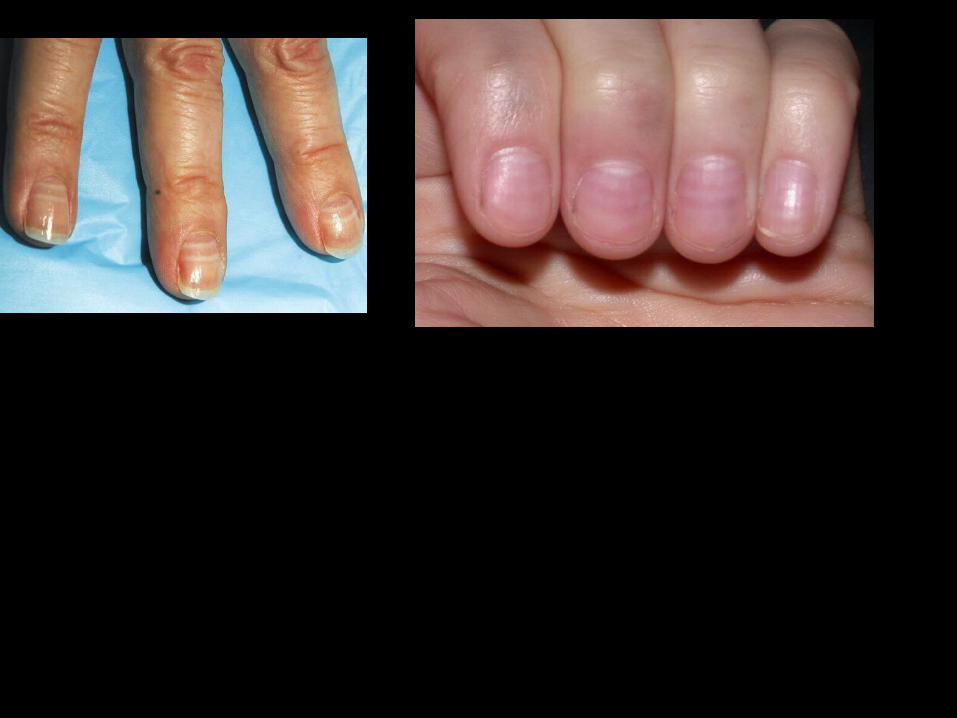

Endocrine manifestations :
Failure of the liver to conjugate or metabolize hormones include diabetes mellitus, which may present as subtle hyperinsulinemia without overt signs; syndrome of inappropriate secretion of antidiuretic hormone, presenting as hyponatremia.
Gynecomastia may be caused by increased production of androstenedione and the increased conversion of estrone to estradiol .
In the mature adolescent, decreased libido, delayed
puberty decreased facial hair, and impotence are caused by reduced testosterone synthesis in the liver .

Neurologic manifestations Changes in consciousness include hypersomnia,
reversal of sleep pattern, apathy, slowed speech, decreased spontaneous movement, and eventually coma.
Personality changes commonly seen in chronic liver disease include irritability, inability to cooperate,, and their true cause may not be understood until frank encephalopathy is present.
Intellectual deterioration with slight or gross confusion may be present. Tests of constructional apraxia such as writing difficulty or the Reitan trail-making test may be difficult to administer if the child is at too early a developmental stage.

Immune function
cirrhosis result in increased susceptibility to bacterial and mycobacterial infections, resulting in an increased rate of pneumonia and spontaneous bacterial peritonitis (SBP).

APPROACH TO CHRONIC LIVERDISEASE

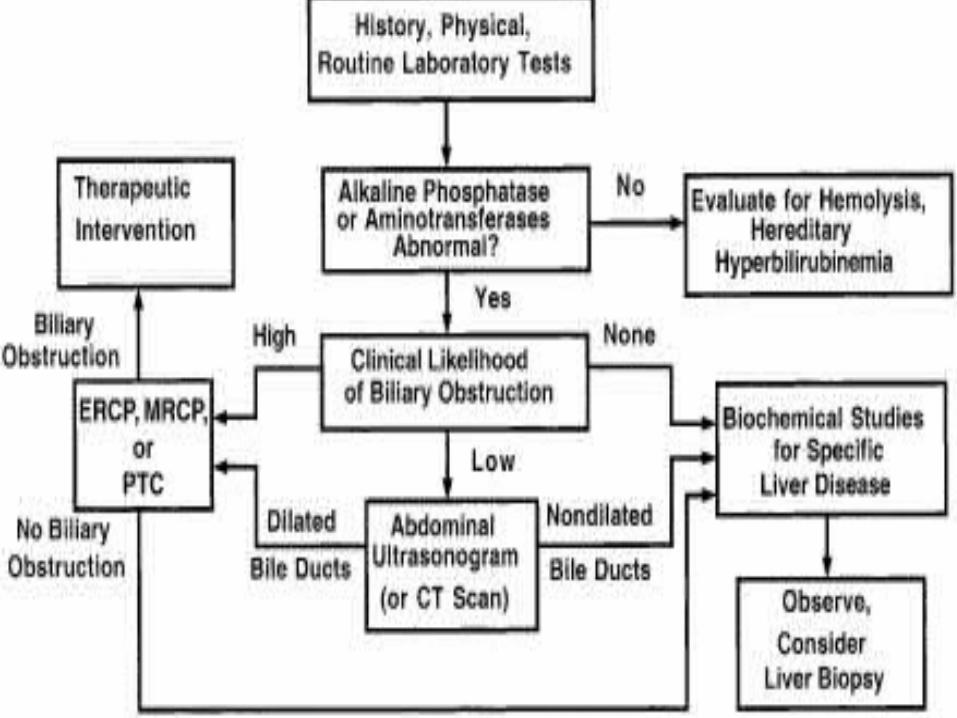
Evaluation of a patient with liver dysfunction and suspected cirrhosis should focus on determining both the cause and the stage of liver disease.
Serologic testing for infectious diseases should include screens for hepatitis B and C.
In appropriate clinical situations (fever in the setting of previous biliary tree surgery), bacterial cultures of blood and possibly liver tissue should be obtained.

Tests for metabolic liver disease should include quantitation of the serum α1-antitrypsin level with determination of levels of protease inhibitor (“π-type”), fasting blood sugar (glycogen storage disease), urinary reducing substances (galactosemia), serum amino acids with urinary organic acids (tyrosinemia), serum iron, and iron binding capacity and ferritin (hemochromatosis), as well as the sweat chloride test (for cystic fibrosis).
The initial evaluation for Wilson’s disease should include serum copper, serum ceruloplasmin, a 24-hour urine collection for copper, and a slit-lamp ophthalmologic exam.

An abdominal ultrasound examination aids in the evaluation of gallstones, choledochal cyst, and Caroli’s disease (cystic dilation of the intrahepatic biliary tree).
The anatomy and blood flow of the hepatic arterial and venous system also should be evaluated. In infants, in whom the consideration of extrahepatic biliary atresia is paramount, a biliary scintiscan should be done.
In patients with suspected extrahepatic biliary tree obstruction, ERCP may be considered. The timing of liver biopsy in the investigtion of suspected cirrhosis in children remains a matter of clinical judgment.
A biopsy may be critical to confirm the presence of cirrhosis suspected on clinical grounds, or if the investigations outlined here fail to reveal the caus of the chronic liver disease

Biochemical tests in liver disease
LIVER FUNCTION TEST : AST , ALT , ALP TOTAL BILIRUBIN / DIRECT BILIRUBIN ALBUMIN TOTAL PROTEIN PT /APTT/ INR COMPLETE BLOOD COUNT / PLATELETS GLUCOSE VIRAL SEROLOGY IRON PROFILE CERULOPLASMIN LEVEL ANA – ASMA-AMA

Hepatocellular • AST/ALT >> ALP• Unconjugated bil >>
conjugated• USG – BILE DUCTS NORMAL
Cholestatic • ALP>>AST/ALT• Conjugated bil>>
unconjugated• ↑GGT, 5’nucleotidase• USG- Intrahepatic biliary
duct dilation

Non hepatic cause of elevated transaminases
• Muscle disease• Thyroid diseases• Bone disease - ALP

Diagnostic Tests in Chronic Liver Disease and Cirrhosis
Disorder Diagnostic Test
HBV HBsAg, E antigen/antibody, HBV DNA
HCV HCV antibody, HCV RNA
CMV CMV serology, urine for CMV antigen
EBV EBV serology, heterophile
Bacterial cholangitis Blood and liver tissue culture
Autoimmune chronic active hepatitis Sedimentation rate, ANA, anti–smooth muscle antibody, antimitochondrial antibody, anti–liver-kidney-microsomal antibody
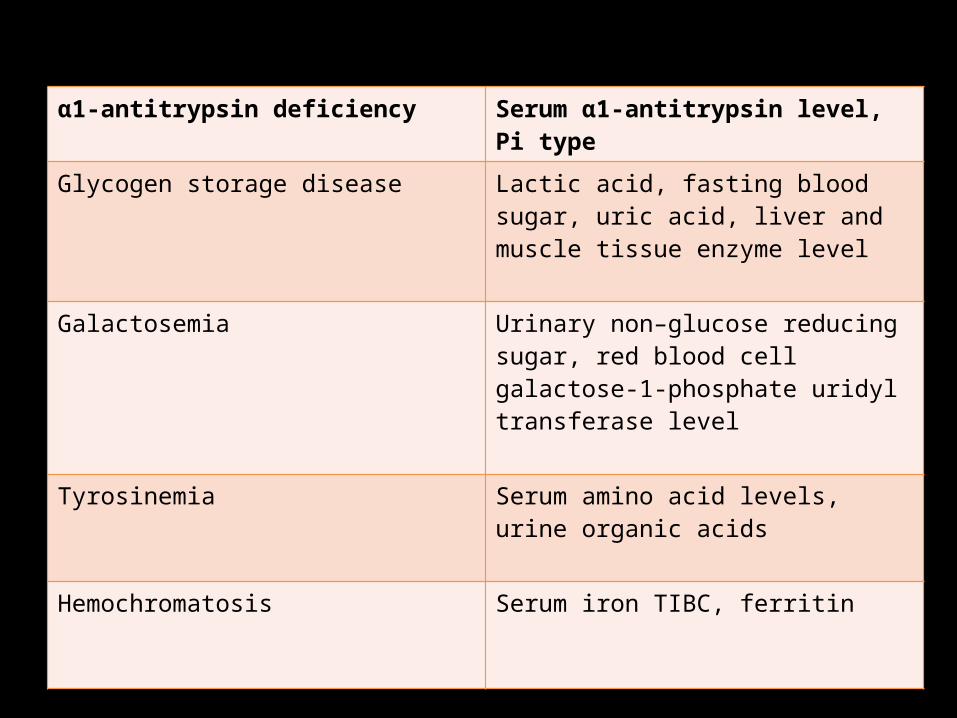
α1-antitrypsin deficiency Serum α1-antitrypsin level, Pi type
Glycogen storage disease Lactic acid, fasting blood sugar, uric acid, liver and muscle tissue enzyme level
Galactosemia Urinary non–glucose reducing sugar, red blood cell galactose-1-phosphate uridyl transferase level
Tyrosinemia Serum amino acid levels, urine organic acids
Hemochromatosis Serum iron TIBC, ferritin
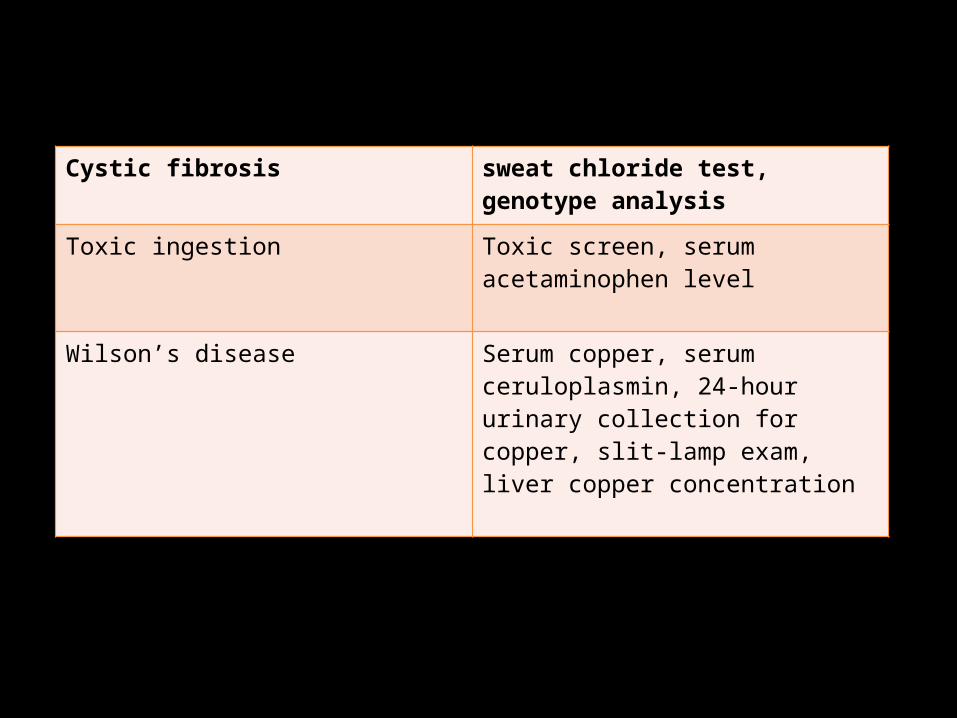
Cystic fibrosis sweat chloride test, genotype analysis
Toxic ingestion Toxic screen, serum acetaminophen level
Wilson’s disease Serum copper, serum ceruloplasmin, 24-hour urinary collection for copper, slit-lamp exam, liver copper concentration


Liver Function Tests: Normal Values & Changes
Tests Normal Values Hepatocellular Jaundice
Uncomplicated Obstructive
Jaundice
Bilirubin Direct Indirect
0.1-0.3 mg/dL0.2-0.7 mg/dL
IncreasedIncreased
IncreasedIncreased
Urine bilirubin None Increased Increased
Serum albumin/ total protein
Alb, 3.5-5.5 g/dLTot, 6.5-8.4 g/dL
Albumin decreased
Unchanged
Alk phos 30-115 IU/L Increased (+) Increased (++++)
Prothrombin time INR of 1.0-1.4; 10% inc. after vit K in 24 hrs
No response to parenteral vit. K; prolonged
Prolonged but responds to parenteral vit. K
ALT, AST ALT: 5-35 IU/LAST: 5-40 IU/L
Inc. in hepato- cellular damage, viral hepatitis
Minimally increased

Differential Diagnosis of Hereditary Jaundice with Normal Liver Chemistries & No Signs or Symptoms of Liver Disease
Unconjugated HyperbilirubinemiaCrigler-Najjar Syndrome
Gilbert’s Type I Type II
Incidence
Inheritance mode
Serum bilirubin usual total (mg/dL)
Defect
Age at onset of jaundice
<7% of pop’n Very rare Uncommon AD AR AR
<3; <6 >20 <20
Mostly B1; inc. All indirect All indirect with fastingHepatic UDP-glucuronyl transferase activityDecreased Absent Marked dec.
Adolescence early neonatal childhood

Differential Diagnosis of Hereditary Jaundice with Normal Liver Chemistries & No Signs or Symptoms of Liver Disease
Unconjugated HyperbilirubinemiaCrigler-Najjar Syndrome
Gilbert’s Type I Type II
Usual clinical features
Liver biopsy
Treatment
Appear in early Jaundice, Asymptomatic adulthood; kernicterus in jaundice, often 1st re- infants, kernicterus cognized w/ 1st 3day of life rare fasting
Normal Normal Normal
Not needed Liver transplant Phenobarbital

Differential Diagnosis of Hereditary Jaundice with Normal Liver Chemistries & No Signs or Symptoms of Liver Disease
Conjugated HyperbilirubinemiaDubin-Johnson Rotor’s Syndrome
IncidenceInheritance modeSerum bilirubin usual total (mg/dL)Defect
Urine total coproporphyrinAge at onset of jaundice
Usual clinical features
Oral cholecystogramLiver biopsyTreatment
UncommonAR2-7; < 25Direct ~ 60%Impaired biliary excretionNormal Childhood, adolescenceAsymptomatic jaundice in young adultsGB not visualizedCharac. pigmentNot needed
Rare AR2-7; < 20Direct ~ 60%Impaired biliary excretionIncreased Adolescence, early adulthoodAsymptomatic jaundice
NormalNo pigmentNone

WILSON DISEASE
• Wilson s disease (WD) is an autosomic recessive Ngenetic disorder affecting gene ATP7B in chromosome 13q . It codes for trans membrane copper transporter in hepatocytes and for excretion into the bile canaliculus as well as for joining copper to ceruloplasmin.
• As a result, there is an abnormal accumulation of copper in liver, brain, kidney, and other organs, mitochondrial damage, and low levels of bile copper and coeruloplasmin.

Prevalence in Europe is 1/30,000, with 1/100 heterozygous. It may present as asymptomatic hypertransaminasaemia or as chronic hepatitis, cirrhosis, or acute liver failure in childhood.
In older children and adults, extrapyramidal neurological symptoms (dysarthria, dystonia, ataxia, tremor, dysphagia), psychiatric manifestations, Kayser–Fleischer ring (due to copper deposits in the Descemet corneal membrane, very specific but not pathognomonic of WD), and renal tubulopathies (with aminoaciduria and phosphaturia) are more frequent.

t is typical to find low plasma ceruloplasmin, although in 5–10% of patients it may be normal (>20 mg/dl), and decreased serum copper levels.
A raised urine copper after penicillamine (higher than 100 μg/day) and an elevated liver copper concentration (higher than 250 μg/g of dry liver tissue) are helpful in diagnosis. Liver biopsy shows the stage of the disease. Genetic tests (>150 mutations) may help.


Alpha-1-antitrypsin deficiency In this autosomal recessive disease there is a chromosome
14 mutation that leads to the production of abnormal and hepatotoxic α1-AT that is retained in the endoplasmic reticulum . Frequency is 1:2000 newborns.
α1-AT glycoprotein may present as 100 variants, with codominant inheritance, which are classified according to the protease inhibitor (Pi) phenotype system based on their electrophoretic moiety: Pi MM variant (with normal serum concentration and normal activity), Pi null-null variant (absence of α1-AT associated with lung disease), defective variants (such as Pi Z and Pi S, with low serum concentration and lung and liver disease).

MM phenotype is found in healthy individuals; ZZ phenotype causes the most severe deficiency and it represents 95% of patients. SZ phenotype may cause liver disease (neonatal cholestasis, mild dysfunction, chronic hepatitis, liver failure, cirrhosis, hepatocellular carcinoma), while MS/MZ phenotypes do not produce liver disease in children but do in adults.
Diagnosis is based on a decrease in the α1 band (electrophoretic gel) , a decrease in α1-AT in blood and Pi phenotype. On the liver biopsy, an abnormal accumulation of α1-AT in liver is present.

Nonalcoholic fatty liver disease
• Nonalcoholic fatty liver disease (NAFLD) includes simple fatty liver (steatosis), nonalcoholic steatohepatitis (NASH) and cirrhosis . Most patients with NAFLD present with obesity, mainly central adiposity, as part of the metabolic syndrome21

The pathology of the NAFLD in children includes:
Type 1, characterized by steatosis, hepatocyte balloonization, and perisinusoidal fibrosis (similar to NAFLD in adults and more frequent in white children in both sexes), and
Type 2 (infantile), characterized by steatosis, portal inflammation, and portal fibrosis (more common in male and children of Asian, Native American, and Hispanic ethnicity).

• Clinical experience in children with NAFLD is limited. NAFLD in childhood and adolescence used to occur in the male obese patient, with AST>ALT , hypertriglyceridaemia , acanthosis nigricans .
• Although most patients remain asymptomatic, other individuals may present with a variety of symptoms: hypertension, dyslipidaemia, insulin resistance, type 2 diabetes, and central obesity.

The gold standard test to distinguish between simple steatosis and NASH is liver biopsy. It confirms diagnosis, assesses steatosis
severity, and serves as a prognostic marker. Patients with simple esteatosis have a benign
course without histological progression, but patients with NAFLD may progress to cirrhosis or hepatocarcinoma .

PROGNOSISWeighting Factor Variable
+15 If cholesterol <100 mg/dL
+15 If positive history of ascites
+13
+11
If indirect bilirubin >6 mg/dL
If indirect bilirubin is 3–6 mg/dL
+10 If PTT is prolonged >29 seconds

Total score of 0–28 places patient in low-risk group (<25% risk of death within 6 months);
score of 28–39 is associated with moderate- risk (25–75% risk of death within 6 months);
score greater than 39 is associated with high risk (>75% of death within 6 months).

































